
Approximation Bounds for Quadratic Optimization with Homogeneous Quadratic Constraints
Upload
independentCategory
view
5download
0
R
Pm
Aa
b
a
ARRAA
KNPPMCS
C
1d
Journal of Photochemistry and Photobiology C: Photochemistry Reviews 11 (2010) 62–72
Contents lists available at ScienceDirect
Journal of Photochemistry and Photobiology C:Photochemistry Reviews
journa l homepage: www.e lsev ier .com/ locate / jphotochemrev
eview
hotophysics of norharmane in solution phase: From homogeneous toicroheterogeneous environments
rabinda Mallicka, Paramita Dasb, Nitin Chattopadhyayb,∗
Department of Chemistry, Kashipur Michael Madhusudan Mahavidyalaya, Purulia - 723132, West Bengal, IndiaDepartment of Chemistry, Jadavpur University, Raja S. C. Mullick Road, Kolkata - 700 032, West Bengal, India
r t i c l e i n f o
rticle history:eceived 11 January 2010eceived in revised form 10 March 2010ccepted 12 March 2010vailable online 19 March 2010
a b s t r a c t
The photobehavior of norharmane (9H-pyrido[3,4-b]-indole) (NHM), one of the vastly used skeleton ofdrugs in therapeutic applications, has recently been the subject of increasing interest due to the finding oftheir phototoxic and photocarcinogenic properties. Its absorption and fluorescence behavior from differ-ent prototropic species show remarkable sensitivity towards the polarity, viscosity and local pH, exhibitedby various microheterogeneous bio and biomimetic environments like micelles, reverse micelles, pro-
eywords:orharmaneroton transferhotophysicsicelle
yclodextrinerum albumin
teins, etc. The significant results obtained for NHM in homogeneous and a series of microheterogeneousenvironments is reviewed in this account. Much attention has been given to the properties of the excitedstates, location and biodistribution of NHM in different biological environments. The results can help inunderstanding the photophysics of the probe in biological environments and in assessing the correlationbetween different prototropic forms and biological activity.
© 2010 Published by Elsevier B.V.
ontents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 632. Homogeneous environments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63
2.1. Singlet state properties . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 632.2. Triplet state properties . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 64
3. Microheterogeneous environments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 643.1. Micelles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 65
3.1.1. Micelle induced prototropic switching: thermodynamic perspective . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 653.1.2. Drug distribution in micelles from time-resolved fluorescence. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 66
3.2. Reverse micelles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 673.3. Cyclodextrins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
3.3.1. NHM–�-CD inclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 693.4. Proteins. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70
3.4.1. Binding with the proteins: assignment of the site . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70
4. Use of photophysical change of NHM for sensing of the inorganic anions5. Concluding remarks and outlook . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
∗ Corresponding author. Tel.: +91 9433948648; fax: +91 33 24146584.E-mail address: [email protected] (N. Chattopadhyay).
389-5567/$20.00 © 2010 Published by Elsevier B.V.oi:10.1016/j.jphotochemrev.2010.03.001
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72

otobio
1
lo[iimdppplasTosbaattmfpwhued
A. Mallick et al. / Journal of Photochemistry and Ph
Arabinda Mallick obtained his M.Sc. and Ph.D. degreesfrom Jadavpur University (Kolkata, India) in 2001 and2006 respectively. As a JSPS Fellow he worked with Profes-sor H. Miyasaka in Osaka University, Japan. Currently heis serving Kashipur Michael Madhusudan Mahavidyalaya,Purulia as a Lecturer. His research interests include flu-orosensing, biophysical chemistry and ultrafast reactiondynamics.
Paramita Das obtained her M.Sc. degree from CalcuttaUniversity in 2004 and Ph.D. degree from Jadavpur Uni-versity in 2008. Her research interest includes biophysicalchemistry, photophysical study of fluorophore in confinedmedia and synthesis and characterization of quantum dotsin polymer shell.
Nitin Chattopadhyay obtained his Ph.D. degree in 1990from Jadavpur University (Kolkata, India). Currently heis a Professor in the Department of Chemistry, JadavpurUniversity. During his post doctoral assignments he vis-ited Katholieke Universiteit Leuven, Belgium and CoimbraUniversity, Portugal. As a visiting scientist, he visitedOsaka University, Japan. He is a Fellow of the IndianAcademy of Sciences. His research interests include fluo-rescence based sensing, photophysical and photochemicalprocesses in bio- and biomimicking environments, laserinduced optoacoustic spectroscopy.
. Introduction
Norharmane (NHM) or �-carboline belongs to the group of alka-oids with a tricyclic pyrido(3,4-b)-indole ring system and is onef the most successful classes of drugs in therapeutic applications1–6]. The pyridine ring in NHM is �-deficient and the indole rings �-excess resulting in two functional sites for its acid–base chem-stry. In the ground state, the electron density is highest in the
id-plane of NHM and on excitation to the S1 state, the electronensity migrates to both ends of the molecule, especially to theyridine nitrogen [7–11]. NHM or �-carboline is found in about 26lant families, cells, animal tissues, human urine and these com-ounds are formed as photoproducts from tryptophan in human
enses [12]. Compounds belonging to this class have gained muchttention in pharmacological sciences as central nervous systemtimulant, hallucinogens and paralysants of cardiac muscle [13,14].he photophysical behavior of NHM has recently been the subjectf increasing fascination due to its novel biological applicationsuch as photosensitization towards a variety of systems includingacteria, fungi, viruses, etc. [15–18]. The use of photosensitizinggents together with light, generally known as photodynamic ther-py (PDT), for the treatment of neoplastic diseases has become aopic of growing medical interest. PDT produces singlet oxygenhat is detrimental to the cancerous cells. This is an established
odality for the cancer treatment. Beljanski and Beljanski haveound that some �-carbolines can destroy selectively and com-letely the proliferative capacity of various types of cancer cells
hich is enhanced upon excitation with UV radiation [19]. NHMas been quite effective in producing singlet oxygen and it can besed as an efficient cancer cell photosensitizer. The extent of PDTfficiency of the photosensitizer depends on the singlet oxygen pro-uction as well as on the biodistribution of the probe moleculelogy C: Photochemistry Reviews 11 (2010) 62–72 63
in different biosystems/organized media. Thus it is important toascertain the probable location of NHM in microheterogeneousenvironments. Study of the binding interaction of NHM with differ-ent biosystems/organized environments thus becomes significantfor studying the net PDT efficiency. In this account, we are goingto demonstrate some photophysical and biophysical studies ofthis biological photosensitizer in homogeneous and some confinedbiological and biomimicking environments like serum albumins,micelles, reverse micelles and cyclodextrins.
