
PDGF and FGF2 pathways regulate distinct oligodendrocyte lineage responses in experimental...
12
PDGF and FGF2 pathways regulate distinct oligodendrocyte lineage responses in experimental demyelination with spontaneous remyelination Joshua C. Murtie, a,1 Yong-Xing Zhou, b Tuan Q. Le, b Adam C. Vana, c and Regina C. Armstrong a,b,c, * a Program in Molecular and Cell Biology, Uniformed Services University of the Health Sciences, Bethesda, MD 20814, USA b Department of Anatomy, Physiology, Genetics, Uniformed Services University of the Health Sciences, Bethesda, MD 20814, USA c Program in Neuroscience, Uniformed Services University of the Health Sciences, Bethesda, MD 20814, USA Received 15 July 2004; revised 28 October 2004; accepted 10 December 2004 Available online 12 February 2005 Repair of myelin damage in the adult CNS requires oligodendrocyte progenitor (OP) proliferation and subsequent differentiation into remyelinating oligodendrocytes. Platelet-derived growth factor (PDGF) and fibroblast growth factor-2 (FGF2) have been predicted to act individually and/or cooperatively to generate remyelinating oligoden- drocytes. Analysis of PDGF alpha receptor (PDGFaR) heterozygous (+/ ) mice indicates that PDGFaR expression modulates oligodendro- cyte density in non-lesioned adult CNS. Analysis of cuprizone demyelination and recovery in PDGFaR+/ mice, FGF2 knockout ( / ) mice, and PDGFaR+/ FGF2 / mice demonstrated that:( 1) OP proliferation and oligodendrocyte regeneration is impaired in PDGFaR heterozygotes, (2) PDGFaR+/ and FGF2 / deletions do not act cooperatively to impair OP amplification, (3) oligodendrocyte differentiation is more frequent in FGF2 / mice, and (4) FGF2 deletion in combination with the PDGFaR+/ genotype rescues impaired oligodendrocyte regeneration of PDGFaR heterozygotes. These findings demonstrate distinct roles for PDGF and FGF2 in vivo in the context of a demyelinating disease with spontaneous remyelination. Published by Elsevier Inc. Keywords: Demyelinating disease; Multiple sclerosis; Remyelination; Demyelination; Oligodendrocyte; Progenitor; Fibroblast growth factor; Platelet-derived growth factor; Cuprizone; Differentiation Demyelination in the CNS, such as in multiple sclerosis lesions, can result in neurological deficits due to impaired nerve conduction and associated axonal damage. Remyelination can occur in the adult CNS; however, the capacity to remyelinate becomes limited with subsequent demyelinating episodes (Bruck et al., 2003). Immature oligodendrocyte lineage cells (OLCs) persist in the adult rodent and human CNS and may have the potential to enhance remyelination if directed to differentiate into oligodendrocytes (Armstrong et al., 1992; Chang et al., 2002; Goldman, 2003). Furthermore, OP proliferation appears to be a prerequisite for extensive remyelination in animal models (Blakemore and Keirstead, 1999). This observation indicates that transient stim- ulation of proliferation of persisting OP cells in human conditions may generate new cells that can more effectively remyelinate lesions. In vitro and in vivo studies have indicated that OLC proliferation and differentiation can be regulated by the activity of specific growth factor signaling pathways. Therefore, the current study examines OLC responses to growth factor signaling in the context of a demyelinating disease model that accomplishes extensive remyelination. Endogenous growth factor pathways that are employed in spontaneous remyelination may provide insight to promote remyelination in human demyelinating diseases. Both PDGF and FGF2 can act individually as mitogens for OPs from neonatal and adult CNS in vitro (Frost et al., 2003; McKinnon et al., 1990; Wolswijk and Noble, 1992). PDGF in combination with FGF2 induces OPs cultured from neonatal and adult rodents to proliferate as self-renewing stem cells and induces adult OPs to cycle more rapidly (Bogler et al., 1990; Wolswijk and Noble, 1992). In addition, FGF2 has been reported to increase OP expression of PDGFaR in vitro (McKinnon et al., 1990). OPs express receptors for PDGF and FGF2 during the proliferative response to demyelination (Redwine and Armstrong, 1998). Further, corresponding expression of PDGF and FGF2 ligands increases in demyelinated lesion areas (Armstrong et al., 2002; Messersmith et al., 2000; Woodruff et al., 2004). It is now important to address the in vivo function of this endogenous PDGF signaling in a demyelinated lesion environment and whether FGF2 signaling alters the PDGFaR response in this context. 0969-9961/$ - see front matter. Published by Elsevier Inc. doi:10.1016/j.nbd.2004.12.006 * Corresponding author. Department of Anatomy, Physiology, and Genetics, Uniformed Services University of the Health Sciences, 4301 Jones Bridge Road, Bethesda, MD 20814-4799, USA. Fax: +1 301 295 1715. E-mail address: [email protected] (R.C. Armstrong). 1 Current address: The Children’s Hospital Boston, Boston, MA 02115, USA. Available online on ScienceDirect (www.sciencedirect.com). www.elsevier.com/locate/ynbdi Neurobiology of Disease 19 (2005) 171 – 182
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of PDGF and FGF2 pathways regulate distinct oligodendrocyte lineage responses in experimental...

www.elsevier.com/locate/ynbdi
Neurobiology of Disease 19 (2005) 171–182
PDGF and FGF2 pathways regulate distinct oligodendrocyte
lineage responses in experimental demyelination with
spontaneous remyelination
Joshua C. Murtie,a,1 Yong-Xing Zhou,b Tuan Q. Le,b
Adam C. Vana,c and Regina C. Armstronga,b,c,*
aProgram in Molecular and Cell Biology, Uniformed Services University of the Health Sciences, Bethesda, MD 20814, USAbDepartment of Anatomy, Physiology, Genetics, Uniformed Services University of the Health Sciences, Bethesda, MD 20814, USAcProgram in Neuroscience, Uniformed Services University of the Health Sciences, Bethesda, MD 20814, USA
Received 15 July 2004; revised 28 October 2004; accepted 10 December 2004
Available online 12 February 2005
Repair of myelin damage in the adult CNS requires oligodendrocyte
progenitor (OP) proliferation and subsequent differentiation into
remyelinating oligodendrocytes. Platelet-derived growth factor (PDGF)
and fibroblast growth factor-2 (FGF2) have been predicted to act
individually and/or cooperatively to generate remyelinating oligoden-
drocytes. Analysis of PDGF alpha receptor (PDGFaR) heterozygous
(+/�) mice indicates that PDGFaR expression modulates oligodendro-
cyte density in non-lesioned adult CNS. Analysis of cuprizone
demyelination and recovery in PDGFaR+/� mice, FGF2 knockout
(�/�) mice, and PDGFaR+/� FGF2�/� mice demonstrated that:( 1)
OP proliferation and oligodendrocyte regeneration is impaired in
PDGFaR heterozygotes, (2) PDGFaR+/� and FGF2�/� deletions do
not act cooperatively to impair OP amplification, (3) oligodendrocyte
differentiation is more frequent in FGF2�/� mice, and (4) FGF2
deletion in combination with the PDGFaR+/� genotype rescues
impaired oligodendrocyte regeneration of PDGFaR heterozygotes.
These findings demonstrate distinct roles for PDGF and FGF2 in vivo in
the context of a demyelinating disease with spontaneous remyelination.
Published by Elsevier Inc.
Keywords: Demyelinating disease; Multiple sclerosis; Remyelination;
Demyelination; Oligodendrocyte; Progenitor; Fibroblast growth factor;
Platelet-derived growth factor; Cuprizone; Differentiation
Demyelination in the CNS, such as in multiple sclerosis lesions,
can result in neurological deficits due to impaired nerve conduction
and associated axonal damage. Remyelination can occur in the
0969-9961/$ - see front matter. Published by Elsevier Inc.
doi:10.1016/j.nbd.2004.12.006
* Corresponding author. Department of Anatomy, Physiology, and
Genetics, Uniformed Services University of the Health Sciences, 4301 Jones
Bridge Road, Bethesda, MD 20814-4799, USA. Fax: +1 301 295 1715.
E-mail address: [email protected] (R.C. Armstrong).1 Current address: The Children’s Hospital Boston, Boston, MA 02115,
USA.
Available online on ScienceDirect (www.sciencedirect.com).
adult CNS; however, the capacity to remyelinate becomes limited
with subsequent demyelinating episodes (Bruck et al., 2003).
Immature oligodendrocyte lineage cells (OLCs) persist in the adult
rodent and human CNS and may have the potential to enhance
remyelination if directed to differentiate into oligodendrocytes
(Armstrong et al., 1992; Chang et al., 2002; Goldman, 2003).
Furthermore, OP proliferation appears to be a prerequisite for
extensive remyelination in animal models (Blakemore and
Keirstead, 1999). This observation indicates that transient stim-
ulation of proliferation of persisting OP cells in human conditions
may generate new cells that can more effectively remyelinate
lesions. In vitro and in vivo studies have indicated that OLC
proliferation and differentiation can be regulated by the activity of
specific growth factor signaling pathways. Therefore, the current
study examines OLC responses to growth factor signaling in the
context of a demyelinating disease model that accomplishes
extensive remyelination. Endogenous growth factor pathways that
are employed in spontaneous remyelination may provide insight to
promote remyelination in human demyelinating diseases.
