
Multiple roles of phosphoinositide-specific phospholipase C ...
Upload
independentCategory
view
2download
0
Protein Kinase C-� Regulates Sphingosine1-Phosphate-mediated Migration of Human LungEndothelial Cells through Activation of Phospholipase D2,Protein Kinase C-�, and Rac1*□S
Received for publication, January 10, 2008, and in revised form, February 21, 2008 Published, JBC Papers in Press, February 22, 2008, DOI 10.1074/jbc.M800250200
Irina Gorshkova‡, Donghong He‡, Evgeny Berdyshev‡, Peter Usatuyk‡, Michael Burns‡, Satish Kalari‡,Yutong Zhao‡, Srikanth Pendyala‡, Joe G. N. Garcia‡, Nigel J. Pyne§, David N. Brindley¶1,and Viswanathan Natarajan‡2
From the ‡Department of Medicine, University of Chicago, Chicago, Illinois 60637, §Department of Physiology and Pharmacology,University of Strathclyde, Glasgow G4 ONR, United Kingdom, and ¶Department of Biochemistry, University of Alberta,Edmonton, Alberta T6G 2S2, Canada
The signaling pathways by which sphingosine 1-phosphate(S1P) potently stimulates endothelial cell migration and angio-genesis are not yet fully defined.We, therefore, investigated therole of protein kinaseC (PKC) isoforms, phospholipaseD (PLD),and Rac in S1P-induced migration of human pulmonary arteryendothelial cells (HPAECs). S1P-induced migration was sensi-tive to S1P1 small interfering RNA (siRNA) and pertussis toxin,demonstrating coupling of S1P1 to Gi. Overexpression of domi-nant negative (dn) PKC-� or -�, but not PKC-� or -�, blockedS1P-inducedmigration. Although S1P activated both PLD1 andPLD2, S1P-induced migration was attenuated by knockingdown PLD2 or expressing dnPLD2 but not PLD1. BlockingPKC-�, but not PKC-�, activity attenuated S1P-mediated PLDstimulation, demonstrating that PKC-�, but not PKC-�, wasupstreamof PLD.Transfection ofHPAECswith dnRac1 or Rac1siRNA attenuated S1P-induced migration. Furthermore, trans-fection with PLD2 siRNA, infection of HPAECs with dnPKC-�,or treatment with myristoylated PKC-� peptide inhibitor abro-gated S1P-induced Rac1 activation. These results establish thatS1P signals through S1P1 and Gi to activate PKC-� and, subse-quently, a PLD2-PKC-�-Rac1 cascade. Activation of this path-way is necessary to stimulate the migration of lung endothelialcells, a key component of the angiogenic process.
Sphingosine 1-phosphate (S1P)3 is a naturally occurring bio-active sphingolipid that elicits multiple cellular responses such
as differentiation, proliferation, survival, and angiogenesis(1–5). S1P acts as an intracellular second messenger. Extracel-lular S1P also activates intracellular signaling pathways throughligation to a family of G-protein-coupled S1P receptors, S1P1–5(previously known as endothelial differentiation gene recep-tors) (6). The S1P-Rs are differentially expressed in differentcell types and are coupled toGi, Gq, orG12/13 (7–9). Coupling ofS1P to S1P1 via Gi activates Rac and Rho (2, 10) and stimulatescell proliferation (4), cortical actin formation (11), assembly ofadherens junction, and angiogenesis (2). Binding of S1P to S1P3induces signaling through Gq or G13 to activate Rho (2, 10, 12),promotes the formation of stress fibers and adherens junctions(2), stimulates phospholipase D (PLD) (13), and activates phos-pholipase C/intracellular Ca2�/protein kinase C (PKC) path-ways (7). Ligation of S1P to S1P1 also initiates cross-talk withother receptors, especially growth factor receptors includingthose for epidermal growth factor (EGF), platelet-derivedgrowth factor, and vascular endothelial growth factor (14). Thefunctional platelet-derived growth factor (PDGF)-�/S1P1 sig-naling complex was postulated to be involved in regulatingmigration ofmouse embryonic fibroblasts in response to PDGF(15). Furthermore, S1P binding to S1P2 inhibits cell migrationvia Gq or G13 (9, 12, 16) and activates adenylate cyclase (17) andmitogen-activated protein kinases (MAPKs) (18). There are fewstudies related to S1P signaling via S1P4 and S1P5; however,these receptorsmay be involved in change in cell shape (19) andneurite retraction (20). In addition to the well described vascu-lar effects of S1P (21), in non-vascular tissues S1P exhibits pro-inflammatory effects such as increased interleukin-6/-8 secre-tion in airway epithelial (22) and ovarian cancer cells (23).In the vasculature, S1P is a key regulator of vascular matura-
tion and angiogenesis under physiological and pathologicalconditions. Angiogenesis, or new blood vessel formation, is
* This work was supported by National Institutes of Health Grant HL RO179396 (to V. N.) and Canadian Institute of Health Research Grant MOP81137 (to D. N. B.). The costs of publication of this article were defrayed inpart by the payment of page charges. This article must therefore be herebymarked “advertisement” in accordance with 18 U.S.C. Section 1734 solely toindicate this fact.
□S The on-line version of this article (available at http://www.jbc.org) containssupplemental Figs. 1– 4 and Table 1.
1 Recipient of a Medical Scientist Award from the Alberta Heritage Founda-tion for Medical Research.
2 To whom correspondence should be addressed: Dept. of Medicine, The Uni-versity of Chicago, CIS Bldg., Rm. W408B, 929 East 57th St., Chicago, IL60637. Tel.: 773-834-2638; Fax: 773-834-2687; E-mail: [email protected].
3 The abbreviations used are: S1P, sphingosine 1-phosphate; S1P1, S1P recep-tor; EC, endothelial cell; HPAEC, human pulmonary artery EC; PLC, phos-
pholipase C; PLD, phospholipase D; PKC, protein kinase C; EGF, epidermalgrowth factor; MAPK, mitogen-activated protein kinase; PA, phosphatidicacid; LPA, lysophosphatidic acid; dn, dominant negative; EGM, endothelialgrowth medium; EBM, endothelial basal medium; ECIS, electrical cell sub-strate impedance sensing; PTx, pertussis toxin; RT, reverse transcription;siRNA, small interfering RNA; BSA, bovine serum albumin; m.o.i., multiplic-ity of infection; PBt, phosphatidylbutanol; TBST, Tris-buffered salineTween; FBS, fetal bovine serum; PBD, p21 binding domain.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 283, NO. 17, pp. 11794 –11806, April 25, 2008© 2008 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
11794 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 17 • APRIL 25, 2008
at University of C
hicago Library on June 11, 2008 w
ww
.jbc.orgD
ownloaded from
http://www.jbc.org/cgi/content/full/M800250200/DC1
Supplemental Material can be found at:

critical for normal embryonic vascular development and intumormetastasis. Although targeted deletion of S1P2 or S1P3 inmice has no adverse effect on embryogenesis, deletion of S1P1caused failure of vascular development leading to a massivehemorrhage and embryonic lethality between E12.5 and E14.5(24). Endothelial cell (EC) migration is an essential componentof angiogenesis that is regulated by growth factors, bioactivemolecules, and intracellular signaling (25). Among the variousagonists, S1P has emerged as a potent angiogenic, and vascularmaturation factor and considerable evidence exists for S1P-induced endothelial cell proliferation (4), migration (26–28),chemotaxis (29), and endothelial cell remodeling (30). Based ona number of studies using inhibitors, siRNA, dn mutants, orgenetically engineered mice, it is becoming evident that severalsignaling pathways including Rho/Rac, phosphatidylinositol3-kinase, Akt, MAPKs, PKC, and changes in intracellular Ca2�
are involved in S1P-induced EC migration (3, 7, 8, 12, 31). Werecently demonstrated that PLD activation by S1P regulatesERK1/2 activation (31) and interleukin-8 secretion in humanbronchial epithelial cells (22, 32). Furthermore, involvement oflipid phosphate phosphatase-1 in regulating lysophosphatidicacid (LPA)-induced phosphatidate (PA) generation and fibro-blastmigration suggests a role for PLD2 in fibroblastmigration,wound healing, and tumor metastasis (33).PA is a bioactive lipid, and its generation by PLD activation
represents an important signaling cascade involved in the reg-ulation of cellular responses including proliferation (34) andcytoskeletal reorganization (35). PA also serves as an immediateprecursor of LPA or diacylglycerol, which is an endogenousactivator of several PKC isoforms (36). PA itself stimulates thePKC-� isoform (37, 38), phosphatidylinositol 4-kinase (39–41),phospholipase C-� (42, 43), and sphingosine kinase-1 (44), andit inhibits protein phosphatase-1 (45).In ECs, very little is known regarding the role of S1P-induced
PLD activation and generation of PA in cell migration, woundhealing, and angiogenesis. Therefore, in the present study weinvestigated the role of S1P on human pulmonary artery endo-thelial cell (HPAEC) and established how the activation of thePKC isoform(s) is involved in upstream and downstream sig-naling of PLD1 and/or PLD2 in relation to the stimulation ofcell migration. Our results show that physiologically relevantconcentrations of S1P markedly stimulated HPAECmigration,which was sensitive to pertussis toxin (PTx) and a S1P1 antag-onist. Furthermore, evidence is provided for the role of PKC-�,but not PKC-�, in S1P-induced PLD activation and the PLD2-mediated stimulation of PKC-�, Rac1, and cell migration.
