
Tightening the large diameter anchor bolts of sign, signal, and ...
Upload
independentCategory
view
3download
0
REVIEW
PDGF: the nuts and bolts of signalling toolbox
Ammad Ahmad Farooqi & Salman Waseem & Asma M. Riaz & Bilal Ahmed Dilawar &
Shahzeray Mukhtar & Sehrish Minhaj & Makhdoom Saad Waseem & Suneel Daniel &Beenish Ali Malik & Ali Nawaz & Shahzad Bhatti
Received: 28 May 2011 /Accepted: 7 July 2011 /Published online: 19 July 2011# International Society of Oncology and BioMarkers (ISOBM) 2011
Abstract PDGF is a growth factor and is extensivelyinvolved in multi-dimensional cellular dynamics. Itswitches on a plethora of molecules other than itsclassical pathway. It is engaged in various transitions ofdevelopment; however, if the unleashed potentials leadastray, it brings forth tumourigenesis. Conventionally, ithas been assumed that the components of this signallingpathway show fidelity and act with a high degree ofautonomy. However, as illustrated by the PDGF signaltransduction, reinterpretation of recent data suggests thatmachinery is often shared between multiple pathways,and other components crosstalk to each other throughmultiple mechanisms. It is important to note thatmetastatic cascade is an intricate process that we haveonly begun to understand in recent years. Many of theearly steps of this PDGF cascade are not readilytargetable in the clinic. In this review, we will unravelthe paradoxes with reference to mitrons and cellularplasticity and discuss how disruption of signalling cascadetriggers cellular proliferation phase transition and metas-tasis. We will also focus on the therapeutic interventionsto counteract resultant molecular disorders.
Keywords PDGF/PDGFR .Mitrons . Cancer . EGCG .
Sulforaphane . Curcumin
Introduction
PDGFs are the growth factors which have a broadspectrum of implications. These factors are involved inmultifaceted mechanistic details. Various activities of thecell are triggered ranging from organogenesis to repair.The amplitude of the signals determines the fate of thecell. In case of derailed or deregulated transduction,error-prone activities are instigated. Recent data suggestthat various isoforms of PDGF bind with differentialaffinities to two related tyrosine kinase receptors,denoted the PDGF α- and β-receptors. The binding ofligand to native receptor induces receptor dimerization,creating receptor homo- or heterodimers. Dimerizationconsequently results in receptor autophosphorylation andkinase activation. It is intriguing to note that receptorautophosphorylation serves to regulate the kinase activityand to generate scaffolds for downstream signallingcomponents [1–9].
Substantial fraction of information has been added inunderstanding signalling events that drive PDGF signaltransduction cascade. Various key players have been wellcharacterized; however, other aspects of the signallingmechanisms involved are still uncertain [10–12]. Thisreview emphasizes on the major determinants of PDGF-mediated transduction cascade and explores how disparatesignalling pathways synergize to regulate cellular activity.Thus, we devote the various sections of this multi-partreview to discussing the undeniable role of PDGF signal-ling in smooth muscle cell regulation, angiogenesis andrespective emerging therapeutic paradigms.
Furthermore, we bring the PDGF-signalling pathwayinto focus, chronicling key concepts and recent advanceswith respect to microRNA (miRNA) signalling in normaland cancerous cells.
A. A. Farooqi (*) : S. Waseem :A. M. Riaz : B. A. Dilawar :S. Mukhtar : S. Minhaj :M. S. Waseem : S. Daniel :B. A. Malik :A. Nawaz : S. Bhatti (*)Institute of Molecular Biology and Biotechnology (IMBB),The University of Lahore,1 km defence road,Lahore, Pakistane-mail: [email protected]: [email protected]
Tumor Biol. (2011) 32:1057–1070DOI 10.1007/s13277-011-0212-3

Differential PDGFR expression in vascular smooth musclecells
It is worth mentioning that serum-starved human arterialand venous smooth muscle cells (SMCs) showed differ-ential proliferative responses to PDGF isoforms. Datasuggested that arterial SMCs were strongly stimulated byPDGF-AA. On the contrary, venous SMCs exhibitedenhanced responsiveness to PDGF-BB. This differentialresponse was accredited to divergence in PDGF receptorexpression. There was a 2.5-fold higher (P<0.05) densityof PDGF receptor-α (PDGF-Rα) and a 6.6-fold lower (P<0.05) density of PDGF-Rβ expression while drawing aparallel between arterial and venous SMCs. Treatment ofarterial SMCs with PDGF-AA resulted in a markedPDGF-Rα activation, escalated phosphorylation ofERK1/2 and Akt and subsequent activation of c-JunNH2-terminal kinase (JNK) and a considerable suppres-sion in expression of the cell cycle inhibitor p27(kip1).However, there was PDGF-Rβ activation and simulta-neous epidermal growth factor receptor (EGFR) trans-activation upon treatment with PDGF-BB in venous SMCs[13].
It has recently been documented that PDGF receptor(PDGFR)-β expression is upregulated in high-grade astro-cytomas. Kaplan–Meier analysis confirmed that the densityof SMA-Vs, the size of SMA-Vs and PDGFR-β expressionwere major prognostic factors [14].
PDGF crosstalks in smooth cell regulation
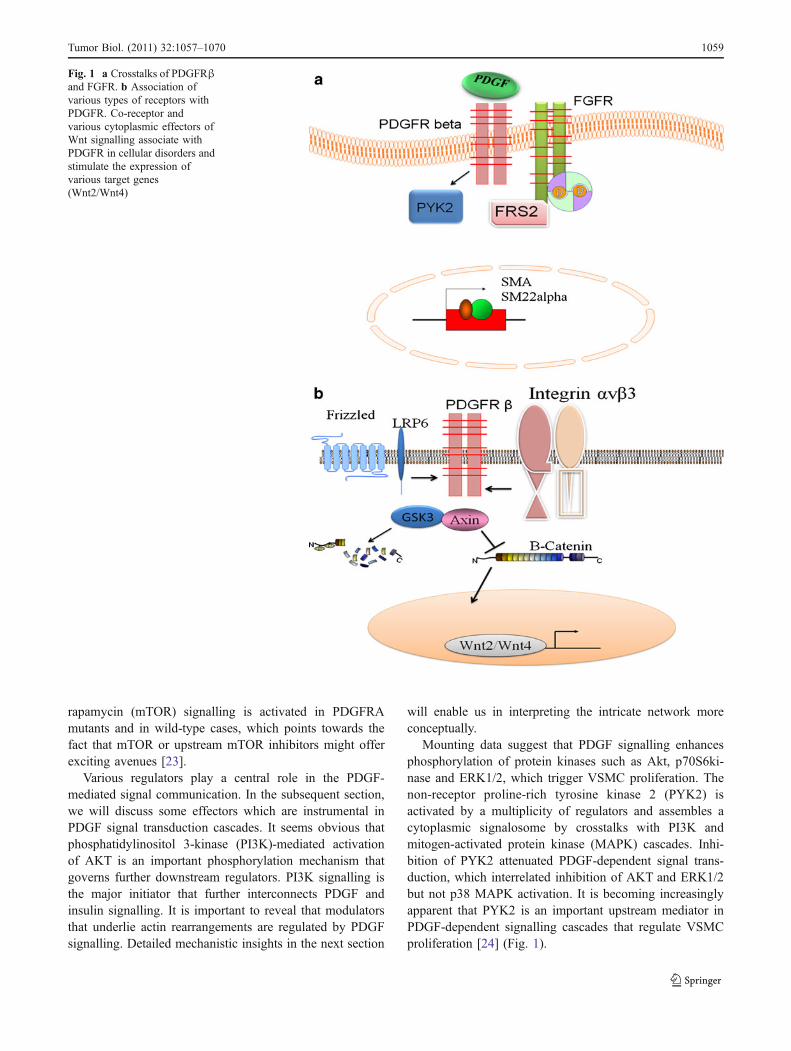
PDGF signalling orchestrates with various signalling whichnegatively regulate PDGF signal dissemination. But thereare various situations in which mutations in the effectors ofvarious signalling cascades potentiate PDGF transductioncascade. Considerable information unfolds a correlationbetween various effectors of Wnt signalling with PDGFtransduction cascade. It has recently been shown thathuman atherosclerotic coronary arteries showed noticeablyenhanced expression of mutant LRP6 (R611C) and coloc-alization with PDGFR-β. It is interesting to note that wild-type LRP6 associates with PDGFR-β and enhances itslysosomal degradation. In accordance with the sameapproach, another receptor that co-existed with PDGFR-βis integrin αvβ3. This receptor offered binding site fortenascin-C. Treatment of smooth muscle cells withtenascin-C and PDGF-BB resulted in an enhanced prolif-eration [15, 16]. A detailed analysis of mutations in Wntsignalling is necessary to evaluate collaboration or conniv-ance with PDGF signalling. Treatment of pulmonaryarterial smooth muscle cell (PASMC) with PDGF inducedGSK3ß inactivation. However, treatment with the PDGFRinhibitor, imatinib, attenuated PDGF-BB-mediated GSK3ß
phosphorylation. This highlights the fact that Wnt signal-ling is positively regulated by PDGF signalling. In supportof this interpretation, a recent documentation suggested thatPDGF-BB treatment resulted in the stimulation of Wnt2and Wnt4 mRNA in proliferating vascular smooth musclecells (VSMCs) (Fig. 1b) [17, 18]. Further investigations areimportant to explore more components of positive feedbackloop that drives cellular disorders
It has lately been explored that PDGFR-β crosstalkswith EGFR upon activation and undergoes cleavage bymolecular scissor (ADAM17). This enzyme is also in-volved in cleavage of EGF from cell surface [19].
