
BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic...
14
Leukemia Research 34 (2010) 1255–1268 Contents lists available at ScienceDirect Leukemia Research journal homepage: www.elsevier.com/locate/leukres Invited review BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: A review Xin An a,b,c,1 , Amit K. Tiwari a,1 , Yibo Sun a,1 , Pei-Rong Ding a,b,c,1 , Charles R. Ashby Jr. a , Zhe-Sheng Chen a,∗ a Department of Pharmaceutical Sciences, College of Pharmacy and Allied Health Professions, St. John’s University, 8000 Utopia Parkway, Jamaica, NY 11439, USA b Department of Medical Oncology, Cancer Center, Sun Yat-Sen University, Guangzhou, China c State Key Laboratory of Oncology in South China, Sun Yat-Sen University, Guangzhou, China article info Article history: Received 21 January 2010 Received in revised form 25 April 2010 Accepted 25 April 2010 Keywords: CML Philadelphia chromosome TKI BCR-ABL abstract Chronic Myeloid Leukemia (CML) is a clonal disease characterized by the presence of the Philadelphia (Ph+) chromosome and its oncogenic product, BCR-ABL, a constitutively active tyrosine kinase, that is present in >90% of the patients. Epidemiologic data indicates that almost 5000 new cases are reported every year and 10% of these patients eventually succumb to the disease. The treatment of CML was revo- lutionized by the introduction of imatinib mesylate (IM, Gleevec ® ), a BCR-ABL tyrosine kinase inhibitor (TKI). The clinical use of specific BCR-ABL inhibitors has resulted in a significantly improved prognosis, response rate, overall survival, and patient outcome in CML patients compared to previous therapeu- tic regimens. However, the complete eradication of CML in patients receiving imatinib was limited by the emergence of resistance mostly due to mutations in the ABL kinase domain and to a lesser extent by molecular residual disease after treatment. The second-generation BCR-ABL TKIs nilotinib (Tasigna ® ) and dasatinib (Sprycel ® ), showed significant activity in clinical trials in patients intolerant or resistant to imatinib therapy, except in those patients with the T315I BCR-ABL mutation. Identifying key components involved in the CML pathogenesis may lead to the exploration of new approaches that might eventually overcome resistance mediated to the BCR-ABL TKIs. Here, we present an overview about the current treatment of Ph+ CML patients with the TKIs and the obstacles to successful treatment with these drugs. © 2010 Elsevier Ltd. All rights reserved. Contents 1. Introduction .......................................................................................................................................... 1256 2. Mechanism of action of BCR-ABL TKIs ............................................................................................................... 1256 3. Monitoring BCR-ABL TKIs therapy: assessment of clinical response ................................................................................ 1257 4. Clinical trials examining the effects of TKIs in Ph+ patients ......................................................................................... 1259 4.1. The first generation of TKI .................................................................................................................... 1259 4.2. The second-generation of TKIs ............................................................................................................... 1259 4.2.1. Dasatinib ............................................................................................................................ 1259 4.2.2. Nilotinib ............................................................................................................................. 1261 5. Resistance to BCR-ABL TKIs .......................................................................................................................... 1262 5.1. BCR-ABL dependent resistance to imatinib .................................................................................................. 1262 5.1.1. Point mutations in the kinase domain of BCR-ABL ................................................................................. 1262 5.1.2. BCR-ABL gene amplification ........................................................................................................ 1262 5.2. BCR-ABL independent mechanisms of resistance ............................................................................................ 1262 5.2.1. Inadequate plasma levels of imatinib .............................................................................................. 1262 5.2.2. Incomplete adherence to the therapeutic regimen ................................................................................ 1263 5.2.3. Pharmacokinetic parameters ....................................................................................................... 1263 5.2.4. Intracellular uptake of imatinib .................................................................................................... 1263 5.2.5. Activation of other signaling pathways ............................................................................................ 1263 ∗ Corresponding author. Tel.: +1 718 990 1432; fax: +1 718 990 1877. E-mail address: [email protected] (Z.-S. Chen). 1 These authors contributed equally to this work. 0145-2126/$ – see front matter © 2010 Elsevier Ltd. All rights reserved. doi:10.1016/j.leukres.2010.04.016
Transcript of BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic...

I
Bp
X
a
b
c
a
ARRA
KCPTB
C
0d
Leukemia Research 34 (2010) 1255–1268
Contents lists available at ScienceDirect
Leukemia Research
journa l homepage: www.e lsev ier .com/ locate / leukres
nvited review
CR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosomeositive chronic myeloid leukemia: A review
in Ana,b,c,1, Amit K. Tiwaria,1, Yibo Suna,1, Pei-Rong Dinga,b,c,1, Charles R. Ashby Jr. a, Zhe-Sheng Chena,∗
Department of Pharmaceutical Sciences, College of Pharmacy and Allied Health Professions, St. John’s University, 8000 Utopia Parkway, Jamaica, NY 11439, USADepartment of Medical Oncology, Cancer Center, Sun Yat-Sen University, Guangzhou, ChinaState Key Laboratory of Oncology in South China, Sun Yat-Sen University, Guangzhou, China
r t i c l e i n f o
rticle history:eceived 21 January 2010eceived in revised form 25 April 2010ccepted 25 April 2010
eywords:MLhiladelphia chromosome
a b s t r a c t
Chronic Myeloid Leukemia (CML) is a clonal disease characterized by the presence of the Philadelphia(Ph+) chromosome and its oncogenic product, BCR-ABL, a constitutively active tyrosine kinase, that ispresent in >90% of the patients. Epidemiologic data indicates that almost 5000 new cases are reportedevery year and 10% of these patients eventually succumb to the disease. The treatment of CML was revo-lutionized by the introduction of imatinib mesylate (IM, Gleevec®), a BCR-ABL tyrosine kinase inhibitor(TKI). The clinical use of specific BCR-ABL inhibitors has resulted in a significantly improved prognosis,response rate, overall survival, and patient outcome in CML patients compared to previous therapeu-tic regimens. However, the complete eradication of CML in patients receiving imatinib was limited bythe emergence of resistance mostly due to mutations in the ABL kinase domain and to a lesser extent
KICR-ABL by molecular residual disease after treatment. The second-generation BCR-ABL TKIs nilotinib (Tasigna®)and dasatinib (Sprycel®), showed significant activity in clinical trials in patients intolerant or resistant toimatinib therapy, except in those patients with the T315I BCR-ABL mutation. Identifying key components
involved in the CML pathogenesis may lead to the exploration of new approaches that might eventuallyovercome resistance mediated to the BCR-ABL TKIs. Here, we present an overview about the currenttreatment of Ph+ CML patients with the TKIs and the obstacles to successful treatment with these drugs.© 2010 Elsevier Ltd. All rights reserved.
ontents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12562. Mechanism of action of BCR-ABL TKIs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12563. Monitoring BCR-ABL TKIs therapy: assessment of clinical response . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12574. Clinical trials examining the effects of TKIs in Ph+ patients . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1259
4.1. The first generation of TKI . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12594.2. The second-generation of TKIs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1259
4.2.1. Dasatinib . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12594.2.2. Nilotinib. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1261
5. Resistance to BCR-ABL TKIs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12625.1. BCR-ABL dependent resistance to imatinib . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1262
5.1.1. Point mutations in the kinase domain of BCR-ABL. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12625.1.2. BCR-ABL gene amplification . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1262
5.2. BCR-ABL independent mechanisms of resistance. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12625.2.1. Inadequate plasma levels of imatinib . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1262
5.2.2. Incomplete adherence to the therapeutic regimen . . . . . .5.2.3. Pharmacokinetic parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . .5.2.4. Intracellular uptake of imatinib . . . . . . . . . . . . . . . . . . . . . . . . . .5.2.5. Activation of other signaling pathways . . . . . . . . . . . . . . . . . .∗ Corresponding author. Tel.: +1 718 990 1432; fax: +1 718 990 1877.E-mail address: [email protected] (Z.-S. Chen).
1 These authors contributed equally to this work.
145-2126/$ – see front matter © 2010 Elsevier Ltd. All rights reserved.oi:10.1016/j.leukres.2010.04.016
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1263. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1263. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1263. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1263

1256 X. An et al. / Leukemia Research 34 (2010) 1255–1268
6. Treatment options for imatinib-resistant CML . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12647. Conclusion. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1265
Conflict of interest . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1265Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1265
. . . . . .
1
dCotwstctiCard
fieacmPttkae3tsadTklodslBrtC
A[vhdpisdcp1
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. Introduction
Chronic Myeloid Leukemia (CML) is a myeloproliferative disor-er due to an acquired mutation affecting hematopoietic stem cells.ML accounts for ∼20% of all leukemias diagnosed in adults andccurs at approximately the same frequency in countries aroundhe world. The annual incidence is 1.6 cases per 100,000 adults,ith a male-to-female ratio of 1.4/1.3. The median age at diagno-
is is approximately 55 years, with less than 10% of patients underhe age of 20 years [1]. There are three distinct phases of CML:hronic, accelerated and blast phase. CML is usually diagnosed inhe chronic phase (CML-CP), which is characterized by an increasen granulocytes and is primarily asymptomatic. However, CML-P ultimately progresses to the accelerated phase (CML-AP), withrapid expansion of granulocytes and blast crisis (CML-BC) that
esembles acute leukemia, leading to metastasis, organ failure, andeath [2].
Two pathologists, Drs. Rudolf Virchow and John Hughes Bennett,rst described CML in 1845 [3,4]. However, in 1960, Drs. Peter Now-ll and David Hungerford, who worked in Philadelphia, describedconsistent chromosomal abnormality in patients with CML; the
hromosome was subsequently called the Philadelphia (Ph) chro-osome [5]. More than 90% of adults with CML are shown to be
h chromosome positive (Ph+) [6]. In 1972, Rowley discoveredhat the Ph chromosome is generated by a reciprocal transloca-ion [t(9;22)(q34;q11)] and fusion between abelson (ABL) tyrosineinase gene at chromosomes 9 and break-point cluster (BCR) genet chromosome 22 [7]. The resulting chimeric BCR-ABL oncogenencodes a 210 kDa protein (in >90% Ph+ CML and approximately0–35% acute lymphocytic leukemia (ALL) patients) with constitu-ive and aberrant ABL tyrosine kinase activity, and this has beenhown to play a causal role in CML [8,9]. A subset of CML patientsre associated with variable 185–230 kDa BCR-ABL oncoproteinepending on the site of the breakpoint in the BCR gene [10].his subtle difference in the cell context and intrinsic tyrosineinase activity arising from each BCR-ABL isoform governs the finaleukemia type [11]. The BCR-ABL fusion protein mediates the devel-pment and maintenance of CML through interaction with multipleownstream signaling partners, resulting in altered cellular adhe-ion, activation of mitogenic signaling, and inhibition of apoptosis,eading to the transformation of hematopoietic stem cells [12].CR-ABL-mediated signaling is also associated with defective DNAepair, resulting in additional chromosomal alterations and muta-ions, which may partly explain the aggressive nature of advancedML [13].
The therapeutic options for CML before the discovery of BCR-BL included cytotoxic drugs such as busulfan and hydroxyurea
14]. Although effective chemotherapy could normalize the ele-ation in white blood cell count, prevent complications due toyperleukocytosis, and control the clinical manifestations of theisease, it did not change the natural history of CML, as mostatients would ultimately progress to the blast phase [2]. The
ntroduction of recombinant interferon-alfa (rIFN-�) provided a
ignificant survival advantage when compared with cytotoxicrugs [15]. The majority of patients using rIFN-� monotherapyould achieve hematologic remission; however, only a minority ofatients achieved a complete cytogenetic remission (ranging from3% to 27%) [16]. Moreover, most patients were unable to tolerate. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1265
rIFN-� because of its adverse effects [13,17]. Allogeneic hematopoi-etic cell transplant (HCT) remains the only established curativeapproach for CML [18]. However, significant morbidity and mor-tality rate, in addition of lack of human leukocyte antigen (HLA)matching donors only allows a small percentage of CML patients touse HCT [19].
As the intricate mechanisms of CML are being detailed, researchis now directed towards developing compounds that selectivelyinhibit the aberrant BCR-ABL tyrosine kinase. Imatinib mesylate(IM, STI-571 or Gleevec), is the first BCR-ABL tyrosine kinaseinhibitor (TKI) to be used for the treatment of CML [20]. However,several subsequent studies reported that point mutations withinthe ABL kinase domain or overexpression of BCR-ABL could leadto imatinib resistance in patients with advanced disease [21,22].Therefore, to surmount imatinib resistance, several other novelTKIs were developed and tested in patients with BCR-ABL positiveCML, such as dasatinib (BMS-354825, Sprycel), nilotinib (AMN-107, Tasigna) and bosutinib (SKI-606) [23–25]. The developmentof these TKIs significantly changed the prognosis and treatmentalgorithms of patients with newly diagnosed CML. Currently, BCR-ABL TKIs are the initial choice of treatment for most patients withCML. Consequently, the use of any type of HCT in patients withCML has experienced a substantial decline. This manuscript willreview the clinical use of BCR-ABL TKIs for the treatment of CML.The mechanisms of action and resistance of TKIs (especially for ima-tinib) will be discussed along with possible treatment options forpatients with imatinib resistance or intolerance.
