
Pd-doped LaCoO3 regenerative catalyst for automotive emissions control
7
Pd-doped LaCoO 3 regenerative catalyst for automotive emissions control Sina Sartipi, Abbas Ali Khodadadi, Yadollah Mortazavi * Catalysis and Nanostructured Materials Research Laboratory, School of Chemical Engineering, University of Tehran, Enghelab Ave., P.O. Box 11365-4563, Tehran, Iran 1. Introduction One of the main sources of air pollution in large cities is automotive exhaust gas emissions. Therefore, catalytic converters have been applied for exhaust gas treatment since the 1970s. Conventional three-way automotive exhaust catalysts contain precious metals (Pt, Pd and Rh) for treatment of pollutants such as CO, unburned hydrocarbons and NO x [1]. One of the reasons of deterioration of these catalysts is considered to be due to the decrease in active surface area of precious metals caused by grain growth in high-temperature redox (reduction–oxidation) fluctua- tions of the exhaust gas. To compensate for this deterioration, conventional catalysts are loaded with excess amounts of precious metals [2]. Due to high prices and limited sources of precious metals, attention has been given to search for an alternative catalytic component to reduce the use or even replace the precious metals. Among the substitute catalysts, perovskite type mixed oxides (ABO 3 ) have shown promising results. The catalytic properties of perovskites for oxidation of pollutants depend on several factors including the nature of B site cations. For oxidation of CO and hydrocarbons, LaCoO 3 is known as one of the most active perovskites [3]. Incorporation of small amounts of precious metals into a perovskite structure can prevent their sintering, reduce losses due to volatilization at high operating temperatures, and avoid reactions with the support that lead to catalyst deactivation [4– 6]. Recent attention has been concentrated on the use of palladium-based catalyst for TWC (three-way catalyst) formula- tion. Pd is well known to have a good resistance to thermal sintering, lower price than Pt and Rh and also have a good activity for oxidation of CO and hydrocarbons [7–10]. The modern gasoline engine has adopted a feedback control system that uses an oxygen sensor for three-way catalysts to function in optimum conditions. Therefore, the environment of automotive exhaust gas fluctuates between oxidative and reductive atmospheres through the operation of catalytic converters. A regenerative catalyst is a catalyst which keeps a high activity through these redox fluctuations by adapting its structure to the environment. This adaptation is a reversible response to the conditions of the atmosphere to which the catalyst is exposed. Due to the regenerative function, the Pd containing perovskites, e.g. LaFe 0.95 Pd 0.05 O 3 , maintain their high activity in the course of their use as the catalytic converter catalysts. It is suggested that in oxide form, Pd may diffuse into the perovskite lattice and replace the B site cation. In reducing atmospheres, Pd segregates out and disperses as metallic nanoparticles on the surface of the perovskites. Transfer of the Pd between the bulk and the surface occurs in response to the redox conditions of the environment [11–21]. It should be mentioned that LaFe 0.95 Pd 0.05 O 3 perovskite has been used industrially as a three-way catalyst for gasoline automotive emission control since October 2002 [19]. Applied Catalysis B: Environmental 83 (2008) 214–220 ARTICLE INFO Article history: Received 14 November 2007 Received in revised form 20 January 2008 Accepted 19 February 2008 Available online 23 February 2008 Keywords: Pollution Perovskite Palladium Regenerative Reduction ABSTRACT The effect of partial substitution of Co by Pd in LaCoO 3 perovskite structure (i.e., LaCo 0.95 Pd 0.05 O 3 ) and the reductive diffusion of Pd from the bulk of perovskite to its surface, thus forming Pd nanoparticles, on CO and C 3 H 8 oxidation present in air (simulated exhaust gas) are reported. X-ray powder diffraction (XRD) analyses