
Polyaniline stabilized highly dispersed Pt nanoparticles: Preparation, characterization and...
13
Polyaniline stabilized highly dispersed Pt nanoparticles: Preparation, characterization and catalytic properties A. Drelinkiewicz a, * , A. Zie ˛ba a , J.W. Sobczak b , M. Bonarowska b , Z. Karpin ´ ski b , A. Waksmundzka-Góra a , J. Stejskal c a Institute of Catalysis and Surface Chemistry, Polish Academy of Sciences, 30-239 Kraków, Niezapominajek 8, Poland b Institute of Physical Chemistry. Polish Academy of Sciences, 01-224 Warszawa, Kasprzaka 44-52, Poland c Institute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic, Heyrovsky Sq. 2, 162 06 Prague, Czech Republic article info Article history: Received 5 February 2009 Received in revised form 3 April 2009 Accepted 10 April 2009 Available online 21 April 2009 Keywords: Polyaniline Platinum Hydrogenation 2-Butyne-1,4-diol Hydrodechlorination CCl 4 Selectivity to C 2 Cl 6 abstract Platinum–polyaniline (2%Pt/PANI) composites are prepared using three precursor solutions of H 2 PtCl 6 differing in the pH and thus in the type of dominating chloro–aqua or chloro–hydroxo complexes of Pt(IV). Various characterization techniques (X-ray diffraction, FTIR and X-ray photoelectron spectroscopy, and electron microscopy; TEM, HRTEM) were employed in order to elucidate the role of platinum precur- sor composition in the state of Pt-species formed in the Pt/PANI composites. In all three as-received Pt/ PANI composites, platinum in two valence states Pt(IV) and Pt(II) was observed thus showing redox reac- tion under preparation of composites. The reduction of Pt(IV) is much easier in the precursor solution of alkaline pH, i.e. when the Pt(IV) complexes comprising aqua and/or hydroxo ligands are the dominating ones. Under these conditions the content of Pt(II)-species is ca. 7 higher than that of Pt(IV) ones. Reduc- tion of as-prepared composites by NaBH 4 gives 2%Pt/PANI catalysts of well-dispersed Pt-metal particles of size ranging from 1 nm up to 4 nm. The average diameters (2.24 nm) of Pt-particles was slightly smal- ler in 2%Pt/PANI catalyst prepared using H 2 PtCl 6 solution of acidic pH. Catalytic properties of Pt/PANI composites are tested in liquid phase hydrogenation of alkyne-type reactant, 2-butyne-1,4-diol (B3-D) and in gas-phase hydrodehalogenation of CCl 4 . In the former, a classical example of alkyne to alkene and finally alkane hydrogenation, a catalytic behavior of Pt/PANI composites in terms of activity and selectivity was found to be similar to that observed for typical inorganic carrier supported Pt-catalysts. CCl 4 hydrodehalogenation on Pt/PANI leads initially to the formation of chloroform, which is the desired reaction product. However, in the course of reaction liberation of HCl brings about large changes, both in overall activity, as well as the selectivity, giving large amounts of dimeric C 2 products, especially C 2 Cl 6 . It is speculated that these changes follow from reprotonation of PANI which acquires Brönsted acidity used in dimerization of CCl 3 radicals. Ó 2009 Elsevier Ltd. All rights reserved. 1. Introduction Polyaniline (PANI) is one of the most extensively studied con- ductive polymer due to its environmental stability, controllable electrical conductivity, and interesting chemical properties [1,2]. The aniline polymers have general formula [(–B–NH–B–NH) Y (B–N@Q@N) 1y ] n where B represents the benzenoid ring, and Q represents the quinoid ring (Scheme 1). The oxidation state of polymer can be continuously varied from the fully reduced (y = 1) to the fully oxidized (y = 0) one and emeraldine (y = 0.5) is the most stable form of polyaniline. Recently, polyaniline with dis- persed metallic nanoparticles has received great attention for many applications including biosensors, electrochemical capaci- tors, and protective coatings against corrosion. A broad field where noble metal–polyaniline composites have been widely studied is electrocatalysis. In particular, platinum–PANI composites obtained by electrochemical procedure have been efficient catalysts in elec- trocatalytic oxidation of methanol and formic acid [3–5]. On the other hand, markedly less attention has been directed to the appli- cability of PANI as the support for heterogenous catalysts, although in recently published papers catalytic properties of Ni, Rh, Os, Pt, Pd–PANI composites have been examined [6–12]. Among noble metal, Pd-containing PANI composites have been most frequently studied. In some cases, their catalytic performances substantially differ or are better than those of their counterpart consisting of classical supports. For instance, Pd–PANI composites exhibited bet- ter activity in the removal of nitrite ions from water than Pd/Al 2 O 3 catalyst [10]. This has been related to the ion-exchange and redox properties of PANI. Pd–PANI composites have been active hetero- geneous catalysts in Suzuki coupling reactions [13], hydrogenation of nitrobenzene [14], carbonyl compounds [15], and 1-hexyne [16]. 1381-5148/$ - see front matter Ó 2009 Elsevier Ltd. All rights reserved. doi:10.1016/j.reactfunctpolym.2009.04.007 * Corresponding author. E-mail address: [email protected] (A. Drelinkiewicz). Reactive & Functional Polymers 69 (2009) 630–642 Contents lists available at ScienceDirect Reactive & Functional Polymers journal homepage: www.elsevier.com/locate/react
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Polyaniline stabilized highly dispersed Pt nanoparticles: Preparation, characterization and...

Reactive & Functional Polymers 69 (2009) 630–642
Contents lists available at ScienceDirect
Reactive & Functional Polymers
journal homepage: www.elsevier .com/ locate / react
Polyaniline stabilized highly dispersed Pt nanoparticles: Preparation,characterization and catalytic properties
A. Drelinkiewicz a,*, A. Zieba a, J.W. Sobczak b, M. Bonarowska b, Z. Karpinski b,A. Waksmundzka-Góra a, J. Stejskal c
a Institute of Catalysis and Surface Chemistry, Polish Academy of Sciences, 30-239 Kraków, Niezapominajek 8, Polandb Institute of Physical Chemistry. Polish Academy of Sciences, 01-224 Warszawa, Kasprzaka 44-52, Polandc Institute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic, Heyrovsky Sq. 2, 162 06 Prague, Czech Republic
a r t i c l e i n f o a b s t r a c t
Article history:Received 5 February 2009Received in revised form 3 April 2009Accepted 10 April 2009Available online 21 April 2009
Keywords:PolyanilinePlatinumHydrogenation2-Butyne-1,4-diolHydrodechlorinationCCl4
Selectivity to C2Cl6
1381-5148/$ - see front matter � 2009 Elsevier Ltd. Adoi:10.1016/j.reactfunctpolym.2009.04.007
* Corresponding author.E-mail address: [email protected] (A. Drelink
Platinum–polyaniline (2%Pt/PANI) composites are prepared using three precursor solutions of H2PtCl6
differing in the pH and thus in the type of dominating chloro–aqua or chloro–hydroxo complexes ofPt(IV). Various characterization techniques (X-ray diffraction, FTIR and X-ray photoelectron spectroscopy,and electron microscopy; TEM, HRTEM) were employed in order to elucidate the role of platinum precur-sor composition in the state of Pt-species formed in the Pt/PANI composites. In all three as-received Pt/PANI composites, platinum in two valence states Pt(IV) and Pt(II) was observed thus showing redox reac-tion under preparation of composites. The reduction of Pt(IV) is much easier in the precursor solution ofalkaline pH, i.e. when the Pt(IV) complexes comprising aqua and/or hydroxo ligands are the dominatingones. Under these conditions the content of Pt(II)-species is ca. 7� higher than that of Pt(IV) ones. Reduc-tion of as-prepared composites by NaBH4 gives 2%Pt/PANI catalysts of well-dispersed Pt-metal particlesof size ranging from 1 nm up to 4 nm. The average diameters (2.24 nm) of Pt-particles was slightly smal-ler in 2%Pt/PANI catalyst prepared using H2PtCl6 solution of acidic pH. Catalytic properties of Pt/PANIcomposites are tested in liquid phase hydrogenation of alkyne-type reactant, 2-butyne-1,4-diol (B3-D)and in gas-phase hydrodehalogenation of CCl4. In the former, a classical example of alkyne to alkeneand finally alkane hydrogenation, a catalytic behavior of Pt/PANI composites in terms of activity andselectivity was found to be similar to that observed for typical inorganic carrier supported Pt-catalysts.CCl4 hydrodehalogenation on Pt/PANI leads initially to the formation of chloroform, which is the desiredreaction product. However, in the course of reaction liberation of HCl brings about large changes, both inoverall activity, as well as the selectivity, giving large amounts of dimeric C2 products, especially C2Cl6. Itis speculated that these changes follow from reprotonation of PANI which acquires Brönsted acidity usedin dimerization of CCl3 radicals.
� 2009 Elsevier Ltd. All rights reserved.
1. Introduction
Polyaniline (PANI) is one of the most extensively studied con-ductive polymer due to its environmental stability, controllableelectrical conductivity, and interesting chemical properties [1,2].The aniline polymers have general formula [(–B–NH–B–NH)Y
(B–N@Q@N)1�y]n where B represents the benzenoid ring, and Qrepresents the quinoid ring (Scheme 1). The oxidation state ofpolymer can be continuously varied from the fully reduced(y = 1) to the fully oxidized (y = 0) one and emeraldine (y = 0.5) isthe most stable form of polyaniline. Recently, polyaniline with dis-persed metallic nanoparticles has received great attention formany applications including biosensors, electrochemical capaci-tors, and protective coatings against corrosion. A broad field where
ll rights reserved.
iewicz).
noble metal–polyaniline composites have been widely studied iselectrocatalysis. In particular, platinum–PANI composites obtainedby electrochemical procedure have been efficient catalysts in elec-trocatalytic oxidation of methanol and formic acid [3–5]. On theother hand, markedly less attention has been directed to the appli-cability of PANI as the support for heterogenous catalysts, althoughin recently published papers catalytic properties of Ni, Rh, Os, Pt,Pd–PANI composites have been examined [6–12]. Among noblemetal, Pd-containing PANI composites have been most frequentlystudied. In some cases, their catalytic performances substantiallydiffer or are better than those of their counterpart consisting ofclassical supports. For instance, Pd–PANI composites exhibited bet-ter activity in the removal of nitrite ions from water than Pd/Al2O3
catalyst [10]. This has been related to the ion-exchange and redoxproperties of PANI. Pd–PANI composites have been active hetero-geneous catalysts in Suzuki coupling reactions [13], hydrogenationof nitrobenzene [14], carbonyl compounds [15], and 1-hexyne [16].

Y 1-Y n
NH NH N N
Scheme 1. Schematic formula of polyaniline.
A. Drelinkiewicz et al. / Reactive & Functional Polymers 69 (2009) 630–642 631
The composites obtained by polymerization of aniline-stabilizedPd nanoparticles have been effective catalysts in Suzuki–Miyauraand Heck reactions of aryl iodides [17]. Markedly less attentionhas been directed to study the applicability of PANI as the supportfor Pt-catalysts although Pt–PANI composites obtained by electro-chemical methods are known to be very efficient electrocatalysts.The interactions in PANI–PtCl4 system (aqueous and nonaqueousconditions) have been studied by Hasik et al. [18,19]. Using spec-troscopic methods (FTIR, UV–VIS, Raman) the changes in PANIstructure i.e. protonation and partial oxidation have been identi-fied. Kinyanjui et al. [20,21] prepared Pt/PANI composites by poly-merization of aniline in the presence of K2PtCl6, as the oxidant byelectrochemical and chemical methods. Upon polymerizationPtCl2�
6 ions were completely reduced to metallic Pt which depos-ited in the form particles of relatively large diameters 0.9–1 lmin size. The Pt-particles of relatively large diameters (30–90 nm)have been also formed after spontaneous reduction of Pt2+ origi-nating from K2PtCl4 by PANI carried out in various solvents (formicacid, N-metylpyrrolidone, dimethyl sulfoxide) [22,23]. Similarly, inthe Pt–PANI composites obtained after reduction of PtCl4 by NaBH4
carried out in the presence of PANI, the Pt nanoparticles of 7–10 nm in size were formed however they strongly aggregated toform large Pt-ensembles 0.2–1.1 lm in size [11]. The activity ofthese Pt–PANI composites has been tested in isopropyl alcoholconversion [7,11]. It has been observed that their redox activitywas ca. 10� higher than the acid–base one. Quite recently, novelPt/PANI catalysts have been synthesized with Pt-particles of 1–2 nm in size [12]. The one is a PANI immobilized Adams catalystprepared by deposition–precipitation of H2PtCl6 onto PANI carriedout in basic medium. The second has been obtained by immobili-zation of a preformed platinum colloid onto PANI matrix [12].When tested in hydrogenation of citral, a,b-unsaturated aldehydethey exhibited the complete reversal of selectivity between thepreferred hydrogenation of the conjugated C@C or C@O groups.The selective hydrogenation of C@O (to form unsaturated alcohols)observed on the second catalyst has been ascribed to the participa-tion of PANI as the charge density donor.