Both PDGF and FGF2 can act individually as mitogens for OPs
from neonatal and adult CNS in vitro (Frost et al., 2003;
McKinnon et al., 1990; Wolswijk and Noble, 1992). PDGF in
combination with FGF2 induces OPs cultured from neonatal and
adult rodents to proliferate as self-renewing stem cells and induces
adult OPs to cycle more rapidly (Bogler et al., 1990; Wolswijk and
Noble, 1992). In addition, FGF2 has been reported to increase OP
expression of PDGFaR in vitro (McKinnon et al., 1990). OPs
express receptors for PDGF and FGF2 during the proliferative
response to demyelination (Redwine and Armstrong, 1998).
Further, corresponding expression of PDGF and FGF2 ligands
increases in demyelinated lesion areas (Armstrong et al., 2002;
Messersmith et al., 2000; Woodruff et al., 2004). It is now
important to address the in vivo function of this endogenous PDGF
signaling in a demyelinated lesion environment and whether FGF2
signaling alters the PDGFaR response in this context.

J.C. Murtie et al. / Neurobiology of Disease 19 (2005) 171–182172
During remyelination, FGF2 may also play an important role
in regulating the timing of terminal differentiation as new
oligodendrocytes are generated. In vitro, FGF2 can inhibit
differentiation of OLCs derived from neonatal rodent brain
(Bansal and Pfeiffer, 1997) or isolated from spinal cords of
mice with demyelinating lesions (Armstrong et al., 2002).
Importantly, when demyelination was induced in FGF2 knock-
out mice, lesion repopulation by oligodendrocytes was signifi-
cantly enhanced without effecting proliferation or survival
(Armstrong et al., 2002). More direct demonstration of the
effect of FGF2 genotype on OLC differentiation in vivo could
determine whether attenuation of FGF2 signaling may improve
the cellular responses to promote remyelination in other
conditions.
The function of endogenous PDGF and FGF2 activity can be
examined in vivo using experimental demyelination of mice with
gene deletions that impair each signaling pathway. PDGF-A
knockout mice have dramatically impaired generation of OPs,
oligodendrocytes, and myelin, yet use in remyelination studies is
precluded due to early postnatal mortality (Fruttiger et al., 1999).
PDGF-A overexpression in experimental demyelination signifi-
cantly increased OP proliferation (Woodruff et al., 2004), but may
not appropriately reflect specific roles of endogenous PDGF
signaling during remyelination in vivo. PDGF ligands directly
regulate OLC responses through activation of PDGFaR, which is
the only PDGFR expressed by OLCs (Hart et al., 1989; McKinnon
et al., 1990). The PDGFaR homozygous null mutation is also
lethal during development; however, PDGFaR heterozygotes
express half the normal amount of PDGFaR without phenotypic
abnormalities (Soriano, 1997). FGF2 knockout mice are also
grossly normal and appropriate for analysis of remyelination
(Armstrong et al., 2002; Zhou et al., 1998).
The current study uses PDGFaR heterozygotes and FGF2
knockout mice to evaluate the contribution of each endogenous
signaling pathway in OLCs in a demyelinating disease model that
undergoes extensive spontaneous remyelination. We find that in
response to demyelination, OP amplification is significantly
reduced and subsequent oligodendroglial repopulation of lesions
is impaired in PDGFaR+/� mice. Using in vivo lineage tracing,
we demonstrate that OLCs differentiate into oligodendrocytes
more frequently in FGF2 knockout mice as compared to wild-type
mice. Our further studies in PDGFaR+/� FGF2�/� mice also
indicate distinct roles of each pathway, and argue against the
predicted cooperation of PDGF and FGF2 in OP amplification in
response to demyelination.
Materials and methods
Animals
Mice were bred and maintained in the USUHS animal housing
facility and all procedures were performed in accordance with
guidelines of the National Institutes of Health, the Society for
Neuroscience, and the USUHS Institutional Animal Care and Use
Committee. PDGFaR-targeted deletion mice on the C57Bl/6
genetic background were obtained from breeding heterozygous
pairs (generously provided by Dr. Soriano, Fred Hutchinson Cancer
Research Center). The PDGFaR targeted deletion replaces a 6.5-kb
fragment corresponding to the signal peptide, first and second Ig
domains (Soriano, 1997). FGF2 knockout mice on the 129 Sv-
Ev:Black Swiss genetic background were obtained from breeding
heterozygous pairs (generously provided by Dr. Doetschman,
University of Cincinnati). This FGF2 knockout was generated by
a targeted deletion replacing a 0.5 kb portion of the FGF2 gene
including 121 bp of the promoter and the entire first exon with an
Hprt mini-gene (Zhou et al., 1998). PDGFaR+/� FGF2�/� mice
and PDGFaR+/+ FGF2+/+ mice were generated by crosses of the
PDGFaR line with FGF2 line and the crosses were maintained on
this mixed genetic background.
Cuprizone experimental demyelination
Cuprizone ingestion results in a reproducible pattern of
extensive corpus callosum demyelination followed by sponta-
neous remyelination, which occurs over a period of weeks after
removal of cuprizone and return to normal chow (Armstrong et
al., 2002; Matsushima and Morell, 2001). For all strains of mice,
cuprizone treatment was started at 8 weeks of age and only male
mice were used. Cuprizone (finely powdered oxalic bis(cyclohex-
ylidenehydrazide); Sigma-Aldrich, St. Louis, MO) doses were
titrated for each of the strains of mice in order to induce a similar
and reproducible extent of demyelination based on histological
analyses (Armstrong et al., 2002). Cuprizone was thoroughly
mixed into milled chow (Harlan Teklad; Madison, WI), which was
available ad libitum. The cuprizone dose used was 0.2% (w/w) for
the PDGFaR line, 0.3% for the FGF2 line, and 0.2% for the
crosses of PDGFaR and FGF2 lines. Mice were maintained on
the cuprizone diet until perfused for analysis or returned to normal
chow pellets after 6 weeks.
Tissue preparation and histopathological analysis
Mice were perfused with 4% paraformaldehyde, and then
brains were dissected prior to overnight post-fixation in 4%
paraformaldehyde (Redwine and Armstrong, 1998). Brain tissue
was cryoprotected overnight at 48C in 30% sucrose and
embedded in OCT compound for immunostaining and in situ
hybridization. For histopathology, tissue was embedded in
paraffin and sections were stained with Luxol fast blue to detect
myelin combined with periodic acid-Schiff reaction for monitor-
ing the macrophage/microglial response (performed by USUHS
histological service).
In situ hybridization
In situ hybridization and preparation of digoxigenin-labeled
riboprobes were performed as previously detailed (Messersmith et
al., 2000; Redwine and Armstrong, 1998). Antisense riboprobes
were used to detect mRNA transcripts for PLP (gift from Dr. Lynn
Hudson; National Institutes of Health; Hudson et al., 1987) and
PDGFaR (gift from Dr. Bill Richardson; University College
London; Fruttiger et al., 1999). The digoxigenin-labeled riboprobes
were hybridized to 15-Am cryosections of brain or spinal cord
tissues. Digoxigenin was detected with an alkaline phosphatase-
conjugated sheep anti-digoxigenin antibody (Boehringer Man-
nheim, Indianapolis, IN), followed by reaction with NBT/BCIP
substrate (DAKO, Carpinteria, CA). For detection of PDGFaR
mRNA, the NBT/BCIP reaction was incubated overnight to
amplify the signal. This protocol allowed grossly similar detection
of PDGFaR mRNA signal from cells in PDGFaR+/� and
PDGFaR+/+ mice.

J.C. Murtie et al. / Neurobiology of Disease 19 (2005) 171–182 173
BrdU incorporation and detection
In situ hybridization combined with bromodeoxyuridine (BrdU)
incorporation was carried out as detailed previously (Redwine and
Armstrong, 1998). Mice were injected intraperitoneally with 200
mg/kg BrdU (Sigma, St. Louis, MO) at 4 h and 2 h prior to sacrifice.
After in situ hybridization detection, sections were treated with HCl
then incubated overnight with a monoclonal anti-BrdU antibody
directly conjugated with horseradish peroxidase (diluted 1:15;
Boehringer Mannheim). Peroxidase activity was detected using
3,3V-diaminobenzidine (DAB; Vector Labs, Burlingame, CA).
Immunohistochemistry
To identify OPs in situ, 15-Am cryosections were immunostained
for NG2 and PDGFaR (Armstrong et al., 2002; Messersmith et al.,
2000). Primary antibodies used were rabbit polyclonal anti-NG2
antibody (1:500; gift from Dr. William Stallcup, La Jolla, CA) and
rat monoclonal anti-PDGFaR antibody (APA5 used at 1:200;
Pharmingen, San Diego, CA). Donkey anti-rabbit IgG F(abV)2conjugated with Cy3 (Jackson Immunoresearch, West Grove, PA)
was used to detect NG2 while the PDGFaR was detected with
biotinylated donkey anti-rat IgG F(abV)2 (Jackson Immunoresearch)
followed by coumarin tyramide amplification (New England
Nuclear, Boston, MA).
Mature oligodendrocytes were identified with CC1, which
immunostains oligodendrocyte cell bodies without labeling myelin
(Fuss et al., 2000). The CC1 antibody (Oncogene Research
Products, Cambridge, MA) was detected with donkey anti-mouse
IgG F(abV)2 conjugated with Cy3 (Jackson Immunoresearch). The
CC1 immunostaining conditions were previously tested to ensure
that CC1 did not label astrocytes or NG2-labeled cells (Messer-
smith et al., 2000).