Apart from the biological aspect, attention has also been focusedon another important and interesting feature of norharmane,namely, its fluorescence from different prototropic species thatshows remarkable sensitivity to some parameters (e.g., polarity,pH) of the microenvironment. Such properties of the �-carbolinecompounds have stimulated the scientists to exploit them forinvestigating the binding sites in different biological targets andalso for analytical purposes. Although a good number of biochemi-cal and biological investigations have been made using norharmane[20–22], a large portion of these studies have been associated withtheir phototoxic effect towards various organisms [21,23–25]. Theirphototoxicity again depends on the diffusion of these moleculesinto different regions of the biological targets or cells [1]. Somerecent literature projects that in aqueous medium, protonationof this molecule is important in excited singlet and triplet states[25,26]. Both triplet state formation and singlet oxygen photo-sensitization decrease on protonation of the molecule [27]. For aparticular photosensitizer molecule, it is often necessary to optfor one prototopic form or a desired composition of the differentprototropic species for achieving better efficiency for a targetedpurpose in a specific environment. Recently, triplet state studieson these molecules by Varela et al. [26] and Becker et al. [27] haverevealed that neutral forms of these compounds have significanttriplet state yield and the long-lived triplet states may play impor-tant role in their photosensitization reactions in vivo in the presenceof oxygen. Photophysical studies of Burrows and co-workers,and our group have independently shown that the neutral formof the fluorophore has strong affinity for hydrophobic domains[22,23,28–32]. In this account, various attempts are presented fromour laboratory as well as others, to study the photophysics of NHMin homogeneous and different confined environments.
2. Homogeneous environments
2.1. Singlet state properties
The photophysical and/or photochemical properties of NHMhave been shown to be strongly dependent on the nature of theenvironment [9,10]. The absorption spectrum of NHM typicallyshows three main absorption bands in the range of 345–390 nmdepending upon the condition of the medium. In aqueous solu-tion at low pH, NHM gives absorption band for cationic species.Hypsochromic shifts corresponding to the transformation from thecation to the neutral species occurs in slightly alkaline solution. Anew red shifted absorption band, ascribed to the NHM anion, isproduced by the release of the pyrrolic proton in strongly alkalinemedium (pH 14). These absorption bands correspond to transitionsfrom the singlet ground states of the different prototropic speciesof NHM (S0) to the corresponding excited singlet states (S1).
The excited state prototropic equilibria of �-carboline systemsin different pure organic solvents and some mixed solvents havebeen studied by Dias et al. [15]. They have shown that excitation
of NHM in its excited state results only in the neutral species inhydrocarbon and nonprotic solvents. Reyman et al. studied the pho-tophysics and kinetics of NHM in binary mixture of acetic acid withdichloromethane and dioxane with benzene [11]. In a neutral pro-tic solvent (methanol), excitation of the neutral species results in
64 A. Mallick et al. / Journal of Photochemistry and Photobiology C: Photochemistry Reviews 11 (2010) 62–72
Sn
mGdotsiteiecfm
2
ttnt�topebisnsb[bsla
TA[
Table 2Low temperature fluorescence and phosphorescence data for NHM in organic glassesat 77 K [26].
Species Fluorescence Phosphorescence ˚F/˚P ˚F + ˚P �phosphorescence
tant molecules [39–43]. Reactants accommodated in molecularassemblies like micelles, reverse micelles, cyclodextrins, vesicles,etc. often achieve a greater degree of organization compared totheir geometries in homogeneous solution, can mimic reactions in
Table 3Triplet state properties of NHM in various solvents [27].
Solvent �T (�s) εT (103 M−1 cm−1) ˚T ˚�
Benzene ≥60 8.2 0.30 0.34ACN ≥110 12.6 . . . 0.40ACN + HCl ≥150 – . . .
cheme 1. Different acid–base equilibria for norharmane; CN: cation–neutral, NA:eutral–anion, ZA: zwitterion–anion and CZ: cation–zwitterion.
ultiple species, e.g., neutral, cation, and zwitterion. Sakurovs andhiggino and Vert et al. have established that NHM exists in fourifferent forms (neutral, cation, anion and zwitterion) dependingn the pH of the medium in aqueous solution [9,10]. At low pH, onlyhe cationic species of NHM emits. The characteristic emission oflightly alkaline NHM solution suggests proton transfer from thenitially excited neutral species. With a further increase in the pH,he intensity of the fluorescence is markedly reduced and two newmission bands are observed. In summary, literature reports thatn aqueous medium between pH 1 and 10, only the cationic speciesmits; at pH ∼12.3 emissions from all the three species, viz., neutral,ation and zwitterion have been recorded. Scheme 1 depicts dif-erent acid–base equilibria for NHM. The absorption and emission
axima for solutions of each species are summarized in Table 1.
.2. Triplet state properties
NHM efficiently passes to the triplet state upon light absorp-ion. Phosphorescence emission has indeed been detected at lowemperature with the energies of the lowest triplet states for theeutral and the cation at 24,400 cm−1 and 21,850 cm−1, respec-ively [26]. The lowest excited triplet state is suggested to have–�* character. Microsecond laser photolysis has shown that the
riplet state undergoes acid–base reaction in water with pKa valuef 5.0 at room temperature. This value is substantially lower com-ared with pKa’s of 7.2 for the ground state and 13.0 for the lowestxcited singlet state of this molecule. The large basicity differencesetween these two excited states have been interpreted by invok-
ng marked differences in the electron distribution in the S1 and T1tates. It is suggested from theoretical calculation [27] and mag-itude of the molar extinction coefficient that the lowest excitedinglet state has (�–�*) character in polar solvents. The separationetween the lowest (�–�*) and (n–�*) is very small (1000 cm−1)33]. A change in the fluorescent state from (�–�*) to (n–�*) haseen reported on decreasing solvent polarity [34]. In polar protic
olvents, however, the excited (�–�*) state always seems to be ofowest energy [35]. Table 2 collects low temperature fluorescencend phosphorescence data for NHM in organic glasses at 77 K.able 1bsorption and emission maxima of different prototropic species of norharmane
10].
Neutral Cation Zwitterion Anion
Absorption 348 370 – 390Emission 385 450 510 –
yield (˚F) yield (˚P) (s)
Neutral 0.30 0.43 0.70 0.73 4.27Cation 0.62 0.16 4.0 0.78 6.89
Table 2 shows that the phosphorescence quantum yield for thecationic form (0.16) of the probe is lower than that of the neu-tral form (0.43) while the difference between the phosphorescencelifetimes of the neutral and cationic forms is less marked. Theseresults indicate that S1 → T1 intersystem crossing in the neutralform is more efficient than that in the cation. The improved effi-ciency of the intersystem crossing from the neutral form has beenrationalized by El-Sayed with the proposition that the S1 → T1 inter-system crossing is favored by spin–orbit coupling [36]. Since thelowest excited singlet and triplet states have predominantly (�–�*)character, the most likely explanation for the efficient intersys-tem crossing is either due to the small energy separation, thereis sufficient population of the (n–�*) state to allow this to becomeinvolved in these processes, or that there is extensive mixing of the(�–�*) and (n–�*) states. Lim has discussed such mixing for closelying states in terms of vibronic coupling [37] and Olba et al. [38]have given good evidence that this type of interaction is involvedin the “proximity effect” observed in the photophysical propertiesof NHM. The flash transient spectra in nonprotic solvents of NHMhas long-lived triplet transient only for the neutral species. Tripletyield (˚T) and singlet oxygen yield (˚�) are quite high. The tran-sient absorption spectra in benzene and ACN are basically same.Both the spectra are quenched in the presence of oxygen [27]. Thetriplet state properties of NHM in various solvents available fromliterature are collected in Table 3.