It is nonetheless recently investigated that PDGFRassociates with various receptors which potentiates anddrives derailed activities of core biological systems. In thefollowing section, we discuss the types of receptors thatresult in integration of linear transduction cascades.Moreover, we also summarize cytoplasmic effectors ofvarious signalling paradigms which coordinate or antago-nize PDGF-mediated signal communication.
It is intriguing to explore that adaptor protein FRS2is necessary for fibroblast growth factor receptor 1(FGFR1)-mediated phenotypic modulation and suppres-sion of VSMC smooth muscle alpha-actin (SMA) geneexpression. PDGF-BB and FGF2 act synchronously totrigger cell proliferation and downregulate SMA andSM22alpha in VSMC. PDGF-BB modulates tyrosinephosphorylation of FGFR1, and this phosphoproteomeactivity is mediated by PDGF receptor-beta. FRS2 co-exists with PDGFRbeta in a complex that requiresFGFR1, and both the extracellular and the intracellulardomains of FGFR1 are required for association withPDGFRbeta, whereas the cytoplasmic domain of FGFR1is required for FRS2 association with the FGFR1-PDGFRbeta complex. Abrogation of FRS2 in VSMC byRNA interference suppressed PDGF-BB-mediated down-regulation of SMA and SM22alpha; however, PDGF-BB-mediated cell proliferation or ERK activation was notdisrupted [20].
Data suggest that there is a tight interaction betweenPDGF and NRP as NRP1 and PDGFR-α co-localised inVSMCs. HCASMC migration induced by PDGF-BB andPDGF-AA was inhibited by NRP1 silencing and byadenoviral overexpression of an NRP1 mutant deficient inintracellular domain (Ad.NRP1ΔC) [21].
FGFR2 stimulates the expression of EGFR andPDGFRalpha via activation of PKCalpha-dependent AP-1transcriptional activity. Furthermore, Cbl-mediated degra-dation of EGFR is inhibited by enhanced association of Cblwith sprout which is triggered by FGFR. It is understand-able that deregulated molecular crosstalks between activat-ed FGFR2, EGFR and PDGFRalpha functionally contributeto the cellular disorders [22]. Mammalian target of
1058 Tumor Biol. (2011) 32:1057–1070
rapamycin (mTOR) signalling is activated in PDGFRAmutants and in wild-type cases, which points towards thefact that mTOR or upstream mTOR inhibitors might offerexciting avenues [23].
Various regulators play a central role in the PDGF-mediated signal communication. In the subsequent section,we will discuss some effectors which are instrumental inPDGF signal transduction cascades. It seems obvious thatphosphatidylinositol 3-kinase (PI3K)-mediated activationof AKT is an important phosphorylation mechanism thatgoverns further downstream regulators. PI3K signalling isthe major initiator that further interconnects PDGF andinsulin signalling. It is important to reveal that modulatorsthat underlie actin rearrangements are regulated by PDGFsignalling. Detailed mechanistic insights in the next section
will enable us in interpreting the intricate network moreconceptually.
Mounting data suggest that PDGF signalling enhancesphosphorylation of protein kinases such as Akt, p70S6ki-nase and ERK1/2, which trigger VSMC proliferation. Thenon-receptor proline-rich tyrosine kinase 2 (PYK2) isactivated by a multiplicity of regulators and assembles acytoplasmic signalosome by crosstalks with PI3K andmitogen-activated protein kinase (MAPK) cascades. Inhi-bition of PYK2 attenuated PDGF-dependent signal trans-duction, which interrelated inhibition of AKT and ERK1/2but not p38 MAPK activation. It is becoming increasinglyapparent that PYK2 is an important upstream mediator inPDGF-dependent signalling cascades that regulate VSMCproliferation [24] (Fig. 1).
Fig. 1 a Crosstalks of PDGFRβand FGFR. b Association ofvarious types of receptors withPDGFR. Co-receptor andvarious cytoplasmic effectors ofWnt signalling associate withPDGFR in cellular disorders andstimulate the expression ofvarious target genes(Wnt2/Wnt4)
Tumor Biol. (2011) 32:1057–1070 1059

Additionally, PDGF enhanced IRS-1/IRS-2 serine phos-phorylation and downregulated IRS-2 expression in aspatio-temporal manner. Outstandingly, abrogation of PI 3-kinase and mTOR attenuated PDGF-modulated Akt andp70S6kinase phosphorylation. Furthermore, IRS-1 serinephosphorylation and IRS-2 downregulation was alsoblocked. Contrary to this, MEK1/ERK inhibitor (U0126)failed to inhibit PDGF-induced IRS-1 serine phosphoryla-tion and IRS-2 downregulation. It is intriguing to note thatPDGF-mediated IRS-1/IRS-2 dysregulation resulted in thedampening of insulin-induced IRS-1/IRS-2-associated PI 3-kinase activity. Purposefully, targeted inhibition of PDGFreceptor tyrosine kinase with imatinib reversed IRS-1/IRS-2dysregulation and retrieved insulin receptor signalling [25].The integration of these multiple layers of new proteins willhave a major impact in translational oncology, leading tosignificant breakthroughs in rational drug design.
It has lately been explored that PDGF induced migrationin rat airway smooth muscle cells by increasing WASP andArp2/3 protein levels along with actin reorganization.However, treatment of cells with Slit2-N inhibited RASMcells migration by suppressing the expressions of WASPand Arp2/3 and consequent actin rearrangement [26]
It has lately been found that SMCs cultured in fibrinhydrogels have a more robust chemotactic response toPDGF-BB compared with FGF-2 [27]. Growth factors(GFs) have remarkable interactions with nonproteoglycanextracellular matrix proteins. In accordance with thisassumption, 12th–14th type three repeats of fibronectin(FN III12-14) were shown to have a higher affinity forPDGF. It is noteworthy that sprouting of human smoothmusclecell spheroids was significantly improved after administrationof FN III12-14 as a carrier of growth factors [28].
Impact of PDGFR fusions on cellular signalling
There are various instances in which there is a genomicrearrangement that results in the formation of fusion/chimericprotein. Focusing on the same concept, a research groupdocumented that occasionally, chimeric receptor, containingthe extracellular domain of hPDGFRbeta, gets fused to thetransmembrane and intracellular regions of discoidin domainreceptor (DDR1). Although chimeric receptors, which arecomposed of various combinations of intracellular regionsfrom DDR1 and TrkA (with the extracellular domain ofhPDGFRbeta), depict ligand (PDGF)-inducible receptorresponses. The DDR1 is characterized by a discoidin I motifin the extracellular domain, a juxtamembrane segment and akinase domain that is 45% similar in structure to that of theNGF receptor, TrkA [29]. Consistently, TEL-PDGFRbeta(TPbeta, also called ETV6-PDGFRB) and FIP1L1-PDGFRalpha (FPalpha) are fusion proteins which circum-vent Cbl-mediated degradation. Ubiquitination of TPbeta and
FPalpha was significantly suppressed compared to that ofwild-type receptors in cells expressing hybrid receptors [30].However, it has lately been documented that Lnkmimetic drugs might provide a novel therapeutic strategyas it is a negative regulator of PDGFR signalling, and itcan bind to the FIP1L1-PDGFRA fusion protein andattenuate signal dissemination [31]. Moreover, fusiononcoproteins (KANK1-PDGFRB) are involved in activatingSTAT independent of JAK. Additionally, phosphorylation ofphospholipase C-ERK1 and ERK2was triggered by this fusedoncoprotein [32].
Role of PDGF in different metastatic and angiogeniccascades
To ensure the correct balance between vascular mainte-nance, division and differentiation, several regulatorymechanisms that modulate the PDGF signal at many stagesof the pathway have been described. Here, we discuss thenature of the PDGF signal and evaluate the catalogue ofmechanisms that adjust it, demonstrating how the finemodulation of PDGF signalling in this context can result inprecise and robust control of vascular remodelling.
It is worth mentioning that quiescent vascular endothelialcells insensitive to PDGF-BB stimulation are re-sensitizedafter stimulation with FGF-2, which transcriptionally switcheson PDGF receptor expression in the activated endothelial cells[33]. Another interesting feature is that PDGF-BB triggeredMAP kinase activity and cell motility of isolated lymphaticendothelial cells. In vivo, PDGF-BB enhanced the growth oflymphatic vessels. Enforced expression of PDGF-BB inmurine fibrosarcoma cells induced tumour lymphangio-genesis, leading to enhanced metastasis in lymph nodes [34].
It is an intriguing feature that PDGFR signallingpotentiated epithelial mesenchymal transition (EMT) andcaused survival in murine and human mammary carcinomacell lines. Moreover, TGF-beta enhanced metastasis ofmammary tumours, induced EMT and escalated PDGFRsignalling [35]. PDGF and PDGFR-α were overexpressedin thymoma, especially in type B2 and B3, in the tumourepithelial cells [36]. Combinatorial blockade of PDGF andVEGFR might offer exciting avenues in management ofadvanced-stage mouse corneal neovascularization andintestinal-type/diffuse-type gastric carcinomas [37, 38].
