
On the Existence of a Possible A2A–D2–β-Arrestin2 Complex: A2A Agonist Modulation of D2...
13
On the Existence of a Possible A 2A –D 2 –β-Arrestin2 Complex: A 2A Agonist Modulation of D 2 Agonist-Induced β-Arrestin2 Recruitment Dasiel O. Borroto-Escuela 1 , Wilber Romero-Fernandez 1 , Alexander O. Tarakanov 2 , Francisco Ciruela 3 , Luigi F. Agnati 4 and Kjell Fuxe 1 ⁎ 1 Department of Neuroscience, Karolinska Institutet. Retzius väg 8, 17177 Stockholm, Sweden 2 St. Petersburg Institute for Informatics and Automation, Russian Academy of Sciences, 193167 Saint Petersburg, Russia 3 Unitat de Farmacologia, Departament Patologia i Terapèutica Experimental, Facultat de Medicina, Universitat de Barcelona, 08907 Barcelona, Spain 4 IRCCS, 41100 Lido Venice, Italy Received 25 November 2010; received in revised form 7 January 2011; accepted 11 January 2011 Available online 20 January 2011 Edited by J. Karn Keywords: heteromerization; G-protein-coupled receptors; allosteric modulation; internalization; receptor–protein interaction Given that coactivation of adenosine A 2A (A 2A R) and dopamine D 2 (D 2 R) receptors results in the coaggregation, cointernalization, and codesensitiza- tion of the A 2A R and D 2 R and the role of scaffolding protein β-arrestin2 in the desensitization, internalization, and signaling of G-protein-coupled receptors, in this study we explored the ability of the A 2A R agonist CGS21680 in A 2A R–D 2 R-coexpressing cells to modulate the D 2 R agonist- induced recruitment of β-arrestin2 to the D 2 R by means of proximity-based bioluminescence resonance energy transfer (BRET 2 ) and co-trafficking analysis. We found evidence that CGS21680 can increase the maximal BRET 2 signal between β-arrestin2 RLuc and D 2L R GFP2 upon D 2 R activation, by increasing the potency of the D 2 R agonist to exert this action. In addition, this change was associated with an increased formation of cytoplasmic clusters containing β-arrestin2 GFP2 and D 2L R YFP as seen from the co- trafficking analysis. Furthermore, the A 2A R agonist advanced the time for the increase in Akt phosphorylation obtained with the D 2 R agonist. Finally, using a novel bioinformatics approach to predict the protein–protein interface, we have also found that amino acid pro-triplets TNY, LLS, RAF, and VSR may be crucial for the -induced β-arrestin2 recruitment by A 2A R– D 2 R heteromers. Taken together, the results indicate that the antagonistic A 2A R–D 2 R allosteric receptor–receptor interaction in A 2A R–D 2 R heteromers favors β-arrestin2 recruitment to the D 2L R protomer with subsequent cointernalization associated with a reduced time onset of Akt phosphor- ylation followed by a rapid dephosphorylation. Thus, β-arrestin2 action becomes more rapid and short-lasting and, in this way, mimics G-protein- mediated signaling. © 2011 Elsevier Ltd. All rights reserved. *Corresponding author. E-mail address: [email protected]. Abbreviations used: A 2A R, adenosine A 2A receptor; D 2 R, dopamine D 2 receptor; BRET, bioluminescence resonance energy transfer; PBS, phosphate-buffered saline. doi:10.1016/j.jmb.2011.01.022 J. Mol. Biol. (2011) 406, 687–699 Contents lists available at www.sciencedirect.com Journal of Molecular Biology journal homepage: http://ees.elsevier.com.jmb 0022-2836/$ - see front matter © 2011 Elsevier Ltd. All rights reserved.
Transcript of On the Existence of a Possible A2A–D2–β-Arrestin2 Complex: A2A Agonist Modulation of D2...

doi:10.1016/j.jmb.2011.01.022 J. Mol. Biol. (2011) 406, 687–699
Contents lists available at www.sciencedirect.com
Journal of Molecular Biologyj ourna l homepage: ht tp : / /ees .e lsev ie r.com. jmb
On the Existence of a Possible A2A–D2–β-Arrestin2Complex: A2A Agonist Modulation of D2Agonist-Induced β-Arrestin2 Recruitment
Dasiel O. Borroto-Escuela1, Wilber Romero-Fernandez1,Alexander O. Tarakanov2, Francisco Ciruela3,Luigi F. Agnati4 and Kjell Fuxe1⁎1Department of Neuroscience, Karolinska Institutet. Retzius väg 8, 17177 Stockholm, Sweden2St. Petersburg Institute for Informatics and Automation, Russian Academy of Sciences, 193167 Saint Petersburg, Russia3Unitat de Farmacologia, Departament Patologia i Terapèutica Experimental, Facultat de Medicina,Universitat de Barcelona, 08907 Barcelona, Spain4IRCCS, 41100 Lido Venice, Italy
Received 25 November 2010;received in revised form7 January 2011;accepted 11 January 2011Available online20 January 2011
Edited by J. Karn
Keywords:heteromerization;G-protein-coupled receptors;allosteric modulation;internalization;receptor–protein interaction
*Corresponding author. E-mail addrAbbreviations used: A2AR, adeno
energy transfer; PBS, phosphate-buf
0022-2836/$ - see front matter © 2011 E
Given that coactivation of adenosine A2A (A2AR) and dopamine D2 (D2R)receptors results in the coaggregation, cointernalization, and codesensitiza-tion of the A2AR and D2R and the role of scaffolding protein β-arrestin2 inthe desensitization, internalization, and signaling of G-protein-coupledreceptors, in this study we explored the ability of the A2AR agonistCGS21680 in A2AR–D2R-coexpressing cells to modulate the D2R agonist-induced recruitment of β-arrestin2 to the D2R by means of proximity-basedbioluminescence resonance energy transfer (BRET2) and co-traffickinganalysis. We found evidence that CGS21680 can increase the maximalBRET2 signal between β-arrestin2RLuc and D2LR
GFP2 upon D2R activation,by increasing the potency of the D2R agonist to exert this action. In addition,this change was associated with an increased formation of cytoplasmicclusters containing β-arrestin2GFP2 and D2LR
YFP as seen from the co-trafficking analysis. Furthermore, the A2AR agonist advanced the time forthe increase in Akt phosphorylation obtained with the D2R agonist. Finally,using a novel bioinformatics approach to predict the protein–proteininterface, we have also found that amino acid pro-triplets TNY, LLS, RAF,and VSR may be crucial for the -induced β-arrestin2 recruitment by A2AR–D2R heteromers. Taken together, the results indicate that the antagonisticA2AR–D2R allosteric receptor–receptor interaction in A2AR–D2R heteromersfavors β-arrestin2 recruitment to the D2LR protomer with subsequentcointernalization associated with a reduced time onset of Akt phosphor-ylation followed by a rapid dephosphorylation. Thus, β-arrestin2 actionbecomes more rapid and short-lasting and, in this way, mimics G-protein-mediated signaling.