confirm the perovskite structure for the catalysts. Scanning electron microscopy (SEM) and BET surface area measurements show that partial substitution of Co by Pd decreases the crystallite size of the perovskite and therefore increases its surface area. H 2 -temperature programmed reduction (TPR) experiments reveal that Pd reduces at 135 8C and facilitates the reduction of Co in the perovskite structure. By partial reduction of the Pd containing catalyst at 180 8C for 30 min, the complete oxidation temperatures of CO and C 3 H 8 decrease by about 70 and 50 8C, respectively. The reduction duration of the Pd containing catalyst strongly affects the T 50 and T 90 temperatures (temperatures at which 50 and 90% conversion occurs, respectively) and has an optimum, where it decreases by increasing the reduction temperature of the catalyst. ß 2008 Elsevier B.V. All rights reserved. * Corresponding author. Tel.: +98 21 6696 7793; fax: +98 21 6696 7793. E-mail address: [email protected] (Y. Mortazavi). Contents lists available at ScienceDirect Applied Catalysis B: Environmental journal homepage: www.elsevier.com/locate/apcatb 0926-3373/$ – see front matter ß 2008 Elsevier B.V. All rights reserved. doi:10.1016/j.apcatb.2008.02.014
-
Upload
independent -
Category
Documents
-
view
5 -
download
0
Transcript of Pd-doped LaCoO3 regenerative catalyst for automotive emissions control

Applied Catalysis B: Environmental 83 (2008) 214–220
Pd-doped LaCoO3 regenerative catalyst for automotive emissions control
Sina Sartipi, Abbas Ali Khodadadi, Yadollah Mortazavi *
Catalysis and Nanostructured Materials Research Laboratory, School of Chemical Engineering, University of Tehran, Enghelab Ave., P.O. Box 11365-4563, Tehran, Iran
A R T I C L E I N F O
Article history:
Received 14 November 2007
Received in revised form 20 January 2008
Accepted 19 February 2008
Available online 23 February 2008
Keywords:
Pollution
Perovskite
Palladium
Regenerative
Reduction
A B S T R A C T
The effect of partial substitution of Co by Pd in LaCoO3 perovskite structure (i.e., LaCo0.95Pd0.05O3) and the
reductive diffusion of Pd from the bulk of perovskite to its surface, thus forming Pd nanoparticles, on CO
and C3H8 oxidation present in air (simulated exhaust gas) are reported. X-ray powder diffraction (XRD)
analyses confirm the perovskite structure for the catalysts. Scanning electron microscopy (SEM) and BET
surface area measurements show that partial substitution of Co by Pd decreases the crystallite size of the
perovskite and therefore increases its surface area. H2-temperature programmed reduction (TPR)
experiments reveal that Pd reduces at 135 8C and facilitates the reduction of Co in the perovskite
structure. By partial reduction of the Pd containing catalyst at 180 8C for 30 min, the complete oxidation
temperatures of CO and C3H8 decrease by about 70 and 50 8C, respectively.
The reduction duration of the Pd containing catalyst strongly affects the T50 and T90 temperatures
(temperatures at which 50 and 90% conversion occurs, respectively) and has an optimum, where it
decreases by increasing the reduction temperature of the catalyst.
� 2008 Elsevier B.V. All rights reserved.
Contents l is ts ava i lab le at ScienceDirec t
Applied Catalysis B: Environmental
journa l homepage: www.e lsev ier .com/ locate /apcatb
1. Introduction
One of the main sources of air pollution in large cities isautomotive exhaust gas emissions. Therefore, catalytic convertershave been applied for exhaust gas treatment since the 1970s.Conventional three-way automotive exhaust catalysts containprecious metals (Pt, Pd and Rh) for treatment of pollutants such asCO, unburned hydrocarbons and NOx [1]. One of the reasons ofdeterioration of these catalysts is considered to be due to thedecrease in active surface area of precious metals caused by graingrowth in high-temperature redox (reduction–oxidation) fluctua-tions of the exhaust gas. To compensate for this deterioration,conventional catalysts are loaded with excess amounts of preciousmetals [2].