Role of PDGF in breast cancer progression
Breast carcinogenesis arises from aberrant decisionmaking by cells concerning their survival or death,proliferation or quiescence, damage repair or bypass.These decisions are made by a plethora of growth factor-mediated molecular signalling networks that process
1060 Tumor Biol. (2011) 32:1057–1070

information from outside and from within the breastcancer cell and commence responses that decide thecellular survival and invasive potential.
PDGF-D is found to be upregulated in invasive breastcancer cell lines; it also correlates with Notch-1 expressionand increases DNA binding activity of NFKB [39, 40].Furthermore phospholipase D is also an important memberof this molecular hierarchy including PDGF-D-inducedNFKB's activation. Two binding sites of NFKB arecritically important for transcriptional activation of PLD-1which is further involved in carcinogenesis [41]. Recentdata suggest that PDGF-BB is significantly higher inpatients with breast cancer compared to the benign breastdisease patients. It is therefore important that analysis ofangiogenesis markers in tumour and serum of breast cancerpatients using multiplex protein assay can enhance diagno-sis and prognosis in this disease [42]. On a similar note,another interesting piece of evidence is that human breastcancers express high levels of PDGF-D. Overexpression ofPDGF-D enhanced tumour growth and lymph nodemetastasis via uncontrolled cellular proliferation and inductionof CXCR4 expression. Abrogation of CXCR4 signallingcompromised PDGF-D-induced lymph node metastasis. Be-sides, enforced expression of PDGF-D increased perivascularcell coverage and normalized tumour blood vessels. Conse-quently, PDGF-D overexpression potentiated drug influx ofdoxorubicin and enhanced its treatment efficacy [43].
It is noteworthy that a considerable number of adiposetissue stem cells (ASCs) migrated towards the tumour-conditioned medium, and more importantly, migration ofhuman ASCs remarkably increased in response to increasedconcentrations of recombinant PDGF-BB. On the contrary,neutralizing antibodies to PDGF receptor-beta decreasedmigration of ASCs towards a breast cancer-conditionedmedium. These data advocate that tumour cell-derivedPDGF-BB is an imperative factor in governing themicroenvironment communication between tumour cellsand local tissue-resident stem cells [44].
MicroRNAs deal with PDGF: opposite sides of the same coin
MicroRNAs are major determinants in the post-transcriptionalcontrol of gene expression. After the classification ofhundreds of miRNAs, the challenge is now to comprehendtheir specific, biologically distinct and differential activity.Signalling pathways are important candidates for miRNA-modulated regulation owing to the sharp dose-sensitive natureof their implications and resultant effects. Undeniably,emerging verification suggests that miRNAs affect theresponsiveness of cells to signalling molecules such as PDGF.Nonetheless, miRNAs serve as nodes of intricate signallingnetwork that guarantee homeostasis and regulate cancer andmetastasis. This is illustrated in Fig. 2.
Growing evidence suggests that transcription of collagenis triggered by miR-29a. Another important facet is thatTGFbeta, PDGF-B or IL-4 reduced the levels of miR-29a innormal fibroblasts. However, abrogation of PDGF-B andTGFbeta pathways by treatment with imatinib recapitulatedthe levels of miR-29a [45].
PDGF is instrumental in cellular migration and oligo-dendrocyte differentiation. Interestingly, PDGF is transcrip-tionally regulated by mitrons which impact various cellularactivities.
It is also captivating to note that loss of miR-143/145results in the formation of podosomes, which are actin-richmembrane protrusions implicated in cellular migrationincluding SMCs. Consistent with the equivalent concept,PDGF modulates podosome formation in SMCs throughthe regulation of miR-143/145 expression. PDGF receptoralpha is the target gene of miR-143 [46]. Furthermore, miR-219 represses the expression of PDGFRalpha, whichnormally helps to promote oligodendrocyte precursor cellproliferation [47].
Another important miRNA believed to be involved inregulation of PDGF receptor is miR-9. It is an activation-induced regulator of PDGFR-β expression in cardiomyo-cytes that negatively regulates PDGFR-β expression uponligand stimulation through direct interaction with the 3′UTR of PDFGR-β [48]. There are various situations inwhich miRNA influences the PDGFR via intermediateproteins. In accordance with this conception, growth factor-induced miR-296 degrades the hepatocyte growth factor-regulated tyrosine kinase substrate (HGS) mRNA, resultingin substantial decrease in levels of HGS and thus suppressingHGS-mediated degradation of the growth factor receptorPDGFRbeta [49]. Additionally, miRNA140 negatively regu-lates PDGF signalling during palatal development; however,disruption of PDGF signalling causes palatal clefting. ThePDGF receptor alpha (PDGFRα) 3′UTR contained amiRNA140 binding site functioning in the negative regula-tion of PDGFRα protein levels in vivo [50].
PDGF intracellular signalling pathways trigger geneexpression during differentiation by regulating microRNAexpression [51]. PDGF negatively regulates proteins andeffectors of TGF transduction cascade via mitrons. Concor-dant with the same approach, PDGF-BB induces microRNA-24 (miR-24), which in turn leads to downregulation ofTribbles-like protein-3 (Trb3). Suppression of Trb3 is tightlycorrelated with reduced expression of Smad proteins anddecrease in BMP and TGFbeta signalling [52]. Evidencesuggest that PDGF-D-induced EMT in prostate cancer,through suppression of miR-200. Therefore, miR-200 recon-stitution offers a promising approach for the treatment ofinvasive prostate cancer [53]. Various oncomirs are inducedby PDGF which potentiate uncontrolled cellular division. Inthis context, miR-221 is transcriptionally induced upon
Tumor Biol. (2011) 32:1057–1070 1061

PDGF treatment in primary VSMCs and enhances cancerprogression [54].
PDGF's dynamics with LRP
PDGF signal transduction has emerged as a governing bodyof biological processes and propagation of information inthe cell. A wealth of information has been gained about thecentral regulation of PDGF pathway in endocytic traffick-ing and protein degradation
Lipoprotein receptor-related protein (LRP) has beenacclaimed as a guardian of PDGF/PDGFR. It crosstalkswith a broad range of proteins which underlie suppressionof PDGF signalling. In support of this notion, factor VII-activating protease (FSAP) can inhibit neointima formationand VSMC proliferation by cleavage of PDGF-BB.Protease nexin-1 inhibits the enzymatic activity of FSAP.FSAP–inhibitor complex is internalized via LRP whichresults in neutralization of its effect on PDGF-BB-mediated
VSMC proliferation [55]. On a similar note, LRP chaper-ones PDGFR from degradation. Cells deficient in LRPshowed an enhanced degradation rate of PDGFR; however,cells reconstituted for LRP restored the levels of PDGFR[56]. Activated PDGFR undergoes tyrosine phosphoryla-tion and consequently interacts with a variety of signallingmolecules, including PI3K as regulation of PI3 kinase byPDGFRbeta is necessary for vascular integrity [57].Captivatingly, MAPK is another downstream substratethat is documented to be modulated by PDGFR, andLRP abrogation resulted in suppression of MAPK-mediated signalling [58]. However, there is a documenta-tion that distinctly highlights the fact that abrogation ofLRP results in an enhanced accumulation of effectors ofTGF signalling in the nucleus and a robust expression ofPDGFRβ [59]. It appears as if it opens another avenue forinvestigation of transcriptome profiling in LRP-deficientenvironment. Diagrammatic representation of the activitiesis shown in Fig. 3.
Fig. 2 Involvement of PDGF in mediating transcriptional responses via miRNA
1062 Tumor Biol. (2011) 32:1057–1070

PTP, PDGF and PKC: referee between two wrestlers
It is significant to note that there is a push and pull betweentwo diametrically opposed enzyme groups in regulatingphosphoproteome. The overexpression, hyperactivation orsuppression of these two proteins dictate endocytosis orrecycling of PDGFR, PKC and T cell protein-tyrosinephosphatase (TC-PTP) works in an anti-parallel manner;PTP attenuates recycling and enhances endocytosis. Con-versely, PKC is involved in the recycling of the receptor viaRab4a [60]. PKC and PTP are two opposing enzymes thatimpact phosphoproteome and resultant cellular activities.Concordant with the same concept, low molecular weightphosphotyrosine phosphatase (LMW-PTP) is involved inplatelet-derived growth factor-mediated cellular activities.In the forthcoming section, we bring to the limelight thefactors which underpin suppression and activation of PTP.