© 2011 Elsevier Ltd. All rights reserved.
ess: [email protected] A2A receptor; D2R, dopamine D2 receptor; BRET, bioluminescence resonancefered saline.
lsevier Ltd. All rights reserved.

688 A2A and D2 Agonists and β-Arrestin2 Recruitment
Introduction
Adenosine A2A receptor (A2AR)–dopamine D2receptor (D2R) heteromerization has been demon-strated by means of biochemical and biophysicalmethods [co-immunoprecipitation, bioluminescenceresonance energy transfer (BRET), and fluorescenceresonance energy transfer analyses] to occur upontransient cotransfection of the two receptors in celllines including HEK293 T cells.1–3 Antagonisticallosteric A2AR–D2R receptor–receptor interactionshave been shown to exist in A2AR–D2R heteromers,reducing the affinity of the D2R agonist bindingsites, Gi/o coupling, and signaling via adenylatecyclase, phospholipase C, and mitogen-activatedprotein kinases.2,4–6
In D2R-cotransfected neuroblastoma cells, coacti-vation of A2AR andD2R results in the coaggregation,cointernalization, and codesensitization of the A2ARand D2R.
7 However, it is unknown how thescaffolding protein β-arrestin2, being recognized toparticipate in the desensitization, internalization,and signaling of G-protein-coupled receptors, isinvolved in these events.8 The sequence amino acid212-IYIV-215 in the N-terminal part of the thirdintracellular loop (IC-3) of the D2R appears criticalfor β-arrestin2 binding.9,10 Furthermore, Lys149 inthe second intracellular loop (IC-2) in the D2R givespreferential β-arrestin2 binding to D2R IC-2 versusD3R IC-2, as shown in studies with glutathione S-transferase fusion proteins of D2R and D3R IC-2.10
β-Arrestin2 is also involved in mediating the Akt/GSK3 signaling of D2Rs.
11,12
In this study, we explored the ability of theA2AR agonist CGS21680 in A2AR–D2R-cotrans-fected cells to modulate the D2R-like agonist-induced recruitment of β-arrestin2 to the D2R bymeans of BRET2 and co-trafficking analyses. Inparallel, the A2AR agonist modulation of the D2R-induced changes in the temporal dynamics ofprotein kinase B (Akt) phosphorylation wasstudied to link changes in signaling to the BRET2
and co-trafficking analyses. Therefore, according tothe experimental results and using a novelbioinformatics approach to predict the protein–protein interface, the amino acid pro-triplets TNY,LLS, RAF, and VSR may be crucial for the agonist-induced β-arrestin2 recruitment by A2AR–D2Rheteromers.
Results
D2R agonist-mediated internalization is increasedupon combined A2AR and D2R agonist treatment
A2AR–D2R heteromers are known to be formedafter transient coexpression of the two receptors in
HEK cells,2,3 resulting in co-trafficking of the D2Rand A2AR.
13 We investigated whether A2AR affectsthe D2R agonist-mediated D2R internalization intransiently cotransfected HEK293T cells using a cellsurface receptor expression assay.HEK293T cells coexpressing 3xHA-D2LR and
A2AR were incubated in the presence of theD2LR agonist quinpirole (0.25 μM) with or withoutthe A2AR agonist CGS21680 (1 μM) at 37 °C for30 min to monitor internalization (Fig. 1a).Notably, quinpirole induced a rapid internaliza-tion of 3xHA-D2LR, resulting in a cell surfaceexpression of approximately 56% of control.However, combined treatment with CGS21680(1 μM) and quinpirole (0.25 μM) further enhanced3xHA-D2LR internalization, reducing the level ofcell surface expression to 28%. Such an increasedinternalization of 3xHA-D2LR was abolished byspecific A2AR and D2R antagonist treatment (datanot shown), suggesting that this effect is mediatedthrough the coactivation of D2LR and A2AR. Asalso seen in Fig. 1a, cotransfected cells preincu-bated with 0.45 M sucrose for 30 min prior toagonist treatment resulted in a marked reductionof 3xHA-D2LR internalization after combinedtreatment as well as D2R agonist treatmentalone. This supports the involvement of clathrin-coated pits in the 3xHA-D2LR internalizationprocess.Internalization may target the D2R and D2R–
