
Propargylic- and chromene-terminated prepolymers. 4 ...
22
High Perform. Polym. 8 (1996) 533–554. Printed in the UK Propargylic- and chromene-terminated prepolymers. 4. Influence of the type of catalyst on reaction mechanisms and kinetics M F Grenier-Loustalot and C Sanglar Laboratoire de physico-chimie des polym` eres, CNRS-URA 1494, H´ elioparc-2, avenue du Pdt Angot, 64000 Pau, France Received 21 May 1996, accepted for publication 10 September 1996 Abstract. The addition of catalyst to polymerize the propargylic function causes a decrease in the reaction onset temperature T 0 . Depending on the nature of the catalyst we demonstrate different reaction mechanisms involving the formation of the chromene group (as in the case of non-catalysed systems), of the Sabourin dimer, or the cyclooctatetraene structure in the molten state. We thus show that the catalytic effect is more determinant towards the propargylic function than towards the chromene group. 1. Introduction Previous work on difunctional propargylic and chromene compounds [1, 2] has shown that the zone of reactivity of these monomers is situated between 200 and 240 ◦ C. In order to broaden the field of application of these systems and to facilitate their use, it appeared interesting to reduce the polymerization temperature by using a catalyst. To this end, we investigated a number of copper-, nickel-, rhodium- and cobalt-based catalysts recommended in the literature [3, 4] to polymerize acetylene triple bonds. The basis of this choice was that a number of transition metals are known to catalyse reactions of acetylene monomers [3], such as cyclotrimerization [5] and the formation of linear polyenes [6]. In the light of published data, two categories of acetylene monomers can be distinguished, whose terminations are: (i) an arylacetylene group; in the absence of catalyst, mechanisms are complex and we have been able to show that in the presence of catalyst, kinetics were modified, while the reaction mechanisms remained comparable [7–9]; (ii) an aryl ether propargylic group (the object of our study). In the absence of catalyst, the reaction mechanism of the latter involves the formation of a stable chromene-type entity (β stage) [1, 10]. Data obtained with non-catalysed systems were compared with those gathered following catalytic action. In the study of catalysed systems, however, we must bear in mind the interest in chromene formation and its homopolymerization, leading to networks with excellent thermal, mechanical and moist aging properties [11, 12]. Thus our study initially involved the analysis of samples of dipropargylic compounds polymerized by heat treatment in the presence of catalyst and which should enable the entities present in the reaction mixture to be identified (chromene 0954-0083/96/040533+22$19.50 c 1996 IOP Publishing Ltd 533 at PENNSYLVANIA STATE UNIV on March 5, 2016 hip.sagepub.com Downloaded from
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of Propargylic- and chromene-terminated prepolymers. 4 ...

High Perform. Polym.8 (1996) 533–554. Printed in the UK
Propargylic- and chromene-terminated prepolymers.4. Influence of the type of catalyst on reaction mechanismsand kinetics
M F Grenier-Loustalot and C SanglarLaboratoire de physico-chimie des polymeres, CNRS-URA 1494, Helioparc-2, avenue du PdtAngot, 64000 Pau, France
Received 21 May 1996, accepted for publication 10 September 1996
Abstract. The addition of catalyst to polymerize the propargylic function causes a decreasein the reaction onset temperatureT0. Depending on the nature of the catalyst we demonstratedifferent reaction mechanisms involving the formation of the chromene group (as in the case ofnon-catalysed systems), of the Sabourin dimer, or the cyclooctatetraene structure in the moltenstate. We thus show that the catalytic effect is more determinant towards the propargylic functionthan towards the chromene group.
1. Introduction
Previous work on difunctional propargylic and chromene compounds [1, 2] has shownthat the zone of reactivity of these monomers is situated between 200 and 240◦C. Inorder to broaden the field of application of these systems and to facilitate their use, itappeared interesting to reduce the polymerization temperature by using a catalyst. To thisend, we investigated a number of copper-, nickel-, rhodium- and cobalt-based catalystsrecommended in the literature [3, 4] to polymerize acetylene triple bonds. The basis of thischoice was that a number of transition metals are known to catalyse reactions of acetylenemonomers [3], such as cyclotrimerization [5] and the formation of linear polyenes [6]. Inthe light of published data, two categories of acetylene monomers can be distinguished,whose terminations are:
(i) an arylacetylene group; in the absence of catalyst, mechanisms are complex and wehave been able to show that in the presence of catalyst, kinetics were modified, while thereaction mechanisms remained comparable [7–9];
(ii) an aryl ether propargylic group (the object of our study).
In the absence of catalyst, the reaction mechanism of the latter involves the formationof a stable chromene-type entity (β stage) [1, 10]. Data obtained with non-catalysedsystems were compared with those gathered following catalytic action. In the study ofcatalysed systems, however, we must bear in mind the interest in chromene formationand its homopolymerization, leading to networks with excellent thermal, mechanical andmoist aging properties [11, 12]. Thus our study initially involved the analysis of samplesof dipropargylic compounds polymerized by heat treatment in the presence of catalyst andwhich should enable the entities present in the reaction mixture to be identified (chromene
0954-0083/96/040533+22$19.50c© 1996 IOP Publishing Ltd 533
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from

534 M F Grenier-Loustalot and C Sanglar
structure or others). If this were not possible, the potential interest in the new structureformed, which could change the expected structures, would be determined.
Subsequently and in light of results obtained with dipropargylic prepolymers in thepresence of catalyst and the interest of dichromene prepolymers, the homopolymerizationreaction of theβ stage in the presence of catalyst was studied. The aim of this was to confirmthat these catalysts act preferentially on the polymerization of the triple bond. Thus, fivecatalysts were investigated in this work (table 1).
Table 1. Chemical formulae of the six catalysts used in this work and chemical structure of thethree compounds studied in this work.
Catalyst Formulae
A (C5H5)Co(CO)2B CuClC CuCl2D (C5H5)2NiE ((C6H5)3P)2NiCl2F RhCl((C6H5)3P)3
2. Experiment
2.1. Samples
The study undertaken and the results presented involve the propargylic compound ofbisphenol-A1 (R = (CH3)2C). We have previously shown that the nature of the bridginggroup R has an effect on reaction rates, but little effect on the mechanisms involved. Sincecompound1 is a solid (as are the catalysts) mixtures were prepared by simple cryogenicgrinding. In order to obtain comparable results from the point of view of reaction reactivityand kinetics, we studied two dichromene compounds (R= (CH3)2C 2, and R= (CF3)2C3), in the presence of catalyst.
These dichromene prepolymers are high-viscosity oils and slight heating was required to
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from

