
Predicting ionic conductivity of solid oxide fuel cell electrolyte from first principles
8
Predicting ionic conductivity of solid oxide fuel cell electrolyte from first principles Rojana Pornprasertsuk a Department of Materials Science and Engineering, Stanford University, Stanford, California 94305 Panchapakesan Ramanarayanan b Department of Mechanical Engineering, Stanford University, Stanford, California 94305 Charles B. Musgrave Department of Chemical Engineering, Stanford University, Stanford, California 94305 Fritz B. Prinz Department of Mechanical Engineering, Stanford University, Stanford, California 94305 Received 1 April 2005; accepted 13 October 2005; published online 22 November 2005 First-principles quantum simulations complemented with kinetic Monte Carlo calculations were performed to gain insight into the oxygen vacancy diffusion mechanism and to explain the effect of dopant composition on ionic conductivity in yttria-stabilized zirconia YSZ. Density-functional theory DFT within the local-density approximation with gradient correction was used to calculate a set of energy barriers that oxygen ions encounter during migration in YSZ by a vacancy mechanism. Kinetic Monte Carlo simulations were then performed using Boltzmann probabilities based on the calculated DFT barriers to determine the dopant concentration dependence of the oxygen self-diffusion coefficient in Y 2 O 3 x ZrO 2 1-2x with x increasing from 6% to 15%. The results from the simulations suggest that the maximum conductivity occurs at 7 – 9 mol % Y 2 O 3 at 600– 1500 K and that the effective activation energy increases at higher Y doping concentrations in good agreement with previously reported literature data. The increase in the effective activation energy for migration arises from the higher-energy barrier for oxygen vacancy diffusion across an Y–Y common edge relative to diffusion across one with a Zr–Y common edge of two adjacent tetrahedra. The binding energies between oxygen vacancies and dopants were extracted up to the fourth nearest-neighbor interaction. Our results reveal that the binding energy is the strongest when the vacancy is in the second nearest-neighbor position relative to the Y dopant atom. The methodology was also applied to scandium-doped zirconia SDZ. Preliminary results from quantum simulations of SDZ suggest that the effective activation energy for vacancy diffusion in SDZ is lower than that of YSZ, in agreement with experimental observations. The agreement with experimental studies on the two systems analyzed in this paper supports the use of this technique as a predictive tool on electrolyte systems not yet characterized experimentally. © 2005 American Institute of Physics. DOI: 10.1063/1.2135889 I. INTRODUCTION One of the key objectives in developing solid oxide fuel cells SOFC is the improvement of ionic conductivity in electrolyte materials. To aid the search for better ionic con- ductivity, we studied oxides with the CaF 2 structure adopting first-principles techniques. Yttria-stabilized zirconia YSZ, the most common SOFC electrolyte, has been extensively studied experimentally and is also the focus of the present study. Our computational results are being compared with previously reported experimental data. To understand the underlying mechanism of oxygen-ion diffusion in oxide materials, several theoretical approaches have been reported previously see Refs. 1–3. Shimojo and Okazaki 1 performed molecular-dynamics MD simulations of oxygen diffusion in YSZ. Their results revealed that the difference in conductivity at different dopant concentrations is not caused by the preference of oxygen vacancies to re- main clustered with dopant atoms but rather originates from the strongly reduced migration probabilities of oxygen ions when the dopant atoms are present in the common edge of the tetrahedra. Meyer et al. 2 used Monte Carlo MC simulations to investigate the effect that vacancy-dopant interactions have on the vacancy transport behavior. They employed MC simu- lations to study the anomalous conductivity of aliovalently doped fluorite oxides. The analysis was based on three inter- action potentials between dopants and vacancies. They con- cluded that the barrier model where the mobility of the va- cancies is reduced in the neighborhood of the dopant ions satisfactory agreed with the experimental results and the MD simulations of Shimojo and Okazaki. 1 Using potentials fit to the experimental data, Shimojo and Okazaki 1 and Meyer et al. 2 were able to describe the behavior of oxygen-ion diffusion in YSZ. Using density- functional theory DFT with generalized gradient approxi- a Electronic mail: [email protected] b Present address: Intel Corporation, Santa Clara, California 95052. JOURNAL OF APPLIED PHYSICS 98, 103513 2005 0021-8979/2005/9810/103513/8/$22.50 © 2005 American Institute of Physics 98, 103513-1 Downloaded 09 Dec 2005 to 171.64.118.168. Redistribution subject to AIP license or copyright, see http://jap.aip.org/jap/copyright.jsp
Transcript of Predicting ionic conductivity of solid oxide fuel cell electrolyte from first principles

JOURNAL OF APPLIED PHYSICS 98, 103513 �2005�
Predicting ionic conductivity of solid oxide fuel cell electrolytefrom first principles
Rojana Pornprasertsuka�
Department of Materials Science and Engineering, Stanford University, Stanford, California 94305
Panchapakesan Ramanarayananb�
Department of Mechanical Engineering, Stanford University, Stanford, California 94305
Charles B. MusgraveDepartment of Chemical Engineering, Stanford University, Stanford, California 94305
Fritz B. PrinzDepartment of Mechanical Engineering, Stanford University, Stanford, California 94305
�Received 1 April 2005; accepted 13 October 2005; published online 22 November 2005�
