
Pumped up: reflections on PfATP6 as the target for artemisinins
8
Pumped up: reflections on PfATP6 as the target for artemisinins Sanjeev Krishna, Serena Pulcini, Catherine M. Moore, Beatrix Huei-Yi Teo, and Henry M. Staines Centre for Infection and Immunity, Division of Clinical Sciences, St George’s, University of London, Cranmer Terrace, London SW17 0RE, UK Sarco/endoplasmic reticulum Ca 2+ -ATPases (SERCAs) are increasingly being studied for therapeutic interven- tions in the fields of cancer, heart disease, and infection. Our suggestion a decade ago that artemisinins (the most important antimalarial class) act by inhibiting parasite SERCAs (PfATP6 and orthologues) expressed in Xenopus oocytes stimulated new directions for research away from conventional site-of-action studies of the food vacuole of the parasite. There is, however, still no con- sensus on how artemisinins act. We have continued to explore the hypothesis that PfATP6 is a key target by confirming that artemisinins inhibit Plasmodium falci- parum PfATP6 when it is expressed in yeast and that it is essential for survival of pathogenic asexual-stage para- sites. These advances are discussed with their implica- tions for our understanding of how parasites regulate calcium in different stages of asexual development and for the global challenge posed by artemisinin resistance. Introduction Extracts of Artemisia annua have been used for several hundred years to treat fevers. Artemisinin (see Glossary), the principal antimalarial compound, was identified in 1972 by Chinese scientists [1], with elucidation of its chemical structure in 1977 [2]. This structure (Figure 1A), with its very unusual arrangement of oxygen atoms in a trioxane configuration located within a sesquiterpene, has stimulated chemists to make fully synthetic compounds (trioxolanes) that retain antimalarial activity (e.g., OZ439 [3]; Figure 1A). Success with this approach has not depended on a detailed knowledge of how artemisinins work as antimalarials, but on whole-cell screening approaches linked to experimentation with chemistry. Artemisinins are now widely used in hundreds of millions of treatment courses for malaria and are extremely versa- tile compounds. There are derivatives (Figure 1A) that vary in their aqueous solubility and can be given orally, intramuscularly [4], intravenously, and intrarectally [5,6]. Artemisinins are used in combination therapies to treat uncomplicated and severe malaria [7]. They are also useful in treating other parasitic infections (e.g., infection with Schistosoma spp.) and opportunistic cytomegalovirus infections and are under active investigation as anti-cancer agents [8]. Surprisingly, artemisinin dosage regimens are still undergoing optimisation for the treatment of malaria [7]. Transport proteins, including cation ATPases, have been very successfully exploited as drug targets, although they remain largely untapped as targets for novel anti- malarials [9] or as antibacterial agents (for P type ATPases) despite recent evidence that they may be selec- tively targeted [10]. In 1993, the first reports of cation ATPases encoded by Plasmodium falciparum were pub- lished and their potential as targets for new therapies was emphasised [11,12]. After we developed new ways to study parasite transporters via heterologous expression in Xeno- pus oocytes [13], we suggested a decade ago that PfATP6, a sarco/endoplasmic reticulum Ca 2+ -ATPase (SERCA) ho- mologue, was a key target for the antimalarial actions of Opinion Glossary Artemisinins: several derivatives are available, including artemether, which is fat-soluble; artesunate, which is a semi-synthetic water-soluble derivative; and dihydroartemisinin, which is the active metabolite of artesunate and arte- mether and is used as an antimalarial in its own right. Intraerythrocytic: within a red blood cell (erythrocyte). Merozoite: parasite stage that is released from host liver cells and invades red blood cells, or is released from red blood cells and invades fresh red blood cells. Microneme: organelle present in all apicomplexa parasites such as Plasmo- dium and Toxoplasma and implicated in invasion mechanisms of these parasites. Parasitophorous vacuole: vacuole formed from membrane layers surrounding an intracellular parasite, for example, after it has invaded a red cell. Plasmodium falciparum: protozoan parasite of the genus Plasmodium and the most ubiquitous and dangerous cause of malaria. Five species naturally infect humans. Ring stage: metabolically relatively quiescent intracellular stage with char- acteristic signet ring-shape that forms very soon after invasion of a red cell. Schizont: trophozoite that has undergone rounds of nuclear division as a prelude to the production of merozoites. SERCA: integral membrane calcium ATPase (from the P type ATPase family) that sequesters calcium within the sarcoplasmic–endoplasmic reticulum using energy supplied by ATP. Toxoplasma gondii: protozoan parasite that is related phylogenetically to malaria parasites (Plasmodium spp.) because it belongs to the apicomplexans. It is more tractable for some cellular and molecular studies. Trophozoite: metabolically active intracellular stage that develops in 18–24 h after invasion, and is much larger and pigmented compared with ring stages. Xenopus laevis: African clawed toad; used to obtain oocytes for heterologous expression studies including those of malarial transport proteins. 0165-6147/$ – see front matter ß 2013 Elsevier Ltd. All rights reserved. http://dx.doi.org/10.1016/j.tips.2013.10.007 Corresponding author: Krishna, S. ([email protected]). Keywords: artemisinin resistance; SERCA; calcium homeostasis; antimalarial; yeast expression; endoperoxides. TIPS-1097; No. of Pages 8 Trends in Pharmacological Sciences xx (2013) 1–8 1
-
Upload
independent -
Category
Documents
-
view
5 -
download
0
Transcript of Pumped up: reflections on PfATP6 as the target for artemisinins

TIPS-1097; No. of Pages 8
Pumped up: reflections on PfATP6 asthe target for artemisininsSanjeev Krishna, Serena Pulcini, Catherine M. Moore, Beatrix Huei-Yi Teo, andHenry M. Staines
Centre for Infection and Immunity, Division of Clinical Sciences, St George’s, University of London, Cranmer Terrace,
London SW17 0RE, UK
Opinion
Glossary
Artemisinins: several derivatives are available, including artemether, which is
fat-soluble; artesunate, which is a semi-synthetic water-soluble derivative; and
dihydroartemisinin, which is the active metabolite of artesunate and arte-
mether and is used as an antimalarial in its own right.
Intraerythrocytic: within a red blood cell (erythrocyte).
Merozoite: parasite stage that is released from host liver cells and invades red
blood cells, or is released from red blood cells and invades fresh red blood
cells.
Microneme: organelle present in all apicomplexa parasites such as Plasmo-
dium and Toxoplasma and implicated in invasion mechanisms of these
parasites.