Due to high prices and limited sources of precious metals,attention has been given to search for an alternative catalyticcomponent to reduce the use or even replace the precious metals.Among the substitute catalysts, perovskite type mixed oxides(ABO3) have shown promising results. The catalytic properties ofperovskites for oxidation of pollutants depend on several factorsincluding the nature of B site cations. For oxidation of CO andhydrocarbons, LaCoO3 is known as one of the most activeperovskites [3].
Incorporation of small amounts of precious metals into aperovskite structure can prevent their sintering, reduce losses due
* Corresponding author. Tel.: +98 21 6696 7793; fax: +98 21 6696 7793.
E-mail address: [email protected] (Y. Mortazavi).
0926-3373/$ – see front matter � 2008 Elsevier B.V. All rights reserved.
doi:10.1016/j.apcatb.2008.02.014
to volatilization at high operating temperatures, and avoidreactions with the support that lead to catalyst deactivation [4–6]. Recent attention has been concentrated on the use ofpalladium-based catalyst for TWC (three-way catalyst) formula-tion. Pd is well known to have a good resistance to thermalsintering, lower price than Pt and Rh and also have a good activityfor oxidation of CO and hydrocarbons [7–10].
The modern gasoline engine has adopted a feedback controlsystem that uses an oxygen sensor for three-way catalysts tofunction in optimum conditions. Therefore, the environment ofautomotive exhaust gas fluctuates between oxidative andreductive atmospheres through the operation of catalyticconverters. A regenerative catalyst is a catalyst which keeps ahigh activity through these redox fluctuations by adapting itsstructure to the environment. This adaptation is a reversibleresponse to the conditions of the atmosphere to which thecatalyst is exposed. Due to the regenerative function, the Pdcontaining perovskites, e.g. LaFe0.95Pd0.05O3, maintain their highactivity in the course of their use as the catalytic convertercatalysts. It is suggested that in oxide form, Pd may diffuse intothe perovskite lattice and replace the B site cation. In reducingatmospheres, Pd segregates out and disperses as metallicnanoparticles on the surface of the perovskites. Transfer of thePd between the bulk and the surface occurs in response to theredox conditions of the environment [11–21]. It should bementioned that LaFe0.95Pd0.05O3 perovskite has been usedindustrially as a three-way catalyst for gasoline automotiveemission control since October 2002 [19].
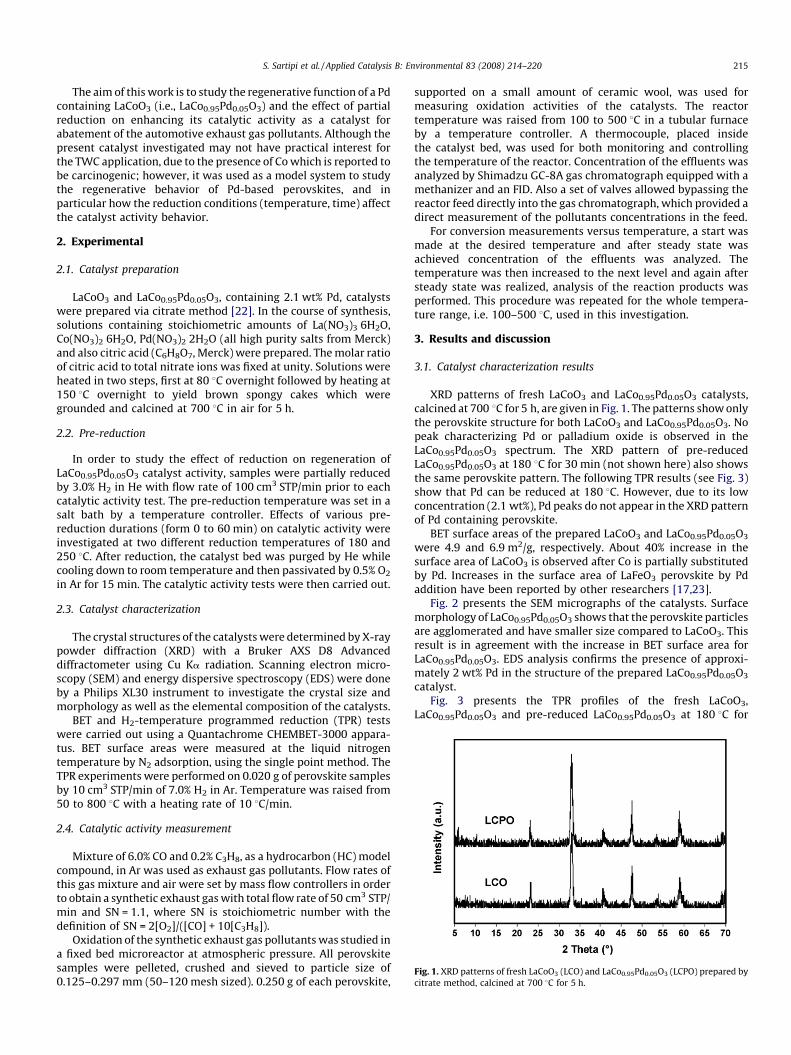
Fig. 1. XRD patterns of fresh LaCoO3 (LCO) and LaCo0.95Pd0.05O3 (LCPO) prepared by
citrate method, calcined at 700 8C for 5 h.