Effect of catalyst on propargylic function 535
homogenize the mixture. Prior work [2] has described their synthesis and physicochemicalnature.
2.2. Experimental techniques
2.2.1. Fourier transform infrared absorption spectroscopy (FT-IR).FT-IR spectra wererecorded with a Bruker IFS 45 spectrometer under the following conditions: spectral width4000–400 cm−1, 32 accumulations, resolution 2 cm−1, signal processing by triangularapodization. Depending on the physical state of the samples, they were recorded eitheras a film between two plates of NaCl or as a potassium bromide pellet.
2.2.2. Nuclear magnetic resonance spectroscopy (NMR).High-resolution liquid state1Hand 13C NMR spectra were recorded with a Bruker AM 400 spectrometer. Spectra wererecorded under the following conditions: pulse angle 90◦ (7 µs for 1H, 4.2 µs for 13C),digital resolution 0.122 Hz/point (1H) and 0.375 Hz/point (13C), corresponding to a spectralwidth of 4000 Hz (1H), 20 000 Hz (13C) and 72 000 Hz (19F), for a memory space of 64 K(1H) and 128 K (13C). Chemical shifts are given with reference to tetramethylsilane (TMS)as internal standard.
Solid phase13C NMR spectra (crossed polarization magic angle spinning) were obtainedwith a Bruker ARX 300 spectrometer operating in quadrupole detection. Samples (finepowder) were placed in a boron nitride sample holder. Contact time was 1000µs, the delaybetween sequences was 5 s and samples were spun at 6500 Hz. A mean of 5000 contactswas required to obtain an interpretable spectrum with a suitable signal/noise ratio. Chemicalshifts were calculated from the line of polyoxymethylene (Delrin) at 80 ppm (internalstandard).
2.2.3. High-performance liquid chromatography (HPLC).HPLC analyses were conductedwith a Varian 5000 spectrometer equipped with a fixed-wavelength UV detector (254 nmin our case) and a VARIAN 9010 spectrometer combined with a photodiode array scanningall wavelengths.
Chromatographic separation was on a column of Spherisorb ODS-2 (C18 bondedsilica) at a flow rate of 1 ml min−1. Solvent conditions varied depending on thesamples for analysis (they are described in the legend corresponding to each chromatogrampresented).
2.2.4. Differential scanning calorimetry (DSC).DSC diagrams were recorded with aSetaram DSC 111G differential scanning calorimeter, programmed at 5 K min−1 (first pass)or 10 K min−1 (second pass).
2.2.5. Operating conditions for monitoring polymerization reactions in isothermalconditions. 400 mg of product were weighed and placed in a glass tube which was sealedunder nitrogen and placed in an oven at a fixed and constant temperature. Samples weretaken at regular intervals (every 30 min).
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from