Myelin was immunostained with monoclonal antibody 8-18C5,
which recognizes MOG (hybridoma cells provided by Dr. Minetta
Gardinier; University of Iowa, Iowa City, IA; Linnington et al.,
1984). MOG immunolabeling was detected with donkey anti-mouse
IgG F(abV)2 conjugated with Cy3 (Jackson Immunoresearch).
Apoptosis
After 6 weeks of cuprizone treatment, adult mouse corpus
callosum was analyzed for cells undergoing apoptosis. Cryosec-
tions (15 Am) were processed using a modified TUNEL assay
(ApopTag Plus peroxidase in situ apoptosis detection kit; Intergen,
Purchase, NY). The 3V-OH DNA ends, generated by DNA
fragmentation typically observed with apoptotic cells, were labeled
with digoxigenin-dUTP using terminal deoxynucleotidyl trans-
ferase (TdT). The digoxigenin tag was then detected with an anti-
digoxigenin antibody conjugated with peroxidase and reacted with
DAB substrate. The sections were lightly counterstained with
methyl green to detect nuclei.
Retrovirus production
A 293-cell line stably transfected with pNIT-GFP, a replication-
incompetent retroviral expression vector encoding GFP, (provided
by Dr. Fred Gage; Salk Institute, La Jolla, CA; Palmer et al., 1999)
was transiently transfected with pMD.G (plasmid containing the
vesicular stomatitis virus glycoprotein) and cultured for 2 days.
Virion-containing supernatants were concentrated 100-fold by
centrifugation at 50,000 � g at 48C for 150 min. Viral pellets
were reconstituted in Hanks balanced salt solution to a final
concentration of 100� the original concentration. Titers (typically
105 cfu/mL) were determined by GFP expression after infection of
NIH 3T3 cells with serial dilutions of concentrated virus.
Replication-incompetent DAP retrovirus, encoding membrane-
associated human placental alkaline phosphatase, was generated
from a stable NIH 3T3 producer cell line (Fields-Berry et al., 1992).
Producer cells were grown to confluence and the supernatant was
collected after 3 days and concentrated 100-fold by centrifugation
at 50,000 � g at 48C for 150 min. Alkaline phosphatase was
detected with NBT/BCIP substrate (DAKO, Carpinteria, CA).
Stereotaxic surgery and retrovirus injection
Three days prior to cuprizone treatment, 8-week-old male mice
were anesthetized with isoflurane and body temperature was
maintained with 378C isothermal heating pads. The head was
stabilized in a stereotaxic apparatus using cushioned ear bars and a
burr hole was drilled into the skull at the following coordinates:
bregma�1.0 mm and 0.25mm lateral to the sagittal suture (Franklin
and Paxinos, 1997). Virus (102 cfu in 1.0 AL) was injected directly
into the corpus callosum at a depth of 1.875 mm and a rate of 0.2 ALper min. After injection, the burr hole was sealed using Gelfoam and
the wound closed with a single wound clip.
After 6 weeks of cuprizone treatment and 3 weeks of recovery,
mice were transcardially perfused with 3% paraformaldehyde in 0.1
M phosphate buffer followed by fixation overnight at 48C in 3%
paraformaldehyde. Tissues to be used for immunohistochemistry
were cryoprotected in 30% sucrose overnight at 48C then
embedded in OCT compound and stored at �808C.
Imaging, quantification, and statistical analysis
Images of in situ hybridization results were captured with a Spot
2 digital camera using Spot Advanced image acquisition software
(Diagnostic Instruments, Sterling Heights, MI) on an Olympus IX-
70 microscope. Fluorescent images were captured using a Laser
Scanning PASCAL 5 Zeiss confocal microscope with a 100�objective. A stack of Z-series images was acquired and the
maximum projection of this image stack was generated using Zeiss
Pascal 5 software. Images were prepared as panels using Adobe
Photoshop.
For comparing cell densities, cells expressing PLP mRNAwere
quantified using unbiased stereological morphometric analysis
(Messersmith et al., 2000; Stereologer System Systems Planning
and Analysis, Inc., Alexandria, VA). Analysis was restricted to the
corpus callosum region, from the midline and extending laterally to
below the cingulum in 15-Am-thick coronal sections. Using the
Stereologer System, the thickness is sampled as part of the
definition of each bdissectorQ volume, so that density measure-
ments reflect cells/mm3. The unbiased stereological method could
not be used appropriately for conditions with relatively few cells of
interest in any particular category. Therefore, quantification of
PDGFR/BrdU single- and double-labeled categories in the corpus
callosum required counting all labeled cells and measuring the area
sampled (Armstrong et al., 2002). Without use of the Stereologer
System, section thickness could not be sampled in the mounted
specimen, and so density units are cells/mm2.
Each category analyzed included 3 or more tissue sections per
mouse and 3 or more mice per condition, except where noted in
J.C. Murtie et al. / Neurobiology of Disease 19 (2005) 171–182174
text and/or figure legends. Unpaired Student’s t tests were used to
identify significant differences between genotypes and/or treat-
ments. No statistical comparisons were made between mice with
different genetic backgrounds (i.e., PDGFaR+/� mice were not
compared to PDGFaR+/� FGF2�/� mice). The significance of
proportions generated in lineage tracing was tested using the v2
statistical test.
Results
Oligodendrocyte and OP densities in non-lesioned white matter of
adult PDGFaR heterozygotes
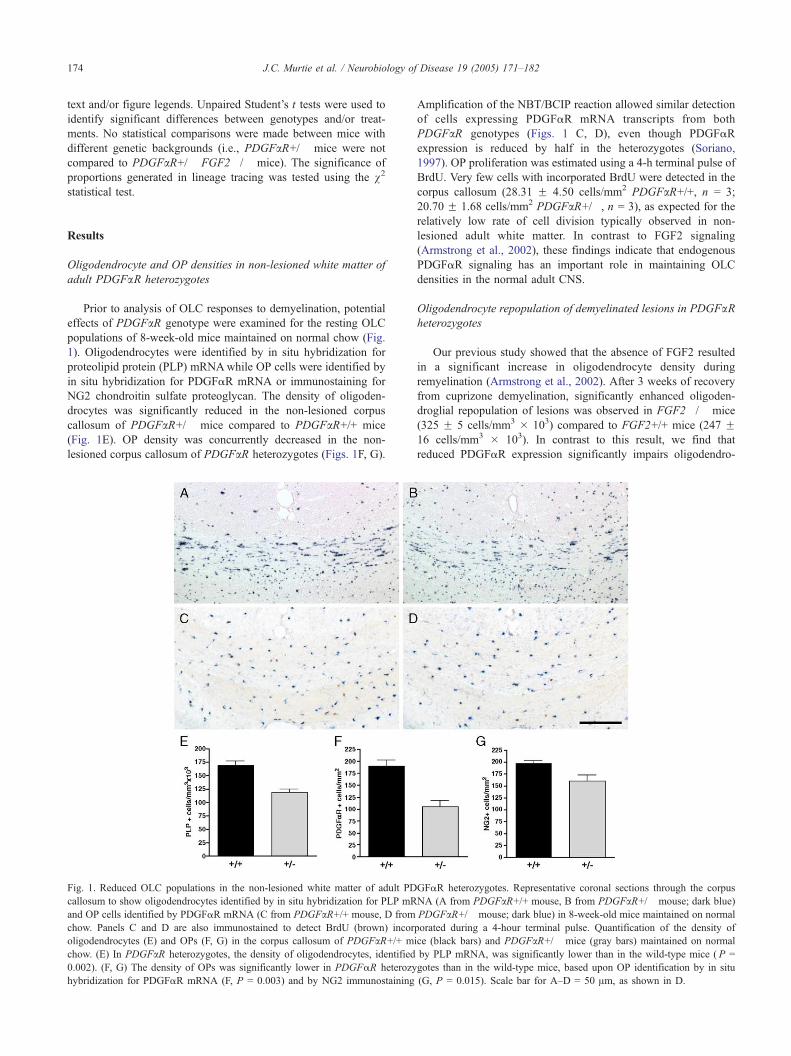
Prior to analysis of OLC responses to demyelination, potential
effects of PDGFaR genotype were examined for the resting OLC
populations of 8-week-old mice maintained on normal chow (Fig.
1). Oligodendrocytes were identified by in situ hybridization for
proteolipid protein (PLP) mRNAwhile OP cells were identified by
in situ hybridization for PDGFaR mRNA or immunostaining for
NG2 chondroitin sulfate proteoglycan. The density of oligoden-
drocytes was significantly reduced in the non-lesioned corpus
callosum of PDGFaR+/� mice compared to PDGFaR+/+ mice
(Fig. 1E). OP density was concurrently decreased in the non-
lesioned corpus callosum of PDGFaR heterozygotes (Figs. 1F, G).