S. Sartipi et al. / Applied Catalysis B: Environmental 83 (2008) 214–220 215
The aim of this work is to study the regenerative function of a Pdcontaining LaCoO3 (i.e., LaCo0.95Pd0.05O3) and the effect of partialreduction on enhancing its catalytic activity as a catalyst forabatement of the automotive exhaust gas pollutants. Although thepresent catalyst investigated may not have practical interest forthe TWC application, due to the presence of Co which is reported tobe carcinogenic; however, it was used as a model system to studythe regenerative behavior of Pd-based perovskites, and inparticular how the reduction conditions (temperature, time) affectthe catalyst activity behavior.
2. Experimental
2.1. Catalyst preparation
LaCoO3 and LaCo0.95Pd0.05O3, containing 2.1 wt% Pd, catalystswere prepared via citrate method [22]. In the course of synthesis,solutions containing stoichiometric amounts of La(NO3)3�6H2O,Co(NO3)2�6H2O, Pd(NO3)2�2H2O (all high purity salts from Merck)and also citric acid (C6H8O7, Merck) were prepared. The molar ratioof citric acid to total nitrate ions was fixed at unity. Solutions wereheated in two steps, first at 80 8C overnight followed by heating at150 8C overnight to yield brown spongy cakes which weregrounded and calcined at 700 8C in air for 5 h.
2.2. Pre-reduction
In order to study the effect of reduction on regeneration ofLaCo0.95Pd0.05O3 catalyst activity, samples were partially reducedby 3.0% H2 in He with flow rate of 100 cm3 STP/min prior to eachcatalytic activity test. The pre-reduction temperature was set in asalt bath by a temperature controller. Effects of various pre-reduction durations (form 0 to 60 min) on catalytic activity wereinvestigated at two different reduction temperatures of 180 and250 8C. After reduction, the catalyst bed was purged by He whilecooling down to room temperature and then passivated by 0.5% O2
in Ar for 15 min. The catalytic activity tests were then carried out.
2.3. Catalyst characterization
The crystal structures of the catalysts were determined by X-raypowder diffraction (XRD) with a Bruker AXS D8 Advanceddiffractometer using Cu Ka radiation. Scanning electron micro-scopy (SEM) and energy dispersive spectroscopy (EDS) were doneby a Philips XL30 instrument to investigate the crystal size andmorphology as well as the elemental composition of the catalysts.
BET and H2-temperature programmed reduction (TPR) testswere carried out using a Quantachrome CHEMBET-3000 appara-tus. BET surface areas were measured at the liquid nitrogentemperature by N2 adsorption, using the single point method. TheTPR experiments were performed on 0.020 g of perovskite samplesby 10 cm3 STP/min of 7.0% H2 in Ar. Temperature was raised from50 to 800 8C with a heating rate of 10 8C/min.