2. Mechanism of action of BCR-ABL TKIs
As described above, cytogenetic abnormalities produce a con-stitutively active BCR-ABL tyrosine kinase, and this has beenimplicated in the pathogenesis of CML. BCR-ABL TKIs can inhibitthe BCR-ABL pathway and significantly reduce the proliferationof BCR-ABL positive CML cells [26]. Imatinib, a 2-phenylaminopyridine-based ATP competitive inhibitor, binds to the inactiveconformation of the ABL protein tyrosine kinase and competitivelyblocks the ATP binding site and prevents its conformational switchto the active form (Fig. 1) [20]. Imatinib inhibits cellular prolif-eration and tumor formation without the induction of apoptosis[27]. An in vitro study reported that imatinib produced a 92–98%decrease in CML colony growth without significantly inhibitingnormal colony growth [27]. Imatinib also inhibits the platelet-derived growth factor receptor (PDGFR), Arg, and c-kit, but notthe Src family kinases [28] (Table 1). Imatinib could also reversebone marrow angiogenesis and decrease the plasma concentra-tion of vascular endothelial growth factor (VEGF) in CML patients[29,30]. Moreover, increasing evidence has emerged indicating thatimatinib exerts profound immunomodulatory effects on T cells andantigen-presenting cells, such as dendritic cells (DC) [31]. However,the exact nature of the effects of imatinib (activation or suppres-sion) on immune cells remains controversial. Some reports showthat imatinib inhibits CD4+ or CD8+ T cell proliferation and acti-
vation, as well as downregulates the Ag-presenting function ofDC [32,33]. In contrast, other studies have shown that imatinibenhances the Ag-presenting function of DC and fosters DC-NKreciprocal activation, thereby promoting the antitumoral functionof NK cells [34,35].
X. An et al. / Leukemia Researc
Fig. 1. Schematic representation of the mechanism of action of the BCR-ABLTKIs.The BCR-ABL tyrosine kinase is a constitutively active kinase that binds ATP andtransfers a phosphate from ATP to tyrosine residues on various substrates. This canproduce an alteration in cellular adhesion, activation of mitogenic signaling andinhibition of apoptosis, resulting in the abnormal proliferation of myeloid cells. TheTKIs, such as imatinib, block the binding of ATP to the BCR-ABL tyrosine kinase andiic
cntSwamb
rCiattvattaetibfiifo
pd
reduction in BCR-ABL/BCR level compared to the median pretreat-
nhibit the tyrosine kinase activity of BCR-ABL-mediated signaling pathways. TKI-nduced inactivation of these pathways significantly reduces the excessive myeloidell proliferation in CML.
Imatinib induces complete hematologic remissions in 97% andomplete cytogenetic response (CCR) in almost 86% of newly diag-osed patients in the chronic phase and thus became the first-lineherapy for CML almost immediately after its introduction [36].ubsequently, it was realized that a durable response to imatinibas only seen in CML-CP patients [21,37]. Some CML-CP and most
dvanced phase patients relapsed after months or years of treat-ent with imatinib due to the development of resistance (this will
e discussed further in the later section of this review) [38,39].Newer BCR-ABL TKIs were developed to overcome imatinib-
esistance and provide more effective treatment for patients withML. Based on the crystal structure of imatinib–ABL complex, sim-
lar ATP-competitive phenylaminopyrimidine class of drugs suchs nilotinib [23] and NS-187 [40] were designed. Similar to ima-inib, nilotinib binds to the inactive conformation of ABL kinase,hereby blocking the substrate binding site proximal to the acti-ation loop, resulting in the inhibition of the ATPase catalyticctivity by disrupting the ATP–phosphate binding site [41]. Crys-allographic studies indicate that nilotinib has a higher affinity forhe ABL kinase domain than imatinib, resulting in greater potencynd selectivity than nilotinib against wild type BCR-ABL [41,42]. Forxample, in vitro, nilotinib was >30-fold more effective than ima-inib in lysing BCR-ABL-expressing cells [43]. In addition, nilotinibs effective against 32 out of 33 imatinib-resistant point mutants,ut had no significant activity against T315I mutants [23,43]. Inact, nilotinib was shown to be significantly effective in prolong-ng survival in imatinib-resistant CML mouse models or in micenjected with both primary marrow cells and/or BCR-ABL trans-ormed hematopoietic cells [25]. Nilotinib also inhibits the activity
f Arg, Kit, and PDGFR, but not Src-family kinases [23,42].Other ATP-competitive, non-phenylpyrimidine-based com-ounds that inihibit BCR-ABL tyrosine kinases were actuallyeveloped as Src-family kinases (SFKs) inhibitors and thus are often
h 34 (2010) 1255–1268 1257
referred to as Src/ABL inhibitors [40]. Dasatinib (BMS-354825;Sprycel) is the only clinically approved Src/ABL inhibitor that isactive against imatinib-resistant or intolerant CML and it inhibitsboth the active and inactive conformations of the ABL domain[24,28,44] (Table 1). An in vitro study showed that dasatinib wasmore potent than imatinib in inhibiting wild type BCR-ABL andmutants with high levels of imatinib resistance, except for thosewith the T315I mutation [43]. Dasatinib also inhibits other tyro-sine kinases, including Src, Lck, YES, EPH receptor A 2 (EPHA2)A2 and PDGFR-� [30,43] and thus inhibits autophosphorylationand downstream phosphorylation of additional targets which maybe involved in mutations in these kinases activation of BCR-ABL-independent pathways in patients with imatinib-intolerant orresistant CML. Other ATP-competitive Src/ABL TKIs under inves-tigation for CML include bosutinib (SKI-606) [25], AP23464 [45],INNO-406 (NS-187) [40] and PD166326 [46].
Since none of the ATP-competitive inhibitors were effectiveagainst BCR-ABL/T315I mutants, targeting the substrate recogni-tion domain rather than ATP-binding site on the kinase domain maybe a promising approach. Several non-ATP-competitive inhibitorssuch as ONO12380 [47], the aurora kinase inhibitor MK-0457(VX-680) [48] and the p-38 inhibitors BIRB-796, SGX-70430 [44]are in clinical development to overcome imatinib-, nilotinib- ordasatinib-resistance in CML patients. Although the clinical signif-icance of these new targets remain to be elucidated, it is possiblethat they may play a role in the efficacy and safety of these drugs.
3. Monitoring BCR-ABL TKIs therapy: assessment of clinicalresponse
Clinical evidence indicates that all CML patients undergoingimatinib treatment should be monitored closely in order to assesstheir response to therapy and to detect early relapse. There arethree different types of responses in CML: (1) a hematologicalresponse (HR), (2) a cytogenetic response (CR) and (3) a molecularresponses (MR). A complete hematologic response (CHR) is definedas the normalization of blood counts and spleen size. Blood countscan be assessed using biweekly testing until a CHR is attained andthen testing can be performed every 3 months. Cytogenetic mon-itoring is the most widely used technique to monitor responsein patients with CML [49]. Typically, it is quantitated by deter-mining the decrease in the number of Ph+ metaphase cells usingboth bone marrow aspiration and cytogenetic evaluation tech-niques. A major cytogenetic response (MCR) is said to be achievedwhen Ph+ metaphase are present in 0–35% of cells. In contrast, theabsence of Ph+ metaphase cells leads to complete CCR. Fluores-cent in situ hybridization (FISH), which analyzes a higher numberof cells (up to 200), can be used instead of conventional cyto-genetic assessment for quantifying cells that are Ph+. However,a significant background level of false-positive results limits theuse of FISH and prevents full correlation with conventional assess-ment. The recent European Leukemia Net (ELN) recommendations[50] suggests cytogenetic testing first at 3 and 6 months, thenevery 6 months until a CCR is achieved and confirmed and sub-sequently every 12 months if regular molecular monitoring cannotbe assured. A MR is determined by a decrease in the amount ofBCR-ABL chimeric mRNA. A complete molecular response (CMR) isachieved when there is no detectable BCR-ABL chimeric mRNA asassessed by reverse transcriptase polymerase chain reaction (RT-PCR). A major molecular response (MMR) is defined as a 3-log
ment level. Hughes and colleagues introduced these definitionsin 2003, while monitoring the response to imatinib in the Inter-national phase III Randomized Study of Interferon versus STI571(IRIS) on previously untreated CML patients. In order to normalize

1258 X. An et al. / Leukemia Research 34 (2010) 1255–1268
Table 1TKIs approved for the treatment of CML.
TKIs Main targets CML indications Recommended dose Administrationrequirements
Main side effects
Imatinib (Gleevec®) Inactive BCR-ABLPDGFRARGKITVEGFDDR1NQO2
Newly diagnosed adultpatients with Ph+ CML-CP;Patients with Ph+ CML (allphases) after failure ofinterferon therapy; pediatricpatients with Ph+ CML-CP whoare newly diagnosed or whosedisease has recurred after HCTor who are resistant tointerferon therapy
CML-CP: 400 mg once dailyCML-AP/-BP: 600 mg oncedaily
Doses should be takenwith a meal and a largeglass of water
Myelosuppression, elevatedliver enzymes, edema/fluidretention, congestive heartfailure/LV dysfunction, bullousdermatologic reactions, NVD,muscle cramps,musculoskeletal pain, fatigue,abdominal pain
Dasatinib (Sprycel®) Active and inactiveBCR-ABLARGKITPDGFRSRCEPHA2DDR1TECBTK
For the treatment of adultswith any phase CML withresistance or intolerance toprior therapy includingimatinib
CML-CP: 100 mg oncedaily;CML-AP/-BP: 140 mg daily.
None Myelosuppression, pleuraleffusions, bleeding, QTprolongation, diarrhea,headache, nausea, rash, fatigue,dyspnea
Nilotinib (Tasigna®) Inactive BCR-ABLPDGFRARGKITVEGFDDR1
For the treatment of Ph+CML-CP/-AP in adult patientsresistant to or intolerant toprior therapy that includedimatinib
CML-CP/-AP: 400 mg twicedaily
Avoid food 2 h beforeand 1 h afterconsumption
Myelosuppression, elevatedliver enzymes, elevated serumlipase, electrolyteabnormalities, QT prolongation(black box warning), rash,pruritus, nausea, fatigue
C iladei
tribtip
tCtmtCt
TC
P
NQO2
ML, chronic myeloid leukaemia; CP, chronic phase; AP, accelerated phase; Ph, Phnhibitors.
he results obtained from three geographically dispersed laborato-ies measuring reductions in BCR-ABL transcripts, the investigatorsntroduced the concept of log10 reduction from a standardizedaseline for untreated CML patients. Consequently, it was deducedhat the imatinib-treated patients had at least a 3 log reductionn the level of BCR-ABL transcripts with negligible risk of diseaserogression over the subsequent 12 months [49,51] (Table 2).
The methodology of RT-PCR is probably the most sensitive assayo detect BCR-ABL chimeric mRNA; in fact, it can detect even oneML cell in a background of more than 100,000 normal cells. Quan-itative RT-PCR (Q-PCR) measures the actual percentage of BCR-ABL
RNA transcripts and shows a significant correlation betweenhe results obtained from peripheral blood and the bone marrow.onsequently, it has emerged as the preferred modality for post-herapeutic monitoring. However, there is a significant and large
able 2riteria for hematological, cytogenic and molecular response.
Response Criteria
HematologicalComplete (CHR) Complete normalization of peripheral blood coun
count < 10 × 109 L−1, platelet count < 450 × 109 L−
no splenomegalyPartial Same as complete hematological except for: pers
cells; platelet count < 50% of pretreatment count,splenomegaly <50% of pretreatment extent but p
CytogenicComplete (CCR) No Ph+ metaphases
Major (MCR) 0–35% Ph+ metaphases (complete + partial)Partial 1–34% Ph+ metaphasesMinor 36–90% Ph+ metaphases
MolecularComplete (CMR) BCR-ABL mRNA undetectable by RT-PCR;Major (MMR) ≥3-log reduction of BCR-ABL mRNA
h+, Philadelphia (Ph) positive chromosome.
headache, constipation,diarrhea, vomiting
lphia (Ph) chromosome; HCT, hematopoietic cell transplant; TKIs, tyrosine kinase
variability in the results obtained from different laboratories, prob-ably due to technical differences in the application of measuringQ-PCR for BCR-ABL in different settings. Thus, it is important tominimize the differences in the Q-PCR methodologies for detectingBCR-ABL transcripts and kinase domain mutations in order to max-imize the reliability of analysis for clinical decisions. One methodto achieve this goal was the recent adoption of an InternationalScale of measurement that was proposed following the consensusmeeting at National Institutes of Health (NIH) in Bethesda in Octo-ber 2005 [52,53]. It is recommended that molecular evaluations beperformed every 3 months in patients with CML [19,50].
The cytogenetic and molecular monitoring of the response toTKIs treatment provides important prognostic information. A 5-year follow-up in the IRIS study showed that no patients progressedto the accelerated blast phase after 12 months if a CCR and a MMR
Monitoring
t: white blood cell1; no immature cells;
Check every 2 weeks until CHRachieved, then monitor every 3 months
istent of immaturebut >450 × 109 L−1;ersistent
Check every 6 months until CCRachieved then every 12–18 months
Check every 3 months

esearc
waaTm
tiAtifooAaesab
arCdbHaScsgcslectgidfuw
ifdhtmm
4p
4
aTitrs
X. An et al. / Leukemia R
ere obtained [54]. The estimated progression free survival (PFS)t 24 months was 100% for patients who received CCR and MMRt 12 months [54]. Monitoring the response to second-generationKIs requires the same tests, but earlier and more frequent testingay be appropriate, because responses are more rapid.On the basis of the magnitude of HR, CR, and MR, and on the
ime when these responses are achieved, the overall response tomatinib can be defined as optimal, suboptimal or failed (Table 3).n optimal response is defined as no indication that a change of
herapy may improve a patient’s survival. A suboptimal responses where the patient may still have a substantial long-term benefitrom continuing the ongoing specific treatment, but the chancesf an optimal outcome are considerably reduced and thus, a sub-ptimal responders may be eligible for an alternative approaches.failed response indicated that a favorable outcome is unlikely
nd that the patient should receive a different treatment when-ver available and applicable. The definition of the response to theecond-generation TKIs dasatinib and nilotinib as second-line ther-py of patients with imatinib-resistant CML-CP was also providedy the new ELN recommendation (Table 4) [50].
