
Positron emission tomography imaging analysis of G2A as a negative modifier of lymphoid...
11
ARTICLE Positron emission tomography imaging analysis of G2A as a negative modifier of lymphoid leukemogenesis initiated by the BCR-ABL oncogene Lu Q. Le, 1,5 Janusz H.S. Kabarowski, 1 Stephane Wong, 1 Khoi Nguyen, 2,3 Sanjiv S. Gambhir, 2,3 and Owen N. Witte 1,2,4,6 1 Department of Microbiology, Immunology, and Molecular Genetics 2 Department of Molecular and Medical Pharmacology 3 Crump Institute for Molecular Imaging 4 Howard Hughes Medical Institute 5 Medical Scientist Training Program University of California, Los Angeles Los Angeles, California 90095 6 Correspondence: [email protected] Summary G2A is a lymphocyte-expressed G protein-coupled receptor whose genetic ablation results in the development of autoimmu- nity. Using HSV-TK reporter gene directed positron emission tomography (PET), we demonstrate that prior to any indication of the onset of illness, mice transplanted with BCR-ABL transduced G2A-deficient bone marrow harbor expanded popula- tions of leukemic cells compared to recipients of wild-type bone marrow. The target cell type and anatomical locations of leukemia development are indistinguishable in animals transplanted with G2A/ or G2A/ cells. Shorter disease latency in the G2A-deficient background is associated with an increased rate of cellular expansion. PET can be successfully applied to the temporal and spatial analysis of Bcr-Abl driven leukemic progression and should have utility for the study of other leukemias and lymphomas. Introduction mouse populations is governed by a complex set of genetic traits which includes modifier loci (Balmain, 2002; Van Dyke The presence of identical disease associated genetic mutations and Jacks, 2002). Several modifier loci have subsequently been within the human population often results in quite different indi- mapped further to define individual modifier genes. Notable vidual outcomes with respect to susceptibility, latency, clinical examples are the association of genetic polymorphisms within features, and severity (Houlston and Tomlinson, 1998). Similarly, the genes encoding secretory phospholipase A 2 (sPLA 2 ) and the mice inheriting the same genetic mutation predisposing to cer- p16 INK4a cyclin-dependent kinase inhibitor products which confer tain diseases may show very different outcomes, attributed to strain dependent susceptibility to intestinal tumorigenesis and differences in genetic background and the influence of one or plasmacytoma induction, respectively (Cormier et al., 1997; more modifier genes. Identification of these modifiers provides Dietrich et al., 1993; Mock et al., 1993; Zhang et al., 2001). Classical approaches to study the impact of candidate mod- important insights into developmental and physiological pro- cesses, and genetic studies remain the most powerful tool with ifier genes upon cancer progression in animal models are limited by the requirement for autopsy to evaluate tumor burden, which which to do so (Nadeau, 2001). The development of cancer results from the progressive necessitates manual exploration of multiple anatomical sites for tumor incidence and imposes a requirement for large experi- accumulation of mutations which ultimately lead to the deregula- tion of normal cellular growth, survival, and differentiative mech- mental groups to control for intersubject variability. Live imaging of cancer progression by techniques such as positron emission anisms. Cancer susceptibility and progression in human and SIGNIFICANCE Expression of the BCR-ABL tyrosine kinase is the initiating etiological event in a subset of human lymphoid leukemias. We have developed a transplantation model for BCR-ABL induced lymphoid tumorigenesis utilizing positron emission tomography to study the modification of leukemogenic progression caused by loss of a novel BCR-ABL transcriptional target, the G protein-coupled receptor G2A. This receptor represents a class of lymphoid growth regulators which modifies the action of oncogenic signals from BCR-ABL. This model allows the kinetic and spatial examination of leukemia initiation and progression in individual animals and the separate contribution of proliferative expansion and leukemic dissemination to these processes. We believe that this system can be further applied to more thoroughly evaluate therapeutic agents targeting specific pathways deregulated by BCR-ABL and other leukemia specific oncoproteins. CANCER CELL : MAY 2002 · VOL. 1 · COPYRIGHT 2002 CELL PRESS 381
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Positron emission tomography imaging analysis of G2A as a negative modifier of lymphoid...

A R T I C L E
Positron emission tomography imaging analysis of G2A as anegative modifier of lymphoid leukemogenesis initiated bythe BCR-ABL oncogene
Lu Q. Le,1,5 Janusz H.S. Kabarowski,1 Stephane Wong,1 Khoi Nguyen,2,3 Sanjiv S. Gambhir,2,3
and Owen N. Witte1,2,4,6
1Department of Microbiology, Immunology, and Molecular Genetics2 Department of Molecular and Medical Pharmacology3 Crump Institute for Molecular Imaging4 Howard Hughes Medical Institute5 Medical Scientist Training ProgramUniversity of California, Los AngelesLos Angeles, California 900956 Correspondence: [email protected]
Summary
G2A is a lymphocyte-expressed G protein-coupled receptor whose genetic ablation results in the development of autoimmu-nity. Using HSV-TK reporter gene directed positron emission tomography (PET), we demonstrate that prior to any indicationof the onset of illness, mice transplanted with BCR-ABL transduced G2A-deficient bone marrow harbor expanded popula-tions of leukemic cells compared to recipients of wild-type bone marrow. The target cell type and anatomical locationsof leukemia development are indistinguishable in animals transplanted with G2A�/� or G2A�/� cells. Shorter diseaselatency in the G2A-deficient background is associated with an increased rate of cellular expansion. PET can be successfullyapplied to the temporal and spatial analysis of Bcr-Abl driven leukemic progression and should have utility for the studyof other leukemias and lymphomas.
Introduction mouse populations is governed by a complex set of genetictraits which includes modifier loci (Balmain, 2002; Van Dyke
The presence of identical disease associated genetic mutations and Jacks, 2002). Several modifier loci have subsequently beenwithin the human population often results in quite different indi- mapped further to define individual modifier genes. Notablevidual outcomes with respect to susceptibility, latency, clinical examples are the association of genetic polymorphisms withinfeatures, and severity (Houlston and Tomlinson, 1998). Similarly, the genes encoding secretory phospholipase A2 (sPLA2) and themice inheriting the same genetic mutation predisposing to cer- p16INK4a cyclin-dependent kinase inhibitor products which confertain diseases may show very different outcomes, attributed to strain dependent susceptibility to intestinal tumorigenesis anddifferences in genetic background and the influence of one or plasmacytoma induction, respectively (Cormier et al., 1997;more modifier genes. Identification of these modifiers provides Dietrich et al., 1993; Mock et al., 1993; Zhang et al., 2001).
Classical approaches to study the impact of candidate mod-important insights into developmental and physiological pro-cesses, and genetic studies remain the most powerful tool with ifier genes upon cancer progression in animal models are limited
by the requirement for autopsy to evaluate tumor burden, whichwhich to do so (Nadeau, 2001).The development of cancer results from the progressive necessitates manual exploration of multiple anatomical sites for
tumor incidence and imposes a requirement for large experi-accumulation of mutations which ultimately lead to the deregula-tion of normal cellular growth, survival, and differentiative mech- mental groups to control for intersubject variability. Live imaging
of cancer progression by techniques such as positron emissionanisms. Cancer susceptibility and progression in human and
S I G N I F I C A N C E
Expression of the BCR-ABL tyrosine kinase is the initiating etiological event in a subset of human lymphoid leukemias. We havedeveloped a transplantation model for BCR-ABL induced lymphoid tumorigenesis utilizing positron emission tomography to studythe modification of leukemogenic progression caused by loss of a novel BCR-ABL transcriptional target, the G protein-coupledreceptor G2A. This receptor represents a class of lymphoid growth regulators which modifies the action of oncogenic signals fromBCR-ABL. This model allows the kinetic and spatial examination of leukemia initiation and progression in individual animals and theseparate contribution of proliferative expansion and leukemic dissemination to these processes. We believe that this system canbe further applied to more thoroughly evaluate therapeutic agents targeting specific pathways deregulated by BCR-ABL and otherleukemia specific oncoproteins.