In this work, the Pt–PANI composites are prepared by reactingPANI with aqueous solutions of hexachloroplatinic acid of variouspH and thus of various types of aquachloro-complexes of Pt(IV).The work aims to investigate the role of the type of Pt-complexesin the precursor solution on the type of Pt-species in the compositesand finally on the morphology of Pt/PANI catalysts. Present studiesare undertaken since in the case of most frequently studied palla-dium–PANI system, the state of Pd-species in palladium–polyani-line composites essentially varied depending on the type of
OHOH CH2CCCH2 OH CHCH2 CHH2
(B3-D)2-butyne-1,4-diol
(B2-D) 2-butene-1,4-d
crotyl alcohol
hydrogenolysis
OHCH2CHH3C CH
Scheme 2. Hydrogenation of
Pd-complexes and the acidity of doping solutions [14,15]. Here,catalytic properties of Pt/PANI composites are examined in thehydrogenation of 2-butyne-1,4-diol (Scheme 2) and in the hydrode-chlorination of CCl4. The first process has been chosen as it is a clas-sical example of alkyne to alkene and alkane hydrogenation andthis reaction is of industrial importance [24,25]. The second reac-tion, hydrodehalogenation reaction may be regarded as the mostpromising method for the treatment and detoxification of organo-halogen waste [26]. In contrast to commonly used chlorocarbonwaste elimination by incineration and catalytic combustion, cata-lytic hydrodechlorination is much less energy consuming processand, what is more important, allows to maintain carbon skeletonsof converted molecules, resulting in formation of useful products.Platinum catalysts, especially when supported on basic carriers likeMgO, are regarded as the most promising in converting carbon tet-rachloride to chloroform [27]. In this respect, usefulness of PANI,which also exhibits strong basic properties [28], as a support forPt-particles appeared of a great interest.
2. Experimental
2.1. Preparation of Pt/PANI composites
Powdered polyaniline in emeraldine oxidation state (y = 0.5)was prepared according to a standard procedure. Aniline was oxi-dized with ammonium peroxodisulphate in 1 M HCl solution at0 �C, followed by deprotonation of the obtained polyaniline–HClsalt in an excess of 0.5 M aqueous NH3 solution. The resulting PANIbase was washed with water and methanol and dried under re-duced pressure.
Commercially available 8 wt% aqueous solution of H2PtCl6 (Al-drich) was used as the basic reagent for the preparation of threeprecursor solutions A, B, and C of the same Pt ions concentrations(1.2 � 10�2 mol/dm3) but various pH (1.8, 5, and 10.8). The solu-tions were prepared according to the procedure reported by Sheli-mov et al. [29,30]. The most acidic solution A (pH = 1.8) wasobtained by diluting a basing 8 wt% aqueous solution of H2PtCl6
with water. In order to prepare less acidic precursor solution B(pH = 5) and alkaline solution C (pH = 10.8) under dilution of a ba-sic 8 wt% H2PtCl6, the NaOH solution (0.01 mol/dm3) was addeddrop wise to attain the appropriate pH values. The pH values ofsolutions were measured by pH-meter with a glass/reference elec-trode. Pt–polyaniline composites were prepared by treatment thepolymers with A, B, or C precursor solutions. The suspension wasstirred at room temperature for 20–30 min when all platinum ionsreacted with the polymer. The completeness of platinum sorptionwas colorimetrically checked by the reaction with KI. The appropri-ate volume of precursor solutions to obtain platinum loadings of2 wt% in final composites was used. Then, the samples were fil-tered off, washed thoroughly with water until the Cl� ions wereeliminated and dried at 80 �C for 2 h. Owing to very short time ofPANI contacting with alkaline solution no destruction of polymerappeared, evidenced by the FTIR and XPS techniques.
The reduction of as-prepared composites was performed byethanolic solution of NaBH4. The sample was suspended in ethanol
OHOH CH2 CH2 CH2 CH2HOCH2H2
iol(B1-D)
butane-1,4-diol
OH CHOCH2CH2 CH2
hydroxybutyraldehydeγ −−
isomerization
2-butyne-1,4-diol (B3-D).

632 A. Drelinkiewicz et al. / Reactive & Functional Polymers 69 (2009) 630–642
and an excess (10� higher than the stoichiometric content) of eth-anolic solution of NaBH4 was added at room temperature. Aftertermination of hydrogen liberation (ca. 1 h) the samples were fil-tered off, thoroughly washed with water and ethanol and driedat 60 �C under reduced pressure.
2.2. Methods of catalysts characterization
The state of Pt-species was characterized by X-ray diffraction(XRD), infrared (FTIR), and X-ray photoelectron (XPS) spectrosco-pies. The surface morphology of reduced samples and the Pt distri-bution were investigated by transmission (TEM) and highresolution electron microscopic (HRTEM) studies.
FTIR studies were carried out using a Bruker-Equinox 55 spec-trometer and a standard KBr pellets technique.
X-ray diffraction studies (XRD) were carried out using the HZG-4 diffractometer with CuKa radiation. XPS spectra were recordedwith the VSW spectrometer (Manchester, model 100) using MgKaradiation. Samples were pressed into pellets for XPS studies. Car-bon C 1s photoelectron peak (BE = 284.6 eV) was used for the bind-ing energy calibration. In this work the XPS studies have beenlimited mainly to the Pt 4f, N 1s, and Cl 2p regions. The other com-ponents of the samples i.e. carbon and oxygen have been also mea-sured however interpretation of those peaks seems to becomplicated and needs more experiments. TEM investigationswere carried out by means of Philips CM-20 instrument operatedat 200 kV. HRTEM analysis was performed using TECNAI G2high-resolution microscope. Samples for TEM and HRTEM studieswere prepared by placing a drop of the suspension of Pt/PANI sam-ple in ethanol onto a carbon-coated copper grid, followed by evap-orating the solvent.
To obtain the particle size distribution diagram and an accurateestimation of the average particle size (d), at least 100 particles (N)for each catalyst were manually counted. The average diameter ofPt-particles was calculated from the formula
d ¼ nidi=N were ni is the number of particles having diameter di:
2.3. Catalytic reactions
2.3.1. Hydrogenation of 2-butyne-1,4-diol (B3-D)Hydrogenation experiments were carried out in an agitated
batch glass reactor at constant atmospheric pressure of hydrogenat temperature 22 �C following the methodology previously de-scribed [31]. Water was used as the solvent. In the initial stage ofhydrogenation experiment the catalyst and the solution containinghydrogenated reagent (2-butyne-1,4-diol) were placed in the reac-tor in such a way that there was no immediate contact between thecatalyst and the solution. Nitrogen (15 min) and subsequentlyhydrogen (30 min) was passed through the reactor. Then, the cat-alyst was contacted with the hydrogenated solution and theexperiment was started. The progress of the hydrogenation wasmonitored by measuring the volume of hydrogen consumedagainst reaction time. Samples of solutions were withdrawn fromthe reactor via a sampling tube at appropriate intervals of timeand they were analysed by GC method. Products analysis was per-formed with a gas chromatograph PE Clarus 500 equipped with aflame ionisation detector in conditions: capillary column Elite-5MS (30 m � 0.25 mm � 0.25 lm coating) with helium as a carriergas (flow rate 1 ml/min) and injection temperature 250 �C. Productseparation was obtained using temperature ramp 80 �C for 1 min,15 �C/min to 220 �C, hold for 3 min. 2-Octanol was used as thestandard.
As a measure of catalyst activity the initial rate of B3-D hydro-genation expressed as a number of B3-D moles reacted per minute
and per gram of Pt in the catalyst (mol min�1 g Pt�1) is assumed.The selectivity towards B2-D is calculated at 85% B3-D conversionform the formula
S½%� ¼ number of B2-D moles formed=number of B3-D moles reacted:
Typically the hydrogenation test was carried out using 20 cm3
of B3-D solution in H2O of c0 B3-D = 0.026 mol/dm3 and catalystconcentration 5 g/dm3. The hydrogenation was carried out up tothe moment when the hydrogen uptake was ceased. Shaking ofthe reactor was carried out at such a speed to ensure that the reac-tion rate does not depend on agitation speed.
2.3.2. Hydrodechlorination of CCl4The reaction of hydrodechlorination (HdCl) of carbon tetrachlo-
ride (analytical reagent from POCh, Gliwice, Poland, purity > 99.6%)was carried out at atmospheric pressure, in a glass flow reactor.The flows of H2 and Ar (all 99.999% pure, further purified by pass-ing through MnO/SiO2 traps), were preset by using mass flow con-trollers (Bronkhorst Hi-Tec). Prior to reaction, the 2% Pt/PANIcatalyst sample (prereduced with ethanolic solution of NaHB4, Sec-tion 2.1) was reduced in flowing 10% H2/Ar (25 cm3/min). Differentreduction temperatures, from 90 to 250 �C, were used. In the reac-tion of carbon tetrachloride (provided from a saturator kept at 0 �C)with hydrogen, the mass of catalyst was �0.01 g. After reduction,the catalyst was cooled to 90 �C, then put into contact with thereaction mixture with a flow of hydrogen (�9 cm3/min) + argon(�20 cm3/min) and CCl4 (�1.3 cm3/min). The partial pressure ratioPH2/PCCl4 was �7:1. The reaction (at 90 �C) was followed by gaschromatography (HP 5890 series II with FID, a 5% Fluorcol/Carbo-pack B column (10 ft) from Supelco). After screening at 90 �C over-night and reaching a steady state, the temperature was graduallydecreased to 80 and 70 �C, and new experimental points were col-lected. Finally, the reactor was reheated to 90 �C, and, in mostcases, the previous results collected at 90 �C were restored. A typ-ical run lasted �24 h.
3. Results and discussion
3.1. Characterization of as-prepared Pt–polyaniline composites
It is well known that H2PtCl6 is a strong acid and [PtCl6]2� an-ions formed due to its dissociation are readily hydrolyzed in anaqueous medium to give a variety of chloroaqua and chlorohy-droxo platinum complexes [32]. This effect has been examinedand discussed by Shelimov et al. under studies dealing withH2PtCl6-alumina systems [29,30]. Using several spectroscopictechniques the speciation and identification of various platinumcomplexes in aqueous solutions of H2PtCl6 as a function of pH havebeen established. The 195Pt-NMR spectroscopy was found to be themost effective method as only this technique enabled the identifi-cation of various chloroaqua and chlorohydroxo Pt(IV) complexes.
The platinum precursor solutions (A, B, and C) studied in thiswork were prepared according to the procedure reported by Sheli-mov et al. [29,30] using the same concentration of Pt ions and thesame values of pH.