2.4. Catalytic activity measurement
Mixture of 6.0% CO and 0.2% C3H8, as a hydrocarbon (HC) modelcompound, in Ar was used as exhaust gas pollutants. Flow rates ofthis gas mixture and air were set by mass flow controllers in orderto obtain a synthetic exhaust gas with total flow rate of 50 cm3 STP/min and SN = 1.1, where SN is stoichiometric number with thedefinition of SN = 2[O2]/([CO] + 10[C3H8]).
Oxidation of the synthetic exhaust gas pollutants was studied ina fixed bed microreactor at atmospheric pressure. All perovskitesamples were pelleted, crushed and sieved to particle size of0.125–0.297 mm (50–120 mesh sized). 0.250 g of each perovskite,
supported on a small amount of ceramic wool, was used formeasuring oxidation activities of the catalysts. The reactortemperature was raised from 100 to 500 8C in a tubular furnaceby a temperature controller. A thermocouple, placed insidethe catalyst bed, was used for both monitoring and controllingthe temperature of the reactor. Concentration of the effluents wasanalyzed by Shimadzu GC-8A gas chromatograph equipped with amethanizer and an FID. Also a set of valves allowed bypassing thereactor feed directly into the gas chromatograph, which provided adirect measurement of the pollutants concentrations in the feed.
For conversion measurements versus temperature, a start wasmade at the desired temperature and after steady state wasachieved concentration of the effluents was analyzed. Thetemperature was then increased to the next level and again aftersteady state was realized, analysis of the reaction products wasperformed. This procedure was repeated for the whole tempera-ture range, i.e. 100–500 8C, used in this investigation.
3. Results and discussion
3.1. Catalyst characterization results