CANCER CELL : MAY 2002 · VOL. 1 · COPYRIGHT 2002 CELL PRESS 381

A R T I C L E
tomography (PET) would circumvent these problems and allow of signaling and biological properties of G2A (Kabarowski et al.,sequential kinetic and spatial measurements of tumor develop- 2000; Le et al., 2001; Weng et al., 1998), this may be due toment in individual animals. altered leukemic cell migration/homing or more rapid cell-cycle
BCR-ABL is a chimeric tyrosine kinase resulting from a re- progression. To address this issue, we utilized positron emissionciprocal translocation of chromosomes 22 and 9, producing a tomography (PET) based on coexpression of the HSV-TK re-fusion product from the cellular genes encoding BCR and c-ABL porter gene to evaluate tumor extent and sites of tumor growth(Chopra et al., 1999; Nowell and Hungerford, 1960; Rowley, in a kinetic manner for individual mice transplanted with BCR-1973). In patients, three fusion products are detected, each ABL transduced wild-type or G2A-deficient bone marrow cells.differing in the amount of BCR encoded sequences they contain. PET is a nuclear imaging technique that combines high im-P185 BCR-ABL is associated with 15%–25% of patients with age resolution with a quantitative method for detecting the loca-aggressive, poor prognostic acute lymphoblastic leukemia tion and levels of radiotracer accumulation in living subjects(ALL), and P210 BCR-ABL is associated with 95% of cases of
(Cherry and Gambhir, 2001). Bone marrow cells were trans-chronic myeloid leukemia (CML). The more recently identified
duced with a bicistronic retroviral vector containing BCR-ABLP230 BCR-ABL is associated with a mild chronic neutrophilicand the HSV-TKsr39 allele as a PET reporter gene whose prod-leukemia (Saglio et al., 1990; Sawyers, 1999). The etiology ofuct will sequester positron-emitting reporter probes (HerschmanBCR-ABL� leukemia has received considerable attention dueet al., 2000). The selective retention of the PET probe in targetto the tremendous efficacy with which the ABL kinase inhibitorytissues is then imaged in living animals, allowing one to generatedrug Gleevec� (STI571) induces disease remission in CML pa-consecutive, quantitative, and spatial measurements showingtients (Druker and Lydon, 2000). However, recent reports ofthe dynamic evolution of tumor development in a noninvasiveclinical resistance to this drug developing in a proportion offashion. We found that prior to any indication of overt illnesspatients have underlined the need for further studies of its patho-(preleukemic phase), mice transplanted with G2A-deficient bonepharmacological actions in vivo (Gorre et al., 2001).marrow harbor significantly expanded populations of BCR-ABLClinical studies and experimental animal models are consis-
tent with BCR-ABL serving as the initiating event in the transfor- positive B lymphoid progenitor cells compared to recipients ofmation/leukemogenic process and with additional genetic, and wild-type bone marrow. However, the spatial pattern of diseaseperhaps epigenetic, events being required for progression to development is similar in all animals, suggesting that the de-overt malignancy (Gishizky and Witte, 1992; Mitelman, 1993; creased latency for leukemogenesis in the context of G2A abla-Sawyers, 1999). The latency for Pre-B lymphoma induction by tion results primarily from increased leukemic cell expansion,the related viral oncoprotein vABL has been shown to be highly rather than altered cellular migration and sites of leukemic dis-mouse strain dependent (Abelson and Rabstein, 1970; Risser semination. These results indicate that G2A is a negative mod-et al., 1978; Rosenberg and Baltimore, 1976). While 90% of ifier of lymphoid leukemogenesis induced by BCR-ABL in vivoneonatal BALB/c mice injected intraperitoneally with Abelson
acting primarily to affect cell proliferation or viability.Murine Leukemia Virus (Abl-MLV) develop lymphomas with amean latent period of 92 days, only 36% of B6 mice develop
Resultsa similar malignancy with a protracted latent period of 120 days(Risser et al., 1985). This implies that genetic context plays a
Effect of G2A ablation on P185 BCR-ABL-mediatedmajor role governing the susceptibility to disease induced bytransformation in vitrothe ABL family of oncoproteins.The transformation activities of P185 BCR-ABL include the abil-We have previously identified a novel G protein-coupled
receptor (GPCR), named G2A, in a search for genes transcrip- ity to eliminate growth factor requirements of bone marrow Btionally regulated by P185 BCR-ABL (Weng et al., 1998). G2A cell progenitors for survival and proliferation (McLaughlin et al.,belongs to a subfamily of GPCR with ligand specificities for 1987, 1989). As G2A is transcriptionally induced in B lymphoidstructurally related lysophospholipids (Im et al., 2001; Kabarow- progenitors by BCR-ABL during this transformation processski et al., 2001; Xu et al., 2000; Zhu et al., 2001). G2A is ex- and suppresses its transformation potency when overexpressedpressed predominantly in hematopoietic cells including T and (Weng et al., 1998), we wished to determine whether inductionB lymphocytes. Forced overexpression of G2A antagonizes the of G2A expression under physiological conditions plays a roletransformation potency of BCR-ABL in vitro (Weng et al., 1998). in BCR-ABL in vitro transformation.G2A-deficient mice develop a late onset, slowly progressive Retrovirally mediated expression of BCR-ABL in primaryautoimmune syndrome, demonstrating a critical role for G2A in
bone marrow results in transformation of Pro/Pre-B cells incontrolling peripheral lymphocyte homeostasis, T lymphocyte
stromal cell-dependent cultures (McLaughlin et al., 1987). Theproliferation and autoimmunity (Le et al., 2001). These observa-growth rate of Pre-B cells from infected marrow can be directlytions support the notion that G2A negatively regulates lympho-monitored and quantitatively reflects BCR-ABL tyrosine kinasecyte growth responses in the context of sustained proliferativeactivity and transformation potency (Lugo et al., 1990). To deter-signals.mine the effect of G2A ablation on P185 BCR-ABL-mediatedIn this study, we have sought to determine by a direct genetictransformation in vitro, bone marrow cells from wild-type andtest whether loss of G2A function modulates the efficiency ofG2A-deficient mice were infected with a retrovirus encodingBCR-ABL leukemogenesis. We found that mice transplantedgreen fluorescent protein (GFP) plus P185 BCR-ABL (MSCV-with BCR-ABL transduced G2A-deficient bone marrow cellsGFP-IRES-BCR-ABL). Liquid cultures were plated in triplicateexhibit earlier tumor onset, more rapid tumor progression, andand monitored for Pre-B cell outgrowth at various time points.shorter survival times compared to those receiving BCR-ABL
transduced wild-type bone marrow. Based on our knowledge All infected cultures, irrespective of genotype, gave rise to mor-
382 CANCER CELL : MAY 2002
A R T I C L E
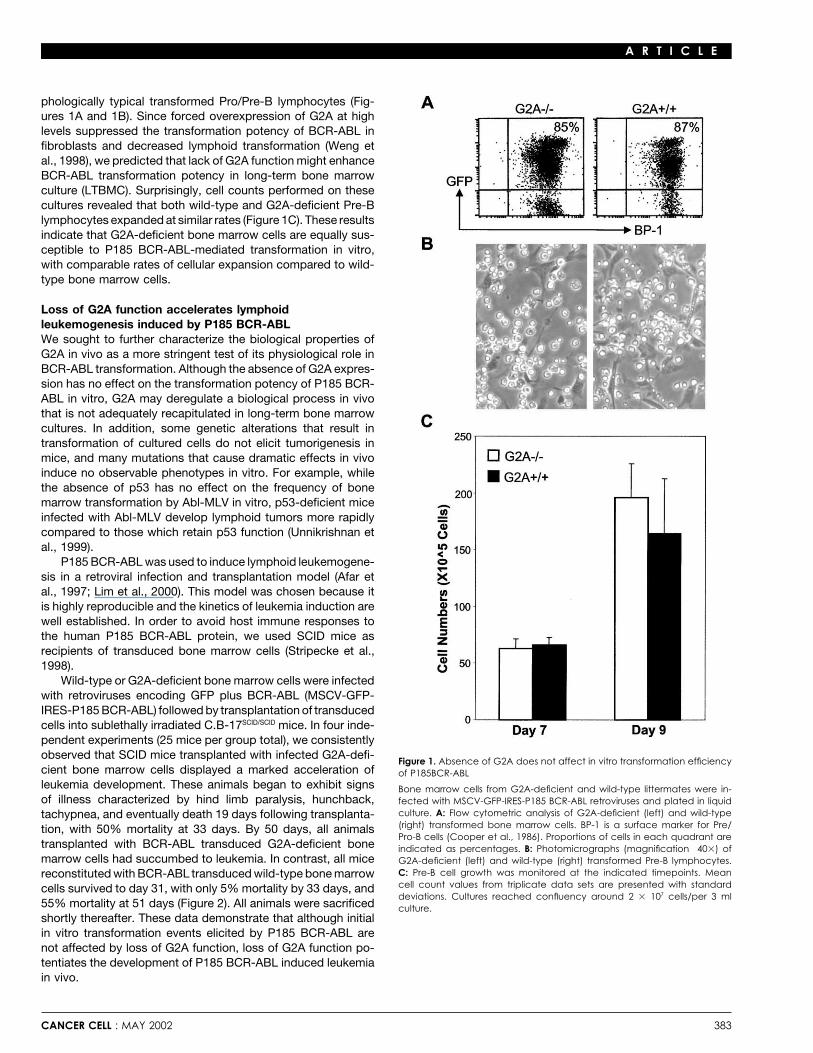
phologically typical transformed Pro/Pre-B lymphocytes (Fig-ures 1A and 1B). Since forced overexpression of G2A at highlevels suppressed the transformation potency of BCR-ABL infibroblasts and decreased lymphoid transformation (Weng etal., 1998), we predicted that lack of G2A function might enhanceBCR-ABL transformation potency in long-term bone marrowculture (LTBMC). Surprisingly, cell counts performed on thesecultures revealed that both wild-type and G2A-deficient Pre-Blymphocytes expanded at similar rates (Figure 1C). These resultsindicate that G2A-deficient bone marrow cells are equally sus-ceptible to P185 BCR-ABL-mediated transformation in vitro,with comparable rates of cellular expansion compared to wild-type bone marrow cells.