Fig. 1. Reduced OLC populations in the non-lesioned white matter of adult PD
callosum to show oligodendrocytes identified by in situ hybridization for PLP mR
and OP cells identified by PDGFaR mRNA (C from PDGFaR+/+ mouse, D from
chow. Panels C and D are also immunostained to detect BrdU (brown) incor
oligodendrocytes (E) and OPs (F, G) in the corpus callosum of PDGFaR+/+ m
chow. (E) In PDGFaR heterozygotes, the density of oligodendrocytes, identified
0.002). (F, G) The density of OPs was significantly lower in PDGFaR heterozy
hybridization for PDGFaR mRNA (F, P = 0.003) and by NG2 immunostaining
Amplification of the NBT/BCIP reaction allowed similar detection
of cells expressing PDGFaR mRNA transcripts from both
PDGFaR genotypes (Figs. 1 C, D), even though PDGFaR
expression is reduced by half in the heterozygotes (Soriano,
1997). OP proliferation was estimated using a 4-h terminal pulse of
BrdU. Very few cells with incorporated BrdU were detected in the
corpus callosum (28.31 F 4.50 cells/mm2 PDGFaR+/+, n = 3;
20.70 F 1.68 cells/mm2 PDGFaR+/�, n = 3), as expected for the
relatively low rate of cell division typically observed in non-
lesioned adult white matter. In contrast to FGF2 signaling
(Armstrong et al., 2002), these findings indicate that endogenous
PDGFaR signaling has an important role in maintaining OLC
densities in the normal adult CNS.
Oligodendrocyte repopulation of demyelinated lesions in PDGFaRheterozygotes
Our previous study showed that the absence of FGF2 resulted
in a significant increase in oligodendrocyte density during
remyelination (Armstrong et al., 2002). After 3 weeks of recovery
from cuprizone demyelination, significantly enhanced oligoden-
droglial repopulation of lesions was observed in FGF2�/� mice
(325 F 5 cells/mm3 � 103) compared to FGF2+/+ mice (247 F16 cells/mm3 � 103). In contrast to this result, we find that
reduced PDGFaR expression significantly impairs oligodendro-
GFaR heterozygotes. Representative coronal sections through the corpus
NA (A from PDGFaR+/+ mouse, B from PDGFaR+/� mouse; dark blue)
PDGFaR+/� mouse; dark blue) in 8-week-old mice maintained on normal
porated during a 4-hour terminal pulse. Quantification of the density of
ice (black bars) and PDGFaR+/� mice (gray bars) maintained on normal
by PLP mRNA, was significantly lower than in the wild-type mice ( P =
gotes than in the wild-type mice, based upon OP identification by in situ
(G, P = 0.015). Scale bar for A–D = 50 Am, as shown in D.

J.C. Murtie et al. / Neurobiology of Disease 19 (2005) 171–182 175
glial lesion repopulation in the same cuprizone model. Myelin
oligodendrocyte glycoprotein (MOG) immunostaining (Fig. 2) and
histological staining (not shown) demonstrated that PDGFaRheterozygotes undergo the same time course of demyelination and
spontaneous remyelination that has been previously reported for
adult mice fed cuprizone (Armstrong et al., 2002; Matsushima and
Morell, 2001). By 3 weeks into the cuprizone treatment,
demyelination is not yet consistently evident in the corpus
callosum (Fig. 2A), but there is already substantial loss of
oligodendrocytes (Fig. 3C). Demyelination of the corpus callosum
is complete by 6 weeks (Fig. 2B). After a further 3 weeks on
normal chow to allow remyelination to proceed, extensive MOG
immunoreactivity is again present throughout the corpus callosum
(Fig. 2C).
Analysis of oligodendrocyte density over the course of
demyelination using PLP mRNA in situ hybridization revealed a
dramatic decrease in lesion repopulation by oligodendrocytes in
PDGFaR heterozygotes (Fig. 3). In PDGFaR+/+ mice, oligoden-
drocyte regeneration is sufficient to regain normal density
throughout the remyelinating corpus callosum after 3 weeks of
Fig. 2. Similar cuprizone disease course in PDGFaR heterozygotes.
Representative coronal views of MOG immunostaining of myelinated
fibers in the corpus callosum over the course of cuprizone treatment in
PDGFaR+/� (A, B, C) and PDGFaR+/+ (not shown) mice. (A) Myelin
is not yet consistently degenerated and phagocytosed after 3 weeks of
cuprizone ingestion so that MOG immunostaining is still present in the
corpus callosum. (B) Maximal demyelination is observed after 6 weeks
of cuprizone ingestion. (C) Remyelination is indicated by MOG
immunoreactivity throughout the corpus callosum after 6 weeks of
cuprizone ingestion followed by 3 weeks of recovery on normal chow.
(m = midline) Scale bar = 100 Am.
Fig. 3. Impaired oligodendrocyte repopulation of demyelinated corpus
callosum in PDGFaR heterozygotes. At specific time points of cuprizone
treatment and recovery, oligodendrocytes were identified by in situ
hybridization for PLP mRNA. Representative images of PLP mRNA in
situ hybridization of coronal brain sections from PDGFaR+/+ (A) and
PDGFaR+/� (B) mice following 6 weeks of continuous cuprizone
ingestion and 3 weeks of recovery on normal chow (A, B; m = midline;
scale bar = 100 Am). (C) Quantitation of PLP mRNA-labeled cell densities
prior to cuprizone ingestion (8 weeks old; no cup), after 3 weeks or 6 weeks
of continuous cuprizone (3 weeks cup and 6 weeks cup), and after a 3-week
recovery period (3 weeks off) with PDGFaR+/� mice (white bars) as
compared to PDGFaR+/+ mice (black bars). Oligodendrocyte regeneration
was significantly impaired in PDGFaR heterozygotes after 6 weeks of
cuprizone ingestion (*P = 0.008) and after 3 weeks of recovery (**P =
0.003). In addition, oligodendrocyte density in PDGFaR heterozygotes
after 3 weeks of recovery did not return to the level found in PDGFaRheterozygotes prior to demyelination (white bars, 3 weeks off vs. no cup;
~P = 0.015).
recovery from cuprizone treatment. In contrast, the oligodendro-
glial repopulation response in PDGFaR+/� mice was impaired so
that the oligodendrocyte density did not return to the level found in
non-lesioned PDGFaR heterozygotes.
Proliferation in PDGFaR heterozygotes and FGF2 knockout mice
Although PDGFaR expression correlates with the vigorous OP
proliferative response to demyelination (Redwine and Armstrong,
1998), a mitogenic function of endogenous PDGFaR signaling has
not been demonstrated in vivo during remyelination. Relative to
cuprizone disease progression, maximal OP proliferation is
observed in the corpus callosum after 5 weeks of cuprizone
treatment (Armstrong et al., 2002). Therefore, the 5-week time
point was chosen to analyze the OP proliferative response in the
corpus callosum using PDGFaR mRNA in situ hybridization in
combination with detection of BrdU (Fig. 4). A 4-h terminal pulse
of BrdU was selected to detect proliferation while minimizing
subsequent time in which differentiation could proceed. NG2
immunostaining was not used to quantify OP density during
remyelination because the intense NG2 immunoreactivity on OP
cell bodies and processes throughout demyelinated lesions makes
accurate cell counts extremely difficult (Keirstead et al., 1998; our
unpublished observations).

Fig. 4. Decreased OP density and proliferation in PDGFaR heterozygotes
and PDGFaR +/� FGF2 �/� mice. (A) Representative image of PDGFaR
mRNA in situ hybridization (blue cytoplasm) combined with BrdU
immunostaining (brown nuclei) in the corpus callosum of a PDGFaRheterozygote. (B) Quantification of OP populations in PDGFaR wild-type
mice (aR +/+, black bars) and heterozygotes (aR +/�, white bars) in
comparison with PDGFaR+/� FGF2�/� mice (aR +/� FGF2 �/�; dark
gray bars). The density of OP cells that had not incorporated BrdU (black
arrowheads in A; aR+ BrdU� in B) is significantly decreased in PDGFaR
heterozygotes (aR +/+ vs. aR +/�; *P = 0.012). A significant decrease was
also observed in the density of dividing OPs (black arrow in A; aR+ BrdU+
in B) (aR +/+ vs. aR +/�; ** P = 0.026). The density of BrdU-labeled cells
that were not identified as OPs (white arrowhead in A; aR- BrdU+ in B) was
also significantly reduced in PDGFaR heterozygotes (aR +/+ vs. aR +/�;f
P = 0.038). The pattern in PDGFaR+/� mice is similar to PDGFaRheterozygotes crossed with FGF2 knockout mice (aR +/� FGF2 �/�).
Scale bar = 25 Am.
Fig. 5. Retroviral infection labels endogenous cycling cells of the adult
white matter that have the capacity to remyelinate. Example of coronal
section of corpus callosum from a mouse injected with DAP replication-
incompetent retrovirus encoding the alkaline phosphatase reporter. DAP
was injected 3 days prior to initiating cuprizone ingestion so that the virus
infected and heritably labeled the endogenous cycling cells of the normal
adult corpus callosum. After 6 weeks of continuous cuprizone followed by
3 weeks on normal chow, membrane-associated alkaline phosphatase
substrate deposition is found on parallel tracks oriented with axons in the
corpus callosum, indicative of remyelination by cells generated from the
endogenous cycling cells. Scale bar = 50 Am.
J.C. Murtie et al. / Neurobiology of Disease 19 (2005) 171–182176
As compared to PDGFaR+/+ mice, the total OP density in
PDGFaR+/� mice was dramatically reduced during the peak of
OP amplification in response to cuprizone demyelination (Fig. 4).
The density of OPs without detectable BrdU (PDGFaR+, BrdU�cell phenotype) was significantly reduced in PDGFaR+/� mice
relative to PDGFaR+/+ mice. The density of proliferating OP cells
(PDGFaR+, BrdU+ cell phenotype) was also significantly reduced
in PDGFaR+/� mice. The labeling index (i.e., the percentage of
PDGFaR+ OP cells with incorporated BrdU) was similar for
PDGFaR+/� (14.46%) and PDGFaR+/+ (14.68%) genotypes.