XRD patterns of fresh LaCoO3 and LaCo0.95Pd0.05O3 catalysts,calcined at 700 8C for 5 h, are given in Fig. 1. The patterns show onlythe perovskite structure for both LaCoO3 and LaCo0.95Pd0.05O3. Nopeak characterizing Pd or palladium oxide is observed in theLaCo0.95Pd0.05O3 spectrum. The XRD pattern of pre-reducedLaCo0.95Pd0.05O3 at 180 8C for 30 min (not shown here) also showsthe same perovskite pattern. The following TPR results (see Fig. 3)show that Pd can be reduced at 180 8C. However, due to its lowconcentration (2.1 wt%), Pd peaks do not appear in the XRD patternof Pd containing perovskite.
BET surface areas of the prepared LaCoO3 and LaCo0.95Pd0.05O3
were 4.9 and 6.9 m2/g, respectively. About 40% increase in thesurface area of LaCoO3 is observed after Co is partially substitutedby Pd. Increases in the surface area of LaFeO3 perovskite by Pdaddition have been reported by other researchers [17,23].
Fig. 2 presents the SEM micrographs of the catalysts. Surfacemorphology of LaCo0.95Pd0.05O3 shows that the perovskite particlesare agglomerated and have smaller size compared to LaCoO3. Thisresult is in agreement with the increase in BET surface area forLaCo0.95Pd0.05O3. EDS analysis confirms the presence of approxi-mately 2 wt% Pd in the structure of the prepared LaCo0.95Pd0.05O3
catalyst.Fig. 3 presents the TPR profiles of the fresh LaCoO3,
LaCo0.95Pd0.05O3 and pre-reduced LaCo0.95Pd0.05O3 at 180 8C for
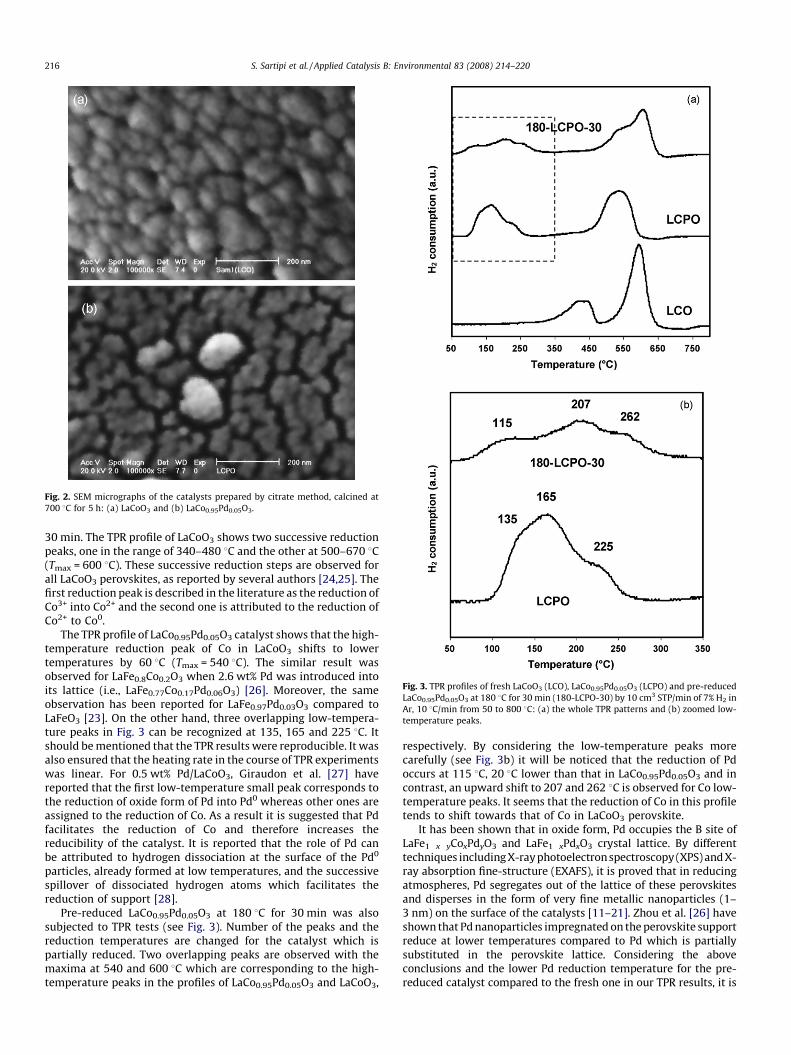
Fig. 2. SEM micrographs of the catalysts prepared by citrate method, calcined at
700 8C for 5 h: (a) LaCoO3 and (b) LaCo0.95Pd0.05O3.
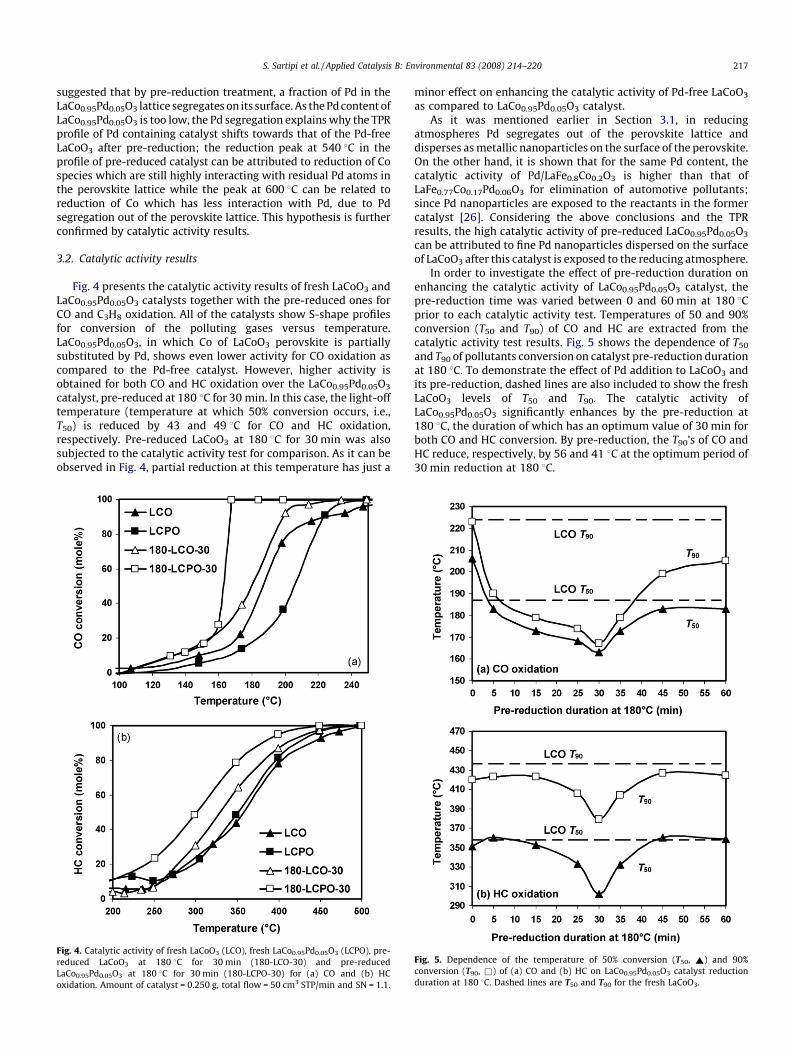
Fig. 3. TPR profiles of fresh LaCoO3 (LCO), LaCo0.95Pd0.05O3 (LCPO) and pre-reduced
LaCo0.95Pd0.05O3 at 180 8C for 30 min (180-LCPO-30) by 10 cm3 STP/min of 7% H2 in
Ar, 10 8C/min from 50 to 800 8C: (a) the whole TPR patterns and (b) zoomed low-
temperature peaks.
S. Sartipi et al. / Applied Catalysis B: Environmental 83 (2008) 214–220216
30 min. The TPR profile of LaCoO3 shows two successive reductionpeaks, one in the range of 340–480 8C and the other at 500–670 8C(Tmax = 600 8C). These successive reduction steps are observed forall LaCoO3 perovskites, as reported by several authors [24,25]. Thefirst reduction peak is described in the literature as the reduction ofCo3+ into Co2+ and the second one is attributed to the reduction ofCo2+ to Co0.