Data reported in [29,30] showed that in strongly acidic H2PtCl6
solution (pH = 1.8) deprotonation reaction of aqua-ligands are fullysuppressed by high concentration of protons. The chloroaqua com-plexes [PtCl6]2�, [PtCl5(H2O)]� and [PtCl4(H2O)2] co-existed in thesolution with predomination of [PtCl5(H2O)]� one. Our platinumprecursor solution A of pH = 1.8 corresponds to these conditions.In less acidic solution of pH ranges from 4.0–6.0, the hydrolysisof [PtCl6]2� anions was easier process and the [PtCl4(OH)(H2O)]�
ion was found as the major one. Our platinum precursor solution
400 600 800 1000 1200 1400 1600 1800
ν [cm-1]
abso
rban
ce
PANI
2%Pt/PANI-A
2%Pt/PANI-C
2%Pt/PANI-B
1142
1164
1497
1580
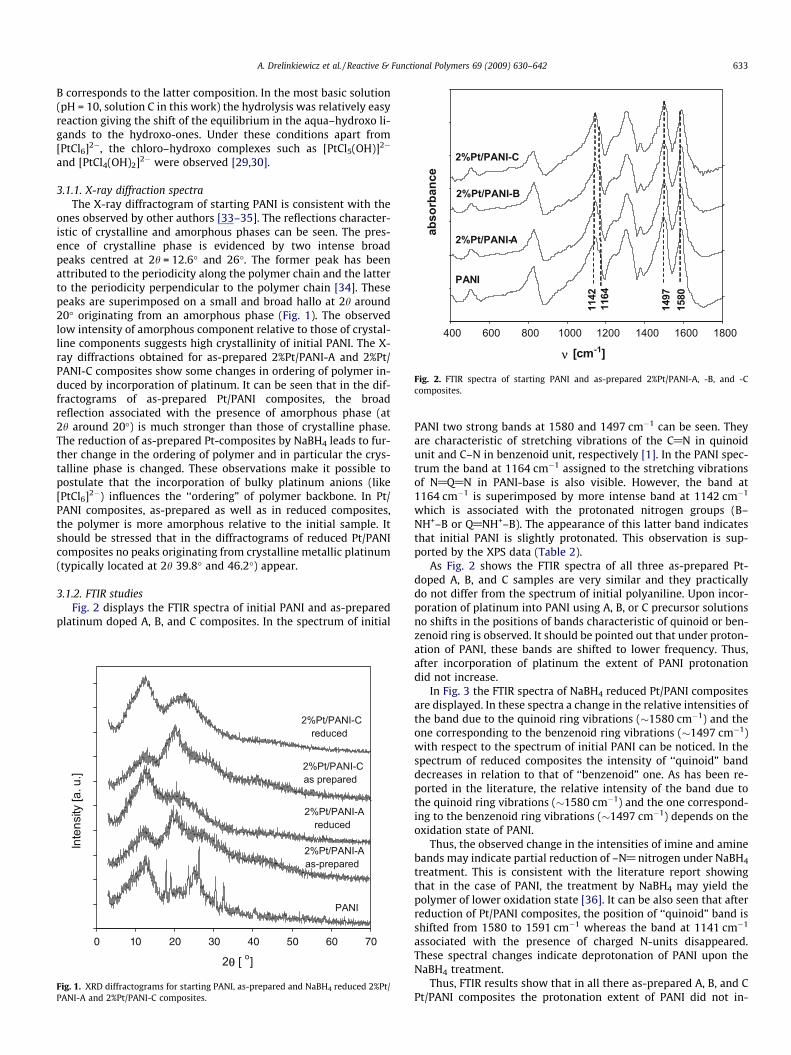
Fig. 2. FTIR spectra of starting PANI and as-prepared 2%Pt/PANI-A, -B, and -Ccomposites.
A. Drelinkiewicz et al. / Reactive & Functional Polymers 69 (2009) 630–642 633
B corresponds to the latter composition. In the most basic solution(pH = 10, solution C in this work) the hydrolysis was relatively easyreaction giving the shift of the equilibrium in the aqua–hydroxo li-gands to the hydroxo-ones. Under these conditions apart from[PtCl6]2�, the chloro–hydroxo complexes such as [PtCl5(OH)]2�
and [PtCl4(OH)2]2� were observed [29,30].
3.1.1. X-ray diffraction spectraThe X-ray diffractogram of starting PANI is consistent with the
ones observed by other authors [33–35]. The reflections character-istic of crystalline and amorphous phases can be seen. The pres-ence of crystalline phase is evidenced by two intense broadpeaks centred at 2h = 12.6� and 26�. The former peak has beenattributed to the periodicity along the polymer chain and the latterto the periodicity perpendicular to the polymer chain [34]. Thesepeaks are superimposed on a small and broad hallo at 2h around20� originating from an amorphous phase (Fig. 1). The observedlow intensity of amorphous component relative to those of crystal-line components suggests high crystallinity of initial PANI. The X-ray diffractions obtained for as-prepared 2%Pt/PANI-A and 2%Pt/PANI-C composites show some changes in ordering of polymer in-duced by incorporation of platinum. It can be seen that in the dif-fractograms of as-prepared Pt/PANI composites, the broadreflection associated with the presence of amorphous phase (at2h around 20�) is much stronger than those of crystalline phase.The reduction of as-prepared Pt-composites by NaBH4 leads to fur-ther change in the ordering of polymer and in particular the crys-talline phase is changed. These observations make it possible topostulate that the incorporation of bulky platinum anions (like[PtCl6]2�) influences the ‘‘ordering” of polymer backbone. In Pt/PANI composites, as-prepared as well as in reduced composites,the polymer is more amorphous relative to the initial sample. Itshould be stressed that in the diffractograms of reduced Pt/PANIcomposites no peaks originating from crystalline metallic platinum(typically located at 2h 39.8� and 46.2�) appear.
3.1.2. FTIR studiesFig. 2 displays the FTIR spectra of initial PANI and as-prepared
platinum doped A, B, and C composites. In the spectrum of initial
0 10 20 30 40 50 60 70
2θ [ o]
Inte
nsity
[a. u
.]
PANI
2%Pt/PANI-A as-prepared
2%Pt/PANI-A reduced
2%Pt/PANI-C as prepared
2%Pt/PANI-C reduced
Fig. 1. XRD diffractograms for starting PANI, as-prepared and NaBH4 reduced 2%Pt/PANI-A and 2%Pt/PANI-C composites.
PANI two strong bands at 1580 and 1497 cm�1 can be seen. Theyare characteristic of stretching vibrations of the C@N in quinoidunit and C–N in benzenoid unit, respectively [1]. In the PANI spec-trum the band at 1164 cm�1 assigned to the stretching vibrationsof N@Q@N in PANI-base is also visible. However, the band at1164 cm�1 is superimposed by more intense band at 1142 cm�1
which is associated with the protonated nitrogen groups (B–NH+–B or Q@NH+–B). The appearance of this latter band indicatesthat initial PANI is slightly protonated. This observation is sup-ported by the XPS data (Table 2).
As Fig. 2 shows the FTIR spectra of all three as-prepared Pt-doped A, B, and C samples are very similar and they practicallydo not differ from the spectrum of initial polyaniline. Upon incor-poration of platinum into PANI using A, B, or C precursor solutionsno shifts in the positions of bands characteristic of quinoid or ben-zenoid ring is observed. It should be pointed out that under proton-ation of PANI, these bands are shifted to lower frequency. Thus,after incorporation of platinum the extent of PANI protonationdid not increase.
In Fig. 3 the FTIR spectra of NaBH4 reduced Pt/PANI compositesare displayed. In these spectra a change in the relative intensities ofthe band due to the quinoid ring vibrations (�1580 cm�1) and theone corresponding to the benzenoid ring vibrations (�1497 cm�1)with respect to the spectrum of initial PANI can be noticed. In thespectrum of reduced composites the intensity of ‘‘quinoid” banddecreases in relation to that of ‘‘benzenoid” one. As has been re-ported in the literature, the relative intensity of the band due tothe quinoid ring vibrations (�1580 cm�1) and the one correspond-ing to the benzenoid ring vibrations (�1497 cm�1) depends on theoxidation state of PANI.
Thus, the observed change in the intensities of imine and aminebands may indicate partial reduction of –N@ nitrogen under NaBH4
treatment. This is consistent with the literature report showingthat in the case of PANI, the treatment by NaBH4 may yield thepolymer of lower oxidation state [36]. It can be also seen that afterreduction of Pt/PANI composites, the position of ‘‘quinoid” band isshifted from 1580 to 1591 cm�1 whereas the band at 1141 cm�1
associated with the presence of charged N-units disappeared.These spectral changes indicate deprotonation of PANI upon theNaBH4 treatment.
Thus, FTIR results show that in all there as-prepared A, B, and CPt/PANI composites the protonation extent of PANI did not in-
400 600 800 1000 1200 1400 1600 1800
ν [cm-1]
abso
rban
ce
2%Pt/PANI-A reduced
2%Pt/PANI-C reduced
1590
1497
1164
Fig. 3. FTIR spectra of NaBH4 reduced 2%Pt/PANI-A and 2%Pt/PANI-C composites.
80 78 76 74 72 70Binding Energy [eV]
Inte
nsity
[a.u
.]
PANI_B
PANI_C
2%Pt/PANI-B
2%Pt/PANI-C
2%Pt/PANI-A
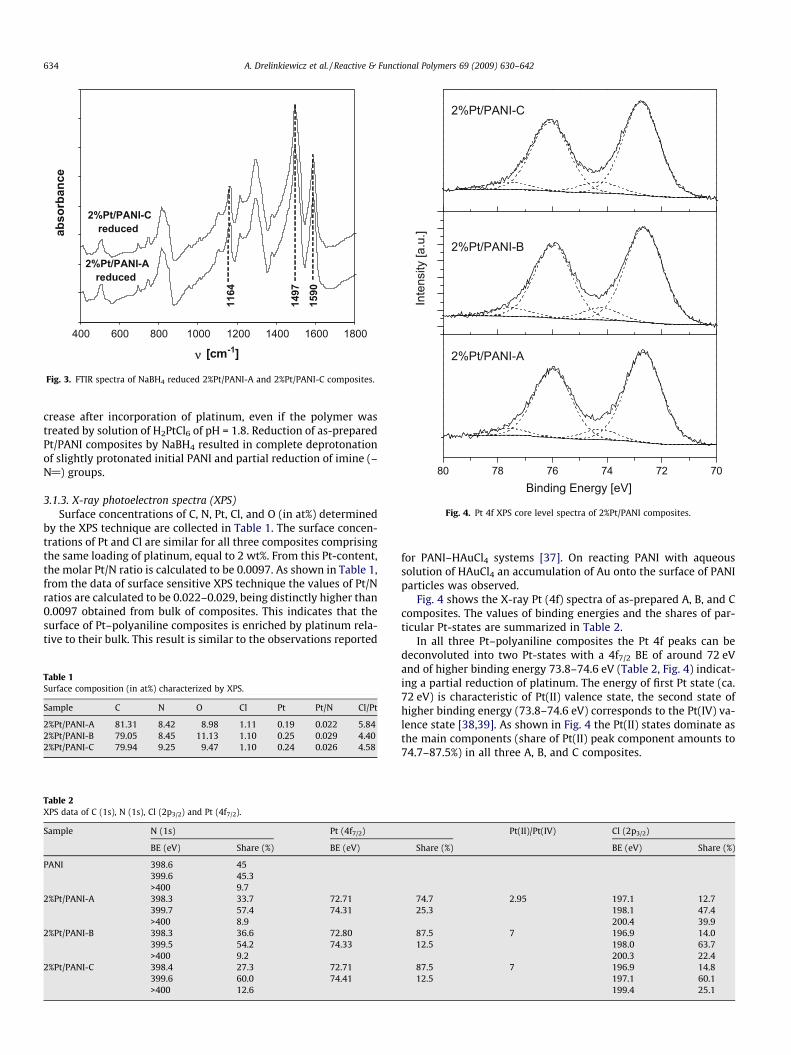
Fig. 4. Pt 4f XPS core level spectra of 2%Pt/PANI composites.
634 A. Drelinkiewicz et al. / Reactive & Functional Polymers 69 (2009) 630–642
crease after incorporation of platinum, even if the polymer wastreated by solution of H2PtCl6 of pH = 1.8. Reduction of as-preparedPt/PANI composites by NaBH4 resulted in complete deprotonationof slightly protonated initial PANI and partial reduction of imine (–N@) groups.
3.1.3. X-ray photoelectron spectra (XPS)Surface concentrations of C, N, Pt, Cl, and O (in at%) determined
by the XPS technique are collected in Table 1. The surface concen-trations of Pt and Cl are similar for all three composites comprisingthe same loading of platinum, equal to 2 wt%. From this Pt-content,the molar Pt/N ratio is calculated to be 0.0097. As shown in Table 1,from the data of surface sensitive XPS technique the values of Pt/Nratios are calculated to be 0.022–0.029, being distinctly higher than0.0097 obtained from bulk of composites. This indicates that thesurface of Pt–polyaniline composites is enriched by platinum rela-tive to their bulk. This result is similar to the observations reported
Table 1Surface composition (in at%) characterized by XPS.
