
Phrases in literary contexts: Patterns and distributions of suspensions in Dickens’s novels
Upload
independentCategory
view
0download
0
00456535(95)0001 l-9
Chemosphere, Vol. 30, No. 6, pp. 1125-l 136, 1995 Copyright 0 1995 Ekvier Science Ltd
Printed in Great Britain. AU rights reserved 0045-6535/95 $9.5O+O.Ofl
photocatalytic lb9hedhtion of2#4&Trlnl~toluene in Aqueous suspensions of l-ltanlum Dioxide
Zhikai Wang and Charles K&al*
Department of Chemistry
The University of Georgia
Athens, Georgia 30602 USA
(Received in USA 24 September 1994; accepted 5 January 1995)
The photocatalytic destruction of 2,4,6+initrotoluene (TNT) in aqueous suspensions of
titanium dioxide (TiOg, Degussa type P25) was investigated. Under aerobic conditions,
irradiation with the Pyrex-filtered output of a 200-W high pressure mercury-arc lamp resulted
in complete (95 f 5%) mineralization of 50 ppm of TNT within a few hours. Rapid
photodegradation of TNT also occurred under anaerobic conditions but without appreciable
mineralization. In the absence of TiOg, direct photolysis of TNT yielded a complex mixture of
colored organic products in both aerobic and anaerobic environments.
INTRQDUCTION
2,4,6-Trinitrotoluene, TNT, is a conventional explosive used by military forces
worldwide. Past manufacturing and disposal practices have resulted in contamination of soil
and groundwater by this toxic nitroaromatic compound. 1 In recent years the remediation of
TNT-contaminated sites has been recognized as a challenging environmental problem, and
several remediation strategies have been implemented with varying degrees of success.
Removal of TNT from water by adsorption onto activated carbon is a proven technology, but it
suffers from problems associated with the regeneration and ultimate disposal of the spent
solid adsorbent. Biological treatmentgf or direct solar photolysis495 of TNT results in complex
mixtures of products, the identities and toxicities of which are not entirely known. Photolysis
of hydrogen peroxide/ferrous ionA!NT solutions (photo-Fenton chemistry) was reported to
1125

1126
decompose TNT,6 but no information about the nature of the photoproducts was provided.
The goal of any remediation strategy is the conversion of targeted pollutants to non-toxic,
environmentally benign products. In the ideal case, organic compounds undergo complete
mineralization to carbon dioxide and other simple inorganic molecules. Several studies have
demonstrated that a wide range of organics can be mineralized when irradiated in an aerated
suspension of an oxide semiconductor such as titanium dioxide, TiOz.7-9 While these
heterogeneous reactions often proceed by complicated mechanisms, it is generally agreed that
the photogenerated holes and electrons which reach the semiconductor surface initiate the
oxidation and reduction, respectively, of adsorbed organic species. Dioxygen, 02, also plays an
important role as an electron scavenger and source of reactive oxygen species such as
superoxide ion, Ox.
We and otherslo- have begun to examine the feasibility of using semiconductor-
mediated photocatalysis for the remediation of TNT-contaminated water. Reported here are
studies of the photolysis of this nitroaromatic compound in aqueous suspensions of TiO2 under
aerobic and anaerobic conditions. For purposes of comparison, a companion study of the direct
photochemistry of TNT in aqueous solution also is described.
-ALDETAIlS
2,4,6-Trinitrotoluene (commercially available from Chem Service with 1020% water
added) was obtained from the Environmental Research Laboratory, U.S. Environmental
Protection Agency and used as received. A 0.198 g sample was stirred with 509 mL of
deionized water (18.0 * 0.1 megaohm resistivity) for 48 hours in the dark, and the resulting
saturated solution, with a TNT concentration of 128 ppm or 5.60 x 10-4 M, was filtered through
a 0.22 pm nylon disk filter. Less concentrated samples were prepared from this stock solution
by dilution. Typically, the pH of these aqueous solutions of TNT was 6.3 + 0.1. Titanium dioxide
powder, type P25, was obtained from Degussa Corporation and used without further
treatment. This material, with a composition of -70% anatase and -30% rutile, has an average
particle size of 21 nm and a specific area of 50 m2/g. Acetonitrile (Baker HPLC grade) and
Ba(OH)2*8HzO (Fisher reagent grade) were used as received.