A2AR heteromers to either a degradative pathway,leading to prolonged attenuation of cell signaling,or to a cell surface recycling pathway, facilitatingreceptor resensitization. We therefore evaluated thetime course of 3xHA-D2LR recycling to the plasmamembrane. Cells were treated with quinpirole(0.25 μM) alone or together with CGS21680(1 μM) during a time course of 0 to 45 min topromote 3xHA-D2LR internalization. The agonistswere then removed by extensive washing withphosphate-buffered saline (PBS), and the reappear-ance of the 3xHA-D2LR at the cell surface wasmonitored over time (from 45 min to 120 min) bychecking its expression level in the surface mem-brane. As shown in Fig. 1b, exposure of transientlycotransfected cells to quinpirole (0.25 μM) for45 min reduced 3xHA-D2LR cell surface expressionby approximately 52%. Thirty minutes after agonistremoval, a highly significant proportion of 3xHA-D2LR returned to the plasma membrane (90%). Inaddition, Fig. 1b shows that combined A2AR andD2R stimulation of the cells resulted in a significantfurther reduction by 23% of the cell surfaceexpression of 3xHA-D2LR after 45 min of incuba-tion compared to the cell surface expressionfollowing quinpirole treatment alone. Furthermore,the time course and rate of return of internalized3xHA-D2LR (i.e., resensitization) to the plasmamembrane after agonist removal were significantly
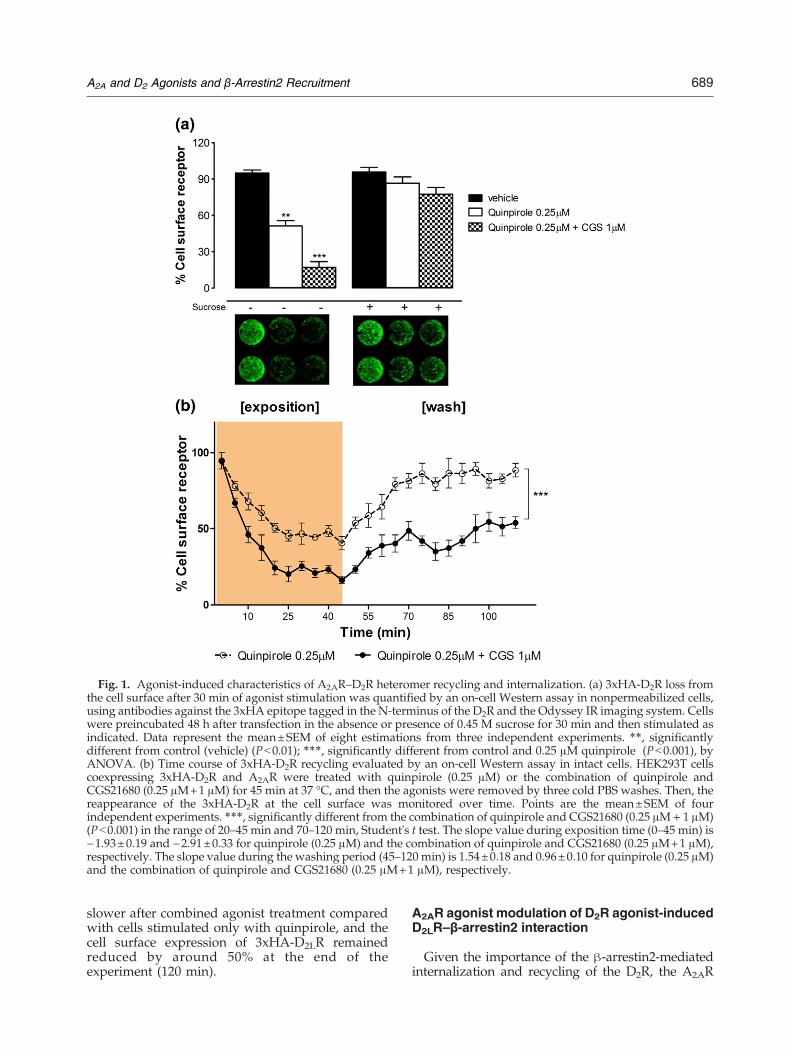
Fig. 1. Agonist-induced characteristics of A2AR–D2R heteromer recycling and internalization. (a) 3xHA-D2R loss fromthe cell surface after 30 min of agonist stimulation was quantified by an on-cell Western assay in nonpermeabilized cells,using antibodies against the 3xHA epitope tagged in the N-terminus of the D2R and the Odyssey IR imaging system. Cellswere preincubated 48 h after transfection in the absence or presence of 0.45 M sucrose for 30 min and then stimulated asindicated. Data represent the mean±SEM of eight estimations from three independent experiments. ⁎⁎, significantlydifferent from control (vehicle) (Pb0.01); ⁎⁎⁎, significantly different from control and 0.25 μM quinpirole (Pb0.001), byANOVA. (b) Time course of 3xHA-D2R recycling evaluated by an on-cell Western assay in intact cells. HEK293T cellscoexpressing 3xHA-D2R and A2AR were treated with quinpirole (0.25 μM) or the combination of quinpirole andCGS21680 (0.25 μM+1 μM) for 45 min at 37 °C, and then the agonists were removed by three cold PBS washes. Then, thereappearance of the 3xHA-D2R at the cell surface was monitored over time. Points are the mean±SEM of fourindependent experiments. ⁎⁎⁎, significantly different from the combination of quinpirole and CGS21680 (0.25 μM+ 1 μM)(Pb0.001) in the range of 20–45 min and 70–120 min, Student's t test. The slope value during exposition time (0–45 min) is−1.93±0.19 and −2.91±0.33 for quinpirole (0.25 μM) and the combination of quinpirole and CGS21680 (0.25 μM+1 μM),respectively. The slope value during the washing period (45–120 min) is 1.54±0.18 and 0.96±0.10 for quinpirole (0.25 μM)and the combination of quinpirole and CGS21680 (0.25 μM+1 μM), respectively.
689A2A and D2 Agonists and β-Arrestin2 Recruitment
slower after combined agonist treatment comparedwith cells stimulated only with quinpirole, and thecell surface expression of 3xHA-D2LR remainedreduced by around 50% at the end of theexperiment (120 min).
A2AR agonist modulation of D2R agonist-inducedD2LR–β-arrestin2 interaction
Given the importance of the β-arrestin2-mediatedinternalization and recycling of the D2R, the A2AR

Fig. 2. β-Arrestin2GFP2 and D2RYFP subcellular distribution after agonist treatment. Fluorescence confocal
microscopy was used to visualize β-arrestin2GFP2 (green) recruitment to human D2RYFP (red) in HEK293T cells
coexpressing β-arrestin2GFP2/D2RYFP/A2AR at 1:1:1 ratio over the ensuing time period with no drug (vehicle), with
quinpirole (0.25 μM), or with the combination of quinpirole and CGS21680 (0.25 μM and 1 μM, respectively). The resultsshown are representative images of three individual experiments. The basal recruitment or translocation ratios (RR)represent the fluorescence area ratio per cell between the area formed from the merger of D2R
YFP–β-arrestin2GFP2 signals(ROI in blue) and the entire D2R
YFP fluorescence area (ROI in green). For each group, at least 30 cells from threeindependent transfections were analyzed by two independent investigators, and the mean and SEM values were used forthe table. The degrees of D2R
YFP–β-arrestin2GFP2 recruitment or translocation are shown as a single z-scan image.
690 A2A and D2 Agonists and β-Arrestin2 Recruitment
agonist modulation of D2LR–β-arrestin2 interactionshas been studied in HEK cells also containing theA2AR, using confocalmicroscopy andBRET assays, in
viewof the existence of allostericA2AR–D2R receptor–receptor interactions.2 The time course of D2LR–β-arrestin2 recruitment and interaction was monitored.