Sample C N O Cl Pt Pt/N Cl/Pt
2%Pt/PANI-A 81.31 8.42 8.98 1.11 0.19 0.022 5.842%Pt/PANI-B 79.05 8.45 11.13 1.10 0.25 0.029 4.402%Pt/PANI-C 79.94 9.25 9.47 1.10 0.24 0.026 4.58
Table 2XPS data of C (1s), N (1s), Cl (2p3/2) and Pt (4f7/2).
Sample N (1s) Pt (4f7/2)
BE (eV) Share (%) BE (eV)
PANI 398.6 45399.6 45.3>400 9.7
2%Pt/PANI-A 398.3 33.7 72.71399.7 57.4 74.31>400 8.9
2%Pt/PANI-B 398.3 36.6 72.80399.5 54.2 74.33>400 9.2
2%Pt/PANI-C 398.4 27.3 72.71399.6 60.0 74.41>400 12.6
for PANI–HAuCl4 systems [37]. On reacting PANI with aqueoussolution of HAuCl4 an accumulation of Au onto the surface of PANIparticles was observed.
Fig. 4 shows the X-ray Pt (4f) spectra of as-prepared A, B, and Ccomposites. The values of binding energies and the shares of par-ticular Pt-states are summarized in Table 2.
In all three Pt–polyaniline composites the Pt 4f peaks can bedeconvoluted into two Pt-states with a 4f7/2 BE of around 72 eVand of higher binding energy 73.8–74.6 eV (Table 2, Fig. 4) indicat-ing a partial reduction of platinum. The energy of first Pt state (ca.72 eV) is characteristic of Pt(II) valence state, the second state ofhigher binding energy (73.8–74.6 eV) corresponds to the Pt(IV) va-lence state [38,39]. As shown in Fig. 4 the Pt(II) states dominate asthe main components (share of Pt(II) peak component amounts to74.7–87.5%) in all three A, B, and C composites.
Pt(II)/Pt(IV) Cl (2p3/2)
Share (%) BE (eV) Share (%)
74.7 2.95 197.1 12.725.3 198.1 47.4
200.4 39.987.5 7 196.9 14.012.5 198.0 63.7
200.3 22.487.5 7 196.9 14.812.5 197.1 60.1
199.4 25.1
404 402 400 398 396
Binding energy [eV]
Inte
nsity
[a.u
.]
PANI
2%Pt/PANI-A
2%Pt/PANI-B
2%Pt/PANI-C
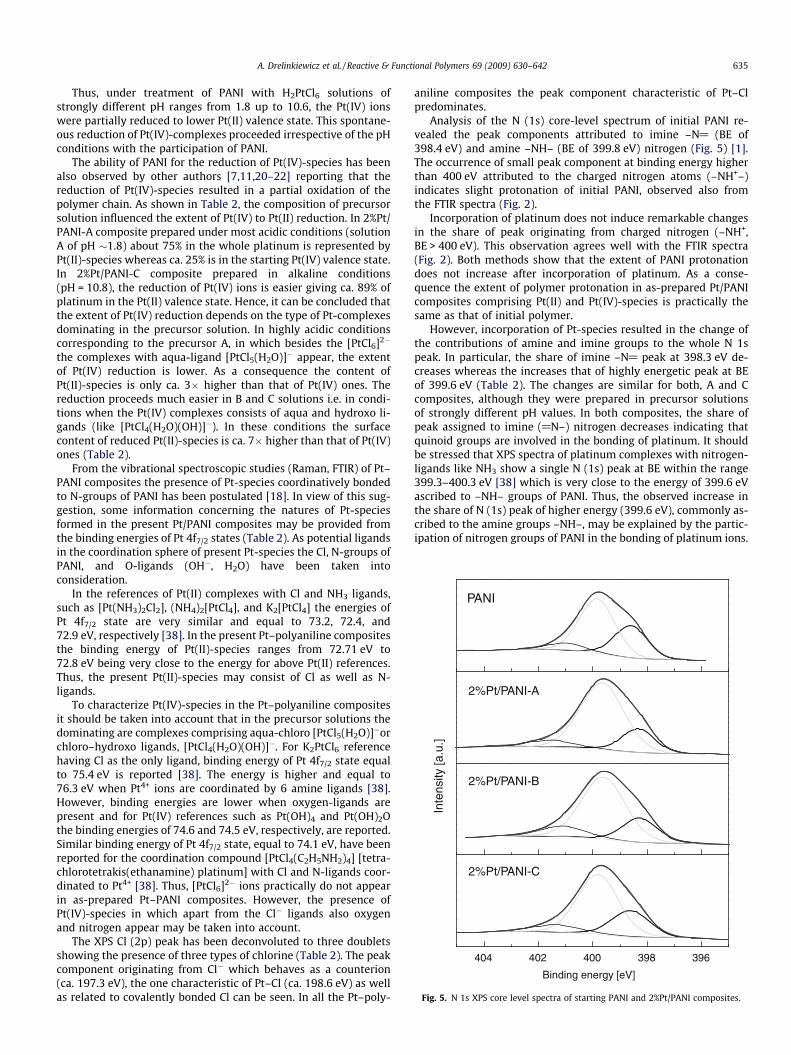
Fig. 5. N 1s XPS core level spectra of starting PANI and 2%Pt/PANI composites.
A. Drelinkiewicz et al. / Reactive & Functional Polymers 69 (2009) 630–642 635
Thus, under treatment of PANI with H2PtCl6 solutions ofstrongly different pH ranges from 1.8 up to 10.6, the Pt(IV) ionswere partially reduced to lower Pt(II) valence state. This spontane-ous reduction of Pt(IV)-complexes proceeded irrespective of the pHconditions with the participation of PANI.
The ability of PANI for the reduction of Pt(IV)-species has beenalso observed by other authors [7,11,20–22] reporting that thereduction of Pt(IV)-species resulted in a partial oxidation of thepolymer chain. As shown in Table 2, the composition of precursorsolution influenced the extent of Pt(IV) to Pt(II) reduction. In 2%Pt/PANI-A composite prepared under most acidic conditions (solutionA of pH �1.8) about 75% in the whole platinum is represented byPt(II)-species whereas ca. 25% is in the starting Pt(IV) valence state.In 2%Pt/PANI-C composite prepared in alkaline conditions(pH = 10.8), the reduction of Pt(IV) ions is easier giving ca. 89% ofplatinum in the Pt(II) valence state. Hence, it can be concluded thatthe extent of Pt(IV) reduction depends on the type of Pt-complexesdominating in the precursor solution. In highly acidic conditionscorresponding to the precursor A, in which besides the [PtCl6]2�
the complexes with aqua-ligand [PtCl5(H2O)]� appear, the extentof Pt(IV) reduction is lower. As a consequence the content ofPt(II)-species is only ca. 3� higher than that of Pt(IV) ones. Thereduction proceeds much easier in B and C solutions i.e. in condi-tions when the Pt(IV) complexes consists of aqua and hydroxo li-gands (like [PtCl4(H2O)(OH)]�). In these conditions the surfacecontent of reduced Pt(II)-species is ca. 7� higher than that of Pt(IV)ones (Table 2).
From the vibrational spectroscopic studies (Raman, FTIR) of Pt–PANI composites the presence of Pt-species coordinatively bondedto N-groups of PANI has been postulated [18]. In view of this sug-gestion, some information concerning the natures of Pt-speciesformed in the present Pt/PANI composites may be provided fromthe binding energies of Pt 4f7/2 states (Table 2). As potential ligandsin the coordination sphere of present Pt-species the Cl, N-groups ofPANI, and O-ligands (OH�, H2O) have been taken intoconsideration.
In the references of Pt(II) complexes with Cl and NH3 ligands,such as [Pt(NH3)2Cl2], (NH4)2[PtCl4], and K2[PtCl4] the energies ofPt 4f7/2 state are very similar and equal to 73.2, 72.4, and72.9 eV, respectively [38]. In the present Pt–polyaniline compositesthe binding energy of Pt(II)-species ranges from 72.71 eV to72.8 eV being very close to the energy for above Pt(II) references.Thus, the present Pt(II)-species may consist of Cl as well as N-ligands.
To characterize Pt(IV)-species in the Pt–polyaniline compositesit should be taken into account that in the precursor solutions thedominating are complexes comprising aqua-chloro [PtCl5(H2O)]�orchloro–hydroxo ligands, [PtCl4(H2O)(OH)]�. For K2PtCl6 referencehaving Cl as the only ligand, binding energy of Pt 4f7/2 state equalto 75.4 eV is reported [38]. The energy is higher and equal to76.3 eV when Pt4+ ions are coordinated by 6 amine ligands [38].However, binding energies are lower when oxygen-ligands arepresent and for Pt(IV) references such as Pt(OH)4 and Pt(OH)2Othe binding energies of 74.6 and 74.5 eV, respectively, are reported.Similar binding energy of Pt 4f7/2 state, equal to 74.1 eV, have beenreported for the coordination compound [PtCl4(C2H5NH2)4] [tetra-chlorotetrakis(ethanamine) platinum] with Cl and N-ligands coor-dinated to Pt4+ [38]. Thus, [PtCl6]2� ions practically do not appearin as-prepared Pt–PANI composites. However, the presence ofPt(IV)-species in which apart from the Cl� ligands also oxygenand nitrogen appear may be taken into account.
The XPS Cl (2p) peak has been deconvoluted to three doubletsshowing the presence of three types of chlorine (Table 2). The peakcomponent originating from Cl� which behaves as a counterion(ca. 197.3 eV), the one characteristic of Pt–Cl (ca. 198.6 eV) as wellas related to covalently bonded Cl can be seen. In all the Pt–poly-
aniline composites the peak component characteristic of Pt–Clpredominates.
Analysis of the N (1s) core-level spectrum of initial PANI re-vealed the peak components attributed to imine –N@ (BE of398.4 eV) and amine –NH– (BE of 399.8 eV) nitrogen (Fig. 5) [1].The occurrence of small peak component at binding energy higherthan 400 eV attributed to the charged nitrogen atoms (–NH+–)indicates slight protonation of initial PANI, observed also fromthe FTIR spectra (Fig. 2).
Incorporation of platinum does not induce remarkable changesin the share of peak originating from charged nitrogen (–NH+,BE > 400 eV). This observation agrees well with the FTIR spectra(Fig. 2). Both methods show that the extent of PANI protonationdoes not increase after incorporation of platinum. As a conse-quence the extent of polymer protonation in as-prepared Pt/PANIcomposites comprising Pt(II) and Pt(IV)-species is practically thesame as that of initial polymer.
However, incorporation of Pt-species resulted in the change ofthe contributions of amine and imine groups to the whole N 1speak. In particular, the share of imine –N@ peak at 398.3 eV de-creases whereas the increases that of highly energetic peak at BEof 399.6 eV (Table 2). The changes are similar for both, A and Ccomposites, although they were prepared in precursor solutionsof strongly different pH values. In both composites, the share ofpeak assigned to imine (@N–) nitrogen decreases indicating thatquinoid groups are involved in the bonding of platinum. It shouldbe stressed that XPS spectra of platinum complexes with nitrogen-ligands like NH3 show a single N (1s) peak at BE within the range399.3–400.3 eV [38] which is very close to the energy of 399.6 eVascribed to –NH– groups of PANI. Thus, the observed increase inthe share of N (1s) peak of higher energy (399.6 eV), commonly as-cribed to the amine groups –NH–, may be explained by the partic-ipation of nitrogen groups of PANI in the bonding of platinum ions.