1127
Continuous photolyses were performed with a 200-W high pressure mercury-arc lamp
situated 40 cm corn the sample. Excitation wavelengths were confined to the region above 290
nm by use of Pyrex reaction vessels. Electronic absorption spectra were recorded on a Varian
DMS 300 spectrophotometer. Solution pH was measured with a Fisher Accumet 25 pH/Ion
meter. Photolyzed solutions were analyzed by high performance liquid chromatography
(HPLC) on an apparatus consisting of an Alltech Associates model 425 pump, a Linear
Instruments UVIS-200 absorbance detector, a Rheodyne sample injector (a 20 PL sample loop
volume was used), and a Hamilton PRP-1 reversed-phase column (25 x 0.41 cm) with an
associated Hamilton PRP-1 guard column. The mobile phase consisted of an 80:20 (v:v) mixture
of acetonitrile/water at a flow rate of 1 mL/min. Prior to use, the mobile phase was passed
through a microfiltration system (0.20 pm pore size) and then sonicated to remove dissolved
gas. Chromatograms were recorded on a Hewlett Packard 3396 Series II integrator. Previously
constructed calibration plots (slightly different plots were obtained in the presence and absence
of TiO2) were used to convert the chromatographic peak height to TNT concentration.
Measurements of total organic carbon (TOC) were performed with a Dohrmann DC-85A TOC
analyzer. Potassium hydrogen phthalate standards (l-50 mg/L) were used for sample
calibration.
For the photocatalytic experiments, a suspension of TiOz was prepared by vigorously
stirring 0.1 g of the semiconductor powder with 1 L of deionized water containing a known
concentration (typically 10 or 50 ppm) of TNT. An aliquot of this mixture was pipetted into a
cylindrical, water-jacketed photolysis cell of 2.0 cm pathlength maintained at 25.0 f 0.5% with
a constant temperature bath. During irradiation, the contents of the cell were continually
purged with water-saturated 02 or N2 gas and stirred with a small magnetic stirring bar. No
effort was made to control solution pH, which typically fell by 2.0-2.5 units for a completely
photolyzed sample. After photolysis, the sample was filtered through a 0.22 Frn nylon disk
filter to remove the larger TiO2 particles. The Iilter was then washed with acetonitrile to
dissolve any substances adhering to the semiconductor surface. The filtrate and washings
were analyzed individually by uv-vis spectrophotometry and HPLC. Similar procedures were
followed in the direct photolysis (TiO2 absent) experiments, except that the photolyzed solution
was not filtered prior to analysis.