Loss of G2A function accelerates lymphoidleukemogenesis induced by P185 BCR-ABLWe sought to further characterize the biological properties ofG2A in vivo as a more stringent test of its physiological role inBCR-ABL transformation. Although the absence of G2A expres-sion has no effect on the transformation potency of P185 BCR-ABL in vitro, G2A may deregulate a biological process in vivothat is not adequately recapitulated in long-term bone marrowcultures. In addition, some genetic alterations that result intransformation of cultured cells do not elicit tumorigenesis inmice, and many mutations that cause dramatic effects in vivoinduce no observable phenotypes in vitro. For example, whilethe absence of p53 has no effect on the frequency of bonemarrow transformation by Abl-MLV in vitro, p53-deficient miceinfected with Abl-MLV develop lymphoid tumors more rapidlycompared to those which retain p53 function (Unnikrishnan etal., 1999).
P185 BCR-ABL was used to induce lymphoid leukemogene-sis in a retroviral infection and transplantation model (Afar etal., 1997; Lim et al., 2000). This model was chosen because itis highly reproducible and the kinetics of leukemia induction arewell established. In order to avoid host immune responses tothe human P185 BCR-ABL protein, we used SCID mice asrecipients of transduced bone marrow cells (Stripecke et al.,1998).
Wild-type or G2A-deficient bone marrow cells were infectedwith retroviruses encoding GFP plus BCR-ABL (MSCV-GFP-IRES-P185 BCR-ABL) followed by transplantation of transducedcells into sublethally irradiated C.B-17SCID/SCID mice. In four inde-pendent experiments (25 mice per group total), we consistentlyobserved that SCID mice transplanted with infected G2A-defi-
Figure 1. Absence of G2A does not affect in vitro transformation efficiencycient bone marrow cells displayed a marked acceleration of of P185BCR-ABLleukemia development. These animals began to exhibit signs Bone marrow cells from G2A-deficient and wild-type littermates were in-of illness characterized by hind limb paralysis, hunchback, fected with MSCV-GFP-IRES-P185 BCR-ABL retroviruses and plated in liquid
culture. A: Flow cytometric analysis of G2A-deficient (left) and wild-typetachypnea, and eventually death 19 days following transplanta-(right) transformed bone marrow cells. BP-1 is a surface marker for Pre/tion, with 50% mortality at 33 days. By 50 days, all animalsPro-B cells (Cooper et al., 1986). Proportions of cells in each quadrant are
transplanted with BCR-ABL transduced G2A-deficient bone indicated as percentages. B: Photomicrographs (magnification 40�) ofmarrow cells had succumbed to leukemia. In contrast, all mice G2A-deficient (left) and wild-type (right) transformed Pre-B lymphocytes.
C: Pre-B cell growth was monitored at the indicated timepoints. Meanreconstituted with BCR-ABL transduced wild-type bone marrowcell count values from triplicate data sets are presented with standardcells survived to day 31, with only 5% mortality by 33 days, anddeviations. Cultures reached confluency around 2 � 107 cells/per 3 ml
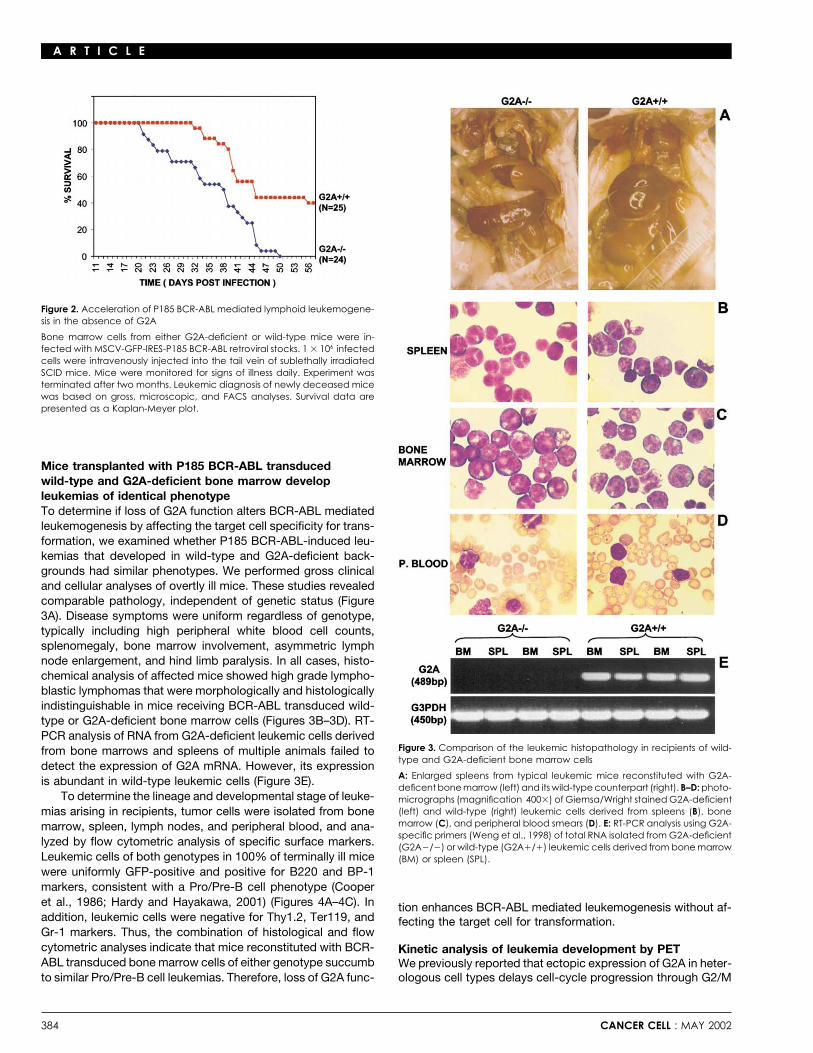
55% mortality at 51 days (Figure 2). All animals were sacrificed culture.shortly thereafter. These data demonstrate that although initialin vitro transformation events elicited by P185 BCR-ABL arenot affected by loss of G2A function, loss of G2A function po-tentiates the development of P185 BCR-ABL induced leukemiain vivo.
CANCER CELL : MAY 2002 383
A R T I C L E
Figure 2. Acceleration of P185 BCR-ABL mediated lymphoid leukemogene-sis in the absence of G2A
Bone marrow cells from either G2A-deficient or wild-type mice were in-fected with MSCV-GFP-IRES-P185 BCR-ABL retroviral stocks. 1 � 106 infectedcells were intravenously injected into the tail vein of sublethally irradiatedSCID mice. Mice were monitored for signs of illness daily. Experiment wasterminated after two months. Leukemic diagnosis of newly deceased micewas based on gross, microscopic, and FACS analyses. Survival data arepresented as a Kaplan-Meyer plot.