636 A. Drelinkiewicz et al. / Reactive & Functional Polymers 69 (2009) 630–642
In conclusion, from the obtained results very high ability ofPANI for reduction of Pt(IV) valence state to lower Pt(II) one is ob-served. This ability depends on the type of Pt(IV)-species in theprecursor solution being higher in the solution of alkaline charac-ter, i.e. when Pt(IV) complexes with hydroxo ligand are present inthe precursor solution. Upon treatment of PANI by high acidicH2PtCl6 solution (i.e. solution A of pH = 1.8) one can expect proton-ation of PANI nitrogen accompanied by the incorporation of[PtCl6]2� in the form of counterions. However, in view of FTIRand XPS data, the extent of PANI protonation practically does notincrease in the Pt/PANI-A composites. It can not be excluded thatplatinum (IV) complexes are introduced as counterions via the pro-tonation reaction, however due to high reductive ability of PANIwith respect to the Pt(IV)-species, they are easily reduced to thePt(II)-ones and the introduced platinum species consist of Cl andO/N ligands.
It should be pointed out that above conclusions are well sup-ported by the results of EXAFS analysis for 2%Pt/PANI compositeswhich were synthesized according to the same procedure as thatused in this study. From the EXAFS analysis the type of neighboratoms around the Pt as well as their coordination numbers weredetermined. However, owing to high complexity of this technique,the data obtained by EXAFS method will be separately published.They showed that first coordination sphere of platinum was formedby Cl atoms and N/O ones in the proportion near to 1:1. Thus, EXAFSdata support the XPS results that platinum species formed in as-prepared Pt/PANI composites consist of Cl and N/O ligands.
3.1.4. Electron microscopic studies (TEM and HRTEM)Since no remarkable differences in properties of A, B, and C
composites were observed by FTIR and XPS techniques, micro-scopic studies were performed for NaBH4 reduced 2%Pt/PANI-A
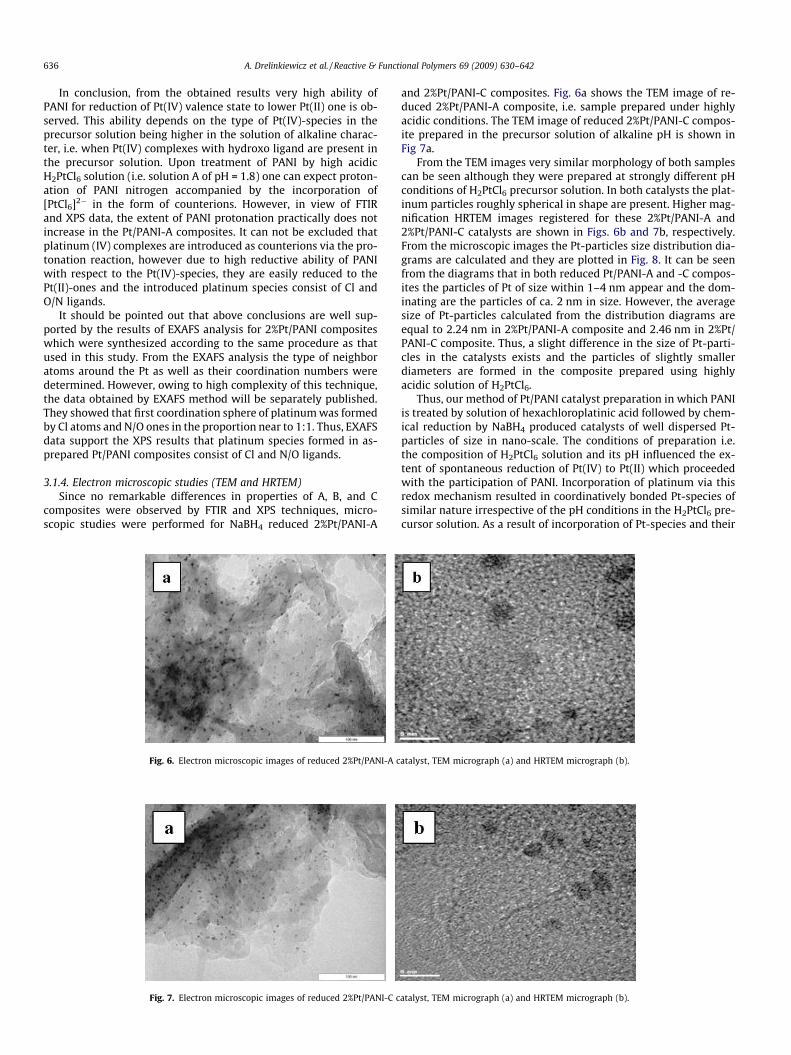
Fig. 6. Electron microscopic images of reduced 2%Pt/PANI-A c
Fig. 7. Electron microscopic images of reduced 2%Pt/PANI-C c
and 2%Pt/PANI-C composites. Fig. 6a shows the TEM image of re-duced 2%Pt/PANI-A composite, i.e. sample prepared under highlyacidic conditions. The TEM image of reduced 2%Pt/PANI-C compos-ite prepared in the precursor solution of alkaline pH is shown inFig 7a.
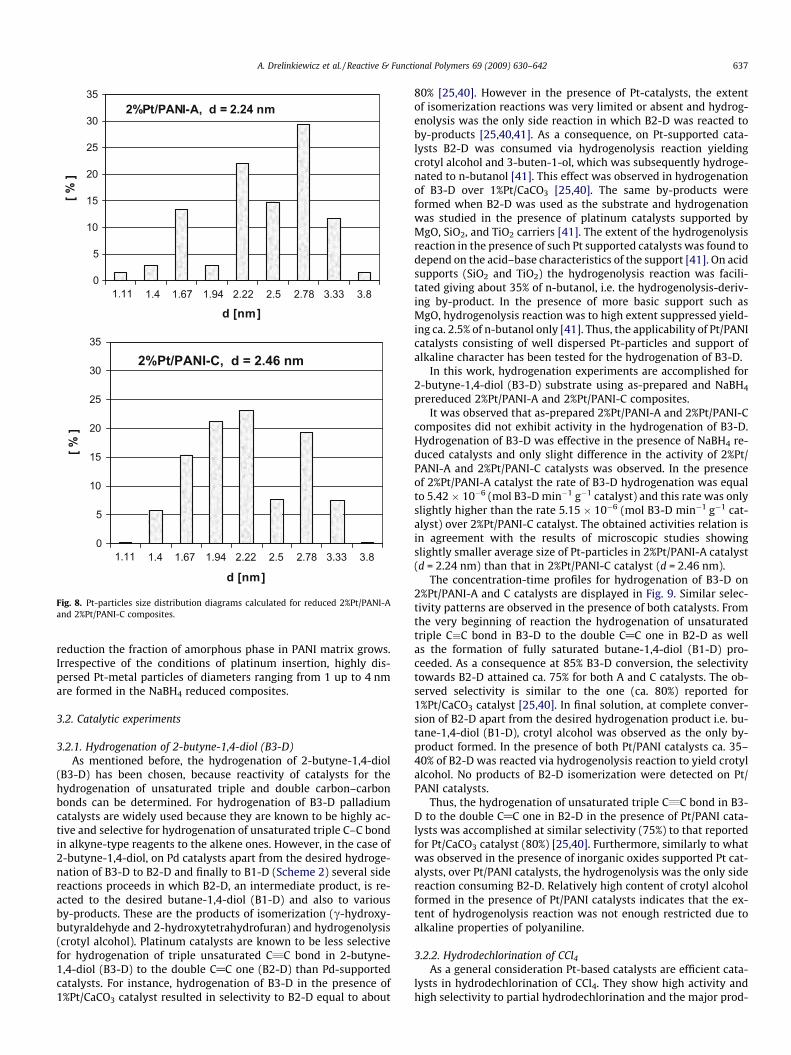
From the TEM images very similar morphology of both samplescan be seen although they were prepared at strongly different pHconditions of H2PtCl6 precursor solution. In both catalysts the plat-inum particles roughly spherical in shape are present. Higher mag-nification HRTEM images registered for these 2%Pt/PANI-A and2%Pt/PANI-C catalysts are shown in Figs. 6b and 7b, respectively.From the microscopic images the Pt-particles size distribution dia-grams are calculated and they are plotted in Fig. 8. It can be seenfrom the diagrams that in both reduced Pt/PANI-A and -C compos-ites the particles of Pt of size within 1–4 nm appear and the dom-inating are the particles of ca. 2 nm in size. However, the averagesize of Pt-particles calculated from the distribution diagrams areequal to 2.24 nm in 2%Pt/PANI-A composite and 2.46 nm in 2%Pt/PANI-C composite. Thus, a slight difference in the size of Pt-parti-cles in the catalysts exists and the particles of slightly smallerdiameters are formed in the composite prepared using highlyacidic solution of H2PtCl6.
Thus, our method of Pt/PANI catalyst preparation in which PANIis treated by solution of hexachloroplatinic acid followed by chem-ical reduction by NaBH4 produced catalysts of well dispersed Pt-particles of size in nano-scale. The conditions of preparation i.e.the composition of H2PtCl6 solution and its pH influenced the ex-tent of spontaneous reduction of Pt(IV) to Pt(II) which proceededwith the participation of PANI. Incorporation of platinum via thisredox mechanism resulted in coordinatively bonded Pt-species ofsimilar nature irrespective of the pH conditions in the H2PtCl6 pre-cursor solution. As a result of incorporation of Pt-species and their
atalyst, TEM micrograph (a) and HRTEM micrograph (b).
atalyst, TEM micrograph (a) and HRTEM micrograph (b).
0
5
10
15
20
25
30
35
d [nm]
[ % ]
2%Pt/PANI-A, d = 2.24 nm
0
5
10
15
20
25
30
35
1.11 1.4 1.67 1.94 2.22 2.5 2.78 3.33 3.8
1.11 1.4 1.67 1.94 2.22 2.5 2.78 3.33 3.8
d [nm]
[ % ]
2%Pt/PANI-C, d = 2.46 nm
Fig. 8. Pt-particles size distribution diagrams calculated for reduced 2%Pt/PANI-Aand 2%Pt/PANI-C composites.
A. Drelinkiewicz et al. / Reactive & Functional Polymers 69 (2009) 630–642 637
reduction the fraction of amorphous phase in PANI matrix grows.Irrespective of the conditions of platinum insertion, highly dis-persed Pt-metal particles of diameters ranging from 1 up to 4 nmare formed in the NaBH4 reduced composites.
3.2. Catalytic experiments
3.2.1. Hydrogenation of 2-butyne-1,4-diol (B3-D)As mentioned before, the hydrogenation of 2-butyne-1,4-diol
(B3-D) has been chosen, because reactivity of catalysts for thehydrogenation of unsaturated triple and double carbon–carbonbonds can be determined. For hydrogenation of B3-D palladiumcatalysts are widely used because they are known to be highly ac-tive and selective for hydrogenation of unsaturated triple C–C bondin alkyne-type reagents to the alkene ones. However, in the case of2-butyne-1,4-diol, on Pd catalysts apart from the desired hydroge-nation of B3-D to B2-D and finally to B1-D (Scheme 2) several sidereactions proceeds in which B2-D, an intermediate product, is re-acted to the desired butane-1,4-diol (B1-D) and also to variousby-products. These are the products of isomerization (c-hydroxy-butyraldehyde and 2-hydroxytetrahydrofuran) and hydrogenolysis(crotyl alcohol). Platinum catalysts are known to be less selectivefor hydrogenation of triple unsaturated C„C bond in 2-butyne-1,4-diol (B3-D) to the double C@C one (B2-D) than Pd-supportedcatalysts. For instance, hydrogenation of B3-D in the presence of1%Pt/CaCO3 catalyst resulted in selectivity to B2-D equal to about
80% [25,40]. However in the presence of Pt-catalysts, the extentof isomerization reactions was very limited or absent and hydrog-enolysis was the only side reaction in which B2-D was reacted toby-products [25,40,41]. As a consequence, on Pt-supported cata-lysts B2-D was consumed via hydrogenolysis reaction yieldingcrotyl alcohol and 3-buten-1-ol, which was subsequently hydroge-nated to n-butanol [41]. This effect was observed in hydrogenationof B3-D over 1%Pt/CaCO3 [25,40]. The same by-products wereformed when B2-D was used as the substrate and hydrogenationwas studied in the presence of platinum catalysts supported byMgO, SiO2, and TiO2 carriers [41]. The extent of the hydrogenolysisreaction in the presence of such Pt supported catalysts was found todepend on the acid–base characteristics of the support [41]. On acidsupports (SiO2 and TiO2) the hydrogenolysis reaction was facili-tated giving about 35% of n-butanol, i.e. the hydrogenolysis-deriv-ing by-product. In the presence of more basic support such asMgO, hydrogenolysis reaction was to high extent suppressed yield-ing ca. 2.5% of n-butanol only [41]. Thus, the applicability of Pt/PANIcatalysts consisting of well dispersed Pt-particles and support ofalkaline character has been tested for the hydrogenation of B3-D.