First-principles quantum simulations complemented with kinetic Monte Carlo calculations wereperformed to gain insight into the oxygen vacancy diffusion mechanism and to explain the effect ofdopant composition on ionic conductivity in yttria-stabilized zirconia �YSZ�. Density-functionaltheory �DFT� within the local-density approximation with gradient correction was used to calculatea set of energy barriers that oxygen ions encounter during migration in YSZ by a vacancymechanism. Kinetic Monte Carlo simulations were then performed using Boltzmann probabilitiesbased on the calculated DFT barriers to determine the dopant concentration dependence of theoxygen self-diffusion coefficient in �Y2O3�x�ZrO2��1−2x� with x increasing from 6% to 15%. Theresults from the simulations suggest that the maximum conductivity occurs at 7–9 mol % Y2O3 at600–1500 K and that the effective activation energy increases at higher Y doping concentrations ingood agreement with previously reported literature data. The increase in the effective activationenergy for migration arises from the higher-energy barrier for oxygen vacancy diffusion across anY–Y common edge relative to diffusion across one with a Zr–Y common edge of two adjacenttetrahedra. The binding energies between oxygen vacancies and dopants were extracted up to thefourth nearest-neighbor interaction. Our results reveal that the binding energy is the strongest whenthe vacancy is in the second nearest-neighbor position relative to the Y dopant atom. Themethodology was also applied to scandium-doped zirconia �SDZ�. Preliminary results fromquantum simulations of SDZ suggest that the effective activation energy for vacancy diffusion inSDZ is lower than that of YSZ, in agreement with experimental observations. The agreement withexperimental studies on the two systems analyzed in this paper supports the use of this technique asa predictive tool on electrolyte systems not yet characterized experimentally. © 2005 AmericanInstitute of Physics. �DOI: 10.1063/1.2135889�
I. INTRODUCTION
One of the key objectives in developing solid oxide fuelcells �SOFC� is the improvement of ionic conductivity inelectrolyte materials. To aid the search for better ionic con-ductivity, we studied oxides with the CaF2 structure adoptingfirst-principles techniques. Yttria-stabilized zirconia �YSZ�,the most common SOFC electrolyte, has been extensivelystudied experimentally and is also the focus of the presentstudy. Our computational results are being compared withpreviously reported experimental data.
To understand the underlying mechanism of oxygen-iondiffusion in oxide materials, several theoretical approacheshave been reported previously �see Refs. 1–3�. Shimojo andOkazaki1 performed molecular-dynamics �MD� simulationsof oxygen diffusion in YSZ. Their results revealed that thedifference in conductivity at different dopant concentrations
a�Electronic mail: [email protected]�
Present address: Intel Corporation, Santa Clara, California 95052.0021-8979/2005/98�10�/103513/8/$22.50 98, 10351
Downloaded 09 Dec 2005 to 171.64.118.168. Redistribution subject to
is not caused by the preference of oxygen vacancies to re-main clustered with dopant atoms but rather originates fromthe strongly reduced migration probabilities of oxygen ionswhen the dopant atoms are present in the common edge ofthe tetrahedra.
Meyer et al.2 used Monte Carlo �MC� simulations toinvestigate the effect that vacancy-dopant interactions haveon the vacancy transport behavior. They employed MC simu-lations to study the anomalous conductivity of aliovalentlydoped fluorite oxides. The analysis was based on three inter-action potentials between dopants and vacancies. They con-cluded that the barrier model where the mobility of the va-cancies is reduced in the neighborhood of the dopant ionssatisfactory agreed with the experimental results and the MDsimulations of Shimojo and Okazaki.1
Using potentials fit to the experimental data, Shimojoand Okazaki1 and Meyer et al.2 were able to describe thebehavior of oxygen-ion diffusion in YSZ. Using density-
functional theory �DFT� with generalized gradient approxi-© 2005 American Institute of Physics3-1
AIP license or copyright, see http://jap.aip.org/jap/copyright.jsp

103513-2 Pornprasertsuk et al. J. Appl. Phys. 98, 103513 �2005�
mation �GGA� to exchange, Eichler3 calculated the diffusionbarriers for oxygen-ion vacancies at several locations in thesupercell of tetragonal YSZ which provide a connected pathfor vacancy migration to positions equivalent to the initialposition. The migration energy for oxygen diffusion was as-sumed to be the difference between the highest energy alongthe path and the lowest energy. Recent research by Rama-narayanan et al.4 suggested that such an approach can serveas an approximation only because the migration energy isobtained from only a specified path.
Independent of the work recently published by Krishna-murthy et al.,5 we have been working �along a similar ap-proach used for SiGe alloys in Ref. 6� to develop an under-standing of ionic conductivity in YSZ using GGA-basedDFT simulations to create a database of migration energybarriers across different nearest-neighbor configurations �tet-rahedra� around the diffusing oxygen ion and oxygen va-cancy. Then, kinetic Monte Carlo �KMC� simulations basedon the DFT barriers are performed to determine the tempera-ture dependence of oxygen-ion diffusion. In contrast to theapproach used by Krishnamurthy et al.,5 we account for abroader range of diffusion barriers in the vicinity of a diffus-ing oxygen ion and an oxygen vacancy. We consider theinfluence of all six cations in two adjacent tetrahedra con-taining diffusing oxygen ion and oxygen vacancy to the mi-gration energy barrier. As a result, interactions between sur-rounding cations and the diffusing oxygen-ion–oxygen-vacancy pair are directly included into the model.Furthermore, the DFT calculations of different cation con-figurations allow us to extract the binding energy of VO
·· YZr�and YZr� YZr� . The resulting binding energy enables us to un-derstand more about the effect of defect interactions on thediffusion mechanism.