Parasitophorous vacuole: vacuole formed from membrane layers surrounding
an intracellular parasite, for example, after it has invaded a red cell.
Plasmodium falciparum: protozoan parasite of the genus Plasmodium and the
most ubiquitous and dangerous cause of malaria. Five species naturally infect
humans.
Ring stage: metabolically relatively quiescent intracellular stage with char-
acteristic signet ring-shape that forms very soon after invasion of a red cell.
Schizont: trophozoite that has undergone rounds of nuclear division as a
Sarco/endoplasmic reticulum Ca2+-ATPases (SERCAs)are increasingly being studied for therapeutic interven-tions in the fields of cancer, heart disease, and infection.Our suggestion a decade ago that artemisinins (the mostimportant antimalarial class) act by inhibiting parasiteSERCAs (PfATP6 and orthologues) expressed in Xenopusoocytes stimulated new directions for research awayfrom conventional site-of-action studies of the foodvacuole of the parasite. There is, however, still no con-sensus on how artemisinins act. We have continued toexplore the hypothesis that PfATP6 is a key target byconfirming that artemisinins inhibit Plasmodium falci-parum PfATP6 when it is expressed in yeast and that it isessential for survival of pathogenic asexual-stage para-sites. These advances are discussed with their implica-tions for our understanding of how parasites regulatecalcium in different stages of asexual development andfor the global challenge posed by artemisinin resistance.
IntroductionExtracts of Artemisia annua have been used for severalhundred years to treat fevers. Artemisinin (see Glossary),the principal antimalarial compound, was identifiedin 1972 by Chinese scientists [1], with elucidation of itschemical structure in 1977 [2]. This structure (Figure 1A),with its very unusual arrangement of oxygen atoms in atrioxane configuration located within a sesquiterpene, hasstimulated chemists to make fully synthetic compounds(trioxolanes) that retain antimalarial activity (e.g., OZ439[3]; Figure 1A). Success with this approach has notdepended on a detailed knowledge of how artemisininswork as antimalarials, but on whole-cell screeningapproaches linked to experimentation with chemistry.Artemisinins are now widely used in hundreds of millionsof treatment courses for malaria and are extremely versa-tile compounds. There are derivatives (Figure 1A) thatvary in their aqueous solubility and can be given orally,intramuscularly [4], intravenously, and intrarectally [5,6].
0165-6147/$ – see front matter
� 2013 Elsevier Ltd. All rights reserved. http://dx.doi.org/10.1016/j.tips.2013.10.007
Corresponding author: Krishna, S. ([email protected]).Keywords: artemisinin resistance; SERCA; calcium homeostasis; antimalarial; yeastexpression; endoperoxides.
Artemisinins are used in combination therapies to treatuncomplicated and severe malaria [7]. They are also usefulin treating other parasitic infections (e.g., infection withSchistosoma spp.) and opportunistic cytomegalovirusinfections and are under active investigation as anti-canceragents [8]. Surprisingly, artemisinin dosage regimensare still undergoing optimisation for the treatment ofmalaria [7].
Transport proteins, including cation ATPases, havebeen very successfully exploited as drug targets, althoughthey remain largely untapped as targets for novel anti-malarials [9] or as antibacterial agents (for P typeATPases) despite recent evidence that they may be selec-tively targeted [10]. In 1993, the first reports of cationATPases encoded by Plasmodium falciparum were pub-lished and their potential as targets for new therapies wasemphasised [11,12]. After we developed new ways to studyparasite transporters via heterologous expression in Xeno-pus oocytes [13], we suggested a decade ago that PfATP6, asarco/endoplasmic reticulum Ca2+-ATPase (SERCA) ho-mologue, was a key target for the antimalarial actions of
prelude to the production of merozoites.
SERCA: integral membrane calcium ATPase (from the P type ATPase family)
that sequesters calcium within the sarcoplasmic–endoplasmic reticulum using
energy supplied by ATP.
Toxoplasma gondii: protozoan parasite that is related phylogenetically to
malaria parasites (Plasmodium spp.) because it belongs to the apicomplexans.
It is more tractable for some cellular and molecular studies.
Trophozoite: metabolically active intracellular stage that develops in 18–24 h
after invasion, and is much larger and pigmented compared with ring stages.
Xenopus laevis: African clawed toad; used to obtain oocytes for heterologous
expression studies including those of malarial transport proteins.
Trends in Pharmacological Sciences xx (2013) 1–8 1

O
O O
O
O
O OO
O
O
O O
O
O O
OO
O
OO
OH
O
O
O
OOH
OHO
O
O
CH3
CH3
CH3
CH3
H3C
H3C
H3CH3C
O
Artemisone
Artemisinin
Cyclopiazonic acid
Thaperoxide 23
OZ439
Thapsigargin
Artemetherbinding site
ADP
S769
K568-C677
Artesunate
Dihydroartemisinin Artemether
(A) (B)
OO O
OO
O
O
OO
O
O
O
O
OO
OO
O
OH H
H
H
H
H H
HH
H
H H H
H
H
H
OH
OHPMBO
OH
H H
N
NH
N
N
S
TRENDS in Pharmacological Sciences
Figure 1. Structures of artemisinins, other inhibitors, and PfATP6. (A) Structures of artemisinin, semi-synthetic derivatives, the fully synthetic ozonide OZ439, cyclopiazonic
acid, thaperoxide, and thapsigargin. (B) A 3D model of PfATP6 showing residues for which field mutations have been associated with decreased artemisinin sensitivity.
S769 is highlighted in red and A623 is found within a disordered region flanked by residues K568 and C677, highlighted in blue. The inset presents a magnification of the N
domain rotated by 1808 to show the position of the disordered insertion, which is not visible in the model on the left. The predicted artemether-binding region is in yellow.
The b-strand connecting the ATP-binding site to S769 is in green. Adapted from Pillai et al. [74].
Opinion Trends in Pharmacological Sciences xxx xxxx, Vol. xxx, No. x
TIPS-1097; No. of Pages 8
artemisinins [14]. SERCAs are critical for calcium homeo-stasis in eukaryotic cells, and their dysregulation hasimportant consequences for cell signalling and function.These properties are already being exploited therapeuti-cally in other contexts. For example, clinical trials of genetherapy aimed at increasing SERCA activity in cardiacmuscles of those in heart failure are under way [15].Thapsigargin (Figure 1A), a specific inhibitor of SERCAs,is also being studied as a treatment for prostate cancer viadelivery as a prodrug coupled to a peptide that is activatedby prostate-specific enzymes [16].