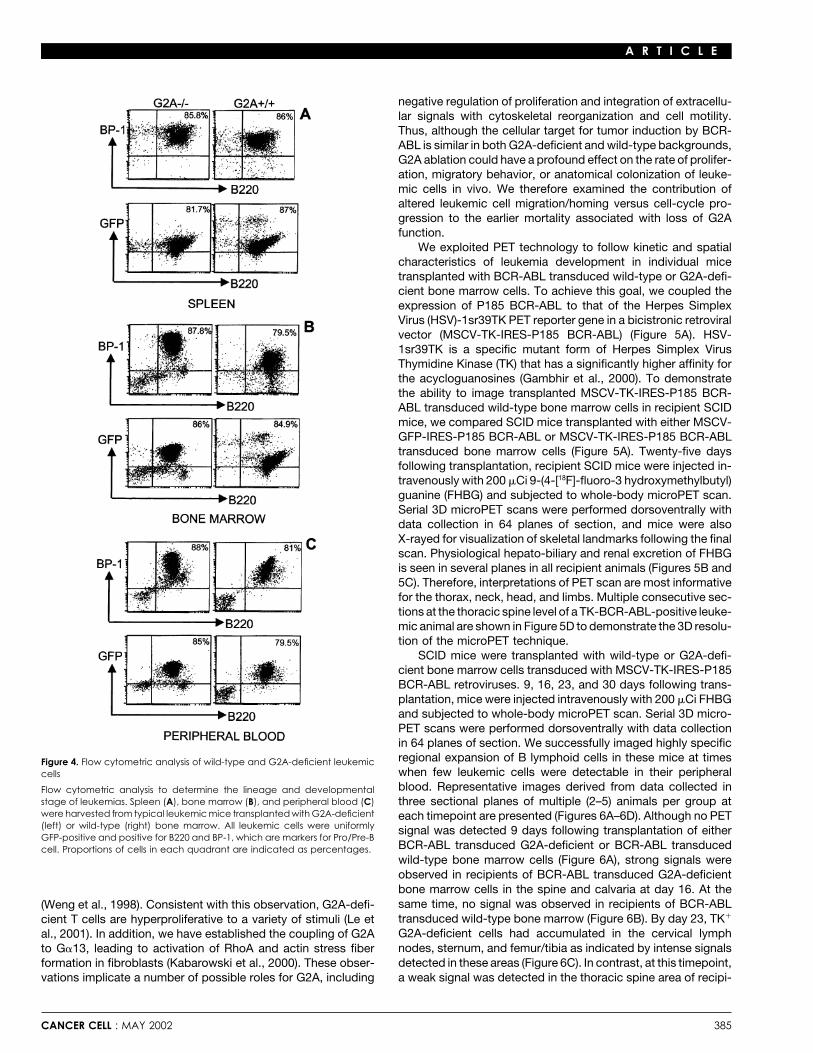
Mice transplanted with P185 BCR-ABL transducedwild-type and G2A-deficient bone marrow developleukemias of identical phenotypeTo determine if loss of G2A function alters BCR-ABL mediatedleukemogenesis by affecting the target cell specificity for trans-formation, we examined whether P185 BCR-ABL-induced leu-kemias that developed in wild-type and G2A-deficient back-grounds had similar phenotypes. We performed gross clinicaland cellular analyses of overtly ill mice. These studies revealedcomparable pathology, independent of genetic status (Figure3A). Disease symptoms were uniform regardless of genotype,typically including high peripheral white blood cell counts,splenomegaly, bone marrow involvement, asymmetric lymphnode enlargement, and hind limb paralysis. In all cases, histo-chemical analysis of affected mice showed high grade lympho-blastic lymphomas that were morphologically and histologicallyindistinguishable in mice receiving BCR-ABL transduced wild-type or G2A-deficient bone marrow cells (Figures 3B–3D). RT-PCR analysis of RNA from G2A-deficient leukemic cells derived
Figure 3. Comparison of the leukemic histopathology in recipients of wild-from bone marrows and spleens of multiple animals failed totype and G2A-deficient bone marrow cells
detect the expression of G2A mRNA. However, its expressionA: Enlarged spleens from typical leukemic mice reconstituted with G2A-is abundant in wild-type leukemic cells (Figure 3E).deficent bone marrow (left) and its wild-type counterpart (right). B–D: photo-
To determine the lineage and developmental stage of leuke- micrographs (magnification 400�) of Giemsa/Wright stained G2A-deficient(left) and wild-type (right) leukemic cells derived from spleens (B), bonemias arising in recipients, tumor cells were isolated from bonemarrow (C), and peripheral blood smears (D). E: RT-PCR analysis using G2A-marrow, spleen, lymph nodes, and peripheral blood, and ana-specific primers (Weng et al., 1998) of total RNA isolated from G2A-deficient
lyzed by flow cytometric analysis of specific surface markers. (G2A�/�) or wild-type (G2A�/�) leukemic cells derived from bone marrowLeukemic cells of both genotypes in 100% of terminally ill mice (BM) or spleen (SPL).were uniformly GFP-positive and positive for B220 and BP-1markers, consistent with a Pro/Pre-B cell phenotype (Cooperet al., 1986; Hardy and Hayakawa, 2001) (Figures 4A–4C). In tion enhances BCR-ABL mediated leukemogenesis without af-addition, leukemic cells were negative for Thy1.2, Ter119, and fecting the target cell for transformation.Gr-1 markers. Thus, the combination of histological and flowcytometric analyses indicate that mice reconstituted with BCR- Kinetic analysis of leukemia development by PETABL transduced bone marrow cells of either genotype succumb We previously reported that ectopic expression of G2A in heter-
ologous cell types delays cell-cycle progression through G2/Mto similar Pro/Pre-B cell leukemias. Therefore, loss of G2A func-
384 CANCER CELL : MAY 2002
A R T I C L E
negative regulation of proliferation and integration of extracellu-lar signals with cytoskeletal reorganization and cell motility.Thus, although the cellular target for tumor induction by BCR-ABL is similar in both G2A-deficient and wild-type backgrounds,G2A ablation could have a profound effect on the rate of prolifer-ation, migratory behavior, or anatomical colonization of leuke-mic cells in vivo. We therefore examined the contribution ofaltered leukemic cell migration/homing versus cell-cycle pro-gression to the earlier mortality associated with loss of G2Afunction.
We exploited PET technology to follow kinetic and spatialcharacteristics of leukemia development in individual micetransplanted with BCR-ABL transduced wild-type or G2A-defi-cient bone marrow cells. To achieve this goal, we coupled theexpression of P185 BCR-ABL to that of the Herpes SimplexVirus (HSV)-1sr39TK PET reporter gene in a bicistronic retroviralvector (MSCV-TK-IRES-P185 BCR-ABL) (Figure 5A). HSV-1sr39TK is a specific mutant form of Herpes Simplex VirusThymidine Kinase (TK) that has a significantly higher affinity forthe acycloguanosines (Gambhir et al., 2000). To demonstratethe ability to image transplanted MSCV-TK-IRES-P185 BCR-ABL transduced wild-type bone marrow cells in recipient SCIDmice, we compared SCID mice transplanted with either MSCV-GFP-IRES-P185 BCR-ABL or MSCV-TK-IRES-P185 BCR-ABLtransduced bone marrow cells (Figure 5A). Twenty-five daysfollowing transplantation, recipient SCID mice were injected in-travenously with 200 �Ci 9-(4-[18F]-fluoro-3 hydroxymethylbutyl)guanine (FHBG) and subjected to whole-body microPET scan.Serial 3D microPET scans were performed dorsoventrally withdata collection in 64 planes of section, and mice were alsoX-rayed for visualization of skeletal landmarks following the finalscan. Physiological hepato-biliary and renal excretion of FHBGis seen in several planes in all recipient animals (Figures 5B and5C). Therefore, interpretations of PET scan are most informativefor the thorax, neck, head, and limbs. Multiple consecutive sec-tions at the thoracic spine level of a TK-BCR-ABL-positive leuke-mic animal are shown in Figure 5D to demonstrate the 3D resolu-tion of the microPET technique.
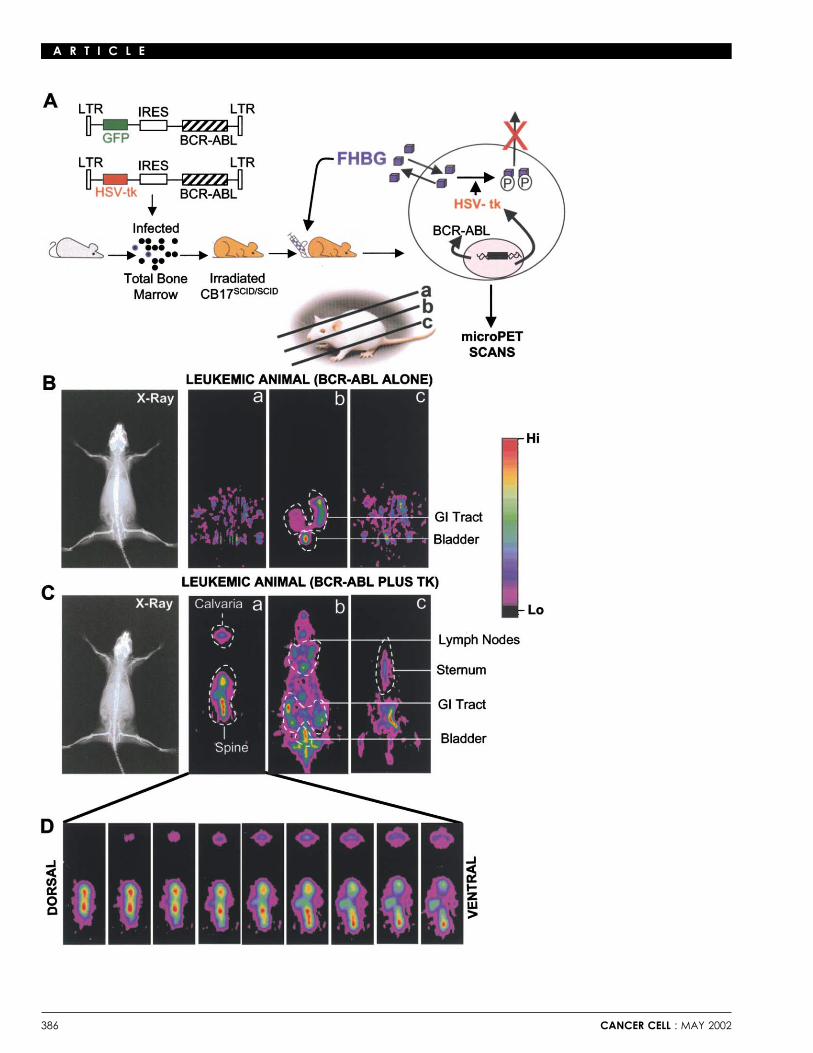
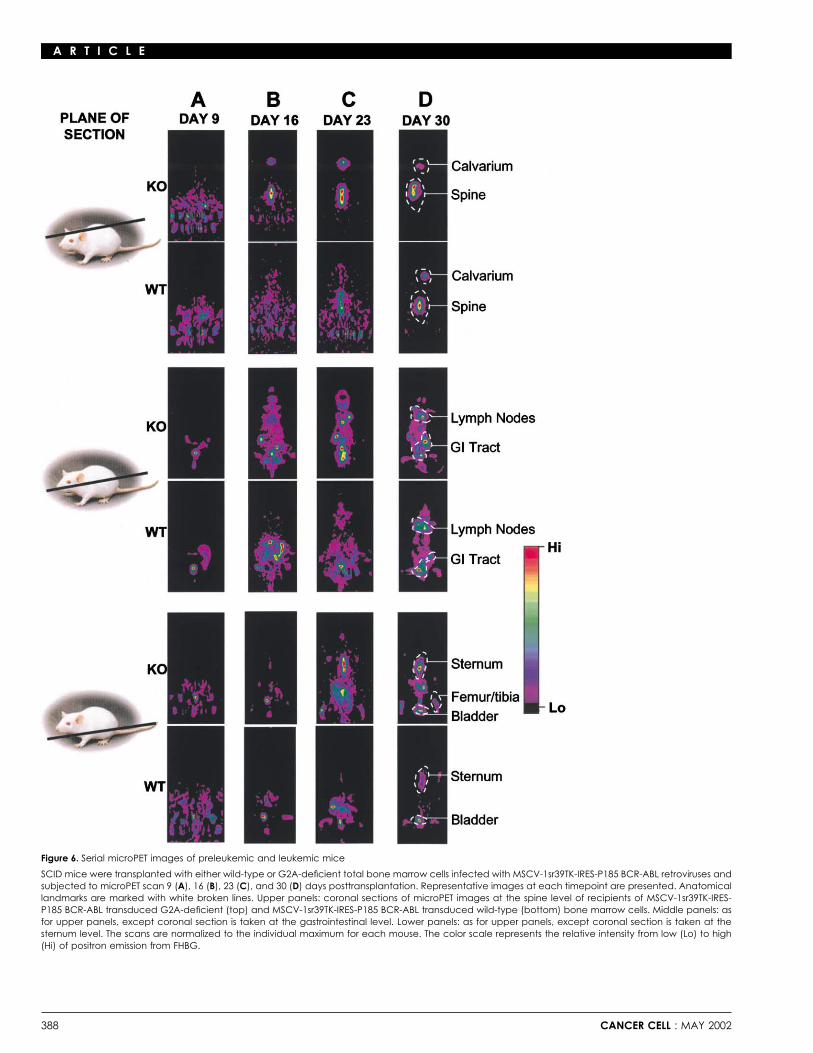
SCID mice were transplanted with wild-type or G2A-defi-cient bone marrow cells transduced with MSCV-TK-IRES-P185BCR-ABL retroviruses. 9, 16, 23, and 30 days following trans-plantation, mice were injected intravenously with 200 �Ci FHBGand subjected to whole-body microPET scan. Serial 3D micro-PET scans were performed dorsoventrally with data collectionin 64 planes of section. We successfully imaged highly specificregional expansion of B lymphoid cells in these mice at timesFigure 4. Flow cytometric analysis of wild-type and G2A-deficient leukemicwhen few leukemic cells were detectable in their peripheralcellsblood. Representative images derived from data collected inFlow cytometric analysis to determine the lineage and developmental
stage of leukemias. Spleen (A), bone marrow (B), and peripheral blood (C) three sectional planes of multiple (2–5) animals per group atwere harvested from typical leukemic mice transplanted with G2A-deficient each timepoint are presented (Figures 6A–6D). Although no PET(left) or wild-type (right) bone marrow. All leukemic cells were uniformly signal was detected 9 days following transplantation of eitherGFP-positive and positive for B220 and BP-1, which are markers for Pro/Pre-B