We assume that the oxygen self-diffusion coefficient �D�for vacancy diffusion consists of temperature-dependent and-independent terms �Eq. �1��:
D = D0� exp�− �HA/kT� = fD0�VO·· �exp�− �HM/kT� , �1�
where �VO·· � is the mole fraction of the oxygen vacancy and f
is the correlation factor. In general, �HA will be composed oftwo terms: the overall migration energy ��HM� and the va-cancy formation energy ��HV�.7 Due to the high vacancyformation energy in zirconia, the number of vacancies ther-mally generated is negligible. Most oxygen-ion vacancies�VO
·· � are generated to satisfy the charge neutrality conditionresulting from the local charge imbalance of aliovalent dop-ants �Y3+� substituting for host-cation sites �Zr4+� �Eq. �2��:
Y2O3 ——→2ZrO2
2YZr� + VO·· + 3OO
x . �2�
The migration energy calculated using DFT is used todetermine the probability that an oxygen ion jumps from itslattice site onto an adjacent vacancy. Since different nearest-neighbor configurations give rise to different migration en-ergy barriers, the overall migration energy ��HM� cannot beassociated with the migration energy of any specificconfiguration.4 Rather, �HM needs to be obtained from sta-tistically averaging a spectrum of migration pathways em-
ploying techniques such as the KMC.Downloaded 09 Dec 2005 to 171.64.118.168. Redistribution subject to
In this study, D0 �containing information of lattice vibra-tional frequency ��0� and jump distance� was assumed to bea constant at all dopant concentrations due to similar vibra-tional entropy contributions and only slight changes in thejump distance at different dopant concentrations:2.57–2.58 Å from 6 to 15 mol % YSZ8 �assuming jump dis-tance is half of lattice parameter�. �The correlation factor isimplicit in the KMC calculations.� After calculating the nor-malized diffusion coefficients �D /D0� at different tempera-tures, the effective activation energy was extracted using anArrhenius plot. This effective activation energy is equivalentto the overall migration energy ��HM� in Eq. �1�. The calcu-lations were repeated by changing the dopant concentration.Thus, the diffusion coefficients and the corresponding effec-tive activation energies were extracted as a function of dop-ant concentration. The results are compared with the experi-mental results at different dopant concentrations anddifferent temperatures.
The present analysis provides useful insights into thevacancy diffusion mechanism and elucidates the influencethat dopant type and concentration have on the ionic conduc-tivity of solid oxide electrolyte materials. It also validates thetechnique as a useful predictive tool.
II. QUANTUM SIMULATIONS: COMPUTATIONALDETAILS AND RESULTS
A. DFT calculations
Cubic ZrO2 has the CaF2 crystal structure: each unit cellconsists of four Zr4+ ions occupying fcc lattice sites and eightO2− ions occupying the tetrahedral sites �Fig. 1�. Dopingwith yttria �Y2O3� replaces Zr by Y atoms and, for every twoY atoms, one oxygen vacancy needs to be created to satisfycharge neutrality. The calculations were performed using theVienna ab initio simulation package �VASP� which employsdensity-functional theory and expands the electronic struc-ture using a plane-wave basis set.9–12 Electron-ion interac-tions are described using the projector-augmented wave�PAW� method13,14 with plane waves up to the energy cutoffat 400 eV �29.4 Ry�. We adopted the PW91 GGA exchange-correlation functional proposed by Perdew and Wang.15 Thek-point sampling was restricted to a single gamma ��� point:
�0,0,0�. The supercell used in the calculations consisted of 30AIP license or copyright, see http://jap.aip.org/jap/copyright.jsp

103513-3 Pornprasertsuk et al. J. Appl. Phys. 98, 103513 �2005�
Zr, 6 Y, and 69 O atoms corresponding to 8.3 mol % YSZ.In principle, there is a range of pathways for an atom to
move to a neighboring vacant site. To determine the migra-tion barrier of the oxygen vacancy, the saddle point was as-sumed to be located on the plane that has the following prop-erties: �i� it is perpendicular to the shortest path and �ii� itcontains the other two cations �Fig. 2�. To calculate the en-ergy at the saddle point, all three ions �two cations and oneoxygen ion� were allowed to relax only within this planewhile the other atoms were allowed to relax in all directions.The corresponding migration energy is the energy differencebetween the saddle-point energy and the initial state energy�Fig. 3�.
Because the diffusion barrier depends on the localatomic environment, migration energies of different configu-rations of neighboring atoms around the diffusing oxygen ionand oxygen vacancy need to be calculated. The number ofdifferent possible arrangements grows unmanageably largeunless approximations are made. We used the observation ofShimojo and Okazaki1 to assume that the barrier dependslargely on the sites’ first nearest neighbors to the diffusioncenter. All migration barrier calculations were performedwith a single dopant concentration of 8.3 mol % YSZ byvarying the location of dopant atoms within the supercell.The results were used as barrier database for kinetic MonteCarlo simulations, which will be discussed in the next sec-tion. �Additionally, we also performed the same calculations
FIG. 2. Positions of cations, diffusing oxygen ion, oxygen vacancy in thetwo common tetrahedra, and the saddle-point plane. ①: cation positions inthe two adjacent tetrahedra, where the number label is used as a referencefor Table I; �: oxygen vacancy in the tetrahedral site �first nearest Zr or Yions in positions 1, 2, 3, and 4�; �: diffusing oxygen ion, which is intetrahedral site �first nearest Zr or Y ion in positions 3, 4, 5, and 6�; - -:saddle-point plane where the diffusing oxygen ion migrates to the sameplane as the common cation sites of the two adjacent tetrahedra �three atomsare allowed to relax in all directions within the plane�.