1128
Detection and quantitation of photoproduced carbon dioxide were accomplished by
passing the 02 or N2 purge stream from the photolysis cell through two consecutive bubblers,
each containing 15 mL of an aqueous barium hydroxide solution of known concentration.
Formation of a white precipitate of barium carbonate confirmed the presence of CO2 (eq. 1) in
the gaseous effluent. The change in pH of the barium hydroxide solution provided a convenient
measure of the number of moles of CO2 liberated. This value was corrected for the small pH
change that occurs in an u&radiated blank sample purged for an identical length of time.
Ba2+ + 20H- + CO2 ----) BaCO&) + H20 (1)
RESULTS AND DISCUSSION
a Dhct Phdolysis of TNT
Figure 1 illustrates the spectral changes undergone by a Na-purged aqueous solution of
TNT (10 ppm) upon irradiation with Pyrex-filtered light. The decrease in intensity of the 234
nm absorption band is accompanied by the appearance of new features at -320 and -510 nm.
The persistence of an isosbestic point at 265 nm throughout the 120 minute photolysis suggests
that a single photochemical reaction dominates. The originally colorless solution turned pink
after 10 minutes of irradiation and retained this color thereafter.
0.80
0.60
Irradiation Times (minutes) 0, 20,40, 60, 80, 120
500 650
WAVELENGTH (IUD)
Firmre Spectral changes observed upon photolysis (lu290 nm) of TNT in a Na-bubbled aqueous solution.
1129
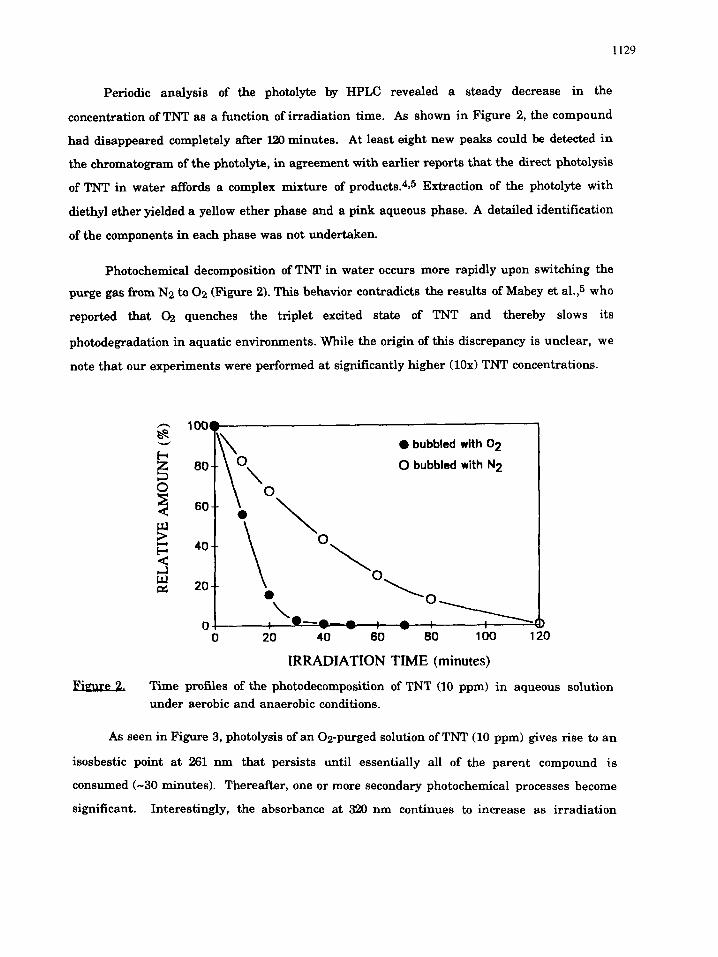
Periodic analysis of the photolyte by HPLC revealed a steady decrease in the
concentration of TNT as a function of irradiation time. As shown in Figure 2, the compound
had disappeared completely after 120 minutes. At least eight new peaks could be detected in
the chromatogram of the photolyte, in agreement with earlier reports that the direct photolysis
of TNT in water affords a complex mixture of products. 415 Extraction of the photolyte with
diethyl ether yielded a yellow ether phase and a pink aqueous phase. A detailed identification
of the components in each phase was not undertaken.
Photochemical decomposition of TNT in water occurs more rapidly upon switching the
purge gas from N2 to 02 (Figure 2). This behavior contradicts the results of Mabey et al.,5 who
reported that 02 quenches the triplet excited state of TNT and thereby slows its
photodegradation in aquatic environments. While the origin of this discrepancy is unclear, we
note that our experiments were performed at significantly higher (10x) TNT concentrations.
1000
\
80-- _\ ‘,
0
6o 0
0 bubbled with 02
0 bubbled with Np
0 20 40 60 80 100 120
IRRADIATION TIME (minutes)
m Time profiles of the photodecomposition of TNT (10 ppm) in aqueous solution under aerobic and anaerobic conditions.
As seen in Figure 3, photolysis of an On-purged solution of TNT (10 ppm) gives rise to an
isosbestic point at 261 nm that persists until essentially all of the parent compound is
consumed (-30 minutes). Thereafter, one or more secondary photochemical processes become
significant. Interestingly, the absorbance at 320 nm continues to increase as irradiation

1130
1.00
I Irradiation Times(minutes)
0.80 J/ 0, 10,20, 30, 60
0.60
0.40
0.20
0.00 . 200 GO 500 650
WAVELBN0TH(nm)
Figure Spectral changes observed upon photolysis (lu29O nm) of TNT in 02-bubbled
aqueous solution.
proceeds while that at 510 nm reaches a constant value. This behavior explains the observation
that the photolyzed solution at first appears pink and then changes to yellow. Extraction of the
photolyte with diethyl ether again yields a yellow ether phase and a pink aqueous phase.
Neither colored solution underwent detectable spectral changes when stored in the dark at
room temperature for two weeks.
Photochemical conversion of TNT to inorganic carbon in an On-purged solution was
monitored by total organic carbon analysis. Figure 4 displays TNT concentration and TOC
content of the solution as a function of irradiation time. Complete disappearance of TNT
occurred within two hours, whereas TOC underwent no perceptible change during the same
period. These results reveal that the direct photolysis of TNT in the absence TiO2 generates
organic products which resist mineralization to CO2. In contrast, addition of TiOz to the yellow
photolyte and further irradiation resulted in decolorization and a continuous decrease in TOC
(Figure 4). As shown in the next section, this ability of illuminated TiO2 to mineralize
refractory compounds forms the basis of an effective remediation strategy for TNT-
contaminated water.