Evidence supports the notion that LMW-PTP docked toand dephosphorylated Tyr-857 of activated PDGFR, thusinhibiting the cell [61]. Another aspect in concordance withthe regulation was documented by a research group whosuggested increment in neointima formation upon PTPabrogation [62]. An additional aspect is that loss of TC-PTPrecapitulates PDGF beta receptor on the cellular surface viarapid recycling, which displays differential traffickingpatterns of PDGF receptor family members. Analysis byconfocal microscopy has shown that in TC-PTP ko mouseembryonic fibroblasts (MEFs), activated PDGF beta receptors
co-existed with Rab4a. Rab4a is a marker for retrieval ofendocytosed entities on the cell membrane. In agreement withthis, cells reconstituted for a dominant-negative Rab4a escalat-ed clearance rate of cell surface receptors on TC-PTP ko MEFs[63]. Analogously, hydrogen peroxide suppressed the activityof PTP [64]. Fascinatingly, hypoxia suppressed expression ofvarious PTPs (T cell PTP, density-enhanced phosphatase-1,PTP1B and SH2 domain-containing phosphatase-2), ensuingin reduced PTP activity. Hypoxia downregulates expressionand activity of PDGFR-β antagonizing PTPs in a HIF-1α-dependent manner [65]. Recent data suggest that lipidperoxides are earlier unrecognized mediators of oxidation ofPTPs. Nonetheless, it is a vital pathway for control of receptortyrosine kinase signalling, which might also underlie diseasesassociated with increased lipid peroxidation [66]. Hypoxiaand hypoxia/reoxygenation (H/R) escalates generation ofreactive oxygen species (ROS). Candidate molecular targetsof ROS are the catalytic site cysteine of PTPs [67, 68].Superoxide dismutase 1 (SOD1) is copper/zinc enzyme foundin the cytoplasm that converts superoxide into hydrogenperoxide and molecular oxygen. In addition, SOD1 inhibitioncauses the downregulation of the PDGF receptor [69].
PDGF and dorsal ruffle formation
The plasma membrane is the supramolecular assembly thatundergoes highly synchronized protrusions and invagina-tions that support the configuration of dorsal ruffles. These
Fig. 3 Effectors involved invarious PDGF-mediated cellularactivities
Tumor Biol. (2011) 32:1057–1070 1063

modellings are triggered by highly conserved actin–mem-brane machinery. These transient surface membrane dis-tortions are discrete. Their role is to help the cell as itmigrates, attaches and invades.
PDGF modulates dorsal ruffle formation in coordinationwith various regulators. In this framework, mammalian actin-binding protein-1 (mAbp1) is localised to dorsal ruffles and isrequired for PDGF-mediated dorsal ruffle formation. Addi-tionally, mAbp1 directly interacts with the actin regulatoryprotein WASp-interacting protein (WIP) through its SH3domain as this interaction between mAbp1 and WIP issignificant in regulating dorsal ruffle formation [70]. Anotherprotein that is central in dorsal ruffle formation is Gab1,located downstream to PDGFR. Gab1 associates constitu-tively with the actin-nucleating factor N-WASP. FollowingPDGFR activation, Gab1 recruits Nck, an activator of N-WASP, resulting in assembly of signalosome, localised todorsal ruffles. Organization of dorsal ruffles involves interac-tion between Gab1 and Nck and also requires functional N-WASP [71]. Particularly, activated Rac1 mediates membranedorsal ruffle formation in response to PDGF. Similarly, Abl-interactor-1 and betaPIX, a guanine nucleotide exchangefactor for Rac1, localise at these Rac1-induced actinstructures and occupy central positions in the induction ofmembrane dorsal ruffling in response to PDGF in fibroblasts[72]. Similarly, a pool of PDGF-activated Src family ofprotein-tyrosine kinases (SFKs) that was insensitive tomembrane cholesterol depletion was found in non-caveolaefractions. Non-caveolin SFK activation was connected to thecompetence of PDGF to induce F-actin conformationalreorientations leading to dorsal ruffle formation [73]. On asimilar note, direct and specific interaction between palladinand Eps8 facilitates rapid and transient remodelling of theactin cytoskeleton, which drives the formation of highlydynamic membrane protrusions after treatment with PDGF[74]. Some proteins suppress the PDGF-mediated dorsalruffle formation. In concordance with this notion, cdc-42interacting protein 4 (CIP4) was observed to suppressPDGFR which blunted ruffle formation. However, knock-down of CIP4 recapitulated dorsal ruffle formation andcellular migration [75].
It is intriguing to note that PDGF induced a threefoldincrease in the proportion of PP2A activity regulated bycholesterol. On the contrary, cholesterol depletion inhibiteddorsal ruffle formation, decreased PP2A levels and increasedthe Hsp27-P to Hsp27 ratio. The documentation highlights thefact that Hsp27 is dephosphorylated by PP2A in dorsal ruffles,in non-caveolar lipid raft microdomains [76].
PDGF interaction with ATM and hetronuclear ribonucleoprotein
PDGF triggered interaction between hnRNP-K and themRNA-encoding myosin regulatory light-chain (MRLC)-
interacting protein (MIR). MIR is an E(3)-ubiquitin ligasethat is documented to be involved in degradation of MRLC.This sequentially rapidly increased MIR expression and ledto ubiquitination and proteasome-mediated degradation ofMRLC and subsequent actin rearrangements [77]. Recentdata suggest that hnRNP-K modulates the expression ofVEGF [78]. A detailed analysis is necessary to pinpoint theinvolvement of PDGF in stimulating the expression ofVEGF via hnRNP-K. Furthermore, attenuation of hnRNP-K suppressed the levels of active MEK and ERK whichalso confirms the fact that hnRNP-K potentiates the “MEK-and ERK-based linear transduction cascade [79].
Emerging evidence points towards an interaction betweenataxia telangiectasia mutated (ATM) and PDGF. It is ofparticular interest that PDGF beta receptor abrogationseverely compromised H2O2-induced ATM activation, indi-cating that ATM lies downstream to the PDGF beta receptorin this signalling cascade [80]. Furthermore, it has recentlybeen cited that PDGFR mediates cytoplasmic activation ofATM [81]. The evidence suggests that a complex barcodeunderlies the heterogeneous response of ATM towardsPDGF/PDGFR.
PDGF and non-invasive EMT
It is compelling to note that correct establishment andmaintenance of cell polarity are decisive for normal cellularphysiological activities. On the other hand, loss of cellpolarity is one of the hallmarks of cancer. In the upcomingsection, we will focus on identifying the stages of tumouraldevelopment that are affected by deregulation of PDGFlinear or integrated signal transduction cascade. Asymmet-ric division is a well-acclaimed regulatory mechanism thattriggers cell numbers and differentiation.
An imperative aspect of PDGF signalling is that it isinvolved in EMT but simultaneously involved in resistingcellular migration, which is an aspect of non-invasivetumour. It is convincing to note that TGFR independentPDGF transduction cascade was instrumental in EMT. EMTwas observed in both SMAD competent and deficient cells,which proves that PDGF-mediated EMT is irrespective ofTGF signalling [82]. The novel findings also show thatPDGF had a role in non-invasive EMT; in this mechanism,there is again no correlation between TGFβ/Smad signal-ling with that of PDGF. The phenomenon was confirmed inmesothelial cells by an increased SNAIL and decreased E-cadherin expression with presence of epithelial and mesen-chymal markers [83]. Scientists have started unveiling thelinks between cell polarity and asymmetric cell division inthe context of cancer. Consistent with same approach,apical–basal polarity and cell–cell adhesion are tightlyinterconnected. Therefore, how loss of cell polarity inepithelial cells may promote epithelial mesenchymal tran-
1064 Tumor Biol. (2011) 32:1057–1070

sition and metastasis needs a detailed investigation.Altogether, it is obvious that PDGF signalling is contribu-tory in loss of epithelial cell polarity and may have animportant role in both the initiation of tumourigenesis andin later stages of tumour development.
PDGF as a major challenge in translational oncology
Recent structural data support the idea that PDGFR is asophisticated machine with multiple signalling outputs. Mis-representation of growth factor signalling is the most impera-tive prerequisite in tumour progression. PDGF signallingmodulates tumour progression by a tumour cell-autonomousmechanism or through tumour–stroma interaction. Doubtlessly,it has either a tumour-suppressing or tumour-promotingfunction depending on cellular context. Many cellularcontext-dependent factors tightly maintain the equilibrium ofPDGF signalling and contribute to the regulation of PDGF-induced cell responses.
Keeping in view some advances in the past few years,the investigation for rational drug design for advancedcancer is far from over. Existing data clearly reveal that thedesigning of new drugs will have minimal, if any,probability of success if it is not guided by comprehensiveknowledge of disease biology. On the other hand, usingbiologic agents to target key molecular pathways, such asthose modulated by PDGF/PDGFR family members, maybe effectual. Without a doubt, there is a necessity toevaluate the efficacy of chemotherapeutic drugs, miRNAand phytonutrients alone and in combinations to explore thepositive results achieved in various phases of clinical trialsin patients that support this approach. Many new anti-PDGF molecules are now under evaluation for thetherapeutic intercession of cancer, but hitherto efforts toidentify reliable predictive factors from phase I and II trialshave produced unconvincing results. This corroborates thesuggestion that refining patient selection is critical to takefull advantage of the benefit of targeted agents, tocircumvent significant toxicities and for the developmentof alternative therapeutic approaches in patients who havenonresponsive disease.
There is a remarkable expression profile of PDGF-α andPDGF-β in endometrial stromal sarcoma [84]. PDGFRactivity is deregulated in human glioblastoma that results inthe activation of downstream kinases such as PI3K, Akt andmTOR [85]. Recent data suggest that synergistic adminis-tration of perifosine and CCI-779 is effective in themanagement of the disease. On a similar note, it has beenrecently documented that imatinib is not efficient in themanagement of glioblastoma [86]. However, some com-pounds have efficacy and offer therapeutic potential. In thiscontext, 2,3,4′,5-tetrahydroxystilbene-2-O-β-d-glucoside(TSG) is suggested to be effective in inhibition of PDGF-
BB-stimulated VSMC proliferation via cell cycle arrest[87]. Similarly, 1,4-naphthoquinone derivative also hasinhibitory effects [88]. Furthermore, benzylideneacetophe-none analogues are also effective in the suppression ofPDGF signal transduction cascade [89].