EXPERIMENTAL PROCEDURES
Materials—S1P, dihydrosphingosine 1-phosphate, and cera-mide 1-phosphate (8:0) were obtained fromAvanti Polar Lipids(Alabaster, AL). LPA, dioleoylglycerol, and brain phosphatidyl-serine were purchased from Sigma-Aldrich. Ceramide1-phos-phate (18:1) was a generous gift from Dr. C. E. Chalfant (Rich-mond, VA). Pertussis toxin was purchased from Calbiochem.SB649146 was from GlaxoSmithKline. Myelin basic proteinwas obtained from Upstate Biotechnology (Lake Placid, NY).Myristoylated PKC-� peptide inhibitor was purchased fromBIOMOL Research Labs Inc. (Plymouth Meeting, PA). Anti-
PKC-� antibody, PKC-� peptide inhibitor, scrambled siRNA,and target siRNA for PLD1, PLD2, and Rac1 were obtainedfrom Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-S1P1 antibody was obtained from Affinity BioReagents(Golden, CO), anti-S1P2, anti-S1P3, anti-S1P4, and anti-S1P5antibodies were purchased from Exalpha Biological Inc. (May-nard, MA), anti-PKC� and anti-PKC-� antibodies were fromBD Transduction Laboratories, and anti-Rac1 antibody wasfrom BD Biosciences Pharmingen. Anti-PKC� and anti-phos-pho-PKC-�(Ser-729) antibodies were purchased from UpstateBiotechnology; anti-phospho-PKC-�/�(Thr-410/403) wasobtained from Cell Signaling Technology Inc. (Danvers, MA).Anti-phosphoserine antibody was from Zymed LaboratoriesInc. (San Francisco, CA). Internal and N-terminal antibodiesfor PLD1 and PLD2 were purchased from BIOSOURCE Inter-national Inc. (Camarillo, CA), and anti-PLD2 antibody waskindly provided byDr. Sylvain Bourgoin (Quebec, PQ,Canada).Anti-�-actin antibody was from Sigma. S1P1 siRNA was fromDharmacon (Lafayette, CO). Rac1 activation assay kit wasobtained from Upstate (Temecula, CA). Lysis buffer was pur-chased from Cell Signaling Technology Inc. (Danvers, MA).Protease inhibitor mixture tablets (EDTA-free Complete) werefrom Roche Diagnostics. Aprotinin and phosphatase inhibitormixture 1 were from Sigma-Aldrich. Ad5CA dominant nega-tive (dn)-PKC-�, dnPKC-�, dnPKC-�, dnPKC-�, and dnPKC�were kindly provided by Dr. Motoi Ohba from Institute ofMolecular Oncology (Showa University, Japan).Cell Culture—HPAECs (passage number 3) were purchased
from Cambrex Inc. (Walkersville, MD) and cultured in com-plete endothelial growth medium (EGM)-2 medium (46). Thecells (passage number 5–8) in 35- or 100-mm dishes or glasscoverslips were used for all the experiments.Endothelial Cell Migration—HPAEC were cultured in 12- or
6-well plates to �95% confluence and then starved in theserum-free EGM-2 medium for 1–3 h or in EBM-2 mediumcontaining 0.1% FBS for 18–24 h. The cell monolayer waswounded by scratching across the monolayer with a 10-�lstandard sterile pipette tip. The scratched monolayer wasrinsed twice with serum-freemedium to remove cell debris andincubated with varying concentrations of S1P. The area (�1cm2 total) in a scratched area was recorded at 0 and 16–24 husing a Hamamatsu digital camera connected to the NikonEclipseTE2000-Smicroscopewith�10 objective andMetaVuesoftware (Universal Imaging Corp.). Images were analyzed bythe Image J software. The effect of S1P and other agents on cellmigration/woundhealingwas quantified by calculating the per-centage of the free area not occupied by cells compared with anarea of the initial wound thatwas defined as closure ofwoundedarea.Electrical Cell Substrate Impedance Sensing (ECIS) Assay—
HPAEC were cultured in 8-well ECIS electrode arrays (8W1E,Applied Biophysics, NY) to�95% confluence and starved in theserum-free EBM-2 medium for 1–3 h. An elevated field (3 V at40,000 Hz for 10 s) was applied to wound the cells on the elec-trode. Either complete medium or medium containing S1P(100–1000 nM) was added, and wound healing was monitoredfor 10–20 h bymeasuring the transendothelial electrical resist-
Regulation of S1P-induced Endothelial Cell Migration
APRIL 25, 2008 • VOLUME 283 • NUMBER 17 JOURNAL OF BIOLOGICAL CHEMISTRY 11795
at University of C
hicago Library on June 11, 2008 w
ww
.jbc.orgD
ownloaded from
ance using the ECIS equipment (11, 47). In all experiments S1Pwas complexed with 0.1% BSA.Infection of HPAECs with Adenoviral Vectors—cDNA for
wild type and catalytically inactivemutants of PLD1, PLD2, anddominant negative Rac1 were subcloned into the pShuttle-CMV vector (32). The recombinant plasmid was linearized andtransfected into HEK293 cells to generate replication-defectiveadenovirus. Generation of purified virus (1010 plaque-formingunits/ml)was carried out by theUniversity of IowaGeneTrans-fer Vector Core. Purified adenovirus (1–10 m.o.i. or plaque-forming units/cell) in complete EGM-2 medium was added toHPAECs grown to �80% confluence in 6-well plates or 60- or100-mm dishes. After 24 h, the virus-containing medium wasreplaced with complete EGM-2 medium. Vector control orinfected cells were subjected to scratch and wound-healingECIS assays, and immunoprecipitates or cell lysates were ana-lyzed by Western blotting.Measurement of PLDActivation by S1P—HPAECs in 35-mm
dishes were labeled with [32P]orthophosphate (5 �Ci/ml) inphosphate-freeDMEMfor 18–24 h at 37 °C in 5%CO2 and 95%air. Cells were then challenged with EBM-2 medium alone orEBM-2 containing S1P plus 0.1% BSA in the presence of 0.1%1-butanol (22, 32). In some experiments incubations were alsocarried out in the presence of 0.1% 3-butanol that served asadditional controls. Incubations were terminated by the addi-tion of 1ml ofmethanol:HCl (100:1 v/v), cells were scraped intoglass tubes, and lipids were extracted by the addition of 1 ml ofmethanol:HCl (100:1 v/v), 2 ml of chloroform, and 0.8 ml of 1 NHCl. [32P]Phosphatidylbutanol (PBt), formed as a result of PLDactivation and transphosphatidylation of [32P]PA to 1-butanol,but not butan-3-ol, was separated from the total lipid extract bythin layer chromatography on 1% potassiumoxalate plates withthe upper phase of ethyl acetate:2,2,4-trimethyl pentane:glacialacetic acid:water (65:10:15:50 v/v) as the developing solventsystem (22, 32). Unlabeled PBt was added as carrier during sep-aration of labeled lipids that were visualized by exposure toiodine vapor. Radioactivity associated with PBt was quantifiedby liquid scintillation counting, and all values were normalizedto 106 dpm in total lipid extract. [32P]PBt formed in control andS1P-challenged samples was expressed as dpm/dish or percentcontrol.Measurement of PKC-� and PKC-� Activation—HPAECs
were cultured in 100-mm dishes to �95% confluence, starvedin EBM-2medium containing 0.1% FBS for 3 h, stimulated withS1P for 5–10 min, washed with cold phosphate-buffered salinecontaining 1 mM vanadate, and lysed with 500 �l of lysis buffercontaining 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mMNa2EDTA, 1mMEGTA, 1%TritonX-100, 2.5mM sodiumpyro-phosphate, 1 mM �-glycerophosphate, 1 mM Na3VO4, 1 mMdithiothreitol, 1 �g/ml leupeptin, 1 �g/ml aprotinin, and pro-tease inhibitors from EDTA-free Complete tablets (RocheApplied Science). Cells were subsequently sonicated twice for15 s and then centrifuged at 10,000� g for 15min. Supernatantswere collected and incubated overnight with polyclonal anti-PKC-� or anti-PKC-� antibody at 4 °C. The immunoprecipi-tates were washed 3 times with lysis buffer and 2 times withkinase buffer (20 mM HEPES (pH 7.4), 25 mM �-glycerophos-phate, 10mMMgCl2, 1mMEGTA, 1mMsodiumorthovanadate,
and 1 mM dithiothreitol) and resuspended in 100 �l of kinasebuffer. The activity of PKC was measured in 100 �l of kinasebuffer containing 25�g ofmyelin basic protein as an exogenoussubstrate towhich 10�MATP, 2�g of dioleoylglycerol, 12�g ofphosphatidylserine, and 20–40 �l of immunoprecipitate wereadded. Incubations were carried out for 10 min at 30 °C andterminated by the addition of 20 �l of Laemmli sample buffer.Samples were then boiled for 5 min and analyzed for phospho-rylation of myelin basic protein by Western blotting with anti-phosphoserine antibody.
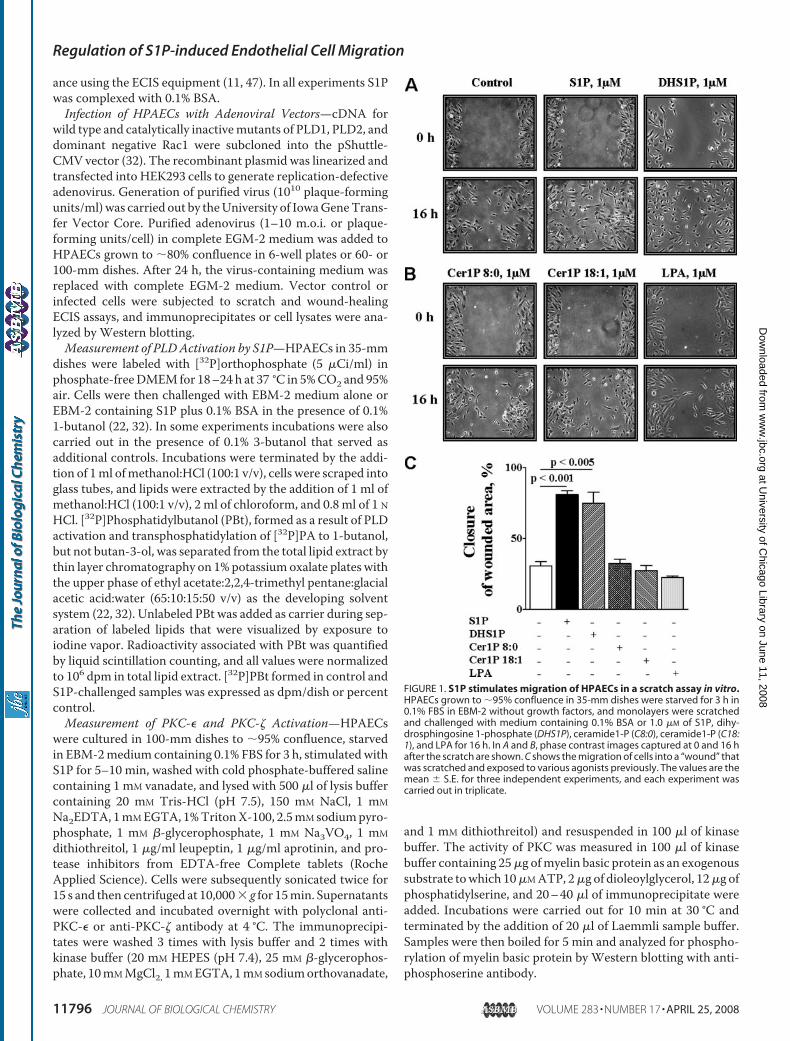
FIGURE 1. S1P stimulates migration of HPAECs in a scratch assay in vitro.HPAECs grown to �95% confluence in 35-mm dishes were starved for 3 h in0.1% FBS in EBM-2 without growth factors, and monolayers were scratchedand challenged with medium containing 0.1% BSA or 1.0 �M of S1P, dihy-drosphingosine 1-phosphate (DHS1P), ceramide1-P (C8:0), ceramide1-P (C18:1), and LPA for 16 h. In A and B, phase contrast images captured at 0 and 16 hafter the scratch are shown. C shows the migration of cells into a “wound” thatwas scratched and exposed to various agonists previously. The values are themean � S.E. for three independent experiments, and each experiment wascarried out in triplicate.
Regulation of S1P-induced Endothelial Cell Migration
11796 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 17 • APRIL 25, 2008
at University of C
hicago Library on June 11, 2008 w
ww
.jbc.orgD
ownloaded from
Rac1 Activation Assay—HPAECs were cultured in 100-mmdishes to �50% confluence for siRNA transfection or to �95%confluence for adenoviral infection or inhibitor treatment.Cells were starved in EBM-2 medium containing 0.1% FBS for3 h before stimulation with S1P for 2–15 min, cell lysates weresubjected to immunoprecipitation with PAK-1 PBD, and Rac1activation was evaluated using the Rac1 Activation assay kit asper the manufacturer’s instruction (Upstate).Western Blot Analysis—HPAECs were cultured in 6-well
plates or 60-mmdishes to�95% confluence and starved for 3 hin EBM-2 medium containing 0.1% FBS. Cells were stimulatedwith S1P (100–1000 nM) for 5–60 min, washed with phos-phate-buffered saline, and lysed with 100–300 �l of lysis buffercontaining 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mMNa2EDTA, 1mMEGTA, 1%TritonX-100, 2.5mM sodiumpyro-phosphate, 1 mM �-glycerophosphate, 1 mM Na3VO4, 1 �g/mlleupeptin, 1 �g/ml aprotinin, and protease inhibitors fromEDTA-free Complete tablets (Roche Applied Science). Celllysates were cleared by centrifugation at 10,000 � g for 10 minand boiled with the Laemmli sample buffer for 5 min. Cell
lysates (20–30 �g protein) wereseparated on 10% or 4–20% SDS-PAGE, transferred to polyvinyli-dene difluoride membranes, andblocked in TBST containing 5%BSAbefore incubationwith primaryantibody (1:1000 dilution) over-night. After blocking, washing, andincubation with appropriate sec-ondary antibody, blots were devel-oped using an ECL chemilumines-cence kit. Western blots werescanned by densitometry, and inte-grated density of pixels in identifiedareas was quantified using Image-Quant Version 5.2 software (GEHealthcare).Immunofluorescence Microscopy—
HPAECs grown on coverslips (18mm) or chamber slides were starvedfor 3 h in EBM-2 containing 0.1%FBS before treatment with S1P(100–1000 nM) for 5–60 min. Cellswere fixed in 3.7% paraformalde-hyde in phosphate-buffered salinefor 10 min, washed 3 times withphosphate-buffered saline, per-meabilized with methanol for 4min at �20 °C, blocked with 2%BSA in TBST, incubated for 1 hwith appropriate primary anti-body (1:200 dilution), washed withTBST, and stained for 1 h with sec-ondary antibody (1:200 dilution)in TBST containing 2% BSA. Cellswere examined using a NikonEclipse TE2000-S immunofluores-cence microscope and a Hamam-
atsu digital camera with �60 oil immersion objective andMeta Vue software.RNA Isolation and Real Time RT-PCR—Total RNA was iso-
lated from HPAECs grown on 35-mm dishes using TRIzol�reagent according to the manufacturer’s instruction. iQ SYBRGreen Supermix was used to do the real time measurementsusing iCycler by Bio-Rad. 18 S (sense, 5�-GTAACCCGTT-GAACCCCATT-3�, and antisense, 5�-CCATCCAATCGG-TAGTAGCG-3�) was used as a housekeeping gene to normal-ize expression. The reactionmixture consisted of 0.3�g of totalRNA (target gene) or 0.03 �g of total RNA (18 S rRNA), 12.5 �lof iQ SYBRGreen, 2�l of cDNA, 1.5�M target primers, or 1�M18 S rRNA primers in a total volume of 25 �l. For all samplesreverse transcriptionwas carried out at 25 °C for 5min followedby cycling to 42 °C for 30 min and 85 °C for 5 min with iScriptcDNA synthesis kit. Amplicon expression in each sample wasnormalized to its 18 S rRNA content. The relative abundance oftarget mRNA in each sample was calculated as 2 raised to thenegative of its threshold cycle value times 106 after being nor-malized to the abundance of its corresponding 18 S rRNA
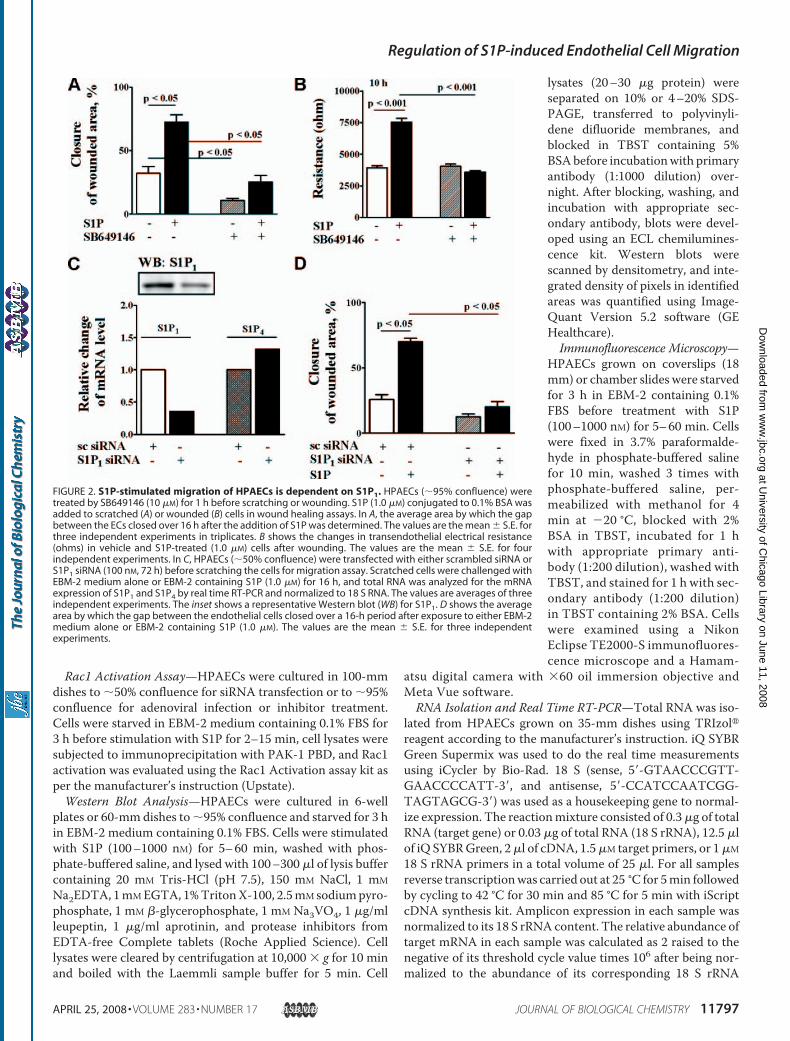
FIGURE 2. S1P-stimulated migration of HPAECs is dependent on S1P1. HPAECs (�95% confluence) weretreated by SB649146 (10 �M) for 1 h before scratching or wounding. S1P (1.0 �M) conjugated to 0.1% BSA wasadded to scratched (A) or wounded (B) cells in wound healing assays. In A, the average area by which the gapbetween the ECs closed over 16 h after the addition of S1P was determined. The values are the mean � S.E. forthree independent experiments in triplicates. B shows the changes in transendothelial electrical resistance(ohms) in vehicle and S1P-treated (1.0 �M) cells after wounding. The values are the mean � S.E. for fourindependent experiments. In C, HPAECs (�50% confluence) were transfected with either scrambled siRNA orS1P1 siRNA (100 nM, 72 h) before scratching the cells for migration assay. Scratched cells were challenged withEBM-2 medium alone or EBM-2 containing S1P (1.0 �M) for 16 h, and total RNA was analyzed for the mRNAexpression of S1P1 and S1P4 by real time RT-PCR and normalized to 18 S RNA. The values are averages of threeindependent experiments. The inset shows a representative Western blot (WB) for S1P1. D shows the averagearea by which the gap between the endothelial cells closed over a 16-h period after exposure to either EBM-2medium alone or EBM-2 containing S1P (1.0 �M). The values are the mean � S.E. for three independentexperiments.