To ascertain the absorption maxima and intensity of the mainT1–Tn transitions of the neutral and cationic forms of NHM, someZINDO/SCI based calculations are also available and the relevantdata from the literature are collected in Table 4.
3. Microheterogeneous environments
The last two decades have witnessed the importance of confinedenvironments on the photophysics of many biologically impor-
Table 4Absorption maxima (�) and oscillator strength (f) of the main T–T transitions ofNHM calculated by the ZINDO/SCI method [27].
Species Transitions � (nm) f
NHM T1–T7 569 0.19T1–T8 496 0.16T1–T13 336 0.07
NHM-H+ T1–T8 533 0.18T1–T12 381 0.04T1–T17 315 0.11

otobio
bispomwfdnoow
3
dniahrm
macaeao
btstttacb
Ftrts
A. Mallick et al. / Journal of Photochemistry and Ph
iosystems and also have potential for energy storage [44]. Thenteriors of these assemblies are often quite different from the bulkolution phase. For aqueous solutions of micelles, cyclodextrins,roteins, the inner core is relatively less polar than the bulk aque-us phase. A reverse picture is found when one deals with reverseicelles. Not only the polarity but also the local viscosity/rigidityithin the organized assemblies appears to be appreciably different
rom the bulk medium; as a result, these can alter the photophysicsrastically. In this section, we will discuss and see how the orga-ized assemblies modify the photophysics of NHM. Since structuresf these organized media have already been discussed in a numberf reviews [45] including the recent one from our own group, so weill skip the detail of this aspect [43].
.1. Micelles
Surfactants can self-organize under specific environmental con-itions to form micelles in aqueous solution. They basically containon-polar hydrocarbon or polyoxyethylenic chain (tail) and an
onic or polar group (head). The core of a micelle is essentially “dry”nd consists of the hydrocarbon chains with polar and/or chargedead groups projecting towards the bulk water. The core is sur-ounded by a polar shell, which is called the Stern layer for an ionicicelle and palisade layer for a non-ionic micelle.Studies from our lab reveal photoswitching of NHM in different
icellar environments [29,32]. It has been shown that addition ofnionic surfactants (sodium alkyl sulfate, SnS, n = 10, 12 and 14)ontaining longer hydrophobic chains of the surfactants to thequeous solution of NHM changes the ground state prototropicquilibrium favorably towards the cationic species. Fig. 1 depictsrepresentative set of absorption spectra of NHM in the presencef varying concentration of sodium tetradecyl sulfate (S14S).
With an increase in the concentration of SnS, the fluorescenceand of NHM, however, shows a different type of variation. An ini-ial decrease followed by an increase along with a hypsochromichift of about 10 nm is observed. In conformity with the absorp-ion study, observation of only the cationic emission band reflectshat the cationic species is stabilized in the S S environments and
nhe effect is ascribed to an electrostatic interaction between thenionic surface charge of the SnS micellar units and the protonatedationic species of the fluorophore. Similar results were reportedy Pal et al. [46] while investigating the photophysical properties
ig. 1. Variation of absorption spectrum of NHM as a function of S14S concentra-ions. Concentrations of S14S in (i) → (vi) are 0.0, 0.78, 0.97, 1.16, 1.35, and 1.53 mM,espectively. Inset shows the variation of absorbance of the cationic band at 372 nmo that of the neutral band at 348 nm as a function of SnS chain length at theiraturation level of interaction ([NHM] = 1 × 10−4 M).
logy C: Photochemistry Reviews 11 (2010) 62–72 65
of rhodamine derivatives in SDS micelle and also by Ghosh et al.[47] during their study of interaction of 1-methylaminopyrene withanionic surfactants. It is important to mention here that the cationicband intensity decreases before micelle formation and it dependson the surfactant chain length [29].
Effect of cationic micelles (nTAB) of different surfactant chainlengths on the photoswitching of NHM has also been studied inthis laboratory [22]. A little change in photoswitching is observedup to critical micellar concentrations (CMC) but after attainmentof CMC, there is a dramatic modulation in the excited state photo-switching, the prototropic equilibrium of NHM is favored towardsthe neutral species of the fluorophore (disfavoring the cationicspecies). This is expected considering the cationic surface chargeof the micelles. Relative enhancements of the neutral emission atthe cost of the cationic emission at the respective saturation levelsof interaction with the micelles appear to be in the order: dode-cyl trimethyl ammonium bromide (DTAB) > tetradecyl trimethylammonium bromide (TTAB) > cetyl trimethyl ammonium bromide(CTAB). Fig. 2 exhibits a set of representative emission spectraof NHM in TTAB environment. The spectral observation has beenrationalized in terms of the polarity and rigidity guided prototropicshift [22].
In the presence of non-ionic surfactant, like triton X-100 (TX-100), there is hardly any change in the absorption spectrum ofNHM. However, addition of TX-100 to the aqueous solution of NHMhas been shown to favor the prototropic equilibrium towards theneutral form [32].
3.1.1. Micelle induced prototropic switching: thermodynamicperspective
A molecular switching is a phenomenon in which a reversibleshifting between two or more stable states takes place in responseto a change in an external trigger like pH, temperature, environ-ment, etc. The photoswitching of NHM is limited between thecationic and the neutral species both in the ground and excitedstates. In the presence of anionic surfactants, the prototropic equi-librium can be represented as
Neutral (N) � Cation (C)
and the equilibrium constant (Keq) can be represented as
Keq = [C][N]
(1)
Fig. 2. Emission spectra of NHM as a function of TTAB concentrations(�exc = 350 nm). Curves (i) → (vii) correspond to 0.0, 2.0, 3.0, 4.0, 5.0, 10.0, 20.0 mM,respectively. Inset shows the variation of relative fluorescence of the neutral bandto that of the cationic band as a function of the chain length of the nTAB surfactantsat the saturation level of interactions ([NHM] = 2 × 10−5 M).

66 A. Mallick et al. / Journal of Photochemistry and Photobiology C: Photochemistry Reviews 11 (2010) 62–72
F s a fuf
waduvffr
etaTam
imiimatooaF
ttdno
Fc
ig. 3. (a) Variation of equilibrium constant (Keq) and free-energy change (�G) aree-energy change (�G) in S10S, S12S and S14S micelles.
here [C] and [N] represent the concentrations of the cationicnd the neutral species, respectively. From the ratio of the opticalensity values of the cation to the neutral species, Keq has been eval-ated at different surfactant concentrations. We have shown theariation of the equilibrium constant (Keq) and the correspondingree-energy change (�G) with increasing surfactant concentrationor the three anionic micellar (SnS) systems [29]. Fig. 3 shows aepresentative plot.