Mounting evidence signifies the fact that multiprongedapproach is more potent and effective in the clinicalmanagement. In support of this notion, two tyrosine kinaseinhibitors, imatinib and vatalanib, are documented toincrease the effects of paclitaxel on PDGF-BB tumours.Consistent with the same concept, imatinib and carboplatinhave enhanced efficiency in suppressing VEGF, PDGF andPDGF-Rα/ß expression in head and neck squamous cellcarcinoma [90, 91]. Docetaxel has been currently acclaimedas an inhibitor of PDGF; however, there are controversialfindings, and future research is necessary to unveilopportunities and challenges in the standardization oftherapy [92, 93]. Bevacizumab has been used in combinationwith (PDGF aptamer, AX102) and displayed therapeuticefficacy [94].
Recently, there is a paradigm shift in the molecularmedicine. To enhance penetration of doxorubicin, it wasconjugated to pPB-HSA through an acid-sensitive hydra-zone linkage. Results suggested that treatment with Dox-HSA-pPB considerably suppressed the C26 tumour growthin mice whereas free doxorubicin-treated mice had lowerresponse to the therapy [95]. Furthermore, pazopanib is asmall-molecule inhibitor of PDGF receptors [96, 97].Baicalin, an herb-derived flavonoid compound, has beenrecently shown in inducing growth arrest of PDGF-stimulated VSMCs [98]. Cediranib is a potent therapeuticintervention for inhibition of VEGF receptor (VEGFR)-2and VEGFR-3 tyrosine kinases. However, recently, itsefficacy has been evaluated in tumour cell lines having anenhanced expression of PDGFR α and PDGFRβ. Further-more, in vivo, ligand-induced PDGFR-β phosphorylationin murine lung tissue was inhibited by 55% followingtreatment with cediranib. Similarly, rat glial tumour xeno-grafts in mice displayed suppression of ligand-inducedphosphorylation of both PDGFR-α and PDGFR-β by 46%to 61% with cediranib [99].
Recently, Affibody molecules have occupied top slot inthe spotlight. They comprise a class of very small bindingproteins that are highly appropriate for in vivo imagingapplications and that can be selected to exclusivelyrecognize a target protein. PDGFRβ-specific Affibodymolecules are effective as they accumulated aroundtumoural blood vessels in a model of spontaneous insuli-noma, confirming a prospective for in vivo targeting [100].
On a similar note, single-chain antibody fragments(scFvs) targeting PDGFRbeta and VEGF-A have currentlygained attention for superior stability. The scFvs were fusedto both termini of human Fc to engineer a bispecific,
Tumor Biol. (2011) 32:1057–1070 1065

tetravalent molecule. This molecule has shown potentactivity, binds both targets concurrently and is stable inserum [101].
Accumulating data points towards that disruption inPDGF signalling and primary dysfunction in the tumourmicroenvironment, in addition to epithelial dysfunction,can be fundamental for carcinogenesis. These currentfindings make a convincing case for a multiprongedapproach for targeting the microenvironment for cancerchemoprevention. It is believed that further investigationswill unravel mechanistic insights of pathophysiology ofthe microenvironment and new approaches to control itwith chemopreventive agents. It is interesting to note thatmicroenvironment of a cancer is an integral part of itsanatomy and physiology, and functionally, one cannotentirely dissociate this microenvironment from what haveusually been called ‘cancer cells’. There is a need foreffective clinical implementation of this knowledge inpreventive strategies.
Impelling evidence points towards the fact that duringhepatic fibrogenesis, suppression of peroxisome proliferator-activated receptor-gamma (PPAR gamma) resulted insimultaneous activation of PDGF and epidermal growthfactor (EGF) signalling in hepatic stellate cells (HSCs).However, curcumin restored the activities of PPARgamma and concomitant suppression of PDGF and EGFsignal transduction cascade.It is also documented to beinvolved in downregulation of PDGF-BB, PDGFRbeta inHSCs [102, 103].
Another attention-grabbing feature is that anthocyaninspresenting a hydroxyl residue at position 3′ suppress PDGF(AB)-induced VEGF expression by inhibiting p38 MAPKand JNK in VSMCs [104]. On a similar note, resveratrolconsiderably suppressed PDGF-stimulated c-Src and Aktkinase activation. It suppressed PDGFR phosphorylation atthe PI 3 kinase and Grb-2 binding sites tyrosine-751 andtyrosine-716, respectively, and remarkably escalated theactivity of PTP1B. This phosphatase is involved indephosphorylation of PDGF-stimulated phosphorylation attyrosine-751 and tyrosine-716 on PDGFR with concomitantreduction in Akt and Erk1/2 kinase activity [105]. Recentdata highlight the fact that piceatannol (a metabolite ofresveratrol) suppressed PI3K activity more effectively thanresveratrol [106]. Similarly, pterostilbene, a natural dime-thylated analogue of resveratrol, suppressed PDGF-BB-mediated phosphorylation of Akt kinase [107].
It is attractive to interpret that lycopene inhibited PDGF-AA-induced SMC and fibroblast migration in a concentration-dependent manner. Mounting data suggest that lycopene-binding region exists within PDGF and is located at loop 2region [108]. Consistent with same line, lycopene restrictsPDGF-BB-induced cellular migration through inhibition ofPI3K/Akt, ERK and p38 activation [109, 110].
Conclusion
In the quest to recognize PDGF signalling, great strideshave been made to comprehend how these proteins controltheir downstream targets. However, scores of mechanismsby which these proteins generate linear or integratedcascades remain obscure.
With an addition of substantial fraction of elucidations tothe pre-existing understandings of PDGF, it is now evidentthat an integrated network of proteins triggers the dynamicsof the cell. Another thing that cannot be overlooked is thecrosstalks of two linear transduction cascades. This inte-grated framework works with striking synergy duringtumour development. It is noteworthy that in cancertherapy, the identification of novel and potent PDGFR/EGFR inhibitors with preferred kinase inhibitory spectrumthat provides superior antitumour efficacy, although withmanageable side effects and toxicities, will continue to bethe key for success. Additionally, interest in targeting PDGFsignalling for intervention of cancer will surely addsufficient information into the existing pool of concepts ofclinical management. On a similar note, clinical successesof these agents are unquestionably based on the broadeninglandscape of knowledge on the molecular mechanisms thatunderpin the development and progression of a malignantphenotype. It is necessary to revisit the existing web ofproteins with reference to PDGF signalling to tailor someeffective clinical outcomes.
References
1. Heldin CH, Eriksson U, Ostman A. New members of theplatelet-derived growth factor family of mitogens. Arch BiochemBiophys. 2002;398(2):284–90.
2. Sun PD, Davies DR. The cystine-knot growth-factor superfamily.Annu Rev Biophys Biomol Struct. 1995;24:269–91.
3. Li X, Eriksson U. Novel PDGF family members: PDGF-C andPDGF-D. Cytokine Growth Factor Rev. 2003;14(2):91–8.
4. Heldin CH, Westermark B. Signal transduction by the receptors forplatelet-derived growth factor. J Cell Sci. 1990;96(Pt 2):193–6.
5. Bowen-Pope DF, Hart CE, Seifert RA. Sera and conditionedmedia contain different isoforms of platelet-derived growthfactor (PDGF) which bind to different classes of PDGF receptor.J Biol Chem. 1989;264(5):2502–8.
6. Gronwald RG, Grant FJ, Haldeman BA, Hart CE, O'Hara PJ,Hagen FS, et al. Cloning and expression of a cDNA coding forthe human platelet-derived growth factor receptor: evidence formore than one receptor class. Proc Natl Acad Sci USA. 1988;85(10):3435–9.
7. Li X, Pontén A, Aase K, Karlsson L, Abramsson A, Uutela M, etal. PDGF-C is a new protease-activated ligand for the PDGFalpha-receptor. Nat Cell Biol. 2000;2(5):302–9.
8. Bergsten E, Uutela M, Li X, Pietras K, Ostman A, Heldin CH, etal. PDGF-D is a specific, protease-activated ligand for the PDGFbeta-receptor. Nat Cell Biol. 2001;3(5):512–6.
9. Changsirikulchai S, Hudkins KL, Goodpaster TA, Volpone J,Topouzis S, Gilbertson DG, et al. Platelet-derived growth factor-
1066 Tumor Biol. (2011) 32:1057–1070

D expression in developing and mature human kidneys. KidneyInt. 2002;62(6):2043–54.
10. Cochran BH, Reffel AC, Stiles CD. Molecular cloning of genesequences regulated by platelet-derived growth factor. Cell.1983;33(3):939–47.
11. Linzer DI, Nathans D. Growth-related changes in specificmRNAs of cultured mouse cells. Proc Natl Acad Sci USA.1983;80(14):4271–5.
12. Almendral JM, Sommer D, Macdonald-Bravo H, Burckhardt J,Perera J, Bravo R. Complexity of the early genetic response togrowth factors in mouse fibroblasts. Mol Cell Biol. 1988;8(5):2140–8.