Finally, the density of BrdU labeled cells that were not identified as
OPs (PDGFaR�, BrdU+ cell phenotype) was also significantly
reduced in PDGFaR heterozygotes compared to PDGFaR wild-
type mice. This population of PDGFaR-negative cells could
include several cell types with responsive yet undetectable levels
of PDGFaR, such as newly formed oligodendrocytes. In addition,
several reports have shown that reactive astrocytes express low
levels of PDGFaR and may also have the potential to respond to
PDGF (Maeda et al., 2001; Redwine and Armstrong, 1998).
To test the potential cooperative interaction of FGF2 and
PDGFaR signaling in the OP proliferative response to demyeli-
nation, OP density and BrdU incorporation was assessed in mice of
the PDGFaR line crossed with mice of the FGF2 line. Our
previous analysis demonstrated that FGF2 knockout alone did not
significantly alter OP density or proliferation (Armstrong et al.,
2002). OP proliferation was significantly impaired in PDGFaR+/�FGF2�/� mice compared to wild-type mice of the same crossed
background (P = 0.006 for comparison of 75.34 F 9.81 cells/mm2
PDGFaR+/� FGF2�/� mice, n = 5, with 119.38 F 42.16 cells/
mm2 PDGFaR+/+ FGF2+/+ mice, n = 4). Importantly, loss of
FGF2 in PDGFaR+/� FGF2�/� mice did not exacerbate the
already reduced OP density or impaired proliferation observed in
PDGFaR+/� mice (Fig. 4). These findings indicate that PDGFaR
signaling is the significant regulator of OP proliferation in response
to demyelination and does not interact with FGF2 signaling to
promote OP proliferation.
Differentiation in PDGFaR heterozygotes and FGF2 knockout
mice
In vivo lineage tracing with replication-incompetent retrovirus
was used to monitor differentiation of endogenous cycling cells
into oligodendrocytes during the course of demyelination and
remyelination. To determine the impact of endogenous PDGF and
FGF2 pathways on OLC differentiation during remyelination,
lineage tracing was performed in PDGFaR heterozygotes and in
FGF2 knockout mice, with comparison to wild-type mice of each
line. Three days prior to initiation of cuprizone treatment, DAP or
NIT-GFP retrovirus was stereotactically injected into the corpus
callosum of 8-week-old mice to infect the endogenous cycling cells
of the adult white matter. Injecting with retrovirus prior to inducing
demyelination allows infection of endogenous cycling cells but not
reactive astrocytes and microglia (Gensert and Goldman, 1997; our
unpublished observations). Retroviral infection of these endoge-
nous cycling cells with a heritable reporter gene can monitor the
generation of remyelinating oligodendrocytes, as demonstrated by
myelin immunolabeling (Gensert and Goldman, 1997) or by
alkaline phosphatase association with myelin membranes (Fig. 5).
After 6 weeks of cuprizone treatment followed by a 3-week
recovery period for remyelination, cells labeled by NIT-GFP

J.C. Murtie et al. / Neurobiology of Disease 19 (2005) 171–182 177
retrovirus were analyzed. Both OPs and oligodendrocytes were
labeled by retroviral GFP after recovery from demyelination.
Oligodendrocytes were identified morphologically by the presence
of bifurcating processes that form bTQ intersections parallel to axonsas well as by immunostaining for the oligodendrocyte marker CC1
(Figs. 6A–C). OPs were identified by double immunostaining for
the OP markers NG2 and PDGFaR (Figs. 6D–G).
In vivo OLC differentiation was assessed by determining the
proportion of total GFP-labeled cells that were identified as OPs or
oligodendrocytes in mice perfused after 3 weeks of recovery from
cuprizone treatment. This quantification indicated that differ-
entiation of OLCs was not changed in PDGFaR+/� mice as
compared to PDGFaR+/+ mice (Figs. 6H, I). Similar proportions
of OPs and oligodendrocytes were found in both genotypes. The
remaining proportion of cells that were not identified by cell type-
specific markers had morphologies consistent with OLCs and this
proportion of cells was also not significantly different between
genotypes.
Fig. 6. Retroviral lineage tracing reveals altered in vivo differentiation with FGF2
into the corpus callosum of 8-week old mice, 3 days later the mice began a 6-week
recovery period on normal chow, then the mice were perfused and GFP expressing
(A–C) A GFP-expressing cell immunolabeled with CC1, a marker of oligodendro
is detectable in the cell body and processes while CC1 immunostaining (B, red) is
points to a bifurcated process forming a bTQ intersection with distal process br
immunolabeled with PDGFaR and NG2, markers of OP cells (representative cell
GFP fluorescence (D, green) and are outlined by NG2 (E, red) immunoreactivity w
Quantitation of the proportion of total GFP+ cells that were morphologically iden
not changed by PDGFaR genotype, in contrast to the significant increase (*P = 0.
(n = 3 mice). (I) Quantitation of the proportion of total GFP+ cells that were ide
immunostained with CC1, or as OP cells, in tissue sections immunostained for PD
in mice of each PDGFaR+ genotype. FGF2�/� mice have a significantly lower
proportion of GFP-labeled oligodendrocytes (~P = 0.003) compared to FGF2+/+
mean F standard error of the proportion.
In contrast, FGF2 genotype clearly influenced OLC differ-
entiation in mice that had recovered from cuprizone demyelina-
tion. The proportion of GFP-positive cells that were
morphologically identified as oligodendrocytes was significantly
higher in FGF2�/� mice, as compared to FGF2+/+ mice
(Fig. 6I). This quantification by morphology was confirmed with
CC1 immunohistochemistry in combination with GFP detection
(Fig. 6K). Consistent with this result, the proportion of GFP-
positive cells identified as OPs (i.e., immunolabeled with NG2 and
PDGFaR) was significantly decreased in FGF2�/� mice
(Fig. 6K). These findings indicate that endogenous FGF2 inhibits
OLC differentiation during remyelination.
Survival in PDGFaR heterozygotes and FGF2 knockout mice
TUNEL analysis was completed on PDGFaR+/+ and
PDGFaR+/� mice after 6 weeks of cuprizone treatment to
determine if OLC survival was affected by a decrease in PDGFaR
genotype but not with PDGFaR genotype. NIT-GFP retrovirus was injected
period of continuous cuprizone ingestion which was followed by a 3-week
cells were analyzed by morphology and cell type-specific immunostaining.
cytes (representative cell example; PDGFaR heterozygote). GFP (A, green)
found in the cell body (C, overlap in cell body appears yellow). The arrow
anches aligned parallel to the axon fibers. (D–G) A GFP-expressing cell
example; PDGFaR heterozygote). An OP cell body and processes contain
hile PDGFaR immunoreactivity (F, blue) is within the soma cytoplasm. (H)
tified (see text) as oligodendrocytes. The proportion of oligodendrocytes is
001) observed in FGF2�/� mice (n = 5 mice) compared to FGF2+/+ mice
ntified by cell type-specific markers as oligodendrocytes, in tissue sections
GFaR and/or NG2. The oligodendrocyte and OP populations are the same
proportion of GFP-labeled OP cells (#P = 0.001) and a significantly higher
mice. (A, D) Scale bars = 10 Am. Values shown represent the proportional

J.C. Murtie et al. / Neurobiology of Disease 19 (2005) 171–182178
expression. Cell death from cuprizone toxicity should be minimal at
this time point (Mason et al., 2000). With this in mind, the 6-week
cuprizone time point should be the appropriate stage at which to test
for a survival effect on OPs and newly formed oligodendrocytes.
TUNEL analysis yielded very few TUNEL+ cells in either
genotype; furthermore, there was no significant genotypic effect
on cell survival (50.76F 6.46 cells/mm2 in PDGFaR+/+ mice, n =
3; 27.83F 6.95 cells/mm2 in PDGFaR+/�mice, n = 3; P = 0.063).
These results are similar to results from TUNEL analysis in FGF2
knockout mice (Armstrong et al., 2002). Together, these data
indicate that neither PDGF nor FGF2 have detectable effects on
OLC survival in vivo.