In this opinion article, we examine recent studies onPfATP6 without discussing in detail competing hypotheses(that have recently been reviewed [17,18]) on how artemi-sinins may act. This may be useful to those interested inthe mechanisms of action of antimalarials, including thosewho may not recognise the possibility that PfATP6 is atarget for artemisinins [19]. Host-cell plasma membranecalcium ATPases may also be important in the pathophys-iology of disease [20].
PfATP6 and stage specificity of artemisinin actionIt is reasonable to suppose that if artemisinins targetPfATP6 in vivo, then the times at which artemisininsare most active should reflect periods when there are
2
important regulatory changes in calcium homeostasis dur-ing the parasite life cycle (Figure 2). This would includestages other than those that occur in erythrocytes, as hasbeen demonstrated in whole life-cycle studies [21]. Othercalcium homeostatic mechanisms may also be able tomodulate artemisinin sensitivity, although it is notewor-thy and perhaps surprising that alternative homeostaticmechanisms remain unclear, particularly given recentfindings that two other reported calcium transportersmay have different physiological roles than originally en-visaged (PfATP4 transports Na+ in parasites) or may notbe essential for parasite blood stages (PfCAX) [22–24].
In the case of intraerythrocytic parasites, the stagespecificity of the action of artemisinins is highly complexand depends on many factors, including the artemisininderivative, the parasites studied, the drug concentration,the treatment duration, the growth assay method, andwhether growth is assessed over the first or subsequentcycles. The first assessment of stage specificity of artemi-sinin suggested that it kills parasites very rapidly, withoutmuch comment on stage specificity [25]. Others demon-strated particular stage specificities of action [26–28].Using artemisinin with a range of different growth assaysand parasite lines, ter Kuile et al. showed that late rings toearly trophozoite stages were relatively more susceptible

Trophozoite18-24 hSchizont
32-36 h
Hepa�c stage
Gametocytes
Ring> 6 h
Erythrocy�c stage Merozoites
1
2
TRENDS in Pharmacological Sciences
Figure 2. Life cycle of the Plasmodium falciparum parasite within its human host. Malarial parasites enter their host during the blood meal of an infected female Anopheles
mosquito and migrate to the liver, where they infect hepatocytes. Over a period of approximately 7 days, the parasites develop and multiply asexually, producing tens of
thousands of new invasive forms called merozoites. These enter the bloodstream, where they infect erythrocytes, entering into a cycle of asexual development. The
invasion process requires extracellular calcium (1, [35]). Following invasion, the parasite (known as the immature ring stage, of which the earliest occurs < 6 h after invasion
and are called tiny rings) remains relatively metabolically inactive. After 18–24 h, the parasite then starts to develop and form into the mature trophozoite stage. As with
invasion, the presence of extracellular calcium is critical during this process, particularly between 20 and 26 h post invasion (2, [35]). During this development phase, the
trophozoites become sticky due to expression of adherence proteins (represented by green shading) at the erythrocyte surface, which cause them to adhere to endothelial
cells in the microvasculature. Over the last 10 h of the intraerythrocytic stage, the parasite divides up to eight times, forming a schizont that contains up to 36 new daughter
merozoites. These are then released from the infected cell and reinvade further erythrocytes, continuing the cycle. Not all parasites remain in this cycle, with a small
proportion differentiating into male and female parasite forms called gametocytes. These remain dormant in the bloodstream until taken up by a feeding mosquito,
allowing transmission between hosts.
Opinion Trends in Pharmacological Sciences xxx xxxx, Vol. xxx, No. x
TIPS-1097; No. of Pages 8
than young rings and schizonts, consistent with the time atwhich parasites increase their metabolic activity mostrapidly [28]. This is broadly supported by a recent studyusing short drug pulses, in which trophozoites (along witha subpopulation of rings) were most sensitive to artemisi-nin and dihydroartemisinin (DHA; Figures 1A and 2) [26].In addition, artemisinin and artesunate have no effect onparasite invasion of erythrocytes, whereas artemisinin(but not artesunate, a semi-synthetic derivative;Figure 1A) is able to stop parasite release from infectederythrocytes [29]. A recent study correlated a relativedecrease in sensitivity of early ring stages of the parasiteto a fixed dose of DHA [30]. Results for 3-h exposure forparasites from patients with decreased parasite clearancetimes suggested that they were less susceptible to thispulsed exposure. However, no mention was made ofpfmdr1 copy number (see below) in the parasites exam-ined, and only one dose (700 nM) and one derivative (DHA)were tested. Some 10% of the parasites demonstrated invitro resistance in this assay, but this proportion of para-sites would be unlikely to influence the prolongation ofparasite clearance times observed in vivo [30]. In any case,the prolonged parasite clearance phenotype cannot be usedto define artemisinin resistance [31], so it cannot be as-sumed that studies that identify particular in vitro phe-notypes or genotypes linked to this phenomenon explainartemisinin resistance.
Wilson et al. argued that the inability of artemisinins toinhibit merozoite invasion is at odds with their targeting ofPfATP6 [29]. This hypothesis is based on the observationthat artemisinin causes microneme protein release inToxoplasma gondii, a parasite closely related to plasmodia,through inhibition of the PfATP6 orthologue TgSERCAand a subsequent increase in cytosolic calcium [32]. How-ever, an increase in cytosolic calcium is part of the processof Plasmodium invasion of erythrocytes [33], so any inter-vention that achieves this may not be inhibitory to thisstage of parasite development. Support for this suggestioncomes from early work that reduced parasite cytosoliccalcium (rather than raising it) using intracellular chela-tors. This had the effect of blocking invasion [34].
Interestingly, parasite invasion requires the presence ofextracellular calcium [35]. One consequence is that calciumtransiently fills the parasitophorous vacuolar space thatsurrounds the newly intracellular parasite [36]. This maybe important for the tiny ring stages of parasites (Figure 2).The uptake of this calcium store into the parasite cytosoland subsequent sequestration in and release by organellessuch as the endoplasmic reticulum, a process modulated byPfATP6, could explain the artemisinin-hypersensitive sub-population of very young ring stage parasites observed byKlonis et al. [26].