Fig. 3. BRET2 studies of β-arrestin2Rluc recruitment toD2LR
GFP2 in living HEK293T cells. BRET2 wasmeasured inHEK293T cells expressing a constant amount of β-arrestin2Rluc (1 μg) and increasing expression levels ofD2LR
GFP2 and treated as indicated. The specificity of theBRET2 signal between D2LR
GFP2 and β-arrestin2Rluc wascontrolled by measuring BRET2 between β-arrestin2Rluc
and GFP2 alone, treated with quinpirole. After theaddition of coelenterazine-400a, the BRET2 signal wasmeasured. Cells expressing the negative control yielded abackground BRET ratio of ∼0.10, while cells coexpressingD2LR
GFP2 and β-arrestin2Rluc yielded higher and saturat-ing BRET2 ratios, indicative of energy transfer andprotein–protein interaction. The BRETmax values areshown as the mean±SEM of three independent experi-ments performed in triplicate. ⁎⁎⁎, significantly differentfrom the vehicle (Pb0.001) by ANOVA.
691A2A and D2 Agonists and β-Arrestin2 Recruitment
As seen in Fig. 2, in the absence of agonisttreatment, the distribution of D2LR
YFP (shown inred) was almost exclusively linked to the plasmamembrane, while β-arrestin2GFP2 protein onlyappeared as a diffuse and uniform distributionof fluorescence throughout the cytosol (shown ingreen). Stimulation of cells with the D2R agonistquinpirole (0.25 μM) for 30–60 min resulted in atime-dependent redistribution of fluorescence(red) from the plasma membrane to cytosoliccompartments, with the appearance of clustersshowing moderate-intensity yellow fluorescencerepresenting colocalization of D2LR
YFP (in red)and β-arrestin2GFP2 (in green). Combined agonisttreatment with quinpirole (as above) and theA2AR agonist CGS21680 (1 μM) for 30–60 mincaused a more reliable β-arrestin2GFP2–D2LR
YFP
body formation, which was seen as an increase inthe amount of small to large clusters having amoderate to very high intensity of yellowishfluorescence.As an additional step to address D2LR–β-
arrestin2 interaction issues, BRET experimentsbetween D2LR
GFP2 and β-arrestin2Rluc were car-ried out in HEK cells transiently cotransfectedwith D2LR
GFP2, A2AR, and β-arrestin2Rluc. Stimu-lation of D2LR
GFP2 with 25 and 100 nM quinpiroleinduced a concentration-dependent increase in themaximal BRET value compared to unstimulatedcells, which is consistent with an active recruit-ment of the β-arrestin2–D2LR complexes (Fig. 3).The basal and D2R agonist-mediated interactionsof the D2LR with β-arrestin2 are specific, since incontrol experiments, a weaker and linear bystanderBRET signal was observed, indicating the nonspe-cific nature of the BRET signal.We next determined the kinetics of β-arrestin2
recruitment to the D2LR after agonist stimulation,using real-time BRET measurements. In cells coex-pressing a fixed ratio (1:2:2) of β-arrestin2Rluc/D2LR
GFP2/A2AR, as seen in Fig. 4a, there was arapid increase of the BRET2 signal after quinpiroletreatment, which was stable for 20 to 50 min. Inthe presence of CGS21680 (50 nM), the BRET2
signal became significantly increased versus quin-pirole treatment alone, and remained increased asa plateau signal from as early as 10 min to120 min after the combined agonist treatment.CGS21680 (50 nM) incubation alone producedonly a small increase in the BRET2 signal duringthe 0- to 120-min time period (Fig. 4a). Figure 4bshows that 30 min of cell incubation withincreasing concentrations of CGS21680 (10–100 nM) caused a concentration-dependent shiftto the left of the quinpirole concentration–responsecurves, indicating an increased potency of quinpiroleto produce an increase in the BRET2 signal in thepresence of increasing concentrations of the A2ARagonist.
Time course and potency of D2R agonist-inducedAKT phosphorylation and its modulation by theA2AR agonist CGS21680
As seen in Fig. 5a, in HEK cells transientlycotransfected with D2LR and A2AR, quinpirole(25 nM) alone produced over the first 30-min periodan increased degree of Akt phosphorylation overbasal activity, followed by a reduction to a plateauvalue starting at the 60-min time point. Treatmentwith CGS21680 alone (50 nM) produced a similartemporal increase in Akt phosphorylation in the first60-min period (Fig. 5a). However, after combinedtreatment with quinpirole (25 nM) and CGS21680(50 nM), a more rapid increase in Akt phosphoryla-tion developed with a peak found already after10 min. This increase in Akt phosphorylation dis-appeared after 40 min, and no clear further changesdeveloped in the period studied up to 110 min.A concentration–response curve with quinpirole
on Akt phosphorylation was made at the 30-mintime interval. As seen in Fig. 5b, CGS21680 (50 nM)shifted this curve to the right, indicating a reduced

Fig. 4. BRET2 studies of β-arrestin2Rluc recruitment to D2LR
GFP2
in living HEK293T cells. BRET2 wasmeasured at a fixed ratio (1:2:2) ofexpression levels of β-arrestin2Rluc/D2LR
GFP2/A2AR and treated as indi-cated. (a) Kinetics of β-arrestin2Rluc
recruitment to D2LRGFP2 after the
addition of the indicated treatmentat time 0. Similar experiments werecarried out on cells without stimu-lation (vehicle). Mean±SEM; n=8in triplicate. ⁎⁎, significantly differentfrom 25 nM quinpirole (Pb0.01) byANOVA. (b)Concentration–responsecurve of quinpirole on β-arrestin2Rluc
recruitment to D2LRGFP2. Cells coex-
pressingβ-arrestin2Rluc andD2LRGFP2
were stimulated in the presence orabsence of different concentrations ofthe A2AR agonist CGS21680. Resultsare expressed as normalized BRET2
ratio as described in Materials andMethods. Data represent the mean±SEM; n= 7 in duplicate.
692 A2A and D2 Agonists and β-Arrestin2 Recruitment
potency of quinpirole to produce Akt phosphoryla-tion in the presence of the A2AR agonist. In addition,we determined and confirmed that the increase inAkt phosphorylation induced by the D2R-likeagonist quinpirole is reduced by 50% with theA2AR-like agonist CGS21680 (50 nM) at 30 min. Thisphenomenon is blocked by lithium chloride (1 mM)and okadaic acid (1 μM) (Fig. 5c).