Oligodendroglial lesion repopulation in PDGFaR+/� FGF2�/�mice
The potential interaction of PDGF and FGF2 pathways in the
OLC response to demyelination was further tested by examining
oligodendrocyte repopulation of lesions in crosses of the PDGFaRand FGF2 lines (Fig. 7). As stated previously, the oligodendroglial
repopulation response to demyelination is hindered in PDGFaRheterozygotes and enhanced in FGF2 knockout mice. If these two
pathways interact as mitogens that promote OP amplification in
this context, then oligodendrocyte regeneration in PDGFaR+/�FGF2�/� mice should be reduced even more dramatically than in
PDGFaR heterozygotes. Alternatively, if PDGF and FGF2 signal-
ing pathways have distinct effects on OLCs during remyelination,
then the oligodendroglial repopulation in PDGFaR+/� FGF2�/�mice should be improved relative to PDGFaR heterozygotes to
reflect the combined effect of the individual mutations. Analysis of
non-lesioned mice revealed initial support of the latter interpreta-
tion. Specifically, the absence of FGF2 in PDGFaR+/� FGF2�/�mice enabled the oligodendrocyte density in non-lesioned white
matter to achieve normal levels (Fig. 7; no cup). Further support of
distinct effects was clear when PDGFaR+/� FGF2�/� mice were
demyelinated by cuprizone ingestion. Mice from the cross of
PDGFaR and FGF2 lines underwent the same characteristic
Fig. 7. Absence of FGF2 rescues PDGFaR heterozygote impairment of
oligodendrocyte repopulation of demyelinated lesions. In situ hybridization
for PLP mRNA to assess oligodendrocyte density over the course of
cuprizone treatment in PDGFaR+/� FGF2�/� mice (white bars) as
compared to PDGFaR+/+ FGF2+/+ mice (black bars) showed no
significant differences between genotypes. The oligodendrocyte density
was quantitated prior to the start of cuprizone treatment (no cup; 8 weeks of
age) to compare normal baseline values, during the period of oligoden-
drocyte loss at 3 weeks of continuous cuprizone treatment (3 weeks cup),
and during the period of oligodendrocyte repopulation after 6 weeks of
continuous cuprizone followed by 3 weeks of recovery on normal chow (6
weeks cup, 3 weeks off).
pattern of oligodendrocyte loss after 3 weeks of continuous
cuprizone treatment (Fig. 7; 3 weeks cup). After 6 weeks of
continuous cuprizone ingestion followed by 3 weeks of recovery
on normal chow, significant oligodendrocyte repopulation occurred
and was similar in PDGFaR+/� FGF2�/� mice as compared to
PDGFaR+/+ FGF2+/+ wild-type mice (Fig. 7; 6 weeks cup, 3
weeks off). These findings are consistent with distinct effects of
endogenous signaling through PDGF and FGF2 on OLCs in the
course of experimental demyelination and remyelination. Specif-
ically, these results support a model (Fig. 8) in which PDGF
signaling enhances OP proliferation during demyelination and
FGF2 predominantly inhibits differentiation of OPs into oligoden-
drocytes during remyelination.
Discussion
In response to demyelination, both PDGF and FGF2 ligand and
receptor expression levels are increased (Armstrong et al., 2002;
Hinks and Franklin, 1999; Messersmith et al., 2000; Woodruff et
al., 2004). These findings in combination with functional in vitro
data indicate that FGF2 and PDGF could potentially act
individually or cooperatively to regulate OLC responses in the
lesion environment. In the current study, we show that PDGFaRheterozygotes have a deficit in proliferation and subsequent lesion
repopulation by oligodendrocytes. This impaired proliferative
response to demyelination in PDGFaR heterozygotes was not
exacerbated by the absence of FGF2 in PDGFaR+/� FGF2�/�mice. In contrast to the current results in PDGFaR+/� mice, FGF2
knockout mice have an enhanced oligodendroglial repopulation
response (Armstrong et al., 2002). We now demonstrate that
removal of endogenous FGF2 from the lesion environment
corresponds with more frequent differentiation of immature cells
into mature oligodendrocytes. This result in FGF2�/� mice is not
likely to result from compensation by other FGF family members
since a similar effect was observed in vitro using a short-term
treatment with anti-FGF2 specific neutralizing antibody (Arm-
strong et al., 2002) and initial studies of these FGF2�/� mice
showed that expression of FGF1, FGF4, and FGF5 was not altered
(Zhou et al., 1998).
PDGF and FGF2 can each act as OP mitogens in vitro (Frost et
al., 2003; McKinnon et al., 1990; Wolswijk and Noble, 1992).
Indeed, the current findings show that PDGF is a critical
endogenous mitogen for OLCs in the adult CNS in vivo. In
contrast, FGF2 is not a significant OP mitogen in this context
(Armstrong et al., 2002). The current in vivo findings are
consistent with the role of PDGFaR signaling in vitro as
stimulating proliferation of OPs from adult CNS (Shi et al.,
1998; Wolswijk and Noble, 1992). In diverse models of
experimental demyelination, amplification of OPs expressing
PDGFaR has been associated with demyelinated lesions (Penderis
et al., 2003; Redwine and Armstrong, 1998) and increased
expression of endogenous PDGF-A ligand (Redwine and Arm-
strong, 1998; Woodruff et al., 2004). Overexpression of PDGF-A
ligand in experimental demyelination further increased OP cell
densities in normal adult and lesioned GFAP-hPDGF-A mice,
demonstrating a functional response of this signaling pathway in
the context of demyelinating disease (Woodruff et al., 2004). The
present results now extend these studies to demonstrate that
endogenous PDGFaR signaling indeed plays a role in promoting
OP proliferation in the context of a demyelinating lesion environ-

Fig. 8. Proposed model for actions of endogenous PDGF and FGF2 on OLCs in the adult CNS. (A) Previous in vitro studies have predicted multiple potential
roles for PDGF and FGF2 in vivo (see text). PDGF and FGF2 can each stimulate OP proliferation, and cooperative interaction of PDGF and FGF2 can induce
OPs from adult CNS to divide more rapidly as a self-renewing line. PDGF can promote appropriately timed OP differentiation while FGF2 strongly inhibits
differentiation. (B) Based on studies using PDGFaR+/� mice (current study), FGF2�/� mice (Armstrong et al., 2002 and current study), and PDGFaR+/�FGF2�/� mice (current study), these growth factors do not act cooperatively in vivo. Reduced PDGFaR expression led to decreased OP proliferation (thin
circular arrow) and fewer oligodendrocytes. Removal of FGF2 inhibition of differentiation (dotted intersecting bars) led to increased OP differentiation into
oligodendrocytes. (C) These results in mice with genetic deletions led to the proposed model of endogenous PDGF and FGF2 functions. PDGF is an important
OP mitogen (thick circular arrow), particularly during the OLC response to demyelination. In contrast, FGF2 predominantly inhibits differentiation (solid
intersecting bars) of OPs into oligodendrocytes, which is critically important during remyelination. (A, B, C) Thickness of arrows corresponds with amplitude
of cellular response; circular arrow indicates OP proliferation; straight arrow indicates promotion of OP differentiation; intersecting bar indicates inhibition of
OP differentiation.
J.C. Murtie et al. / Neurobiology of Disease 19 (2005) 171–182 179
ment. Both PDGF-A ligand overexpression (Woodruff et al., 2004)
and PDGFaR deficiency (current study) altered OP densities in
demyelinated lesions without altering the BrdU-labeling index, as
compared to wild-type mice. Therefore, in response to demyeli-
nation, PDGF may stimulate conversion of resting OP cells into
rapidly proliferating OP cells without subsequently altering the OP
cell cycle length during the disease progression.
Surprisingly, we also show that PDGFaR genotype plays a
significant role in the maintenance of OPs and oligodendrocytes in
the non-lesioned adult white matter. The current findings in non-
lesioned adult CNS are consistent with previous in vivo
manipulations of PDGF ligand levels that demonstrated the
importance of PDGF in regulation of OP and oligodendrocyte cell
density during development (Barres and Raff, 1993; Calver et al.,
1998; Fruttiger et al., 1999). However, these reports conclude that
axon number is the overriding factor that controls the final
oligodendrocyte density in the non-lesioned white matter at the
end of myelination. Our findings indicate that PDGFaR signaling
continues to contribute to OP and oligodendrocyte cell densities in
the adult CNS. Presumably, PDGFaR signaling then acts in
concert with axonal signals and other growth factor pathways.
Several studies have implicated PDGF and FGF2 as influencing
OLC differentiation (Allamargot et al., 2001; Bansal and Pfeiffer,
1997; Bogler et al., 1990; Wilson et al., 2003). We used retroviral
lineage tracing over the course of demyelination and remyelination
to demonstrate that FGF2 deletion, but not PDGFaR hetero-
zygosity, altered OLC differentiation. This direct analysis of the
progression from cycling cell through differentiated oligodendro-
cyte indicates that endogenous FGF2 inhibits OLC differentiation.
This effect of FGF2 was also predicted from our previous analysis
of OLC population densities in FGF2 knockout mice undergoing
demyelination and remyelination (Armstrong et al., 2002). This
role of FGF2 as an inhibitor of OLC differentiation in vivo during
remyelination contrasts with a report of developmental myelination
in FGFR3 null mice in which endogenous FGFR3 signalling was
predicted to promote OLC differentiation (Oh et al., 2003). These
results may reflect differential expression of FGFR1, FGFR2, and/
or FGFR3 at different OLCs stages in development as compared to
in remyelinating lesions (Bansal et al., 1996; Messersmith et al.,
2000). Alternatively, these findings may be explained by preferred
activation of FGFR3 by FGF family members that may be more
optimal ligands than FGF2 (Chellaiah et al., 1999; Ornitz and
Leder, 1992). Furthermore, future studies will need to address
whether FGF2 acts directly on OLCs in this context, since FGFRs
are expressed by multiple cell types in demyelinated lesions (Liu et
al., 1998; Messersmith et al., 2000).
PDGF can promote survival of OP cells and newly generated
oligodendrocytes (Barres et al., 1992; Ebner et al., 2000), while
FGF2 has been reported to be detrimental for oligodendrocyte
viability (Muir and Compston, 1996). However, an effect on cell
survival was not detected using TUNEL analysis during the
transition from demyelination to remyelination in PDGFaRheterozygotes (current study) or in FGF2 knockout mice (Arm-
strong et al., 2002). TUNEL analysis and other assays of apoptosis
identify cells actively undergoing apoptosis that have not yet been
removed by phagocytosis. Therefore, it is possible that subtle
changes in cell survival are difficult to detect in this interval.