Regulation of S1P-induced Endothelial Cell Migration
APRIL 25, 2008 • VOLUME 283 • NUMBER 17 JOURNAL OF BIOLOGICAL CHEMISTRY 11797
at University of C
hicago Library on June 11, 2008 w
ww
.jbc.orgD
ownloaded from
(housekeeping gene) (2�(primer threshold cycle)2�(18 S threshold cycle) �106). All primers were designed by inspection of the genes ofinterest using Primer 3 software. Negative controls consistingof reaction mixtures containing all components except targetRNA were included with each of the RT-PCR runs. To verifythat amplified products were derived from mRNA and did notrepresent genomic DNA contamination, representative PCRmixtures for each gene were run in the absence of the RTenzyme after first being cycled to 95 °C for 15 min. In theabsence of reverse transcription, no PCR products wereobserved.siRNATransfection—HPAECs grown to�50% confluence in
6-well plates or chamber slides were transfected with GeneSilencer� (Gene Therapy System, Inc. San Diego, CA)-trans-fecting agent containing scrambled siRNA (50–100 nM) orsiRNA for target proteins (50–100 nM) in serum-free EBM-2medium according to themanufacturer’s recommendation. Tooptimize conditions for efficient transfection, HPAECs weretransfected with Fl-Luciferase GL2 Duplex siRNA (targetsequence. 5�-CGTACGCGGAATACTTCGA-3�, Dharmacon)as a positive control. After 3 h of transfection, 1 ml of freshcomplete EGM-2 medium containing 10% FBS was added, andcells were cultured for additional 72 h and analyzed for mRNAlevels by real time PCR or protein expression by Western blot-ting. Scrambled siRNA control or siRNA-transfected cells weresubjected to scratch or wound healing experiments asdescribed earlier.Statistical Analysis—Analysis of variance and Student-New-
man-Keul’s test were used to compare the means of two ormore different treatment groups. The level of significance wasset to p� 0.05 unless otherwise stated. Results are expressed asmean � S.E.
RESULTS
S1P, but Not LPA or Ceramide-1-phosphate, StimulatesMigration of HPAECs—To determine the role of exogenousS1P, dihydro-S1P, LPA, or ceramide 1-phosphate in EC motil-ity, we employed two complimentary assays of wound closurethat measure the non-directional, chemokinetic movement.Among the four lipid phosphates tested, S1P was the mostpotent stimulator of HPAECmigration in both the scratch andECIS wound healing assays (Fig. 1, A–C). Furthermore, thewound closure was dose-dependent, with �50% of thewounded area remaining free of cells after exposure to S1P (10nM) for 16 h as compared with �25% with 100 nM S1P (supple-
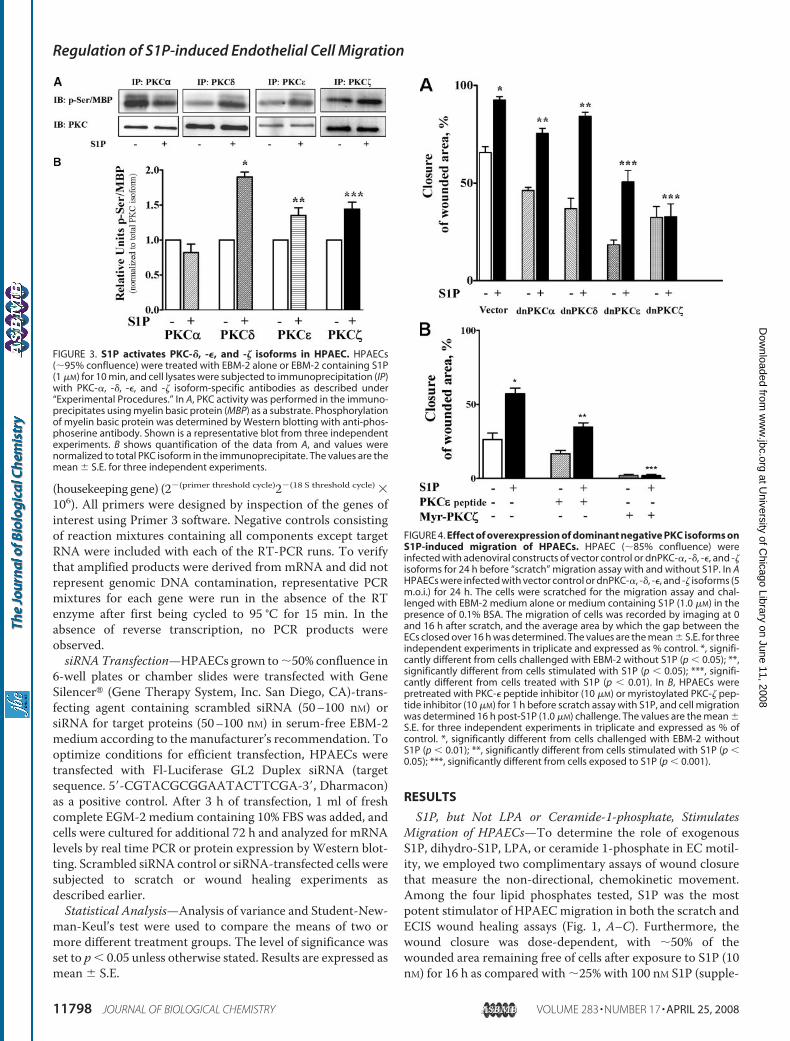
FIGURE 3. S1P activates PKC-�, -�, and -� isoforms in HPAEC. HPAECs(�95% confluence) were treated with EBM-2 alone or EBM-2 containing S1P(1 �M) for 10 min, and cell lysates were subjected to immunoprecipitation (IP)with PKC-�, -�, -�, and -� isoform-specific antibodies as described under“Experimental Procedures.” In A, PKC activity was performed in the immuno-precipitates using myelin basic protein (MBP) as a substrate. Phosphorylationof myelin basic protein was determined by Western blotting with anti-phos-phoserine antibody. Shown is a representative blot from three independentexperiments. B shows quantification of the data from A, and values werenormalized to total PKC isoform in the immunoprecipitate. The values are themean � S.E. for three independent experiments.
FIGURE 4. Effect of overexpression of dominant negative PKC isoforms onS1P-induced migration of HPAECs. HPAEC (�85% confluence) wereinfected with adenoviral constructs of vector control or dnPKC-�, -�, -�, and -�isoforms for 24 h before “scratch” migration assay with and without S1P. In AHPAECs were infected with vector control or dnPKC-�, -�, -�, and -� isoforms (5m.o.i.) for 24 h. The cells were scratched for the migration assay and chal-lenged with EBM-2 medium alone or medium containing S1P (1.0 �M) in thepresence of 0.1% BSA. The migration of cells was recorded by imaging at 0and 16 h after scratch, and the average area by which the gap between theECs closed over 16 h was determined. The values are the mean � S.E. for threeindependent experiments in triplicate and expressed as % control. *, signifi-cantly different from cells challenged with EBM-2 without S1P (p � 0.05); **,significantly different from cells stimulated with S1P (p � 0.05); ***, signifi-cantly different from cells treated with S1P (p � 0.01). In B, HPAECs werepretreated with PKC-� peptide inhibitor (10 �M) or myristoylated PKC-� pep-tide inhibitor (10 �M) for 1 h before scratch assay with S1P, and cell migrationwas determined 16 h post-S1P (1.0 �M) challenge. The values are the mean �S.E. for three independent experiments in triplicate and expressed as % ofcontrol. *, significantly different from cells challenged with EBM-2 withoutS1P (p � 0.01); **, significantly different from cells stimulated with S1P (p �0.05); ***, significantly different from cells exposed to S1P (p � 0.001).