BCR-ABL transduced G2A-deficient or BCR-ABL transducedcell. Proportions of cells in each quadrant are indicated as percentages.wild-type bone marrow cells (Figure 6A), strong signals wereobserved in recipients of BCR-ABL transduced G2A-deficientbone marrow cells in the spine and calvaria at day 16. At thesame time, no signal was observed in recipients of BCR-ABL(Weng et al., 1998). Consistent with this observation, G2A-defi-transduced wild-type bone marrow (Figure 6B). By day 23, TK�cient T cells are hyperproliferative to a variety of stimuli (Le etG2A-deficient cells had accumulated in the cervical lymphal., 2001). In addition, we have established the coupling of G2Anodes, sternum, and femur/tibia as indicated by intense signalsto G�13, leading to activation of RhoA and actin stress fiberdetected in these areas (Figure 6C). In contrast, at this timepoint,formation in fibroblasts (Kabarowski et al., 2000). These obser-
vations implicate a number of possible roles for G2A, including a weak signal was detected in the thoracic spine area of recipi-
CANCER CELL : MAY 2002 385
A R T I C L E
386 CANCER CELL : MAY 2002

A R T I C L E
ents of BCR-ABL transduced wild-type bone marrow cells. a dramatic effect on the kinetics of leukemogenesis in vivo,loss of G2A function has no discernable effect on B lymphoidThirty days posttransplantation, TK� wild-type leukemic cells
eventually extended to the calvaria, cervical lymph nodes, ster- transformation by BCR-ABL in LTBMCs in vitro. These resultssuggest that G2A regulates a biological process in vivo that isnum, and femur (Figure 6D). However, at this timepoint, the PET
signal was weaker compared to that observed in recipients of not adequately recapitulated in long-term bone marrow cultures.Although the precise molecular basis underlying the modifi-BCR-ABL transduced G2A-deficient bone marrow cells.
These observations indicate that the initiation and progres- cation of BCR-ABL mediated leukemogenesis by G2A is pres-ently unknown, several possible explanations can be proposedsion of disease is significantly accelerated in mice receiving
MSCV-TK-IRES-P185 BCR-ABL transduced G2A-deficient based on our knowledge of the biological properties of thisreceptor. We recently identified the inflammatory lysophospho-bone marrow cells. However, the pattern and anatomical loca-
tions of leukemia development were indistinguishable in all ani- lipid lysophosphatidylcholine (LPC) as a ligand for G2A (Kaba-rowski et al., 2001), and it is possible that high concentrationsmals tested, suggesting that increased mortality of mice trans-
planted with BCR-ABL transduced G2A-deficient bone marrow of LPC or other cofactors in vivo work through G2A to limitlymphocyte growth. G2A may thereby constitute a transcription-is associated with increased cellular expansion and resultant
leukemic burden, rather than an altered route of leukemic dis- ally regulated negative feedback loop utilized by cells as a globalprotective mechanism to balance sustained proliferative stimuli.semination and organ/tissue involvement.Transcriptional induction of Smad 7 by TGF� and SuppressorOf Cytokine Signaling (Socs) proteins in reponse to cytokineDiscussionreceptor activation serve as paradigms for the activation of anegative growth regulatory pathway by positive growth signalsThe ability to noninvasively visualize tumor development in ani-
mal models has become a major area of cancer research. We (Krebs and Hilton, 2001; Nakao et al., 1997).It is possible that transcriptional induction of G2A in BCR-have developed a novel transplantation model for BCR-ABL
induced leukemia in which expression of BCR-ABL is coupled ABL expressing B lymphoid progenitors elicits signals that spe-cifically antagonize one or more of those mediating key patho-to that of the Herpes Simplex Virus 1sr39 thymidine kinase
PET reporter gene. This model allows the kinetic and spatial physiological processes downstream of BCR-ABL. In this re-spect, the ability of BCR-ABL to decrease the adhesive capacityexamination of leukemia initiation and progression in individual
animals and the separate contribution of proliferative expansion of hematopoietic progenitors to stromal elements is believedto contribute to their escape from normal growth inhibitory in-and leukemic dissemination to these processes. Our ability to
successfully monitor leukemia development in individual recipi- fluences within the bone marrow microenvironment and abnor-mal homing to extramedullary sites (Gordon et al., 1987a,ent mice by microPET imaging validates the use of this model of
BCR-ABL induced leukemia for the study of candidate modifiers 1987b). This property is associated with the ability of BCR-ABLto associate with the actin cytoskeleton (McWhirter and Wang,and effector pathways. We therefore applied this model to ad-
dress whether the G protein-coupled receptor G2A, a transcrip- 1993) and target the Rho family GTPase RAC (Skorski et al.,1998). G2A also modifies cytoskeletal organization and cellulartional target of BCR-ABL, modifies leukemogenesis induced by
this oncoprotein. adhesion by activating RhoA leading to assembly of actin stressfibers and focal adhesion contacts (Kabarowski et al., 2000).Although G2A function is dispensable for normal T and B
lymphopoiesis throughout young adulthood, G2A-deficient T Coordinate regulation of Rho family GTPases (CDC42, RAC,RhoA) is critical for the normal regulation of cellular adhesivelymphocytes exhibit hyperproliferative responses in vitro, and
G2A-deficient mice develop a slowly progressive systemic auto- and migratory behavior (Nobes and Hall, 1995; Sander et al.,1999), suggesting that G2A may modify the leukemogenic po-immune syndrome (Le et al., 2001). While these observations
have supported a role for G2A as a negative regulator of lympho- tency of BCR-ABL by altering cellular migratory/homing or pro-liferative properties through the concurrent deregulation of RACcyte growth responses, our present studies extend the biologi-
cal context in which this property is manifest to include onco- and RhoA activities.To address these possibilities, we utilized PET to simultane-genic transformation. G2A is a powerful negative modifier of
P185 BCR-ABL mediated leukemogenesis. Mice transplanted ously examine cellular expansion and migratory characteristicsin individual preleukemic mice. We observed that prior to anywith BCR-ABL transduced G2A-deficient bone marrow cells
exhibit earlier tumor onset, more rapid tumor progression, and indication of overt illness, mice transplanted with G2A-deficientbone marrow harbor significantly expanded populations of leu-shorter survival times compared to those receiving BCR-ABL
transduced wild-type bone marrow cells. Interestingly, despite kemic cells compared to recipients of wild-type bone marrow.