Furthermore, even though PDGFaR expression is reduced to
approximately half the wild-type expression level in PDGFaRheterozygotes (Soriano, 1997), we cannot yet rule out the
possibility that this reduced expression of PDGFaR could still be
sufficient to provide the necessary survival signaling.

J.C. Murtie et al. / Neurobiology of Disease 19 (2005) 171–182180
PDGFaR heterozygotes and FGF2 knockout mice provided an
excellent opportunity to test the potential in vivo interaction
between PDGF and FGF2 signaling pathways. Cooperation
between PDGF and FGF2 has been reported to convert slowly
dividing OPs from adult CNS into rapidly dividing cells in vitro,
indicative of an important potential role in repair of demyelinated
lesions in vivo (Chari and Blakemore, 2002; Wolswijk and Noble,
1992). Accordingly, absence of FGF2 combined with decreased
expression of PDGFaR may have been predicted to exacerbate the
PDGFaR heterozygote effect of impaired OP proliferation and
lesion repopulation by oligodendrocytes. In fact, our results do not
support a role for FGF2 cooperating with PDGFaR signaling for a
mitogenic effect on OPs in vivo. Using PDGFaR+/� FGF2�/�mice, there was no observed interaction between PDGF and FGF2
as OP mitogens (Fig. 4). In addition, oligodendroglial repopulation
in response to demyelination was not further impaired as compared
to PDGFaR+/� mice but was instead improved in PDGFaR+/�FGF2�/� mice (Fig. 7).
Based on our in vivo analyses of genetic deletions in
experimental demyelination (current study and Armstrong et al.,
2002), we propose a model in which endogenous PDGF and FGF2
predominantly mediate distinct effects on OLCs in demyelinated
white matter (Fig. 8). Specifically, in vivo PDGFaR signaling is a
predominant regulator of OP proliferation in response to demyeli-
nation while FGF2 is a predominant inhibitor of OP differentiation
during remyelination. These effects have been observed within the
CNS white matter and in the course of experimental demyelination
with spontaneous remyelination. This model does not address
potential PDGF and/or FGF2 regulation of neural stem cell
responses that could occur in distinct sites. Furthermore, the
genetic deletions utilized here address removal of endogenous
PDGFaR and FGF2 signaling. Additional potential neural stem
cell and OLC responses may be elicited by genetic overexpression
or exogenous administration to elevate PDGF and FGF2 levels
(Lachapelle et al., 2002; Ruffini et al., 2001; Woodruff et al.,
2004). This model contrasts with PDGF and FGF2 roles predicted
from mainly in vitro studies that indicated a more significant
contribution of endogenous FGF2 in the OP proliferative response
to demyelination (Chari and Blakemore, 2002; Wolswijk and
Noble, 1995). Our previous in vitro analysis of OLC cells derived
from demyelinated lesions of wild type mice used pharmacological
reagents to transiently impair PDGFaR and/or FGF2 activity, with
comparison to administration of exogenous PDGF-AA and/or
FGF2 (Frost et al., 2003). This in vitro work also indicated an
FGF2 effect on both OLC differentiation and OP proliferation as
well as a cooperative effect of PDGF and FGF2 signaling (Frost et
al., 2003). Therefore, we speculate that the current difference
between previous in vitro studies and the current in vivo model
reflects a difference of signaling in the complexity of the in vivo
context. However, we cannot rule out the possibility that other
differences in the PDGFaR+/� or FGF2�/� mice could have
occurred during development and play a role in the current results.
We show that PDGFaR signaling is critical for normal OP
amplification in response to demyelination. However, generation
of oligodendrocytes from this reduced OP pool was improved by
removal of FGF2 inhibition of OLC differentiation. This finding is
extremely important with respect to potential treatment of diseases
in which oligodendrocyte regeneration is needed for remyelination.
To promote remyelination, transient attenuation of FGF2 signaling
might be sufficient for overcoming reduced OP density and/or a
lesion environment that lacks adequate support for fully exploiting
the potential for oligodendrocyte regeneration. In the current
disease model, as in most rodent experimental models, the
oligodendrocyte density is sufficient and the environment is
favorable for remyelination so that these cellular effects do not
appear to markedly alter remyelination (data not shown for
PDGFaR heterozygotes, current study; Armstrong et al., 2002).
Further studies will be needed to determine the extent to which the
cellular responses identified in this study can be exploited to
improve remyelination and recovery of function in less favorable
lesion contexts that may mimic human disease conditions.
With direct significance to the human context, OPs from human
fetal tissue are responsive to PDGF and express PDGFaR (Wilson
et al., 2003; Zhang et al., 2000). Furthermore, OPs that express
PDGFaR can be abundant in MS lesions (Maeda et al., 2001),
indicating the potential to respond to changes in PDGF expression.
FGF2 may also be present in MS lesions (Holley et al., 2003) and
may limit successful remyelination through inhibition of endoge-
nous OLC differentiation. PDGF and FGF2 are likely to regulate
OLC responses in vivo in the context of multiple signals in the
lesion environment, such as other growth factors, cytokine, and cell
adhesion molecules. Understanding the interactions between
various signals and the resulting OLC responses will be necessary
for the success of growth factor-based treatments to promote repair
in demyelinating diseases.
Acknowledgments
We thank Dr. Philippe Soriano for providing breeding pairs of
the PDGFaR targeted deletion mice, Dr. Thomas Doetschman for
providing breeding pairs of the FGF2 targeted deletion mice as
heterozygotes, Drs. William Stallcup and Minetta Gardinier for
antibodies, Dr. Fred Gage for providing pNIT-GFP 293 cells and
pMD.G plasmid, Dr. Steve Levison for advice on the retroviral
lineage tracing, and Drs. Lynn Hudson and William Richardson for
plasmids. We appreciate the comments of Dr. Nicole Dobson and
Dr. Joe Nielsen. This work was supported by NIH grant NS39293.
References
Allamargot, C., Pouplard-Barthelaix, A., Fressinaud, C., 2001. A single
intracerebral microinjection of platelet-derived growth factor (PDGF)
accelerates the rate of remyelination in vivo. Brain Res. 918 (1–2),
28–39.
Armstrong, R.C., Dorn, H.H., Kufta, C.V., Friedman, E., Dubois-Dalcq,
M.E., 1992. Pre-oligodendrocytes from adult human CNS. J. Neurosci.
12 (4), 1538–1547.
Armstrong, R.C., Le, T.Q., Frost, E.E., Borke, R.C., Vana, A.C., 2002.
Absence of fibroblast growth factor 2 promotes oligodendroglial
repopulation of demyelinated white matter. J. Neurosci. 22 (19),
8574–8585.
Bansal, R., Pfeiffer, S.E., 1997. Regulation of oligodendrocyte differentiation
by fibroblast growth factors. Adv. Exp. Med. Biol. 429, 69–77.
Bansal, R., Kumar, M., Murray, K., Morrison, R.S., Pfeiffer, S.E., 1996.
Regulation of FGF receptors in the oligodendrocyte lineage. Mol. Cell.
Neurosci. 7 (4), 263–275.
Barres, B.A., Raff, M.C., 1993. Proliferation of oligodendrocyte
precursor cells depends on electrical activity in axons. Nature 361
(6409), 258–260.
Barres, B.A., Hart, I.K., Coles, H.S., Burne, J.F., Voyvodic, J.T.,
Richardson, W.D., Raff, M.C., 1992. Cell death and control of cell
survival in the oligodendrocyte lineage. Cell 70 (1), 31–46.

J.C. Murtie et al. / Neurobiology of Disease 19 (2005) 171–182 181
Blakemore, W.F., Keirstead, H.S., 1999. The origin of remyelinating cells
in the central nervous system. J. Neuroimmunol. 98 (1), 69–76.
Bogler, O., Wren, D., Barnett, S.C., Land, H., Noble, M., 1990.
Cooperation between two growth factors promotes extended self-
renewal and inhibits differentiation of oligodendrocyte-type-2 astro-
cyte (O-2A) progenitor cells. Proc. Natl. Acad. Sci. U. S. A. 87 (16),
6368–6372.
Bruck, W., Kuhlmann, T., Stadelmann, C., 2003. Remyelination in multiple
sclerosis. J. Neurol. Sci. 206 (2), 181–185.
Calver, A.R., Hall, A.C., Yu, W.P., Walsh, F.S., Heath, J.K., Betsholtz, C.,
Richardson, W.D., 1998. Oligodendrocyte population dynamics and the
role of PDGF in vivo. Neuron 20 (5), 869–882.
Chang, A., Tourtellotte, W.W., Rudick, R., Trapp, B.D., 2002. Premyeli-
nating oligodendrocytes in chronic lesions of multiple sclerosis.
N. Engl. J. Med. 346 (3), 165–173.
Chari, D.M., Blakemore, W.F., 2002. New insights into remyelination
failure in multiple sclerosis: implications for glial cell transplantation.
Mult. Scler. 8 (4), 271–277.
Chellaiah, A., Yuan, W., Chellaiah, M., Ornitz, D.M., 1999. Mapping
ligand binding domains in chimeric fibroblast growth factor receptor
molecules. Multiple regions determine ligand binding specificity.
J. Biol. Chem. 274 (49), 34785–34794.
Ebner, S., Dunbar, M., McKinnon, R.D., 2000. Distinct roles for PI3K in
proliferation and survival of oligodendrocyte progenitor cells. J. Neuro-
sci. Res. 62 (3), 336–345.
