
Production of meso- and giga-porous zirconia particles — An improved multi-step particle...
8
Production of meso- and giga-porous zirconia particles — An improved multi-step particle aggregation process Abhinandan Pattanayak, Anuradha Subramanian ⁎ Department of Chemical and Biomolecular Engineering, University of Nebraska-Lincoln, 207L Othmer Hall, Lincoln, NE, Zip-68588, USA abstract article info Article history: Received 28 April 2008 Received in revised form 12 January 2009 Accepted 26 January 2009 Available online 10 February 2009 Keywords: Catalyst Giga-porous Macro-porous Oil emulsion Polymer induced colloidal aggregation Zirconia Macro- and giga-porous zirconia supports were prepared from a 20% colloidal sol of zirconia (ZrO 2 ) by a combination of a polymer-induced colloid aggregation (PICA) process and the oil emulsion (OE) method. The effect of the pH of the initial sol on the size of the PICA particles, and subsequently on the final product, made by oil-emulsion assisted aggregation of the PICA particles was thoroughly investigated. Both the PICA and the OE methods were further optimized for performance. Particle morphology and porosity of the resultant particles were characterized by scanning electron microscopy, mercury intrusion–extrusion porosimetry, and nitrogen adsorption–desorption sorptometry. The supports were comprised of stable aggregates of 50– 250 μm in size. The pore and throat size distributions showed narrow bi-modal distributions over two distinct size scales: 10–100 nm and 600–3000 nm. In addition, different combinations of aggregation techniques and porous supports prepared in previous steps for use in a subsequent aggregation were evaluated. Optimal amounts of zirconia sol and 10–100 micron porous spherical particles produced by the OE method in an earlier step were combined in an additional OE process to yield stable giga-porous supports. Porous zirconia particles obtained after calcination and sintering had particle sizes of 0.15–3.5 mm and multimodal pore and throat distributions over a range of 50 nm–8 μm. © 2009 Elsevier B.V. All rights reserved. 1. Introduction The popularity of chromatographic processes in bioseparations has grown with advances in the fabrication and development of support materials, resulting in increased resolution in less time [1]. Supports- based on zirconia, which has high-density particles and excellent thermal and chemical stability, can offer increased flexibility relative to silica and polymeric phases. While the surface chemistry of zirconia has been studied in detail, the widespread applicability of zirconia particles in chromatography has been limited by the lack of availability of varying particle and pore sizes. Our previous work documented a spray-drying technique for production of 25–40 μm zirconia particles, with a pore size of 22 nm, from a 100 nm colloid suspension [2]. In addition, the utility of EDTPA-modified zirconia particles in the purification of immunoglobulins from cell culture supernatant and serum has also been demonstrated [2–4]. An analysis of the mass transport fluxes governing the transport of hIgG in the first generation of zirconia particles (particle diameter 22–35 μm and pore size 22 nm) suggested that pore diffusion was the rate-limiting transport mechanism for EDTPA-modified zirconia particles [5,6]. This was supported by the analysis that the support pore diameter should be in the range of 43 nm for hIgG (MW 155,000 Da) with a diameter of 8.5 nm, (the Renkin equation recommends a pore diameter that is at least five times the diameter of the solute to avoid severely restricting the rates of diffusion). The pore size requirements for supports intended for use in the separation of biomolecules (ranging from albumin [MW 56,000 Da] to fibrinogen [450,000 Da]) are in the range of 40 to 80 nm. Based on our research findings, the next logical step was then to produce zirconia supports with particle diameters in the range of 50 to 200 μm with pore sizes in the range of 35 to 100 nm. An alternative route to generate large pore sizes also includes the use of colloidal zirconia with larger particle sizes (i.e., 200 nm to 400 nm). However, it is worth noting that current techniques to produce colloid zirconia particles with sizes larger than 100 nm are tedious and uneconomical and are not commercially available. Thus, sol–gel and surfactant emulsion processes have been combined with organic and inorganic templates to fabricate macro/meso-porous structures of oxides like silica, titania, and zirconia. Successful and selective removal of these templates replaces the template sites with hollowness [7]. Initial attempts to control pore diameters and volumes of zirconia beads, prepared by the sol–gel process [8], involved incubation of the beads in a solution containing NaCl, the porogen [9,10]. This prevented the collapse of pore structure during the essential sintering step. The obtained zirconia particles were 1.3± 0.5 μm with a pore size of 20–35 nm. The literature cites the use of Powder Technology 192 (2009) 359–366 ⁎ Corresponding author. Tel.: +1 402 472 3463; fax: +1 402 472 6989. E-mail address: [email protected] (A. Subramanian). 0032-5910/$ – see front matter © 2009 Elsevier B.V. All rights reserved. doi:10.1016/j.powtec.2009.01.023 Contents lists available at ScienceDirect Powder Technology journal homepage: www.elsevier.com/locate/powtec
-
Upload
northwestern -
Category
Documents
-
view
2 -
download
0
Transcript of Production of meso- and giga-porous zirconia particles — An improved multi-step particle...