In this work, hydrogenation experiments are accomplished for2-butyne-1,4-diol (B3-D) substrate using as-prepared and NaBH4
prereduced 2%Pt/PANI-A and 2%Pt/PANI-C composites.It was observed that as-prepared 2%Pt/PANI-A and 2%Pt/PANI-C
composites did not exhibit activity in the hydrogenation of B3-D.Hydrogenation of B3-D was effective in the presence of NaBH4 re-duced catalysts and only slight difference in the activity of 2%Pt/PANI-A and 2%Pt/PANI-C catalysts was observed. In the presenceof 2%Pt/PANI-A catalyst the rate of B3-D hydrogenation was equalto 5.42 � 10�6 (mol B3-D min�1 g�1 catalyst) and this rate was onlyslightly higher than the rate 5.15 � 10�6 (mol B3-D min�1 g�1 cat-alyst) over 2%Pt/PANI-C catalyst. The obtained activities relation isin agreement with the results of microscopic studies showingslightly smaller average size of Pt-particles in 2%Pt/PANI-A catalyst(d = 2.24 nm) than that in 2%Pt/PANI-C catalyst (d = 2.46 nm).
The concentration-time profiles for hydrogenation of B3-D on2%Pt/PANI-A and C catalysts are displayed in Fig. 9. Similar selec-tivity patterns are observed in the presence of both catalysts. Fromthe very beginning of reaction the hydrogenation of unsaturatedtriple C�C bond in B3-D to the double C@C one in B2-D as wellas the formation of fully saturated butane-1,4-diol (B1-D) pro-ceeded. As a consequence at 85% B3-D conversion, the selectivitytowards B2-D attained ca. 75% for both A and C catalysts. The ob-served selectivity is similar to the one (ca. 80%) reported for1%Pt/CaCO3 catalyst [25,40]. In final solution, at complete conver-sion of B2-D apart from the desired hydrogenation product i.e. bu-tane-1,4-diol (B1-D), crotyl alcohol was observed as the only by-product formed. In the presence of both Pt/PANI catalysts ca. 35–40% of B2-D was reacted via hydrogenolysis reaction to yield crotylalcohol. No products of B2-D isomerization were detected on Pt/PANI catalysts.
Thus, the hydrogenation of unsaturated triple C„C bond in B3-D to the double C@C one in B2-D in the presence of Pt/PANI cata-lysts was accomplished at similar selectivity (75%) to that reportedfor Pt/CaCO3 catalyst (80%) [25,40]. Furthermore, similarly to whatwas observed in the presence of inorganic oxides supported Pt cat-alysts, over Pt/PANI catalysts, the hydrogenolysis was the only sidereaction consuming B2-D. Relatively high content of crotyl alcoholformed in the presence of Pt/PANI catalysts indicates that the ex-tent of hydrogenolysis reaction was not enough restricted due toalkaline properties of polyaniline.
3.2.2. Hydrodechlorination of CCl4As a general consideration Pt-based catalysts are efficient cata-
lysts in hydrodechlorination of CCl4. They show high activity andhigh selectivity to partial hydrodechlorination and the major prod-
30 32 34 36 38 40 42 440
500
1000
1500
2000
2500
3000
reduced at 295oC
Inte
nsi
ty, a
.u.
2 theta (CuKα), deg.
reduced at 250oC
reduced at 150oC
reduced at 90oC
Pt (111)
Pt crystallites ~3 nm
Pt crystallites ~5 nm
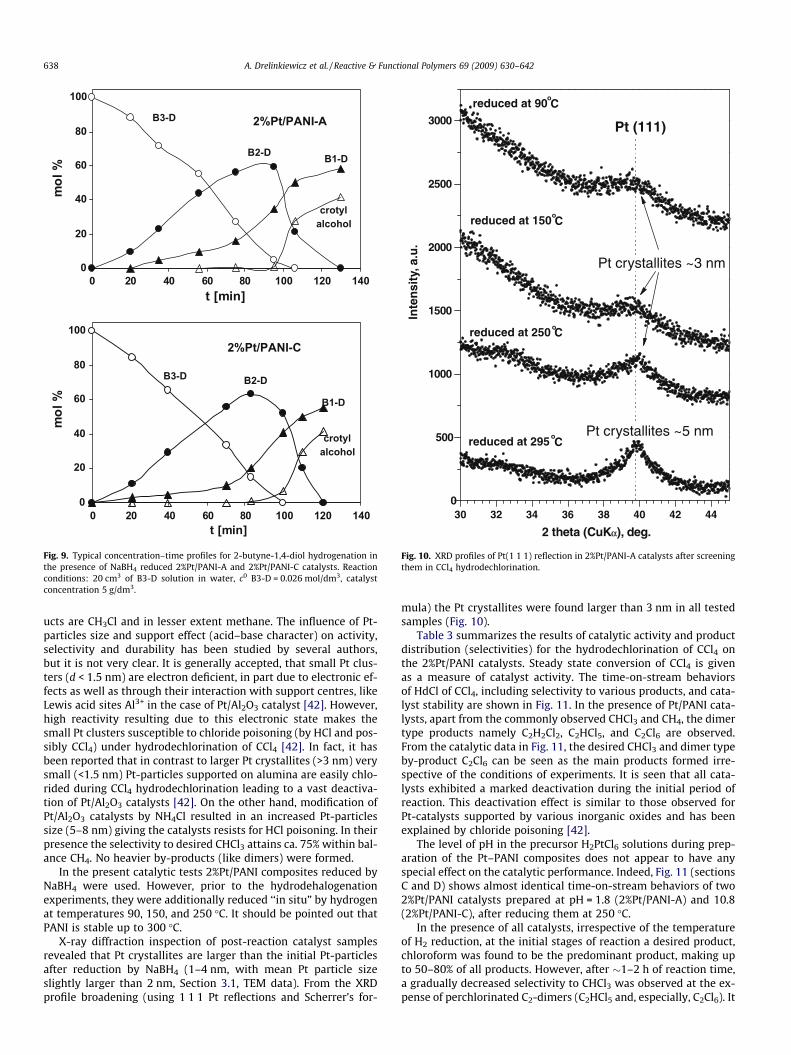
Fig. 10. XRD profiles of Pt(1 1 1) reflection in 2%Pt/PANI-A catalysts after screeningthem in CCl4 hydrodechlorination.
0
20
40
60
80
100
0 20 40 60 80 100 120 140
0 20 40 60 80 100 120 140
t [min]
mol
%
2%Pt/PANI-AB3-D
B2-D B1-D
crotylalcohol
0
20
40
60
80
100
t [min]
mol
%
2%Pt/PANI-C
B3-D B2-D
B1-D
crotylalcohol
Fig. 9. Typical concentration–time profiles for 2-butyne-1,4-diol hydrogenation inthe presence of NaBH4 reduced 2%Pt/PANI-A and 2%Pt/PANI-C catalysts. Reactionconditions: 20 cm3 of B3-D solution in water, c0 B3-D = 0.026 mol/dm3, catalystconcentration 5 g/dm3.
638 A. Drelinkiewicz et al. / Reactive & Functional Polymers 69 (2009) 630–642
ucts are CH3Cl and in lesser extent methane. The influence of Pt-particles size and support effect (acid–base character) on activity,selectivity and durability has been studied by several authors,but it is not very clear. It is generally accepted, that small Pt clus-ters (d < 1.5 nm) are electron deficient, in part due to electronic ef-fects as well as through their interaction with support centres, likeLewis acid sites Al3+ in the case of Pt/Al2O3 catalyst [42]. However,high reactivity resulting due to this electronic state makes thesmall Pt clusters susceptible to chloride poisoning (by HCl and pos-sibly CCl4) under hydrodechlorination of CCl4 [42]. In fact, it hasbeen reported that in contrast to larger Pt crystallites (>3 nm) verysmall (<1.5 nm) Pt-particles supported on alumina are easily chlo-rided during CCl4 hydrodechlorination leading to a vast deactiva-tion of Pt/Al2O3 catalysts [42]. On the other hand, modification ofPt/Al2O3 catalysts by NH4Cl resulted in an increased Pt-particlessize (5–8 nm) giving the catalysts resists for HCl poisoning. In theirpresence the selectivity to desired CHCl3 attains ca. 75% within bal-ance CH4. No heavier by-products (like dimers) were formed.
In the present catalytic tests 2%Pt/PANI composites reduced byNaBH4 were used. However, prior to the hydrodehalogenationexperiments, they were additionally reduced ‘‘in situ” by hydrogenat temperatures 90, 150, and 250 �C. It should be pointed out thatPANI is stable up to 300 �C.
X-ray diffraction inspection of post-reaction catalyst samplesrevealed that Pt crystallites are larger than the initial Pt-particlesafter reduction by NaBH4 (1–4 nm, with mean Pt particle sizeslightly larger than 2 nm, Section 3.1, TEM data). From the XRDprofile broadening (using 1 1 1 Pt reflections and Scherrer’s for-
mula) the Pt crystallites were found larger than 3 nm in all testedsamples (Fig. 10).
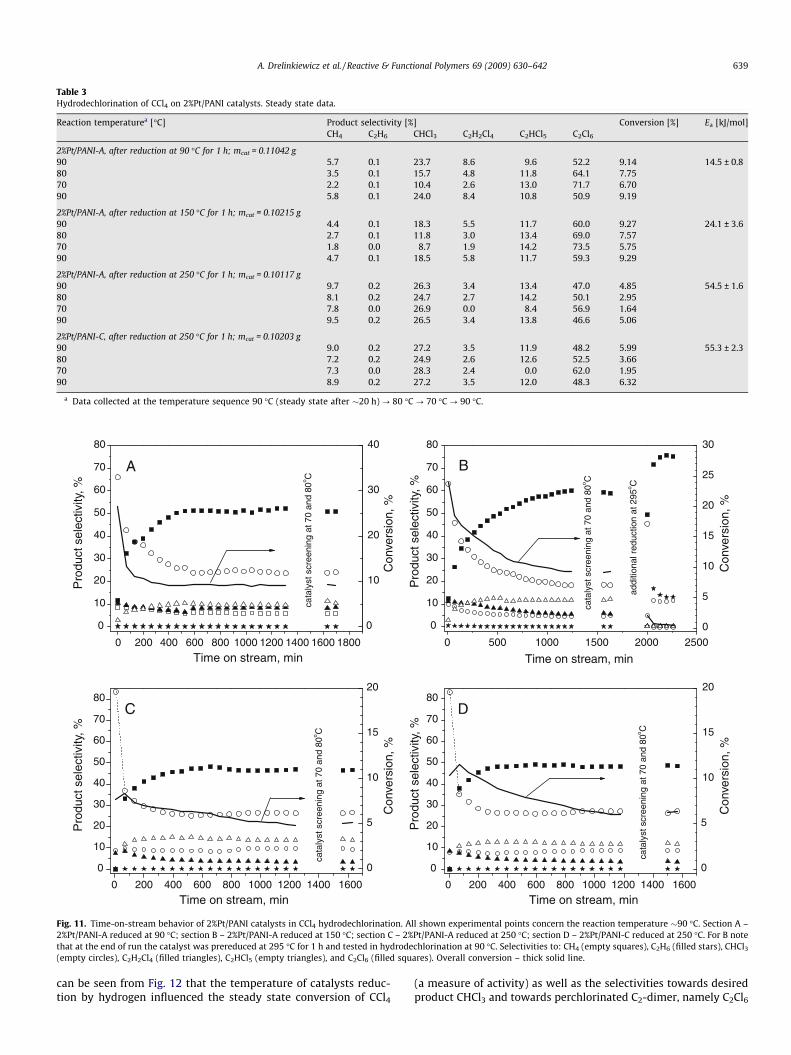
Table 3 summarizes the results of catalytic activity and productdistribution (selectivities) for the hydrodechlorination of CCl4 onthe 2%Pt/PANI catalysts. Steady state conversion of CCl4 is givenas a measure of catalyst activity. The time-on-stream behaviorsof HdCl of CCl4, including selectivity to various products, and cata-lyst stability are shown in Fig. 11. In the presence of Pt/PANI cata-lysts, apart from the commonly observed CHCl3 and CH4, the dimertype products namely C2H2Cl2, C2HCl5, and C2Cl6 are observed.From the catalytic data in Fig. 11, the desired CHCl3 and dimer typeby-product C2Cl6 can be seen as the main products formed irre-spective of the conditions of experiments. It is seen that all cata-lysts exhibited a marked deactivation during the initial period ofreaction. This deactivation effect is similar to those observed forPt-catalysts supported by various inorganic oxides and has beenexplained by chloride poisoning [42].