Fields-Berry, S.C., Halliday, A.L., Cepko, C.L., 1992. A recombinant
retrovirus encoding alkaline phosphatase confirms clonal boundary
assignment in lineage analysis of murine retina. Proc. Natl. Acad. Sci.
U. S. A. 89 (2), 693–697.
Franklin, K.B.J., Paxinos, G., 1997. The Mouse Brain in Stereotaxic
Coordinates. Academic Press, San Diego.
Frost, E.E., Nielsen, J.A., Le, T.Q., Armstrong, R.C., 2003. PDGF and
FGF2 regulate oligodendrocyte progenitor responses to demyelination.
J. Neurobiol. 54 (3), 457–472.
Fruttiger, M., Karlsson, L., Hall, A.C., Abramsson, A., Calver, A.R.,
Bostrom, H., Willetts, K., Bertold, C.H., Heath, J.K., Betsholtz, C.,
et al., 1999. Defective oligodendrocyte development and severe
hypomyelination in PDGF-A knockout mice. Development 126 (3),
457–467.
Fuss, B., Mallon, B., Phan, T., Ohlemeyer, C., Kirchhoff, F., Nishiyama, A.,
Macklin, W.B., 2000. Purification and analysis of in vivo-differentiated
oligodendrocytes expressing the green fluorescent protein. Dev. Biol.
218 (2), 259–274.
Gensert, J.M., Goldman, J.E., 1997. Endogenous progenitors remyelinate
demyelinated axons in the adult CNS. Neuron 19 (1), 197–203.
Goldman, J.E., 2003. What are the characteristics of cycling cells in the
adult central nervous system? J. Cell. Biochem. 88 (1), 20–23.
Hart, I.K., Richardson, W.D., Heldin, C.H., Westermark, B., Raff, M.C.,
1989. PDGF receptors on cells of the oligodendrocyte-type-2 astrocyte
(O-2A) cell lineage. Development 105 (3), 595–603.
Hinks, G.L., Franklin, R.J., 1999. Distinctive patterns of PDGF-A, FGF-2,
IGF-I, and TGF-beta1 gene expression during remyelination of
experimentally-induced spinal cord demyelination. Mol. Cell. Neurosci.
14 (2), 153–168.
Holley, J.E., Gveric, D., Newcombe, J., Cuzner, M.L., Gutowski, N.J.,
2003. Astrocyte characterization in the multiple sclerosis glial scar.
Neuropathol. Appl. Neurobiol. 29 (5), 434–444.
Hudson, L.D., Berndt, J.A., Puckett, C., Kozak, C.A., Lazzarini, R.A.,
1987. Aberrant splicing of proteolipid protein mRNA in the dysmyeli-
nating jimpy mutant mouse. Proc. Natl. Acad. Sci. U. S. A. 84 (5),
1454–1458.
Keirstead, H.S., Levine, J.M., Blakemore, W.F., 1998. Response of the
oligodendrocyte progenitor cell population (defined by NG2 labelling)
to demyelination of the adult spinal cord. Glia 22 (2), 161–170.
Lachapelle, F., Avellana-Adalid, V., Nait-Oumesmar, B., Baron-Van
Evercooren, A., 2002. Fibroblast growth factor-2 (FGF-2) and
platelet-derived growth factor AB (PDGF AB) promote adult SVZ-
derived oligodendrogenesis in vivo. Mol. Cell. Neurosci. 20 (3),
390–403.
Linnington, C., Webb, M., Woodhams, P.L., 1984. A novel myelin-
associated glycoprotein defined by a mouse monoclonal antibody.
J. Neuroimmunol. 6 (6), 387–396.
Liu, X., Mashour, G.A., Webster, H.F., Kurtz, A., 1998. Basic FGF and
FGF receptor 1 are expressed in microglia during experimental
autoimmune encephalomyelitis: temporally distinct expression of
midkine and pleiotrophin. Glia 24 (4), 390–397.
Maeda, Y., Solanky, M., Menonna, J., Chapin, J., Li, W., Dowling, P., 2001.
Platelet-derived growth factor-alpha receptor-positive oligodendroglia
are frequent in multiple sclerosis lesions. Ann. Neurol. 49 (6), 776–785.
Mason, J.L., Ye, P., Suzuki, K., D’Ercole, A.J., Matsushima, G.K., 2000.
Insulin-like growth factor-1 inhibits mature oligodendrocyte apoptosis
during primary demyelination. J. Neurosci. 20 (15), 5703–5708.
Matsushima, G.K., Morell, P., 2001. The neurotoxicant, cuprizone, as a
model to study demyelination and remyelination in the central nervous
system. Brain Pathol. 11 (1), 107–116.
McKinnon, R.D., Matsui, T., Dubois-Dalcq, M., Aaronson, S.A., 1990.
FGF modulates the PDGF-driven pathway of oligodendrocyte develop-
ment. Neuron 5 (5), 603–614.
Messersmith, D.J., Murtie, J.C., Le, T.Q., Frost, E.E., Armstrong, R.C.,
2000. Fibroblast growth factor 2 (FGF2) and FGF receptor expression
in an experimental demyelinating disease with extensive remyelination.
J. Neurosci. Res. 62 (2), 241–256.
Muir, D.A., Compston, D.A., 1996. Growth factor stimulation triggers
apoptotic cell death in mature oligodendrocytes. J. Neurosci. Res. 44
(1), 1–11.
Oh, L.Y., Denninger, A., Colvin, J.S., Vyas, A., Tole, S., Ornitz, D.M.,
Bansal, R., 2003. Fibroblast growth factor receptor 3 signaling regulates
the onset of oligodendrocyte terminal differentiation. J. Neurosci. 23
(3), 883–894.
Ornitz, D.M., Leder, P., 1992. Ligand specificity and heparin dependence of
fibroblast growth factor receptors 1 and 3. J. Biol. Chem. 267 (23),
16305–16311.
Palmer, T.D., Markakis, E.A., Willhoite, A.R., Safar, F., Gage, F.H., 1999.
Fibroblast growth factor-2 activates a latent neurogenic program in
neural stem cells from diverse regions of the adult CNS. J. Neurosci. 19
(19), 8487–8497.
Penderis, J., Shields, S.A., Franklin, R.J., 2003. Impaired remyelination and
depletion of oligodendrocyte progenitors does not occur following
repeated episodes of focal demyelination in the rat central nervous
system. Brain 126 (Pt. 6), 1382–1391.
Redwine, J.M., Armstrong, R.C., 1998. In vivo proliferation of oligoden-
drocyte progenitors expressing PDGFalphaR during early remyelina-
tion. J. Neurobiol. 37 (3), 413–428.
Ruffini, F., Furlan, R., Poliani, P.L., Brambilla, E., Marconi, P.C., Bergami,
A., Desina, G., Glorioso, J.C., Comi, G., Martino, G., 2001. Fibroblast
growth factor-II gene therapy reverts the clinical course and the
pathological signs of chronic experimental autoimmune encephalomye-
litis in C57BL/6 mice. Gene Ther. 8 (16), 1207–1213.
Shi, J., Marinovich, A., Barres, B.A., 1998. Purification and character-
ization of adult oligodendrocyte precursor cells from the rat optic nerve.
J. Neurosci. 18 (12), 4627–4636.
Soriano, P., 1997. The PDGF alpha receptor is required for neural crest cell
development and for normal patterning of the somites. Development
124 (14), 2691–2700.
Wilson, H.C., Onischke, C., Raine, C.S., 2003. Human oligodendrocyte
precursor cells in vitro: phenotypic analysis and differential response to
growth factors. Glia 44 (2), 153–165.
Wolswijk, G., Noble, M., 1992. Cooperation between PDGF and FGF
converts slowly dividing O-2Aadult progenitor cells to rapidly dividing
cells with characteristics of O-2Aperinatal progenitor cells. J. Cell Biol.
118 (4), 889–900.
Wolswijk, G., Noble, M., 1995. In vitro studies of the development,
maintenance and regeneration of the oligodendrocyte-type-2 astrocyte
(O-2A) lineage in the adult central nervous system. In: Kettenmann, H.,

J.C. Murtie et al. / Neurobiology of Disease 19 (2005) 171–182182
Sutliff, R.L. (Eds.), Neuroglia. Oxford Univ. Press, New York,
pp. 149–161.
Woodruff, R.H., Fruttiger, M., Richardson, W.D., Franklin, R.J., 2004.
Platelet-derived growth factor regulates oligodendrocyte progenitor
numbers in adult CNS and their response following CNS demyelina-
tion. Mol. Cell. Neurosci. 25 (2), 252–262.
Zhang, S.C., Ge, B., Duncan, I.D., 2000. Tracing human oligodendroglial
development in vitro. J. Neurosci. Res. 59 (3), 421–429.
Zhou, M., Sutliff, R.L., Paul, R.J., Lorenz, J.N., Hoying, J.B.,
Haudenschild, C.C., Yin, M., Coffin, J.D., Kong, L., Kranias, E.G.,
et al., 1998. Fibroblast growth factor 2 control of vascular tone. Nat.
Med. 4 (2), 201–207.








![PET imaging of demyelination and remyelination in the cuprizone mouse model for multiple sclerosis: A comparison between [11C]CIC and [11C]MeDAS](https://static.fdokumen.com/doc/165x107/63419d7d8768bcaafb01b673/pet-imaging-of-demyelination-and-remyelination-in-the-cuprizone-mouse-model-for.jpg)