Regulation of S1P-induced Endothelial Cell Migration
11798 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 17 • APRIL 25, 2008
at University of C
hicago Library on June 11, 2008 w
ww
.jbc.orgD
ownloaded from
mental Fig. 1,A–D). Our results on S1P-induced cell migrationare in agreement with a previous report (26). In contrast, LPAand ceramide 1-phosphate did not stimulate cell migration sig-nificantly in HPAECs, and this finding differs from an earlierreport on LPA-induced migration of bovine aortic and humanumbilical vein ECs (27).S1P1 Is Required for S1P-induced HPAEC Migration—S1P
exerts its action through G-protein-coupled S1P1–5 receptors(7–9). To determine which S1P receptor subtype(s) is involvedin S1P-induced cellmigration, we first examined the expressionof S1P receptors by real time RT-PCR using specific primersderived from human sequences (supplemental Table 1). Anal-yses of total RNA by real time RT-PCR showed that all the fiveS1P receptors were expressed in the order of S1P1 � S1P3 �S1P2 � S1P4 � S1P5 (supplemental Fig. 1A). Complementingthe mRNA expression profile, Western blot analyses showedS1P1 to be abundantly expressed inHPAECs (supplemental Fig.2B). Because S1P receptors are coupled to heterotrimeric Gproteins (7–9), we examined the effect of treatingHPAECswith100 ng/ml of PTx for 3 h. This significantly blocked basal as wellas S1P-mediated cell migration and wound healing (supple-mental Fig. 2, C and D). These results suggest that S1P1 is the
predominant receptor expressed inHPAECs and the involvement of Giin transducing signals from S1Pligation to its receptors in cellmigration.To obtain evidence that S1P1 is
involved in S1P-induced cell migra-tion and wound healing, we testedthe ability of SB649146, whichreduces constitutively active S1P1and blocks S1P binding (48), onS1P-mediated cell motility. Pre-treatment of HPAECs withSB649146 (10 �M) for 1 h reducedS1P-induced migration (Fig. 2A).Furthermore, SB649146 decreasedthe basal migration of cells in theabsence of added S1P (Fig. 2A). Asimilar effect of SB649146 on S1P-induced EC migration was ob-served in ECIS wound healingassay (Fig. 2B).To further characterize the role
of S1P1 in S1P-mediated cell migra-tion,HPAECswere transfectedwithS1P1 siRNA (100 nM) for 72 h, cellswere wounded and challenged withS1P (1 �M) for 16 h, and migrationwas evaluated. S1P1 siRNA effec-tively down-regulated mRNA ex-pression of S1P1, but not S1P4, andprotein expression of S1P1 (�60%)(Fig. 2C). Furthermore, the S1P-in-duced migration of HPAECs wasalmost completely attenuated byknockdown of S1P1 (Fig. 2D). These
combined results establish the role for S1P1 in regulating S1P-induced cell migration in HPAECs.Involvement of PKC-� and PKC-� in S1P-induced HPAEC
Migration—It has been reported that PKC-� regulates theaction of S1P on ECmotility (49); however, the PKC isoform(s)involved in S1P-induced migration has not been well defined.Analysis of total cell lysates by Western blotting revealed thatPKC-�, -�, -�, and -� are the predominant isoforms present inHPAECs (results not shown). To investigate which isoforms ofPKC are activated by S1P, serum-deprived HPAECs were chal-lenged with S1P (1 �M), and activation of PKC isoforms wasdetermined by immunoprecipitating each PKC isoform sepa-rately and measuring the activity. Immunoprecipitates ofPKC-�, -�, and -�, but not PKC-�, from S1P-challenged cellsexhibited enhanced serine phosphorylation of myelin basicprotein (Fig. 3). Next, we investigated the role of PKC-�, -�, -�,and -� isoforms in S1P-stimulated HPAECmigration by infect-ing cells with dominant negative isoforms of each PKC. Infec-tion of HPAECs with adenoviral vectors encoding fordnPKC-�, -�, -�, and -� (5 m.o.i.) for 24 h resulted in overex-pression of each protein (�5-fold) (results not shown). Over-expression of dnPKC-�, and -� isoforms (5 m.o.i.), but not
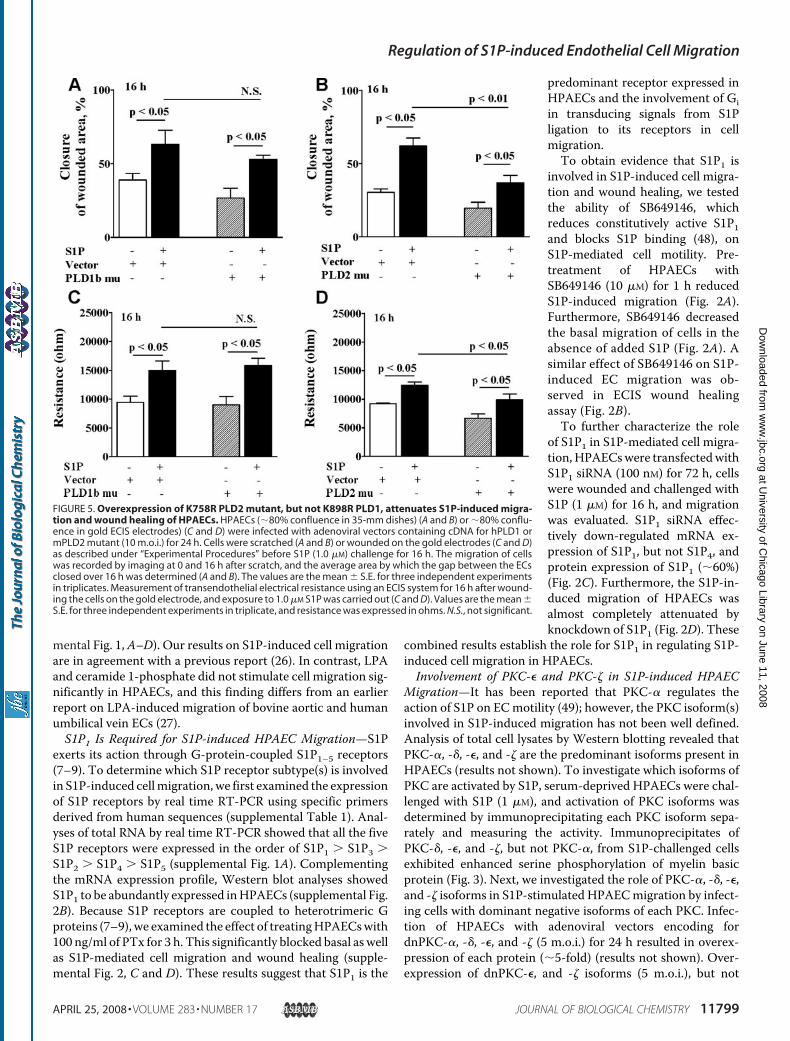
FIGURE 5. Overexpression of K758R PLD2 mutant, but not K898R PLD1, attenuates S1P-induced migra-tion and wound healing of HPAECs. HPAECs (�80% confluence in 35-mm dishes) (A and B) or �80% conflu-ence in gold ECIS electrodes) (C and D) were infected with adenoviral vectors containing cDNA for hPLD1 ormPLD2 mutant (10 m.o.i.) for 24 h. Cells were scratched (A and B) or wounded on the gold electrodes (C and D)as described under “Experimental Procedures” before S1P (1.0 �M) challenge for 16 h. The migration of cellswas recorded by imaging at 0 and 16 h after scratch, and the average area by which the gap between the ECsclosed over 16 h was determined (A and B). The values are the mean � S.E. for three independent experimentsin triplicates. Measurement of transendothelial electrical resistance using an ECIS system for 16 h after wound-ing the cells on the gold electrode, and exposure to 1.0 �M S1P was carried out (C and D). Values are the mean �S.E. for three independent experiments in triplicate, and resistance was expressed in ohms. N.S., not significant.
Regulation of S1P-induced Endothelial Cell Migration
APRIL 25, 2008 • VOLUME 283 • NUMBER 17 JOURNAL OF BIOLOGICAL CHEMISTRY 11799
at University of C
hicago Library on June 11, 2008 w
ww
.jbc.orgD
ownloaded from
PKC-� and -� isoforms, significantly reduced S1P-induced cellmigration compared with vector-infected cells (Fig. 4A). Fur-thermore, the effect of dnPKC-� and -� overexpression on S1P-induced wound closure was dependent onmultiplicity of infec-tion with maximum inhibition observed at 10 m.o.i. (data notshown). The role of PKC-� and -� in cell migration was alsotested using specific peptide inhibitors and is shown in Fig. 4B.Pretreatment of HPAECs with either PKC-� peptide inhibitor(10 �M) or myristoylated PKC-� peptide (10 �M) attenuatedS1P-induced cell migration. These results establish that S1P-induced wound closure is dependent on activation of PKC-�and -� isoforms in HPAECs.S1P Activates PLD1 and PLD2 in HPAECs—PLD is activated
by S1P in bronchial epithelial cells and fibroblasts (22), and wehave previously showed that PLD2 regulated the LPA-mediatedmigration of rat fibroblasts (33). Here, we investigated the roleof PLD isoforms in S1P-induced cell migration. Cell lysatesfrom HPAECs infected with adenoviral vectors encoding wildtype and catalytically inactive mutants of hPLD1 and mPLD2(10 m.o.i.) for 24 h showed increased protein expression (sup-plemental Fig. 3, A and B). The functional significance of wildtype and mutants of hPLD1 and mPLD2 overexpression wastested by quantifying the accumulation of [32P]PBt in the pres-ence of 1-butanol (32). In HPAECs infected with wild typehPLD1 andmPLD2constructs, S1P stimulated accumulation of[32P]PBt. In vector-infected cells, S1P stimulated [32P]PBt accu-mulation by 2.5-fold (vehicle, 1164 � 76 dpm; S1P, 2588 � 132dpm). Overexpression of hPLD1 (vehicle, 850 � 89 dpm; S1P,5061 � 280 dpm) or mPLD2 (vehicle, 896 � 74 dpm; S1P,7656 � 410 dpm) enhanced S1P-mediated [32P]PBt accumula-tion compared with vector controls (supplemental Fig. 3,C and
D). However, overexpression of thecatalytically inactive mutants ofhPLD1-K898R (vehicle, 766 � 171dpm; S1P, 1272 � 61 dpm) andmPLD2-K758R (vehicle, 1140 �136 dpm; S1P, 1543 � 116 dpm)partially blocked S1P-induced[32P]PBt formation without affect-ing the basal activity (supplementalFig. 3,C andD). To examine the roleof S1P1 in S1P-induced PLD activa-tion, we investigated the effects ofSB 649146 and PTx on S1P-inducedPLD activity. SB649146, attenuatedPLD activation by S1P (vehicle,1267 � 123 dpm; S1P, 4330 � 286dpm; SB649146 � vehicle, 1229 �92 dpm; SB649146 � S1P, 2640 �568 dpm) showing the involvementof S1P1 receptor (supplemental Fig.3E). Also, S1P activation oftransphosphorylation of [32P]PA to1-butanol was PTx-sensitive (vehi-cle, 881 � 146 dpm; S1P, 1889 � 77dpm; PTx� vehicle, 810� 169 dpm;PTx � S1P, 1177 � 146 dpm), indi-cating coupling of S1P1 to Gi (sup-
plemental Fig. 3F). This supports the hypothesis that S1P-in-duced activation of PLD is via S1P1 coupled to Gi in HPAECs.PLD2, but Not PLD1, Is Involved in S1P-induced HPAEC
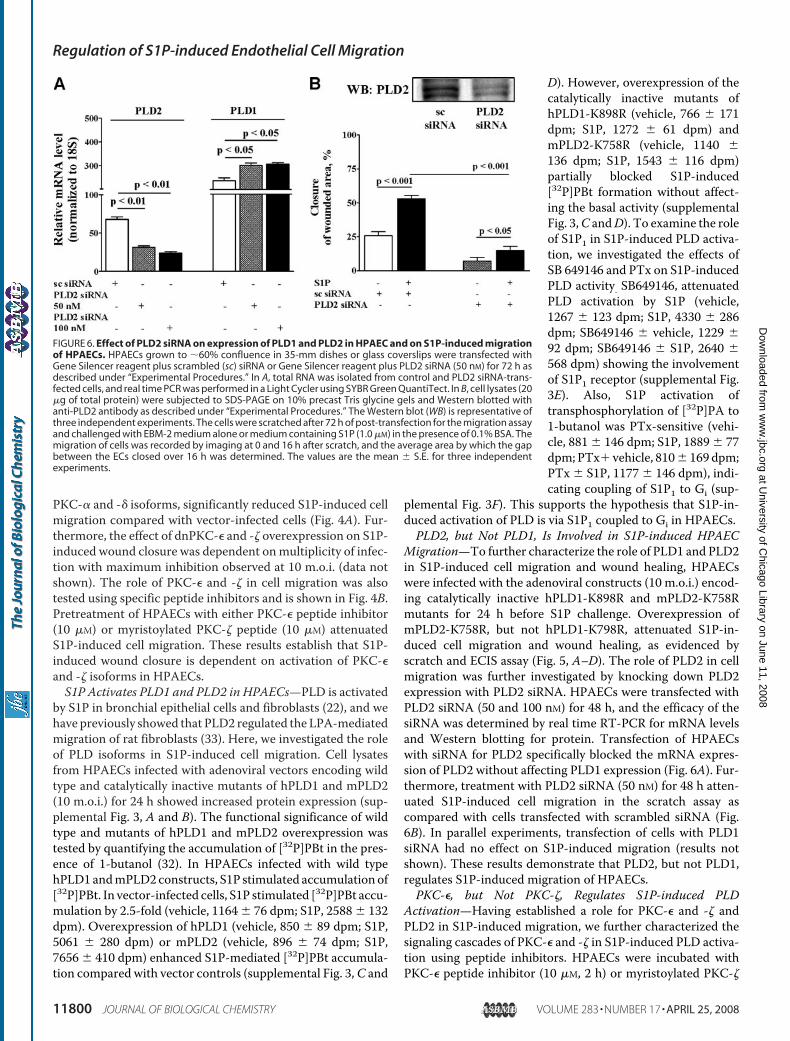
Migration—To further characterize the role of PLD1 and PLD2in S1P-induced cell migration and wound healing, HPAECswere infected with the adenoviral constructs (10 m.o.i.) encod-ing catalytically inactive hPLD1-K898R and mPLD2-K758Rmutants for 24 h before S1P challenge. Overexpression ofmPLD2-K758R, but not hPLD1-K798R, attenuated S1P-in-duced cell migration and wound healing, as evidenced byscratch and ECIS assay (Fig. 5, A–D). The role of PLD2 in cellmigration was further investigated by knocking down PLD2expression with PLD2 siRNA. HPAECs were transfected withPLD2 siRNA (50 and 100 nM) for 48 h, and the efficacy of thesiRNA was determined by real time RT-PCR for mRNA levelsand Western blotting for protein. Transfection of HPAECswith siRNA for PLD2 specifically blocked the mRNA expres-sion of PLD2 without affecting PLD1 expression (Fig. 6A). Fur-thermore, treatment with PLD2 siRNA (50 nM) for 48 h atten-uated S1P-induced cell migration in the scratch assay ascompared with cells transfected with scrambled siRNA (Fig.6B). In parallel experiments, transfection of cells with PLD1siRNA had no effect on S1P-induced migration (results notshown). These results demonstrate that PLD2, but not PLD1,regulates S1P-induced migration of HPAECs.PKC-�, but Not PKC-�, Regulates S1P-induced PLD
Activation—Having established a role for PKC-� and -� andPLD2 in S1P-induced migration, we further characterized thesignaling cascades of PKC-� and -� in S1P-induced PLD activa-tion using peptide inhibitors. HPAECs were incubated withPKC-� peptide inhibitor (10 �M, 2 h) or myristoylated PKC-�
FIGURE 6. Effect of PLD2 siRNA on expression of PLD1 and PLD2 in HPAEC and on S1P-induced migrationof HPAECs. HPAECs grown to �60% confluence in 35-mm dishes or glass coverslips were transfected withGene Silencer reagent plus scrambled (sc) siRNA or Gene Silencer reagent plus PLD2 siRNA (50 nM) for 72 h asdescribed under “Experimental Procedures.” In A, total RNA was isolated from control and PLD2 siRNA-trans-fected cells, and real time PCR was performed in a Light Cycler using SYBR Green QuantiTect. In B, cell lysates (20�g of total protein) were subjected to SDS-PAGE on 10% precast Tris glycine gels and Western blotted withanti-PLD2 antibody as described under “Experimental Procedures.” The Western blot (WB) is representative ofthree independent experiments. The cells were scratched after 72 h of post-transfection for the migration assayand challenged with EBM-2 medium alone or medium containing S1P (1.0 �M) in the presence of 0.1% BSA. Themigration of cells was recorded by imaging at 0 and 16 h after scratch, and the average area by which the gapbetween the ECs closed over 16 h was determined. The values are the mean � S.E. for three independentexperiments.