536 M F Grenier-Loustalot and C Sanglar
3. Analysis of the results
3.1. Study of polymerization mechanisms of dipropargylic prepolymers in the presence ofcatalyst
Compound1 (R = (CH3)2C) was thermally polymerized in the presence of five catalystswhose proportions are listed in table 2. Each sample was analysed by DSC in the temperaturerange of 50 to 350◦C (5 K min−1 rise). The resulting DSC diagrams (figure 1) show thatall catalysts with the exception of (A) considerably decreased the reaction onset temperatureT0 (50 < 1T < 150◦C) (table 2). Depending on the nature of the catalyst, one or twopolymerization exotherms were seen on the DSC diagrams.
Table 2. Influence of the catalyst on the polymerization onset temperatureT0.
Catalyst Quantity (pcr) T0(◦C)
A 0.5 240B 1 194C 1 185D 0.1 161E 0.3 120F 1 90
0 240
Thus, cuprous (B) and cupric (C) chlorides reduced the reaction temperature by 45 and55◦C respectively. In the case of CuCl (B) there was an intense peak at 194◦C attributableto catalysed polymerization reactions, and a residual of non-catalysed polymerization at240◦C. The latter was absent when the same experiment was run in the presence of cupricchloride (C). This compound is apparently the best adapted to these systems, since forthe same percentage of catalyst, polymerization temperature was lower and polymerizationresiduals were absent.
In the case of the Wilkinson catalyst (F: RhCl((C6H5)3P)3) reported in the literature[13], there were two polymerization exotherms. This confirms that both catalysed and non-catalysed reactions were occurring. The exotherm at the higher temperature is attributableto non-catalysed reactions of residual propargylic functions, in light of the shape of theDSC diagram of compound1, whereas catalysed reactions occurred at a lower temperature(T0 = 90◦C). A catalyst content of 1 pcr was not sufficient to totally shift the polymerizationexotherm at 90◦C. Beyond this temperature, we are no longer in the framework of catalyticdoses, and because of the thermal stability of these systems this may cause a degradationof the final properties of the material.
If only reaction onset temperatures (T0) are taken into account in order to improve theutilization of these polymers (without taking into account the reactions involved and thefinal properties of the crosslinked network), we should essentially study three catalysts.In particular, nickeloceneD (C5H5)2Ni (T0 = 161◦C) and catalystE ((C5H5)3P)2NiCl2(T0 = 120◦C) used in small quantities (0.1 and 0.3 pcr) in comparison to catalystsA andC, which catalyse all the propargylic functions present in the reaction medium at relativelylow percentages. CatalystF at high concentration began to catalyse the systems when thedipropargylic monomer melted. In order to catalyse the entire reaction medium, however,high concentrations were necessary (%> 4 pcr).
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from
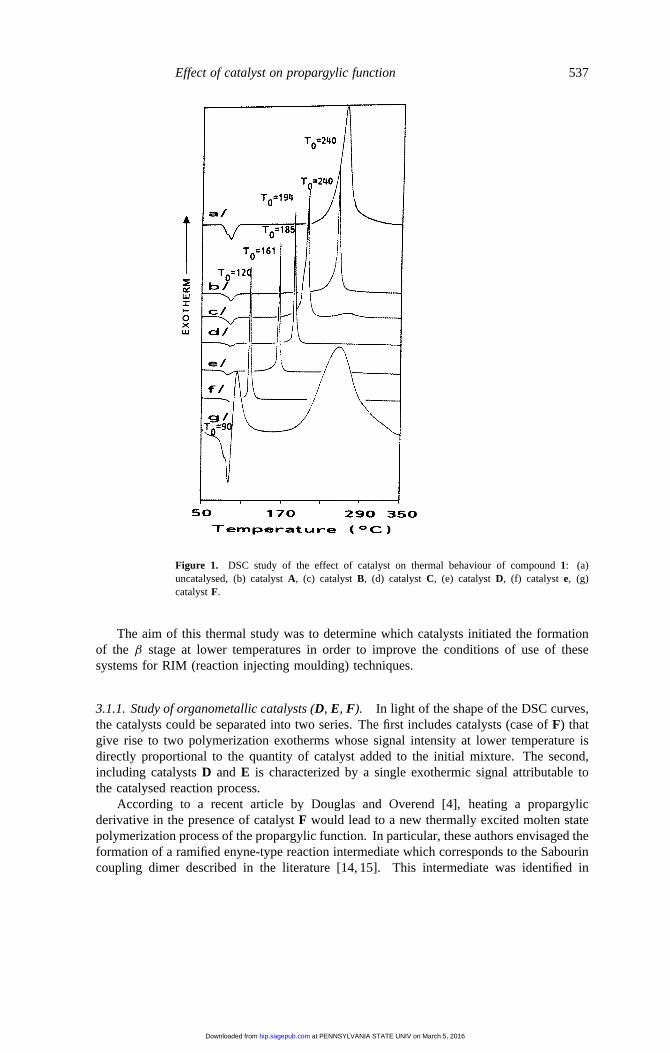
Effect of catalyst on propargylic function 537
Figure 1. DSC study of the effect of catalyst on thermal behaviour of compound1: (a)uncatalysed, (b) catalystA, (c) catalystB, (d) catalystC, (e) catalystD, (f) catalyst e, (g)catalystF.
The aim of this thermal study was to determine which catalysts initiated the formationof the β stage at lower temperatures in order to improve the conditions of use of thesesystems for RIM (reaction injecting moulding) techniques.
3.1.1. Study of organometallic catalysts (D, E, F). In light of the shape of the DSC curves,the catalysts could be separated into two series. The first includes catalysts (case ofF) thatgive rise to two polymerization exotherms whose signal intensity at lower temperature isdirectly proportional to the quantity of catalyst added to the initial mixture. The second,including catalystsD and E is characterized by a single exothermic signal attributable tothe catalysed reaction process.
According to a recent article by Douglas and Overend [4], heating a propargylicderivative in the presence of catalystF would lead to a new thermally excited molten statepolymerization process of the propargylic function. In particular, these authors envisaged theformation of a ramified enyne-type reaction intermediate which corresponds to the Sabourincoupling dimer described in the literature [14, 15]. This intermediate was identified in
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from

538 M F Grenier-Loustalot and C Sanglar
Figure 2. Structure of the Sabourin coupling dimer according to [14].
work on the mechanisms and kinetics of molten state thermal polymerization of telechelicoligomers with acetylene terminations [7–9]. The Sabourin coupling dimer [14, 15] resultsfrom head to tail chain coupling between two acetylene functions (figure 2) according to [16].
We believed it necessary to confirm this structure by the simultaneous1H and13C NMRanalysis of two rings, which would furnish data on the presence of C≡C−C=C links inthe chain. In particular, we were interested in catalysed reactions occurring at the exothermappearing at 90◦C. In order to do this and to analyse the first species formed, we treatedsamples at temperatures varying from 50 to 130◦C (5 K min−1 rise). Temperature conditionswere chosen in order to reproduce the conditions of the DSC study and to be situatedbetween the two polymerization exotherms, since beyond this point the polymerizationof non-catalysed residual propargylic groups was expected (formation of theβ stage andhomopolymerization of the latter) as already reported [1, 2].
The1H NMR spectroscopic analysis of the reaction mixture (figure 3) showed new linesindicating the presence of an entity different from theβ stage. In particular, between 4.3 and5.8 ppm, four relatively broad singlets appeared in the region where the methylene groupsof residual dipropargylic prepolymers resonate (compound1) [3]. In addition two singletswere observed at 1.6 ppm, a region in which methyl group protons resonate, correspondingto the two methyl groups of residual monomer1 and of the Sabourin dimer formed. Thesignals at 4.58 and 4.83 could be attributed to the methylene groups appearing on both sidesof the C≡C−C=C link. The signal at 4.83 ppm, closer to that characteristic of compound1 (doublet at 4.6 ppm), is in fact attributable to the two equivalent 3′ protonsα to the triplebond (table 3), whereas protons 3α to the double bond also resonated as a singlet butaround 4.6 ppm. Protons 1 and 1′ α to the double bond that blocked all rotation movementresonated as a singlet because of their magnetic inequality. They appeared at a lower field(5.6 ppm) than protons 3 and 3′.
Simultaneous analysis of the sample by liquid state13C NMR confirmed the formationof the Sabourin dimer (figure 4). In the 30–90 ppm region, carbons 3 and 3’ (methylenegroups) resonated, appearing at 70 and 56 ppm respectively, as well as those of acetylenecarbons characteristic of C≡C−C=C links. The latter resonated around 85 ppm whereasthe acetylene carbons characteristic of the residual propargylic group appeared at 75 and78 ppm [2].
Similarly, between 120 and 127 ppm there were two signals characteristic of theprotruding carbon group〉C=CH2.
The structure of this reaction intermediate could be identified throughout the entirespectral range (0–200 ppm) (table 4).
In agreement with published data [7–9, 16], we have shown that in the presence ofcatalystF compound1 led to the formation of a principal Sabourin coupling dimer product.Considering the small proportion of this compound in the medium after heat treatment,we attempted to determine if it could be obtained in larger proportions. After varying the
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from