The amino acid pro-triplet homologies TNY, LLS,RAF, and VSR may be crucial for the D2Ragonist-induced β-arrestin2 recruitment toA2AR–D2R heteromers
On the basis of a bioinformatics approach,Tarakanov and Fuxe14 have deduced a set of triplet
homologies that may be responsible for receptor–receptor interactions. This set consists of twononintersecting subsets: “pro-triplets” and “contra-triplets.” Any pro-triplet appears as a homology inat least one heterodimer but does not appear as ahomology in any non-heterodimer. Conversely, anycontra-triplet appears in at least one non-hetero-dimer but does not appear in any heterodimer. Forexample (see Fig. 6), the triplet of amino acidresidues TNY (Thr-Asn-Tyr) appear as a homologyin four heterodimers: A2A–D2, ADRA1B–ADRA1D,A2A–D3, and D2–D3, which is true also for thetriplet LLS (Leu-Leu-Ser), as seen in four hetero-dimers: TLR1–TLR2, ADRA1B–ADRA1D, MHC2–CD4, and MHC1–CD8. Thus, both triplets TNY andLLS appear together as homologies in the

Fig. 5. A2AR agonist modulation of D2R-induced Akt phosphorylation. (a) An on-cell Western assay was used to measure Akt phosphorylation in HEK293T cellscoexpressing A2AR and D2R and treated with different agonists at durations ranging from 0 to 120 min. Similar experiments were carried out on cells without stimulation(vehicle). (Top) The data are plotted as a sixth-order polynomial equation and represent the mean±SEM; n= 6 in triplicate; (bottom) the data for longer time intervals areplottedwithin bars. The quinpirole group is significantly different from the combined quinpirole and CGS21680 group in the range of 5–10 and 25–45min (⁎⁎, Pb0.01) and85–100min (⁎⁎⁎,Pb0.001). TheCGS21680 group is significantly different from the combined quinpirole andCGS21680 group in the range of 5–10 and 20–25min (+,Pb0.05)and 25–45min (++, Pb0.01) byANOVA. (b) Concentration–response curve of quinpirole-inducedAkt phosphorylation. Cells coexpressingD2R andA2ARwere stimulatedin the presence or absence of the A2AR agonist CGS21680 (50 nM) during 30 min. CGS21680 at 50 nM shifts the concentration–response curve of quinpirole on Aktphosphorylation to the right. Data represent the mean±SEM; n= 6 in duplicate. (c) AKT phosphorylation response after agonist-induced A2AR and D2LR activation.HEK293T cells were transiently cotransfected with 1 μg of both (D2LR and A2AR) expression vectors and treated for 30 min with different inhibitors (lithium chloride andokadaic acid), agonists or antagonists. The data represent themean±SEM of three independent experiments performed in triplicate. ⁎⁎⁎, significantly different from 50 nMCGS21680 (Pb0.001); +, significantly different from25 nMquinpirole (Pb0.05); ###, significantly different from the combination of 25 nMquinpirole and 50 nMCGS21680(Pb0.001), by ANOVA. quinp, quinpirol (25 nM); racl, raclopride (1 μM); CGS, CGS21680 (50 nM); ZM, ZM241385 (1 μM); LiCl, lithium chloride (1 mM).
693A2Aand
D2Agonists
andβ-A
rrestin2Recruitm
ent

Fig. 6. In silicoanalysis and sequence alignment ofA2AR,D2R, andβ-arrestin2 receptor–receptor–scaffoldproteinhomologiescontaining the triplets TNY, LLS, RAF, and VSR in the complex interface. The amino acid codes of the receptors were obtainedfrom the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/, 2009), where all the receptors havebeen considered human. (a) The triplet TNY (grey shaded) in the first intracellular loops (IC-1) of theA2AR andD2R between thefirst and second transmembrane α-helices (TM1–TM2) as well as in the C-tail of β-arrestin2. (b) The triplet LLS (grey shaded) inthe C-tail of the A2AR and in β-arrestin2. (c) The triplet RAF (grey shaded) in the third intracellular loop (IL3) between the TM5andTM6of theD2Rand inβ-arrestin2. (d) The tripletVSR (grey shaded) in the IC-1 of theD2Rand inβ-arrestin2.Note the tripletTNY (grey shaded) close to VSR in the IC-1 of the D2R. Bottom, a summary of the pro-triplet homology analysis results.