Powder Technology 192 (2009) 359–366
Contents lists available at ScienceDirect
Powder Technology
j ourna l homepage: www.e lsev ie r.com/ locate /powtec
Production of meso- and giga-porous zirconia particles — An improved multi-stepparticle aggregation process
Abhinandan Pattanayak, Anuradha Subramanian ⁎Department of Chemical and Biomolecular Engineering, University of Nebraska-Lincoln, 207L Othmer Hall, Lincoln, NE, Zip-68588, USA
⁎ Corresponding author. Tel.: +1 402 472 3463; fax: +E-mail address: [email protected] (A. Subram
0032-5910/$ – see front matter © 2009 Elsevier B.V. Adoi:10.1016/j.powtec.2009.01.023
a b s t r a c t
a r t i c l e i n f oArticle history:Received 28 April 2008Received in revised form 12 January 2009Accepted 26 January 2009Available online 10 February 2009
Keywords:CatalystGiga-porousMacro-porousOil emulsionPolymer induced colloidal aggregationZirconia
Macro- and giga-porous zirconia supports were prepared from a 20% colloidal sol of zirconia (ZrO2) by acombination of a polymer-induced colloid aggregation (PICA) process and the oil emulsion (OE) method. Theeffect of the pH of the initial sol on the size of the PICA particles, and subsequently on the final product, madeby oil-emulsion assisted aggregation of the PICA particles was thoroughly investigated. Both the PICA and theOE methods were further optimized for performance. Particle morphology and porosity of the resultantparticles were characterized by scanning electron microscopy, mercury intrusion–extrusion porosimetry, andnitrogen adsorption–desorption sorptometry. The supports were comprised of stable aggregates of 50–250 μm in size. The pore and throat size distributions showed narrow bi-modal distributions over twodistinct size scales: 10–100 nm and 600–3000 nm. In addition, different combinations of aggregationtechniques and porous supports prepared in previous steps for use in a subsequent aggregation wereevaluated. Optimal amounts of zirconia sol and 10–100 micron porous spherical particles produced by the OEmethod in an earlier step were combined in an additional OE process to yield stable giga-porous supports.Porous zirconia particles obtained after calcination and sintering had particle sizes of 0.15–3.5 mm andmultimodal pore and throat distributions over a range of 50 nm–8 μm.
© 2009 Elsevier B.V. All rights reserved.
1. Introduction
The popularity of chromatographic processes in bioseparations hasgrown with advances in the fabrication and development of supportmaterials, resulting in increased resolution in less time [1]. Supports-based on zirconia, which has high-density particles and excellentthermal and chemical stability, can offer increased flexibility relativeto silica and polymeric phases. While the surface chemistry of zirconiahas been studied in detail, the widespread applicability of zirconiaparticles in chromatography has been limited by the lack ofavailability of varying particle and pore sizes. Our previous workdocumented a spray-drying technique for production of 25–40 μmzirconia particles, with a pore size of 22 nm, from a 100 nm colloidsuspension [2]. In addition, the utility of EDTPA-modified zirconiaparticles in the purification of immunoglobulins from cell culturesupernatant and serum has also been demonstrated [2–4]. An analysisof themass transport fluxes governing the transport of hIgG in the firstgeneration of zirconia particles (particle diameter 22–35 μm and poresize 22 nm) suggested that pore diffusion was the rate-limitingtransport mechanism for EDTPA-modified zirconia particles [5,6]. This
1 402 472 6989.anian).
ll rights reserved.
was supported by the analysis that the support pore diameter shouldbe in the range of 43 nm for hIgG (MW155,000 Da) with a diameter of8.5 nm, (the Renkin equation recommends a pore diameter that is atleast five times the diameter of the solute to avoid severely restrictingthe rates of diffusion). The pore size requirements for supportsintended for use in the separation of biomolecules (ranging fromalbumin [MW 56,000 Da] to fibrinogen [450,000 Da]) are in the rangeof 40 to 80 nm. Based on our research findings, the next logical stepwas then to produce zirconia supports with particle diameters in therange of 50 to 200 μm with pore sizes in the range of 35 to 100 nm.
An alternative route to generate large pore sizes also includes theuse of colloidal zirconia with larger particle sizes (i.e., 200 nm to400 nm). However, it is worth noting that current techniques toproduce colloid zirconia particles with sizes larger than 100 nm aretedious and uneconomical and are not commercially available. Thus,sol–gel and surfactant emulsion processes have been combined withorganic and inorganic templates to fabricate macro/meso-porousstructures of oxides like silica, titania, and zirconia. Successful andselective removal of these templates replaces the template sites withhollowness [7]. Initial attempts to control pore diameters and volumesof zirconia beads, prepared by the sol–gel process [8], involvedincubation of the beads in a solution containing NaCl, the porogen[9,10]. This prevented the collapse of pore structure during theessential sintering step. The obtained zirconia particles were 1.3±0.5 µm with a pore size of 20–35 nm. The literature cites the use of

360 A. Pattanayak, A. Subramanian / Powder Technology 192 (2009) 359–366
diverse templates such as latex [11], cellulose acetate, cellulose nitrate,polyamide, polyethersulfone, polypropylene [12], acryl amide, glyci-dylmethacrylate [13], polystyrene cross-linked with divinyl benzene(beads) [14], polymethylmethacrylate [15], and others. In addition,surfactants like cationic dodecyltrimethylammoniumbromide andcetyltrimethylammoniumbromide, anionic sodium dodecylsulphate[16,17], nonionic polyethylene oxide and poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) block copolymer [18,19], aswell as macroemulsion and microemulsion [20,21] techniques, havefrequently been used to produce porous zirconia powders. The overalloutcome, however, yielded particle sizes of 0.1–3 µm, pore sizes 2.5–700 nm, and surface areas of 25–600 m2/g, depending on theprecursor, template, and method being used.
Previously, researchers have documented the application andsynthesis of stable zirconia chromatographic supports via polymer-induced colloid aggregation (PICA) [22–25] and spray drying (SD) [2]methods. In a recent, related study [26,27], the pore characteristics ofzirconia spheres were manipulated by the inclusion of a “leachableporogen” during the PICA step. Porogens at specific ratios were ableto enhance pore sizes, areas and, volumes, however; a reduction in themechanical strength of particles due to pore wall-thinning and porebody collapse was observed at higher porogen ratios. In addition,when porogens are included in the PICA method, irregular particles of3–15 µm are produced [26,27]. Other techniques such as oil emulsion(OE) and spray drying (SD) are commonly used to overcome theirregularities caused by the PICA method. This paper incorporates theOE method [28,29], which typically produces 10–120 µm sphericalparticles into the standard PICA process. This eliminates the draw-backs of both the PICA and OE processes, while retaining theirrespective advantages in production of meso- and giga-pores. In thisreport, both the PICA and OE methods were used, at each stephierarchically adding larger pores, maintaining mechanical stability,and enlarging the overall particle size. Fig. 1 depicts the proposedparticle preparation technique, where Panel-I represents experimentsalready discussed in our previous report on the use of porogens andPanel-II shows utilization of primary aggregates from experiments inPanel-I in combination with either or both of the OE and PICAmethods. This process may be repeated several times until the desiredpore and particle sizes have been obtained. None of the experimentsin Panel-II involved porogens, and only zirconia sol or its modifiedforms were used tomake the final products. Furthermore, the effect ofpH and concentrations of the starting material, mode and duration ofsedimentation, and presence of free colloids on the quality of the finalproduct were evaluated. The final products were characterized usingSEM, N2 sorptometry, and mercury porosimetry.