Effect of catalyst on propargylic function 539
Figure 3. Identification of the Sabourin dimer by1H NMR (compound 1 + F 1 pcr;50 → 130◦C; solvent CDCl3).
proportion of catalyst in the reaction mixture, it was found that the quantity of Sabourindimer formed was directly proportional to the quantity of catalystF added. Catalyst shouldreach practically 10 pcr in order to obtain 100% dimer, but in this case it is not a catalyticrole that is shown, but rather that compoundF initiates the formation of the Sabourin dimerwithout leading to the formation of higher oligomers.
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from

540 M F Grenier-Loustalot and C Sanglar
Table 3. Identification of Sabourin dimer by1H NMR.
Atomsa Chemical Shifts (ppm)
1 5.59
1′ 5.65
3 4.47
3′ 4.77
5–9 6.796–8 7.10
a Designation of atoms:
Table 4. Identification of the Sabourin dimer by liquid state13C NMR.
Carbonsa Chemical shifts (ppm)
1 123.251′ 84.68
2 126.432′ 85.55
3 70.12
3′ 56.54
4 156.044′ 155.60
5–9 114.315′–9′ 114.31
6–8 143.477 143.947′ 143.84
a Designation of atoms:
In the case of the nickel-based catalystsD andF, the mechanisms of polymerization ofpropargylic derivatives had been previously studied with1H NMR spectroscopy [4]. Newresonance lines were detectable in the region where methylene groups appeared and wouldbe characteristic of the cyclotrimerization reaction leading to 1,3,5- and 1,2,3-trisubstitutedtrimers (figure 5).
Considering the complexity of the expected structures, and in the absence of modelcompounds characteristic of each structure that may form, we undertook a precise anddetailed mechanistic study in order to confirm the formation of the structures proposedabove. CatalystE had a polymerization exotherm at 120◦C and so it was decided to workat 100◦C in order to identify the initial species formed. The sample subjected to this
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from

Effect of catalyst on propargylic function 541
Figure 4. Identification of the Sabourin dimer by13C NMR (compound1 + F 1 pcr;50 → 130◦C; solvent CDCl3).
temperature for 45 min was analysed by HPLC. There were two major peaks that couldbe attributed to residual compound1 (retention time, rt= 15 min) and to a compound ofhigher mass (rt= 45 min) whose structure could correspond to the condensation of threemonomers as described in the literature [4].
The simultaneous1H NMR analysis of the reaction mixture enabled new signals to bedetected (figure 6):
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from

542 M F Grenier-Loustalot and C Sanglar
Figure 5. Formation of 1,3,5 and 1,2,4 trimers according to [4].
(i) in particular the appearance of two singlets at 5 and 5.1 ppm that could correspondto methylene groups after a ring formation reaction;
(ii) two singlets around 7.4 and 7.6 ppm indicating the substitution of aromatic rings;(iii) two doublets around 7.4 and 7.5 ppm attributable to a propargylic–chromene system.
Considering the multiplicity of the signals found in the spectrum between 7.4 and7.6 ppm (figure 6) the intermolecular ring formation reaction by addition of three acetylenegroups leading to the structures proposed in figure 5 was not possible. Simultaneous studyof the same sample by liquid state13C NMR indicated the presence of new resonancelines in very specific regions in comparison to the initial monomer. Spectra recorded fromselective DEPT pulse sequences enabled us to determine the class of carbons, facilitatingthe assignment of the different signals shown in the spectrum of figure 7(a). Thus, between30 and 45 ppm, carbons characteristic of the(CH3)2C pivot resonated. Beyond that, carbonresonance lines of the residual propargylic function of compound1 were present between50 and 80 ppm, as well as at four signals corresponding to methylene group carbons. Thesecarbons, two by two, have very similar chemical environments which would appear to showthat there was an addition reaction between four propargylic ether groups.
The DEPT spectrum between 105 and 140 ppm (figure 7(b)) also enabled us tocharacterize the signals characteristic of four tertiary carbons (114–127 ppm) and fourquaternary carbons (135–137 ppm) of equal intensity. As in the case of the methylenecarbons, two quaternary carbons resonated at very similar frequencies (δ: 134.9 and135.5 ppm) and the others at lower field (δ: 137.4 and 138 ppm) (table 5). Thus, themethylene carbons in the 50–80 ppm region were directly bound to these quaternary carbons(table 5). Similarly the presence of tertiary carbon (CH) resonance lines supports thehypothesis of a reaction between four OCH2C≡CH groups, leading to the formation of aneight-membered ring.
NMR spectroscopy results obtained with this sample suggested a cyclooctatetraenestructure with four substituents. In order to verify its formation, a structural study wasconducted, based on inter- and intramolecular addition mechanisms. Considering the
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from