1131
80
\
TOC
0
IRRADIATION TIME (hours)
lm?iamA Time profile of the photodecomposition of TNT (50 ppm) in OS-bubbled aqueous solution and the accompanying change in total organic carbon (TOC). At the time indicated by the asterisk, TiO2 was added to the sample.
Irradiating an Og-bubbled heterogeneous mixture of TiOz (0.1 g/L) and aqueous TNT (10
ppm) causes the spectral changes depicted in Figure 5. The 264-m-n absorption of TNT steadily
decreases in intensity and, significantly, no new features are evident in the near ultraviolet or
visible wavelength region. Likewise, no new bands are observed in the spectrum of the
acetonitrile used to wash the TiO2 after it was separated from the photolyzed solution. These
results establish that the photodegradation of TNT in the presence of TiOz proceeds via a
pathway that differs from the one followed in direct photolysis.
Disappearance of TNT in the irradiated TiO2 slurry obeyed first-order kinetics
(correlation coefficient = 0.998) over several half lives. As seen in Figure 6, >98% of the organic
substrate had decomposed after 69 minutes of photolysis. Most importantly, loss of TNT was
accompanied by a decrease in the TOC of the sample, albeit at a much slower rate. This
behavior suggests that TiOz-catalyzed photolysis of TNT in On-purged water initially generates
organic intermediates which subsequently undergo mineralization to CO2 and other inorganic

1132
1.20
Irradiation Times(minutea) 1.00
0,2.5,5, 10,30
8 0.80
9
8 0.60
v1 : 0.40
0.20
0.00 200 250 300 330 400 450
WAVELENGTH
Fieure Spectral changes observed upon photolysis (10290 nm) of an 02-bubbled aqueous slurry of TiO2 containing TNT (10 ppm). Spectra were measured after removing TiO2 by filtration.
compounds. The amount of CO2 produced was determined by bubbling the effluent gas from
the photolysis cell through a barium hydroxide solution and measuring the resulting change
A
\ TOC A
\
A.A\A_ 01 ??4 : 1 1 I
0.0 1 .o 2.0 3.0 4.0 5.0
IRRADIATION TIME (hours)
Fieure Time profile of the photodecomposition of TNT (50 ppm) in an Oa-bubbled aqueous slurry of TiO2 and the accompanying change in total organic carbon (TOC).

1133
in pH (recall eq. 1). Within our limits of error (* 5%), we find that TNT undergoes
stoichiometric conversion to CO2 (eq. 2). Presumably, mineralization converts the nitrogen in
TNT+ 702 hv ) 7Ca + nitrogen-containing (2) TiO, inorganic compounds
TNT to NOi and/or NH: . Recent studies of the TiOa-photocatalytic destruction of
nitroaromatic compounds identified both species among the products and demonstrated that
their relative concentrations depended upon the parent compound, its concentration, and the
degree of conversion.13114
c. Mechanistic considerations
Irradiating TiO2 with light of energy equal to or greater than the band gap excites an
electron from the valence band to the conduction band, thereby creating a separated electron-
hole (e--h+ ) pair (eq. 3).7-s These photogenerated charge carriers can recombine within the
bulk semiconductor or migrate to the surface where they also can recombine or undergo redox
processes with adsorbed species. In aqueous media, the holes can oxidize water molecules (eq.
4) or hydroxide ions (eq. 5) to form hydroxyl radicals. Conduction band electrons can be
TiO2 x e- + h+ (3)
Ti02-OH2 + h+ ---) Ti02-OH’ + H+ (4)
TiOz-OH- + h+ - Ti02-OH’ (5)
02 + e- --) 0: (6)
scavenged by 02 to produce the superoxide ion, 0: (eq. 6), which can undergo further reactions
such as reduction to O”,-, protonation to HO; , or disproportionation to 02 and Oi-.
Considerable evidence supports the assignment of the hydroxyl radical, either bound to
the semiconductor surface or diffusing freely in solution, as the active oxidizing species in the
TiOa-catalyzed photooxidation of organic substrates. 7-o In the case of TNT, *OH attack on the
methyl group and/or the aromatic ring initiates a sequence of reactions that ultimately results
in complete mineralization. Dioxygen can enhance the quantum efficiency of this process by
scavenging photogenerated electrons (eq. 6) and thereby hinder unproductive e--h+
recombination. Moreover, 02 appears to be the logical source of the reactive oxygen species