Fig. 3 projects that as the surfactant concentration increases thequilibrium moves towards the cationic species for all the surfac-ant systems studied. This has been ascribed to the electrostaticttraction between the cation of NHM and the anionic micelles.he interesting finding of this work has been that the �G value,t the saturation level of the probe–micelle interaction, becomesore negative as the surfactant chain length increases (Fig. 3b).In cationic micelles the photoswitching in the excited state
s just of the reverse pattern, i.e., the cationic form is convertedore to its neutral species as the surfactant concentration is
ncreased [22]. As mentioned above, the photoswitching of NHMn the excited state can be readily understood from the measure-
ent of the equilibrium constants and the free-energy changest various concentrations of the surfactants from the ratios ofhe fluorescence intensities and/or fluorescence quantum yieldsf the neutral and the cationic species (considering a constancyf the molar extinction coefficient as the fluorophore is excitedt the isosbestic point, 350 nm). A representative plot is shown inig. 4.
Figs. 3 and 4 demonstrate that the equilibrium for the pro-
otropic reaction of NHM moves towards the cationic species inhe presence of the anionic micelles, while it moves in the oppositeirection in cationic micellar environments. The trends are ratio-alized simply in the light of the stabilization and destabilizationf the cationic species of NHM formed through the protonationig. 4. (a) Variation of equilibrium constant and free-energy change as a function of CThange (�G) in DTAB, TTAB and CTAB micellar media.
nction of S14S concentration and (b) variation of equilibrium constant (Keq) and
reaction, as mentioned above, in anionic and cationic micelles,respectively, because of the surface charge characteristics of themicellar units. The studies of the prototropic reaction of the fluo-rophore in different micelles further reveal that with a variation inthe chain length of the surfactants comprising the micelles affectsthe equilibrium remarkably. While an increase in the surfactantchain length in the SnS surfactant series leads to a shift of the equi-librium more towards the protonated form, a similar variation inthe nTAB series disfavors the protonation process. This can be ratio-nalized from the following argument. As the surfactant chain lengthincreases in both the series the aggregation number of the micel-lar units increases [29,22]. This leads to an increase in the micellarsurface charge as well as the surface charge density. This basicallyenhances the effect of the electrostatic interaction as it is observedin an individual ionic micelle. The nature of the surface charge beingopposite (negative in the case of SnS series and positive in nTABseries), the observation goes in the opposite directions in the twoseries.
3.1.2. Drug distribution in micelles from time-resolvedfluorescence
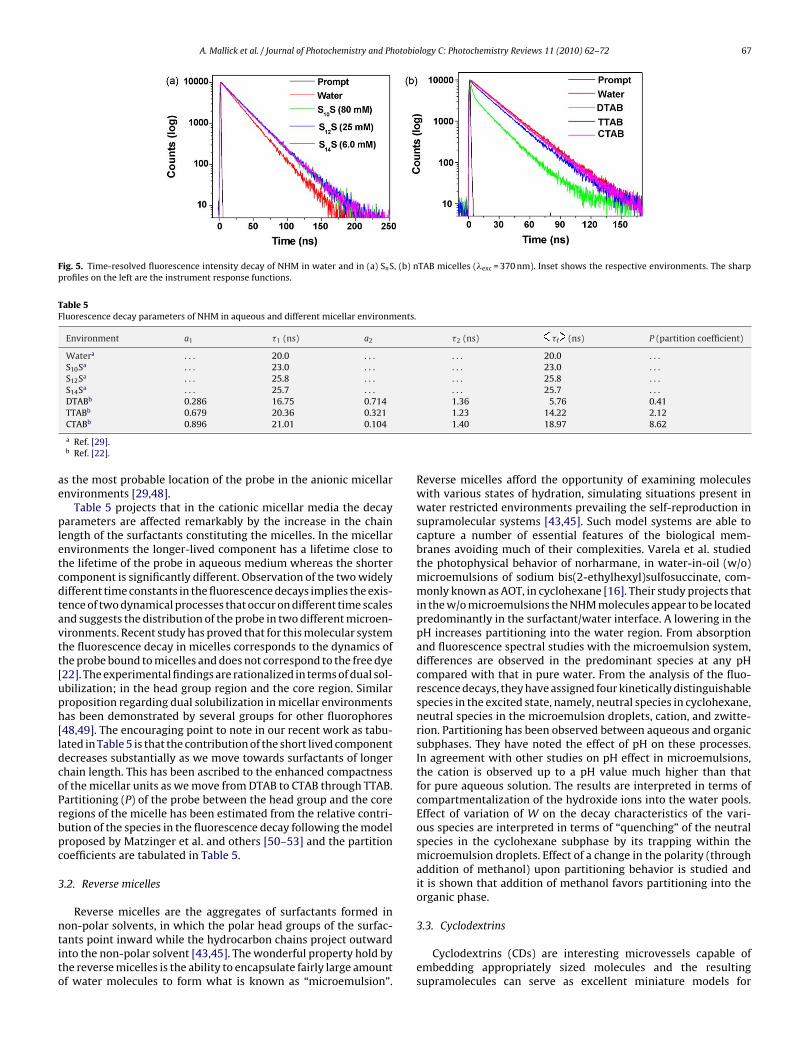
Time-resolved fluorescence technique can provide valuableinformation regarding the location and distribution of a probein complex microheterogeneous environments in a better way.In water and anionic micelles NHM exhibits a single exponentialdecay. However the decay becomes biexponential in cationic micel-lar media [29,22]. Typical decay profiles of NHM in aqueous anddifferent micellar environments (SnS and nTAB) are shown in Fig. 5.
The fluorescence lifetimes of NHM in different micelles (anionicand cationic) are tabulated in Table 5 along with the lifetime ofthe probe in pure water. The single exponential decay behavior isinvoked for the single site solubilization of the probe molecule inthe anionic micellar environments. The studies propose stern layer
AB concentrations and (b) variation of equilibrium constant (Keq) and free-energy
A. Mallick et al. / Journal of Photochemistry and Photobiology C: Photochemistry Reviews 11 (2010) 62–72 67
Fig. 5. Time-resolved fluorescence intensity decay of NHM in water and in (a) SnS, (b) nTAB micelles (�exc = 370 nm). Inset shows the respective environments. The sharpprofiles on the left are the instrument response functions.
Table 5Fluorescence decay parameters of NHM in aqueous and different micellar environments.
Environment a1 �1 (ns) a2 �2 (ns) �f (ns) P (partition coefficient)
Watera . . . 20.0 . . . . . . 20.0 . . .S10Sa . . . 23.0 . . . . . . 23.0 . . .S12Sa . . . 25.8 . . . . . . 25.8 . . .S14Sa . . . 25.7 . . . . . . 25.7 . . .DTABb 0.286 16.75 0.714 1.36 5.76 0.41
ae
pletcdtavtt[uph[ldcoPrbpc
3
ntito
TTABb 0.679 20.36 0.321CTABb 0.896 21.01 0.104
a Ref. [29].b Ref. [22].
s the most probable location of the probe in the anionic micellarnvironments [29,48].