Effect of catalyst on propargylic function 543
Figure 6. Identification of the cyclooctatetraene structure by1H NMR using compound1 in thepresence of 0.3 pcr of catalystE (T = 100◦C, t = 45 min, solvent CDCl3).
Table 5. Chemical shifts (ppm) of the compound formed in the presence of catalystE(T = 100◦C, t = 45 min, CDCl3).
Class of carbons Chemical shifts (ppm)
CH2 67.65, 67.84, 69.53, 69.68
CH 114.68, 125.97, 127.32, 129.17
− |C|− 134.94, 135.55, 137.40, 137.96
143.40 (C7)156.40 (C4)
polymerization of simple arylacetylene, the formation of benzene structures [17] is proposedaccording to a ring formation reaction involving a growing polyene radical. The reactionmay be transposed to propargylic ethers, which in the presence of catalystE and under heattreatment, yield conjugated polyene chains. Subsequently, the cyclooctatetraene structuremay form by intramolecular ring formation of the growing polyene radical with fourunsaturated bonds. The most probable reaction path is shown in figure 8. Dependingon the head-to-head or tail-to-tail links of the four propargylic functions, cyclooctatetraenestructures are obtained with differing substitution. In the present case, among the threepossible structures (figure 9) resulting from preferential addition of dienes, only one will bechosen based on the results of NMR results. Only the asymmetrical structure3 enables the
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from

544 M F Grenier-Loustalot and C Sanglar
Figure 7. Identification of the class of carbons and the cyclooctatetraene structure by13C NMR(T = 100◦C, t = 45 min, solvent CDCl3). (a) Decoupled, liquid state13C NMR protonspectrum. (b) DEPT spectrum (θ = 135◦).
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from

Effect of catalyst on propargylic function 545
Figure 8. Reaction path for the formation of the cyclo-octatetraene structure.
explanation of the multiplicity of1H NMR spectroscopy signals and the class of carbonsfound in the13C NMR spectrum.
In order to clarify the structure of 1,4,6,8-cyclooctatetraene, we considered the twopossible configurations on each unsaturated bond:
(i) the cis configuration;(ii) the trans configuration, thermally favoured.
In the case of conjugated polyene sequences, four possible configurations wereconsidered (figure 10). The configurational study of these structures showed that ringclosure was possible only in the case of thecis configuration, enabling us to propose eithera plane structure (cis–cisoidal configuration) or a tub structure (cis–transoidal configuration)for this compound.
Results obtained with1H NMR spectroscopy, electron diffraction and x-ray diffraction[18] showed that equilibrium strongly favours the tub structure at room temperature, witha very small quantity of planar structure. If we take ring tension into account andgiven that the C–C bond angle in the plane structure is about 130◦, the tub structure islogically favoured. Furthermore, in our case we should take into account the particularlyvoluminous structures on the cyclooctatetraene ring. They may assume preferential spatialorientations, thereby leading to a change in equilibrium and shift toward the tub structurewhich thus seems to be favoured. Since liquid state NMR spectroscopy enabled us toidentify a different reaction mechanism, it was interesting to analyse the samples (after 16to 24 h of heat treatment at 100◦C), insoluble in the usual solvents, in order to determinewhether crosslinking was related only to the formation of cyclooctatetraene structures orwhether several propargylic functions subsisted at chain ends, yielding theβ stage and itshomopolymerization.
This was done by carrying out the same study of samples with catalysts, based on resultsobtained with FT-IR and solid state13C NMR. There was noν C=C band in the infraredspectrum characteristic of chromene at 1634 cm−1 [2]. Several absorption bands disappearedbetween 1000 and 1200 cm−1 and only vibrations from the aromatic ring and the isopropylgroup remained unchanged, since they were not involved in the reaction mechanism. Inorder to define the network structure formed in presence of catalyst, we compared the solid
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from
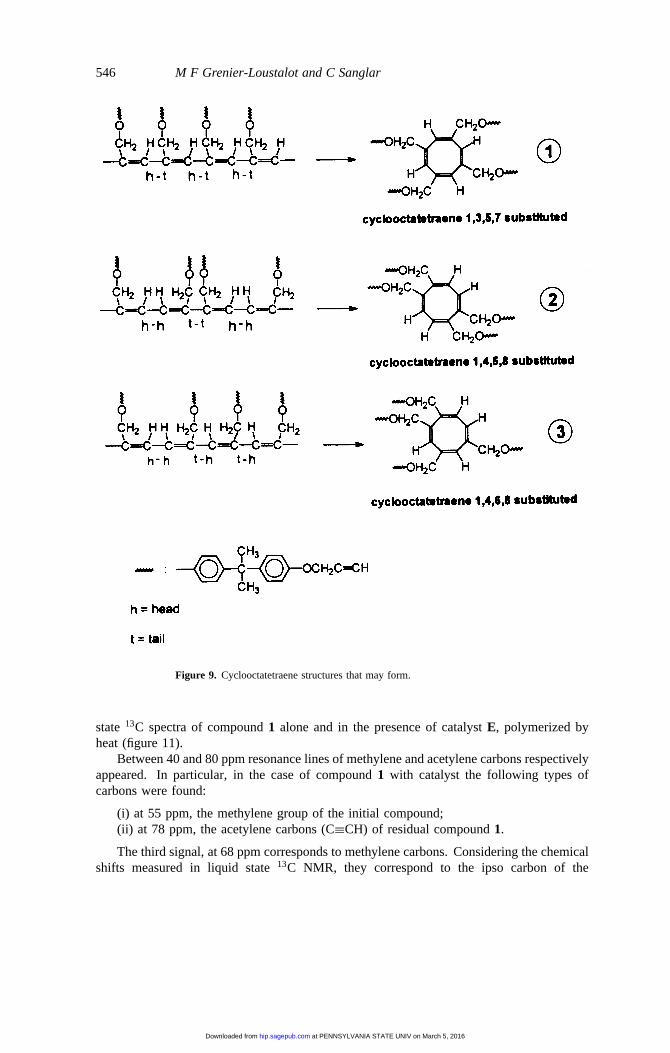
546 M F Grenier-Loustalot and C Sanglar
Figure 9. Cyclooctatetraene structures that may form.
state13C spectra of compound1 alone and in the presence of catalystE, polymerized byheat (figure 11).
Between 40 and 80 ppm resonance lines of methylene and acetylene carbons respectivelyappeared. In particular, in the case of compound1 with catalyst the following types ofcarbons were found:
(i) at 55 ppm, the methylene group of the initial compound;(ii) at 78 ppm, the acetylene carbons (C≡CH) of residual compound1.
The third signal, at 68 ppm corresponds to methylene carbons. Considering the chemicalshifts measured in liquid state13C NMR, they correspond to the ipso carbon of the
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from