FIG. 3. Illustration of the migration energy barrier. The middle point is theenergy corresponding to the saddle point where three atoms �diffusing oxy-gen ion and two cations that need to be diffused across� align in the same
plane.Downloaded 09 Dec 2005 to 171.64.118.168. Redistribution subject to
with 14.3 mol % YSZ. Another migration energy databasefor 6–15 mol % YSZ was then established by linearly inter-polating between the 8.3 and 14.3 mol % YSZ data sets. Theresults of the interpolation database turn out to be similar tothe 8.3 mol % YSZ database.�
In this study, the migration energy was assumed to de-pend only on the arrangement of cations in the two adjacenttetrahedra containing the diffusing atom and the vacancy �at-oms 1–6, Fig. 1�. Consequently, the interactions of oxygen-ion vacancies and the other interactions beyond the compo-nents in the two tetrahedra were neglected. To further reducethe computational complexity, we assumed that there wereno more than three Y ions in the six cation sites. Further-more, the binding energies of VO
·· YZr� and YZr� YZr� up to fourthnearest-neighbor interactions were extracted from the energydifferences between different defect arrangements modeledby the various supercells considered. Because all supercellshad oxygen vacancies which were at least fifth nearest neigh-bors to each other, the binding energy of VO
·· VO·· was not ob-
tained.To investigate the dependence of the effective activation
energy on the type of dopants, migration energy calculationswere performed on scandium-doped zirconia �SDZ� by em-ploying the same approach as described above for YSZ.
B. Results and discussion
The lattice parameter of the 8.3 mol % YSZ obtained byvolume relaxation was 5.14 Å which is in good agreementwith the experimental value of 5.14–5.16 Å.8,16 Diffusionbarriers of each combination of cations in the two adjacenttetrahedra are summarized in Table I. The highest diffusionbarrier was found for oxygen-vacancy motion between twoadjacent tetrahedra containing Y–Y common edge, which isin agreement with the results by Shimojo and Okazaki1 andKrishnamurthy et al.5 The reason for the higher migrationenergy across two tetrahedra containing Y–Y versus Zr–Zrcommon edge could arise from two main factors: the smallerspace for the oxygen ion to move �due to the bigger ionicradius of Y3+ compared with Zr4+� and the binding energybetween YZr� and VO
·· . Furthermore, the results �Table I� sug-gest that the migration energy barriers are sensitive to thesurrounding atoms beyond the two common edge cations dueto an association effect between defects. With the same Y–Ycommon edge, the activation changes from 1.23 to 1.40 eVif an additional Y atoms is present at atom position 5 or 6�Fig. 2�. This result suggests that the barriers are sensitive tothe surrounding cations and the difference in the migrationenergy barrier partly comes from the association between thedefects.
The binding energy of VO·· YZr� and YZr� YZr� depends on the
identity of the other atoms surrounding these sites. We obtainthe average binding energy by using a least-squares fit to thebinding energies of the various cases where the identities ofthe other atoms surrounding these sites are different. Thebinding energies of VO
·· YZr� and YZr� YZr� at distance from firstto fourth nearest neighbors obtained from the least-squaressolution are listed in Table II. The plot of the binding energy
with respect to the distance between the defects �in terms ofAIP license or copyright, see http://jap.aip.org/jap/copyright.jsp

103513-4 Pornprasertsuk et al. J. Appl. Phys. 98, 103513 �2005�
neighborhood position� is shown in Fig. 4. A contribution tothe error in the calculated binding energy may arise fromneglecting of other possible defect complexes such asYZr� VO
·· YZr� . The negative binding energy of VO·· YZr� suggests
that VO·· and YZr� are attracted to each other and, on the other
hand, the positive binding energy shows that YZr� and YZr�repel each other. The highest binding energies occur whenVO
·· is in the second nearest-neighbor position relative to Y,which is in good agreement with the experimentalobservations.17–19 Mott-Littleton defect calculations byZacate et al.20 using Born-like pairwise potentials also sug-gest that oversized dopants �such as Y, Gd, and Sm� prefer acluster geometry in which the oxygen vacancy resides in thesecond nearest-neighbor site.
The results for both VO·· YZr� and YZr� YZr� show lower bind-
ing energies beyond the second nearest neighbors and ap-proach zero at the fourth nearest-neighbor interaction. The
TABLE I. Summary of the DFT migration energy barriers in yttria-stabilizedin the two tetrahedra containing the diffusing oxygen ion and the oxygen vacthe oxygen vacancy and 3, 4, 5, and 6 are the positions of cations in the tra
1 2 3 4 5 6Migration
energy
Zr Zr Zr Zr Zr Zr 0.67Y Zr Zr Zr Zr Zr 0.20Zr Y Zr Zr Zr Zr 0.20Zr Zr Y Zr Zr Zr 1.19Zr Zr Zr Y Zr Zr 1.19Zr Zr Zr Zr Y Zr 0.80Zr Zr Zr Zr Zr Y 0.80Y Y Zr Zr Zr Zr 0.23Y Zr Y Zr Zr Zr 0.88Y Zr Zr Y Zr Zr 0.88Y Zr Zr Zr Y Zr 0.39Y Zr Zr Zr Zr Y 0.62Zr Y Y Zr Zr Zr 0.88Zr Y Zr Y Zr Zr 0.88Zr Y Zr Zr Y Zr 0.62Zr Y Zr Zr Zr Y 0.39Zr Zr Y Y Zr Zr 1.23Zr Zr Y Zr Y Zr 0.71Zr Zr Y Zr Zr Y 0.71Zr Zr Zr Y Y Zr 0.71Zr Zr Zr Y Zr Y 0.71
TABLE II. List of calculated binding energies of VO·· YZr� and YZr� YZr� from the
first nearest-neighbor �1st NN� to the fourth nearest-neighbor �4th NN� in-teractions. The binding energy is the least-squares solution of the combina-tions of total energies obtained from quantum calculations.