Fig. 1. Proposed pathways to synthesize porous zirconia particles. Panel-I depicts the synthesuse of porogens), Panel-II utilizes primary aggregates to form secondary macroporous/mes
2. Experimental section
2.1. Materials
Isopropyl alcohol, nitric acid, hydrochloric acid, formaldehyde(37%), sodium chloride and granulated urea were purchased fromFisher Scientific (Hanover Park, IL, USA). Aqueous zirconia sol (pH=3.0,100 nm), containing 20wt.% ZrO2 particles, was purchased from NyacolProducts Inc. (Ashland, MA, USA). Additionally, oleyl alcohol (70%) waspurchased from Sigma Aldrich and peanut oil (Lou Ana) was purchasedfromWal-Mart. Twomulti-speed medium capacity peristaltic pumps, anon-contact tachometer, a digital thermometer, an oil bath, and a benchtop ultra-centrifuge (Fisher Scientific Marathon 21000R, Thomas IEC)were some of the miscellaneous equipment involved in this study.
2.2. Methods
Broadly, the experiments belonged to the following two categories:
1) Oil Emulsion method of aggregation of particles that had beenfabricated by Polymer Induced Colloid Aggregation in a prior step.The final product or the method may be conveniently termed OE-of-PICA.
2) Oil Emulsion method of aggregation assisted by Polymer InducedColloid Aggregation of particles prepared by the Oil Emulsionmethod in a prior step (OE+PICA-of-OE).
The OE procedure detailed elsewhere [28,29] was adopted andfollowed. Briefly, each OE batch was pumped with 100–125 ml ofzirconia sol at 12ml/min, along with peanut oil at 36ml/min, throughan inline mixture. The dispersion was discharged in a plastic tumblercontaining 375 ml of a stirred (400 rpm) and preheated (100–105 °C)mixture of 1:2 oleyl alcohol and peanut oil. The surrounding oil bathwas maintained at 110–114 °C. After a batch time of 120–150 min, themixture was cooled, the oil decanted, and the particles were washedin 100–125 ml of 2-propanol. Finally, the particles were heat-cured ina muffle furnace at 115 °C/10–16 h, 175 °C/6–12 h, 375 °C/2 h, 750 °C/6 h, 900 °C/3 h, and 1200 °C/3 h.
2.2.1. OE-of-PICAThe PICA particles were produced, in bulk scale as multiple
batches, by the PICA method detailed elsewhere [22–25]. Pre-sonicated zirconia sol, with a pH ranging from 1.5 to 1.75, was mixedwith urea and 37% formaldehyde was added, followed by vigorousstirring. After 2 h of polymerization, the reaction was quenched withsix times the initial solution volume and allowed to settle. Sediments
is of micro/macro porous primary aggregates from zirconia colloid (with or without theoporous aggregates (with or without porogens).

361A. Pattanayak, A. Subramanian / Powder Technology 192 (2009) 359–366
were collected and re-subjected to sedimentation in a fresh volume ofDI water. The residuewas dispersed in a calculated amount of DI watersuch that the PICA sol (solution containing PICA particles) was 16–18%(w/w) of particles. This solution was stirred continuously to preventsedimentation. Subsequently, the PICA particles were aggregated bythe general OE procedure already discussed for making particles fromzirconia sol.
2.2.2. OE+PICA-of-OEAbout 15 g of previously sintered OE particles (10–100 µm) were
soaked in 30 ml (pH=1.5) nitric acid and 7–10 ml of 20% zirconia solwas added such that the colloid content in the overall mixturewas 10–20%. Necessary amounts of urea (1.25 M) and formaldehyde (2.4 M)were added and the preparation was stirred for 20–25 min. After anincrease in viscosity due to polymerization, the mixture was pumpedto the OE setup. Contact of the slurry with the hot oil phase wasexpected to increase the urea-formaldehyde polymerization rate [24]and the fine zirconia colloid particles stabilized the larger OE particles,thereby preventing the slurry droplets from dissociating to individualparticles upon drying. The rest of the curing process was similar to thegeneral OE procedure. The OE particles being used already containedlarge pores, which needed protection from the fresh colloid insolution. Two pore filling methods were formulated:
a) Pore saturation with NaCl: Prior to the preparation of themixture for the secondary OE step, the primary OE particles weresaturated with an aqueous slurry of NaCl (10–15 wt.%) and dried tofree flowing powder in an oven at 110 °C for 24 h. This pore fillingidea was similar to that of Shalliker et al. [9,10], where 5% NaClsolution was used. However, the use of NaCl also calls for itsremoval after sintering of the larger secondary OE particles, whichmay be achieved by overnight rocking of the particles in DI waterand then heating the particles to dryness at 375 °C.b) Pore saturation with water: The OE particles were initiallyboiled in DI water for 2–3 h followed by a slow cooling before the“secondary OE particle-making” step took place. The particles werenever allowed to dry or leave the aqueous phase.
3. Material characterization
3.1. SEM imaging
SEM imaging was carried out with the Hitachi S7400 on all thePICA samples (Electron microscopy facility, University of Nebraska-Lincoln) for accelerating voltages between 5 and 15 kV. Photographswere taken of a representative sample of each powder by sparinglysprinkling the powder onto double-sided sticky carbon conductive
Fig. 2. SEM images showing the effect of sol concentration on OE spherules. Sol concentratiorefined after sintering.
tape (Ted Pella Inc.) that was attached to an SEM stub. The particleswere then coated with a chromium film of thickness 0.2 nm by DCsputtering at 125 mA for 60 s. SEM micrographs were acquired asshown in Figs. 2, 4, 5, 6, and 8.