Table 5 projects that in the cationic micellar media the decayarameters are affected remarkably by the increase in the chain
ength of the surfactants constituting the micelles. In the micellarnvironments the longer-lived component has a lifetime close tohe lifetime of the probe in aqueous medium whereas the shorteromponent is significantly different. Observation of the two widelyifferent time constants in the fluorescence decays implies the exis-ence of two dynamical processes that occur on different time scalesnd suggests the distribution of the probe in two different microen-ironments. Recent study has proved that for this molecular systemhe fluorescence decay in micelles corresponds to the dynamics ofhe probe bound to micelles and does not correspond to the free dye22]. The experimental findings are rationalized in terms of dual sol-bilization; in the head group region and the core region. Similarroposition regarding dual solubilization in micellar environmentsas been demonstrated by several groups for other fluorophores48,49]. The encouraging point to note in our recent work as tabu-ated in Table 5 is that the contribution of the short lived componentecreases substantially as we move towards surfactants of longerhain length. This has been ascribed to the enhanced compactnessf the micellar units as we move from DTAB to CTAB through TTAB.artitioning (P) of the probe between the head group and the coreegions of the micelle has been estimated from the relative contri-ution of the species in the fluorescence decay following the modelroposed by Matzinger et al. and others [50–53] and the partitionoefficients are tabulated in Table 5.
.2. Reverse micelles
Reverse micelles are the aggregates of surfactants formed in
on-polar solvents, in which the polar head groups of the surfac-ants point inward while the hydrocarbon chains project outwardnto the non-polar solvent [43,45]. The wonderful property hold byhe reverse micelles is the ability to encapsulate fairly large amountf water molecules to form what is known as “microemulsion”.1.23 14.22 2.121.40 18.97 8.62
Reverse micelles afford the opportunity of examining moleculeswith various states of hydration, simulating situations present inwater restricted environments prevailing the self-reproduction insupramolecular systems [43,45]. Such model systems are able tocapture a number of essential features of the biological mem-branes avoiding much of their complexities. Varela et al. studiedthe photophysical behavior of norharmane, in water-in-oil (w/o)microemulsions of sodium bis(2-ethylhexyl)sulfosuccinate, com-monly known as AOT, in cyclohexane [16]. Their study projects thatin the w/o microemulsions the NHM molecules appear to be locatedpredominantly in the surfactant/water interface. A lowering in thepH increases partitioning into the water region. From absorptionand fluorescence spectral studies with the microemulsion system,differences are observed in the predominant species at any pHcompared with that in pure water. From the analysis of the fluo-rescence decays, they have assigned four kinetically distinguishablespecies in the excited state, namely, neutral species in cyclohexane,neutral species in the microemulsion droplets, cation, and zwitte-rion. Partitioning has been observed between aqueous and organicsubphases. They have noted the effect of pH on these processes.In agreement with other studies on pH effect in microemulsions,the cation is observed up to a pH value much higher than thatfor pure aqueous solution. The results are interpreted in terms ofcompartmentalization of the hydroxide ions into the water pools.Effect of variation of W on the decay characteristics of the vari-ous species are interpreted in terms of “quenching” of the neutralspecies in the cyclohexane subphase by its trapping within themicroemulsion droplets. Effect of a change in the polarity (throughaddition of methanol) upon partitioning behavior is studied andit is shown that addition of methanol favors partitioning into theorganic phase.
3.3. Cyclodextrins
Cyclodextrins (CDs) are interesting microvessels capable ofembedding appropriately sized molecules and the resultingsupramolecules can serve as excellent miniature models for

68 A. Mallick et al. / Journal of Photochemistry and Photobiology C: Photochemistry Reviews 11 (2010) 62–72
imen
ent�las
ct[titu4tst�p
Scheme 2. Model structures and d
nzyme–substrate complexes. The CD molecules have an inter-al cavity accessible to the guest molecules of proper dimensionhrough an opening of 4.5–5.3 Å, 6.0–7.0 Å, and 7.5–8.5 Å for �-CD,-CD and �-CD, respectively; the depths of all remaining more or
ess the same (7.9 Å) [54,55]. Thus, depending on the cavity size, CDsre capable of encapsulating guest molecules of different dimen-ions, with various guest:CD stoichiometries (Scheme 2).
We have studied the interaction of NHM with differentyclodextrins. A remarkable modification in the ground state pro-otropic transformation of NHM is noted upon addition of �-CD30]. Formation of 1:1 NHM–�-CD complex at lower �-CD concen-ration and 1:2 NHM–�-CD complex at higher �-CD concentrations established. Gradual addition of �-CD to the aqueous solu-ion of NHM changes the emission spectra dramatically. Initially,p to ∼2 mM �-CD concentration the cationic band (peak at50 nm) increases with increasing concentration of �-CD. On fur-
her increase in the concentration of the �-CD in the aqueousolution of NHM the cationic emission decreases with a concomi-ant development of a new band at around 380 nm. Addition of-CD and �-CD has been found to have no perceptible effect on thehotophysics of NHM either in the ground or in the excited states.Fig. 6. Double reciprocal plots for complexation between NHM and �-CD
sions of �-, �- and �-cyclodextrin.
Prados et al. have studied the excited state photoswitching reac-tion of norharmane in the presence of modified CDs in solutions atdifferent pH values [56]. They have shown that the spectral shapeand the species present in the equilibrium depend on both the CDand the pH of the medium. Their study reveals that at pH 4.0 theemission corresponds to the cationic form of NHM. At pH 7.8 theemission spectra show the bands corresponding to the cationicand the neutral forms in the case of the complexes with �-CDand (2-hydroxypropyl)cyclodextrin (HP-CD), and only the neutralband for the complexes of heptakis(2,3-di-O-methyl)cyclodextrin(DM-CD) and heptakis(2,3,6-tri-O-methyl)cyclodextrin (TM-CD).At pH 13.0 all the complexes exhibit the neutral form togetherwith the zwitterionic form of the probe. The photoswitching hasbeen explained by proposing that the low polarity environmentafforded by the CDs significantly alters the proton transfer pho-toreaction of norharmane. The differences observed in the spectral
shape and the fluorescence intensities obtained for titration ofthe inclusion complexes with HCl and NaOH allow to concludethat in the cyclodextrin cavities the excited state prototropicequilibrium is not attained within the lifetime of the excitedstate.in aqueous solution considering (a) 1:1 and (b) 1:2 stoichiometries.

A. Mallick et al. / Journal of Photochemistry and Photobiology C: Photochemistry Reviews 11 (2010) 62–72 69
F -CD ir
3
fTBtar
wcifl(icr1ppr
Naoa�cop
amenable to complex formation with another �-CD leading tothe formation of a 1:2 inclusion complex between NHM and �-
ig. 7. Linear fits for double reciprocal plots for complexation between NHM and �ange of �-CD.