Effect of catalyst on propargylic function 547
Figure 10. Configurations of 1,4,6,8 cyclooctatetraene.
cyclooctatetraene structure. Only compound1, which after heat treatment led to theβstage, furnished a signal at 65 ppm characteristic of the OCH2 group of the chromenegroup. In order to confirm the absence of theβ stage in presence of catalystE, the zonebetween 100 and 140 ppm was studied, where carbons 1 and 2 of the chromene groupappeared (at 122 and 123 ppm). The comparison of the spectra (figure 11) shows that thestructure of the network was modified in this zone and that the 123 ppm signal was absent.This led to the conclusion that compound1 in the presence of catalystE never yielded theβ stage or the three-dimensional network obtained by chromene homopolymerization [2].On the other hand, the cyclooctatetraene structure already described in solution could beconfirmed as a result of the signals at 68 ppm (OCH2) and 137 ppm, corresponding to allthe quaternary carbons of the eight-membered ring.
In conclusion, the dipropargylic compound1 in the presence of catalystE led to theformation of a network (figure 12), excluding the formation of the chromene group and itshomopolymerization in spite of the residual propargylic functions.
The thermal study with catalystD showed a single reaction isotherm starting at 160◦C.In order to identify the species formed and to determine the reaction mechanism, we workedin isothermal conditions at 140◦C. The first samples taken after 30 and 45 min of heattreatment were analysed by liquid state13C and1H NMR according to previously describedmethodology. The1H NMR signal characteristic of the methyl group (δ = 1–1.8 ppm)demonstrates the presence of two signals (singlets). Residual compound1 at 1.62 ppm wasin the vast majority in comparison to the signal at 1.57 ppm, characteristic of the compoundformed during catalysed processes.
In comparison to spectra of the non-catalysed product (chromene+phenol), the presenceof catalystD modified the reaction mechanism and yielded neither the formation of theβ
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from
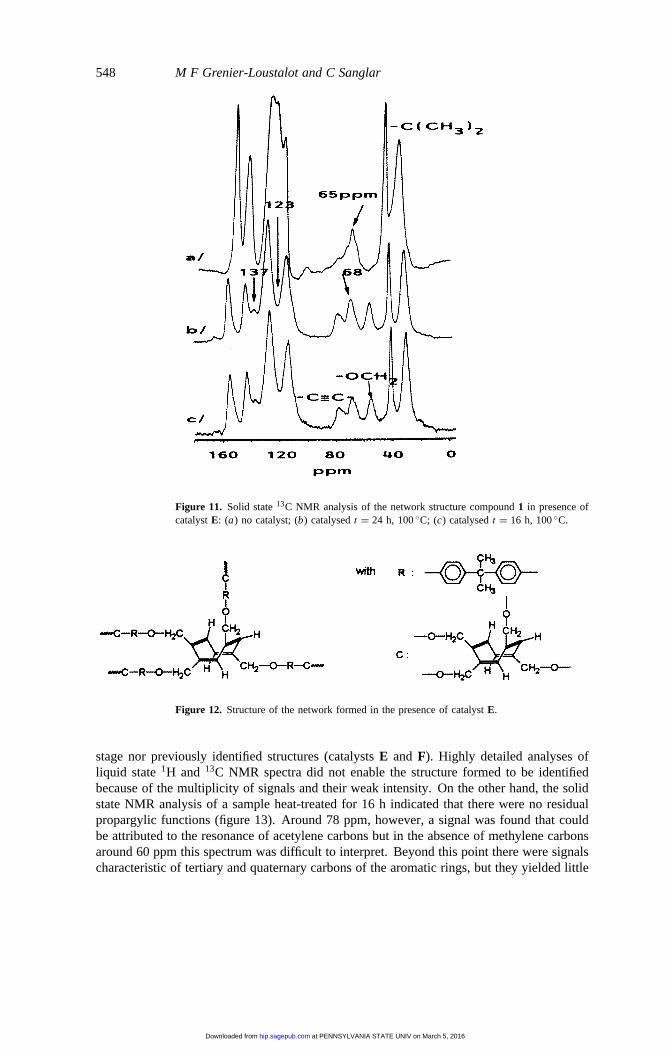
548 M F Grenier-Loustalot and C Sanglar
Figure 11. Solid state13C NMR analysis of the network structure compound1 in presence ofcatalystE: (a) no catalyst; (b) catalysedt = 24 h, 100◦C; (c) catalysedt = 16 h, 100◦C.
Figure 12. Structure of the network formed in the presence of catalystE.
stage nor previously identified structures (catalystsE and F). Highly detailed analyses ofliquid state1H and 13C NMR spectra did not enable the structure formed to be identifiedbecause of the multiplicity of signals and their weak intensity. On the other hand, the solidstate NMR analysis of a sample heat-treated for 16 h indicated that there were no residualpropargylic functions (figure 13). Around 78 ppm, however, a signal was found that couldbe attributed to the resonance of acetylene carbons but in the absence of methylene carbonsaround 60 ppm this spectrum was difficult to interpret. Beyond this point there were signalscharacteristic of tertiary and quaternary carbons of the aromatic rings, but they yielded little
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from