However, the presence of a small number of point mutationsnd amplification of BCR-ABL kinase level lead to the emergence ofesistance, resulting in relapse episodes in patients who were in aCR on imatinib therapy [55]. Point mutations in BCR-ABL kinaseomain are frequently involved in imatinib-resistance, and maye an important determinant in clinical decisions (discussed later).owever, current evidence does not support mutation screening onroutine basis unless there is an indication for a loss of response.
everal recent reports suggested that routine mutation screeningould provide valuable information, such as predicting response topecific TKIs and identifying patients at high risk of disease pro-ression [56,57]. In addition, BCR-ABL Q-PCR monitoring resultsan be used to select patients with an increased risk of mutation,ince some studies showed that elevations in BCR-ABL transcriptevels might indicate a potential for BCR-ABL gene mutations andmergence of imatinib resistance [58–60]. However, the optimalut-off of increased BCR-ABL mRNA to predict mutation is still con-roversial. The National Comprehensive Cancer Network (NCCN)uidelines have provisionally recommended mutation screeningn cases with a 10-fold or greater increase of BCR-ABL mRNA isetected [49]. However, a study by Press et al. showed that a 10-old threshold to trigger mutation screening was insensitive and notniversally applicable, and a 2.6-fold increase in BCR-ABL mRNAas the optimal cutoff for predicting a concomitant mutation [61].
TKIs-related adverse events (AEs) are another important factormpacting the tolerability and safety of CML therapy. The success-ul monitoring and management of AEs is critical in maximizing theosing and therefore may optimize the response to imatinib. Bothematological and non-hematological AEs (i.e. gastrointestinal dis-urbances, cardiotoxicity, liver toxicity, pleural effusion, edema,
uscle cramps) should be monitored closely throughout treat-ent.
. Clinical trials examining the effects of TKIs in Ph+atients
.1. The first generation of TKI
Imatinib was the first FDA (Food and Drug Administration)pproved TKI for the treatment of advanced stage Ph+ CML patients.
his was based information from two successful phase II studiesn CML-AP and CML-BC patients [21,62] using 600 mg/day of ima-inib. Previous phase I and phase II clinical studies in CML patientsesistant or intolerant to rIFN-� confirmed its clinical efficacy andafety [38,63]. Imatinib is also approved by the FDA as a first-lineh 34 (2010) 1255–1268 1259
treatment for patients with CML-CP based on data from a ran-domized phase III trial known as IRIS [36] that compared imatinib400 mg daily with IFN-� plus cytosine arabinoside (Ara-C) in 1106previously untreated CML-CP patients. Crossover was allowed inpatients that experienced treatment failure or intolerance. After 19months (median) of imatinib treatment, the estimated CHR, MCRand CCR were 96%, 87% and 76%, respectively; A 5-year follow-upresult showed that the estimated cumulative best rates of CHR andCCR were 98% and 87%, respectively [54]. The most recent 6-yearupdate data reported that 63% of all patients randomized to receiveimatinib were still on study treatment and showed a CCR at thelast assessment. The best cumulative CCR rate was 82%; the esti-mated event-free survival at 6 years was 83%. In 95% of patients,imatinib was well tolerated, as only 5% of patients discontinuedtreatment due to adverse effects. Moreover, no new adverse eventswere reported following long-term use of imatinib [64]. However,due to the relatively high rate of crossovers (∼90%) from rIFN-�plus Ara-C group to imatinib group within a year of the study, per-plexes the true determination of the survival benefit with imatinibversus IFN-� plus Ara-C in the IRIS study.
Until now, the maximum tolerated dose of imatinib had not beenestablished and an initial dose of 400 mg daily for adult CML-CPpatients was recommended. Several prospective, non-randomizedstudies reported that responses to imatinib were more rapid andsuperior in the patients who were treated with doses of 600 or800 mg daily [65,66]. The TOPS trial (tyrosine kinase inhibitoroptimization and selectivity) [67] was a prospective, open-label,randomized phase III trial (476 patients at 103 sites in 19 countries)that compared the efficacy of a high dose of imatinib (800 mg) toa standard dose (400 mg) of imatinib for the first-line treatment ofCML-CP patients. The results indicated that a significantly greaternumber of patients receiving 800 mg/day of imatinib, achieved anMMR at 3 and 6 months, but not at 12 months, when comparedto 400 mg/day of imatinib. Another similar randomized study bythe ELN (mentioned earlier) compared the efficacy of 400–800 mgof imatinib as a first-line therapy for CML patients [50]. The studyenrolled 216 high-risk (as defined by the Sokal index) Ph+ CMLpatients. The 1-year CCR rates were 64% and 58% for the highdose (800 mg) and the standard dose (400 mg) arm, respectively.No significant differences were observed in the CR rates, MR ratesor in the rates of other events at 3 and 6 months. Therefore, theresults from these studies do not support the use of high-doseimatinib (800 mg daily) as a front-line treatment in high-risk CMLpatients.
Although first-line imatinib treatment achieved an extremelyhigh response rate and a low relapse rate in CML patients, somepatients do experience imatinib resistance or intolerance. In theabove-mentioned IRIS trial, 31% of patients did not achieve a CCRduring 12 months of treatment and 13% still had not achieved CCRafter 5 years of treatment. In addition, 3–7% of patients in thattrial experienced treatment failure during the first 3 years, and 5%of patients discontinued imatinib because of intolerable adverseeffects [54]. Therefore, many patients require alternative treatmentin the case of imatinib failure or intolerance.
4.2. The second-generation of TKIs
4.2.1. DasatinibAs mentioned above, dasatinib inhibits a number of kinases,
including BCR-ABL and Src and is more potent than imatinib againstBCR-ABL TKs [68]. Dasatinib has been clinically evaluated in phases
I, II, and III trials in adult’s Ph+ CML patients, who are intolerant orresistant to imatinib-therapy.A phase I study [69] reported the effects of dasatinib(15–240 mg/day) once or twice daily in 4 weeks treatment cyclesin the patients with various phases of CML or with Ph+ ALL that
1260 X. An et al. / Leukemia Research 34 (2010) 1255–1268
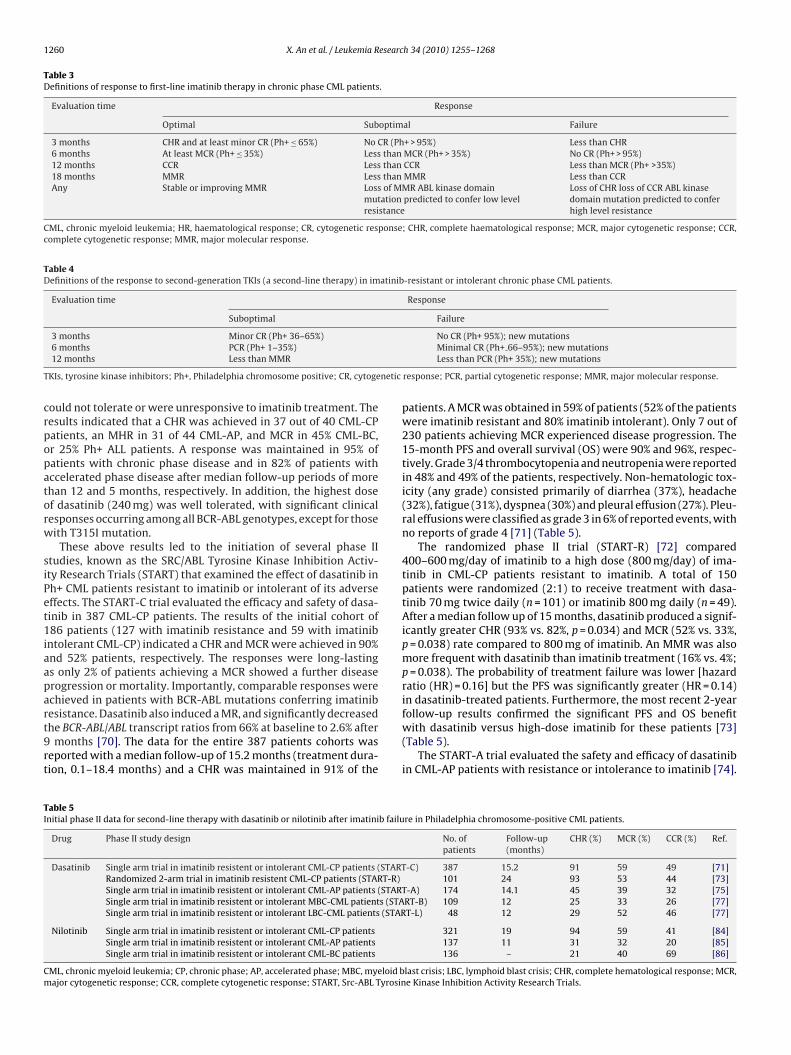
Table 3Definitions of response to first-line imatinib therapy in chronic phase CML patients.
Evaluation time Response
Optimal Suboptimal Failure
3 months CHR and at least minor CR (Ph+ ≤ 65%) No CR (Ph+ > 95%) Less than CHR6 months At least MCR (Ph+ ≤ 35%) Less than MCR (Ph+ > 35%) No CR (Ph+ > 95%)12 months CCR Less than CCR Less than MCR (Ph+ >35%)18 months MMR Less than MMR Less than CCRAny Stable or improving MMR Loss of MMR ABL kinase domain
mutation predicted to confer low levelresistance
Loss of CHR loss of CCR ABL kinasedomain mutation predicted to conferhigh level resistance
CML, chronic myeloid leukemia; HR, haematological response; CR, cytogenetic response; CHR, complete haematological response; MCR, major cytogenetic response; CCR,complete cytogenetic response; MMR, major molecular response.
Table 4Definitions of the response to second-generation TKIs (a second-line therapy) in imatinib-resistant or intolerant chronic phase CML patients.
Evaluation time Response
Suboptimal Failure
3 months Minor CR (Ph+ 36–65%) No CR (Ph+ 95%); new mutations
T netic
crpopatorw
siPet1iaapart9rt
TI
Cm
6 months PCR (Ph+ 1–35%)12 months Less than MMR
KIs, tyrosine kinase inhibitors; Ph+, Philadelphia chromosome positive; CR, cytoge
ould not tolerate or were unresponsive to imatinib treatment. Theesults indicated that a CHR was achieved in 37 out of 40 CML-CPatients, an MHR in 31 of 44 CML-AP, and MCR in 45% CML-BC,r 25% Ph+ ALL patients. A response was maintained in 95% ofatients with chronic phase disease and in 82% of patients withccelerated phase disease after median follow-up periods of morehan 12 and 5 months, respectively. In addition, the highest dosef dasatinib (240 mg) was well tolerated, with significant clinicalesponses occurring among all BCR-ABL genotypes, except for thoseith T315I mutation.
These above results led to the initiation of several phase IItudies, known as the SRC/ABL Tyrosine Kinase Inhibition Activ-ty Research Trials (START) that examined the effect of dasatinib inh+ CML patients resistant to imatinib or intolerant of its adverseffects. The START-C trial evaluated the efficacy and safety of dasa-inib in 387 CML-CP patients. The results of the initial cohort of86 patients (127 with imatinib resistance and 59 with imatinib
ntolerant CML-CP) indicated a CHR and MCR were achieved in 90%nd 52% patients, respectively. The responses were long-lastings only 2% of patients achieving a MCR showed a further diseaserogression or mortality. Importantly, comparable responses werechieved in patients with BCR-ABL mutations conferring imatinibesistance. Dasatinib also induced a MR, and significantly decreased
he BCR-ABL/ABL transcript ratios from 66% at baseline to 2.6% aftermonths [70]. The data for the entire 387 patients cohorts waseported with a median follow-up of 15.2 months (treatment dura-ion, 0.1–18.4 months) and a CHR was maintained in 91% of the
able 5nitial phase II data for second-line therapy with dasatinib or nilotinib after imatinib failu
Drug Phase II study design
Dasatinib Single arm trial in imatinib resistent or intolerant CML-CP patients (STARTRandomized 2-arm trial in imatinib resistent CML-CP patients (START-R)Single arm trial in imatinib resistent or intolerant CML-AP patients (STARSingle arm trial in imatinib resistent or intolerant MBC-CML patients (STASingle arm trial in imatinib resistent or intolerant LBC-CML patients (STAR
Nilotinib Single arm trial in imatinib resistent or intolerant CML-CP patientsSingle arm trial in imatinib resistent or intolerant CML-AP patientsSingle arm trial in imatinib resistent or intolerant CML-BC patients
ML, chronic myeloid leukemia; CP, chronic phase; AP, accelerated phase; MBC, myeloid bajor cytogenetic response; CCR, complete cytogenetic response; START, Src-ABL Tyrosin
Minimal CR (Ph+ 66–95%); new mutationsLess than PCR (Ph+ 35%); new mutations
response; PCR, partial cytogenetic response; MMR, major molecular response.
patients. A MCR was obtained in 59% of patients (52% of the patientswere imatinib resistant and 80% imatinib intolerant). Only 7 out of230 patients achieving MCR experienced disease progression. The15-month PFS and overall survival (OS) were 90% and 96%, respec-tively. Grade 3/4 thrombocytopenia and neutropenia were reportedin 48% and 49% of the patients, respectively. Non-hematologic tox-icity (any grade) consisted primarily of diarrhea (37%), headache(32%), fatigue (31%), dyspnea (30%) and pleural effusion (27%). Pleu-ral effusions were classified as grade 3 in 6% of reported events, withno reports of grade 4 [71] (Table 5).
The randomized phase II trial (START-R) [72] compared400–600 mg/day of imatinib to a high dose (800 mg/day) of ima-tinib in CML-CP patients resistant to imatinib. A total of 150patients were randomized (2:1) to receive treatment with dasa-tinib 70 mg twice daily (n = 101) or imatinib 800 mg daily (n = 49).After a median follow up of 15 months, dasatinib produced a signif-icantly greater CHR (93% vs. 82%, p = 0.034) and MCR (52% vs. 33%,p = 0.038) rate compared to 800 mg of imatinib. An MMR was alsomore frequent with dasatinib than imatinib treatment (16% vs. 4%;p = 0.038). The probability of treatment failure was lower [hazardratio (HR) = 0.16] but the PFS was significantly greater (HR = 0.14)in dasatinib-treated patients. Furthermore, the most recent 2-yearfollow-up results confirmed the significant PFS and OS benefit
with dasatinib versus high-dose imatinib for these patients [73](Table 5).The START-A trial evaluated the safety and efficacy of dasatinibin CML-AP patients with resistance or intolerance to imatinib [74].
re in Philadelphia chromosome-positive CML patients.
No. ofpatients
Follow-up(months)
CHR (%) MCR (%) CCR (%) Ref.