.3.1. NHM–ˇ-CD inclusionDepending on the concentration of �-CD, NHM has been
ound to form either 1:1 or 1:2 NHM–�-CD complexes [30].he stoichiometry of the complexes are established applyingenesi–Hildebrand equation using the fluorescence intensities ofhe probe in the absence and the presence of CD [57]. For 1:1nd 1:2 complexation the said equation takes the following forms,espectively:
1
I0f − If
= 1
I0f − I′f
+ 1
K(I0f − I′f)[CD]
(2)
1
I0f − If
= 1
I0f − I′f
+ 1
K(I0f − I′f)[CD]2
(3)
here K is the equilibrium constant for the complexation pro-ess. I0
f , the fluorescence intensity of free NHM, I′f, the fluorescencentensity of the NHM–�-CD inclusion complex and If, the observeduorescence intensity. Fig. 6 reveals that neither Eq. (2) nor Eq.3) is valid for the entire range of concentration of �-CD studiedndicating that the NHM–�-CD stoichiometry in the lower CD con-entration range is different from that at higher CD concentrationange [30]. However, at lower �-CD concentration (<2 mM) plot of/(I0
f − If) against 1/[CD] and at higher �-CD concentration (>2 mM)lot of 1/(I0
f − If) against 1/[CD]2 are found to be linear [30]. Fig. 7resents the respective plots in the segmented �-CD concentrationanges.
On the basis of the above discussion it is concluded that a 1:1HM–�-CD complex is formed at lower �-CD concentration while1:2 complex is formed at higher �-CD concentration. On the basisf molecular size, it is apparent that NHM (calculated length 8.7 Å
nd transverse cross-section 5 Å) is too bulky to fit entirely in the-CD cavity (diameter 7.0 Å and depth 7.9 Å). Hence in case of 1:1omplexation a part of the probe remains exposed to the bulk aque-us phase (Scheme 3). During the process of inclusion of NHM tworobable orientations can be considered. Either the phenyl ring canScheme 3. Inclusion of NHM in �-CD cavity and cooperative proton transfer.
n aqueous solution for (a) lower concentration range and (b) higher concentration
dip inside the �-CD cavity keeping the pyridine nitrogen of NHMexposed to bulk water or the other way round. In either case thepyrrolic NH group has to reside at the periphery of the rim ofthe �-CD. Had the fluorophore been oriented in the latter patternthere would have been no scope of enhancement of cationic emis-sion because in that situation the pyridine N remains deep insideof cyclodextrin cavity where protonation is not feasible becauseof the non-polar character of the microenvironment. Since, thecationic emission of NHM (at 450 nm) increases with the forma-tion of 1:1 complex, the pyridine N is believed to remain exposedto the protic environment. Enhancement of the cationic emission ofNHM with gradual increase in the concentration of �-CD (<2 mM)is rationalized from the cooperative proton transfer mechanism asproposed by Chattopadhyay [58] based on the flip-flop motion ofthe alcoholic hydrogen present at the rim of the cyclodextrin assuggested by Saenger [59] (Scheme 3). High local concentration ofalcoholic –OH groups of �-CD at its rim undergoing cooperativeproton transfer process enables NHM to acquire proton facilitatingthe formation of the cationic species and justifies the enhancementof the cationic emission.
At higher �-CD concentrations (>2 mM), the fluorescence inten-sity of the neutral emission is found to increase at the cost ofthe cationic band. The decrease in the cationic emission indicatesthat either the stabilization of the cation decreases at higher �-CDconcentrations or increased �-CD concentration leads to a lesseravailability of water molecules (proton donor) or both happeningtogether. When a NHM molecule is entrapped by a single �-CDmolecule, a portion of the fluorophore remains exposed outsidethe cavity due to the relative size factor. This open part is also
CD (Scheme 4). This type of complexation basically encapsulatesthe probe from all around removing the solvent water molecules,
Scheme 4. 1:2 complexation between NHM and �-CD in aqueous medium.

70 A. Mallick et al. / Journal of Photochemistry and Photobiology C: Photochemistry Reviews 11 (2010) 62–72
F s (�exc = 350 nm). In (a) curves (i) → (vii) correspond to 0, 10, 20, 40, 60, 80, 100 �M HSA,r BSA, respectively.
tHtltc1i(
3
sibI[o
cwnodas
3
ossiaiaccmo
(mttts
Table 6Micropolarity values in terms of ET(30) for different forms of BSA and HSA [28].
Different states of proteins HSA BSA
ig. 8. Emission spectra of NHM as a function of (a) HSA and (b) BSA concentrationespectively, and in (b) curves (i) → (vii) correspond to 0, 20, 40, 65, 90, 140, 190 �M
he latter being essential for the formation of the cationic species.ence formation of the protonated species is restricted because of
he non-availability of the proton donor (water). At the same time, aowering in the polarity of the microenvironment also destabilizeshe cationic species. The marked lowering in the polarity and proticharacter of the microenvironment of the fluorophore within the:2 complex is thus assigned to be responsible for an enhancement
n the neutral emission (380 nm) at the cost of the cationic emission450 nm).
.4. Proteins
Serum albumins (bovine serum albumin (BSA) and humanerum albumin (HSA)) are most widely studied abundant proteinsn plasma, and are identified as the major transport proteins inlood plasma for many compounds including fatty acids [60,61].nteraction of NHM with BSA and HSA has been studied recently28,31]. The variation of the profile of NHM is modified as a functionf BSA and HSA concentrations (Fig. 8).
A greater extent of drop of the fluorescence intensity of itsationic species has been observed in HSA compared to BSAhereas the appearance of the neutral band is rather less promi-ent in HSA. The exact cause of low fluorescence quantum yieldf the neutral species of NHM in HSA environment is unknown tillate; might be originating from an energy transfer between NHMnd albumins through some unidentified mechanism [28]. Furthertudies are invited before offering an unequivocal rationalization.
.4.1. Binding with the proteins: assignment of the siteTo assess the binding site of NHM in the protein environments
ne can couple together the denaturation, micropolarity mea-urement and the fluorescence resonance energy transfer (FRET)tudies. We have determined the micropolarity around the drugn different states of the proteins, i.e., at native (N), intermedi-te (Int) and unfolded (U) states. Literature reports reveal that thentermediate state (in the presence of 4.8–5.2 M urea) [62] is char-cterized by the unfolding of domain III and partial loss of the nativeonformation of the domain I [62,63]. The unfolded (U) state isharacterized mainly by the unfolding of domain II [63–65]. Theeasured micropolarity values around the drug at different states
f both the proteins are given in Table 6.It is clear from the above table that, for the N to Int transition
involving domains I and III) there is a marked difference in the
icropolarity values and for Int to U transition (involving domain II)here is relatively smaller polarity difference in HSA. This observa-ion leads to the point that there is a possibility of the drug moleculeo be in either of the domains (I, II or III). At this situation the FRETtudy provides an effective way to check the proximity of the probe
Native (N) 54.9 57.1Intermediate (Int) 58.1 57.9Unfolded (U) 58.9 58.2
molecule to the tryptophan (Trp) residue of the proteins. In spite ofthe fact that there is a very good overlap between the emission bandof the tryptophan and the absorption band of NHM, occurrence ofthe FRET is noticed neither in HSA nor in BSA. Lack of observationof the FRET phenomenon from HSA to NHM suggests that the drugmolecule is not located near the Trp moiety in HSA; and excludesthe possibility of the localization of the drug in domain II (whereTrp-214 is situated). Measured micropolarity near domain II in HSAis around 50.1 [66]. With the present probe molecule the measuredmicropolarity at the binding site is found to be 54.1, much higherthan the normal value at domain II. This suggests that the bindingsite is domain I.