694 A2A and D2 Agonists and β-Arrestin2 Recruitment

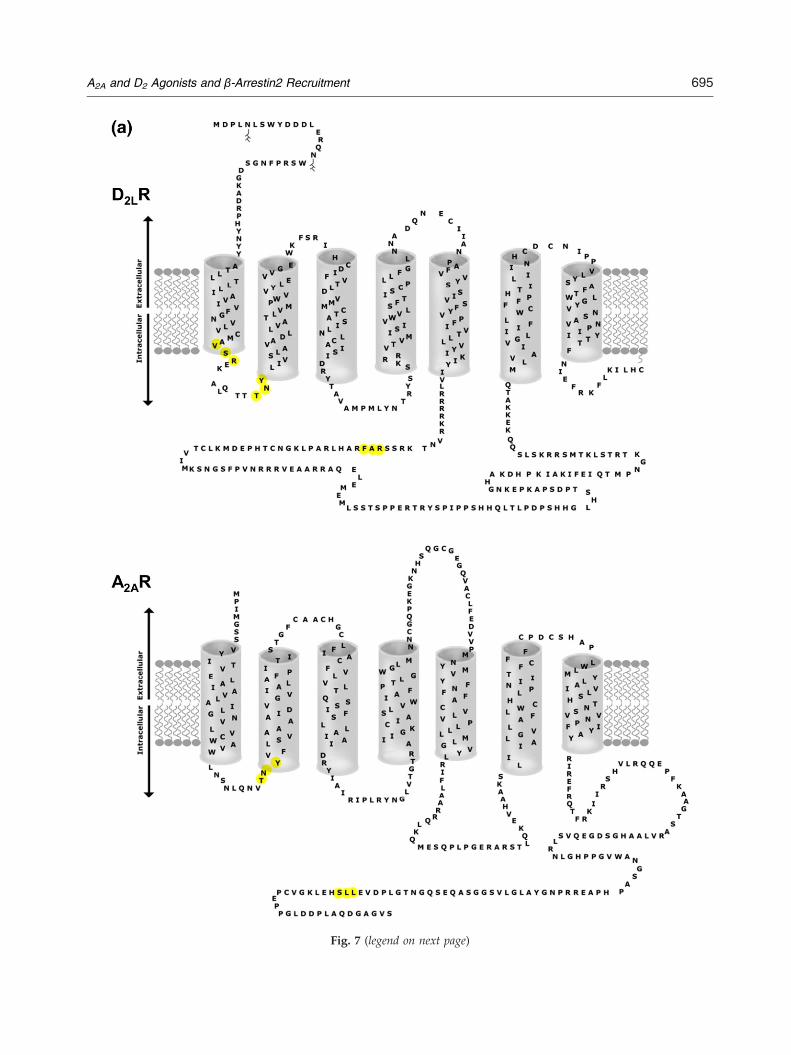
Fig. 7. (a) A two-dimensional topology of the human D2LR sequence (top) and the human A2AR sequence (bottom) ispresented (extracellular space at the top and the intracellular space at the bottom). Filled circles (in yellow) represent thetriplets of amino acid residues determined by bioinformatic analysis to be implicated on the D2LR–A2AR–β-arrestin2heteromer–scaffold protein complex interface. (b) Three-dimensional molecular models of A2AR–D2R heteromer andhuman β-arrestin2 were built based on their crystal structures (PDB code 3EML), the crystal structure of rhodopsin (PDBcode 2Z73), and the crystal structure of arrestin2 (PDB code 2WTR), respectively, as structural templates, by means of thehomology modeling program Accelrys Discovery Studio 2.5 (San Diego, CA, USA). The model with the highestPROCHECK score was selected and subjected to molecular dynamic simulation. Molecular dynamic simulations wereperformed using the GROMACS package. The predicted interface interactions bymeans of the triplet homology approach(see Materials and Methods) are shown in red spheres. Blue spheres represent the putative PKC phosphorylation sites(NetPhosK v1.0). Left, lateral view of A2AR–D2R heteromer and β-arrestin2, with arrows indicating how β-arrestin2 mayapproach the A2AR–D2R heteromer. Right, view of A2AR–D2R heteromer from the intracellular side.
696 A2A and D2 Agonists and β-Arrestin2 Recruitment
heterodimer ADRA1B–ADRA1D. Moreover, thetriplets LLS, RAF, and VSR may play a role in HIVentry, according to Tarakanov and Fuxe. Takentogether with the experimental results, these bioin-formatic predictions suggest the existence of a basicset of common triplets in β-arrestin as well as in thetwo participating receptors that may participate inthe A2AR–D2R–β-arrestin2 complex interactioninterfaces (Fig. 7a and b).
Discussion
Previousworks have shown that transient cotrans-fection of the A2AR and D2R in HEK cells results in
the formation of A2AR–D2R heteromers.2,3 There-fore, the observation that the A2AR agonistCGS21680 can increase the maximal BRET2 signalbetween β-arrestin2RLuc and D2LR
GFP2 upon D2Ractivation with quinpirole by increasing the potencyof the D2R agonist to exert this action is of substantialinterest. It indicates that the A2AR agonist-inducedactivation of the A2AR in the heteromer can favor thebinding of β-arrestin2 to the D2LR protomer. Thischange was associated with an increased formationof cytoplasmic clusters containing β-arrestin2GFP2
andD2LRYFP, as indicated by the yellow fluorescence
in the clusters. These structures likely represent theinternalized vesicles containing the A2A–D2L
YFP

697A2A and D2 Agonists and β-Arrestin2 Recruitment
heteromer to which β-arrestin2GFP2 has attached. Itseems possible that the mechanism for these actionsof the A2AR agonist may be the known antagonisticA2AR–D2R interaction,15 which reduces the Gi/ocoupling of the D2R. Thus, the A2AR agonist willfavor the removal of Gi/o from the D2R and, in thisway, facilitate the binding of β-arrestin2 to the D2R.The results from the β-arrestin2-mediated D2R
signaling as seen from Akt phosphorylation are inline with the BRET2 signaling and β-arrestin2GFP2
and D2LRYFP co-trafficking results. The A2AR ago-
nist advanced the time for the increase of Aktphosphorylation obtained with the D2R agonist,which can partly be explained by the facilitation ofβ-arrestin2 binding on the D2LR and in part by theindication that the A2AR agonist itself increases RTKsignaling, leading to the increased Akt phosphory-lation observed with the A2AR agonist alone (seeRefs. 11,16). The subsequent rapid decrease in Aktphosphorylation can be caused by the recruitment ofPP2A to the D2R–GRK–β-arrestin2–Akt complex.11
This interpretation is also supported by the demon-stration that when studied at the time of Aktdephosphorylation, the A2AR agonist markedlyreduced the potency of the D2R agonist to producean increase in Akt phosphorylation.The bioinformatic analysis has allowed to postulate
one possible model of the A2AR–D2R–β-arrestin2complex (Fig. 7b) based inter alia on the demonstra-tion of TNY/RAF/VSR pro-triplet homologies be-tween the D2LR and β-arrestin2 involving the IC-3and IC-1 of the D2LR and of LLS/TNY pro-triplethomologies between the A2AR and β-arrestin2involving the C-terminal tail and IC-1 of the A2AR(Fig. 6).Taken together, the results indicate that the antag-
onistic A2AR–D2R allosteric receptor–receptor interac-tion in A2AR–D2R heteromers favors β-arrestin2recruitment to the D2LR protomer with subsequentcointernalization associated with a reduced timeonset of Akt phosphorylation, followed by a rapiddephosphorylation most likely through the recruit-ment of PP2A. Thus, the time course of β-arrestin2action becomes more rapid and short-lasting and, inthis way, mimics G-protein-mediated signaling.11
Materials and Methods
Plasmid constructs
β-Arrestin2Rluc and β-arrestin2GFP2 were generated byPCR amplification of the coding sequences of human β-arrestin2 (kindly provided by M. Pérez-Alea, Covalab,France) without their stop codons and subcloned inpGFP2-N3 (Perkin-Elmer, Madrid, Spain) and pRluc-N3(Packard Bioscience, Spain). On the other hand, the cDNAencoding the D2LR without its stop codon was subcloned
in pGFP2-N1 (Perkin-Elmer, Spain) and pEYFP-N1 (Clon-tech, Germany).