13. Li L, Blumenthal DK, Terry CM, He Y, Carlson ML, CheungAK. PDGF-induced proliferation in human arterial and venoussmooth muscle cells: molecular basis for differential effects ofPDGF isoforms. J Cell Biochem. 2011;112:289–98.
14. Sato S, Sato Y, Hatakeyama K, Marutsuka K, Yamashita A,Takeshima H, et al. Quantitative analysis of vessels with smoothmuscle layer in astrocytic tumors: correlation with histological gradeand prognostic significance. Histol Histopathol. 2011;26:497–504.
15. Ishigaki T, Imanaka-Yoshida K, Shimojo N, Matsushima S, TakiW, Yoshida T. Tenascin-C enhances crosstalk signaling of integrinαvβ3/PDGFR-β complex by SRC recruitment promotingPDGF-induced proliferation and migration in smooth musclecells. J Cell Physiol. 2011 (in press)
16. Tsaousi A,Williams H, Lyon CA, Taylor V, SwainA, Johnson JL, etal. Wnt4/β-catenin signaling induces VSMC proliferation and isassociated with intimal thickening. Circ Res. 2011;108:427–36.
17. Sklepkiewicz P, Schermuly RT, Tian X, Ghofrani HA, WeissmannN, Sedding D, et al. Glycogen synthase kinase 3beta contributesto proliferation of arterial smooth muscle cells in pulmonaryhypertension. PLoS One. 2011;6:e18883.
18. Keramati AR, Singh R, Lin A, Faramarzi S, Ye ZJ, Mane S, et al.Wild-type LRP6 inhibits, whereas atherosclerosis-linkedLRP6R611C increases PDGF-dependent vascular smooth musclecell proliferation. Proc Natl Acad Sci USA. 2011;108:1914–8.
19. Mendelson K, Swendeman S, Saftig P, Blobel CP. Stimulationof platelet-derived growth factor receptor beta (PDGFRbeta)activates ADAM17 and promotes metalloproteinase-dependentcross-talk between the PDGFRbeta and epidermal growthfactor receptor (EGFR) signaling pathways. J Biol Chem.2010;285:25024–32.
20. Chen PY, Simons M, Friesel R. FRS2 via fibroblast growth factorreceptor 1 is required for platelet-derived growth factor receptorbeta-mediated regulation of vascular smooth muscle marker geneexpression. J Biol Chem. 2009;284(23):15980–92.
21. Pellet-Many C, Frankel P, Evans IM, Herzog B, Jünemann-Ramírez M, Zachary IC. Neuropilin-1 mediates PDGF stimula-tion of vascular smooth muscle cell migration and signalling viap130Cas. Biochem J. 2011;435:609–18.
22. Miraoui H, Ringe J, Häupl T, Marie PJ. Increased EFG- andPDGFalpha-receptor signaling by mutant FGF-receptor 2 con-tributes to osteoblast dysfunction in Apert craniosynostosis. HumMol Genet. 2010;19:1678–89.
23. Sápi Z, Füle T, Hajdu M, Matolcsy A, Moskovszky L, Márk A,et al. The activated targets of mTOR signaling pathway arecharacteristic for PDGFRA mutant and wild-type rather than KITmutant GISTs. Diagn Mol Pathol. 2011;20:22–33.
24. Pérez J, Torres RA, Rocic P, Cismowski MJ, Weber DS, Darley-Usmar VM, et al. PYK2 signaling is required for PDGF-dependent vascular smooth muscle cell proliferation. Am JPhysiol Cell Physiol. 2011;301:C242–51.
25. Zhao Y, Biswas SK, McNulty PH, Kozak M, Jun JY, Segar L.PDGF-induced vascular smooth muscle cell proliferation isassociated with dysregulation of insulin receptor substrates. AmJ Physiol Cell Physiol. 2011;300:C1375–85.
26. Ning Y, Sun Q, Dong Y, Xu W, Zhang W, Huang H, et al. Slit2-Ninhibits PDGF-induced migration in rat airway smooth musclecells: WASP and Arp2/3 involved. Toxicology. 2011;283:32–40.
27. Ucuzian AA, Brewster LP, East AT, Pang Y, Gassman AA,Greisler HP. Characterization of the chemotactic and mitogenicresponse of SMCs to PDGF-BB and FGF-2 in fibrin hydrogels. JBiomed Mater Res A. 2010;94:988–96.
28. Martino MM, Hubbell JA. The 12th-14th type III repeats offibronectin function as a highly promiscuous growth factor-binding domain. FASEB J. 2010;24:4711–21.
29. Foehr ED, Tatavos A, Tanabe E, Raffioni S, Goetz S, Dimarco E,et al. Discoidin domain receptor 1 (DDR1) signaling in PC12cells: activation of juxtamembrane domains in PDGFR/DDR/TrkA chimeric receptors. FASEB J. 2000;14:973–81.
30. Toffalini F, Kallin A, Vandenberghe P, Pierre P, Michaux L, CoolsJ, et al. The fusion proteins TEL-PDGFRbeta and FIP1L1-PDGFRalpha escape ubiquitination and degradation. Haemato-logica. 2009;94:1085–93.
31. Gueller S, Hehn S, Nowak V, Gery S, Serve H, Brandts CH, et al.Adaptor protein Lnk binds to PDGF receptor and inhibits PDGF-dependent signaling. Exp Hematol. 2011;39:591–600.
32. Medves S, Noel LA, Montano-Almendras CP, Albu RI, SchoemansH, Constantinescu SN, Demoulin JB. Multiple oligomerizationdomains of KANK1-PDGFRB are required for JAK2-independenthematopoietic cell proliferation and signaling via STAT5 and ERK.Haematologica. 2011 (in press) AOP 32
33. Cao Y, Cao R, Hedlund EM. R Regulation of tumor angiogenesisand metastasis by FGF and PDGF signaling pathways. J MolMed. 2008;86(7):785–9.
34. Cao R, Björndahl MA, Religa P, Clasper S, Garvin S, Galter D,et al. PDGF-BB induces intratumoral lymphangiogenesis andpromotes lymphatic metastasis. Cancer Cell. 2004;6(4):333–45.
35. Jechlinger M, Sommer A, Moriggl R, Seither P, Kraut N,Capodiecci P, et al. Autocrine PDGFR signaling promotesmammary cancer metastasis. J Clin Invest. 2006;116(6):1561–70.
36. Cimpean AM, Ceauşu R, Encică S, Gaje PN, Ribatti D, Raica M.Platelet-derived growth factor and platelet-derived growth factorreceptor-α expression in the normal human thymus andthymoma. Int J Exp Pathol. 2011 (in press) AOP
37. Suzuki S, Dobashi Y, Hatakeyama Y, Tajiri R, Fujimura T, HeldinCH, et al. Clinicopathological significance of platelet-derivedgrowth factor (PDGF)-B and vascular endothelial growth factor-A expression, PDGF receptor-β phosphorylation, and micro-vessel density in gastric cancer. BMC Cancer. 2010;10:659.
38. Chaoran Z, Zhirong L, Gezhi X. Combination of vascularendothelial growth factor receptor/platelet-derived growth factorreceptor inhibition markedly improves the antiangiogenic effica-cy for advanced stage mouse corneal neovascularization. GraefesArch Clin Exp Ophthalmol. 2011. (in press) AOP
39. Ahmad A, Wang Z, Kong D, Ali R, Ali S, Banerjee S, et al.Platelet-derived growth factor-D contributes to aggressiveness ofbreast cancer cells by up-regulating Notch and NF-κB signalingpathways. Breast Cancer Res Treat. 2011;126(1):15–25.
40. Wang Z, Kong D, Banerjee S, Li Y, Adsay NV, Abbruzzese J, etal. Down-regulation of platelet-derived growth factor-D inhibitscell growth and angiogenesis through inactivation of Notch-1and nuclear factor-kappaB signaling. Cancer Res. 2007;67(23):11377–85.
41. Kang DW, Min do S. Platelet derived growth factor increasesphospholipase D1 but not phospholipase D2 expression viaNFkappaB signaling pathway and enhances invasion of breastcancer cells. Cancer Lett. 2010;294(1):125–33.
42. Rykala J, Przybylowska K, Majsterek I, Pasz-Walczak G, SygutA, Dziki A, Kruk-Jeromin J. Angiogenesis markers quantificationin breast cancer and their correlation with clinicopathologicalprognostic variables. Pathol Oncol Res. 2011 (in press).
Tumor Biol. (2011) 32:1057–1070 1067

43. Liu J, Liao S, Huang Y, Samuel R, Shi T, Naxerova K, et al.PDGF-D improves drug delivery and efficacy via vascularnormalization, but promotes lymphatic metastasis by activatingCXCR4 in breast cancer. Clin Cancer Res. 2011;17:3638–48.
44. Gehmert S, Gehmert S, Prantl L, Vykoukal J, Alt E, Song YH.Breast cancer cells attract the migration of adipose tissue-derivedstem cells via the PDGF-BB/PDGFR-beta signaling pathway.Biochem Biophys Res Commun. 2010;398:601–5.
45. Maurer B, Stanczyk J, Jüngel A, Akhmetshina A, Trenkmann M,Brock M, et al. MicroRNA-29, a key regulator of collagenexpression in systemic sclerosis. Arthritis Rheum. 2010;62:1733–43.