The TPR profile of LaCo0.95Pd0.05O3 catalyst shows that the high-temperature reduction peak of Co in LaCoO3 shifts to lowertemperatures by 60 8C (Tmax = 540 8C). The similar result wasobserved for LaFe0.8Co0.2O3 when 2.6 wt% Pd was introduced intoits lattice (i.e., LaFe0.77Co0.17Pd0.06O3) [26]. Moreover, the sameobservation has been reported for LaFe0.97Pd0.03O3 compared toLaFeO3 [23]. On the other hand, three overlapping low-tempera-ture peaks in Fig. 3 can be recognized at 135, 165 and 225 8C. Itshould be mentioned that the TPR results were reproducible. It wasalso ensured that the heating rate in the course of TPR experimentswas linear. For 0.5 wt% Pd/LaCoO3, Giraudon et al. [27] havereported that the first low-temperature small peak corresponds tothe reduction of oxide form of Pd into Pd0 whereas other ones areassigned to the reduction of Co. As a result it is suggested that Pdfacilitates the reduction of Co and therefore increases thereducibility of the catalyst. It is reported that the role of Pd canbe attributed to hydrogen dissociation at the surface of the Pd0
particles, already formed at low temperatures, and the successivespillover of dissociated hydrogen atoms which facilitates thereduction of support [28].
Pre-reduced LaCo0.95Pd0.05O3 at 180 8C for 30 min was alsosubjected to TPR tests (see Fig. 3). Number of the peaks and thereduction temperatures are changed for the catalyst which ispartially reduced. Two overlapping peaks are observed with themaxima at 540 and 600 8C which are corresponding to the high-temperature peaks in the profiles of LaCo0.95Pd0.05O3 and LaCoO3,
respectively. By considering the low-temperature peaks morecarefully (see Fig. 3b) it will be noticed that the reduction of Pdoccurs at 115 8C, 20 8C lower than that in LaCo0.95Pd0.05O3 and incontrast, an upward shift to 207 and 262 8C is observed for Co low-temperature peaks. It seems that the reduction of Co in this profiletends to shift towards that of Co in LaCoO3 perovskite.