3.2. PSD/TSD from N2 sorption
PSD/TSD from nitrogen adsorption and desorption isotherm dataat 77 K were obtained using Micromeritics ASAP 2010 sorptometer.About 250 mg of each sample was analyzed at 42 different P/P0(relative pressure) points on both the adsorption and desorptionbranches. The isotherms obtained from a plot of “volume adsorbed”(cm3/g STP) versus “relative pressure” (P/P0) were compared againstthe following: 1) type IV isotherm with a type H1 hysteresis loop(indicating the presence of “cylindrical” pores), 2) type IV isothermwith a type H2 hysteresis loop (indicating the presence of “ink-bottleshaped” pores) and 3) type IV isotherm with an intermediate typeH1–H2 hysteresis loop (indicated by a combination of “ink-bottleshaped” and “cylindrical” pores) [9,10].
3.3. PSD/TSD from mercury porosimetry
Mercury porosimetry was performed for selected samples pre-sented in this report. All the samples were sent for analysis to theParticle Engineering Research Center at the University of Florida. TheQuantachrome Autoscan 60Mercury Porosimeter was used to analyzethe samples and Quantachrome Autoscan Poro2PC (Version 3.0) soft-ware was used to generate pore related data. Each analysis requiredabout 1 cm3 of sample and the pore size calculations and surface areaswere based on a contact angle of 140°.
4. Results and discussion
4.1. Effect of sol concentration
The effect of sol concentration on the morphology of particlesmade by the OE process was studied. The sol was concentrated to 20%,40%, and 50% by centrifugation of the commercially available 20% soland then diluted to the desired concentration. The feed rates of sol andoil and other process parameters of an OE batch were unaltered.Typically, as shown in Fig. 2 (a, b, c), all particles were near-spherical.The samples studied were not subjected to size classification byscreening, and all samples were imaged randomly. At a low solconcentration of 20%, the diameter ranged between 15 and 50 μm,with an average particle size of about 25 μm (Fig. 2 a). Each particlegave the impression of a smooth surface. At 40% sol concentration, theaverage particle size increased to 100 μm. The diameter ranged from40–150 μm (Fig. 2 b). However, another interesting feature was
n (wt.%) used: (a) 20%; (b) 40%; and (c) 50%. The particle size distribution has not been
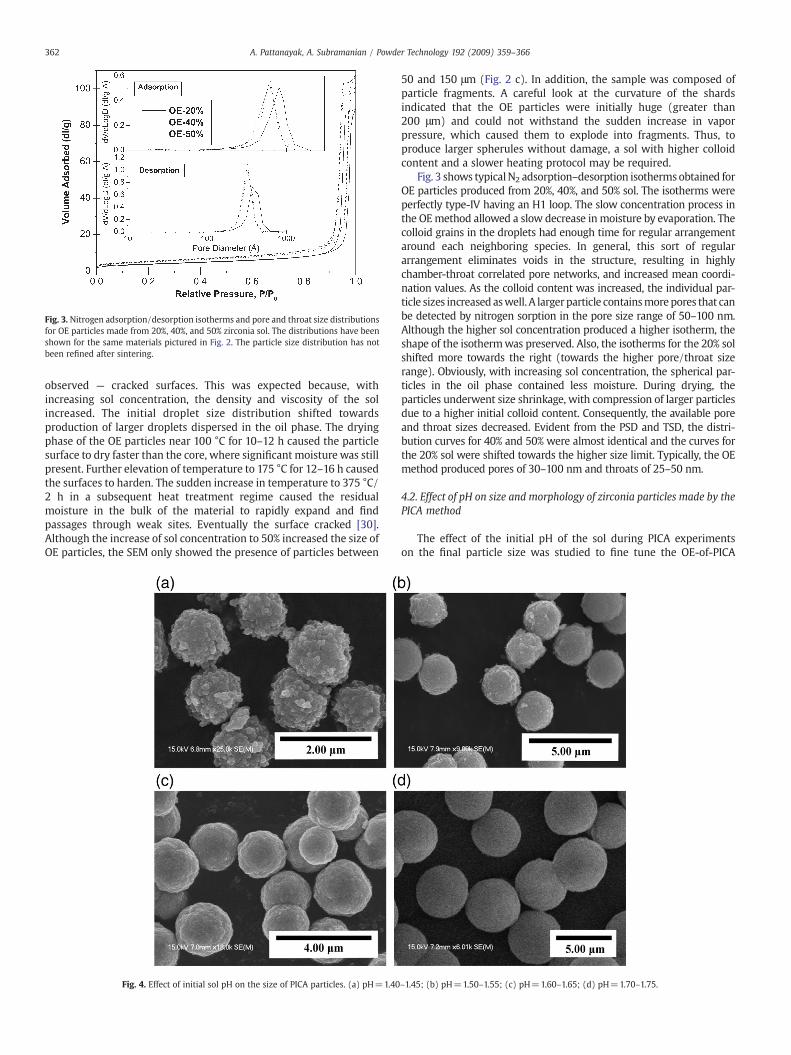
Fig. 3. Nitrogen adsorption/desorption isotherms and pore and throat size distributionsfor OE particles made from 20%, 40%, and 50% zirconia sol. The distributions have beenshown for the same materials pictured in Fig. 2. The particle size distribution has notbeen refined after sintering.
362 A. Pattanayak, A. Subramanian / Powder Technology 192 (2009) 359–366
observed — cracked surfaces. This was expected because, withincreasing sol concentration, the density and viscosity of the solincreased. The initial droplet size distribution shifted towardsproduction of larger droplets dispersed in the oil phase. The dryingphase of the OE particles near 100 °C for 10–12 h caused the particlesurface to dry faster than the core, where significant moisture was stillpresent. Further elevation of temperature to 175 °C for 12–16 h causedthe surfaces to harden. The sudden increase in temperature to 375 °C/2 h in a subsequent heat treatment regime caused the residualmoisture in the bulk of the material to rapidly expand and findpassages through weak sites. Eventually the surface cracked [30].Although the increase of sol concentration to 50% increased the size ofOE particles, the SEM only showed the presence of particles between
Fig. 4. Effect of initial sol pH on the size of PICA particles. (a) pH=1.40
50 and 150 μm (Fig. 2 c). In addition, the sample was composed ofparticle fragments. A careful look at the curvature of the shardsindicated that the OE particles were initially huge (greater than200 μm) and could not withstand the sudden increase in vaporpressure, which caused them to explode into fragments. Thus, toproduce larger spherules without damage, a sol with higher colloidcontent and a slower heating protocol may be required.