46. Quintavalle M, Elia L, Condorelli G, Courtneidge SA. Micro-RNA control of podosome formation in vascular smooth musclecells in vivo and in vitro. J Cell Biol. 2010;189:13–22.
47. Dugas JC, Cuellar TL, Scholze A, Ason B, Ibrahim A, Emery B,et al. Dicer1 and miR-219 are required for normal oligodendro-cyte differentiation and myelination. Neuron. 2010;65:597–611.
48. Zhang J, Chintalgattu V, Shih T, Ai D, Xia Y, Khakoo AY.MicroRNA-9 is an activation-induced regulator of PDGFR-betaexpression in cardiomyocytes. J Mol Cell Cardiol. 2011 (inpress) AOP.
49. Würdinger T, Tannous BA, Saydam O, Skog J, Grau S,Soutschek J, et al. miR-296 regulates growth factor receptoroverexpression in angiogenic endothelial cells. Cancer Cell.2008;14:382–93.
50. Eberhart JK, He X, Swartz ME, Yan YL, Song H, Boling TC, etal. MicroRNA Mirn140 modulates Pdgf signaling during palato-genesis. Nat Genet. 2008;40:290–8.
51. Goff LA, Boucher S, Ricupero CL, Fenstermacher S, SwerdelM, Chase LG, et al. Differentiating human multipotent mesen-chymal stromal cells regulate microRNAs: prediction of micro-RNA regulation by PDGF during osteogenesis. Exp Hematol.2008;36(10):1354–69.
52. Chan MC, Hilyard AC, Wu C, Davis BN, Hill NS, Lal A, et al.Molecular basis for antagonism between PDGF and the TGFbetafamily of signalling pathways by control of miR-24 expression.EMBO J. 2010;29(3):559–73.
53. Kong D, Li Y, Wang Z, Banerjee S, Ahmad A, Kim HR, et al.miR-200 regulates PDGF-D-mediated epithelial-mesenchymaltransition, adhesion, and invasion of prostate cancer cells. StemCells. 2009;27(8):1712–21.
54. Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A. Inductionof microRNA-221 by platelet-derived growth factor signaling iscritical for modulation of vascular smooth muscle phenotype. JBiol Chem. 2009;284(6):3728–38.
55. Muhl L, Nykjaer A, Wygrecka M, Monard D, Preissner KT,Kanse SM. Inhibition of PDGF-BB by Factor VII-activatingprotease (FSAP) is neutralized by protease nexin-1, and theFSAP-inhibitor complexes are internalized via LRP. Biochem J.2007;404(2):191–6.
56. Takayama Y, May P, Anderson RG, Herz J. Low densitylipoprotein receptor-related protein 1 (LRP1) controls endocyto-sis and c-CBL-mediated ubiquitination of the platelet-derivedgrowth factor receptor beta (PDGFR beta). J Biol Chem.2005;280(18):18504–10.
57. Zhou L, Takayama Y, Boucher P, Tallquist MD, Herz J. LRP1regulates architecture of the vascular wall by controllingPDGFRbeta-dependent phosphatidylinositol 3-kinase activation.PLoS One. 2009;4(9):e6922.
58. Muratoglu SC, Mikhailenko I, Newton C, Migliorini M,Strickland DK. Low density lipoprotein receptor-relatedprotein 1 (LRP1) forms a signaling complex with platelet-derived growth factor receptor-beta in endosomes andregulates activation of the MAPK pathway. J Biol Chem.2010;285(19):14308–17.
59. Boucher P, Li WP, Matz RL, Takayama Y, Auwerx J, AndersonRG, et al. LRP1 functions as an atheroprotective integrator ofTGFbeta and PDFG signals in the vascular wall: implications forMarfan syndrome. PLoS One. 2007;2(5):e448.
60. Hellberg C, Schmees C, Karlsson S, Ahgren A, Heldin CH.Activation of protein kinase C alpha is necessary for sorting thePDGF beta-receptor to Rab4a-dependent recycling. Mol BiolCell. 2009;20(12):2856–63.
61. Chiarugi P, Cirri P, Taddei ML, Giannoni E, Fiaschi T, Buricchi F,et al. Insight into the role of low molecular weight phosphotyr-osine phosphatase (LMW-PTP) on platelet-derived growth factorreceptor (PDGF-r) signaling. LMW-PTP controls PDGF-r kinaseactivity through TYR-857 dephosphorylation. J Biol Chem.2002;277(40):37331–8.
62. Kappert K, Paulsson J, Sparwel J, Leppänen O, Hellberg C,Ostman A, et al. Dynamic changes in the expression ofDEP-1 and other PDGF receptor-antagonizing PTPs duringonset and termination of neointima formation. FASEB J.2007;21:523–34.
63. Karlsson S, Kowanetz K, Sandin A, Persson C, Ostman A,Heldin CH, et al. Loss of T-cell protein tyrosine phosphataseinduces recycling of the platelet-derived growth factor (PDGF)beta-receptor but not the PDGF alpha-receptor. Mol Biol Cell.2006;17:4846–55.
64. Kanda M, Ihara Y, Murata H, Urata Y, Kono T, Yodoi J, et al.Glutaredoxin modulates platelet-derived growth factor-dependent cell signaling by regulating the redox status oflow molecular weight protein-tyrosine phosphatase. J BiolChem. 2006;281:28518–28.
65. Freyhaus H, Dagnell M, Leuchs M, Vantler M, Berghausen EM,Caglayan E, et al. Hypoxia enhances platelet-derived growthfactor signaling in the pulmonary vasculature by down-regulation of protein tyrosine phosphatases. Am J Respir CritCare Med. 2011;183:1092–102.
66. Conrad M, Sandin A, Förster H, Seiler A, Frijhoff J, DagnellM, et al. Hooft van Huijsduijnen R, Aspenström P, BöhmerF, Ostman A. 12/15-lipoxygenase-derived lipid peroxidescontrol receptor tyrosine kinase signaling through oxidationof protein tyrosine phosphatases. Proc Natl Acad Sci USA.2010;107:15774–9.
67. Boivin B, Tonks NK. Analysis of the redox regulation of proteintyrosine phosphatase superfamily members utilizing a cysteinyl-labeling assay. Methods Enzymol. 2010;474:35–50.
68. Sandin A, Dagnell M, Gonon A, Pernow J, Stangl V, AspenströmP, et al. Hypoxia followed by re-oxygenation induces oxidationof tyrosine phosphatases. Cell Signal. 2011;23:820–6.
69. Juarez JC, Manuia M, Burnett ME, Betancourt O, Boivin B,Shaw DE, et al. Superoxide dismutase 1 (SOD1) is essentialfor H2O2-mediated oxidation and inactivation of phospha-tases in growth factor signaling. Proc Natl Acad Sci USA.2008;105:7147–52.
70. Cortesio CL, Perrin BJ, Bennin DA, Huttenlocher A. Actin-binding protein-1 interacts with WASp-interacting protein toregulate growth factor-induced dorsal ruffle formation. Mol BiolCell. 2010;21(1):186–97.
71. Abella JV, Vaillancourt R, Frigault MM, Ponzo MG, Zuo D,Sangwan V, et al. The Gab1 scaffold regulates RTK-dependentdorsal ruffle formation through the adaptor Nck. J Cell Sci.2010;123:1306–19.
72. Campa F, Machuy N, Klein A, Rudel T. A new interactionbetween Abi-1 and betaPIX involved in PDGF-activated actincytoskeleton reorganisation. Cell Res. 2006;16:759–70.
73. Veracini L, Franco M, Boureux A, Simon V, Roche S, BenistantC. Two distinct pools of Src family tyrosine kinases regulatePDGF-induced DNA synthesis and actin dorsal ruffles. J CellSci. 2006;119:2921–34.
1068 Tumor Biol. (2011) 32:1057–1070

74. Goicoechea S, Arneman D, Disanza A, Garcia-Mata R, Scita G,Otey CA. Palladin binds to Eps8 and enhances the formation ofdorsal ruffles and podosomes in vascular smooth muscle cells. JCell Sci. 2006;119:3316–24.
75. Toguchi M, Richnau N, Ruusala A, Aspenström P. Membersof the CIP4 family of proteins participate in the regulationof platelet-derived growth factor receptor-beta-dependentactin reorganization and migration. Biol Cell. 2010;102(4):215–30.
76. Berrou E, Bryckaert M. Recruitment of protein phosphatase 2Ato dorsal ruffles by platelet-derived growth factor in smoothmuscle cells: dephosphorylation of Hsp27. Exp Cell Res.2009;315:836–48.
77. Nagano K, Bornhauser BC, Warnasuriya G, Entwistle A, CramerR, Lindholm D, et al. PDGF regulates the actin cytoskeletonthrough hnRNP-K-mediated activation of the ubiquitin E3-ligaseMIR. EMBO J. 2006;25(9):1871–82.
78. Uribe DJ, Guo K, Shin YJ, Sun D. Heterogeneous nuclearribonucleoprotein K and nucleolin as transcriptional activatorsof the vascular endothelial growth factor promoter throughinteraction with secondary DNA structures. Biochemistry.2011;50:3796–806.