Associated defect Binding energy
V–Y 1st NN −0.2988V–Y 2nd NN −0.3531V–Y 3rd NN −0.1859V–Y 4th NN −0.1328Y–Y 1st NN 0.0335Y–Y 2nd NN 0.1451Y–Y 3rd NN 0.0048Y–Y 4th NN −0.0973
Downloaded 09 Dec 2005 to 171.64.118.168. Redistribution subject to
binding energies of VO·· YZr� are 0.13–0.35 eV which are at
least 25% of the migration energy barrier range of0.2–1.4 eV and, therefore, can influence the oxygen migra-tion in YSZ. To obtain accurate results for the migrationenergy barriers, surrounding cations around diffusing oxygenand oxygen-vacancy pair up to the third nearest neighborsshould be taken into account. However, due to impracticalcomputational times for the DFT calculations, in this study,we consider interactions only up to the second nearest-neighbor VO
·· YZr� �in the diffusing direction�, which representsthe strongest association energy, in the migration energy bar-rier calculations.
For the SDZ calculations, the lattice parameter of8.3 mol % SDZ obtained was 5.09 Å which is in good agree-
nia �YSZ�. The numbers in the first row indicate the positions of the cations�1, 2, 3, and 4 are the positions of the cations in the tratrahedron containingdron containing the diffusion oxygen ion, as shown in Fig. 2�.
1 2 3 4 5 6Migration
energy
Zr Zr Zr Zr Y Y 0.45Y Y Y Zr Zr Zr 0.70Y Y Zr Y Zr Zr 0.70Y Y Zr Zr Y Zr 0.28Y Y Zr Zr Zr Y 0.28Y Zr Y Y Zr Zr 1.20Y Zr Y Zr Y Zr 0.79Y Zr Y Zr Zr Y 1.09Y Zr Zr Y Y Zr 0.79Y Zr Zr Y Zr Y 1.09Y Zr Zr Zr Y Y 0.70Zr Y Y Y Zr Zr 1.20Zr Y Y Zr Y Zr 1.09Zr Y Y Zr Zr Y 0.79Zr Y Zr Y Y Zr 1.09Zr Y Zr Y Zr Y 0.79Zr Y Zr Zr Y Y 0.70Zr Zr Y Y Y Zr 1.40Zr Zr Y Y Zr Y 1.40Zr Zr Y Zr Y Y 0.94Zr Zr Zr Y Y Y 0.94
FIG. 4. Plot of the binding energy from first nearest-neighbor to the fourthnearest-neighbor YZr� YZr� and VO
·· YZr� interaction extracted by a least-squares··
zircoancytrahe
fit of the total energies of the different arrangements of VO and YZr� .
AIP license or copyright, see http://jap.aip.org/jap/copyright.jsp

103513-5 Pornprasertsuk et al. J. Appl. Phys. 98, 103513 �2005�
ment with the experimental value of 5.09 Å �at 10 mol %SDZ�.21 The migration energy barriers of SDZ comparedwith those of YSZ are shown in Table III. The calculationssuggest that without any dopants in the two adjacent tetrahe-dra, the migration energy of oxygen ions is slightly higher inSDZ than that in YSZ because of the smaller lattice param-eter of SDZ. However, the migration energy of the oxygenion in SDZ becomes lower than that in YSZ due to the pres-ence of dopants in the two tetrahedra; the smaller radius ofSc relative to Y provides for a larger space for the oxygenion—this effect wins over the smaller lattice parameter ofSDZ.
Experimental results also showed that the activation en-ergy of 11 mol % SDZ is smaller than that in 11% YSZ�0.7 eV in SDZ versus 0.9 eV in YSZ�.22 Sc3+-substitutedZr4+ in SDZ results in the same defect chemistry as Y3+ inYSZ; this implies that both systems have the same oxygen-vacancy concentrations. Furthermore, assuming that D0 ofboth YSZ and SDZ are approximately the same, the diffusioncoefficient of SDZ is higher than YSZ at high dopant con-centrations which was observed experimentally in10–15 mol % doped zirconia.21,22 The calculations of Zacateet al.20 show that VO
·· ScZr� has the lowest binding energy com-pared with other trivalent dopants, which may be one of thereasons for the lower migration energy and higher oxygenself-diffusion coefficients in SDC compared with YSZ.
Although the migration energy barrier database givessome insight into the dependence of the migration energy onthe ionic radius of the dopant and the lattice parameter, it isonly representative of atomistic processes at 0 K. We per-formed a KMC simulation using the database to study thetemperature dependence of the diffusivity and hence deter-mine an effective activation energy for the entire diffusionprocess.
III. KINETIC MONTE CARLO SIMULATIONS:COMPUTATIONAL DETAILS AND RESULTS
A. Kinetic Monte Carlo simulations
The KMC technique attempts to capture the effects ofrare atomic processes that directly contribute to changes inmacroscopic properties. Therefore, the results of the simula-tions, after they are statistically averaged, are likely to reflectmacroscopic behavior. In this study, KMC was used to simu-late an activated random-walk process in a randomly distrib-uted landscape of vacancy and Y atoms. The KMC procedure
TABLE III. Comparison of DFT migration energiezirconia �YSZ�. The numbers in the first row indicataining the diffusion oxygen ion and the oxygen vacatetrahedron containing the oxygen vacancy and 3, 4,containing the diffusion oxygen ion, as shown in Fig
1 2 3 4 5
Zr Zr Zr Zr ZrZr Zr Sc/Y Zr ZrZr Zr Sc/Y Sc/Y Zr
consists of a series of KMC moves. Each move consists of
Downloaded 09 Dec 2005 to 171.64.118.168. Redistribution subject to
the following steps.23 �i� Identify all possible events from thecurrent configuration. In fluorite oxides, there are six pos-sible events for each oxygen vacancy �six nearest-neighboroxygen sites�, except in the case where any one of the firstnearest neighbors is also a vacancy. �ii� Obtain the rates foreach of the events ��i�. Rate is proportional to exp�−�Em /kT�, where the migration energy ��Em� is obtainedfrom the ab initio calculations �Table I�. �iii� Generate apseudorandom number � between 0 and 1. �iv� Advance thetime of each step ��t� by −ln��� /�i�i.