-C) 387 15.2 91 59 49 [71]101 24 93 53 44 [73]
T-A) 174 14.1 45 39 32 [75]RT-B) 109 12 25 33 26 [77]T-L) 48 12 29 52 46 [77]
321 19 94 59 41 [84]137 11 31 32 20 [85]136 – 21 40 69 [86]
last crisis; LBC, lymphoid blast crisis; CHR, complete hematological response; MCR,e Kinase Inhibition Activity Research Trials.

esearc
Ai6oIgbrdnAstmroistr
rbs8ataptr3
tlindC1oo0rd1tadtcydctr1iTad
fioit
X. An et al. / Leukemia R
n 8-month follow-up of the first 107 patients enrolled in the studyndicated an overall, major, or complete HR was obtained in 81%,4%, and 39% of patients, respectively. In addition, 33% and 24%f these patients attained a major or complete CR, respectively.n the 69 patients who achieved a MHR, only 7 relapsed and pro-ressed to the disease, however, 76% of patients were estimated toe alive and were progression-free after 10 months. The responseates in 60% of patients with baseline BCR-ABL mutations did notiffer from the total population. Dasatinib was well tolerated ando imatinib-intolerant patients discontinued dasatinib because ofEs. The follow-up data from all 174 patients have confirmed theafety and efficacy of dasatinib for CMP-AP patients with resis-ance or intolerance to imatinib. After a median follow-up of 14.1
onths, a MHR and CHR were attained in 64% and 45% of patients,espectively and a MCR and CCR were achieved in 39% and 32%f patients, respectively (Table 5). These responses were achievedrrespective of the imatinib status (resistance or intolerance), priortem-cell transplantation or the presence of a prior BCR-ABL muta-ion. The 12-month PFS and OS in these patients were 66% and 82%,espectively [75].
The efficacy of dasatinib for CML-BC patients with imatinibesistance or intolerance was evaluated in the START-B (myeloidlast crisis, MBC-CML patients) and START-L (lymphoid blast cri-is, LBC-CML patients) trials [76,77]. In the MBC-CML patients, theand 12 months follow-up results indicated MHR rates to be 34%
nd 34%, respectively, and the MCR rates were 31% and 33%, respec-ively. Rapid and durable responses were obtained with median PFSnd OS being 6.7 and 11.8 months, respectively. In the LBC-CMLatients, the 8-month and 12-month follow-up results indicatedhat the MHR rates were 31% and 35%, respectively, and the MCRates were 50% and 52%, respectively. The median PFS and OS were.0 and 5.3 months, respectively.
The initial approved dose of dasatinib for CML patients resis-ant or intolerant to imatinib was 70 mg twice daily. However, aonger follow-up of the data from the phase I trial of dasatinibndicated that the once-a-day treatment group experienced sig-ificantly fewer adverse effects compared to those treated twiceaily [69]. In addition, the median daily doses for patients withML-CP during the START-C and START-R trials were 101 mg and03 mg, respectively [70,72]. These results prompted the initiationf two phase III, dose optimization trials in patients with resistancer intolerance to imatinib, designated CA180-034 [78] and CA180-35 [79]. In the CA180-034 trial, 670 CML-CP patients with imatinibesistance or intolerance were randomly assigned (1:1:1:1) to fourasatinib treatment groups: 100 mg once daily, 50 mg twice daily,40 mg once daily, or 70 mg twice daily. The results indicatedhat after a minimum follow-up of 6 months, similar efficacy waschieved across all the four groups. However, the 100 mg onceaily dasatinib treatment resulted in significantly lower rates ofoxicity and lower frequencies of dose interruptions or reductionsompared to the 70 mg twice-daily regimen. The most recent 3-ear follow-up results have confirmed the efficacy and safety ofaily dose of 100 mg dasatinib [79]. In the CA180-035 trial, the effi-acy and safety of 140 mg dasatinib once daily was compared tohe 70 mg twice-daily regimen in CML-AP patients with imatinibesistance or intolerance. After a median follow-up of 15 months,40 mg dasatinib daily doses demonstrated equivalent efficacy but
mproved safety compared to the 70 mg twice-daily regimen [80].herefore, based on data from these phase III studies, the FDA haspproved dasatinib 100 mg daily for CML-CP patients and 140 mgaily for CML-AP and CML-BC patients [49].
Currently, the efficacy of dasatinib is being evaluated as arst-line treatment in previously untreated CML-CP patients in anngoing phase II trial by Borthakur and Kantarjian [81]. The resultsndicated a rapid CCR and a favorable toxicity profile with dasatinibreatment. A large, randomized trial comparing dasatinib 100 mg
h 34 (2010) 1255–1268 1261
once daily with imatinib 400 mg daily in newly diagnosed patientsis also being undertaken [49].
4.2.2. NilotinibThe FDA approved nilotinib, another second-generation BCR-
ABL tyrosine kinase inhibitor, in 2007 for the treatment of imatinibresistance or intolerance in CML patients. In a phase I dose-escalation study, nilotinib was found to be significantly moreeffective than imatinib-resistant CML patients, with a relativelyfavorable safety profile compared to imatinib [82]. Subsequently,a phase II trial was conducted [83] where 280 imatinib resistantor intolerant patients were enrolled and treated with 400 mg b.i.d.of nilotinib. The MCR and CCR rates after a 6 months follow-upwere 48%, and 31%, respectively. Survival past 12 months was esti-mated to be 95% in the nilotinib treated patients. Nilotinib waseffective in patients harboring BCR-ABL mutations associated withimatinib resistance (except T315I), and also in patients with a resis-tance mechanism independent of BCR-ABL mutations. The reportedAEs were mostly mild to moderate and there was minimal cross-intolerance to imatinib. A minimum 19-month follow-up result ofall 321 patients enrolled in the study showed that nilotinib pro-duced a rapid and sustained CHR and MCR rates. A CHR and MCRwere observed in 94% and 59% of patients, respectively, and 78%of patients maintained a MCR after 24 months. The estimated OSrate was 88% at 24 months. The safety profile of dasatinib did notchange with longer follow-up intervals [84].
Le Coutre et al. [85] conducted a phase II trial evaluating the effi-cacy and safety of nilotinib in CML-AP patients with resistance orintolerance to imatinib. Initially, 119 patients were enrolled withmedian treatment duration of 202 days. The HR and MCR rateswere 47% and 29%, respectively, and the 12 months OS rate was79%. Non-hematologic AEs reported were mostly mild to moder-ate in severity. The most common grade 3 or higher hematologicAEs were thrombocytopenia (35%) and neutropenia (21%). Grade3 or higher bilirubin and lipase elevations occurred in 9% and 18%of patients, respectively, resulting in treatment discontinuation in1 patient. Long-term follow-up results of the total 137 patientsconfirmed that nilotinib induces a rapid and sustained response inCML-AP patients who do not respond to prior imatinib treatmentand there was a significantly favorable risk/benefit ratio. The HR,CHR, MCR and CCR rates were 56%, 31%, 32% and 20%, respectively,and the median time to HR was 1 month. After 24 months, 54% and70% of patients maintained HR and MCR, respectively, and the esti-mated OS at 24 months was 67%. Only 9% of patients discontinuedtherapy due to drug-related AEs [85]. Thus, based on the resultsfrom the above studies, the FDA, in 2007, approved nilotinib forthe treatment of Ph+ CML-CP or CML-AP patients with resistanceor intolerance to imatinib.
Nilotinib has also been shown to have efficacy in CML-BCpatients. A phase II study reported the safety and efficacy data in136 CML-BC patients treated with nilotinib. The results showedthat the HR, MCR, CCR rates were 21%, 40% and 29%, respectivelyand the OS was 42% at 12 months [86]. However, more than half ofthe patients discontinued treatment due to disease progression andtherefore, the FDA has not approved nilotinib for the treatment ofCML-BC patients at this time. The efficacy of nilotinib was evaluatedin another phase II trial (GIMEMA CML Working Party) as a first-linetreatment in previously untreated CML-CP patients [87]. A total of73 patients were enrolled and followed up for 210 days. In the ITT(intent to treatment) analysis, the CHR rates at 3 and 6 months were100% and 98%, respectively, and the CCR rates were 78% and 96%,
respectively. The cytogenetic and molecular responses to nilotinibwere significantly faster than that of imatinib. Only 1 patient withthe T315I mutation progressed to the accelerated or blast phasesat 6 months. All AEs (grade 3/4) were manageable by appropriatedose adjustments.
1 esearc
5
trtraIoa6spcomR
rrsBtaBitem
5
mot
5
dittabtaobtFm2awsdpttod
5m
262 X. An et al. / Leukemia R
. Resistance to BCR-ABL TKIs
Although the majority of Ph+ CML patients benefit from ima-inib treatment, a substantial number of patients are either initiallyefractory to treatment (primary resistance) [88] or develop resis-ance during the course of treatment (secondary or acquiredesistance) [89]. Imatinib-resistance is defined as the failure tochieve CHR at 3 months, CR at 6 months and MCR at 12 months.n patients with advanced-stage disease, there are frequent reportsf both primary and secondary resistance [22,52,90,91]. Hochhausnd La Rosee [92], reported primary resistance in 5%, 4%, 24% and6% of patients in early CP, late CP, AP and BC, respectively, andecondary resistance were observed in 4%, 13%, 51% and 88% ofatients in early CP, late CP, AP and BC, respectively. Resistancean be further categorized as hematologic (lack of normalizationf peripheral blood counts), cytogenetic (persistence of Ph+ chro-osome), and molecular (persistence of BCR-ABL transcripts by
T-PCR) resistance [93].There are a number of mechanisms that can elicit imatinib
esistance. In addition, the mechanisms for primary and secondaryesistance often overlap. In general, imatinib resistance can beubdivided in BCR-ABL-dependent and -independent mechanisms.CR-ABL-dependent mechanism includes point mutations withinhe BCR-ABL kinase domain that interfere with imatinib bindingnd the over-expression or amplification of the BCR-ABL gene.CR-ABL-independent mechanisms include factors influencing the
ntracellular concentration of imatinib (e.g. binding of imatinibo serum �-1 acid glycoprotein or alterations in drug influx andfflux), and activation of BCR-ABL independent pathways, such asembers of the Src kinase family.
.1. BCR-ABL dependent resistance to imatinib
The reactivation of BCR-ABL signaling is the most commonechanism of imatinib resistance and this is frequently the result
f point mutations in BCR-ABL TK domain, and less commonly dueo BCR-ABL gene amplification or over-expression [22].
.1.1. Point mutations in the kinase domain of BCR-ABLPoint mutations cause amino acid substitutions inside the kinase
omain of the BCR-ABL protein and disrupt the binding site ofmatinib on the tyrosine kinase, resulting in a loss of sensitivityo the drug. Mutations can either directly interrupt critical con-act points between the drug and the BCR-ABL protein or induceconformational change, resulting in a protein that is unable to
ind imatinib [94]. There are al least 33-point mutations knowno occur within the ABL kinase and these may lead to secondary orcquired resistance after imatinib treatment [42,95]. The frequencyf BCR-ABL mutations in patients with resistance to imatinib haseen ranged from 40% to 90%, depending on the definition of resis-ance, the methodology of detection, and the CML phase [89,90,96].or example, point mutations rarely occur in early CML-CP but areore frequent with disease progression. There are approximately
0 ABL kinase domain residues involved in imatinib binding [9]nd a mutation could significantly alter the interaction of imatinibith the target. Moreover, different mutations can occur at the
ame position, resulting in a different amino acid substitution and aifferent level of resistance. Currently, approximately 90 differentoint mutations have been identified [44,52,91] and the majority ofhese mutations are relatively rare. The two most common muta-ions are T315I and P-binding phosphate loop (P-loop) mutations,
ccurs, especially in CML patients in the advanced phase of theisease [44,96]..1.1.1. T315I mutation. The first documentation of the T315Iutation was by Gorre et al. [22], who found that 6 of 9 advanced
h 34 (2010) 1255–1268
stage CML or Ph+ ALL patients with imatinib resistance had thismutation. The T315I mutation represents the replacement of athreonine by an isoleucine at amino acid position 315 in the ABLcomponent of the kinase. This mutation decreases the probabilityof a critical hydrogen bond between imatinib and BCR-ABL [93]. Inaddition, isoleucine creates steric hindrance, preventing the bind-ing of imatinib, thereby conferring resistance [92]. In a more recentstudy, the T315I mutation was detected in 15% of 112 patients withCML who initially failed imatinib therapy [95]. The T315I mutationproduces the highest magnitude of resistance to not only imatinibbut also to other second-generation TKIs such as dasatinib and nilo-tinib, compared to other mutations. This occurs because all of thethree TKIs need to form a hydrogen bond with threonine at 315positions to bind to their targets. A considerable effort has beenmade to find treatments to overcome the T315I mutation.
5.1.1.2. P-loop mutations. The structure of BCR-ABL contains twoflexible loops, the ATP-binding P-loop and the activation loop,which have specific arrangements in the inactive conformationof BCR-ABL that stabilize the basal conformation [93]. Mutationsin these loops destabilize their arrangement such that the kinasedomain cannot assume the inactive conformation required for ima-tinib binding. Mutations in the P-loop region are the most common,accounting for 36–48% of all mutations [94,97]. The frequency ofP-loop mutations clearly increases in the accelerated phase andblast crisis as well as with disease duration [98]. There are clinicaldata indicating that BCR-ABL mutations in the P-loop is 70-fold to100-fold less sensitive to imatinib compared with native BCR-ABL,leading to poor prognosis in patients receiving imatinib [94,99].A study of 319 CML-CP patients found that mutations, even with-out evidence of imatinib resistance, was significantly predictive forloss of CCR and progressed to the advanced phase [56]. However,another similar study by Jabbour et al. could not confirm this finding[97]. The prognosis was similar as patients carrying P-loop muta-tions or other mutations when receiving treatment with the newsecond-line TKIs, especially dasatinib. In the START trials, dasatinibinduced similar rates of MHR and MCR in imatinib resistance CML-AP and CML-BC patients independent of the presence of P-loop orother mutations [74,80].