1134
required for the stoichiometric production of CO2 (eq. 2).
Given the importance of dioxygen in the photomineralization process,15 we were
surprised to find that the disappearance of TNT also occurs rapidly in an illuminated TiO2
slurry under anaerobic conditions (Figure 7). In an attempt to understand this behavior, we
photolyzed a N2-bubbled suspension of TiO2 and aqueous TNT (56 ppm) for 6 hours and
analyzed the effluent gas. Less than 6% of the TOC was converted to CO2. By comparison, an
OS-bubbled sample irradiated for 4 hours underwent 60% conversion to CO2. These results
demonstrate that the rapid loss of TNT under anaerobic conditions does not result from
mineralization. As an alternative explanation, we suggest that conduction band electrons are
captured by the electronegative nitro groups of TNT. This process should be particularly
favorable in the absence of competitive electron scavenging by 02. Precedent for a reductive
degradation pathway can be found in a variety of biological systems, where the initial steps in
0 bubbled with 02
0 bubbled with Np
IRRADIATION TIME (minutes)
Firmre Time profiles of the photodecomposition of TNT (10 ppm) in an aqueous slurry of TiO2 under aerobic and anaerobic conditions.
the metabolism of TNT effect the sequential reduction of the nitro groups, via nitroso and
hydroxylamine intermediates, to the amine. 1s Further studies are needed to determine
whether a similar pathway exists in the presence of illuminated TiO2.

1135
CONCLUDING REMARKS
Direct photolysis of TNT in water under an Og or Ng atmosphere yields a complex
mixture of colored organic products. In contrast, complete (95 f 5%) mineralization of TNT
occurs in an irradiated aqueous slurry of TiOg under aerobic conditions. While the detailed
mechanism of TiOg-catalyzed photomineralization has yet to be elucidated, a pathway
involving hydroxyl radicals as the active oxidizing species seems likely. Dioxygen can function
as a scavenger of conduction band electrons and as a source of reactive oxygen species such as
superoxide ion. Under anaerobic conditions, TiOz-catalyzed photolysis results in the rapid
degradation of TNT but without appreciable mineralization. Reduction of the nitro groups of
TNT by conduction band electrons is suggested to be responsible for this behavior. Our results
indicate that TiOg photocatalysis deserves further study as a strategy for the destruction of
TNT.
Acknowledgments. We thank the U.S. Environmental Protection Agency for financial support
and Drs. Richard G. Zepp, Eric J. Weber, William L. Miller, and Michael S. Elovitz for
technical assistance and advice.
1.
2.
3.
4.
5.
6.
7.
8.
REFERENCES
Garg, R., Grasso, D., and Hoag, G. Hazardous waste and Hazardous Mat.4?rIals 8,319
(19911.
Spiker, J. K., Crawford, D. L., and Crawford, R. L. Appl. Euviron. MicrobioL as, 3199
(1992).
Michels, J. and Gottschalk, G. Appl. Environ. Microbial. IB(T, 187 (1994).
Burlinson, N. E., Sitzman, M. E., Kaplan, L. A., and Kayser, E. J. Org. Chem. && 3695
(1979).
Mabey, W. R., Tse, D., Baraze, A., and Mill, T. Chemosphere X& 3 (1983).
Cater, S. R., Brown, P. M., Buckley, J. A., Stevens, R. D. S. U. S. Patent 5,043,080 (1991).
Turchi, C. S. and Ollis, D. F. J. Catal. m, 178 (1990).
Matthews, R. W. Pure AppL Chem. f& 1285 (1992).

1136
9. Pehzzetti, E. and Minero, C. Electrochim. Acta. a,47 (1993).
10. Schmelling, D. C. and Gray, K. A., In PhotocatalyUc purification and Tnzatment d
Water and Air, D. F. Ollis and H. AI-Ekabi, Eds., Elsevier, Amsterdam, 1993, p. 625.
11. Prairie, M. R., Stange, B. M., Rodacy, P. R., Leslie, P. K., Huang, M., and Datye, A. K.,
Abstract for First In&national tinfervmce m Advanced Oxidation Technologies #or
Water and Air F&mediation, June 2530,1994, London, Ontario, Canada, p. 194.
12. Sierka, R. A., Abstract for First In&national Co- on Advanced oxidation
Technolo@s finr Water and Air Remediation, June 25-30, 1994, London, Ontario,
Canada, p. 199.
13. Low, G. K.-C., McEvoy, S. R., and Matthews, R. W. Environ. Sci. Technol. f&, 460 (1991).
14. Pelizzetti, E., Minero, C., Piccinini, P., and Vincenti, M. Coord. Chem. Rev. L&L 183
(1993).
15. Barbeni, M., Pramauro, E., Pelizzetti, E., Borgarello, E., Gratzel, M., and Serpone, N.
NOUV. J. Chim. 8,547 (1964).
16. McCormick, N. G., Feeherry, F. E., and Levinson, H. S. Appl. Enviroa Micxwbiol 3,
949 (1976).
Copyright © 2022 FDOKUMEN




