Wasserman et al. also demonstrated that extracellularcalcium is essential as the parasite develops, especially
3

Opinion Trends in Pharmacological Sciences xxx xxxx, Vol. xxx, No. x
TIPS-1097; No. of Pages 8
between 20 and 26 h post invasion [35], the point at whichsome have reported that the parasite becomes most sensi-tive to artemisinins [26,28]. It is tempting to speculate thatthe extracellular calcium is again sourced by the parasitein a process that requires PfATP6 to be functional. PfATP6may then control calcium oscillations in the parasite cyto-sol that can critically manipulate the cell cycle, againsuggesting that PfATP6 might be targeted by artemisinins.Nagamune et al. have already demonstrated that artemi-sinin affects calcium oscillations in T. gondii [32], suggest-ing that parallel experiments should be attempted in P.falciparum over the entire intraerythrocytic life cycle.
PfATP6: heterologous expression and field mutationsExamination of PfATP6 as a possible target for artemisi-nins was provoked by their chemical similarity to thapsi-gargin, a potent and previously well-characterisedinhibitor of SERCAs, including mammalian SERCAs.Experiments established that artemisinins inhibit PfATP6relatively specifically and potently when expressed inXenopus laevis oocytes [14]. This was followed by confir-matory mutagenesis studies that identified some of themolecular determinants of the specificity of artemisininsfor malarial calcium ATPases and raised the possibilitythat mutations in this target sequence in parasites couldalso make them more resistant to artemisinin [37]. Thiswarning about one way in which artemisinin resistancecould arise was soon followed by publication of ex vivostudies on parasites from patients in French Guiana thatrelated mutations in PfATP6 (mainly S769N) to decreasedsensitivity to artemether [38]. Since then, extensive geno-typing studies from geographically diverse regions haveshown that PfATP6 is a highly polymorphic sequence[39,40], which complicates the assessment of any function-al consequences that may arise. In other areas, evidencecontinues to accrue that the S769N mutation may beselected for by artemisinins [41]. These findings also sug-gest that not all polymorphisms (nor indeed the majority)may be expected to influence sensitivity to artemisinins[42]. An added difficulty is that there may be fitness costs tothe parasite of these mutations in PfATP6 (as discussedbelow). A more detailed review of mutations in PfATP6 andsensitivity to artemisinins indicates, at least at present,that artemisinin drug selection in southeast Asia has notresulted in strong genetic associations with mutations inPfATP6 [43].
Geographical structuring of the frequency of some ami-no acid polymorphisms in PfATP6 also makes it harder todiscern associations with artemisinin resistance. In someregions of the world, genomic analysis has establishedsignificant relatedness for parasites with the clinical phe-notype of slower parasite clearance times after treatmentwith artemisinin-based combination therapies [44]. Wehave argued elsewhere, with some support, that thesephenotypes should not be taken to represent artemisininresistance per se [31,45]. In these areas, increased pfmdr1copy number is associated with increased risk of treatmentfailure after artemisinin combination therapies (TFACT)[46,47]. An increase in pfmdr1 copy number leads to pro-duction of more of the P glycoprotein orthologue Pgh1,which diminishes drug concentrations at their targets. If
4
this mechanism is associated with treatment failures, thenit is logical that there will not be selection pressure forparticular sequences at their targets, making redundantthe expectation that there will be increased frequency ofparticular sequences in PfATP6. This mechanism of resis-tance may be especially important in areas that alreadyhave parasites with increased pfmdr1 copy numbers atrelatively high frequencies [48–50]. The ease of selectingfor parasites with increased pfmdr1 copy number by arte-misinin derivatives has been well shown in in vitro experi-ments [51] and the link to pfmdr1 itself was established intransfection studies [52].
PFATP6 transfection studiesTransfection studies enable us to assess if an encodinggene is essential for parasite survival at particular life-cycle stages, as well as the functional impact of sequencepolymorphisms on parasites. Using methodology success-fully applied to study other malarial transport proteins[53], knockout via single homologous crossover recombina-tion of PfATP6 (using sequence at the 30 end) wasattempted in P. falciparum 3D7 strain [54]. When disrup-tion was impossible, complementation experiments withan episomal plasmid carrying the entire open readingframe of the wild type or a mutated pfatp6 gene wereattempted, but did not rescue the parasites. Tagging ofthe gene at the 30 end with GFP, c-myc, or double hae-magglutinin epitopes also failed to label pfatp6. Theseresults demonstrate that the gene is indispensable forasexual blood-stage parasite survival and that modifica-tions at the 30 end of the gene are not feasible. As confir-matory experiments, we used the genetically moreamenable Plasmodium berghei mouse model and trans-fected parasites with linearised plasmids to attempt adouble crossover recombination experiment, equally with-out success [54].
Different animal models have been used in recent yearsto understand mechanisms of artemisinin resistance, andhave exploited the ability to carry out genetic crosses inthese systems. The applicability of results from animalmodels to human infections has been debated, in thecontext of both studies on the pathophysiology of infection[55] and drug treatment and resistance studies. For exam-ple, chloroquine resistance mechanisms differ betweenanimal [56] and human infections [57]. Several studieshave identified new mechanisms of resistance to artemi-sinins in animal infections [58,59], although a commontheme seems to be amplification in the mdr1 orthologues ofboth animal and human infections [60].