24 �v� Choose one ofthe events depending on the random number �, consistentwith the relative rate �i of all the events. �vi� Reconfigure thesystem according to the chosen event. �vii� Update andrecord the new position of the vacancy and time.
The diffusion coefficient of oxygen vacancy �Dv� wascalculated as given by Eq. �3�:
Dv = limt→� x2/6t , �3�
where t is the time calculated as the sum of all �t of eachjump and x2 is the mean-squared displacement.7 Subse-quently, the oxygen self-diffusion coefficient �D� can be cal-culated as given by Eq. �4�.
D = �VO·· �Dv. �4�
According to Eq. �1�, the overall migration energy or theeffective activation energy of the oxygen self-diffusion coef-ficient ��HM� can subsequently be obtained from the slopeof the Arrhenius plot of diffusion coefficients with respect tothe inverse temperature.
B. Simulation results and discussion
The periodic supercells used in the KMC simulationswere 5�5�5 unit cells of cubic ZrO2. Y atoms and oxygen-ion vacancies were introduced randomly according to theconcentration of Y2O3 dopants with the following restriction:Y ions are made to be distributed such that there are no morethan three Y ions within the six nearest-neighbor positions ofthe diffusing oxygen ion and the oxygen vacancy �Figs. 1and 2�. �The energy of the system which has more than threeY ions will be so large that such configurations will be rare,thus justifying this restriction.� The KMC simulation was rununtil a total of 2 000 000 jumps were performed. Twelve dif-ferent random initial distributions were used for each con-
scandia-doped zirconia �SDZ� and yttria-stabilizedpositions of the cations in the two tetrahedra con-
�1, 2, 3, and 4 are the positions of the cations in the6 are the positions of the cations in the tetrahedron
Migration energy�SDZ�
Migration energy�YSZ�
r 0.75 0.67r 0.93 1.19r 1.10 1.23
s inte thency
5, and. 2�.
6
ZZZ
centration between 6 and 15 mol % YSZ and at each tem-
AIP license or copyright, see http://jap.aip.org/jap/copyright.jsp

103513-6 Pornprasertsuk et al. J. Appl. Phys. 98, 103513 �2005�
perature between 600 and 1500 K. The calculated valuesusing 10�10�10 unit cells with 3 000 000 jumps givesimilar results.
The effective activation energy for the 8 mol % YSZ is0.7 eV, which is slightly lower than the experimental valueof 0.83–1.05 eV.25,26 This is probably caused by the exclu-sion of vacancy-vacancy interactions, the interactions withextended neighbors beyond the two adjacent tetrahedra, andinaccuracies in DFT calculations. However, at higher dopingconcentrations, the activation energy obtained from KMCsplits into two regions at around 900–1050 K �Fig. 5�. At15 mol % YSZ, the activation energies at low-temperatureand high-temperature regions are 0.8 and 1.0 eV, respec-tively. This indicates that with decreasing temperatures, ratecontrolling processes with lower activation energy becomestatistically more prevalent, as expected.
The dependence of the effective activation energy withrespect to doping concentration is summarized in Fig. 6. Thebehavior predicted by KMC at high temperatures �T�1050 K� qualitatively agrees with the experimental obser-vations though 0.06–0.25 eV is lower than the experimentalvalues.26 The calculations of Krishnamurthy et al.5 yield aneffective activation energy of 0.62 eV at concentration levels
FIG. 5. Arrhenius plot of normalized oxygen self-diffusion coefficients�D /D0� obtained from kinetic Monte Carlo simulations with respect to tem-perature from 600 to 1500 K. �a� 8 mol % YSZ: the effective activationenergy at T�1050 K is 0.68 eV and at T1050 K is 0.70 eV. �b�15 mol % YSZ: the effective activation energy at T�1050 K is 0.8 eV andat T1050 K is 1.0 eV.
as high as the 15 mol % YSZ and appear to differ from
Downloaded 09 Dec 2005 to 171.64.118.168. Redistribution subject to
observations26 by more than 0.5 eV. The higher effectivebarrier our simulations predicted is likely due to the inclu-sion of pathways involving differing arrangements in all sixnearest-neighbor cations in the vicinity of the diffusing oxy-gen and vacancy pair. At finite temperatures, the additionalpathways we consider will have nonzero probabilities andthus increase the effective barrier over models which pre-clude these paths.