Table 6 reveals that the difference in the micropolarity values forN to Int transition of BSA (involving domains I and III) is relativelylarge compared to the polarity difference for Int to U transition(involving domain II). This implies a greater probability for theNHM molecule to reside in either of domains I or III rather thanin domain II. Again a reasonably higher micropolarity value aroundthe drug in the native BSA compared to that in HSA proposes thatthe drug is not situated in domain III since this domain is known tobe more hydrophobic. Hence the most probable binding site of thedrug in BSA is supposed to be domain I. Non-occurrence of FRET inBSA environment, further, indicates that the drug is situated awayfrom the Trp-134. So the most probable binding site of the drug inboth BSA and HSA is near domain I; and the drug is more exposedto the aqueous environment in BSA environment compared to thesituation in HSA.
4. Use of photophysical change of NHM for sensing of theinorganic anions
Analyte recognition by a molecular fluorescent sensor by anymechanism remains a challenge in the sensor research. In thisregard use of a pH sensitive probe like NHM demands attention.Recently a strategy has been developed from our school by couplinga choice of a proper pH sensitive fluorophore and the ratiomet-
ric variation of the absorption, excitation or emission responses ofthe probe for the detection of selected inorganic anions includingcyanide ion, one of the most fatal anions for the living beings, downto the micromolar level of concentrations [67]. In this work we haveexploited norharmane (NHM), to serve as an effective sensor. This
A. Mallick et al. / Journal of Photochemistry and Photobiology C: Photochemistry Reviews 11 (2010) 62–72 71
F M as( ef. [67
wtcor
iuKsm
Feb
ig. 9. Absorption (a), fluorescence (b) and fluorescence excitation (c) spectra of NHc), the emission was monitored at 450 nm. The figure has been reproduced from R
ork shows that the absorption and fluorescence excitation spec-ral patterns of NHM are highly sensitive to the presence of theyanide ion (CN−) in the solution. Presence of trace amount of KCNr NaCN leads to a remarkable ratiometric change in these spectralesponses of the sensor molecule (Fig. 9).
To verify the applicability of exploiting the molecule as inorganicon sensor in aqueous solution, some assays have been performed
sing NHM with various salts like KCl, KBr, KI, KCN, CH3COOK,H2PO4, KHSO4, KNO3, KHCO3, etc. with varying dissociation con-tants of their conjugate acids. Plot of the ratio of the absorbanceseasured at 348 and 372 nm (A348/A372) in the presence of theig. 10. Chemosensory absorption spectral response (A348/A372) of NHM in the pres-nce of 150 �M of different inorganic anions in aqueous solutions. The figure haseen reproduced from Ref. [67].
a function of KCN concentration in aqueous solution. For (b), �exc. = 348 nm and for].
same amount (150 �M) of different salts demonstrates the selec-tivity factor of the sensor molecule (Fig. 10). The spectroscopicresponses remain essentially unaffected upon addition of most ofthe salts. However, positive and graded responses are obtained for afew anions only; more prominent responses being obtained for theanions whose conjugate acids have lower dissociation constants.
The observation of the ratiometric chemosensory responsetowards some selected ions has been established to be exclusivelydue to the pH effect. This is particularly significant and prominentfor cyanide ion, because of the extremely low Ka value of its con-jugate acid (HCN). It has also been explored that the ratiometricspectral response of NHM can be utilized for the detection andestimation of the cyanide ion down to the level of micromolarconcentration [67].
5. Concluding remarks and outlook
Norharmane belonging to the indole family has a generous andexploitable photochemistry. We have summarized the photophys-ical behavior of this important molecular system in homogeneousand different confined microaggregates and pointed out their pho-tophysical and biological implications. This account shows that insome confined environments NHM has enhanced yield of the neu-tral species that has significant triplet state yield; the long-livedtriplet states, playing an important role in their photosensitiza-tion reactions in vivo. A deeper knowledge of the photophysics ofnorharmane allows a better understanding of the role of this class
of compounds in different bioenvironments and this is importantfrom a biomedical point of view since the probe is known to bean efficient photosensitizer. The review also reveals that the fluo-rophore can be successfully used for the detection and estimation ofcyanide ion, one of the most detrimental ions for the living beings.
7 otobio
A
fraa
R
[[
[[
[
[
[
[[
[[
[
[
[
[
[
[
[
[
[
[
[[[
[[[[
[[
[
[[
[
[[[
[
[[[
[[
[[[[
[[[
[[
[
[
[
2 A. Mallick et al. / Journal of Photochemistry and Ph
cknowledgements
Financial support from DBT and CSIR, Govt. of India, is grate-ully acknowledged. The authors are indebted to many laboratoriesound the globe for their research works, contributing much for thisccount. We owe to Drs. B. Haldar, P. Purkayastha, A. Chakrabartynd D. Sarkar for their investigations from our own group.
eferences
[1] R.A. Abrimovitch, I.D. Spencer, Adv. Heterocycl. Chem. 3 (1964) 79–207.[2] D. Reyman, A. Pardo, J.M.L. Poyato, J. Phys. Chem. 98 (1994) 10408–10411.[3] J.B. Hudson, G.H.N. Towers, Photochem. Photobiol. 48 (1988) 289–296.[4] G.H.N. Towers, Z. Abramowski, J. Nat. Prod. 46 (1993) 576–581.[5] H.S. Ham, K.H. Chae, Bull. Korean Chem. Soc. 7 (1986) 478–479.[6] M.G. Miguel, H.D. Burrows, M.A.E. Pereira, A.P. Varela, Colloids Surfaces A 176
(2001) 85–99.[7] S. Draxler, M.E. Lippitsch, J. Phys. Chem. 97 (1993) 11493–11496.[8] J.R.F. Allen, B.R. Holmstedt, Phytochemistry 19 (1980) 1573–1582.[9] R. Sakurovs, K.P. Ghiggino, J. Photochem. 18 (1982) 1–8.10] F.T. Vert, I.Z. Sanchez, A.O. Torrent, J. Photochem. 23 (1983) 355–368.11] D. Reyman, M.H. Vinas, J.M.L. Poyato, A. Pardo, J. Phys. Chem. A 101 (1997)
768–775.12] J. Dillon, A. Spector, K. Nakanishi, Nature 259 (1976) 422–423.13] E. Schilitter, H.J. Bein (Eds.), Medicinal Chemistry, vol. 11, Academic Press, New
York, 1967.14] V. Sniecks, R.H.F. Manske (Eds.), The Alkaloids, vol. 11, Academic Press, New
York, 1968.15] A. Dias, A.P. Varela, M.G. Miguel, A.L. Macanita, R.S. Becker, J. Phys. Chem. 96
(1992) 10290–10296.16] A.P. Varela, M.G. Miguel, A.L. Macanita, H.D. Burrows, R.S. Becker, J. Phys. Chem.
99 (1995) 16093–16100.17] G. Duportail, Int. J. Macromol. 3 (1981) 188–192.18] M.A. Munoz, C. Carmona, J. Hidalgo, P. Guardado, M. Balon, Bioorg. Med. Chem.