Transfection approaches are also effective in establish-ing if genes are involved in drug resistance. PfATP6 hasbeen mutated to test if particular single nucleotide poly-morphisms decrease sensitivity to artemisinin derivatives.On the basis of prediction after modelling studies (seebelow) and results from Xenopus oocytes, the gene encod-ing PfATP6 protein carrying the L263E mutation wasefficiently introduced in two different wild type back-grounds, 7G8 (Brazil) and D10 (Papua New Guinea),and susceptibility to artemisinins was evaluated [61].IC50 values in individual experiments were variable, andpooled analysis revealed increased resistance in the 263E
0
50
100
150
200
Parasite clone
Nor
mal
ised
IC50
val
ues (
%)
0/36 7/54
p = 0.039(A) (B)
0 50 100 150 200
0
50
100
150
200
250
300
PfATP6 (µM)
IC50
(µM
)
ATM S769 ATM 769N
TRENDS in Pharmacological Sciences
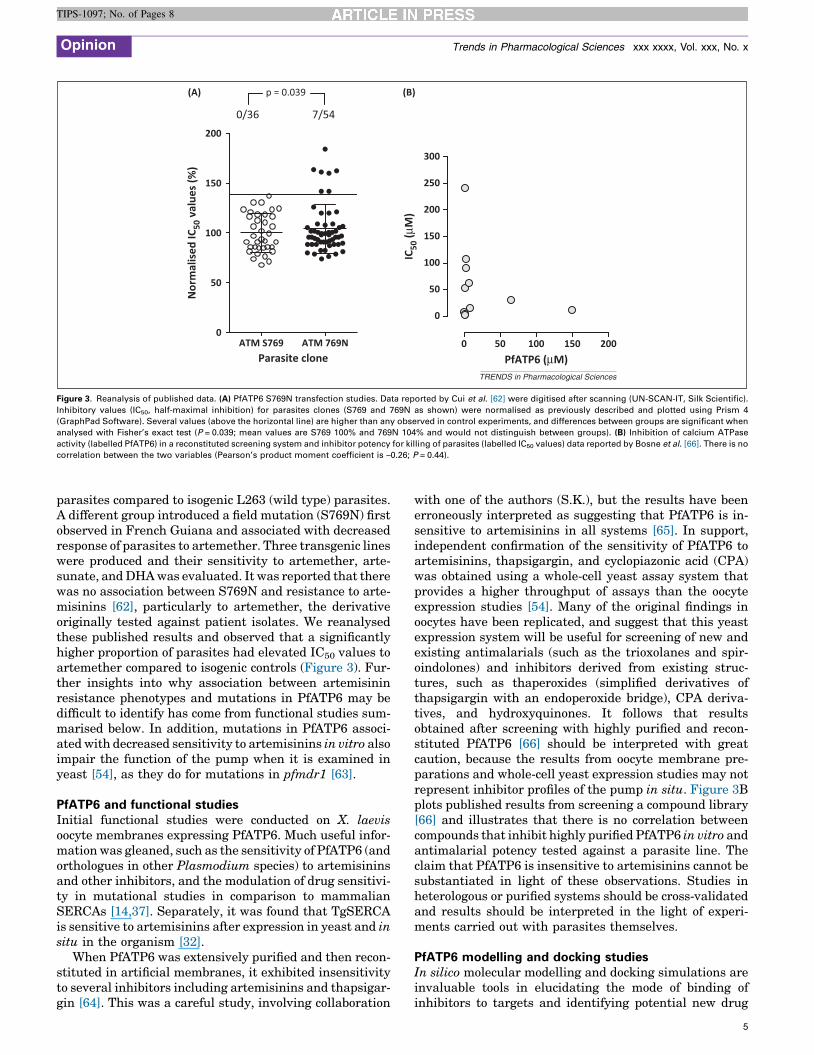
Figure 3. Reanalysis of published data. (A) PfATP6 S769N transfection studies. Data reported by Cui et al. [62] were digitised after scanning (UN-SCAN-IT, Silk Scientific).
Inhibitory values (IC50, half-maximal inhibition) for parasites clones (S769 and 769N as shown) were normalised as previously described and plotted using Prism 4
(GraphPad Software). Several values (above the horizontal line) are higher than any observed in control experiments, and differences between groups are significant when
analysed with Fisher’s exact test (P = 0.039; mean values are S769 100% and 769N 104% and would not distinguish between groups). (B) Inhibition of calcium ATPase
activity (labelled PfATP6) in a reconstituted screening system and inhibitor potency for killing of parasites (labelled IC50 values) data reported by Bosne et al. [66]. There is no
correlation between the two variables (Pearson’s product moment coefficient is –0.26; P = 0.44).
Opinion Trends in Pharmacological Sciences xxx xxxx, Vol. xxx, No. x
TIPS-1097; No. of Pages 8
parasites compared to isogenic L263 (wild type) parasites.A different group introduced a field mutation (S769N) firstobserved in French Guiana and associated with decreasedresponse of parasites to artemether. Three transgenic lineswere produced and their sensitivity to artemether, arte-sunate, and DHA was evaluated. It was reported that therewas no association between S769N and resistance to arte-misinins [62], particularly to artemether, the derivativeoriginally tested against patient isolates. We reanalysedthese published results and observed that a significantlyhigher proportion of parasites had elevated IC50 values toartemether compared to isogenic controls (Figure 3). Fur-ther insights into why association between artemisininresistance phenotypes and mutations in PfATP6 may bedifficult to identify has come from functional studies sum-marised below. In addition, mutations in PfATP6 associ-ated with decreased sensitivity to artemisinins in vitro alsoimpair the function of the pump when it is examined inyeast [54], as they do for mutations in pfmdr1 [63].
PfATP6 and functional studiesInitial functional studies were conducted on X. laevisoocyte membranes expressing PfATP6. Much useful infor-mation was gleaned, such as the sensitivity of PfATP6 (andorthologues in other Plasmodium species) to artemisininsand other inhibitors, and the modulation of drug sensitivi-ty in mutational studies in comparison to mammalianSERCAs [14,37]. Separately, it was found that TgSERCAis sensitive to artemisinins after expression in yeast and insitu in the organism [32].
When PfATP6 was extensively purified and then recon-stituted in artificial membranes, it exhibited insensitivityto several inhibitors including artemisinins and thapsigar-gin [64]. This was a careful study, involving collaboration
with one of the authors (S.K.), but the results have beenerroneously interpreted as suggesting that PfATP6 is in-sensitive to artemisinins in all systems [65]. In support,independent confirmation of the sensitivity of PfATP6 toartemisinins, thapsigargin, and cyclopiazonic acid (CPA)was obtained using a whole-cell yeast assay system thatprovides a higher throughput of assays than the oocyteexpression studies [54]. Many of the original findings inoocytes have been replicated, and suggest that this yeastexpression system will be useful for screening of new andexisting antimalarials (such as the trioxolanes and spir-oindolones) and inhibitors derived from existing struc-tures, such as thaperoxides (simplified derivatives ofthapsigargin with an endoperoxide bridge), CPA deriva-tives, and hydroxyquinones. It follows that resultsobtained after screening with highly purified and recon-stituted PfATP6 [66] should be interpreted with greatcaution, because the results from oocyte membrane pre-parations and whole-cell yeast expression studies may notrepresent inhibitor profiles of the pump in situ. Figure 3Bplots published results from screening a compound library[66] and illustrates that there is no correlation betweencompounds that inhibit highly purified PfATP6 in vitro andantimalarial potency tested against a parasite line. Theclaim that PfATP6 is insensitive to artemisinins cannot besubstantiated in light of these observations. Studies inheterologous or purified systems should be cross-validatedand results should be interpreted in the light of experi-ments carried out with parasites themselves.