The flat region at the beginning of the plot indicatesfewer interactions between oxygen vacancy and Y ions atlow doping concentrations. The activation energies at lowconcentrations are close to the migration energy of the twotetrahedra containing all Zr atoms. However, in the calcula-tion, the flat region of the activation energy ranges from6 to 8 mol % YSZ, while the experimental results show in-creasing activation energies above 6 mol % YSZ. This dis-parity may be the result of neglecting other interactions be-tween oxygen vacancy and their outlying Y neighbors. Athigh concentrations of YSZ, the KMC calculations indicatean increase in the activation energy, similar to the experi-mental results.8,26
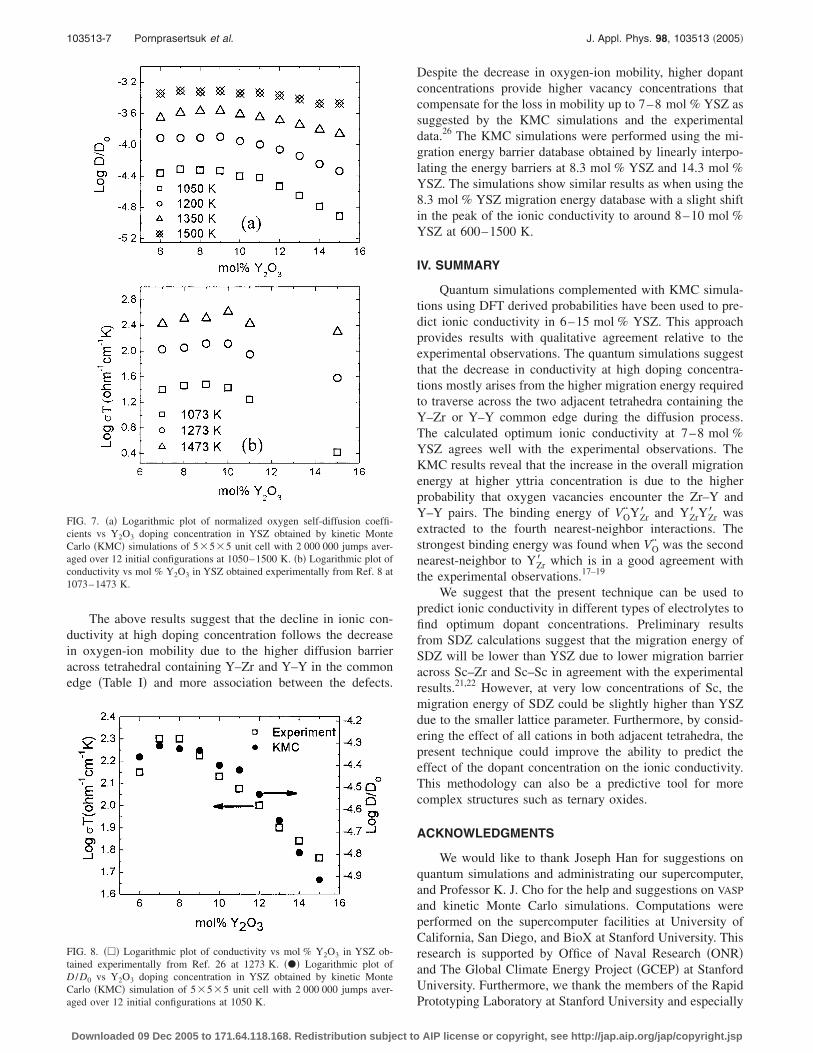
The logarithmic plot of normalized oxygen self-diffusioncoefficients at concentrations ranging from 6 to 15 mol %YSZ �5�5�5 unit cells, 2 000 000 jumps, and 12 configu-rations� at 1050–1500 K is shown in Fig. 7�a�. The highestoxygen self-diffusion coefficients obtained from the KMCsimulations are between 7 and 8 mol % YSZ at 750–1200 Kand slightly shift to 8 and 9 mol % at 1350–1500 K. Theshift of the maximum conductivity has also been observedexperimentally8 �Fig. 7�b�� and is also in good agreementwith the results of Krishnamurthy et al.5 Because of the gen-eral underestimation of the migration energy barriers by theDFT calculations the magnitude of the KMC simulations ap-pears to match the experimental values at higher tempera-ture. The magnitude of the oxygen diffusivity at 1050 K is invery good agreement with the experimental results at 1273 K
FIG. 6. Summary of the effective activation energy with respect to yttriadoping concentration in YSZ. ��� the experimental results obtained fromRef. 26 at 950–1000 °C, ��� KMC High T is the effective activation energyat T1050 K, and ��� KMC Low T is the effective activation energy atT�1050 K obtained from kinetic Monte Carlo simulations using 5�5�5unit cell with 2 000 000 jumps averaged over 12 initial configurations.
�Fig. 8�.
AIP license or copyright, see http://jap.aip.org/jap/copyright.jsp
103513-7 Pornprasertsuk et al. J. Appl. Phys. 98, 103513 �2005�
The above results suggest that the decline in ionic con-ductivity at high doping concentration follows the decreasein oxygen-ion mobility due to the higher diffusion barrieracross tetrahedral containing Y–Zr and Y–Y in the commonedge �Table I� and more association between the defects.
FIG. 7. �a� Logarithmic plot of normalized oxygen self-diffusion coeffi-cients vs Y2O3 doping concentration in YSZ obtained by kinetic MonteCarlo �KMC� simulations of 5�5�5 unit cell with 2 000 000 jumps aver-aged over 12 initial configurations at 1050–1500 K. �b� Logarithmic plot ofconductivity vs mol % Y2O3 in YSZ obtained experimentally from Ref. 8 at1073–1473 K.
FIG. 8. ��� Logarithmic plot of conductivity vs mol % Y2O3 in YSZ ob-tained experimentally from Ref. 26 at 1273 K. ��� Logarithmic plot ofD /D0 vs Y2O3 doping concentration in YSZ obtained by kinetic MonteCarlo �KMC� simulation of 5�5�5 unit cell with 2 000 000 jumps aver-
aged over 12 initial configurations at 1050 K.Downloaded 09 Dec 2005 to 171.64.118.168. Redistribution subject to
Despite the decrease in oxygen-ion mobility, higher dopantconcentrations provide higher vacancy concentrations thatcompensate for the loss in mobility up to 7–8 mol % YSZ assuggested by the KMC simulations and the experimentaldata.26 The KMC simulations were performed using the mi-gration energy barrier database obtained by linearly interpo-lating the energy barriers at 8.3 mol % YSZ and 14.3 mol %YSZ. The simulations show similar results as when using the8.3 mol % YSZ migration energy database with a slight shiftin the peak of the ionic conductivity to around 8–10 mol %YSZ at 600–1500 K.