Cell culture and transfection
HEK293T cells (American Type Culture Collection,USA) were grown in Dulbecco's modified Eagle's mediumsupplemented with 2 mM L-glutamine, 100 units/mlpenicillin/streptomycin, and 10% (v/v) fetal bovineserum at 37 °C and in an atmosphere of 5% CO2. Fortransfection, the cells were plated in six-well dishes at aconcentration of 1×106 cells/well or in 75-cm2 flasks andcultured overnight before transfection. The cells weretransiently transfected using either linear polyethyleni-mine reagent (Polysciences Inc., USA) or TransFectin (Bio-Rad, USA).
Cell surface receptor expression assays
The cell surface expression of the D2R was evaluatedby on-cell Western analysis experiments performed ontransiently cotransfected 3xHA-D2R/A2AR HEK293Tcells.17 The cells were seeded onto poly-D-lysine-coated96-well plates (Corning, Corning, NY), grown for 48 h,and then treated for 5 to 120 min (quinpirole, 0.25 μM;CGS21680, 1 μM) in medium containing 1.0 mg/mlbovine serum albumin. When specified, the agonist wasremoved by extensive washing with PBS. After the drugtreatment, the cells were immediately fixed with 4%paraformaldehyde for 10 min at room temperature,washed (without detergent to avoid membrane permea-bilization), blocked for 90 min in LI-COR OdysseyBlocking Buffer® (LI-COR Biosciences, UK), and thenincubated overnight at 4 °C with monoclonal anti-HAantibody (1:1000; Sigma Aldrich). After further washingwith LI-COR Odyssey Blocking Buffer®, the cells wereincubated with an infrared secondary antibody (goatanti-rabbit, 1:1500; LI-COR Biosciences, UK) in LI-COROdyssey Blocking Buffer®, and plates were scannedwith an Odyssey infrared scanner. The integrity of theplasma membrane was evaluated with and without theaddition of detergent (0.1% Triton X-100). Total cellnumber was normalized using the DRAQ5/Sapphire700 staining agent. Results are presented as thepercentage of total protein detected at the cell surface.
Receptor–β-arrestin recruitment analysis byfluorescence confocal microscopy
β-Arrestin recruitment and translocation by the D2Rwere evaluated by fluorescence confocal microscopy usingtransiently cotransfected HEK293T cells with constant(1 μg) amounts of plasmids encoding for β-arrestin2GFP2,D2LR
YFP, and A2AR. The cells were incubated with D2Ragonist (quinpirole, 0.25 μM) in the presence or absence ofA2AR agonist (CGS21680, 1 μM) at different time intervals.Then, the cells were fixed in 4% paraformaldehyde for10 min, washed with PBS containing 20 mM glycine, andmounted in Vectashield immunofluorescence medium(Vector Laboratories, UK). Microscope observations wereperformed with a 63× oil immersion objective in a LeicaTCS-SL confocal microscope (Leica, USA). The amounts ofD2R
YFP–β-arrestin2GFP2 recruited or translocated are

698 A2A and D2 Agonists and β-Arrestin2 Recruitment
shown as a single z-scan image. The basal recruitment ortranslocation ratios (RR) represent the fluorescence arearatio between the merger of D2R
YFP–β-arrestin2GFP2
signals and the entire D2RYFP fluorescence area per cell
signal.
Monitoring D2R–β-arrestin2 interaction usingBRET2assay
A BRET2 saturation assay was performed as previ-ously described.18 Briefly, 48 h after transfection,HEK293T cells transiently transfected with constant(1 μg) or increasing amounts (0.25–9 μg) of plasmidsencoding for β-arrestin2Rluc and D2LR
GFP2, respectively,were rapidly washed twice in PBS, detached, andresuspended in the same buffer. For BRET2 measure-ment, coelenterazine-400a, also known as DeepBlue™Csubstrate, was added at a final concentration of 5 μM,and readings were performed 1 min after analysis withthe POLARstar Optima plate reader (BMG Labtechnol-ogies, Offenburg, Germany). For dose–response andkinetic BRET2 experiments, HEK293T cells were tran-siently transfected at a constant ratio (1:2:2) of β-arrestin2Rluc/D2LR
GFP2/A2AR. Agonist-promoted BRET2
was calculated by subtracting the BRET2 ratio obtainedin the absence of the agonist from that obtained in thepresence of the agonist. In the case of kinetic measure-ments, coelenterazine-400a was added simultaneouslywith the agonist, followed by BRET2 measurements. Ineach experiment, the specificities of D2LR–β-arrestin2interactions were assessed by comparison with cellsexpressing D2LR
GFP2 alone.
AKT phosphorylation assay
HEK293T cells transiently coexpressing D2LR and A2ARwere treated with the indicated concentration of eachagonist or inhibitor at the indicated time periods and thenfixed in a final concentration of 4% paraformaldehyde.After fixing, the cells were permeabilized by washing fivetimes with 0.1% Triton X-100 in PBS, blocked for 90 min atroom temperature in LI-COR Odyssey Blocking Buffer®,and then incubated overnight at 4 °C with primarymonoclonal rabbit anti-phospho-Akt-Thr308 antibody(diluted 1/500; Cell Signaling Technology, Stockholm,Sweden. After extensive washing, the cells were incubatedin the dark with secondary infrared probe-labelled goatanti-rabbit antibodies (diluted 1/1500) for 1 h at roomtemperature, washed, and scanned with an Odysseyinfrared scanner (LI-COR Biosciences).
Bioinformatic analysis of the heteromer–β-arrestin2interface interaction
On the basis of a bioinformatics approach, a set of aminoacid triplet homologies have been deduced in receptorheterodimers that may be responsible for receptor–receptor interactions.14 It has been indicated how suchtriplets of amino acid residues and their “teams” may beutilized to construct a “kind of code” that determines(and/or predicts) which receptors should or should notform heterodimers. In this study, the A2AR–D2LR hetero-dimer has been analyzed for the existence of a basic set of
common triplets between these two participating recep-tors and the β-arrestin2 scaffold protein that may beresponsible for the A2AR–D2R/β-arrestin2 interactions.