Regulation of S1P-induced Endothelial Cell Migration
11800 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 17 • APRIL 25, 2008
at University of C
hicago Library on June 11, 2008 w
ww
.jbc.orgD
ownloaded from
peptide (10 �M, 2 h). In parallelexperiments, cells were labeledwith [32P]orthophosphate over-night before stimulation with S1Pfor activation of PLD. Pretreatmentof cells with either PKC-� peptideinhibitor or myristoylated PKC-�peptide attenuated translocation ofthe respective isoform to cellperiphery as determined by immu-nofluorescence microscopy (sup-plemental Fig. 4, A and B). Addi-tionally, as shown in supplementalFig. 4, C and D, the S1P stimulationof [32P]PBt formation by treatmentwith S1P for 5 min was blocked bydnPKC-� and PKC-� peptide inhib-itor but not by dnPKC-� and myris-toylated PKC-� peptide. Theseresults demonstrate that S1P acti-vated PKC-� upstream of PLD andthat PKC-� is downstreamof PLD inHPAECs.PLD2 siRNA Attenuates S1P-in-
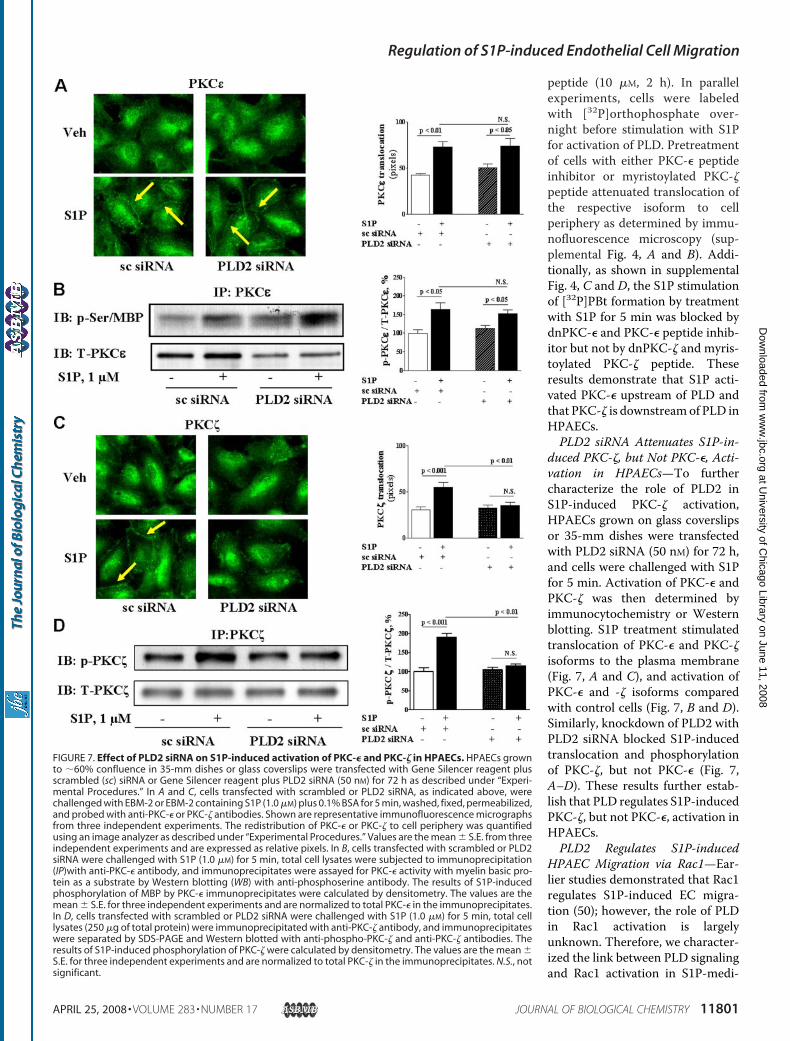
duced PKC-�, but Not PKC-�, Acti-vation in HPAECs—To furthercharacterize the role of PLD2 inS1P-induced PKC-� activation,HPAECs grown on glass coverslipsor 35-mm dishes were transfectedwith PLD2 siRNA (50 nM) for 72 h,and cells were challenged with S1Pfor 5 min. Activation of PKC-� andPKC-� was then determined byimmunocytochemistry or Westernblotting. S1P treatment stimulatedtranslocation of PKC-� and PKC-�isoforms to the plasma membrane(Fig. 7, A and C), and activation ofPKC-� and -� isoforms comparedwith control cells (Fig. 7, B and D).Similarly, knockdown of PLD2 withPLD2 siRNA blocked S1P-inducedtranslocation and phosphorylationof PKC-�, but not PKC-� (Fig. 7,A–D). These results further estab-lish that PLD regulates S1P-inducedPKC-�, but not PKC-�, activation inHPAECs.PLD2 Regulates S1P-induced
HPAEC Migration via Rac1—Ear-lier studies demonstrated that Rac1regulates S1P-induced EC migra-tion (50); however, the role of PLDin Rac1 activation is largelyunknown. Therefore, we character-ized the link between PLD signalingand Rac1 activation in S1P-medi-
FIGURE 7. Effect of PLD2 siRNA on S1P-induced activation of PKC-� and PKC-� in HPAECs. HPAECs grownto �60% confluence in 35-mm dishes or glass coverslips were transfected with Gene Silencer reagent plusscrambled (sc) siRNA or Gene Silencer reagent plus PLD2 siRNA (50 nM) for 72 h as described under “Experi-mental Procedures.” In A and C, cells transfected with scrambled or PLD2 siRNA, as indicated above, werechallenged with EBM-2 or EBM-2 containing S1P (1.0 �M) plus 0.1% BSA for 5 min, washed, fixed, permeabilized,and probed with anti-PKC-� or PKC-� antibodies. Shown are representative immunofluorescence micrographsfrom three independent experiments. The redistribution of PKC-� or PKC-� to cell periphery was quantifiedusing an image analyzer as described under “Experimental Procedures.” Values are the mean � S.E. from threeindependent experiments and are expressed as relative pixels. In B, cells transfected with scrambled or PLD2siRNA were challenged with S1P (1.0 �M) for 5 min, total cell lysates were subjected to immunoprecipitation(IP)with anti-PKC-� antibody, and immunoprecipitates were assayed for PKC-� activity with myelin basic pro-tein as a substrate by Western blotting (WB) with anti-phosphoserine antibody. The results of S1P-inducedphosphorylation of MBP by PKC-� immunoprecipitates were calculated by densitometry. The values are themean � S.E. for three independent experiments and are normalized to total PKC-� in the immunoprecipitates.In D, cells transfected with scrambled or PLD2 siRNA were challenged with S1P (1.0 �M) for 5 min, total celllysates (250 �g of total protein) were immunoprecipitated with anti-PKC-� antibody, and immunoprecipitateswere separated by SDS-PAGE and Western blotted with anti-phospho-PKC-� and anti-PKC-� antibodies. Theresults of S1P-induced phosphorylation of PKC-� were calculated by densitometry. The values are the mean �S.E. for three independent experiments and are normalized to total PKC-� in the immunoprecipitates. N.S., notsignificant.