PfATP6 modelling and docking studiesIn silico molecular modelling and docking simulations areinvaluable tools in elucidating the mode of binding ofinhibitors to targets and identifying potential new drug
5

Opinion Trends in Pharmacological Sciences xxx xxxx, Vol. xxx, No. x
TIPS-1097; No. of Pages 8
candidates. Because the crystal structure of PfATP6 has notyet been solved, all in silico models are based on the crystalstructure of SERCA1a, which was provided by Toyoshimaet al. in 2000 [67] and has subsequently included structuresof several conformations, including active (E1) and inactive(E2) states, and those complexed with inhibitors such asthapsigargin [68]. Figure 1B shows a recent example of aPfATP6 model. The PfATP6 sequence contains many low-complexity insertions that are not present in other ortholo-gues, which may make it harder to crystallise.
The first in silico mutagenesis study on PfATP6 was byUhlemann et al. and provided insights into the functionalconsequences of mutations in the thapsigargin/artemisi-nin-binding pocket [37]. Additional studies supported thehypothesis that artemisinins and thaperoxides interactwith PfATP6 in the same binding pocket as thapsigargindoes [69–72]. Only one study has found no correlationbetween the activity of artemisinins and their simulateddocking with PfATP6 [73]. This could be explained by theunusual approach adopted for orientating artemisinins,with their peroxide bond positioned towards the inner partof the binding cavity. All other studies orientated theperoxide bond towards the solvent, which implies thatanomalous results reported by Garah et al. may be dueto multiple methodological factors and that subsequentinterpretations should be viewed with caution.
The effects of the field mutations S769N and A623E onthe binding of artemisinins have also been studied in silico[74]. These are both distant from the binding site(Figure 1), so modulation of artemisinin binding wouldhave to be through allosteric effects, which are still poorlyunderstood for this pump. A recent computational analysishas provided some clarification by modelling PfATP6 inseveral conformations. Results suggest that an Fe–arte-misinin adduct has the highest calculated affinity forbinding to PfATP6 (even compared to artemisinin itself),confirm interactions with several previously identifiedresidues, identify some new interacting residues, anddemonstrate a mechanism by which allosteric effectsmay be mediated [75]. However, the existence of the Feadduct in vivo requires experimental verification, and thismay be difficult to achieve if artemisinins do not primarilyact on the food vacuole.
Another argument raised against PfATP6 as a target forartemisinins is the observation that enantiomeric 1,2,4-trioxanes do not differ much in their antimalarial potency[43]. However, the assays used to assess potency involveinfected cells (whole cell assays), in which many variablesunrelated to enantioselectivity may influence potency(such as differential delivery of the compounds to the targetand the relative stability of the compounds in vivo). Thestereoselectivity argument can also be applied to quinineand its diastereomer quinidine, which have similar anti-malarial potency [76].
Concluding remarksPfATP6 has been chemically and genetically validated as akey drug target. Artemisinins are one chemical class thatinhibit PfATP6, but other druggable classes that caninhibit mutated versions of PfATP6 relatively resistantto artemisinins should now be identified. Monitoring of
6
polymorphisms in PfATP6 in molecular epidemiology stud-ies should continue. Functional consequences (both fortolerance of calcium and drug sensitivity) can be pursuedrapidly using yeast expression and more slowly with para-site transfection technologies. The whole-cell yeast expres-sion system can be turned to screening in higher-throughput approaches. Elucidation of a structure forPfATP6 will prove both challenging and rewarding, butshould use materials that preserve function and suscepti-bility to inhibitors.
Although transfection studies (above) have demonstrat-ed that PfATP6 is likely to be essential during the asexualblood stage of the parasite life cycle, more detailed studiesare required (with conditional knockdowns) to resolve withgreater accuracy the point(s) at which PfATP6 plays acritical role. As the elucidation of PfATP6 function inthe cell biology of infection is being explored, new waysto exploit PfATP6 as a drug target should also be vigorous-ly pursued.
References1 Zhang, J.F., ed. (2013) A Detailed Chronological Record of Project 523
and the Discovery and Development of Qinghaosu (Artemisinin),Strategic Book Publishing
2 Coordinating Research Group for the Structure of Artemisinin (1977) Anew sesquiterpene lactone – artemisinin. Kuo Xue Tung Bao 22, 142
3 Charman, S.A. et al. (2011) Synthetic ozonide drug candidate OZ439offers new hope for a single-dose cure of uncomplicated malaria. Proc.Natl. Acad. Sci. U.S.A. 108, 4400–4405
4 Nealon, C. et al. (2002) Intramuscular bioavailability and clinicalefficacy of artesunate in Gabonese children with severe malaria.Antimicrob. Agents Chemother. 46, 3933–3939
5 Gomes, M.F. et al. (2009) Pre-referral rectal artesunate to preventdeath and disability in severe malaria: a placebo-controlled trial.Lancet 373, 557–566
6 Krishna, S. et al. (2001) Bioavailability and preliminary clinical efficacyof intrarectal artesunate in Ghanaian children with moderate malaria.Antimicrob. Agents Chemother. 45, 509–516
7 Kremsner, P.G. et al. (2012) A simplified intravenous artesunateregimen for severe malaria. J. Infect. Dis. 205, 312–319
8 Krishna, S. et al. (2008) Artemisinins: their growing importance inmedicine. Trends Pharmacol. Sci. 29, 520–527
9 Staines, H.M. et al. (2010) Exploiting the therapeutic potential ofPlasmodium falciparum solute transporters. Trends Parasitol. 26,284–296