Effect of catalyst on propargylic function 549
Figure 13. Solid state13C NMR spectrum of compound1 + catalystD (T = 140◦C, t = 16 h).
data because of the width of the lines and the absence of model compounds enabling thespectrum to be interpreted precisely.
It was found that the use of nickelocene is not totally uninteresting in terms of theactivation of the molten state reaction by heat, even though we could not define the precisestructure of the network formed. The use of the material at 140◦C, however, remainsan interesting temperature for which, if this catalyst is to be used in the future, it will benecessary to conduct a more detailed study of the final properties of the crosslinked material(mechanical resistance, thermal stability, etc).
3.1.2. Study of copper-based catalysts (D, C). The dynamic DSC thermal study(50–350◦C) showed that catalystsB and C reduced reaction temperature by 45 and55◦C respectively. In order to control the reaction mechanism involved during heattreatment we followed changes in the heat-treated reaction mechanism under isothermalconditions at 180◦C with time. Monitoring the reaction with1H NMR indicated theformation of propargylic–chromene and dichromene prepolymers, previously identified whenpolymerization kinetics were followed without catalyst [1]. As a first approximation, it maybe said that catalystsB and C did not modify the molten state polymerization mechanismof propargylic prepolymers.
The solid state13C NMR analysis of polymerized samples (T = 190◦C for 16 h)(figure 14) shows that the structure of the network formed was very similar from a structuralpoint of view, whether or not a catalyst was used. Thus in the 0–80 ppm region, carbons ofthe isopropyl group not affected by the reaction mechanism resonated (at 31 and 42 ppm),as did methylene carbons of chromene and of the polymer formed by homopolymerizationof the β stage. The study of the different samples showed a change in the 65 ppm signalwhich was slightly less shielded when the polymer became predominant. It was in fact seenthat for the same heat treatment, compound1 in the presence of CuCl2 exhibited greaterreaction advancement.
Since catalystC was more interesting from the point of view of reaction onsettemperature and reaction progression, we compared the curves derived from data obtainedby isothermal calorimetry (figure 15) for compound1 alone, at 220◦C and for compound
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from

550 M F Grenier-Loustalot and C Sanglar
Figure 14. Influence of catalystsB and C on the structure of polymerized dipropargylicprepolymer: solid state13C NMR (T = 180◦C): (a) Compound1 no catalyst; (b) compound1+ 1% B, (c) compound1 + 1% C.
1 + catalystC at 180◦C. For identical reaction times but 40◦C lower, catalystC led to asignificant increase in both reaction advancement and rate. As an example, it was seen thatafter 1 h ofreaction, the advancement of compound1 at 220◦C was 0.5, which was 0.92for the same compound in the presence of catalystC at 180◦C. The use of a copper-basedcatalyst thus enables the reaction temperature to be lowered at the same time as acceleratingthe reaction process, leading to the formation of the three-dimensional structure describedin the literature [2].
3.2. Study of the homopolymerization reaction of theβ stage in the presence of catalyst
Considering the results obtained for theβ stage in the presence of a catalyst, four catalystswere chosen (B,C, E, F), based on the reaction temperature and the structures formed afterheat treatment. Compound2 was mixed with different catalysts at 1 phr, and after grindingeach sample was analysed by DSC in high-pressure stainless steel capsules. The DSCdiagrams (figure 16) show that regardless of the catalysts used there was an exothermalpolymerization peak, showing that in fact homopolymerization had occurred. Cupricchloride C, on the other hand, was the only catalyst that had an effect on the reactiononset temperatureT0.
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from

Effect of catalyst on propargylic function 551
Figure 15. Influence of catalystC on reaction advancement:�, 1, 220◦C; �, 1 + C, 180◦C.
Since the polymerization onset temperatureT0 of compound 2 was 235◦C, thetemperature decrease observed was about 95◦C. This shows that this catalyst had a veryspecific effect on intramolecular ring formation of the propargylic function and also had acertain influence on the opening of the double bond of theβ stage.
Based on thermal analysis results, the homopolymerization of theβ stage in the presenceof catalystC was studied in order to evaluate its influence on polymerization advancementand on the structure of the network at the end of the reaction. As an example figure 17shows the solid state13C NMR spectra of compound3 after 5 h ofheat treatment at 210◦Cfor the non-catalysed system and at 160◦C for the catalysed system in the presence of 1%catalystC.
We chose compound3 for this study since the hexafluoropropane pivot did not maskthe carbon signals at 30 ppm, corresponding to crosslinking by an ene–ene reaction of thechromene groups. At the limit of detection of solid state13C NMR, the presence of catalystC did not modify network structure, but for the same reaction time 50◦C lower, it furnisheda lower advancement in the presence of cupric chloride (based on the spectra shown infigure 17). The results obtained after quantifying the bands around 30 ppm are shown infigure 18. It was found that the 160◦C heat treatment was significant since it led to apolymer progression of 66% after 8 h instead of 84% without catalyst at 210◦C.
Overall, catalystC furnished good results, particularly concerning reaction temperature.The temperature reduction will lead to easier use and a broader range of applications.CatalystC, on the other hand, apparently limited the crosslinking rate to 70% at the chosenpolymerization temperature. It was thus found necessary to develop a thermal cycle leadingto the total crosslinking of the material in order to obtain better thermal properties.
4. Conclusion
The use of catalysts to polymerize the propargylic function in the molten state initiallypresented two findings of interest:
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from