5.1.2. BCR-ABL gene amplificationAnother well described, but less common mechanism of clinical
resistance to imatinib, is BCR/ABL gene amplification or an increasein BCR/ABL mRNA levels. It has been shown that both mechanismproduces an increase in BCR-ABL protein levels, and this restoresoncogenic signaling in presence of a given drug concentration [96].Increasing the imatinib dose could surmount this kind of resistance,provided that severe or intolerable side effects are not produced.
5.2. BCR-ABL independent mechanisms of resistance
5.2.1. Inadequate plasma levels of imatinibSeveral studies have reported significant variability in plasma
levels of imatinib among CML patients receiving standard dailydose of 400 mg imatinib [100,101]. The trough plasma levels ofimatinib were significantly higher in patients achieving a CCR andMMR at 12 months [102]. A sub-analysis of the IRIS study indi-cated that plasma level of imatinib following the first month oftreatment was a significant prognostic factor for long-term clinicalresponse [103]. However, a study by Forrest et al. [104] showedthat neither CCR nor MMR at 1 year was significantly correlated
with the mean plasma trough level of imatinib after a median of1298 days of standard therapy with imatinib. There are a num-ber of potential explanations for the low trough plasma levels ofimatinib, such as incomplete adherence of patients to the thera-peutic regimen, individual variations in the pharmacokinetics of
esearc
ici
5
ftC[cioadsc
5
iataid[teiitAfco
5
mo
5pmta[iavpssAiaHccooaAAt
X. An et al. / Leukemia R
matinib and intracellular uptake of imatinib. Therefore, the asso-iation between clinical response and the plasma levels of imatinibs still controversial and remains to be determined.
.2.2. Incomplete adherence to the therapeutic regimenThe continuous and adequate dosing of imatinib is essential
or achieving optimal outcomes in CML patients. Early retrospec-ive data showed a high incidence of imatinib non-adherence inML patients and this could lead to undesired clinical outcomes105,106]. The ADAGIO (adherence assessment with gleevec: indi-ators and outcomes) study [107] evaluated adherence to imatinibn 169 CML patients and found that during the initial 90-day periodf imatinib treatment, 1/3rd of patients were considered to be non-dherent. Only 14.2% of patients were adherent to the prescribedose of imatinib. Overall, patients with a suboptimal response wereignificantly less likely to have been adherent (23.2%) to imatinibompared to those that had an optimal response (7.3%) [107].
.2.3. Pharmacokinetic parametersThe cytochrome P450 enzymes metabolize imatinib, primar-
ly by the CYP3A4 isoform [108,109]. The variability in CYP3A4ctivity among individuals is substantial [110], and may contributeo some of the variation in imatinib levels between patients. Inddition, other drugs taken by patients can alter CYP3A4 activ-ty. For example, drugs that inhibit CYP3A4 may increase, whereasrugs that induce CYP3A4 may decrease plasma levels of imatinib111]. Further polymorphism studies are mandated to understandhe significance of cytochrome P450 enzymes on the plasma lev-ls of imatinib and response to therapy in CML patients. Anothermportant pharmacokinetic factor involved in imatinib-resistances alpha-1 acid glycoprotein (AGP), which is an acute-phase reac-ant protein found in the plasma [112,113]. It has been reported thatGP binds imatinib in a 1:1 molar ratio and this could reduce the
ree concentration of imatinib and attenuate its entry into leukemicells [112]. However, there is controversy regarding the associationf increased AGP levels and imatinib resistance [113].
.2.4. Intracellular uptake of imatinibThe amount of imatinib that is present in the target cells is deter-
ined by balance between proteins that mediate the inward andutward (efflux) transport of imatinib.
.2.4.1. ATP-binding cassette (ABC) transporters. The ABC trans-orters are ATP-dependent multidrug efflux pumps and encompassultidrug resistant gene product ABCB1 (P-glycoprotein; MDR1),
he breast cancer resistant protein ABCG2 (BCRP; MXR; ABCP)nd members of multiple resistant proteins ABCC (MRP) family114]. The ABC transporters utilize ATP and actively efflux var-ous endogenous and exogenous substance from cells, and thusttenuate their concentration intracellularly [114]. In vitro and inivo studies have shown that the over-expression of ABC trans-orters can produce resistance to drugs used in the treatment ofolid and non-solid cancers, including CML. For example, initialtudies in BCR-ABL TKI resistance revealed over-expression of theBCB1 transporter in cells from patient in CML-BC. This resulted
n imatinib resistance as a result of suboptimal bioavailabilitynd reduced efficacy in advanced-phase CML patients [100,115].owever, the imatinib efflux activity of ABCB1 is less pronouncedompared to that of other classical cytotoxic drugs and there isontroversy in terms of correlating imatinib resistance with ABCB1verexpression [116,117]. Furthermore, in another clinical study,
ver-expression of ABCB1 lead to progression of disease to thedvanced phase with a lack of MCR [118], although inhibition ofBCB1 did not sensitize CML CD34+ cells to imatinib [119]. AnotherBC efflux transporter, ABCG2, which is expressed in the gas-rointestinal epithelium and in the blood-brain barrier, was also
h 34 (2010) 1255–1268 1263
implicated in the active efflux of imatinib from cells [120]. Thereare conflicting data regarding the role of ABCG2 in imatinib sensitiv-ity, where imatinib is a substrate of ABCG2 at lower concentration[121,122] and an inhibitor at clinically relevant concentrations[123]. Therefore, imatinib is an inhibitor of, but not a substrate forABCG2 [116]. Nilotinib, like imatinib, has a high affinity for bothABCB1 and ABCG2 and was shown to be a substrate of ABCB1 [124]and ABCG2 [121,125]. However, at clinically relevant concentra-tion of nilotinib, resistance was not observed in the presence ofABCG2 [121,125]. Nonetheless, we recently showed that nilotinibsignificantly inhibited the efflux activity of both ABCB1 and ABCG2when combined with their respective substrates [125]. Dasatinib,unlike imatinib, enters cells via passive diffusion but it can alsobe actively effluxed by both ABCB1 [126] and ABCG2 [127]. There-fore, a potential benefit of combining a low-dose of dasatinib andnilotinib for CML cells was suggested, which may provide an addi-tive/synergistic antileukemic effect, particularly to leukemic stemcells that are considered to be both refractory to the TKI therapyand expressing ABCB1. Interestingly, no significant difference wasobserved in dasatinib accumulation inside the gastrointestinal tractof ABCB1 wild type or knockout mice [128]. Recently, our resultsindicate that both imatinib and nilotinib, but not dasatinib, couldinhibit the active transport function of the ABCC10 transporter[129]. However, the role of ABCC10 in BCR-ABL TKIs has not yetbeen established.
5.2.4.2. Organic cation transporter 1 (OCT1; SLC22A1). The influxtransporter OCT1 plays a significant role in imatinib resistant byinhibiting imatinib influx and thus decreasing the intracellularbioavailability of imatinib [130]. Patients with a low expression,activity or polymorphisms of OCT1 had significantly lower intra-cellular levels of imatinib and were shown to have a significantlylower probability of achieving a cytogenetic or molecular remission[131]. In addition, data from the cohort study of TIDEL (therapeuticintensification in de novo leukemia) patients revealed that OCT-1activity is an important determinant in the molecular response toimatinib [131]. Patients with high OCT-1 activity achieved an excel-lent molecular response regardless of dose, whereas the response ofpatients with low OCT-1 activity was significantly dose-dependent.For example, 82% of patients with low OCT-1 activity who received<600 mg imatinib failed to achieve a MMR by 18 months, comparedto 17% for patients with high OCT-1 activity and a dose of imatinib<600 mg/day [131]. However, the uptake of both dasatinib [127]and nilotinib is not highly dependent upon OCT1 [42,77]. Therefore,these two drugs may achieve adequate intracellular concentrationseven in patients with low OCT1 expression. Prospective studiesare currently investigating whether the expression of this influxtransporter can be used to guide treatment decisions.
5.2.5. Activation of other signaling pathwaysIn a few patient groups, resistance may be caused by the acti-
vation of other signaling pathways, particularly the SFKs. In fact,there is accumulating preclinical and clinical evidence indicat-ing that the SFKs are involved in mediating imatinib resistance.The SFKs have been implicated in BCR-ABL signaling and medi-ate imatinib resistance by stabilizing the active conformation ofBCR-ABL, a conformation that does not bind imatinib [133]. Fur-thermore, increasing evidence suggests that SFKs are also involvedin BCR-ABL-independent forms of imatinib resistance. The SFKsmembers, such as Lyn and Hck, are over-expressed in CML cell lineswith BCR-ABL-independent imatinib resistance, and inhibition of
both SFKs and BCR-ABL (mutants and wild-type) in these cellsresulted in an enhanced apoptotic response [134,135]. The trans-fection of imatinib-sensitive cell lines with a constitutively activeform of Lyn confers resistance to imatinib [136]. Furthermore, theexpression of p53/56 Lyn kinase was significantly up-regulated
1 esearc
i[LctfoA(t
iCrCCaCCatacfotisiiittopbe
6
ptn
saa(dad3thtC
irpttt(
264 X. An et al. / Leukemia R
n imatinib-resistant [137] as well as nilotinib-resistant cell lines124]. Sensitivity was restored in these resistant cell lines whenyn was silenced using small interference RNA or when theseells were incubated with specific Src inhibitors [124,137]. In addi-ion, dasatinib, an inhibitor of both BCR-ABL and Src kinases, wasound to be effective in the treatment of CML patients resistantr intolerant to imatinib or nilotinib [72,73]. Novel dual SFK/BCR-BL inhibitors, such as bosutinib (SKI-606) [138,139] and NS-187
INNO-406) [132] are under development and may provide addedherapeutic advantages.
Imatinib resistance can also be produced by other mechanisms,ncluding clonal evolution and CML stem cell quiescence [140].lonal evolution is defined as the acquisition of additional, non-andom cytogenetic abnormalities in the metaphase of Ph+ [102].lonal evolution is considered a feature of the accelerated phase ofML and is significantly correlated with BCR-ABL mutations [97]nd these mechanisms may play a pivotal role in CML progression.ML stem cells (Lin-CD34+) account for approximately 0.5% of theD34+ population and are characterized by resistance to TKIs ther-py [140]. The persistence of TKI-resistant leukemic stem cells ishought to play a role in disease recurrence after TKIs therapy. Inddition, a recent study has shown that TKIs could induce CML stemells arrest by re-activation of Forkhead box, a sub-group O (FOXO)amily of transcription factors (TFs), which makes the eradicationf CML stem cells more difficult. FOXOs are a family of transcrip-ion factors that play an important role in cell cycle arrest throughnduction of genes involved in the cell cycle in normal haemopoietictem cells. FOXO transcription factor activity is partially retainedn quiescent CML stem cells and is induced by tyrosine kinasenhibitors in CML progenitor cells [141]. BCR-ABL signaling couldnhibit the transcriptional activity of the FOXOs through activatinghe PI3K/AKT pathway [142] and contribute to CML cell prolifera-ion and malignant transformation. The TKIs could inhibit the effectf BCR-ABL by re-activation of FOXOs, thus maintaining the anti-roliferative property of stem cells. Hence, newer strategies maye needed to target CML stem cells by preventing or reversing thisffect of TKIs on FOXOs.
. Treatment options for imatinib-resistant CML
The treatment options for imatinib-resistant or intolerant CMLatients may include strategies such as increasing the dose of ima-inib, the use of second-generation TKIs such as dasatinib andilotinib and HCT or other investigational compounds.
Increasing the dose of imatinib has been shown to overcomeome cases of primary imatinib resistance, but the response is usu-lly short acting [143]. Kantarjian et al. [144] recently conductedretrospective analysis of 106 newly diagnosed CML-CP patients
IRIS trial) who were initially treated with 400 mg of imatinib aay, and then received either 600 or 800 mg daily because of eithersuboptimal response or resistance to imatinib. The rates of free-om from progression to the accelerated phase or blast phase and-year OS after 600 or 800 mg of imatinib were 89% and 84%, respec-ively. Similarly, other studies (reviewed previously in this article)ave also reported that escalation of the dose of imatinib had onlyransient or no significant benefit in patients who did not achieveCR on conventional doses [66–68].
The second-generation TKIs dasatinib and nilotinib offermproved potency and a greater likelihood of success in imatinib-esistant patients. As previously stated, many clinical trials have
roven the efficacy and safety of dasatinib and nilotinib forhe treatment of patients with resistance or intolerance to ima-inib [70–72,75,83,145]. The phase II START-R trial [73] comparedhe therapeutic response to dasatinib with high-dose imatinib800 mg) in CML-CP patients resistant to low dose imatinibh 34 (2010) 1255–1268
(400–600 mg). The initial and subsequent 2-year follow-up resultsindicated that dasatinib produced a significantly greater therapeu-tic response than high-dose imatinib for the patients in their study[75,76]. Currently, there are no direct reports of comparative trialsbetween dasatinib and nilotinib. Overall, only the minority of BCR-ABL mutations appears to be less responsive to either nilotinib ordasatinib or other investigational drugs in clinical trial. Therefore,precaution should be taken when deciding what drug to prescribefor which patient as different resistance factors that may be present.
Allogeneic HCT (AlloHCT) is another potential therapeuticmodality for CML patients, particularly in those who are intoler-ant to TKIs or have mutations such as the T315I mutation, whichcan produce significant resistance to all of the clinically availableTKIs. Also, AlloHCT should be considered in patients who have pro-gressed to the advanced forms of CML and in those with molecularlyand cytogenetically unfavorable disease, since responses to BCR-ABL TKIs are unlikely to be sustained [146]. In addition, it seemsthat in high-risk or advanced-phase patients, a more aggressiveapproach would be to combine second-generation TKIs initially,followed by AlloHCT [77]. The updated ELN guidelines recommendsAlloHCT for patients in CML-AP or BP or with the T315I mutationand also for the patients who fail or have suboptimal response tosecond-line TKIs such as nilotinib or dasatinib [50].