Regulation of S1P-induced Endothelial Cell Migration
APRIL 25, 2008 • VOLUME 283 • NUMBER 17 JOURNAL OF BIOLOGICAL CHEMISTRY 11801
at University of C
hicago Library on June 11, 2008 w
ww
.jbc.orgD
ownloaded from
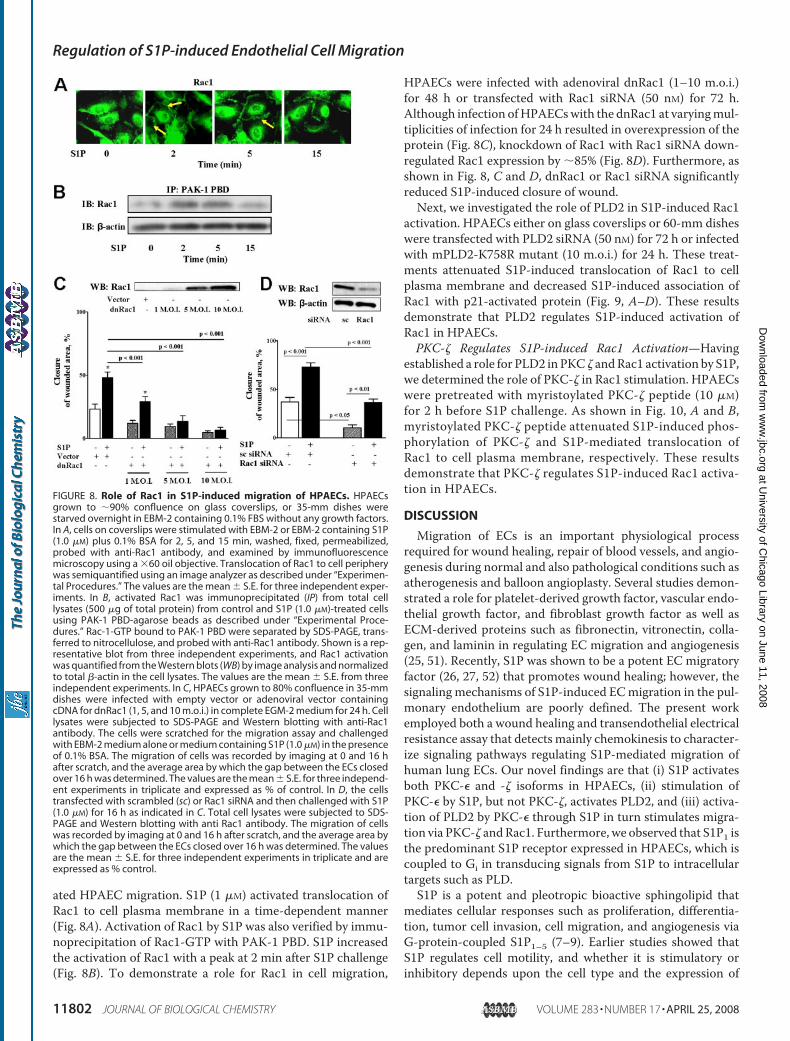
ated HPAEC migration. S1P (1 �M) activated translocation ofRac1 to cell plasma membrane in a time-dependent manner(Fig. 8A). Activation of Rac1 by S1P was also verified by immu-noprecipitation of Rac1-GTP with PAK-1 PBD. S1P increasedthe activation of Rac1 with a peak at 2 min after S1P challenge(Fig. 8B). To demonstrate a role for Rac1 in cell migration,
HPAECs were infected with adenoviral dnRac1 (1–10 m.o.i.)for 48 h or transfected with Rac1 siRNA (50 nM) for 72 h.Although infection ofHPAECswith the dnRac1 at varyingmul-tiplicities of infection for 24 h resulted in overexpression of theprotein (Fig. 8C), knockdown of Rac1 with Rac1 siRNA down-regulated Rac1 expression by �85% (Fig. 8D). Furthermore, asshown in Fig. 8, C and D, dnRac1 or Rac1 siRNA significantlyreduced S1P-induced closure of wound.Next, we investigated the role of PLD2 in S1P-induced Rac1
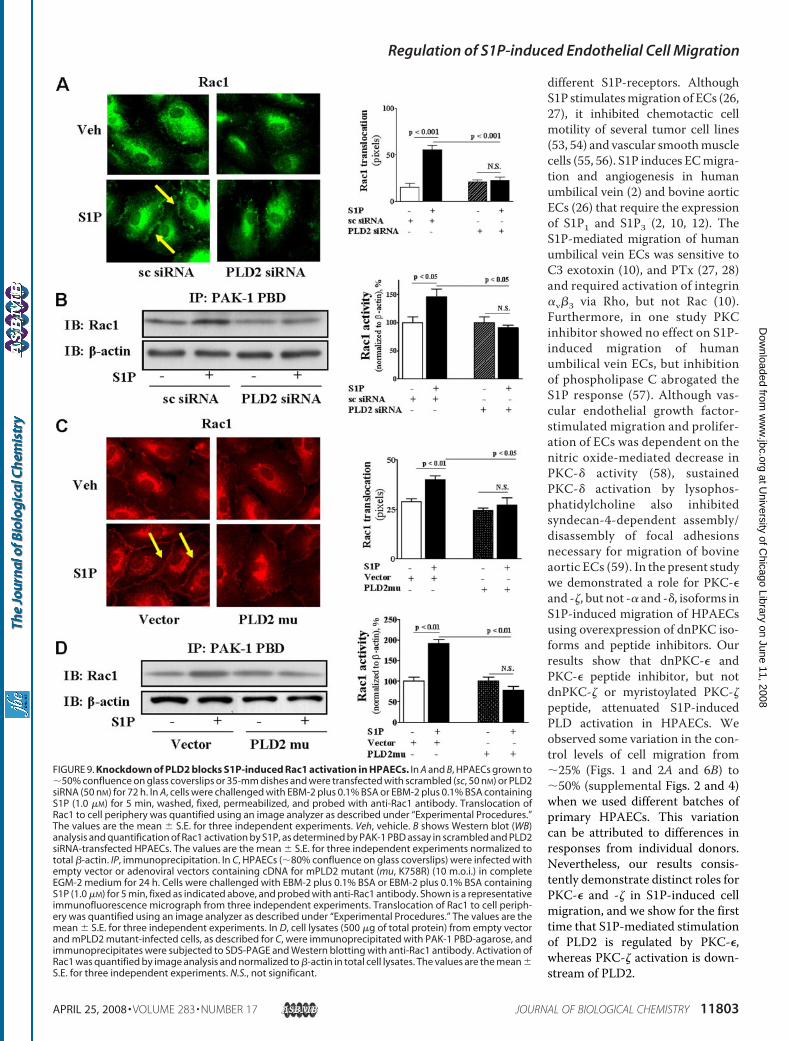
activation. HPAECs either on glass coverslips or 60-mm disheswere transfected with PLD2 siRNA (50 nM) for 72 h or infectedwith mPLD2-K758R mutant (10 m.o.i.) for 24 h. These treat-ments attenuated S1P-induced translocation of Rac1 to cellplasma membrane and decreased S1P-induced association ofRac1 with p21-activated protein (Fig. 9, A–D). These resultsdemonstrate that PLD2 regulates S1P-induced activation ofRac1 in HPAECs.PKC-� Regulates S1P-induced Rac1 Activation—Having
established a role for PLD2 inPKC � andRac1 activation by S1P,we determined the role of PKC-� in Rac1 stimulation. HPAECswere pretreated with myristoylated PKC-� peptide (10 �M)for 2 h before S1P challenge. As shown in Fig. 10, A and B,myristoylated PKC-� peptide attenuated S1P-induced phos-phorylation of PKC-� and S1P-mediated translocation ofRac1 to cell plasma membrane, respectively. These resultsdemonstrate that PKC-� regulates S1P-induced Rac1 activa-tion in HPAECs.
DISCUSSION
Migration of ECs is an important physiological processrequired for wound healing, repair of blood vessels, and angio-genesis during normal and also pathological conditions such asatherogenesis and balloon angioplasty. Several studies demon-strated a role for platelet-derived growth factor, vascular endo-thelial growth factor, and fibroblast growth factor as well asECM-derived proteins such as fibronectin, vitronectin, colla-gen, and laminin in regulating EC migration and angiogenesis(25, 51). Recently, S1P was shown to be a potent EC migratoryfactor (26, 27, 52) that promotes wound healing; however, thesignaling mechanisms of S1P-induced ECmigration in the pul-monary endothelium are poorly defined. The present workemployed both a wound healing and transendothelial electricalresistance assay that detects mainly chemokinesis to character-ize signaling pathways regulating S1P-mediated migration ofhuman lung ECs. Our novel findings are that (i) S1P activatesboth PKC-� and -� isoforms in HPAECs, (ii) stimulation ofPKC-� by S1P, but not PKC-�, activates PLD2, and (iii) activa-tion of PLD2 by PKC-� through S1P in turn stimulates migra-tion via PKC-� andRac1. Furthermore, we observed that S1P1 isthe predominant S1P receptor expressed in HPAECs, which iscoupled to Gi in transducing signals from S1P to intracellulartargets such as PLD.S1P is a potent and pleotropic bioactive sphingolipid that
mediates cellular responses such as proliferation, differentia-tion, tumor cell invasion, cell migration, and angiogenesis viaG-protein-coupled S1P1–5 (7–9). Earlier studies showed thatS1P regulates cell motility, and whether it is stimulatory orinhibitory depends upon the cell type and the expression of
FIGURE 8. Role of Rac1 in S1P-induced migration of HPAECs. HPAECsgrown to �90% confluence on glass coverslips, or 35-mm dishes werestarved overnight in EBM-2 containing 0.1% FBS without any growth factors.In A, cells on coverslips were stimulated with EBM-2 or EBM-2 containing S1P(1.0 �M) plus 0.1% BSA for 2, 5, and 15 min, washed, fixed, permeabilized,probed with anti-Rac1 antibody, and examined by immunofluorescencemicroscopy using a �60 oil objective. Translocation of Rac1 to cell peripherywas semiquantified using an image analyzer as described under “Experimen-tal Procedures.” The values are the mean � S.E. for three independent exper-iments. In B, activated Rac1 was immunoprecipitated (IP) from total celllysates (500 �g of total protein) from control and S1P (1.0 �M)-treated cellsusing PAK-1 PBD-agarose beads as described under “Experimental Proce-dures.” Rac-1-GTP bound to PAK-1 PBD were separated by SDS-PAGE, trans-ferred to nitrocellulose, and probed with anti-Rac1 antibody. Shown is a rep-resentative blot from three independent experiments, and Rac1 activationwas quantified from the Western blots (WB) by image analysis and normalizedto total �-actin in the cell lysates. The values are the mean � S.E. from threeindependent experiments. In C, HPAECs grown to 80% confluence in 35-mmdishes were infected with empty vector or adenoviral vector containingcDNA for dnRac1 (1, 5, and 10 m.o.i.) in complete EGM-2 medium for 24 h. Celllysates were subjected to SDS-PAGE and Western blotting with anti-Rac1antibody. The cells were scratched for the migration assay and challengedwith EBM-2 medium alone or medium containing S1P (1.0 �M) in the presenceof 0.1% BSA. The migration of cells was recorded by imaging at 0 and 16 hafter scratch, and the average area by which the gap between the ECs closedover 16 h was determined. The values are the mean � S.E. for three independ-ent experiments in triplicate and expressed as % of control. In D, the cellstransfected with scrambled (sc) or Rac1 siRNA and then challenged with S1P(1.0 �M) for 16 h as indicated in C. Total cell lysates were subjected to SDS-PAGE and Western blotting with anti Rac1 antibody. The migration of cellswas recorded by imaging at 0 and 16 h after scratch, and the average area bywhich the gap between the ECs closed over 16 h was determined. The valuesare the mean � S.E. for three independent experiments in triplicate and areexpressed as % control.
Regulation of S1P-induced Endothelial Cell Migration
11802 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 17 • APRIL 25, 2008
at University of C
hicago Library on June 11, 2008 w
ww
.jbc.orgD
ownloaded from
different S1P-receptors. AlthoughS1P stimulatesmigration of ECs (26,27), it inhibited chemotactic cellmotility of several tumor cell lines(53, 54) and vascular smoothmusclecells (55, 56). S1P induces ECmigra-tion and angiogenesis in humanumbilical vein (2) and bovine aorticECs (26) that require the expressionof S1P1 and S1P3 (2, 10, 12). TheS1P-mediated migration of humanumbilical vein ECs was sensitive toC3 exotoxin (10), and PTx (27, 28)and required activation of integrin�v�3 via Rho, but not Rac (10).Furthermore, in one study PKCinhibitor showed no effect on S1P-induced migration of humanumbilical vein ECs, but inhibitionof phospholipase C abrogated theS1P response (57). Although vas-cular endothelial growth factor-stimulated migration and prolifer-ation of ECs was dependent on thenitric oxide-mediated decrease inPKC-� activity (58), sustainedPKC-� activation by lysophos-phatidylcholine also inhibitedsyndecan-4-dependent assembly/disassembly of focal adhesionsnecessary for migration of bovineaortic ECs (59). In the present studywe demonstrated a role for PKC-�and -�, but not -� and -�, isoforms inS1P-induced migration of HPAECsusing overexpression of dnPKC iso-forms and peptide inhibitors. Ourresults show that dnPKC-� andPKC-� peptide inhibitor, but notdnPKC-� or myristoylated PKC-�peptide, attenuated S1P-inducedPLD activation in HPAECs. Weobserved some variation in the con-trol levels of cell migration from�25% (Figs. 1 and 2A and 6B) to�50% (supplemental Figs. 2 and 4)when we used different batches ofprimary HPAECs. This variationcan be attributed to differences inresponses from individual donors.Nevertheless, our results consis-tently demonstrate distinct roles forPKC-� and -� in S1P-induced cellmigration, and we show for the firsttime that S1P-mediated stimulationof PLD2 is regulated by PKC-�,whereas PKC-� activation is down-stream of PLD2.
FIGURE 9. Knockdown of PLD2 blocks S1P-induced Rac1 activation in HPAECs. In A and B, HPAECs grown to�50% confluence on glass coverslips or 35-mm dishes and were transfected with scrambled (sc, 50 nM) or PLD2siRNA (50 nM) for 72 h. In A, cells were challenged with EBM-2 plus 0.1% BSA or EBM-2 plus 0.1% BSA containingS1P (1.0 �M) for 5 min, washed, fixed, permeabilized, and probed with anti-Rac1 antibody. Translocation ofRac1 to cell periphery was quantified using an image analyzer as described under “Experimental Procedures.”The values are the mean � S.E. for three independent experiments. Veh, vehicle. B shows Western blot (WB)analysis and quantification of Rac1 activation by S1P, as determined by PAK-1 PBD assay in scrambled and PLD2siRNA-transfected HPAECs. The values are the mean � S.E. for three independent experiments normalized tototal �-actin. IP, immunoprecipitation. In C, HPAECs (�80% confluence on glass coverslips) were infected withempty vector or adenoviral vectors containing cDNA for mPLD2 mutant (mu, K758R) (10 m.o.i.) in completeEGM-2 medium for 24 h. Cells were challenged with EBM-2 plus 0.1% BSA or EBM-2 plus 0.1% BSA containingS1P (1.0 �M) for 5 min, fixed as indicated above, and probed with anti-Rac1 antibody. Shown is a representativeimmunofluorescence micrograph from three independent experiments. Translocation of Rac1 to cell periph-ery was quantified using an image analyzer as described under “Experimental Procedures.” The values are themean � S.E. for three independent experiments. In D, cell lysates (500 �g of total protein) from empty vectorand mPLD2 mutant-infected cells, as described for C, were immunoprecipitated with PAK-1 PBD-agarose, andimmunoprecipitates were subjected to SDS-PAGE and Western blotting with anti-Rac1 antibody. Activation ofRac1 was quantified by image analysis and normalized to �-actin in total cell lysates. The values are the mean �S.E. for three independent experiments. N.S., not significant.