552 M F Grenier-Loustalot and C Sanglar
Figure 16. DSC diagrams of compound2 in the presence of catalyst (1 pcr):B = CuCl;C = CuCl2; E = ((C6H5)3P)2NiCl2; F = RhCl((C6H5)3P)3.
(i) the decreased reaction temperature;(ii) the appearance of different reaction mechanisms leading to new chemical structures.
Among the catalysts studied in this work two categories can be distinguished:
(i) those modifying the reaction mechanism (F, E and D) in comparison to the non-catalysed system;
(ii) those auto-accelerating the formation of chromene and its homopolymerization (CandB).
Nevertheless, in order to better define the influence of the catalyst on the triple bondand on dichromene formed, we undertook a molten state thermal polymerization studyof difunctional compounds (dipropargylic and dichromene prepolymers) in the presence ofcatalyst. This study showed that, depending on their chemical nature, several polymerizationreaction mechanisms of the acetylene function may be involved. We were in fact able toshow that among the five catalysts reducing the reaction onset temperatureT0 of compound1, only one (C) acted on the exothermal peak characteristic of the homopolymerizationof compound 2 (dichromene prepolymer). Cupric chloride (C) tends to lower thereactivity zone of dipropargylic and dichromene prepolymers without changing the reactionmechanism. This confirms published data [10], i.e. catalystB used to prepare chromeneprepolymers in solvent, and catalystC, which is also copper based, would be better adapted
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from
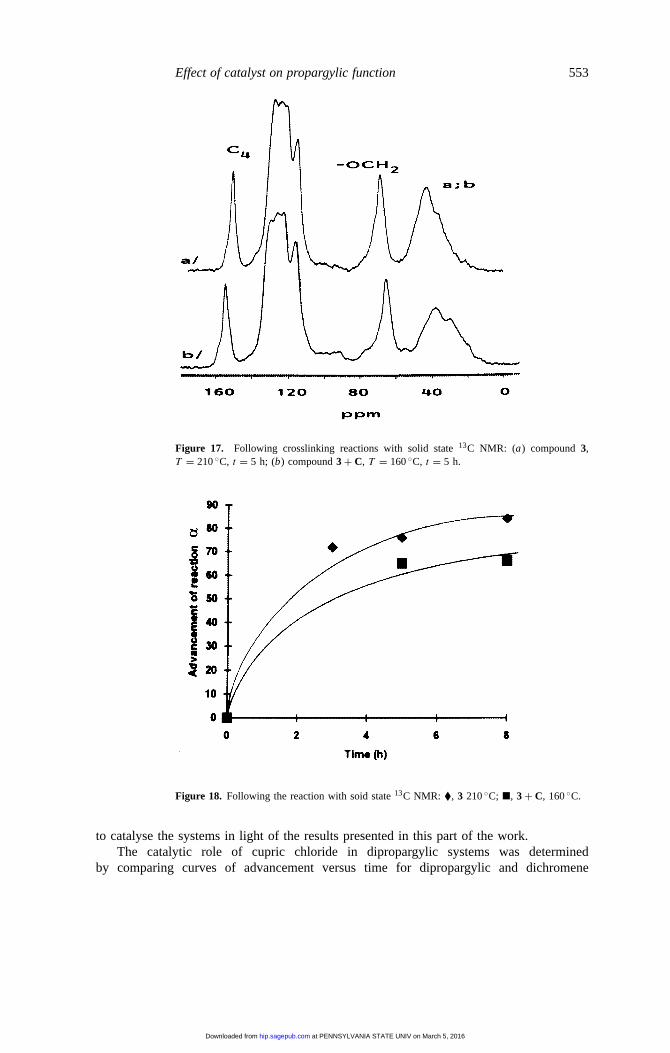
Effect of catalyst on propargylic function 553
Figure 17. Following crosslinking reactions with solid state13C NMR: (a) compound3,T = 210◦C, t = 5 h; (b) compound3 + C, T = 160◦C, t = 5 h.
Figure 18. Following the reaction with soid state13C NMR: �, 3 210◦C; �, 3 + C, 160◦C.
to catalyse the systems in light of the results presented in this part of the work.The catalytic role of cupric chloride in dipropargylic systems was determined
by comparing curves of advancement versus time for dipropargylic and dichromene
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from

554 M F Grenier-Loustalot and C Sanglar
prepolymers in the absence and presence of catalystC; in the case of the dichromeneprepolymer, however, the reaction is limited to 70% advancement. CatalystC may actpreferentially on the triple bond by activating the reaction and less on the double bond ofcompound3. In the case of compound3, the reaction is limited by gelling which leads toa reduced mobility of reaction functions and reduces the reaction rate, since after gellingonly diffusion mechanisms subsist.
Overall, the catalysts studied in this paper (nickel-, copper- and rhodium-based) areparticularly well adapted to the polymerization reactions of the acetylene group. We wereable to show that various mechanisms may occur, depending upon the nature of the catalyst.If we desire to go further in these catalysed systems, it is of fundamental interest to studythe networks formed (cyclooctatetraene, structures, etc), in terms of mechanical propertiesand thermal stability in order to evaluate the interest of these structures. In the present case,the utilization of catalysts would appear to be more appropriate for the polymerization ofdipropargylic prepolymers in comparison to dichromene prepolymers.
References
[1] Grenier-Loustalot M F and Sanglar C 1996High Perform. Polym.8 315[2] Grenier-Loustalot M F and Sanglar C 1996High Perform. Polym.8 341[3] Douglas W E and Overend A S 1991Polym. Commun.32 16[4] Douglas W E and Overend A S 1993Polymer34 7[5] Sefcik M D, Stejskal E O, McKay R A and Schaefer J 1979Macromolecules12 423[6] Collman J P, Hegedus L S, Norton J R and Finke R G 1987Principles and Applications of Organotransition
Metal Chemistry2nd edn (Mill Valley: University Science Books)[7] Grenier-Loustalot M F 1994High Perform. Polym.6 347[8] Grenier-Loustalot M F and Grenier P 1994Polym. Preprints351 33[9] Grenier-Loustalot M F 1995Makromol. Chem.93 237
[10] Grenier-Loustalot M F and Sanglar C 1996Eur. Polym. J.submitted[11] Dirlikov S K 1990 High Perform. Polym.2 1[12] Godschaix J P, Inbasekaran M N, Bartos B P, Scheck D M and Laman S A 199022nd Int. SAMPE Technical
Conf. (6–8 November)[13] Edwards H G M,Johnson A F and Lewis I R 1993Spectrochim. Acta49A 707–14[14] Sabourin E T 1984J. Mol. Cat.26 362[15] Sabourin E T 1979Am. Chem. Soc. Div. Petr. Prepr.24 233[16] Gandon S, Mison P, Bartholin M, Mercier R, Sillion B, Geneve E, Grenier P and Grenier-Loustalot M F
1996Polymerat press[17] Chauser M G, Rodionov Y U M and Cherhashin M I 1977J. Macromol. Sci. Chem.A11 1113[18] Roberts J D and Caserio M C 1965Basic Principles of Organic Chemistry(New York: Elsevier)243 826
at PENNSYLVANIA STATE UNIV on March 5, 2016hip.sagepub.comDownloaded from




