The level of pH in the precursor H2PtCl6 solutions during prep-aration of the Pt–PANI composites does not appear to have anyspecial effect on the catalytic performance. Indeed, Fig. 11 (sectionsC and D) shows almost identical time-on-stream behaviors of two2%Pt/PANI catalysts prepared at pH = 1.8 (2%Pt/PANI-A) and 10.8(2%Pt/PANI-C), after reducing them at 250 �C.
In the presence of all catalysts, irrespective of the temperatureof H2 reduction, at the initial stages of reaction a desired product,chloroform was found to be the predominant product, making upto 50–80% of all products. However, after �1–2 h of reaction time,a gradually decreased selectivity to CHCl3 was observed at the ex-pense of perchlorinated C2-dimers (C2HCl5 and, especially, C2Cl6). It
0 200 400 600 800 1000 1200 1400 1600 1800
0
10
20
30
40
50
60
70
80
0
10
20
30
40
cata
lyst
scr
eeni
ng a
t 70
and
80o C
Con
vers
ion,
%
Pro
duct
sel
ectiv
ity, %
Time on stream, min
A
0 200 400 600 800 1000 1200 1400 1600 0 200 400 600 800 1000 1200 1400 1600
0
10
20
30
40
50
60
70
80
0
5
10
15
20
cata
lyst
scr
eeni
ng a
t 70
and
80o C
Pro
duct
sel
ectiv
ity, %
Time on stream, min
Con
vers
ion,
%
C
0 500 1000 1500 2000 2500
0
10
20
30
40
50
60
70
80
0
5
10
15
20
25
30
cata
lyst
scr
eeni
ng a
t 70
and
80o C
Pro
duct
sel
ectiv
ity, %
Time on stream, min
addi
tiona
l red
uctio
n at
295
o C
Con
vers
ion,
%
B
0
10
20
30
40
50
60
70
80
0
5
10
15
20
cata
lyst
scr
eeni
ng a
t 70
and
80o C
Pro
duct
sel
ectiv
ity, %
Time on stream, min
Con
vers
ion,
%D
Fig. 11. Time-on-stream behavior of 2%Pt/PANI catalysts in CCl4 hydrodechlorination. All shown experimental points concern the reaction temperature �90 �C. Section A –2%Pt/PANI-A reduced at 90 �C; section B – 2%Pt/PANI-A reduced at 150 �C; section C – 2%Pt/PANI-A reduced at 250 �C; section D – 2%Pt/PANI-C reduced at 250 �C. For B notethat at the end of run the catalyst was prereduced at 295 �C for 1 h and tested in hydrodechlorination at 90 �C. Selectivities to: CH4 (empty squares), C2H6 (filled stars), CHCl3
(empty circles), C2H2Cl4 (filled triangles), C2HCl5 (empty triangles), and C2Cl6 (filled squares). Overall conversion – thick solid line.
Table 3Hydrodechlorination of CCl4 on 2%Pt/PANI catalysts. Steady state data.
Reaction temperaturea [�C] Product selectivity [%] Conversion [%] Ea [kJ/mol]CH4 C2H6 CHCl3 C2H2Cl4 C2HCl5 C2Cl6
2%Pt/PANI-A, after reduction at 90 �C for 1 h; mcat = 0.11042 g90 5.7 0.1 23.7 8.6 9.6 52.2 9.14 14.5 ± 0.880 3.5 0.1 15.7 4.8 11.8 64.1 7.7570 2.2 0.1 10.4 2.6 13.0 71.7 6.7090 5.8 0.1 24.0 8.4 10.8 50.9 9.19
2%Pt/PANI-A, after reduction at 150 �C for 1 h; mcat = 0.10215 g90 4.4 0.1 18.3 5.5 11.7 60.0 9.27 24.1 ± 3.680 2.7 0.1 11.8 3.0 13.4 69.0 7.5770 1.8 0.0 8.7 1.9 14.2 73.5 5.7590 4.7 0.1 18.5 5.8 11.7 59.3 9.29
2%Pt/PANI-A, after reduction at 250 �C for 1 h; mcat = 0.10117 g90 9.7 0.2 26.3 3.4 13.4 47.0 4.85 54.5 ± 1.680 8.1 0.2 24.7 2.7 14.2 50.1 2.9570 7.8 0.0 26.9 0.0 8.4 56.9 1.6490 9.5 0.2 26.5 3.4 13.8 46.6 5.06
2%Pt/PANI-C, after reduction at 250 �C for 1 h; mcat = 0.10203 g90 9.0 0.2 27.2 3.5 11.9 48.2 5.99 55.3 ± 2.380 7.2 0.2 24.9 2.6 12.6 52.5 3.6670 7.3 0.0 28.3 2.4 0.0 62.0 1.9590 8.9 0.2 27.2 3.5 12.0 48.3 6.32
a Data collected at the temperature sequence 90 �C (steady state after �20 h) ? 80 �C ? 70 �C ? 90 �C.
A. Drelinkiewicz et al. / Reactive & Functional Polymers 69 (2009) 630–642 639
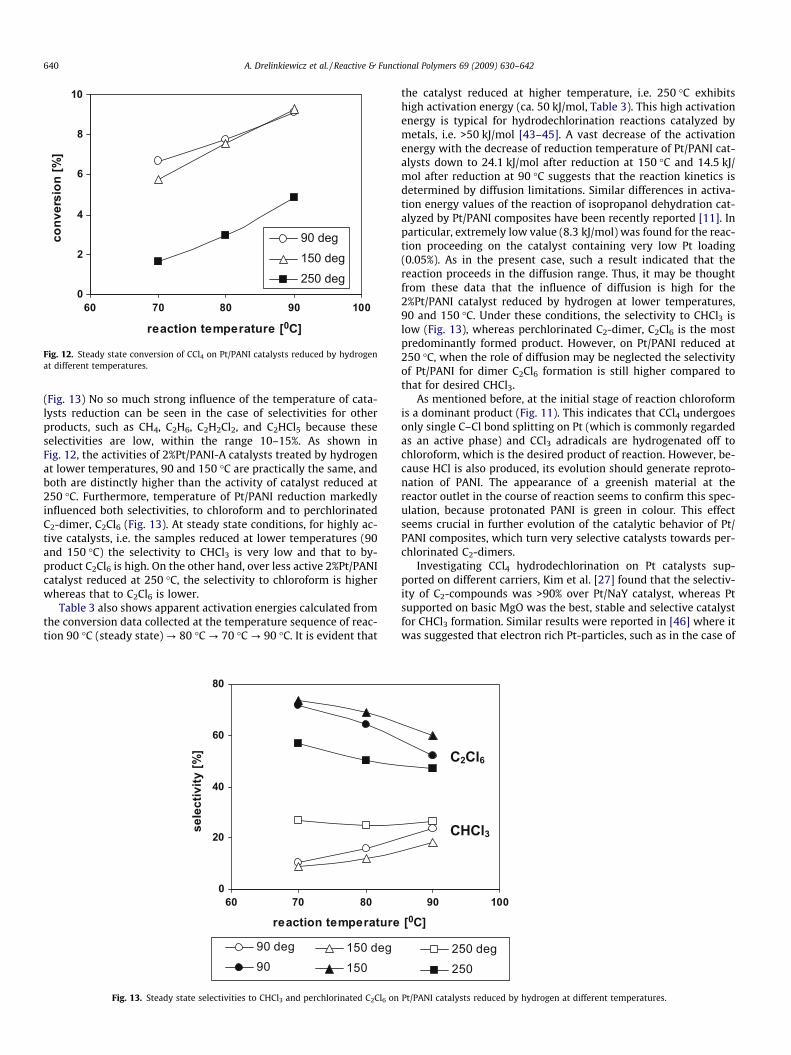
can be seen from Fig. 12 that the temperature of catalysts reduc-tion by hydrogen influenced the steady state conversion of CCl4
(a measure of activity) as well as the selectivities towards desiredproduct CHCl3 and towards perchlorinated C2-dimer, namely C2Cl6
0
2
4
6
8
10
60 70 80 90 100
reaction temperature [0C]
conv
ersi
on [%
]
90 deg150 deg250 deg
Fig. 12. Steady state conversion of CCl4 on Pt/PANI catalysts reduced by hydrogenat different temperatures.
640 A. Drelinkiewicz et al. / Reactive & Functional Polymers 69 (2009) 630–642
(Fig. 13) No so much strong influence of the temperature of cata-lysts reduction can be seen in the case of selectivities for otherproducts, such as CH4, C2H6, C2H2Cl2, and C2HCl5 because theseselectivities are low, within the range 10–15%. As shown inFig. 12, the activities of 2%Pt/PANI-A catalysts treated by hydrogenat lower temperatures, 90 and 150 �C are practically the same, andboth are distinctly higher than the activity of catalyst reduced at250 �C. Furthermore, temperature of Pt/PANI reduction markedlyinfluenced both selectivities, to chloroform and to perchlorinatedC2-dimer, C2Cl6 (Fig. 13). At steady state conditions, for highly ac-tive catalysts, i.e. the samples reduced at lower temperatures (90and 150 �C) the selectivity to CHCl3 is very low and that to by-product C2Cl6 is high. On the other hand, over less active 2%Pt/PANIcatalyst reduced at 250 �C, the selectivity to chloroform is higherwhereas that to C2Cl6 is lower.
Table 3 also shows apparent activation energies calculated fromthe conversion data collected at the temperature sequence of reac-tion 90 �C (steady state) ? 80 �C ? 70 �C ? 90 �C. It is evident that
0
20
40
60
80
60 70 80
reaction temperature
sele
ctiv
ity [%
]
90 deg90
150 deg150
Fig. 13. Steady state selectivities to CHCl3 and perchlorinated C2Cl6 on
the catalyst reduced at higher temperature, i.e. 250 �C exhibitshigh activation energy (ca. 50 kJ/mol, Table 3). This high activationenergy is typical for hydrodechlorination reactions catalyzed bymetals, i.e. >50 kJ/mol [43–45]. A vast decrease of the activationenergy with the decrease of reduction temperature of Pt/PANI cat-alysts down to 24.1 kJ/mol after reduction at 150 �C and 14.5 kJ/mol after reduction at 90 �C suggests that the reaction kinetics isdetermined by diffusion limitations. Similar differences in activa-tion energy values of the reaction of isopropanol dehydration cat-alyzed by Pt/PANI composites have been recently reported [11]. Inparticular, extremely low value (8.3 kJ/mol) was found for the reac-tion proceeding on the catalyst containing very low Pt loading(0.05%). As in the present case, such a result indicated that thereaction proceeds in the diffusion range. Thus, it may be thoughtfrom these data that the influence of diffusion is high for the2%Pt/PANI catalyst reduced by hydrogen at lower temperatures,90 and 150 �C. Under these conditions, the selectivity to CHCl3 islow (Fig. 13), whereas perchlorinated C2-dimer, C2Cl6 is the mostpredominantly formed product. However, on Pt/PANI reduced at250 �C, when the role of diffusion may be neglected the selectivityof Pt/PANI for dimer C2Cl6 formation is still higher compared tothat for desired CHCl3.
As mentioned before, at the initial stage of reaction chloroformis a dominant product (Fig. 11). This indicates that CCl4 undergoesonly single C–Cl bond splitting on Pt (which is commonly regardedas an active phase) and CCl3 adradicals are hydrogenated off tochloroform, which is the desired product of reaction. However, be-cause HCl is also produced, its evolution should generate reproto-nation of PANI. The appearance of a greenish material at thereactor outlet in the course of reaction seems to confirm this spec-ulation, because protonated PANI is green in colour. This effectseems crucial in further evolution of the catalytic behavior of Pt/PANI composites, which turn very selective catalysts towards per-chlorinated C2-dimers.