Figure 5. Overview of PET technique
A: Schematic representation of the protocol for PET imaging of preleukemic mice. SCID mice were transplanted with wild-type total bone marrow cellsinfected with MSCV-GFP-IRES-P185 BCR-ABL or MSCV-1sr39TK-IRES-P185 BCR-ABL retroviral stocks. 25 days following transplantation, recipient SCID micewere administered 200 �Ci of [18F] FHBG fluoropenciclovir and subsequently subjected to serial microPET scans dorsoventrally. Representative examples ofmicroPET scan images of 3 sectional planes a, b, and c from (B) a control leukemic SCID mouse (BCR-ABL alone) and (C) a TK� leukemic SCID mouse(BCR-ABL plus TK). Anatomical landmarks are marked with white broken lines. Following the final scan, mice were X-rayed for identification of anatomicallandmarks. Physiological bowel and renal excretions of the radiolabeled probe represent background and are seen in both control and TK� animals.Signals of TK-labeled tumor cells are detected in the spine, calvarium, and lymph node areas of leukemic animals. Multiple consecutive dorsoventralsections at the thoracic spine level of a TK� leukemic animal are shown in D. The scans are normalized to the individual maximum for each mouse. Thecolor scale represents the relative intensity from low (Lo) to high (Hi) of positron emission from FHBG.
CANCER CELL : MAY 2002 387
A R T I C L E
Figure 6. Serial microPET images of preleukemic and leukemic mice
SCID mice were transplanted with either wild-type or G2A-deficient total bone marrow cells infected with MSCV-1sr39TK-IRES-P185 BCR-ABL retroviruses andsubjected to microPET scan 9 (A), 16 (B), 23 (C), and 30 (D) days posttransplantation. Representative images at each timepoint are presented. Anatomicallandmarks are marked with white broken lines. Upper panels: coronal sections of microPET images at the spine level of recipients of MSCV-1sr39TK-IRES-P185 BCR-ABL transduced G2A-deficient (top) and MSCV-1sr39TK-IRES-P185 BCR-ABL transduced wild-type (bottom) bone marrow cells. Middle panels: asfor upper panels, except coronal section is taken at the gastrointestinal level. Lower panels: as for upper panels, except coronal section is taken at thesternum level. The scans are normalized to the individual maximum for each mouse. The color scale represents the relative intensity from low (Lo) to high(Hi) of positron emission from FHBG.
388 CANCER CELL : MAY 2002

A R T I C L E
retroviral stock was used to infect bone marrow cells from either G2A-However, the anatomical sites of disease development are simi-deficient or wild-type mice.lar in all animals, suggesting that the increased mortality of mice
transplanted with BCR-ABL transduced G2A-deficient boneBone marrow transformation and in vivo leukemogenesis assay
marrow is likely associated with increased cellular expansion Bone marrow cells were freshly isolated from the tibias and femurs of 4–8and resultant leukemic burden, rather than altered leukemic week old G2A-deficient or wild-type littermates. Bone marrow cells weredissemination and organ/tissue involvement. subsequently infected with MSCV-GFP-IRES-BCR-ABL retroviruses and
plated in triplicate in 6 cm dishes as previously described (McLaughlin etG2A exhibits a high degree of homology to several otheral., 1987). The numbers of nonadherent Pre-B cells were counted 7–9 daysGPCRs, most notably TDAG8, GPR4, and OGR1, which arepostinfection.coexpressed in lymphocytes (Choi et al., 1996; Heiber et al.,
For in vivo leukemogenesis assays, 6–8 week old severe combined1995; Xu and Casey, 1996) and share ligand specificities towardimmunodeficient (SCID) mice were sublethally irradiated with 275 rads one
structurally related lysophospholipids (Im et al., 2001; Kabarow- day prior to reconstitution. Whole bone marrow from G2A-deficient or wild-ski et al., 2001; Xu et al., 2000; Zhu et al., 2001). Interestingly, type littermates was isolated and infected with retroviruses as describedthe human genes encoding G2A, TDAG8, and OGR1 are located above. Three hours after infection, bone marrow cells were injected intrave-
nously by tail vein into recipient SCID mice. Animals were monitored dailyon chromosome 14q32, a region in which abnormalities arefor signs of illness for two months as previously described (Afar et al., 1997;frequently found in lymphoid proliferative disorders and lympho-Lim et al., 2000). For PET imaging of leukemic animals, whole bone marrowmas (Kyaw et al., 1998; Weng et al., 1998). We detect a highfrom G2A-deficient or wild-type littermates was infected with MSCV-HSV-
level of G2A mRNA expression in multiple human Ph-positive 1sr39TK-IRES-P185 BCR-ABL retroviral stocks and transplanted into SCIDcell lines (unpublished data). Future studies will address the mice as described above.question of whether alterations in expression or specific muta-
Histological analysistions, translocation, and/or rearrangements within the G2A lo-Spleen, bone marrow, and blood were harvested from overtly ill animals andcus are associated with the progression of human Ph-positivesingle-cell suspensions from these tissues were depleted of red blood cellslymphoid leukemias.by hypotonic lysis and cytospun onto microscope slides (Fisher Scientific).In conclusion, G2A functions as a negative modifier of BCR-Peripheral blood smears and single-cell suspensions from spleen and bone
ABL induced leukemogenesis. By adapting a murine trans- marrow were then analyzed by Giemsa/Wright staining using the HEMAplantation model of BCR-ABL induced leukemia for microPET solution as per manufacturer’s protocol (Biochemical Sciences, Inc., Newimaging, we have developed a system with the potential to study Jersey).
preleukemic events and resolve the contribution of candidateRNA and PCR analysiscellular and biological processes otherwise inseparable in ani-An RT-PCR method was used to measure the G2A RNA levels as previouslymal model systems evaluated by autopsy alone. In this regard,described (Weng et al., 1998). In brief, total RNA was isolated from either
loss of G2A function primarily affects the early proliferative wild-type or G2A-deficient BCR-ABL-transformed leukemic cells from boneexpansion of B lymphoid cells in the bone marrow. Such models marrow or spleen. First-strand cDNA was synthesized from 5 �g of totalshould allow more thorough evaluation of therapeutic agents RNA, using the Superscript preamplification system (GIBCO/BRL). Using
G2A-specific primers P1 (TAGCGGTCGCAGGAAATGCAG) and P2 (CAGwith respect to their impact on both cell expansion and sitesGACTGGCTTGGGTCATT), 10% of the first-strand cDNA synthesis productof leukemia development.was subsequently used for PCR. Glyceraldehyde 3-phosphate dehydroge-nase (G3PDH) control amplimer set (CLONTECH) was included as a controlExperimental proceduresto ensure equivalent template levels and quality of RNA.