IV. SUMMARY
Quantum simulations complemented with KMC simula-tions using DFT derived probabilities have been used to pre-dict ionic conductivity in 6–15 mol % YSZ. This approachprovides results with qualitative agreement relative to theexperimental observations. The quantum simulations suggestthat the decrease in conductivity at high doping concentra-tions mostly arises from the higher migration energy requiredto traverse across the two adjacent tetrahedra containing theY–Zr or Y–Y common edge during the diffusion process.The calculated optimum ionic conductivity at 7–8 mol %YSZ agrees well with the experimental observations. TheKMC results reveal that the increase in the overall migrationenergy at higher yttria concentration is due to the higherprobability that oxygen vacancies encounter the Zr–Y andY–Y pairs. The binding energy of VO
·· YZr� and YZr� YZr� wasextracted to the fourth nearest-neighbor interactions. Thestrongest binding energy was found when VO
·· was the secondnearest-neighbor to YZr� which is in a good agreement withthe experimental observations.17–19
We suggest that the present technique can be used topredict ionic conductivity in different types of electrolytes tofind optimum dopant concentrations. Preliminary resultsfrom SDZ calculations suggest that the migration energy ofSDZ will be lower than YSZ due to lower migration barrieracross Sc–Zr and Sc–Sc in agreement with the experimentalresults.21,22 However, at very low concentrations of Sc, themigration energy of SDZ could be slightly higher than YSZdue to the smaller lattice parameter. Furthermore, by consid-ering the effect of all cations in both adjacent tetrahedra, thepresent technique could improve the ability to predict theeffect of the dopant concentration on the ionic conductivity.This methodology can also be a predictive tool for morecomplex structures such as ternary oxides.
ACKNOWLEDGMENTS
We would like to thank Joseph Han for suggestions onquantum simulations and administrating our supercomputer,and Professor K. J. Cho for the help and suggestions on VASP
and kinetic Monte Carlo simulations. Computations wereperformed on the supercomputer facilities at University ofCalifornia, San Diego, and BioX at Stanford University. Thisresearch is supported by Office of Naval Research �ONR�and The Global Climate Energy Project �GCEP� at StanfordUniversity. Furthermore, we thank the members of the Rapid
Prototyping Laboratory at Stanford University and especiallyAIP license or copyright, see http://jap.aip.org/jap/copyright.jsp

103513-8 Pornprasertsuk et al. J. Appl. Phys. 98, 103513 �2005�
the fuel cell team for their support and suggestions. Thiswork was supported by a scholarship from Thai government�R.P.�.
1F. Shimojo and H. Okazaki, J. Phys. Soc. Jpn. 61, 4106 �1992�.2M. Meyer, N. Nicoloso, and V. Jaenisch, Phys. Rev. B 56, 5961 �1997�.3A. Eichler, Phys. Rev. B 64, 174103 �2001�.4P. Ramanarayanan, B. Srinivasan, K. Cho, and B. M. Clemens, J. Appl.Phys. 96, 7095 �2004�.
5R. Krishnamurthy, Y. G. Yoon, D. J. Srolovitz, and R. Car, J. Am. Ceram.Soc. 87, 1821 �2004�.
6P. Ramanarayanan, K. Cho, and B. M. Clemens, J. Appl. Phys. 94, 174�2003�.
7P. Shewmon, Diffusion in Solids �The Minerals, Metals and MaterialsSociety, USZ, 1989�, Chap. 2.
8D. W. Stickler and W. G. Carlson, J. Am. Ceram. Soc. 47, 122 �1964�.9G. Kresse and J. Hafner, Phys. Rev. B 47, 558 �1993�.
10G. Kresse and J. Hafner, Phys. Rev. B 49, 14251 �1994�.11G. Kresse and J. Furthmüller, Comput. Mater. Sci. 6, 15 �1996�.12G. Kresse and J. Furthmüller, Phys. Rev. B 54, 11169 �1996�.13
P. E. Blöchl, Phys. Rev. B 50, 17953 �1994�.Downloaded 09 Dec 2005 to 171.64.118.168. Redistribution subject to
14G. Kresse and D. Joubert, Phys. Rev. B 59, 1758 �1998�.15J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson,
D. J. Singh, and C. Fiolhais, Phys. Rev. B 46, 6671 �1992�.16M. Putkonen, T. Sajavaara, J. Niinistö, L. Johansson, and L. Niinistö, J.
Mater. Chem. 12, 442 �2002�.17P. Li, I. Chen, and J. E. Penner-Hahn, Phys. Rev. B 48, 10074 �1993�.18P. Li and I. Chen, J. Am. Ceram. Soc. 77, 118 �1994�.19C. R. A. Catlow, A. V. Chadwick, G. N. Greaves, and L. M. Moroney, J.
Am. Ceram. Soc. 69, 272 �1986�.20M. O. Zacate, L. Minervini, D. J. Bradfield, R. W. Grimes, and K. E.
Sickafus, Solid State Ionics 128, 243 �2000�.21J. M. Dixon, L. D. LaGrange, U. Merten, C. F. Miller, and J. T. Porter II,
J. Electrochem. Soc. 110, 276 �1963�.22T. I. Politova and J. T. S. Irvine, Solid State Ionics 168, 153 �2004�.23A. F. Voter, Phys. Rev. B 34, 6819 �1986�.24A. B. Bortz, M. H. Kalos, and J. L. Lebowitz, J. Comput. Phys. 17, 10
�1975�.25R. Pornprasertsuk, J. Cheng, Y. Saito, T. M. Gür, and F. Prinz, Proc.-
Electrochem. Soc. �in press�.26A. I. Ioffe, D. S. Rutman, and S. V. Karpachov, Electrochim. Acta 23, 141
�1978�.
AIP license or copyright, see http://jap.aip.org/jap/copyright.jsp




