Fig. 3 shows typicalN2 adsorption–desorption isothermsobtained forOE particles produced from 20%, 40%, and 50% sol. The isotherms wereperfectly type-IV having an H1 loop. The slow concentration process inthe OEmethod allowed a slow decrease in moisture by evaporation. Thecolloid grains in the droplets had enough time for regular arrangementaround each neighboring species. In general, this sort of regulararrangement eliminates voids in the structure, resulting in highlychamber-throat correlated pore networks, and increased mean coordi-nation values. As the colloid content was increased, the individual par-ticle sizes increased aswell. A largerparticle containsmorepores that canbe detected by nitrogen sorption in the pore size range of 50–100 nm.Although the higher sol concentration produced a higher isotherm, theshape of the isothermwas preserved. Also, the isotherms for the 20% solshifted more towards the right (towards the higher pore/throat sizerange). Obviously, with increasing sol concentration, the spherical par-ticles in the oil phase contained less moisture. During drying, theparticles underwent size shrinkage, with compression of larger particlesdue to a higher initial colloid content. Consequently, the available poreand throat sizes decreased. Evident from the PSD and TSD, the distri-bution curves for 40% and 50% were almost identical and the curves forthe 20% sol were shifted towards the higher size limit. Typically, the OEmethod produced pores of 30–100 nm and throats of 25–50 nm.
4.2. Effect of pH on size and morphology of zirconia particles made by thePICA method
The effect of the initial pH of the sol during PICA experimentson the final particle size was studied to fine tune the OE-of-PICA
–1.45; (b) pH=1.50–1.55; (c) pH=1.60–1.65; (d) pH=1.70–1.75.

Fig. 5. (a–d) SEM images showing typical size and morphology of particles prepared from PICA particles by the OE method (OE-of-PICA). The particle size distribution has not beenrefined after sintering (a).
363A. Pattanayak, A. Subramanian / Powder Technology 192 (2009) 359–366
experiments. Separate experiments were conducted at pH 1.40–1.45,1.50–1.55, 1.60–1.65, and 1.70–1.75. Fig. 4 shows particle morphologyafter sintering and polymer removal. At a low pH (1.40–1.45), theparticles formed coarse spheres between 0.9 and 1.2 µm. The primaryparticles were aggregates of 100 nm spherical zirconia colloids. Withincreasing pH (Fig. 4 b, c, d), the morphology of the primary particleschanged from smaller textured spheres to larger smooth spherules.The registered diameter ranges for the remaining pH regimes were1.5–2.0 µm, 1.8–2.4 µm, and 4.0–4.5 µm, respectively. As detailedelsewhere [22], the mechanical strength of PICA particles improvessufficiently to withstand HPLC column packing for a sol pH above 1.4.For the present study, the pH was restricted between 1.40 and 1.75. Atthe lower end of this range, the particles were largely composed ofloosely packed sub-micrometer aggregates, whereas, towards theupper limit, the particles were rigid shells. The dependence ofaggregate size on the solution pH and the bridging flocculationmechanism have been elaborated by Annen et al. [22] and have been
Fig. 6. SEM images of OE-of-PICA particles made at different initial
found to agree well with the present study. Above pH=1.75, thecontribution of small pores to the total porosity is very significant [22],which is undesirable for many applications.
4.3. OE-of-PICA particles — general morphology and pore characteristics
Macro-porous zirconia supports were successfully synthesizedusing the OE-of-PICA method. The process was better controlled byusing optimum pH and sedimentation conditions discussed in theprevious sections. Fig. 5 (a–f) presents general morphologies of theOE-of-PICA particles at various magnifications. Typically, theseparticles were spherical and ranged between 75 and 250 μm (Fig. 5a). A closer look on the surface revealed a dense and random packingof the constituent PICA spheres (Fig. 5 b, c and d). Roughly thousandsof micron-sized spheres were arranged in a single OE particle, and thearrangement of these particles revealed a thermodynamically stablesystem in which at least three spheres enclosed a void on a two-
sol pH. (a) pH=1.45–1.50; pH=1.55–1.65; (c) pH=1.70–1.75.
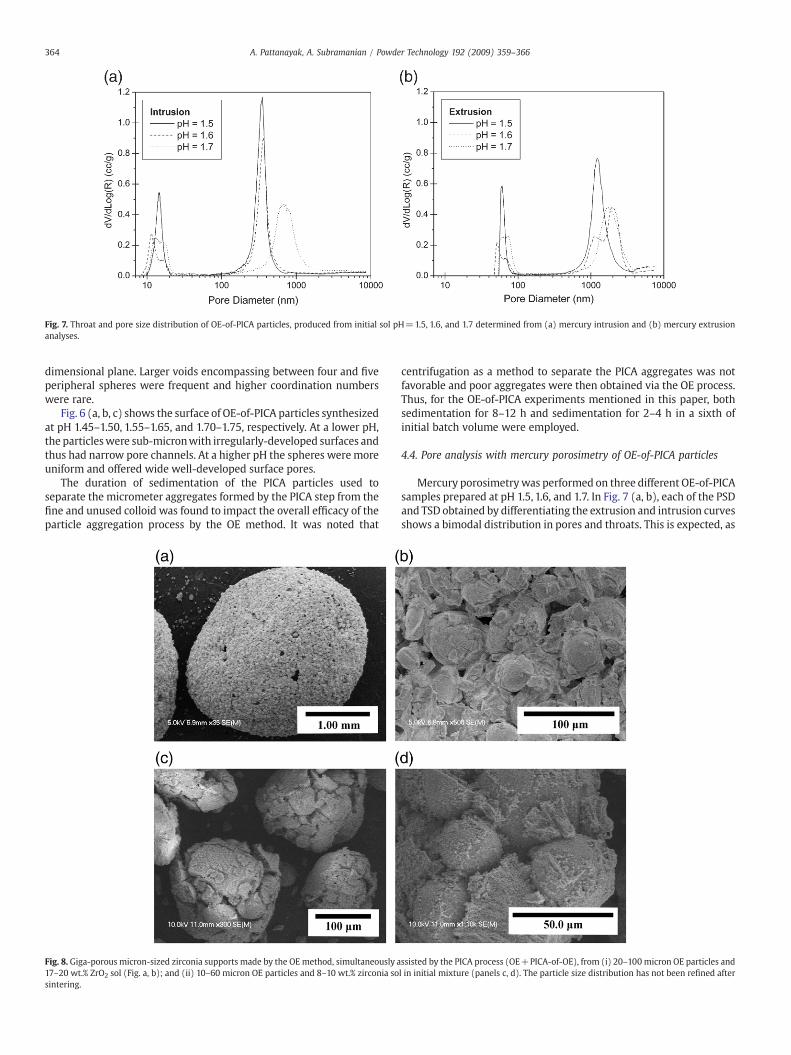
Fig. 7. Throat and pore size distribution of OE-of-PICA particles, produced from initial sol pH=1.5, 1.6, and 1.7 determined from (a) mercury intrusion and (b) mercury extrusionanalyses.