Investigating CCl4 hydrodechlorination on Pt catalysts sup-ported on different carriers, Kim et al. [27] found that the selectiv-ity of C2-compounds was >90% over Pt/NaY catalyst, whereas Ptsupported on basic MgO was the best, stable and selective catalystfor CHCl3 formation. Similar results were reported in [46] where itwas suggested that electron rich Pt-particles, such as in the case of
90 100
[0C]
250 deg250
C2Cl6
CHCl3
Pt/PANI catalysts reduced by hydrogen at different temperatures.

A. Drelinkiewicz et al. / Reactive & Functional Polymers 69 (2009) 630–642 641
Pt on MgO support owing to the interaction with basic O2� sites,favour the formation of desired CHCl3. In contrast, Pt catalysts sup-ported on zeolite or silica–alumina (acidic supports) appeared notappropriate for hydrodechlorination of CCl4 because of their highselectivity to C2-compounds and rapid deactivation by coke. Kimet al. [27] suggested that coke is formed by an oligomerizationreaction catalyzed by acid sites on the supports. The most rapidlydeactivating catalysts are supported on acid supports and, simulta-neously, show high selectivity for C2-compounds that are also theproducts of oligomerization of CCl4. A similar situation seems toexist in the present case. Initially very selective towards CHCl3
Pt/PANI catalysts quickly lose this ability by a large change in thestructure of PANI caused by HCl evolution in the course of hyd-rodechlorination. Reprotonation of PANI brings about a buildupof acidity, responsible for observed changes in the catalyticbehavior.
An attempt to sinter Pt-particles by the reduction of Pt/PANIcomposites at 295 �C (Fig. 10, section B) brought about a ratherdrastic decrease of overall activity, although the mean size of Ptcrystallites increased only to >5 nm (XRD profile in Fig. 10). Simul-taneously, the selectivity towards perchlorinated dimer, C2Cl6 in-creased to 75% (at the cost of C2HCl5 and CHCl3), Fig. 11, sectionB. Such a dramatic change in the catalytic behavior would followfrom an extensive encapsulation of Pt-particles. For this reason, avery limited exposed surface area of platinum is capable of produc-ing a limited flux of CCl3 radicals, which are instaneously dimer-ized to C2Cl6.
Thus, although PANI exhibits basic character, the reactivity ofPt/PANI catalysts under hydrodechlorination of CCl4 is similar tothose of catalysts with Brönsted acidity. However, in the case ofPt/PANI the acidic centres are formed under the catalytic test dueto the protonation of the PANI by reaction product, HCl. Further-more, our HdCl results clearly show that Pt/PANI composites whenin protonated form may be used as bi-functional catalysts exhibit-ing both redox (Pt) and Brönsted acid (H+) properties.
4. Conclusions
Platinum–polyaniline composites are prepared using threeaqueous solutions of H2PtCl6 differing in the pH and thus in thetype of dominating chloro–aqua or chloro–hydroxo complexes ofPt(IV). In all the Pt–polyaniline composites, platinum in two va-lence states Pt(IV) and Pt(II) appear indicating redox mechanismunder platinum incorporation. The composition of Pt(IV) precursorsolution influences the extent of Pt(IV) to Pt(II) reduction. Thereduction of Pt(IV) proceeds much easier in alkaline solutions i.e.when the Pt(IV) complexes consist of aqua and hydroxo ligandslike [PtCl4(H2O)(OH)]�. XPS spectra show that under these condi-tions the content of reduced Pt(II)-species is ca. 7� higher thanthat of Pt(IV) ones. The nature of Pt-species formed in the PANI ma-trix is similar irrespective of the composition of precursor solution.They consist of Cl and N/O ligands.
Upon reduction of as-prepared composites by NaBH4 the Pt-me-tal nanoparticles are formed of size within 2–4 nm independentlyof the composition of precursor solution. However, HRTEM studiesshows slightly lower average diameter of Pt-particles (2.24 nm) in2%Pt/PANI-A catalyst prepared using precursor solution of acidicpH. Catalytic performance of Pt/PANI composites in hydrogenationof 2-butyne-1,4-diol is similar to that observed for typical inor-ganic carrier supported Pt-catalysts. Pt centres present in the re-duced form of Pt/PANI composites are reactive towards thehydrogenation of unsaturated triple C„C and double C@C bonds.They are also able to catalyze the hydrogenolysis reaction of 2-bu-tene-1,4-diol to give crotyl alcohol. In the reaction of CCl4 hydrode-halogenation on Pt/PANI composites, CHCl3 appears a dominant
product, but only at the initial stage of reaction. Unavoidable for-mation of HCl brings about large changes, both in overall activity,as well as the selectivity. Dimeric C2 products, especially C2Cl6,start to dominate. It is speculated that these changes are not pri-marily due to changes in platinum component of the catalyst butrather follow from reprotonation of PANI which acquires Brönstedacidity used in dimerization of CCl3 radicals.
Acknowledgments
This work was financially supported by Polish Committee forScientific Research grant PBZ-KBN-116/T09/2004. A. Ziebaacknowledges a Ph.D. grant of the Polish Academy of Sciences. Itwas also supported in part by the Polish EKOKAT network.
References
[1] E.T. Kang, K.G. Neoh, K.L. Tan, Prog. Polym. Sci. 23 (1998) 277.[2] A. Malinauskas, Polymer 42 (2001) 3957.[3] H. Laborde, Appl. Electrochem. 24 (1994) 219.[4] E.C. Venancio, W.T. Napporn, A.J. Motheo, Electrochim. Acta 47 (2002) 1495.[5] L. Niu, Q. Li, F. Wei, X. Chen, H. Wang, Synth. Met. 139 (2003) 271.[6] A. Houdayer, R. Schneider, D. Billaud, J. Ghanbaja, J. Lambert, Synth. Met. 151
(2005) 165.[7] M. Hasik, W. Turek, A. Nyczyk, E. Stochmal, A. Bernasik, A. Sniechota, A.
Sołtysek, Catal. Lett. 127 (2009) 304.[8] B.M. Choudary, M. Roy, S. Roy, M.L. Kantam, B. Sreedhar, K.V. Kumar, Adv.
Synth. Catal. 348 (2006) 1734.[9] M.L. Kantam, M. Roy, S. Roy, B. Sreedhar, R.L. De, Catal. Commun. 9 (2008)
2226.[10] I. Dodouche, F. Epron, Appl. Catal. B 76 (2007) 291.[11] A. Nyczyk, A. Sniechota, A. Adamczyk, A. Bernasik, W. Turek, M. Hasik, Eur.
Polym. J. 44 (2008) 1594.[12] M. Steffan, F. Klasovsky, J. Arras, Ch. Roth, J. Radnik, H. Hofmeister, P. Claus,
Adv. Synth. Catal. 350 (2008) 1337.[13] B.J. Gallon, R.W. Kojima, R.B. Kaner, P.L. Diaconescu, Angew. Chem. Int. Ed. 46
(2007) 7251.[14] S.W. Huang, K.G. Neoh, C.W. Shih, D.S. Lim, E.T. Kang, H.S. Han, K.L. Tan, Synth.
Met. 96 (1998) 117.[15] A. Drelinkiewicz, M. Hasik, M. Choczynski, Mater. Res. Bull. 33 (1998) 739.[16] J.W. Sobczak, A. Kosinski, A. Bilinski, J. Pielaszek, W. Palczewska, Adv. Mater.
Opt. Electron. 8 (1998) 295.[17] A. Houdayer, R. Schneider, D. Billaud, J. Ghanbaja, J. Lambert, Appl. Organom.
Chem. 19 (2005) 1239.[18] M. Hasik, Cz. Paluszkiewicz, E. Wenda, Vibrat. Spectrosc. 29 (2002) 191.[19] M. Hasik, Cz. Paluszkiewicz, E. Bielanska, J. Mol. Struct. 744-747 (2005) 677.[20] J.M. Kinyanjui, N.R. Wijeratne, J. Hanks, D.W. Hatchett, Electrochim. Acta 51
(2006) 2825.[21] J.M. Kinyanjui, R. Harris-Bur, J.G. Wagner, N.R. Wijeratne, D.W. Hatchett,
Macromolecules 37 (2004) 8745.[22] A.P. O‘Mullane, S.E. Dale, J.V. Macpherson, P.R. Unwin, Chem. Commun. (2006)
1606.[23] A.P. O‘Mullane, S.E. Dale, T.M. Day, N.R. Wilson, J.V. Macpherson, P.R. Unwin, J.
Solid State Electrochem. 10 (2006) 792.[24] J.M. Winterbottom, H. Marwan, J. Viladevall, S. Sharma, S. Raymashasay, Stud.
Surf. Sci. Catal. 108 (1997) 59.[25] M.M. Telkar, C.V. Rode, R. Jaganathan, V.H. Rane, R.V. Chaudhari, J. Mol. Catal.
A: Chem. 187 (2002) 81.[26] F.J. Urbano, J.M. Marinas, J. Mol. Catal. A: Chem. 173 (2001) 329.[27] S.Y. Kim, H.C. Choi, O.B. Yanga, K.H. Lee, J.S. Lee, Y.G. Kim, J. Chem. Soc., Chem.
Commun. (1995) 2169.[28] M.M. Chehimi, M.-L. Abel, C. Perruchot, M. Delamar, S.F. Lascelles, S.P. Armes,
Synth. Met. 104 (1999) 51.[29] B. Shelimov, J.F. Lambert, M. Che, B. Didillon, J. Catal. 185 (1999) 462.[30] B. Shelimov, J.F. Lambert, M. Che, B. Didillon, J. Am. Chem. Soc. 121 (1999) 545.[31] A. Drelinkiewicz, J. Stejskal, A. Waksmundzka, J.W. Sobczak, Synth. Met. 140
(2004) 233.[32] F.R. Hartley, The Chemistry of Platinum and Palladium, Applied Science
Publishers Ltd., London, 1973.[33] J.P. Pouget, M.E. Józefowicz, A.J. Epstein, X. Tang, A.G. MacDiarmid,
Macromolecules 24 (1991) 779.[34] Y.B. Moon, Y. Cao, P. Smith, A.J. Heeger, Polym. Commun. 30 (1989) 196.[35] W. Łu _zny, M. Sniechowski, J. Laska, Synth. Met. 126 (2002) 27.[36] A.G. McDiarmid, A.J. Epstein, Faraday Discuss. Chem. Soc. 88 (1989) 317.[37] J. Wang, K.G. Neoh, E.T. Kang, J. Coll. Interf. Sci. 239 (2001) 78.[38] NIST Standard Reference Database 20, Version 3.2 (Web Version), National
Institute of Standards and Technology, Gaithersburg, 2001.[39] J.H. Lunsford, D.S. Treybig, J. Catal. 68 (1981) 192.[40] M.G. Musolino, P. De Maio, A. Donato, R. Pietropaolo, Appl. Catal. A: Gen. 285
(2005) 50.[41] M.M. Telkar, C.V. Rode, V.H. Rane, R.V. Chaudhari, Catal. Commun. 6 (2005) 725.

642 A. Drelinkiewicz et al. / Reactive & Functional Polymers 69 (2009) 630–642
[42] Z.C. Zhang, B.C. Beard, Appl. Catal. A 174 (1998) 33.[43] M. Legawiec-Jarzyna, W. Juszczyk, M. Bonarowska, Z. Kaszkur, L. Kepinski, Z.
Kowalczyk, Z. Karpinski, Topics Catal., in press, doi:10.1007/s11244-009-9258-5.
[44] A.L.D. Ramos, M. Schmal, D.A.G. Aranda, G.A. Somorjai, J. Catal. 192 (2000) 423.[45] Z. Karpinski, K. Early, J.L. d’Itri, J. Catal. 164 (1996) 378.[46] V. Dal Santo, C. Dossi, S. Recchia, P.E. Colavita, G. Vlaic, R. Psaro, J. Mol. Catal. A:
Chem. 182–183 (2002) 157.




