MiceCell surface staining and flow cytometric analysisG2A-deficient mice were generated via disruption of the G2A locus by homol-Single-cell suspensions from spleen, bone marrow and blood, were preparedogous recombination in 129SvJ embryonic stem (ES) cells as previouslyas described above and stained with combinations of the following antibod-described (Le et al., 2001). Heterozygotes carrying the G2A null mutationies from PharMingen (San Diego, CA), unless otherwise stated: PE-conju-were intercrossed, and offspring were genotyped by Southern blot analysisgated anti-BP-1, PE-conjugated anti-CD43, PE-conjugated anti-Thy1.2, PE-of tail DNA to identify homozygous mutant mice. All studies reported in thisconjugated anti-Ter119, PE-conjugated anti-Gr1, and Tri-color-conjugatedmanuscript were conducted with mice backcrossed 5 generations onto theanti-B220 (CALTAG, Burlingame, CA). Data was acquired on a FACScanBALB/c background. C.B-17SCID/SCID mice (Custer et al., 1985) were obtained(Becton Dickinson) and analyzed using CellQuest software. Live cells werefrom Taconic (Germantown, New York) and housed in isolated barrier facili-gated based on forward and side scatter (Le et al., 2001).ties containing autoclaved food, water, bedding, and cages. All other animals
were housed in the conventional animal facility at UCLA (Los Angeles, Califor-PET imagingnia). Animal care and use is in compliance with regulations of the Chancellor’s9-(4-[18F]-fluoro-3 hydroxymethylbutyl)guanine (FHBG), a substrate for theAnimal Research Committee (ARC) at UCLA.HSV1-sr39TK enzyme, was synthesized as previously described (Yaghoubiet al., 2001). At day 9, 16, 23, and 30 posttransplantation, mice were imagedRetroviral vectors and generation of virus stocksby using the microPET scanner developed at the Crump Institute for Biologi-The retroviral vector pMSCV was used (Hawley et al., 1994). P185 BCR-cal Imaging (UCLA) (also available through Concorde Microsystems, Knox-ABL cDNA was ligated with the encephalomyocarditis virus Internal Ribo-ville, TN) (Cherry and Gambhir, 2001). Mice were tail-vein injected with �200some Entry Site (IRES) (CLONTECH, Palo Alto, CA) and then cloned into�Ci of FHBG 30 min prior to anesthetization by intraperitoneal injection ofpMSCV. Green Fluorescent Protein (CLONTECH) or HSV-1sr39TK was in-40 �l of a ketamine and xylazine (4:1) solution. Mice were then placed in aserted into pMSCV at a unique EcoRI site between the 3� LTR and IRES-spread-prone position on a 8 � 12 cm board and scanned by microPET.P185 BCR-ABL encoding sequences to generate MSCV-GFP-IRES-BCR-Acquisition time was 56 min (8 min per bed position, seven bed positionsABL and MSCV-1sr39TK-IRES-P185 BCR-ABL, respectively. HSV-1sr39TKtotal), and images were reconstructed by using an interative three-dimen-was a gift from Dr. Margaret Black (Washington State University) (Gambhirsional reconstruction technique as described previously (Qi et al., 1998). Atet al., 2000).the end of the microPET scan series, animals were sacrificed and detectionRetroviral stocks were prepared by transient cotransfection of 293T
cells with retroviral vectors and Psi� ecotropic packaging vector (Muller et of leukemic cells in different organs verified the locations of the signals onmicroPET.al., 1991). Supernatants were collected 24–48 hr posttransfection. The same
CANCER CELL : MAY 2002 389

A R T I C L E
(1987a). Altered adhesive interactions with marrow stroma of haematopoieticAcknowledgmentsprogenitor cells in chronic myeloid leukaemia. Nature 328, 342–344.
We thank Dr. S. Larry Zipursky and members of the Witte lab and Gambhir Gordon, M.Y., Riley, G.P., Watt, S.M., and Greaves, M.F. (1987b). Compart-lab for helpful discussions; James Johnson, Jami McLaughlin, and Shirley mentalization of a haematopoietic growth factor (GM-SCF) by glycosamino-Quan for expert technical help; and J.C. White for assistance in the prepara- glycans in the bone marrow microenvironment. Nature 326, 403–405.tion of this manuscript. We are grateful to Dr. Michael Phelps and Dr. Harvey
Gorre, M.E., Mohammed, M., Ellwood, K., Hsu, N., Paquette, R., Rao, P.N.,Herschman for introducing us to the benefits of PET reporter gene analysis.and Sawyers, C.L. (2001). Clinical resistance to STI-571 cancer therapyL.Q.L. is a Fellow of the University of California, Los Angeles, Medical Scien-caused by BCR-ABL gene mutation or amplification. Science 293, 876–880.tist Training Program, and supported by a USHHS Institutional National
Research Service Award #T32 CA090576 and the Medical Scientist Training Hardy, R.R., and Hayakawa, K. (2001). B cell development pathways. Annu.Program training grant. J.H.S.K. is a Fellow of the Leukemia and Lymphoma Rev. Immunol. 19, 595–621.Society (formerly the Leukemia Society of America). O.N.W. is an Investigator
Hawley, R.G., Lieu, F.H.L., Fong, Z.C., and Hawley, T.S. (1994). Versatileof the Howard Hughes Medical Institute. This work was partially supportedretroviral vectors for potential use in gene therapy. Gene Ther. 1, 136–138.by NIH grant CA76204 to O.N.W. Imaging described in this paper was
supported by funding from DOE contract DE-FC03-87ER60615 (SSG), NIH Heiber, M., Docherty, J.M., Shah, G., Nguyen, T., Cheng, R., Heng, H.H.,RO1 CA82214-01 (SSG), and SAIRP R24 CA92865 (SSG). Marchese, A., Tsui, L.C., Shi, X., George, S.R., and O’Dowd, B.F. (1995).
Isolation of three novel human genes encoding G protein-coupled receptors.DNA Cell Biol. 14, 25–35.
Herschman, H.R., MacLaren, D.C., Iyer, M., Namavari, M., Bobinski, K.,Received: April 9, 2002 Green, L.A., Wu, L., Berk, A.J., Toyokuni, T., Barrio, J.R., et al. (2000). SeeingRevised: April 25, 2002 is believing: Non-invasive, quantitative and repetitive imaging of reporter
gene expression in living animals, using positron emission tomography. J.References Neurosci. Res. 59, 699–705.
Houlston, R.S., and Tomlinson, I.P. (1998). Modifier genes in humans: strate-Abelson, H.T., and Rabstein, L.S. (1970). Lymphosarcoma: Virus-induced gies for identification. Eur. J. Hum. Genet. 6, 80–88.thymic-independent disease in mice. Cancer Res. 30, 2213–2222.
Im, D.S., Heise, C.E., Nguyen, T., O’Dowd, B.F., and Lynch, K.R. (2001).Afar, D.E.H., Han, L., McLaughlin, J., Wong, S., Dhaka, A., Parmar, K., Identification of a molecular target of psychosine and its role in globoid cellRosenberg, N., Witte, O.N., and Colicelli, J. (1997). Regulation of the onco- formation. J. Cell Biol. 153, 429–434.genic activity of BCR-ABL by a tightly bound substrate protein RIN1. Immu-nity 6, 773–782. Kabarowski, J.H.S., Feramisco, J.D., Le, L.Q., Gu, J.L., Luoh, S.W., Simon,
M.I., and Witte, O.N. (2000). Direct genetic demonstration of G alpha 13Balmain, A. (2002). Cancer as a complex genetic trait. Tumor susceptibility coupling to the orphan G protein-coupled receptor G2A leading to Rho-A-in humans and mouse models. Cell 108, 145–152. dependent actin rearrangement. Proc. Natl. Acad. Sci. USA 97, 12109–
12114.Cherry, S.R., and Gambhir, S.S. (2001). Use of positron emission tomographyin animal research. ILAR J. 42, 219–232. Kabarowski, J.H.S., Zhu, K., Le, L.Q., Witte, O.N., and Xu, Y. (2001). Lyso-
phosphatidylcholine is a high affinity ligand for the immunoregulatory recep-Choi, J.W., Lee, S.Y., and Choi, Y. (1996). Identification of a putative Gtor G2A. Science 293, 702–705.protein-coupled receptor induced during activation-induced apoptosis of T
cells. Cell. Immunol. 168, 78–84. Krebs, D.L., and Hilton, D.J. (2001). SOCS proteins: negative regulators ofcytokine signaling. Stem Cells 19, 378–387.Chopra, R., Pu, Q.Q., and Elefanty, A.G. (1999). Biology of BCR-ABL. Blood
Rev. 13, 211–229. Kyaw, H., Zeng, Z., Su, K., Fan, P., Shell, B.K., Carter, K.C., and Li, Y. (1998).Cloning, characterization, and mapping of human homolog of mouse T-cellCooper, M.D., Mulvaney, D., Coutinho, A., and Cazenave, P.A. (1986). Adeath-associated gene. DNA Cell Biol. 17, 493–500.novel cell surface molecule on early B-lineage cells. Nature 321, 616–618.
Le, L.Q., Kabarowski, J.H.S., Weng, Z., Satterthwaite, A.B., Harvill, E.T.,Cormier, R.T., Hong, K.H., Halberg, R.B., Hawkins, T.L., Richardson, P.,Jensen, E.R., Miller, J.F., and Witte, O.N. (2001). Mice lacking the orphan GMulherkar, R., Dove, W.F., and Lander, E.S. (1997). Secretory phospholipaseprotein-coupled receptor, G2A, develop a late-onset autoimmune syndrome.Pla2g2a confers resistance to intestinal tumorigenesis. Nat. Genet. 17,Immunity 14, 561–571.88–91.