364 A. Pattanayak, A. Subramanian / Powder Technology 192 (2009) 359–366
dimensional plane. Larger voids encompassing between four and fiveperipheral spheres were frequent and higher coordination numberswere rare.
Fig. 6 (a, b, c) shows the surface of OE-of-PICA particles synthesizedat pH 1.45–1.50, 1.55–1.65, and 1.70–1.75, respectively. At a lower pH,the particleswere sub-micronwith irregularly-developed surfaces andthus had narrow pore channels. At a higher pH the spheres weremoreuniform and offered wide well-developed surface pores.
The duration of sedimentation of the PICA particles used toseparate the micrometer aggregates formed by the PICA step from thefine and unused colloid was found to impact the overall efficacy of theparticle aggregation process by the OE method. It was noted that
Fig. 8. Giga-porous micron-sized zirconia supports made by the OEmethod, simultaneously a17–20 wt.% ZrO2 sol (Fig. a, b); and (ii) 10–60 micron OE particles and 8–10 wt.% zirconia sosintering.
centrifugation as a method to separate the PICA aggregates was notfavorable and poor aggregates were then obtained via the OE process.Thus, for the OE-of-PICA experiments mentioned in this paper, bothsedimentation for 8–12 h and sedimentation for 2–4 h in a sixth ofinitial batch volume were employed.
4.4. Pore analysis with mercury porosimetry of OE-of-PICA particles
Mercury porosimetrywas performed on three different OE-of-PICAsamples prepared at pH 1.5, 1.6, and 1.7. In Fig. 7 (a, b), each of the PSDand TSD obtained by differentiating the extrusion and intrusion curvesshows a bimodal distribution in pores and throats. This is expected, as
ssisted by the PICA process (OE+PICA-of-OE), from (i) 20–100micron OE particles andl in initial mixture (panels c, d). The particle size distribution has not been refined after
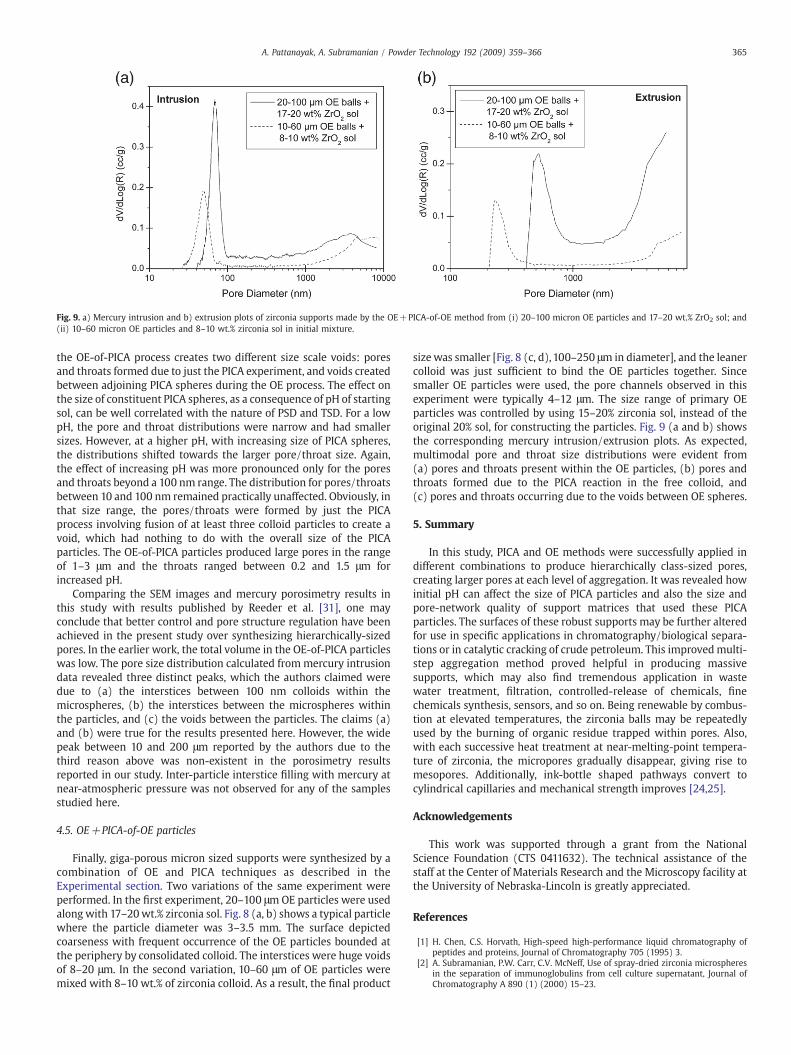
Fig. 9. a) Mercury intrusion and b) extrusion plots of zirconia supports made by the OE+PICA-of-OE method from (i) 20–100 micron OE particles and 17–20 wt.% ZrO2 sol; and(ii) 10–60 micron OE particles and 8–10 wt.% zirconia sol in initial mixture.