Autologous stem cell transplantation (auto-SCT) could alsoeliminate a Ph+ clone bearing a BCR-ABL kinase domain mutation,thus could be a promising therapeutic option for imatinib resis-tance. Recent studies have shown that peripheral blood stem cells(PBSCs) can be successfully mobilized and harvested in patientspretreated with imatinib and or second-generation TKIs [147,148].However, at this time, only a few studies have been published aboutthe successful treatment of imatinib-resistant CML patients withauto-SCT alone or in combination with dasatinib [147,148]. Futurestudies are warranted in order to determine the efficacy and safetyof auto-SCT alone or in combination with the newly developed TKIsin imatinib resistant patients.
Patients with the T315I mutation are also candidates forclinical trials with new investigative drugs, such as the ATPnon-competitive ABL TKI, ON012380, the aurora kinase inhibitorMK-0457, and the p38 MAP kinase inhibitor BIRB796 [44,47,137].All of these compounds have been reported to be effective in CMLpatients with the T315I mutation [149]. In addition, a more recentreport indicate that a selective allosteric BCR-ABL inhibitor, GNF-2, is able to bind to the myristate-binding site of ABL, leading tostructural dynamics changes at the ATP-binding site. Additionally,GNF-5, an analogue of GNF-2, which has improved pharmacokineticproperties when used in combination with the ATP-competitiveinhibitors imatinib or nilotinib, suppressed the emergence of resis-tant mutations in vitro [150]. In addition, GNF-5 displayed additiveinhibitory activity in biochemical and cellular assays against theT315I human BCR-ABL mutant and displayed in vivo efficacy againstthis recalcitrant mutant in a murine bone-marrow transplantationmodel [150].In summary, there are a number of treatment optionsin CML patients that fail to respond to imatinib. In order to optimizethe therapeutic benefit without an unnecessary delay, the charac-teristics of the disease and clinical profile of the patient should bethoroughly evaluated upon the occurrence of imatinib failure priorto choosing a specific therapeutic regimen. For example, patientsshould be questioned in detail to determine if they are adherent tothe all aspects of imatinib treatment. Furthermore, patients shouldbe asked questions about all other medications (prescription andover-the-counter) they are taking so as to minimize the possibil-
ity of problematic drug–drug interactions at the pharmacokineticand pharmacodynamic level. The determination of the plasma levelof imatinib should be considered if there is uncertainty about anindividual’s compliance with the medications. Mutation analysiscan provide additional information regarding the current treat-
esearc
mitiwttaad
7
mtsptAdsanfsadinfao
C
A
(vRtGaC
wr
R
X. An et al. / Leukemia R
ent regimen in use. Thus, patients with the T315I mutant, whonduces a broad degree of resistance, will not benefit from any ofhe currently approved TKIs. Subsequently, the treatment optionsn these patients should include investigational drugs that interact
ith the active site of the BCR-ABL kinase or the use of HCT in ordero produce a therapeutic response. A patient’s history of safety andolerability may also impact the subsequent choice of treatmentnd one should consider the potential impact of the drug’s AEs onny of the patient’s preexisting conditions before making a finalecision.
. Conclusion
The introduction of the BCR-ABL TKIs has revolutionized theanagement of Ph+ CML patients. Patients diagnosed with CML
oday, as opposed to 10–20 years ago, are expected to have a sub-tantially longer survival rate. However, further improvements inrognosis and complete disease eradication are goals that have yeto be reached in CML treatment. The use of first generation BCR-BL TKI imatinib, as reviewed in this article, has yielded someegree of successes as a first-line therapy. However, there areome patients who are resistant or intolerant to imatinib ther-py. The currently approved second-generation TKIs, dasatinib andilotinib, are effective in most patients who experience treatment
ailure with imatinib. However, these agents are not efficacious inmall group of resistant patients as a consequence of the inter-ction of multiple factors including T315I mutation. Indeed, theiverse and complex body of knowledge that we have gained
n understanding chemotherapeutic failure has provided us withovel strategies to overcome CML disease. It seems pertinent to
urther explore and determine the resistance mechanisms and toscertain the resistance profiles of these known BCR-ABL TKIs inrder to provide the optimal treatment regimens.
onflict of interest
All authors have no conflict of interest regarding this paper.
cknowledgements
We thank Chun-Ling Dai, Kamlesh Sodani, and Chung LeeCollege of Pharmacy and Allied Health Professions, St. John’s Uni-ersity), and Yangmin Chen (Ernest Mario School of Pharmacy,utgers University) for the editorial assistance. This work was par-ially supported by fund from St. John’s University Research Seedrant (579-1110-7002, Z.S. Chen). Pei-Rong Ding is a recipient offellowship for overseas study supported by China Scholarship
ouncil CSC).Contributions. X.A., Y.S., A.K.T. and P.-R.D. contributed equally in
riting different sections of this review. C.R.A. and Z-S.C. edited theeview.
eferences
[1] Cortes J. Natural history and staging of chronic myelogenous leukemia. Hema-tol Oncol Clin North Am 2004;18:569–84, viii.
[2] Faderl S, Talpaz M, Estrov Z, Kantarjian HM. Chronic myelogenous leukemia:biology and therapy. Ann Intern Med 1999;131:207–19.
[3] Bennett JH. Case of hypertrophy of the spleen and liver in which death tookplace from suppuration of the blood. Edinb Med Surg J 1845;64:413–23.
[4] Virchow RWB. Frorieps Notizen; 1845. p. 36.[5] Nowell PCHD. A minute chromosome in human chronic granulocytic
leukemia. Science 1960;132:1497.
[6] Erikson J, Griffin CA, ar-Rushdi A, Valtieri M, Hoxie J, Finan J, et al. Hetero-geneity of chromosome 22 breakpoint in Philadelphia-positive (Ph+) acutelymphocytic leukemia. Proc Natl Acad Sci USA 1986;83:1807–11.
[7] Rowley JD. Letter: a new consistent chromosomal abnormality in chronicmyelogenous leukaemia identified by quinacrine fluorescence and Giemsastaining. Nature 1973;243:290–3.
h 34 (2010) 1255–1268 1265
[8] Deininger MW, Goldman JM, Melo JV. The molecular biology of chronicmyeloid leukemia. Blood 2000;96:3343–56.
[9] Kantarjian HM, Talpaz M, Giles F, O’Brien S, Cortes J. New insights into thepathophysiology of chronic myeloid leukemia and imatinib resistance. AnnIntern Med 2006;145:913–23.
[10] Sawyers CL. Chronic myeloid leukemia. N Engl J Med 1999;340(April(17)):1330–40.
[11] Kantarjian HM, Giles F, Quintas-Cardama A, Cortes J. Important therapeutictargets in chronic myelogenous leukemia. Clin Cancer Res 2007;13(February(4)):1089–97.
[12] Sawyers CL. Signal transduction pathways involved in BCR-ABL transforma-tion. Baillieres Clin Haematol 1997;10:223–31.
[13] Calabretta B, Perrotti D. The biology of CML blast crisis. Blood2004;103:4010–22.
[14] Bolin RW, Robinson WA, Sutherland J, Hamman RF. Busulfan versus hydrox-yurea in long-term therapy of chronic myelogenous leukemia. Cancer1982;50:1683–6.
[15] Interferon alfa versus chemotherapy for chronic myeloid leukemia: a meta-analysis of seven randomized trials: Chronic Myeloid Leukemia Trialists’Collaborative Group. J Natl Cancer Inst 1997;89:1616–20.
[16] Bonifazi F, de Vivo A, Rosti G, Guilhot F, Guilhot J, Trabacchi E, et al. Chronicmyeloid leukemia and interferon-alpha: a study of complete cytogeneticresponders. Blood 2001;98:3074–81.
[17] Ohnishi K, Ohno R, Tomonaga M, Kamada N, Onozawa K, Kuramoto A,et al. A randomized trial comparing interferon-alpha with busulfan fornewly diagnosed chronic myelogenous leukemia in chronic phase. Blood1995;86:906–16.
[18] Silver RT, Woolf SH, Hehlmann R, Appelbaum FR, Anderson J, Bennett C, etal. An evidence-based analysis of the effect of busulfan, hydroxyurea, inter-feron, and allogeneic bone marrow transplantation in treating the chronicphase of chronic myeloid leukemia: developed for the American Society ofHematology. Blood 1999;94:1517–36.
[19] Baccarani M, Saglio G, Goldman J, Hochhaus A, Simonsson B, Appelbaum F, etal. Evolving concepts in the management of chronic myeloid leukemia: rec-ommendations from an expert panel on behalf of the European LeukemiaNet.Blood 2006;108:1809–20.
[20] Savage DG, Antman KH. Imatinib mesylate—a new oral targeted therapy. NEngl J Med 2002;346:683–93.
[21] Sawyers CL, Hochhaus A, Feldman E, Goldman JM, Miller CB, Ottmann OG, etal. Imatinib induces hematologic and cytogenetic responses in patients withchronic myelogenous leukemia in myeloid blast crisis: results of a phase IIstudy. Blood 2002;99:3530–9.
[22] Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinicalresistance to STI-571 cancer therapy caused by BCR-ABL gene mutation oramplification. Science 2001;293:876–80.
[23] Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A,et al. Characterization of AMN107, a selective inhibitor of native and mutantBcr-Abl. Cancer Cell 2005;7:129–41.
[24] Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinibresistance with a novel ABL kinase inhibitor. Science 2004;305:399–401.
[25] Golas JM, Arndt K, Etienne C, Lucas J, Nardin D, Gibbons J, et al. SKI-606, a4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is apotent antiproliferative agent against chronic myelogenous leukemia cells inculture and causes regression of K562 xenografts in nude mice. Cancer Res2003;63:375–81.
[26] Kantarjian HM, Giles F, Quintas-Cardama A, Cortes J. Important therapeutictargets in chronic myelogenous leukemia. Clin Cancer Res 2007;13:1089–97.
[27] Holtz MS, Slovak ML, Zhang F, Sawyers CL, Forman SJ, Bhatia R. Imatinib mesy-late (STI571) inhibits growth of primitive malignant progenitors in chronicmyelogenous leukemia through reversal of abnormally increased prolifera-tion. Blood 2002;99:3792–800.
[28] Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effectsof a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Ablpositive cells. Nat Med 1996;2:561–6.
[29] Legros L, Bourcier C, Jacquel A, Mahon FX, Cassuto JP, Auberger P, et al.Imatinib mesylate (STI571) decreases the vascular endothelial growth fac-tor plasma concentration in patients with chronic myeloid leukemia. Blood2004;104:495–501.
[30] Kvasnicka HM, Thiele J, Staib P, Schmitt-Graeff A, Griesshammer M, KloseJ, et al. Reversal of bone marrow angiogenesis in chronic myeloid leukemiafollowing imatinib mesylate (STI571) therapy. Blood 2004;103:3549–51.
[31] Appel S, Boehmler AM, Grunebach F, Muller MR, Rupf A, Weck MM, et al.Imatinib mesylate affects the development and function of dendritic cells gen-erated from CD34+ peripheral blood progenitor cells. Blood 2004;103(January(2)):538–44.
[32] Taieb J, Maruyama K, Borg C, Terme M, Zitvogel L. Imatinib mesylate impairsFlt3L-mediated dendritic cell expansion and antitumor effects in vivo. Blood2004;103(March (5)):1966–7 [author reply 1967].
[33] Appel S, Rupf A, Weck MM, Schoor O, Brummendorf TH, Weinschenk T, et
al. Effects of imatinib on monocyte-derived dendritic cells are mediated byinhibition of nuclear factor-kappaB and Akt signaling pathways. Clin CancerRes 2005;11(March (5)):1928–40.[34] Wang H, Cheng F, Cuenca A, Horna P, Zheng Z, Bhalla K, et al. Imatinib mesylate(STI-571) enhances antigen-presenting cell function and overcomes tumor-induced CD4+ T-cell tolerance. Blood 2005;105(February (3)):1135–43.

1 esearc
266 X. An et al. / Leukemia R[35] Wehner R, Wendisch M, Schakel K, Bornhauser M, Platzbecker U, Mohr B, etal. Imatinib mesylate does not impair the immunogenicity of human myeloidblood dendritic cells. Leukemia 2006;20(September (9)):1629–32.
[36] O’Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, CervantesF, et al. Imatinib compared with interferon and low-dose cytarabine fornewly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med2003;348:994–1004.
[37] Goldman JM, Melo JV. Chronic myeloid leukemia—advances in biology andnew approaches to treatment. N Engl J Med 2003;349:1451–64.
[38] Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacyand safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronicmyeloid leukemia. N Engl J Med 2001;344:1031–7.
[39] Ottmann OG, Druker BJ, Sawyers CL, Goldman JM, Reiffers J, Silver RT, et al. Aphase 2 study of imatinib in patients with relapsed or refractory Philadelphiachromosome-positive acute lymphoid leukemias. Blood 2002;100:1965–71.
[40] Kimura S, Naito H, Segawa H, Kuroda J, Yuasa T, Sato K, et al. NS-187, a potentand selective dual Bcr-Abl/Lyn tyrosine kinase inhibitor, is a novel agent forimatinib-resistant leukemia. Blood 2005;106:3948–54.
[41] Manley PW, Cowan-Jacob SW, Mestan J. Advances in the structural biology,design and clinical development of Bcr-Abl kinase inhibitors for the treatmentof chronic myeloid leukaemia. Biochim Biophys Acta 2005;1754:3–13.
[42] Weisberg E, Manley P, Mestan J, Cowan-Jacob S, Ray A, Griffin JD.AMN107 (nilotinib): a novel and selective inhibitor of BCR-ABL. Br J Cancer2006;94:1765–9.
[43] O’Hare T, Walters DK, Stoffregen EP, Jia T, Manley PW, Mestan J, et al. Invitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clin-ically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res2005;65:4500–5.
[44] Quintas-Cardama A, Kantarjian H, Cortes J. Flying under the radar: the newwave of BCR-ABL inhibitors. Nat Rev Drug Discov 2007;6:834–48.
[45] O’Hare T, Pollock R, Stoffregen EP, Keats JA, Abdullah OM, Moseson EM,et al. Inhibition of wild-type and mutant Bcr-Abl by AP23464, a potentATP-based oncogenic protein kinase inhibitor: implications for CML. Blood2004;104:2532–9.