10 Kotsubei, A. et al. (2013) Probing determinants of cyclopiazonic acidsensitivity of bacterial Ca2+-ATPases. FEBS J. 280, 5441–5449
11 Kimura, M. et al. (1993) Cloning of a Ca2+-ATPase gene of Plasmodiumfalciparum and comparison with vertebrate Ca2+-ATPases. J. Cell Sci.104, 1129–1136
12 Krishna, S. et al. (1993) A family of cation ATPase-like molecules fromPlasmodium falciparum. J. Cell Biol. 120, 385–398
13 Krishna, S. and Woodrow, C.J. (1999) Expression of parasitetransporters in Xenopus oocytes. Novartis Found. Symp. 226, 126–139
14 Eckstein-Ludwig, U. et al. (2003) Artemisinins target the SERCA ofPlasmodium falciparum. Nature 424, 957–961
15 Mariani, J.A. et al. (2011) Augmentation of left ventricular mechanicsby recirculation-mediated AAV2/1-SERCA2a gene delivery inexperimental heart failure. Eur. J. Heart Fail. 13, 247–253
16 Denmeade, S.R. et al. (2012) Engineering a prostate-specific membraneantigen-activated tumor endothelial cell prodrug for cancer therapy.Sci. Transl. Med. 4, 140ra86
17 Klonis, N. et al. (2013) Iron and heme metabolism in Plasmodiumfalciparum and the mechanism of action of artemisinins. Curr. Opin.Microbiol. http://dx.doi.org/10.1016/j.mib.2013.07.005
18 O’Neill, P.M. et al. (2010) The molecular mechanism of action ofartemisinin – the debate continues. Molecules 15, 1705–1721
19 Miller, L.H. et al. (2013) Malaria biology and disease pathogenesis:insights for new treatments. Nat. Med. 19, 156–167

Opinion Trends in Pharmacological Sciences xxx xxxx, Vol. xxx, No. x
TIPS-1097; No. of Pages 8
20 Bedu-Addo, G. et al. (2013) An ATP2B4 polymorphism protects againstmalaria in pregnancy. J. Infect. Dis. 207, 1600–1603
21 Delves, M. et al. (2012) The activities of current antimalarial drugs onthe life cycle stages of Plasmodium: a comparative study with humanand rodent parasites. PLoS Med. 9, e1001169
22 Guttery, D.S. et al. (2013) The Plasmodium berghei Ca2+/H+ exchanger,PbCAX, is essential for tolerance to environmental Ca2+ during sexualdevelopment. PLoS Pathog. 9, e1003191
23 Krishna, S. et al. (2001) Expression and functional characterization of aPlasmodium falciparum Ca2+-ATPase (PfATP4) belonging to asubclass unique to apicomplexan organisms. J. Biol. Chem. 276,10782–10787
24 Spillman, N.J. et al. (2013) Na+ regulation in the malaria parasitePlasmodium falciparum involves the cation ATPase PfATP4 and is atarget of the spiroindolone antimalarials. Cell Host Microbe 13, 227–237
25 Geary, T.G. et al. (1989) Stage specific actions of antimalarial drugs onPlasmodium falciparum in culture. Am. J. Trop. Med. Hyg. 40, 240–244
26 Klonis, N. et al. (2013) Altered temporal response of malaria parasitesdetermines differential sensitivity to artemisinin. Proc. Natl. Acad.Sci. U.S.A. 110, 5157–5162
27 Skinner, T.S. et al. (1996) In vitro stage-specific sensitivity ofPlasmodium falciparum to quinine and artemisinin drugs. Int. J.Parasitol. 26, 519–525
28 ter Kuile, F. et al. (1993) Plasmodium falciparum: in vitro studies of thepharmacodynamic properties of drugs used for the treatment of severemalaria. Exp. Parasitol. 76, 85–95
29 Wilson, D.W. et al. (2013) Defining the timing of action of antimalarialdrugs against Plasmodium falciparum. Antimicrob. Agents Chemother.57, 1455–1467
30 Witkowski, B. et al. (2013) Novel phenotypic assays for the detection ofartemisinin-resistant Plasmodium falciparum malaria in Cambodia:in-vitro and ex-vivo drug–response studies. Lancet Infect. Dis. http://dx.doi.org/10.1016/S1473-3099(13)70252-4
31 Krishna, S. and Kremsner, P.G. (2013) Antidogmatic approaches toartemisinin resistance: reappraisal as treatment failure withartemisinin combination therapy. Trends Parasitol. 29, 313–317
32 Nagamune, K. et al. (2007) Artemisinin induces calcium-dependentprotein secretion in the protozoan parasite Toxoplasma gondii.Eukaryot. Cell 6, 2147–2156
33 Singh, S. et al. (2010) Distinct external signals trigger sequentialrelease of apical organelles during erythrocyte invasion by malariaparasites. PLoS Pathog. 6, e1000746
34 Wasserman, M. and Chaparro, J. (1996) Intraerythrocytic calciumchelators inhibit the invasion of Plasmodium falciparum. Parasitol.Res. 82, 102–107
35 Wasserman, M. et al. (1982) Effects of Ca2+ depletion on the asexual cellcycle of Plasmodium falciparum. Am. J. Trop. Med. Hyg. 31, 711–717
36 Gazarini, M.L. et al. (2003) Calcium signaling in a low calciumenvironment: how the intracellular malaria parasite solves theproblem. J. Cell Biol. 161, 103–110
37 Uhlemann, A.C. et al. (2005) A single amino acid residue can determinethe sensitivity of SERCAs to artemisinins. Nat. Struct. Mol. Biol. 12,628–629
38 Jambou, R. et al. (2005) Resistance of Plasmodium falciparum fieldisolates to in-vitro artemether and point mutations of the SERCA-typePfATPase6. Lancet 366, 1960–1963
39 Miao, M. et al. (2013) Genetic diversity and lack of artemisininselection signature on the Plasmodium falciparum ATP6 in theGreater Mekong subregion. PLoS ONE 8, e59192
40 Tanabe, K. et al. (2011) Spontaneous mutations in the Plasmodiumfalciparum sarcoplasmic/endoplasmic reticulum Ca2+-ATPase(PfATP6) gene among geographically widespread parasitepopulations unexposed to artemisinin-based combination therapies.Antimicrob. Agents Chemother. 55, 94–100
41 Zakeri, S. et al. (2012) Molecular assessment of atpase6 mutationsassociated with artemisinin resistance among unexposed and exposedPlasmodium falciparum clinical isolates to artemisinin-basedcombination therapy. Malar. J. 11, 373
42 Wang, Z. et al. (2012) In vitro sensitivity of Plasmodium falciparumfrom China–Myanmar border area to major ACT drugs andpolymorphisms in potential target genes. PLoS ONE 7, e30927
43 Ding, X.C. et al. (2011) Plasmodium sensitivity to artemisinins: magicbullets hit elusive targets. Trends Parasitol. 27, 73–81
44 Miotto, O. et al. (2013) Multiple populations of artemisinin-resistantPlasmodium falciparum in Cambodia. Nat. Genet. 45, 648–655