Statistical analysis
The number of samples (n) in each experimentalcondition is indicated in the figure legends. When twoexperimental conditions were compared, a statisticalanalysis was performed using an unpaired t test.Otherwise, a statistical analysis was performed byANOVA followed by Tukey's multiple comparison posthoc test. A P value of ≤0.05 was considered significant.
Acknowledgements
This work was supported by grants from theSwedish Research Council (04X-715), the Torstenand Ragnar Söderberg Foundation, the Hjärnfondenand Marianne and Marcus Wallenberg Foundation(to K.F.), and Ministerio de Ciencia e Innovación(SAF2008-01462 and Consolider-Ingenio CSD2R008-00005 to F.C.). A.O.T. did not receive any supportfor this work.
References
1. Kamiya, T., Saitoh, O., Yoshioka, K. & Nakata, H.(2003). Oligomerization of adenosine A2A and dopa-mine D2 receptors in living cells. Biochem. Biophys. Res.Commun. 306, 544–549.
2. Borrota-Escuela, D. O., Marcellino, D., Narvaez, M.,Flajolet, M., Heintz, N., Agnati, L. et al. (2010). A serinepoint mutation in the adenosine A2AR C-terminal tailreduces receptor heteromerization and allostericmodulation of the dopamine D2R. Biochem. Biophys.Res. Commun. 394, 222–227.
3. Canals, M., Marcellino, D., Fanelli, F., Ciruela, F., deBenedetti, P., Goldberg, S. R. et al. (2003). AdenosineA2A–dopamine D2 receptor–receptor heteromerization:qualitative and quantitative assessment by fluorescenceand bioluminescence energy transfer. J. Biol. Chem. 278,46741–46749.
4. Ferre, S., von Euler, G., Johansson, B., Fredholm, B. B.& Fuxe, K. (1991). Stimulation of high-affinity aden-osine A2 receptors decreases the affinity of dopamineD2 receptors in rat striatal membranes. Proc. Natl Acad.Sci. USA, 88, 7238–7241.
5. Fuxe, K., Ferre, S., Zoli, M. & Agnati, L. F. (1998).Integrated events in central dopamine transmission asanalyzed at multiple levels. Evidence for intramem-brane adenosine A2A/dopamine D2 and adenosineA1/dopamine D1 receptor interactions in the basalganglia. Brain Res. Brain Res. Rev. 26, 258–273.
6. Fuxe, K., Ferre, S., Genedani, S., Franco, R. & Agnati,L. F. (2007). Adenosine receptor–dopamine receptorinteractions in the basal ganglia and their relevance forbrain function. Physiol. Behav. 92, 210–217.

699A2A and D2 Agonists and β-Arrestin2 Recruitment
7. Hillion, J., Canals, M., Torvinen, M., Casado, V., Scott,R., Terasmaa, A. et al. (2002). Coaggregation, coin-ternalization, and codesensitization of adenosine A2Areceptors and dopamine D2 receptors. J. Biol. Chem.277, 18091–18097.
8. Pierce, K. L. & Lefkowitz, R. J. (2001). Classical andnew roles of β-arrestins in the regulation of G-protein-coupled receptors. Nat. Rev. Neurosci. 2, 727–733.
9. Lan, H., Liu, Y., Bell, M. I., Gurevich, V. V. & Neve,K. A. (2009). A dopamine D2 receptor mutant capableof G protein-mediated signaling but deficient inarrestin binding. Mol. Pharmacol. 75, 113–123.
10. Lan, H., Teeter, M. M., Gurevich, V. V. & Neve, K. A.(2009). An intracellular loop 2 amino acid residuedetermines differential binding of arrestin to thedopamine D2 and D3 receptors. Mol. Pharmacol. 75,19–26.
11. Beaulieu, J. M., Gainetdinov, R. R. & Caron, M. G.(2007). The Akt-GSK-3 signaling cascade in the actionsof dopamine. Trends Pharmacol. Sci. 28, 166–172.
12. Beaulieu, J. M., Gainetdinov, R. R. & Caron, M. G.(2009). Akt/GSK3 signaling in the action of psycho-tropic drugs.Annu. Rev. Pharmacol. Toxicol. 49, 327–347.
13. Torvinen, M., Torri, C., Tombesi, A., Marcellino, D.,Watson, S., Lluis, C. et al. (2005). Trafficking of
adenosine A2A and dopamine D2 receptors. J. Mol.Neurosci. 25, 191–200.
14. Tarakanov, A. O. & Fuxe, K. G. (2010). Triplet puzzle:homologies of receptor heteromers. J. Mol. Neurosci.41, 294–303.
15. Fuxe, K., Marcellino, D., Genedani, S. & Agnati, L.(2007). Adenosine A2A receptors, dopamine D2 recep-tors and their interactions in Parkinson's disease.Mov.Disord. 22, 1990–2017.
16. Golder, F. J., Ranganathan, L., Satriotomo, I., Hoffman,M., Lovett-Barr, M. R., Watters, J. J. et al. (2008). Spinaladenosine A2a receptor activation elicits long-lastingphrenic motor facilitation. J. Neurosci. 28, 2033–2042.
17. Delisle, B. P., Underkofler, H. A., Moungey, B. M.,Slind, J. K., Kilby, J. A., Best, J. M. et al. (2009). SmallGTPase determinants for the Golgi processing andplasmalemmal expression of human ether-a-go-gorelated (hERG) K+ channels. J. Biol. Chem. 284,2844–2853.
18. Borroto-Escuela, D. O., Correia, P. A., Perez Alea, M.,Narvaez, M., Garriga, P., Fuxe, K. et al. (2010).Impaired M(3) muscarinic acetylcholine receptorsignal transduction through blockade of binding ofmultiple proteins to its third intracellular loop. Cell.Physiol. Biochem. 25, 397–408.












![In vivo vulnerability to competition by endogenous dopamine: Comparison of the D2 receptor agonist radiotracer (-)-N-[11C]propyl-norapomorphine ([11C]NPA) with the D2 receptor antagonist](https://static.fdokumen.com/doc/165x107/631cb975a1cc32504f0c9f3c/in-vivo-vulnerability-to-competition-by-endogenous-dopamine-comparison-of-the-d2-1675155902.jpg)