3 (1995) 41–47.19] M. Beljanski, M.S. Beljanski, I.R.C.S. Med. Sci. 50 (1984) 587–588.20] M. Nagao, T. Yahagi, T. Sugimura, Biochem. Biophys. Res. Commun. 83 (1978)
373–378.21] R.A. Larson, K.A. Marley, R.W. Tuveson, M.R. Berenbaum, Photochem. Photobiol.
48 (1988) 665–674.22] A. Chakrabarty, P. Das, A. Mallick, N. Chattopadhyay, J. Phys. Chem. B 112 (2008)
3684–3692.23] A. Dias, A.P. Varela, M.G. Miguel, R.S. Becker, H.D. Burrows, A.L. Macanita, J.
Phys. Chem. 100 (1996) 17970–17977.24] M. Balon, J. Hidalgo, P. Guardado, M.A. Mufioz, C. Carmona, J. Chem. Soc. Perkin
Trans. 2 (1993) 99.25] A.P. Varela, A. Dias, M.G. Miguel, R.S. Becker, A.L. Macanita, J. Phys. Chem. 99
(1995) 2239–2240.26] A.P. Varela, H.D. Burrows, P. Douglas, M.G. Miguel, J. Photochem. Photobiol. A:
Chem. 146 (2001) 29–36.
27] R.S. Becker, L.F.V. Ferreirar, F. Elisei, I. Machado, L. Latterini, Photochem. Pho-tobiol. 81 (2005) 1195–1204.28] A. Chakrabarty, A. Mallick, B. Haldar, P. Das, N. Chattopadhyay, Biomacro-
molecules 8 (2007) 920–927.29] A. Chakrabarty, A. Mallick, B. Haldar, P. Purakayastha, P. Das, N. Chattopadhyay,
Langmuir 23 (2007) 4842–4848.
[
[
[
logy C: Photochemistry Reviews 11 (2010) 62–72
30] A. Mallick, B. Haldar, N. Chattopadhyay, J. Photochem. Photobiol. B: Biol. 78(2005) 215–221.
31] A. Mallick, N. Chattopadhyay, Photochem. Photobiol. 81 (2005) 419–424.32] A. Mallick, N. Chattopadhyay, Biophys. Chem. 109 (2004) 261–270.33] R.S. Becker, M. Kasha, in: F.H. Johnson (Ed.), The Luminescence of Biological
Systems, American Association for the Advancement of Science, Washington,DC, 1995, p. 25.
34] A. Olba, F. Tomas, I. Zabala, J. Luminescence 47 (1990) 27–34.35] M.A. El-Sayed, M. Kasha, Spectrochim. Acta 15 (1960) 758–759.36] M.A. El-Sayed, J. Chem. Phys. 38 (1963) 2834–2838.37] E.C. Lim, Vibronic interactions and luminescence in aromatic molecules with
nonbonding electrons, in: E.C. Lim (Ed.), Excited States, vol. 3, Academic Press,New York, 1977, pp. 305–337.
38] A. Olba, S. Monzo, I. Zabala, J. Fluoresc. 8 (1998) 133–138.39] A. Mallick, B. Haldar, S. Maiti, S.C. Bera, N. Chattopadhyay, J. Phys. Chem. B 109
(2005) 14675–14683.40] P. Das, A. Chakrabarty, A. Mallick, N. Chattopadhyay, J. Phys. Chem. B 111 (2007)
11169–11176.41] B. Sengupta, P.K. Sengupta, Biopolymers 72 (2003) 427–434.42] J. Guharay, B. Sengupta, P.K. Sengupta, Proteins: Struct., Funct. Genet. 43 (2001)
75–81.43] A. Mallick, P. Purkayastha, N. Chattopadhyay, J. Photochem. Photobiol. C: Pho-
tochem. Rev. 8 (2007) 109–127.44] S. Hashimoto, J.K. Thomas, J. Am. Chem. Soc. 107 (1985) 4655–4659.45] K. Bhattachryya, M. Chowdhury, Chem. Rev. 93 (2003) 507–535.46] P. Pal, H. Zeng, G. Durocher, D. Girard, R. Giasson, L. Blanchard, L. Gaboury, L.
Villeneuve, J. Photochem. Photobiol. A: Chem. 98 (1996) 65–72.47] S.K. Ghosh, A. Pal, S. Kundu, M. Mandal, S. Nath, T.S. Pal, Langmuir 20 (2004)
5209–5213.48] H. Cang, D.D. Brace, M.D. Fayer, J. Phys. Chem. B 105 (2001) 10007–10015.49] M. Shannigrahi, S. Bagchi, Chem. Phys. Lett. 396 (2004) 367–371.50] S. Matzinger, D.M. Hussey, M.D. Fayer, J. Phys. Chem. B 102 (1998) 7216–
7224.51] H. Chakraborty, M. Sarkar, Langmuir 20 (2004) 3551–3558.52] H. Chakraborty, R. Banerjee, M. Sarkar, Biophys. Chem. 104 (2003) 315–
325.53] C. Rottman, D. Avnir, J. Am. Chem. Soc. 123 (2001) 5730–5734.54] V.T. D’Souza, M.L. Bender, Acc. Chem. Res. 20 (1987) 146–152.55] S. Li, W.C. Purdy, Chem. Rev. 92 (1992) 1457–1470.56] J.L. Prados, A.G. León, A.I. Olives, M.A. Martín, B.D. Castillo, J. Photochem. Pho-
tobiol. A: Chem. 173 (2005) 287–295.57] M.L. Benesi, J.H. Hildebrand, J. Am. Chem. Soc. 71 (1949) 2703–2707.58] N. Chattopadhyay, J. Photochem. Photobiol. A: Chem. 58 (1990) 31–36.59] W. Saenger, in: J.L. Atwood, J.E.D. Davis, D.D. Macnicol (Eds.), Inclusion Com-
pounds, Vol. 2, Academic Press, London, 1984, p. 231.60] H.X. Min, D.C. Carter, Nature 358 (1992) 209–215.61] T. Peters, Serum Albumin, Advances in Protein Chemistry, vol. 37, Academic
Press, New York, 1985, pp. 161–245.62] S. Tayyab, N. Sharma, M.M. Khan, Biochem. Biophys. Res. Commun. 277 (2000)
83–88.63] B. Ahmad, M.K.A. Khan, S.K. Haq, R.H. Khan, Biochem. Biophys. Res. Commun.
314 (2004) 166–173.64] M. Dockal, D.C. Carter, F. Ruker, J. Biol. Chem. 275 (2000) 3042–3050.
65] B. Ahmad, S. Parveen, R.H. Khan, Biomacromolecules 7 (2006) 1350–1356.66] A. Mallick, B. Haldar, N. Chattopadhyay, J. Phys. Chem. B 109 (2005)
14683–14690.67] D. Sarkar, A. Mallick, B. Haldar, N. Chattopadhyay, Chem. Phys. Lett. 484 (2010)
168–172.
Copyright © 2022 FDOKUMEN




