45 Ferreira, P.E. et al. (2013) Artemisinin resistance in Plasmodiumfalciparum: what is it really? Trends Parasitol. 29, 318–320
46 Na-Bangchang, K. et al. (2013) Identification of resistance ofPlasmodium falciparum to artesunate–mefloquine combination inan area along the Thai–Myanmar border: integration of clinico–parasitological response, systemic drug exposure, and in vitroparasite sensitivity. Malar. J. 12, 263
47 Price, R.N. et al. (2004) Mefloquine resistance in Plasmodiumfalciparum and increased pfmdr1 gene copy number. Lancet 364,438–447
48 Muhamad, P. et al. (2011) Polymorphisms of molecular markers ofantimalarial drug resistance and relationship with artesunate–mefloquine combination therapy in patients with uncomplicatedPlasmodium falciparum malaria in Thailand. Am. J. Trop. Med.Hyg. 85, 568–572
49 Phompradit, P. et al. (2011) Molecular analysis of pfatp6 and pfmdr1polymorphisms and their association with in vitro sensitivity inPlasmodium falciparum isolates from the Thai–Myanmar border.Acta Trop. 120, 130–135
50 Uhlemann, A.C. et al. (2007) Intrahost selection of Plasmodiumfalciparum pfmdr1 alleles after antimalarial treatment on thenorthwestern border of Thailand. J. Infect. Dis. 195, 134–141
51 Chavchich, M. et al. (2010) Role of pfmdr1 amplification and expressionin induction of resistance to artemisinin derivatives in Plasmodiumfalciparum. Antimicrob. Agents Chemother. 54, 2455–2464
52 Sidhu, A.B. et al. (2006) Decreasing pfmdr1 copy number inPlasmodium falciparum malaria heightens susceptibility tomefloquine, lumefantrine, halofantrine, quinine, and artemisinin. J.Infect. Dis. 194, 528–535
53 Slavic, K. et al. (2010) Life cycle studies of the hexose transporter ofPlasmodium species and genetic validation of their essentiality. Mol.Microbiol. 75, 1402–1413
54 Pulcini, S. et al. (2013) Expression in yeast links field polymorphisms inPfATP6 to in vitro artemisinin resistance and identifies new inhibitorclasses. J. Infect. Dis. 208, 468–478
55 Craig, A.G. et al. (2012) The role of animal models for research onsevere malaria. PLoS Pathog. 8, e1002401
56 Hunt, P. et al. (2004) Chloroquine resistance in Plasmodium chabaudi:are chloroquine-resistance transporter (crt) and multi-drug resistance(mdr1) orthologues involved? Mol. Biochem. Parasitol. 133, 27–35
57 Ecker, A. et al. (2012) PfCRT and its role in antimalarial drugresistance. Trends Parasitol. 28, 504–514
58 Henriques, G. et al. (2013) Artemisinin resistance in rodent malaria –mutation in the AP2 adaptor m-chain suggests involvement ofendocytosis and membrane protein trafficking. Malar. J. 12, 118
59 Hunt, P. et al. (2007) Gene encoding a deubiquitinating enzyme ismutated in artesunate- and chloroquine-resistant rodent malariaparasites. Mol. Microbiol. 65, 27–40
60 Borges, S. et al. (2011) Genomewide scan reveals amplification of mdr1as a common denominator of resistance to mefloquine, lumefantrine,and artemisinin in Plasmodium chabaudi malaria parasites.Antimicrob. Agents Chemother. 55, 4858–4865
61 Valderramos, S.G. et al. (2010) Investigations into the role of thePlasmodium falciparum SERCA (PfATP6) L263E mutation inartemisinin action and resistance. Antimicrob. Agents Chemother.54, 3842–3852
62 Cui, L. et al. (2012) Lack of association of the S769N mutation inPlasmodium falciparum SERCA (PfATP6) with resistance toartemisinins. Antimicrob. Agents Chemother. 56, 2546–2552
63 Froberg, G. et al. (2013) Assessing the cost–benefit effect of aPlasmodium falciparum drug resistance mutation on parasitegrowth in vitro. Antimicrob. Agents Chemother. 57, 887–892
64 Cardi, D. et al. (2010) Purified E255L mutant SERCA1a and purifiedPfATP6 are sensitive to SERCA-type inhibitors but insensitive toartemisinins. J. Biol. Chem. 285, 26406–26416
65 Arnou, B. et al. (2011) The Plasmodium falciparum Ca2+-ATPasePfATP6: insensitive to artemisinin, but a potential drug target.Biochem. Soc. Trans. 39, 823–831
7

Opinion Trends in Pharmacological Sciences xxx xxxx, Vol. xxx, No. x
TIPS-1097; No. of Pages 8
66 David-Bosne, S. et al. (2013) Antimalarial screening via large-scalepurification of Plasmodium falciparum Ca2+-ATPase 6 and in vitrostudies. FEBS J. 280, 5419–5429
67 Toyoshima, C. et al. (2000) Crystal structure of the calcium pump ofsarcoplasmic reticulum at 2.6 A resolution. Nature 405, 647–655
68 Toyoshima, C. and Cornelius, F. (2013) New crystal structures of PII-typeATPases: excitement continues. Curr. Opin. Struct. Biol. 23, 507–514
69 Helal, M.A. and Avery, M.A. (2012) Combined receptor-based and ligand-based approach to delineate the mode of binding of guaianolide-endoperoxides to PfATP6. Bioorg. Med. Chem. Lett. 22, 5410–5414
70 Jung, M. et al. (2005) Three-dimensional structure of Plasmodiumfalciparum Ca2+-ATPase (PfATP6) and docking of artemisininderivatives to PfATP6. Bioorg. Med. Chem. Lett. 15, 2994–2997
71 Naik, P.K. et al. (2010) The binding modes and binding affinities ofartemisinin derivatives with Plasmodium falciparum Ca2+-ATPase(PfATP6). J. Mol. Model. 17, 333–357
8
72 Sun, L. et al. (2010) Design, synthesis, and development of novelguaianolide-endoperoxides as potential antimalarial agents. J. Med.Chem. 53, 7864–7868
73 Garah, F.B. et al. (2009) Docking studies of structurally diverseantimalarial drugs targeting PfATP6: no correlation between insilico binding affinity and in vitro antimalarial activity.ChemMedChem 4, 1469–1479
74 Pillai, D.R. et al. (2012) Artemether resistance in vitro is linked tomutations in PfATP6 that also interact with mutations in PfMDR1 intravellers returning with Plasmodium falciparum infections. Malar. J.11, 131
75 Shandilya, A. et al. (2013) A plausible mechanism for the antimalarialactivity of artemisinin: a computational approach. Sci. Rep. 3, 2513
76 Krishna, S. and White, N.J. (1996) Pharmacokinetics of quinine,chloroquine and amodiaquine. Clinical implications. Clin.Pharmacokinet. 30, 263–299




