Lim, Y.-M., Wong, S., Lau, G., Witte, O.N., and Colicelli, J. (2000). BCR/ABLCuster, R.P., Bosma, G.C., and Bosma, M.J. (1985). Severe combined immu-inhibition by an escort/phosphatase fusion protein. Proc. Natl. Acad. Sci.nodeficiency (SCID) in the mouse. Pathology, reconstitution, neoplasms.USA 97, 12233–12238.Am. J. Pathol. 120, 464–477.Lugo, T.G., Pendergast, A., Muller, A.J., and Witte, O.N. (1990). Tyrosine
Dietrich, W.F., Lander, E.S., Smith, J.S., Moser, A.R., Gould, K.A., Luongo,kinase activity and transformation potency of bcr-abl oncogene products.
C., Borenstein, N., and Dove, W. (1993). Genetic identification of Mom-1, aScience 247, 1079–1082.
major modifier locus affecting Min-Induced intestinal neoplasia in the mouse.Cell 75, 631–639. McLaughlin, J., Chianese, E., and Witte, O.N. (1987). In vitro transformation
of immature hematopoietic cells by the P210 BCR/ABL oncogene productDruker, B.J., and Lydon, N.B. (2000). Lessons learned from the development
of the Philadelphia chromosome. Proc. Natl. Acad. Sci. USA 84, 6558–6562.of an abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J. Clin.Invest. 105, 3–7. McLaughlin, J., Chianese, E., and Witte, O.N. (1989). Alternative forms of the
BCR-ABL oncogene have quantitatively different potencies for stimulation ofGambhir, S.S., Bauer, E., Black, M.E., Liang, Q., Kokoris, M.S., Barrio, J.R., immature lymphoid cells. Mol. Cell. Biol. 9, 1866–1874.Iyer, M., Namavari, M., Phelps, M.E., and Herschman, H.R. (2000). A mutantherpes simplex virus type 1 thymidine kinase reporter gene shows improved McWhirter, J.R., and Wang, J.Y.J. (1993). An actin-binding function contrib-sensitivity for imaging reporter gene expression with positron emission to- utes to transformation by the bcr-abl oncoprotein of philadelphia chromo-mography. Proc. Natl. Acad. Sci. USA 97, 2785–2790. some-positive human leukemias. EMBO J. 12, 1533–1546.
Gishizky, M.L., and Witte, O.N. (1992). Initiation of deregulated growth of Mitelman, F. (1993). The cytogenetic scenario of chronic myeloid leukemia.multipotent progenitor cells by bcr-abl in vitro. Science 256, 836–839. Leuk. Lymphoma 11, 11–15.
Mock, B.A., Krall, M.M., and Dosik, J.K. (1993). Genetic mapping of tumorGordon, M.Y., Dowding, C.R., Riley, G.P., Goldman, J.M., and Greaves, M.F.
390 CANCER CELL : MAY 2002

A R T I C L E
susceptibility genes involved in mouse plasmacytomagenesis. Proc. Natl. between both GTPases determines cellular morphology and migratory be-havior. J. Cell Biol. 147, 1009–1022.Acad. Sci. USA 90, 9499–9503.
Sawyers, C.L. (1999). Chronic myeloid leukemia. N. Engl. J. Med. 340, 1330–Muller, A.J., Young, J.C., Pendergast, A.-M., Pondel, M., Landau, N.R.,1340.Littman, D.R., and Witte, O.N. (1991). BCR first exon sequences specifically
activate Bcr/Abl tyrosine kinase oncogene of Philadelphia chromosome posi- Skorski, T., Wlodarski, P., Daheron, L., Salomoni, P., Nieborowska-Skorska,tive human leukemias. Mol. Cell. Biol. 11, 1785–1792. M., Majewski, M., Wasik, M., and Calabretta, B. (1998). BCR/ABL-mediated
leukemogenesis requires the activity of the small GTP-binding protein Rac.Nadeau, J.H. (2001). Modifier genes in mice and humans. Nat. Rev. Genet.
Proc. Natl. Acad. Sci. USA 95, 11858–11862.2, 165–174.
Stripecke, R., Skelton, D.C., Gruber, T., Afar, D., Pattengale, P.K., Witte,Nakao, A., Afrakhte, M., Moren, A., Nakayama, T., Christian, J.L., Heuchel, O.N., and Kohn, D.B. (1998). Immune response to Philadelphia chromosome-R., Itoh, S., Kawabata, M., Heldin, N.E., Heldin, C.H., and ten Dijke, P. positive acute lymphoblastic leukemia induced by expression of CD80, in-(1997). Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta terleukin 2, and granulocyte-macrophage colony-stimulating factor. Hum.signalling. Nature 389, 631–635. Gene Ther. 9, 2049–2062.
Unnikrishnan, I., Radfar, A., Jenab-Wolcott, J., and Rosenberg, N. (1999).Nobes, C.D., and Hall, A. (1995). Rho, Rac, and Cdc42 GTPases regulatep53 mediates apoptotic crisis in primary Abelson virus-transformed pre-Bthe assembly of multimolecular focal complexes associated with actin stresscells. Mol. Cell. Biol. 19, 4825–4831.fibers, lamellipodia, and filopodia. Cell 81, 53–62.
Van Dyke, T., and Jacks, T. (2002). Cancer modeling in the modern era.Nowell, P.C., and Hungerford, D.A. (1960). A minute chromosome in humanProgress and challenges. Cell 108, 135–144.chronic granulocytic leukemia. Science 132, 1497.
Weng, Z., Fluckiger, A.C., Nisitani, S., Wahl, M.I., Le, L.Q., Hunter, C.A.,Qi, J., Leahy, R.M., Cherry, S.R., Chatziioannou, A., and Farquhar, T.H.Fernal, A.A., Le Beau, M.M., and Witte, O.N. (1998). A DNA damage and
(1998). High-resolution 3D Bayesian image reconstruction using the micro-stress inducible G protein-coupled receptor blocks cells in G2/M. Proc. Natl.
PET small-animal scanner. Phys. Med. Biol. 43, 1001–1013. Acad. Sci. USA 95, 12334–12339.
Risser, R., Potter, M., and Rowe, W.P. (1978). Abelson virus-induced lympho- Xu, Y., and Casey, G. (1996). Identification of human OGR1, a novel Gmagenesis in mice. J. Exp. Med. 148, 714–726. protein-coupled receptor that maps to chromosome 14. Genomics 5, 397–
402.Risser, R., Kaehler, D., and Lamph, W.W. (1985). Different genes control the
Xu, Y., Zhu, K., Hong, G., Wu, W., Baudhuin, L.M., Xiao, Y., and Damron,susceptibility of mice to Moloney or Abelson murine leukemia viruses. J.D.S. (2000). Sphingosylphosphorylcholine is a ligand for ovarian cancerVirol. 55, 547–553.G-protein-coupled receptor 1. Nat. Cell Biol. 2, 261–267.
Rosenberg, N., and Baltimore, D. (1976). A quantitative assay for transforma-Yaghoubi, S., Barrio, J.R., Dahlbom, M., Iyer, M., Namavari, M., Satyamurthy,tion of bone marrow cells by Abelson murine leukemia virus. J. Exp. Med.N., Goldman, R., Herschman, H.R., Phelps, M.E., and Gambhir, S.S. (2001).143, 1453–1463.Human pharmacokinetic and dosimetry studies of [(18)F]FHBG: a reporterprobe for imaging herpes simplex virus type-1 thymidine kinase reporterRowley, J.D. (1973). A new consistent chromosomal abnormality in chronicgene expression. J. Nucl. Med. 42, 1225–1234.myelogeneous leukemia identified by quinacrine fluorescence and giemsa
staining. Nature 243, 290–293. Zhang, S.L., DuBois, W., Ramsay, E.S., Bliskovski, V., Morse, H.C., Tad-desse-Heath, L., Vass, W.C., DePinho, R.A., and Mock, B.A. (2001). Effi-
Saglio, G., Guerrasio, A., Rosso, C., Zaccaria, A., Tassinari, A., Serra, A., ciency alleles of the Pctr1 modifier locus for plasmacytoma susceptibility.Rege-Cambrin, G., Mazza, U., and Gavosto, F. (1990). New type of Bcr/Abl Mol. Cell. Biol. 21, 310–318.junction in Philadelphia chromosome-positive chronic myelogenous leuke-
Zhu, K., Baudhuin, L.M., Hong, G., Williams, F.S., Cristina, K.L., Kabarowski,mia. Blood 76, 1819–1824.J.H.S., Witte, O.N., and Xu, Y. (2001). Sphingosylphosphorylcholine and
Sander, E.E., ten Klooster, J.P., van Delft, S., van der Kammen, R.A., and Lysophosphatidylcholine are ligands for the G Protein-coupled receptorGPR4. J. Biol. Chem. 276, 41325–41335.Collard, J.G. (1999). Rac downregulates Rho activity: reciprocal balance
CANCER CELL : MAY 2002 391


















![Design, synthesis, and biological evaluation of pyrazolo [3, 4-d] pyrimidines active in vivo on the Bcr-Abl T315I mutant](https://static.fdokumen.com/doc/165x107/6333d434b94d623842026483/design-synthesis-and-biological-evaluation-of-pyrazolo-3-4-d-pyrimidines-active.jpg)