[46] Wisniewski D, Lambek CL, Liu C, Strife A, Veach DR, Nagar B, et al. Charac-terization of potent inhibitors of the Bcr-Abl and the c-kit receptor tyrosinekinases. Cancer Res 2002;62:4244–55.
[47] Gumireddy K, Baker SJ, Cosenza SC, John P, Kang AD, Robell KA, et al. A non-ATP-competitive inhibitor of BCR-ABL overrides imatinib resistance. Proc NatlAcad Sci USA 2005;102:1992–7.
[48] Giles FJ, Cortes J, Jones D, Bergstrom D, Kantarjian H, Freedman SJ. MK-0457,a novel kinase inhibitor, is active in patients with chronic myeloid leukemiaor acute lymphocytic leukemia with the T315I BCR-ABL mutation. Blood2007;109:500–2.
[49] O’Brien S. NCCN clinical practice guidelines in oncology: chronic myel-ogenous leukemia, version 2.2010. http://www.nccn.org/professionals/physician gls/PDF/cml.pdf [accessed July 2009].
[50] Baccarani M, Cortes J, Pane F, Niederwieser D, Saglio G, Apperley J, et al.Chronic myeloid leukemia: an update of concepts and management recom-mendations of European Leukemia Net. J Clin Oncol 2009;27:6041–51.
[51] Hughes TP, Kaeda J, Branford S, Rudzki Z, Hochhaus A, Hensley ML, et al.Frequency of major molecular responses to imatinib or interferon alfa pluscytarabine in newly diagnosed chronic myeloid leukemia. N Engl J Med2003;349(October (15)):1423–32.
[52] Hughes T, Branford S. Molecular monitoring of BCR-ABL as a guide to clinicalmanagement in chronic myeloid leukaemia. Blood Rev 2006;20:29–41.
[53] Branford S, Cross NC, Hochhaus A, Radich J, Saglio G, Kaeda J, et al. Rationalefor the recommendations for harmonizing current methodology for detectingBCR-ABL transcripts in patients with chronic myeloid leukaemia. Leukemia2006;20(November (11)):1925–30.
[54] Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N,et al. Five-year follow-up of patients receiving imatinib for chronic myeloidleukemia. N Engl J Med 2006;355:2408–17.
[55] Press RD, Galderisi C, Yang R, Rempfer C, Willis SG, Mauro MJ, et al. A half-logincrease in BCR-ABL RNA predicts a higher risk of relapse in patients withchronic myeloid leukemia with an imatinib-induced complete cytogeneticresponse. Clin Cancer Res 2007;13:6136–43.
[56] Khorashad JS, de Lavallade H, Apperley JF, Milojkovic D, Reid AG, Bua M, et al.Finding of kinase domain mutations in patients with chronic phase chronicmyeloid leukemia responding to imatinib may identify those at high risk ofdisease progression. J Clin Oncol 2008;26:4806–13.
[57] Hughes T, Saglio G, Branford S, Soverini S, Kim DW, Muller MC, et al. Impact ofbaseline BCR-ABL mutations on response to nilotinib in patients with chronicmyeloid leukemia in chronic phase. J Clin Oncol 2009;27:4204–10.
[58] Barnes DJ, Palaiologou D, Panousopoulou E, Schultheis B, Yong AS, WongA, et al. Bcr-Abl expression levels determine the rate of development ofresistance to imatinib mesylate in chronic myeloid leukemia. Cancer Res2005;65:8912–9.
[59] Polakova KM, Polivkova V, Rulcova J, Klamova H, Jurcek T, Dvorakova D, etal. Constant BCR-ABL transcript level >or =0.1% (IS) in patients with CML
responding to imatinib with complete cytogenetic remission may indicatemutation analysis. Exp Hematol 2010;38(January (1)):20–6.[60] Wang L, Knight K, Lucas C, Clark RE. The role of serial BCR-ABL transcriptmonitoring in predicting the emergence of BCR-ABL kinase mutations inimatinib-treated patients with chronic myeloid leukemia. Haematologica2006;91:235–9.
h 34 (2010) 1255–1268
[61] Press RD, Willis SG, Laudadio J, Mauro MJ, Deininger MW. Determining the risein BCR-ABL RNA that optimally predicts a kinase domain mutation in patientswith chronic myeloid leukemia on imatinib. Blood 2009;114(September(13)):2598–605.
[62] Talpaz M, Silver RT, Druker BJ, Goldman JM, Gambacorti-Passerini C, GuilhotF, et al. Imatinib induces durable hematologic and cytogenetic responses inpatients with accelerated phase chronic myeloid leukemia: results of a phase2 study. Blood 2002;99:1928–37.
[63] Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, Gambacorti-Passerini C, et al. Hematologic and cytogenetic responses to imatinib mesylatein chronic myelogenous leukemia. N Engl J Med 2002;346:645–52.
[64] Hochhaus A, O’Brien SG, Guilhot F, Druker BJ, Branford S, Foroni L, et al. Six-year follow-up of patients receiving imatinib for the first-line treatment ofchronic myeloid leukemia. Leukemia 2009;23:1054–61.
[65] Cortes J, Giles F, O’Brien S, Thomas D, Garcia-Manero G, Rios MB, et al. Resultof high-dose imatinib mesylate in patients with Philadelphia chromosome-positive chronic myeloid leukemia after failure of interferon-alpha. Blood2003;102:83–6.
[66] Kantarjian H, Talpaz M, O’Brien S, Garcia-Manero G, Verstovsek S, Giles F,et al. High-dose imatinib mesylate therapy in newly diagnosed Philadel-phia chromosome-positive chronic phase chronic myeloid leukemia. Blood2004;103:2873–8.
[67] Cortes J, Baccarani M, Guilhot F. A phase III, randomized, open-label studyof 400 mg versus 800 mg of imatinib mesylate (IM) in patients (pts) withnewly diagnosed, previously untreated chronic myeloid leukemia in chronicphase (CML-CP) using molecular endpoints: 1-year results of TOPS (tyrosinekinase inhibitor optimization and selectivity) study. In: ASH Annual MeetingAbstracts, vol. 112. 2008. p. 335.
[68] Baranska M, Lewandowski K, Gniot M, Iwola M, Lewandowska M, Komar-nicki M. Dasatinib treatment can overcome imatinib and nilotinib resistancein CML patient carrying F359I mutation of BCR-ABL oncogene. J Appl Genet2008;49:201–3.
[69] Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, et al. Dasatinibin imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl JMed 2006;354:2531–41.
[70] Hochhaus A, Kantarjian HM, Baccarani M, Lipton JH, Apperley JF, Druker BJ,et al. Dasatinib induces notable hematologic and cytogenetic responses inchronic-phase chronic myeloid leukemia after failure of imatinib therapy.Blood 2007;109:2303–9.
[71] Hochhaus A, Baccarani M, Deininger M, Apperley JF, Lipton JH, Goldberg SL, etal. Dasatinib induces durable cytogenetic responses in patients with chronicmyelogenous leukemia in chronic phase with resistance or intolerance toimatinib. Leukemia 2008;22:1200–6.
[72] Kantarjian H, Pasquini R, Hamerschlak N, Rousselot P, Holowiecki J, JootarS, et al. Dasatinib or high-dose imatinib for chronic-phase chronic myeloidleukemia after failure of first-line imatinib: a randomized phase 2 trial. Blood2007;109:5143–50.
[73] Kantarjian H, Pasquini R, Levy V, Jootar S, Holowiecki J, Hamerschlak N, et al.Dasatinib or high-dose imatinib for chronic-phase chronic myeloid leukemiaresistant to imatinib at a dose of 400 to 600 milligrams daily: two-year follow-up of a randomized phase 2 study (START-R). Cancer 2009;115:4136–47.
[74] Guilhot F, Apperley J, Kim DW, Bullorsky EO, Baccarani M, Roboz GJ, et al. Dasa-tinib induces significant hematologic and cytogenetic responses in patientswith imatinib-resistant or -intolerant chronic myeloid leukemia in acceler-ated phase. Blood 2007;109:4143–50.
[75] Apperley JF, Cortes JE, Kim DW, Roy L, Roboz GJ, Rosti G, et al. Dasatinib in thetreatment of chronic myeloid leukemia in accelerated phase after imatinibfailure: the START a trial. J Clin Oncol 2009;27:3472–9.
[76] Cortes J, Rousselot P, Kim DW, Ritchie E, Hamerschlak N, Coutre S, et al. Dasa-tinib induces complete hematologic and cytogenetic responses in patientswith imatinib-resistant or -intolerant chronic myeloid leukemia in blast crisis.Blood 2007;109:3207–13.
[77] Cortes J, Kim DW, Raffoux E, Martinelli G, Ritchie E, Roy L, et al. Efficacy andsafety of dasatinib in imatinib-resistant or -intolerant patients with chronicmyeloid leukemia in blast phase. Leukemia 2008;22:2176–83.
[78] Shah NP, Kantarjian HM, Kim DW, Rea D, Dorlhiac-Llacer PE, Milone JH,et al. Intermittent target inhibition with dasatinib 100 mg once daily pre-serves efficacy and improves tolerability in imatinib-resistant and -intolerantchronic-phase chronic myeloid leukemia. J Clin Oncol 2008;26:3204–12.
[79] Stone RM, Kim DW, Kantarjian HM, Rousselot Aea P. Dasatinib dose-optimization study in chronic phase chronic myeloid leukemia (CML-CP):three-year follow-up with dasatinib 100 mg once daily and landmark analy-sis of cytogenetic response and progression-free survival (PFS). J Clin Oncol2009;27:7007 (Meeting Abstracts).
[80] Kantarjian H, Cortes J, Kim DW, Dorlhiac-Llacer P, Pasquini R, DiPersio J, etal. Phase 3 study of dasatinib 140 mg once daily versus 70 mg twice dailyin patients with chronic myeloid leukemia in accelerated phase resistant orintolerant to imatinib: 15-month median follow-up. Blood 2009;113:6322–9.
[81] Borthakur G, Kantarjian HM. SMOBe, efficacy of dasatinib in patients (pts)with previously untreated chronic myelogenous leukemia (CML) in early
chronic phase (CML-CP). J Clin Oncol 2008;26:7013 [abstracts].[82] Kantarjian H, Giles F, Wunderle L, Bhalla K, O’Brien S, Wassmann B, et al.Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positiveALL. N Engl J Med 2006;354:2542–51.
[83] Kantarjian HM, Giles F, Gattermann N, Bhalla K, Alimena G, Palandri F, etal. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase

esearc
[
[
[
[
[
[
[
X. An et al. / Leukemia R
inhibitor, is effective in patients with Philadelphia chromosome-positivechronic myelogenous leukemia in chronic phase following imatinib resis-tance and intolerance. Blood 2007;110:3540–6.
[84] Kantarjian H, Giles F, Bhalla K. Nilotinib in chronic myeloid leukemia patientsin chronic phase (CML-CP) with imatinib (IM) resistance or intolerance:longer follow-up results of a phase II study. J Clin Oncol 2009;27:7029 [meet-ing abstract].
[85] Le Coutre PD, Giles F, Hochhaus A, et al. Nilotinib in chronic myeloid leukemiapatients in accelerated phase (CML-AP) with imatinib (IM) resistance or intol-erance: longer follow-up results of a phase II study. J Clin Oncol 2009;27:7057[meeting abstracts].
[86] Giles FJ, Larson RA, Kantarjian HM. Nilotinib in patients with Philadelphiachromosome-positive chronic myelogenous leukemia in blast crisis (CML-BC) who are resistant or intolerant to imatinib. J Clin Oncol 2008:7017 (ASCOMeeting Abstracts).
[87] Martinelli G, Castagnetti F, Poerio A, et al. Molecular responses with nilo-tinib 800 mg daily as first-line treatment of chronic myeloid leukemia inchronic phase: results of a phase II trial of the GIMEMA CML WP. J Clin Oncol2009;27:7074 [meeting abstracts].
[88] Litzow MR. Imatinib resistance: obstacles and opportunities. Arch Pathol LabMed 2006;130:669–79.
[89] Shah NP. Medical management of CML. Hematol Am Soc Hematol Educ Prog2007:371–5.
[90] Lowenberg B. Minimal residual disease in chronic myeloid leukemia. N EnglJ Med 2003;349:1399–401.
[91] Azam M, Latek RR, Daley GQ. Mechanisms of autoinhibition andSTI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell2003;112:831–43.
[92] Hochhaus A, La Rosee P. Imatinib therapy in chronic myelogenous leukemia:strategies to avoid and overcome resistance. Leukemia 2004;18:1321–31.
[93] Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, et al. Mul-tiple BCR-ABL kinase domain mutations confer polyclonal resistance to thetyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisischronic myeloid leukemia. Cancer Cell 2002;2:117–25.
[94] Branford S, Rudzki Z, Walsh S, Parkinson I, Grigg A, Szer J, et al. Detectionof BCR-ABL mutations in patients with CML treated with imatinib is virtu-ally always accompanied by clinical resistance, and mutations in the ATPphosphate-binding loop (P-loop) are associated with a poor prognosis. Blood2003;102:276–83.
[95] Cortes J, Jabbour E, Kantarjian H, Yin CC, Shan J, O’Brien S, et al. Dynam-ics of BCR-ABL kinase domain mutations in chronic myeloid leukemiaafter sequential treatment with multiple tyrosine kinase inhibitors. Blood2007;110:4005–11.
[96] Hochhaus A, Kreil S, Corbin AS, La Rosee P, Muller MC, Lahaye T, et al. Molecu-lar and chromosomal mechanisms of resistance to imatinib (STI571) therapy.Leukemia 2002;16:2190–6.
[97] Jabbour E, Kantarjian H, Jones D, Talpaz M, Bekele N, O’Brien S, etal. Frequency and clinical significance of BCR-ABL mutations in patientswith chronic myeloid leukemia treated with imatinib mesylate. Leukemia2006;20:1767–73.
[98] Soverini S, Colarossi S, Gnani A, Rosti G, Castagnetti F, Poerio A, et al. Con-tribution of ABL kinase domain mutations to imatinib resistance in differentsubsets of Philadelphia-positive patients: by the GIMEMA Working Party onChronic Myeloid Leukemia. Clin Cancer Res 2006;12:7374–9.