79. Mikula M, Bomsztyk K. Direct recruitment of ERK cascadecomponents to inducible genes is regulated by heteroge-neous nuclear ribonucleoprotein (hnRNP) K. J Biol Chem.2011;286:9763–75.
80. Chen K, Albano A, Ho A, Keaney Jr JF. Activation of p53 byoxidative stress involves platelet-derived growth factor-betareceptor-mediated ataxia telangiectasia mutated (ATM) kinaseactivation. J Biol Chem. 2003;278:39527–33.
81. Kim TS, Kawaguchi M, Suzuki M, Jung CG, Asai K, ShibamotoY, et al. The ZFHX3 (ATBF1) transcription factor inducesPDGFRB, which activates ATM in the cytoplasm to protectcerebellar neurons from oxidative stress. Dis Model Mech.2010;3:752–62.
82. Ikushima H, Komuro A, Isogaya K, Shinozaki M, Hellman U,Miyazawa K, et al. An Id-like molecule, HHM, is a synexpres-sion group-restricted regulator of TGF-beta signalling. EMBO J.2008;27(22):2955–65.
83. Patel P, West-Mays J, Kolb M, Rodrigues JC, Hoff CM, MargettsPJ. Platelet derived growth factor B and epithelial mesenchymaltransition of peritoneal mesothelial cells. Matrix Biol. 2010;29(2):97–106.
84. Cheng X, Yang G, Schmeler KM, Coleman RL, Tu X, Liu J, etal. Recurrence patterns and prognosis of endometrial stromalsarcoma and the potential of tyrosine kinase-inhibiting therapy.Gynecol Oncol. 2011;121:323–7.
85. Pitter KL, Galbán CJ, Galbán S, Tehrani OS, Li F, Charles N, etal. Perifosine and CCI 779 co-operate to induce cell death anddecrease proliferation in PTEN-intact and PTEN-deficientPDGF-driven murine glioblastoma. PLoS One. 2011;6:e14545.AOP.
86. Dong Y, Jia L, Wang X, Tan X, Xu J, Deng Z, et al. Selectiveinhibition of PDGFR by imatinib elicits the sustained activationof ERK and downstream receptor signaling in malignant gliomacells. Int J Oncol. 2011;38:555–69.
87. Xu XL, Huang YJ, Wang YQ, Chen XF, Zhang W. 2,3,4′,5-Tetrahydroxystilbene-2-O-β-d-glucoside inhibits platelet-derivedgrowth factor-induced proliferation of vascular smooth musclecells by regulating the cell cycle. Clin Exp Pharmacol Physiol.2011;38:307–13.
88. Kim TJ, Yun YP. Antiproliferative activity of NQ304, a synthetic1,4-naphthoquinone, is mediated via the suppressions of thePI3K/Akt and ERK1/2 signaling pathways in PDGF-BB-stimulated vascular smooth muscle cells. Vascul Pharmacol.2007;46:43–51.
89. Kim TJ, Han HJ, Kim YJ, Jung JC, Yu JY, Lee JJ, et al.Inhibitory effects of BST406, a newly synthesized benzylide-neacetophenone derivative, on abnormal vascular smooth musclecell proliferation. Biol Pharm Bull. 2010;33:900–4.
90. Schultz JD, Rotunno S, Riedel F, Anders C, Erben P, HofheinzRD, et al. Synergistic effects of imatinib and carboplatin onVEGF, PDGF and PDGF-Rα/ß expression in squamous cellcarcinoma of the head and neck in vitro. Int J Oncol.2011;38:1001–12.
91. Kłosowska-Wardęga A, Hasumi Y, Ahgren A, Heldin CH,Hellberg C. Combination therapy using imatinib and vatalanibimproves the therapeutic efficiency of paclitaxel towards amouse melanoma tumor. Melanoma Res. 2011 (in press) AOP.
92. Park ES, Yoo JM, Lim Y, Tudev M, Yoo HS, Hong JT, et al.Inhibitory effects of docetaxel on platelet-derived growth factor(PDGF)-BB-induced proliferation of vascular smooth musclecells through blocking PDGF-receptor β phosphorylation. JPharmacol Sci. 2011;116:204–13.
93. Mathew P, Thall PF, Wen S, Bucana C, Jones D, Horne E, et al.Dynamic change in phosphorylated platelet-derived growthfactor receptor in peripheral blood leukocytes following doce-taxel therapy predicts progression-free and overall survival inprostate cancer. Br J Cancer. 2008;99:1426–32.
94. Lu C, Shahzad MM, Moreno-Smith M, Lin YG, JenningsNB, Allen JK, et al. Targeting pericytes with a PDGF-Baptamer in human ovarian carcinoma models. Cancer BiolTher. 2010;9:176–82.
95. Prakash J, de Jong E, Post E, Gouw AS, Beljaars L, Poelstra K.A novel approach to deliver anticancer drugs to key cell types intumors using a PDGF receptor-binding cyclic peptide containingcarrier. J Control Release. 2010;145:91–101.
96. Tailor TD, Hanna G, Yarmolenko PS, Dreher MR, Betof AS,Nixon AB, et al. Effect of pazopanib on tumor microenviron-ment and liposome delivery. Mol Cancer Ther. 2010;9:1798–808.
97. Hutson TE, Davis ID, Machiels JP, De Souza PL, Rottey S, HongBF, et al. Efficacy and safety of pazopanib in patients withmetastatic renal cell carcinoma. J Clin Oncol. 2010;28:475–80.
98. Dong LH, Wen JK, Miao SB, Jia Z, Hu HJ, Sun RH, et al.Baicalin inhibits PDGF-BB-stimulated vascular smooth musclecell proliferation through suppressing PDGFRβ-ERK signalingand increase in p27 accumulation and prevents injury-inducedneointimal hyperplasia. Cell Res. 2010;20:1252–62.
99. Brave SR, Ratcliffe K, Wilson Z, James NH, Ashton S,Wainwright A, et al. Assessing the activity of cediranib, aVEGFR-2/3 tyrosine kinase inhibitor, against VEGFR-1 andmembers of the structurally related PDGFR family. Mol CancerTher. 2011;10:861–73.
100. Lindborg M, Cortez E, Höidén-Guthenberg I, Gunneriusson E,von Hage E, Syud F, et al. Engineered high-affinity affibodymolecules targeting platelet-derived growth factor receptor β invivo. J Mol Biol. 2011;407:298–315.
101. Mabry R, Gilbertson DG, Frank A, Vu T, Ardourel D, OstranderC, et al. A dual-targeting PDGFRbeta/VEGF-A moleculeassembled from stable antibody fragments demonstrates anti-angiogenic activity in vitro and in vivo. MAbs. 2010;2:20–34.
102. Lin J, Chen A. Activation of peroxisome proliferator-activatedreceptor-gamma by curcumin blocks the signaling pathways forPDGF and EGF in hepatic stellate cells. Lab Invest.2008;88:529–40.
103. Zhao ZD, Huang ZS. Study on effects of curcumin onexpressions of PDGF-BB, PDGFRbeta and ERK1 of HSC.Zhong Yao Cai. 2009;32:732–5.
104. Oak MH, Bedoui JE, Madeira SV, Chalupsky K, Schini-KerthVB. Delphinidin and cyanidin inhibit PDGF(AB)-induced VEGFrelease in vascular smooth muscle cells by preventing activationof p38 MAPK and JNK. Br J Pharmacol. 2006;149:283–90.
Tumor Biol. (2011) 32:1057–1070 1069

105. Venkatesan B, Ghosh-Choudhury N, Das F, Mahimainathan L,Kamat A, Kasinath BS, et al. Resveratrol inhibits PDGF receptormitogenic signaling in mesangial cells: role of PTP1B. FASEB J.2008;22:3469–82.
106. Choi KH, Kim JE, Song NR, Son JE, Hwang MK, Byun S, etal. Phosphoinositide 3-kinase is a novel target of piceatannolfor inhibiting PDGF-BB-induced proliferation and migrationin human aortic smooth muscle cells. Cardiovasc Res.2010;85:836–44.
107. Park ES, Lim Y, Hong JT, Yoo HS, Lee CK, Pyo MY, et al.Pterostilbene, a natural dimethylated analog of resveratrol, inhibitsrat aortic vascular smooth muscle cell proliferation by blockingAkt-dependent pathway. Vascul Pharmacol. 2010;53:61–7.
108. Chen CP, Hung CF, Lee SC, Lo HM, Wu PH, Wu WB. Lycopenebinding compromised PDGF-AA/-AB signaling and migration insmooth muscle cells and fibroblasts: prediction of the possiblelycopene binding site within PDGF. Naunyn SchmiedebergsArch Pharmacol. 2010;381:401–14.
109. Chan CM, Fang JY, Lin HH, Yang CY, Hung CF. Lycopeneinhibits PDGF-BB-induced retinal pigment epithelial cell migra-tion by suppression of PI3K/Akt and MAPK pathways. BiochemBiophys Res Commun. 2009;388:172–6.
110. Chiang HS, Wu WB, Fang JY, Chen DF, Chen BH, Huang CC, etal. Lycopene inhibits PDGF-BB-induced signaling and migrationin human dermal fibroblasts through interaction with PDGF-BB.Life Sci. 2007;81:1509–17.
1070 Tumor Biol. (2011) 32:1057–1070
Copyright © 2022 FDOKUMEN




