365A. Pattanayak, A. Subramanian / Powder Technology 192 (2009) 359–366
the OE-of-PICA process creates two different size scale voids: poresand throats formed due to just the PICA experiment, and voids createdbetween adjoining PICA spheres during the OE process. The effect onthe size of constituent PICA spheres, as a consequence of pH of startingsol, can be well correlated with the nature of PSD and TSD. For a lowpH, the pore and throat distributions were narrow and had smallersizes. However, at a higher pH, with increasing size of PICA spheres,the distributions shifted towards the larger pore/throat size. Again,the effect of increasing pH was more pronounced only for the poresand throats beyond a 100 nm range. The distribution for pores/throatsbetween 10 and 100 nm remained practically unaffected. Obviously, inthat size range, the pores/throats were formed by just the PICAprocess involving fusion of at least three colloid particles to create avoid, which had nothing to do with the overall size of the PICAparticles. The OE-of-PICA particles produced large pores in the rangeof 1–3 μm and the throats ranged between 0.2 and 1.5 μm forincreased pH.
Comparing the SEM images and mercury porosimetry results inthis study with results published by Reeder et al. [31], one mayconclude that better control and pore structure regulation have beenachieved in the present study over synthesizing hierarchically-sizedpores. In the earlier work, the total volume in the OE-of-PICA particleswas low. The pore size distribution calculated frommercury intrusiondata revealed three distinct peaks, which the authors claimed weredue to (a) the interstices between 100 nm colloids within themicrospheres, (b) the interstices between the microspheres withinthe particles, and (c) the voids between the particles. The claims (a)and (b) were true for the results presented here. However, the widepeak between 10 and 200 μm reported by the authors due to thethird reason above was non-existent in the porosimetry resultsreported in our study. Inter-particle interstice filling with mercury atnear-atmospheric pressure was not observed for any of the samplesstudied here.
4.5. OE+PICA-of-OE particles
Finally, giga-porous micron sized supports were synthesized by acombination of OE and PICA techniques as described in theExperimental section. Two variations of the same experiment wereperformed. In the first experiment, 20–100 μmOE particles were usedalongwith 17–20wt.% zirconia sol. Fig. 8 (a, b) shows a typical particlewhere the particle diameter was 3–3.5 mm. The surface depictedcoarseness with frequent occurrence of the OE particles bounded atthe periphery by consolidated colloid. The interstices were huge voidsof 8–20 μm. In the second variation, 10–60 μm of OE particles weremixed with 8–10 wt.% of zirconia colloid. As a result, the final product
size was smaller [Fig. 8 (c, d), 100–250 μm in diameter], and the leanercolloid was just sufficient to bind the OE particles together. Sincesmaller OE particles were used, the pore channels observed in thisexperiment were typically 4–12 μm. The size range of primary OEparticles was controlled by using 15–20% zirconia sol, instead of theoriginal 20% sol, for constructing the particles. Fig. 9 (a and b) showsthe corresponding mercury intrusion/extrusion plots. As expected,multimodal pore and throat size distributions were evident from(a) pores and throats present within the OE particles, (b) pores andthroats formed due to the PICA reaction in the free colloid, and(c) pores and throats occurring due to the voids between OE spheres.
5. Summary
In this study, PICA and OE methods were successfully applied indifferent combinations to produce hierarchically class-sized pores,creating larger pores at each level of aggregation. It was revealed howinitial pH can affect the size of PICA particles and also the size andpore-network quality of support matrices that used these PICAparticles. The surfaces of these robust supports may be further alteredfor use in specific applications in chromatography/biological separa-tions or in catalytic cracking of crude petroleum. This improvedmulti-step aggregation method proved helpful in producing massivesupports, which may also find tremendous application in wastewater treatment, filtration, controlled-release of chemicals, finechemicals synthesis, sensors, and so on. Being renewable by combus-tion at elevated temperatures, the zirconia balls may be repeatedlyused by the burning of organic residue trapped within pores. Also,with each successive heat treatment at near-melting-point tempera-ture of zirconia, the micropores gradually disappear, giving rise tomesopores. Additionally, ink-bottle shaped pathways convert tocylindrical capillaries and mechanical strength improves [24,25].
Acknowledgements
This work was supported through a grant from the NationalScience Foundation (CTS 0411632). The technical assistance of thestaff at the Center of Materials Research and the Microscopy facility atthe University of Nebraska-Lincoln is greatly appreciated.
References
[1] H. Chen, C.S. Horvath, High-speed high-performance liquid chromatography ofpeptides and proteins, Journal of Chromatography 705 (1995) 3.
[2] A. Subramanian, P.W. Carr, C.V. McNeff, Use of spray-dried zirconia microspheresin the separation of immunoglobulins from cell culture supernatant, Journal ofChromatography A 890 (1) (2000) 15–23.

366 A. Pattanayak, A. Subramanian / Powder Technology 192 (2009) 359–366
[3] Anuradha Subramanian, Sabyasachi Sarkar, Use of a modified zirconia support inthe separation of immunoproteins, Journal of Chromatography A 944 (2002)179–187.
[4] Anuradha Subramanian, Sabyasachi Sarkar, Interaction of immunoglobulin G withN,N,N′,N′-ethylenediaminetetramethylenephosphonic acid-modified zirconia,Journal of Chromatography A 989 (2003) 131–138.
[5] Anuradha Subramanisn, Sabyasachi Sarkar, Modeling of immunoglobulin uptakeby N,N,N′N′ ethylenediaminetetramethylenephosphonic acid-modified zirconiaparticles under static and dynamic conditions, Journal of Chromatography B 821(2005) 81–87.
[6] Sabyasachi Sarkar, Peter W. Carr, Anuradha Subramanian, Identification of themass transfer mechanisms involved in the transport of human immunoglobulin-Gin N,N,N′,N′-ethylenediaminetetramethylenephosphonic acid modified zirconia,Journal of Chromatography B 821 (2) (2005) 124–131.