Regulation of S1P-induced Endothelial Cell Migration
APRIL 25, 2008 • VOLUME 283 • NUMBER 17 JOURNAL OF BIOLOGICAL CHEMISTRY 11803
at University of C
hicago Library on June 11, 2008 w
ww
.jbc.orgD
ownloaded from
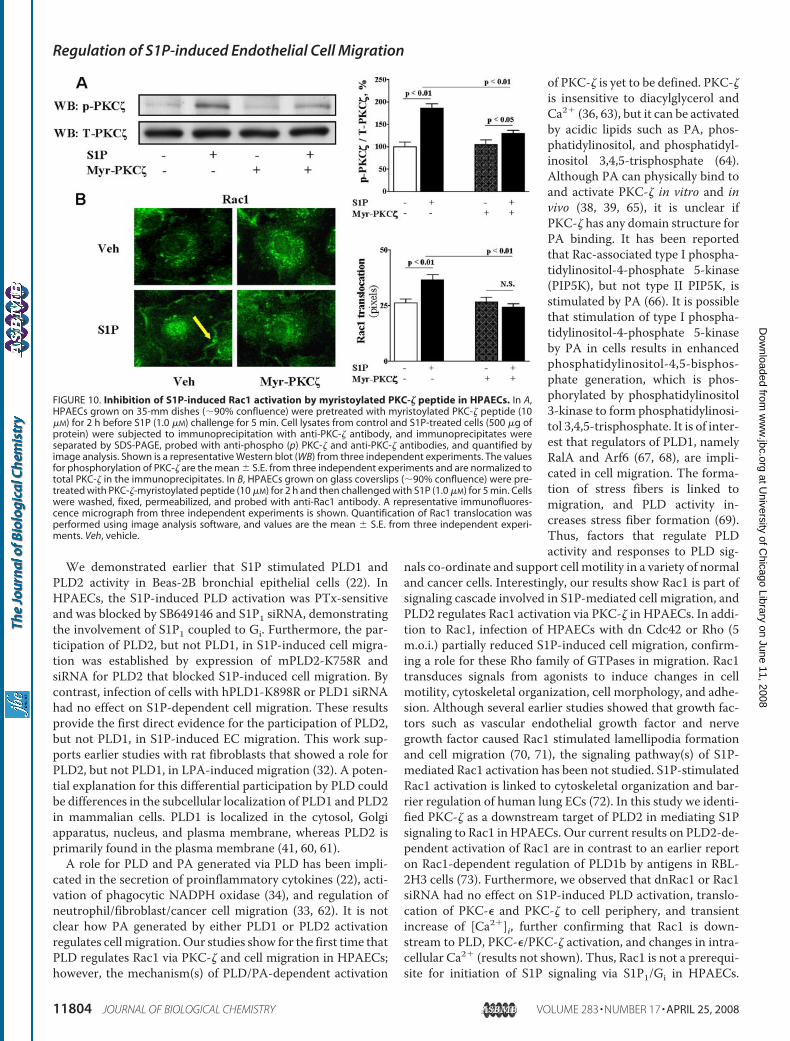
We demonstrated earlier that S1P stimulated PLD1 andPLD2 activity in Beas-2B bronchial epithelial cells (22). InHPAECs, the S1P-induced PLD activation was PTx-sensitiveand was blocked by SB649146 and S1P1 siRNA, demonstratingthe involvement of S1P1 coupled to Gi. Furthermore, the par-ticipation of PLD2, but not PLD1, in S1P-induced cell migra-tion was established by expression of mPLD2-K758R andsiRNA for PLD2 that blocked S1P-induced cell migration. Bycontrast, infection of cells with hPLD1-K898R or PLD1 siRNAhad no effect on S1P-dependent cell migration. These resultsprovide the first direct evidence for the participation of PLD2,but not PLD1, in S1P-induced EC migration. This work sup-ports earlier studies with rat fibroblasts that showed a role forPLD2, but not PLD1, in LPA-induced migration (32). A poten-tial explanation for this differential participation by PLD couldbe differences in the subcellular localization of PLD1 and PLD2in mammalian cells. PLD1 is localized in the cytosol, Golgiapparatus, nucleus, and plasma membrane, whereas PLD2 isprimarily found in the plasma membrane (41, 60, 61).A role for PLD and PA generated via PLD has been impli-
cated in the secretion of proinflammatory cytokines (22), acti-vation of phagocytic NADPH oxidase (34), and regulation ofneutrophil/fibroblast/cancer cell migration (33, 62). It is notclear how PA generated by either PLD1 or PLD2 activationregulates cell migration. Our studies show for the first time thatPLD regulates Rac1 via PKC-� and cell migration in HPAECs;however, the mechanism(s) of PLD/PA-dependent activation
of PKC-� is yet to be defined. PKC-�is insensitive to diacylglycerol andCa2� (36, 63), but it can be activatedby acidic lipids such as PA, phos-phatidylinositol, and phosphatidyl-inositol 3,4,5-trisphosphate (64).Although PA can physically bind toand activate PKC-� in vitro and invivo (38, 39, 65), it is unclear ifPKC-� has any domain structure forPA binding. It has been reportedthat Rac-associated type I phospha-tidylinositol-4-phosphate 5-kinase(PIP5K), but not type II PIP5K, isstimulated by PA (66). It is possiblethat stimulation of type I phospha-tidylinositol-4-phosphate 5-kinaseby PA in cells results in enhancedphosphatidylinositol-4,5-bisphos-phate generation, which is phos-phorylated by phosphatidylinositol3-kinase to form phosphatidylinosi-tol 3,4,5-trisphosphate. It is of inter-est that regulators of PLD1, namelyRalA and Arf6 (67, 68), are impli-cated in cell migration. The forma-tion of stress fibers is linked tomigration, and PLD activity in-creases stress fiber formation (69).Thus, factors that regulate PLDactivity and responses to PLD sig-
nals co-ordinate and support cell motility in a variety of normaland cancer cells. Interestingly, our results show Rac1 is part ofsignaling cascade involved in S1P-mediated cell migration, andPLD2 regulates Rac1 activation via PKC-� in HPAECs. In addi-tion to Rac1, infection of HPAECs with dn Cdc42 or Rho (5m.o.i.) partially reduced S1P-induced cell migration, confirm-ing a role for these Rho family of GTPases in migration. Rac1transduces signals from agonists to induce changes in cellmotility, cytoskeletal organization, cell morphology, and adhe-sion. Although several earlier studies showed that growth fac-tors such as vascular endothelial growth factor and nervegrowth factor caused Rac1 stimulated lamellipodia formationand cell migration (70, 71), the signaling pathway(s) of S1P-mediated Rac1 activation has been not studied. S1P-stimulatedRac1 activation is linked to cytoskeletal organization and bar-rier regulation of human lung ECs (72). In this study we identi-fied PKC-� as a downstream target of PLD2 in mediating S1Psignaling to Rac1 in HPAECs. Our current results on PLD2-de-pendent activation of Rac1 are in contrast to an earlier reporton Rac1-dependent regulation of PLD1b by antigens in RBL-2H3 cells (73). Furthermore, we observed that dnRac1 or Rac1siRNA had no effect on S1P-induced PLD activation, translo-cation of PKC-� and PKC-� to cell periphery, and transientincrease of [Ca2�]i, further confirming that Rac1 is down-stream to PLD, PKC-�/PKC-� activation, and changes in intra-cellular Ca2� (results not shown). Thus, Rac1 is not a prerequi-site for initiation of S1P signaling via S1P1/Gi in HPAECs.
FIGURE 10. Inhibition of S1P-induced Rac1 activation by myristoylated PKC-� peptide in HPAECs. In A,HPAECs grown on 35-mm dishes (�90% confluence) were pretreated with myristoylated PKC-� peptide (10�M) for 2 h before S1P (1.0 �M) challenge for 5 min. Cell lysates from control and S1P-treated cells (500 �g ofprotein) were subjected to immunoprecipitation with anti-PKC-� antibody, and immunoprecipitates wereseparated by SDS-PAGE, probed with anti-phospho (p) PKC-� and anti-PKC-� antibodies, and quantified byimage analysis. Shown is a representative Western blot (WB) from three independent experiments. The valuesfor phosphorylation of PKC-� are the mean � S.E. from three independent experiments and are normalized tototal PKC-� in the immunoprecipitates. In B, HPAECs grown on glass coverslips (�90% confluence) were pre-treated with PKC-�-myristoylated peptide (10 �M) for 2 h and then challenged with S1P (1.0 �M) for 5 min. Cellswere washed, fixed, permeabilized, and probed with anti-Rac1 antibody. A representative immunofluores-cence micrograph from three independent experiments is shown. Quantification of Rac1 translocation wasperformed using image analysis software, and values are the mean � S.E. from three independent experi-ments. Veh, vehicle.
Regulation of S1P-induced Endothelial Cell Migration
11804 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 17 • APRIL 25, 2008
at University of C
hicago Library on June 11, 2008 w
ww
.jbc.orgD
ownloaded from

Furthermore, S1P-induced transient [Ca2�]i was sensitive toPTx (�85% inhibition) and SB649146 (�80% inhibition) treat-ment, suggesting participation of S1P1 linked to Gi in theresponse (results not shown). Although, we have not studiedthe essential role of [Ca2�]i in endothelial cell motility, studiescarried out in HUVECs indicate that [Ca2�]i signal is linked toS1P-induced cell migration via tyrosine phosphorylation offocal adhesion kinase (2, 57).S1P-induced cell migration is not completely attenuated by
blocking PLD2 with catalytically inactive mutant/siRNA (Figs.5B and 6B), which may reflect an incomplete inhibition ofPLD2.However, we predict that additional pathways independ-ent of PLD2 might be involved in mediating cell motility inHPAECs. For example, in HPAECs S1P also promotes Tiam1/Rac1 activation via phosphatidylinositol 3-kinase (PI3K) (74),suggesting a potential role for PI3K signaling in cell motility.Earlier, we demonstrated a role for phosphatidylinositol 3-ki-nase (PI3K) in S1P-induced EC migration (75); however, it isunclear if PI3K signals via PKC-� and/or Rac1 in cell migration.Interestingly, blocking phosphatidylinositol 3-kinase withLY294002 (1–25 �M) did not attenuate S1P-induced PLD acti-vation, indicating the absence of phosphatidylinositol 3-kinase-dependent activation of PLD signaling by S1P inHPAECs.4 Fur-thermore, specific guanine nucleotide exchange factors couplegrowth factor signaling to the Rho family of GTPases (71, 76).The effect of vascular endothelial growth factor on Rac1-de-pendent motility of human umbilical vein ECs is regulated byVav2 guanine nucleotide exchange factor (GEF) (69), and ourpreliminary results show that S1P-induced Rac1 activation ispartly dependent onTiam1, anotherGEF for Rac1.4 However, therole of Tiam1 in PLD2-dependent regulation of Rac1 via PKC-� isunclear, and future studies will address the role of guanine nucle-otide exchange factors in S1P-induced cell motility.S1P is a natural component of plasma and is present in nM to
�M levels (7, 46). Although stimulation of human lung ECmigration by S1P is important for normal blood vessel function,S1P also stimulates neovascularization and tumor cell metasta-sis. Precise understanding of S1P-mediated signaling is impor-tant in developing novel therapeutic agents against targets reg-ulating ECmotility. Our identification of PKC-� as an activatorof PLD2 regulating S1P-induced migration and coupling PLD2to Rac1 via PKC-� indicates that PKC-� and PLD2 could bepotential targets for inhibiting excessive angiogenesis.
Acknowledgments—We thank Dr. C. E. Chalfant from the VirginiaCommonwealth University School of Medicine (Richmond, VA) forthe C18:1 ceramide 1-phosphate and Dr. Motoi Ohba from the Insti-tute of Molecular Oncology (Showa University, Japan) for kindly pro-viding aliquots of adenoviral constructs of vector and dominant neg-ative PKC�, -�, -�, -�, and -� isoforms. We thank the services of theUniversity of Iowa Gene Transfer Vector Core, supported in part bythe National Institutes of Health and Roy J. Carver Foundation, forviral amplification and generation of purified dominant negativePKC isoform adenoviral constructs.
REFERENCES1. Zhang, H., Desai, N. N., Olivera, A., Seki, T., Brooker, G., and Spiegel, S.
(1991) J. Cell Biol. 114, 155–1672. Lee, M. J., Thangada, S., Claffey, K. P., Ancellin, N., Liu, C. H., Kluk, M.,
Volpi, M., Sha’afi, R. I., and Hla, T. (1999) Cell 99, 301–3123. Pyne, S., and Pyne, N. (2000) Pharmacol. Ther. 88, 115–1314. Kluk, M. J., and Hla, T. (2001) Circ. Res. 89, 496–5025. Ozaki, H., Hla, T., and Lee, M. J. (2003) J. Atheroscler. Thromb. 10,
125–1316. Spiegel, S., and Milstien, S. (2003) Biochem. Soc. Trans. 31, 1216–12197. Pyne, S., and Pyne, N. J. (2000) Biochem. J., 349, 385–4028. Spiegel, S., and Milstien, S. (2002) J. Biol. Chem. 277, 25851–258549. Sanchez, T., and Hla, T. (2004) J. Cell. Biochem. 92, 913–92210. Paik, J. H., Chae, S. S., Lee, M. J., Thangada, S., and Hla, T. (2001) J. Biol.