[99] Soverini S, Martinelli G, Rosti G, Bassi S, Amabile M, Poerio A, et al. ABLmutations in late chronic phase chronic myeloid leukemia patients withup-front cytogenetic resistance to imatinib are associated with a greaterlikelihood of progression to blast crisis and shorter survival: a study by theGIMEMA Working Party on Chronic Myeloid Leukemia. J Clin Oncol 2005;23:4100– 9.
100] le Coutre P, Kreuzer KA, Pursche S, Bonin M, Leopold T, Baskaynak G, et al.Pharmacokinetics and cellular uptake of imatinib and its main metaboliteCGP74588. Cancer Chemother Pharmacol 2004;53:313–23.
101] Peng B, Hayes M, Resta D, Racine-Poon A, Druker BJ, Talpaz M, et al. Pharma-cokinetics and pharmacodynamics of imatinib in a phase I trial with chronicmyeloid leukemia patients. J Clin Oncol 2004;22:935–42.
102] Picard S, Titier K, Etienne G, Teilhet E, Ducint D, Bernard MA, et al. Troughimatinib plasma levels are associated with both cytogenetic and molecu-lar responses to standard-dose imatinib in chronic myeloid leukemia. Blood2007;109:3496–9.
103] Larson RA, Druker BJ, Guilhot F, O’Brien SG, Riviere GJ, Krahnke T, et al.Imatinib pharmacokinetics and its correlation with response and safety inchronic-phase chronic myeloid leukemia: a subanalysis of the IRIS study.Blood 2008;111:4022–8.
104] Forrest DL, Trainor S, Brinkman RR, Barnett MJ, Hogge DE, Nevill TJ, et al.Cytogenetic and molecular responses to standard-dose imatinib in chronicmyeloid leukemia are correlated with Sokal risk scores and duration of ther-apy but not trough imatinib plasma levels. Leuk Res 2009;33:271–5.
105] Darkow T, Henk HJ, Thomas SK, Feng W, Baladi JF, Goldberg GA, et al.Treatment interruptions and non-adherence with imatinib and associated
healthcare costs: a retrospective analysis among managed care patientswith chronic myelogenous leukaemia. Pharmacoeconomics 2007;25:481–96.106] Halpern R, Barghout V, Mody-Patel N, Williams D. Relationship betweencompliance, costs, hospitalizations for CML and GIST patients using imatinibmesylate [abstract]. J Clin Oncol 2008;26:5S [abstract 6598].
h 34 (2010) 1255–1268 1267
[107] Noens L, van Lierde MA, De Bock R, Verhoef G, Zachee P, Berneman Z, et al.Prevalence, determinants, and outcomes of nonadherence to imatinib ther-apy in patients with chronic myeloid leukemia: the ADAGIO study. Blood2009;113:5401–11.
[108] van Erp NP, Gelderblom H, Karlsson MO, Li J, Zhao M, Ouwerkerk J, et al. Influ-ence of CYP3A4 inhibition on the steady-state pharmacokinetics of imatinib.Clin Cancer Res 2007;13:7394–400.
[109] Gréen H, Skoglund K, Rommel F, Mirghani RA, Lotfi K. CYP3A activity influ-ences imatinib response in patients with chronic myeloid leukemia: a pilotstudy on in vivo CYP3A activity. Eur J Clin Pharmacol 2010;66:383–6.
[110] Wilkinson GR. Cytochrome P4503A (CYP3A) metabolism: prediction of in vivoactivity in humans. J Pharmacokinet Biopharm 1996;24:475–90.
[111] eng B, Lloyd P, Schran H. Clinical pharmacokinetics of imatinib. Clin Pharma-cokinet 2005;44:879–94.
[112] Gambacorti-Passerini C, Zucchetti M, Russo D, Frapolli R, Verga M, BungaroS, et al. Alpha1 acid glycoprotein binds to imatinib (STI571) and substantiallyalters its pharmacokinetics in chronic myeloid leukemia patients. Clin CancerRes 2003;9:625–32.
[113] Jørgensen HG, Elliott MA, Allen EK, Carr CE, Holyoake TL, Smith KD. Alpha1-acid glycoprotein expressed in the plasma of chronic myeloid leukemiapatients does not mediate significant in vitro resistance to STI571. Blood2002;5:713–5.
[114] Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med2002;53:615–27.
[115] Mahon FX, Belloc F, Lagarde V, Chollet C, Moreau-Gaudry F, Reiffers J, etal. MDR1 gene overexpression confers resistance to imatinib mesylate inleukemia cell line models. Blood 2003;101:2368–73.
[116] Ferrao PT, Frost MJ, Siah SP, Ashman LK. Overexpression of P-glycoproteinin K562 cells does not confer resistance to the growth inhibitory effects ofimatinib (STI571) in vitro. Blood 2003;102:4499–503.
[117] Assef Y, Rubio F, Colo G, del Monaco S, Costas MA, Kotsias BA. Imatinibresistance in multidrug-resistant K562 human leukemic cells. Leuk Res2009;33:710–6.
[118] Galimberti S, Cervetti G, Guerrini F, Testi R, Pacini S, Fazzi R, et al. Quanti-tative molecular monitoring of BCR-ABL and MDR1 transcripts in patientswith chronic myeloid leukemia during Imatinib treatment. Cancer GenetCytogenet 2005;162:57–62.
[119] Hatziieremia S, Jordanides NE, Holyoake TL, Mountford JC, Jorgensen HG. Inhi-bition of MDR1 does not sensitize primitive chronic myeloid leukemia CD34+cells to imatinib. Exp Hematol 2009;37:692–700.
[120] Mao Q, Unadkat JD. Role of the breast cancer resistance protein (ABCG2) indrug transport. AAPS J 2005;7:E118–33.
[121] Brendel C, Scharenberg C, Dohse M, Robey RW, Bates SE, Shukla S, et al. Ima-tinib mesylate and nilotinib (AMN107) exhibit high-affinity interaction withABCG2 on primitive hematopoietic stem cells. Leukemia 2007;21:1267–75.
[122] Burger H, van Tol H, Boersma AW, Brok M, Wiemer EA, Stoter G, et al. Ima-tinib mesylate (STI571) is a substrate for the breast cancer resistance protein(BCRP)/ABCG2 drug pump. Blood 2004;104:2940–2.
[123] Shukla S, Sauna ZE, Ambudkar SV. Evidence for the interaction of imatinibat the transport-substrate site(s) of the multidrug-resistance-linked ABCdrug transporters ABCB1 (P-glycoprotein) and ABCG2. Leukemia 2008;22:445–7.
[124] Mahon FX, Hayette S, Lagarde V, Belloc F, Turcq B, Nicolini F, et al. Evidencethat resistance to nilotinib may be due to BCR-ABL, Pgp, or Src kinase over-expression. Cancer Res 2008;68:9809–16.
[125] Tiwari AK, Sodani K, Wang SR, Kuang YH, Ashby Jr CR, Chen X, et al. Nilotinib(AMN107, Tasigna) reverses multidrug resistance by inhibiting the activityof the ABCB1/Pgp and ABCG2/BCRP/MXR transporters. Biochem Pharmacol2009;78:153–61.
[126] Chen Y, Agarwal S, Shaik NM, Chen C, Yang Z, Elmquist WF. P-glycoproteinand breast cancer resistance protein influence brain distribution of dasatinib.J Pharmacol Exp Ther 2009;330:956–63.
[127] Hiwase DK, Saunders V, Hewett D, Frede A, Zrim S, Dang P, et al. Dasatinibcellular uptake and efflux in chronic myeloid leukemia cells: therapeuticimplications. Clin Cancer Res 2008;14:3881–8.
[128] Kamath AV, Wang J, Lee FY, Marathe PH. Preclinical pharmacokinetics and invitro metabolism of dasatinib (BMS-354825): a potent oral multi-targetedkinase inhibitor against SRC and BCR-ABL. Cancer Chemother Pharmacol2008;61:365–76.
[129] Shen T, Kuang YH, Ashby CR, Lei Y, Chen A, Zhou Y, et al. Imatinib and nilotinibreverse multidrug resistance in cancer cells by inhibiting the efflux activityof the MRP7 (ABCC10). PLoS One 2009;4:e7520.
[130] Thomas J, Wang L, Clark RE, Pirmohamed M. Active transport of imatinib intoand out of cells: implications for drug resistance. Blood 2004;104:3739–45.
[131] White DL, Saunders VA, Dang P, Engler J, Venables A, Zrim S, et al. Most CMLpatients who have a suboptimal response to imatinib have low OCT-1 activity:higher doses of imatinib may overcome the negative impact of low OCT-1activity. Blood 2007;110:4064–72.
[132] Niwa T, Asaki T, Kimura S. NS-187 (INNO-406), a Bcr-Abl/Lyn dual tyrosinekinase inhibitor. Anal Chem Insights 2007;2:93–106.
[133] Meyn III MA, Wilson MB, Abdi FA, Fahey N, Schiavone AP, Wu J, et al. Srcfamily kinases phosphorylate the Bcr-Abl SH3-SH2 region and modulate Bcr-Abl transforming activity. J Biol Chem 2006;281:30907–16.
[134] Donato NJ, Wu JY, Stapley J, Gallick G, Lin H, Arlinghaus R, et al. BCR-ABL inde-pendence and LYN kinase overexpression in chronic myelogenous leukemiacells selected for resistance to STI571. Blood 2003;101:690–8.

1 esearc
268 X. An et al. / Leukemia R[135] Donato NJ, Wu JY, Stapley J, Lin H, Arlinghaus R, Aggarwal BB, et al. Imatinibmesylate resistance through BCR-ABL independence in chronic myelogenousleukemia. Cancer Res 2004;64:672–7.
[136] Dai Y, Rahmani M, Corey SJ, Dent P, Grant S. A Bcr/Abl-independent, Lyn-dependent form of imatinib mesylate (STI-571) resistance is associated withaltered expression of Bcl-2. J Biol Chem 2004;279:34227–39.
[137] Wu J, Meng F, Kong LY, Peng Z, Ying Y, Bornmann WG, et al. Associationbetween imatinib-resistant BCR-ABL mutation-negative leukemia and per-sistent activation of LYN kinase. J Natl Cancer Inst 2008;100:926–39.
[138] Redaelli S, Piazza R, Rostagno R, Magistroni V, Perini P, Marega M, et al. Activityof bosutinib, dasatinib, and nilotinib against 18 imatinib-resistant BCR/ABLmutants. J Clin Oncol 2009;27:469–71.
[139] Puttini M, Coluccia AM, Boschelli F, Cleris L, Marchesi E, Donella-Deana A, etal. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, againstimatinib-resistant Bcr-Abl+ neoplastic cells. Cancer Res 2006;66:11314–22.
[140] Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L,et al. Primitive, quiescent, Philadelphia-positive stem cells from patientswith chronic myeloid leukemia are insensitive to STI571 in vitro. Blood2002;99:319–25.
[141] Burgering BM. A brief introduction to FOXOlogy. Oncogene 2008;27(April(16)):2258–62.
[142] Komatsu N, Watanabe T, Uchida M, Mori M, Kirito K, Kikuchi S, et al. Amember of Forkhead transcription factor FKHRL1 is a downstream effectorof STI571-induced cell cycle arrest in BCR-ABL-expressing cells. J Biol Chem2003;278(February (8)):6411–9.
[143] Marin D, Goldman JM, Olavarria E, Apperley JF. Transient benefit only fromincreasing the imatinib dose in CML patients who do not achieve com-
h 34 (2010) 1255–1268
plete cytogenetic remissions on conventional doses. Blood 2003;102:2702–3[author reply 2703–4].
[144] Kantarjian HM, Larson RA, Guilhot F, O’Brien SG, Mone M, Rudoltz M, et al.Efficacy of imatinib dose escalation in patients with chronic myeloid leukemiain chronic phase. Cancer 2009;115:551–60.
[145] Le Coutre P, Ottmann OG, Giles F, Kim DW, Cortes J, Gattermann N,et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosinekinase inhibitor, is active in patients with imatinib-resistant or -intolerantaccelerated-phase chronic myelogenous leukemia. Blood 2008;111:1834–9.
[146] Weisser M, Schleuning M, Haferlach C, Schwerdtfeger R, Kolb HJ. Allo-geneic stem-cell transplantation provides excellent results in advanced stagechronic myeloid leukemia with major cytogenetic response to pre-transplantimatinib therapy. Leuk Lymphoma 2007;48:295–301.
[147] Kreuzer KA, Kluhs C, Baskaynak G, Movassaghi K, Dorken B, le Coutre P.Filgrastim-induced stem cell mobilization in chronic myeloid leukaemiapatients during imatinib therapy: safety, feasibility and evidence for an effi-cient in vivo purging. Br J Haematol 2004;124(January (2)):195–9.
[148] Rea D, Raffoux E, Cayuela JM, Maarek O, Dombret H. Sustained majormolecular response in the absence of any antileukaemic therapy after dasa-tinib treatment and autologous peripheral blood stem cell transplantationin a patient with imatinib-resistant myeloblastic-phase chronic myeloidleukaemia. Leukemia 2009;23(June (6)):1158–9.
[149] Mughal TI, Goldman JM. Emerging strategies for the treatment of mutantBcr-Abl T315I myeloid leukemia. Clin Lymphoma Myeloma 2007;7(Suppl.2):S81–4.
[150] Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, et al. Target-ing Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature2010;463(January (7280)):501–6.

![Design, synthesis, and biological evaluation of pyrazolo [3, 4-d] pyrimidines active in vivo on the Bcr-Abl T315I mutant](https://static.fdokumen.com/doc/165x107/6333d434b94d623842026483/design-synthesis-and-biological-evaluation-of-pyrazolo-3-4-d-pyrimidines-active.jpg)
















![Course: Biostatistics Lecture No: [ 2 ] - Philadelphia University](https://static.fdokumen.com/doc/165x107/633a2618749bc7c55d0d5094/course-biostatistics-lecture-no-2-philadelphia-university.jpg)