[7] Galo J. de, A.A. Soler-Illia, Clement Sanchez, Benedicte Lebeau and Joel Patarin,Chemical strategies to design textured materials: from microporous andmesoporous oxides to nanonetworks and hierarchical structures, ChemicalReviews 102 (2002) 4093–4138.
[8] U. Trudinger, G. Muller, K.K. Unger, Porous zirconia and titania as packingmaterialsfor HPLC, Journal of Chromtography 535 (1990) 111–125.
[9] R.A. Shalliker, G.K. Douglas, P.R. Comino, P.E. Kavanagh, Examination of variouspore size zirconias for potential chromatographic applications, Journal of PowderTechnology 91 (1997) 17–23.
[10] R.A. Shalliker, G.K. Douglas, Controlling the pore structure of zirconia for chro-matographic applications, Journal of Liquid Chromatography & Related Technol-ogies 20 (11) (1997) 1651–1666.
[11] Carla Verissimo, Oswaldo L. Alves, Microstructural modifications in macroporousoxides prepared via latex templating: synthesis and thermal stability of porousmicrostructure, Journal of the American Ceramic Society 89 (7) (2006) 2226–2231.
[12] Jan H. Schattka, Edeline H.-M. Wong, Markus Antonietti, Rachel A. Caruso, Sol–geltemplating of membranes to form thick, porous titania, titania/zirconia andtitania/silica films, Journal of Materials Chemistry 16 (2006) 1414–1420.
[13] Jan H. Schattka, Dmitry G. Shchukin, Jianguang Jia, Markus Antonietti, Rachel A.Caruso, Photocatalytic activities of porous titania and titania/zirconia structuresformed by using a polymer gel templating technique, Chemistry of Materials 14(2002) 5103–5108.
[14] Dmitry G. Shchukin, Rachel A. Caruso, Template synthesis and photocatalyticproperties of porous metal oxide spheres formed by nanoparticle infiltration,Chemistry of Materials 16 (2004) 2287–2292.
[15] Fengqiu Tang, Hiroshi Fudouzi, Tetsuo Uchikoshi, Yoshio Sakka, Preparation ofporous materials with controlled pore size and porosity, Journal of the EuropeanCeramic Society 24 (2004) 341–344.
[16] A. Imhof, D.J. Pine, Uniform macroporous ceramics and plastics by emulsiontemplating, Advanced Materials 10 (9) (1998).
[17] R. Linacero, J. Aguado-Serrano, M.L. Rojas-cervantes, Preparation of mesoporousTiO2 by the sol–gel method assisted by surfactants, Journal of Materials Science 41(2006) 2457–2464.
[18] Young Kyu Hwang, Kashinath Rangu Patil, Sung Hwa Jhung, Jong-San Chang,Young June Ko, Sang-Eon Park, Control of pore size and condensation rate of cubicmesoporous silica thin films using a swelling agent, Microporous and MesoporousMaterials 78 (2005) 245–253.
[19] Tie-Zhen Ren, Zhong-Yong Yuan, Bao-Lian Su, Hierarchical microtubular nanopor-ous zirconia with an extremely high surface area and pore volume, ChemicalPhysics Letters 388 (2004) 46–49.
[20] Clifford Y. Tai, Mei-Hwa Lee, Yu-ChunWu, Control of zirconia particle size by usingtwo-emulsion precipitation technique, Chemical Engineering Science 56 (2001)2389–2398.
[21] Jintao He, Tian Ma, Yong Huang, Jinlong Yang, Study on preparation of sphericalzirconia powder in microemulsion system and its surface characteristics, KeyEngineering Materials 280–283 (2005) 977–980 Vols.
[22] M.J. Annen, R. Kizhappali, P.W. Carr, A.V. McCormick, Development of porouszirconia spheres by polymerization-induced colloid aggregation, Journal ofMaterials Science 29 (1994) 6123–6130.
[23] A.M. Clausen, A. Subramanian, P.W. Carr, Purification of Mabs using a modifiedcation-exchange support, Journal of Chromatography 831 (1999) 63–72.
[24] Peter W. Carr, Alon V. Mccormick, Michael J. Annen, Lifang Sun, Jason R. Brown,Synthesis of porous inorganic particles by polymerization-induced colloidaggregation (PICA), United States 42 (1996).
[25] A.N. Sathyagal, P.W. Carr, A.V. McCormick, Synthesis of porous zirconia spheres —mechanism and prospects for multistep processing, Journal of Colloid andInterface Science 219 (1) (1999) 20–30.
[26] Abhinandan Pattanayak, Effect of porogens on particle morphology, pore size, poregeometry and pore architecture of zirconia supports. Thesis (M.S.), University ofNebraska-Lincoln, 2007.
[27] Abhinandan Pattanayak and Anuradha Subramanian, Impact of porogens on thepore characteristics of zirconia particles made by Polymer Induced Colloid Aggre-gation (PICA), International Journal of Applied Ceramic Technology — submittedfor publication.
[28] M.J. Robichaud, A.N. Sathyagal, P.W. Carr, A.V. McCormick, M.C. Flickinger, Animproved oil emulsion synthesis method for large, porous zirconia particles forpacked- or fluidized-bed protein chromatography, Separation Science andTechnology 32 (15) (1997) 2547–2559.
[29] A. Mullick, C.M. Griffith, M.C. Flickinger, Expanded and packed bed albuminadsorption on fluoride modified zirconia, Biotechnology and Bioengineering 60(1998) 333–340.
[30] Robert E. Treybal,Mass Transfer Operations, International 3rdRevised Ed.McGraw-Hill Publishing Co., 1980
[31] D.H. Reeder, A.M. Clausen, M.J. Annen, P.W. Carr, M.C. Flickinger, A.V. Mccormick,An approach to hierarchically structured porous zirconia aggregates, Journal ofColloid and Interface Science 184 (1) (1996) 328–330.




