Chem. 276, 11830–1183711. Garcia, J. G., Liu, F., Verin, A. D., Birukova, A., Dechert, M. A., Gerthoffer,
W. T., Bamberg, J. R., and English, D. (2001) J. Clin. Investig. 108, 689–70112. Donati, C., and Bruni, P. (2006) Biochim. Biophys. Acta 1758, 2037–204813. Banno, Y., Takuwa, Y., Akao, Y., Okamoto, H., Osawa, Y., Naganawa, T.,
Nakashima, S., Suh, P. G., and Nozawa, Y. (2001) J. Biol. Chem. 276,35622–35628
14. Pyne, N. J., Waters, C., Moughal, N. A., Sambi, B. S., and Pyne, S. (2003)Biochem. Soc. Trans. 31, 1220–1225
15. Long, J. S., Natarajan, V., Tigyi, G., Pyne, S., and Pyne, N. J. (2006) Pros-taglandins Other Lipid Mediat. 80, 74–78
16. Okamoto, H., Takuwa, N., Yokomizo, T., Sugimoto, N., Sakurada, S.,Shigematsu, H., and Takuwa, Y. (2000)Mol. Cell. Biol. 20, 9247–9261
17. Kon, J., Sato, K., Watanabe, T., Tomura, H., Kuwabara, A., Kimura, T.,Tamama, K., Ishizuka, T., Murata, N., Kanda, T., Kobayashi, I., Ohta, H.,Ui, M., and Okajima, F. (1999) J. Biol. Chem. 274, 23940–23947
18. Gonda, K., Okamoto, H., Takuwa, N., Yatomi, Y., Okazaki, H., Sakurai, T.,Kimura, S., Sillard, R., Harii, K., and Takuwa, Y. (1999) Biochem. J. 337,67–75
19. Graler, M. H., Grosse, R., Kusch, A., Kremmer, E., Gudermann, T., andLipp, M. (2003) J. Cell. Biochem. 89, 507–519
20. Jaillard, C., Harrison, S., Stankoff, B., Aigrot, M. S., Calver, A. R., Duddy,G., Walsh, F. S., Pangalos, M. N., Arimura, N., Kaibuchi, K., Zalc, B., andLubetzki, C. (2005) J. Neurosci. 25, 1459–1469
21. Michel, M. C., Mulders, A. C., Jongsma, M., Alewijnse, A. E., and Peters,S. L. (2007) Acta Paediatr. Suppl. 96, 44–48
22. Cummings, R. J., Parinandi, N. L., Zaiman, A., Wang, L., Usatyuk, P. V.,Garcia, J. G., and Natarajan, V. (2002) J. Biol. Chem. 277, 30227–30235
23. Schwartz, B. M., Hong, G., Morrison, B. H., Wu, W., Baudhuin, L. M.,Xiao, Y. J., Mok, S. C., and Xu, Y. (2001) Gynecol. Oncol. 81, 291–300
24. Kono, M., Mi, Y., Liu, Y., Sasaki, T., Allende, M. L., Wu, Y. P., Yamashita,T., and Proia, R. L. (2004) J. Biol. Chem. 279, 29367–29373
25. Lamalice, L., Le Boeuf, F., and Huot, J. (2007) Circ. Res. 100, 782–79426. Panetti, T. S., Nowlen, J., and Mosher, D. F. (2000) Arterioscler. Thromb.
Vasc. Biol. 20, 1013–101927. Lee, H., Goetzl, E. J., and An, S. (2000) Am. J. Physiol. Cell Physiol. 278,
612–61828. English, D., Welch, Z., Kovala, A. T., Harvey, K., Volpert, O. V., Brindley,
D. N., and Garcia, J. G. (2002) FASEB J. 14, 2255–226529. Lee, M. J., Thangada, S., Paik, J. H., Sapkota, G. P., Ancellin, N., Chae, S. S.,
Wu, M., Morales-Ruiz, M., Sessa, W. C., Alessi, D. R., and Hla, T. (2001)Mol. Cell 8, 693–704
30. Li, Y., Uruno, T., Haudenschild, C., Dudek, S. M., Garcia, J. G., and Zhan,X. (2004) Exp. Cell Res. 298, 107–121
31. Alvarez, S. E., Milstien, S., and Spiegel, S. (2007) Trends Endocrinol.Metab. 18, 300–307
32. Wang, L., Cummings, R., Usatyuk, P., Morris, A., Irani, K., and Natarajan,V. (2002) Biochem. J. 367, 751–760
33. Pilquil, C., Dewald, J., Cherney, A., Gorshkova, I., Tigyi, G., English, D.,Natarajan, V., and Brindley, D. N. (2006) J. Biol. Chem. 281, 38418–38429
34. Cummings, R., Parinandi, N., Wang, L., Usatyuk, P., and Natarajan, V.(2002)Mol. Cell. Biochem. 234–235, 99–109
35. Cross, M. J., Roberts, S., Ridley, A. J., Hodgkin, M. N., Stewart, A.,4 I. Gorshkova and V. Natarajan, unpublished results.
Regulation of S1P-induced Endothelial Cell Migration
APRIL 25, 2008 • VOLUME 283 • NUMBER 17 JOURNAL OF BIOLOGICAL CHEMISTRY 11805
at University of C
hicago Library on June 11, 2008 w
ww
.jbc.orgD
ownloaded from

Claesson-Welsh, L., and Wakelam, M. J. (1996) Curr. Biol. 6, 588–59736. Ono, Y., Fujii, T., Ogita, K., Kikkawa, U., Igarashi, K, and Nishizuka, Y.
(1989) Proc. Natl. Acad. Sci. U. S. A. 86, 3099–310337. Stasek, J. E., Jr., Natarajan, V., and Garcia, J. G. (1993) Biochem. Biophys.
Res. Commun. 191, 1341–134138. Limatola, C., Schaap, D., Moolenaar, W. H., and van Blitterswijk, W. J.
(1994) Biochem. J. 304, 1001–100839. Moritz, A., De Graan, P. N., Gispen,W. H., andWirtz, K.W. (1992) J. Biol.
Chem. 267, 7207–721040. Jenkins, G. H., Fisette, P. L., and Anderson, R. A. (1994) J. Biol. Chem. 269,
11547–1155441. Jenkins, G. M., and Frohman, M. A. (2005) Cell. Mol. Life Sci. 62,
2305–231642. Jones, G. A., and Carpenter, G. (1993) J. Biol. Chem. 268, 20845–2085043. Cockcroft, S. (2001) Cell. Mol. Life Sci. 58, 1674–168744. Delon, C.,Manifava,M.,Wood, E., Thompson, D., Krugmann, S., Pyne, S.,
and Ktistakis, N. T. (2004) J. Biol. Chem. 279, 44763–4477445. Kishikawa, K., Chalfant, C. E., Perry, D. K., Bielawska, A., and Hannun,
Y. A. (1999) J. Biol. Chem. 274, 21335–2134146. Zhao, Y., Kalari, S. K., Usatyuk, P. V., Gorshkova, I., He, D., Watkins, T.,
Brindley, D. N., Sun, C., Bittman, R., Garcia, J. G., Berdyshev, E. V., andNatarajan, V. (2007) J. Biol. Chem. 282, 14165–14177
47. Usatyuk, P. V., Parinandi, N. L., and Natarajan, V. (2006) J. Biol. Chem.281, 35554–35566
48. Waters, C.M., Long, J., Gorshkova, I., Fujiwara, Y., Connell,M., Belmonte,K. E., Tigyi, G., Natarajan, V., Pyne, S., and Pyne, N. J. (2006) FASEB J. 20,509–511
49. Thompson, B., Ancellin, N., Fernandez, S. M., Hla, T., and Sha’afi, R. I.(2006) Prostaglandins Other Lipid Mediat. 80, 15–27
50. Li, Z., Paik, J. H., Wang, Z., Hla, T., and Wu, D. (2005) ProstaglandinsOther Lipid Mediat. 76, 95–104
51. Davis, G. E., and Senger, D. R. (2005) Circ. Res. 97, 1093–110752. English, D., Kovala, A. T., Welch, Z., Harvey, K. A., Siddiqui, R. A., Brind-
ley, D.N., andGarcia, J. G. (1999) J. Hematother. StemCell Res. 8, 627–63453. Sadahira, Y., Ruan, F., Hakomori, S., and Igarashi, Y. (1992) Proc. Natl.
Acad. Sci. U. S. A. 89, 9686–969054. Wang, F., Nohara, K., Olivera, A., Thompson, E.W., and Spiegel, S. (1999)
Exp. Cell Res. 247, 17–2855. Ryu, Y., Takuwa, N., Sugimoto, N., Sakurada, S., Usui, S., Okamoto, H.,
Matsui, O., and Takuwa, Y. (2002) Circ. Res. 90, 325–33256. Boguslawski, G., Grogg, J. R., Welch, Z., Ciechanowicz, S., Sliva, D.,
Kovala, A. T.,McGlynn, P., Brindley, D. N., Rhoades, R. A., and English, D.(2002) Exp. Cell Res. 274, 264–274
57. Lee, O.-H., Lee, D.-J., Kim, Y.-M., Kim, Y. S., Kwon, H. J., Kim, K.-W., andKwon, Y.-G. (2000) Biochem. Biophys. Res. Commun. 268, 47–53
58. Shizukuda, Y., Tang, S., Yokota, R., and Ware, J. A. (1999) Circ. Res. 85,247–256
59. Chaudhuri, P., Colles, S.M., Fox, P. L., andGraham, L.M. (2005)Circ. Res.97, 674–681
60. Freyberg, Z., Sweeney, D., Siddhanta, A., Bourgoin, S., Frohman, M., andShields, D. (2001)Mol. Biol. Cell 12, 943–955
61. Du, G., Huang, P., Liang, B. T., and Frohman, M. A. (2004)Mol. Biol. Cell15, 1024–1030
62. Lehman, N., Di Fulvio, M., McCray, N., Campos, I., Tabatabaian, F., andGomez-Cambronero, J. (2006) Blood 108, 3564–3572
63. Ohno, S., and Nishizuka, Y. (2002) J. Biochem. (Tokyo) 132, 509–51164. Hirai, T., and Chida, K. (2003) J. Biochem. (Tokyo) 133, 1–765. Kanaho, Y., Nakayama, K., Frohman,M.A., andYokozeki, T. (2007)Meth-
ods Enzymol. 434, 155–16966. Tolias, K. F., Couvillon, A. D., Cantley, L. C., and Carpenter, C. L. (1998)
Mol. Cell. Biol. 18, 762–77067. Kim, J. H., Lee, S. D., Han, J. M., Lee, T. G., Kim, Y., Park, J. B., Lambeth,
J. D., Suh, P. G., and Ryu, S. H. (1998) FEBS Lett. 430, 231–23568. Santy, L. C., and Casanova, J. E. (2001) J. Cell Biol. 154, 599–61069. Porcelli, A.M., Ghelli, A., Hrelia, S., and Rugolo,M. (2002)Cell. Signal. 14,
75–8170. Yasui, H., Katoh, H., Yamaguchi, Y., Aoki, J., Fujita, H., Mori, K., and
Negishi, M. (2001) J. Biol. Chem. 276, 15298–1530571. Garrett, T. A., Van Buul, J. D., and Burridge, K. (2007) Exp. Cell Res. 313,
3285–329772. Shikata, Y., Birukov, K. G., and Garcia, J. G. (2003) J. Appl. Physiol. 94,
1193–120373. Powner, D. J., Hodgkin, M. N., and Wakelam, M. J. (2002)Mol. Biol. Cell
13, 1252–126274. Singleton, P. A., Dudek, S.M., Chiang, E. T., andGarcia, J. G. (2005)FASEB
J. 19, 1646–165675. Gorshkova, I. A., Berdyshev, E. V., Saatian, B., Garcia, J. G. N., and Nat-
arajan, V. (2005) FASEB J. 19, (Abstr. 282)76. Overbeck, A. F., Brtva, T. R., Cox, A. D., Graham, S. M., Huff, S. Y., Khos-
ravi-Far, R., Quilliam, L. A., Solski, P. A., and Der, C. J. (1995)Mol. Reprod.Dev. 42, 468–476
Regulation of S1P-induced Endothelial Cell Migration
11806 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 17 • APRIL 25, 2008
at University of C
hicago Library on June 11, 2008 w
ww
.jbc.orgD
ownloaded from
Copyright © 2022 FDOKUMEN















![In vivo vulnerability to competition by endogenous dopamine: Comparison of the D2 receptor agonist radiotracer (-)-N-[11C]propyl-norapomorphine ([11C]NPA) with the D2 receptor antagonist](https://static.fdokumen.com/doc/165x107/631cb975a1cc32504f0c9f3c/in-vivo-vulnerability-to-competition-by-endogenous-dopamine-comparison-of-the-d2-1675155902.jpg)