It has been shown that in oxide form, Pd occupies the B site ofLaFe1�x�yCoxPdyO3 and LaFe1�xPdxO3 crystal lattice. By differenttechniques including X-ray photoelectron spectroscopy (XPS) and X-ray absorption fine-structure (EXAFS), it is proved that in reducingatmospheres, Pd segregates out of the lattice of these perovskitesand disperses in the form of very fine metallic nanoparticles (1–3 nm) on the surface of the catalysts [11–21]. Zhou et al. [26] haveshown that Pd nanoparticles impregnated on the perovskite supportreduce at lower temperatures compared to Pd which is partiallysubstituted in the perovskite lattice. Considering the aboveconclusions and the lower Pd reduction temperature for the pre-reduced catalyst compared to the fresh one in our TPR results, it is
S. Sartipi et al. / Applied Catalysis B: Environmental 83 (2008) 214–220 217
suggested that by pre-reduction treatment, a fraction of Pd in theLaCo0.95Pd0.05O3 lattice segregates on its surface. As the Pd content ofLaCo0.95Pd0.05O3 is too low, the Pd segregation explains why the TPRprofile of Pd containing catalyst shifts towards that of the Pd-freeLaCoO3 after pre-reduction; the reduction peak at 540 8C in theprofile of pre-reduced catalyst can be attributed to reduction of Cospecies which are still highly interacting with residual Pd atoms inthe perovskite lattice while the peak at 600 8C can be related toreduction of Co which has less interaction with Pd, due to Pdsegregation out of the perovskite lattice. This hypothesis is furtherconfirmed by catalytic activity results.
3.2. Catalytic activity results
Fig. 4 presents the catalytic activity results of fresh LaCoO3 andLaCo0.95Pd0.05O3 catalysts together with the pre-reduced ones forCO and C3H8 oxidation. All of the catalysts show S-shape profilesfor conversion of the polluting gases versus temperature.LaCo0.95Pd0.05O3, in which Co of LaCoO3 perovskite is partiallysubstituted by Pd, shows even lower activity for CO oxidation ascompared to the Pd-free catalyst. However, higher activity isobtained for both CO and HC oxidation over the LaCo0.95Pd0.05O3
catalyst, pre-reduced at 180 8C for 30 min. In this case, the light-offtemperature (temperature at which 50% conversion occurs, i.e.,T50) is reduced by 43 and 49 8C for CO and HC oxidation,respectively. Pre-reduced LaCoO3 at 180 8C for 30 min was alsosubjected to the catalytic activity test for comparison. As it can beobserved in Fig. 4, partial reduction at this temperature has just a
Fig. 4. Catalytic activity of fresh LaCoO3 (LCO), fresh LaCo0.95Pd0.05O3 (LCPO), pre-
reduced LaCoO3 at 180 8C for 30 min (180-LCO-30) and pre-reduced
LaCo0.95Pd0.05O3 at 180 8C for 30 min (180-LCPO-30) for (a) CO and (b) HC
oxidation. Amount of catalyst = 0.250 g, total flow = 50 cm3 STP/min and SN = 1.1.
minor effect on enhancing the catalytic activity of Pd-free LaCoO3
as compared to LaCo0.95Pd0.05O3 catalyst.As it was mentioned earlier in Section 3.1, in reducing
atmospheres Pd segregates out of the perovskite lattice anddisperses as metallic nanoparticles on the surface of the perovskite.On the other hand, it is shown that for the same Pd content, thecatalytic activity of Pd/LaFe0.8Co0.2O3 is higher than that ofLaFe0.77Co0.17Pd0.06O3 for elimination of automotive pollutants;since Pd nanoparticles are exposed to the reactants in the formercatalyst [26]. Considering the above conclusions and the TPRresults, the high catalytic activity of pre-reduced LaCo0.95Pd0.05O3
can be attributed to fine Pd nanoparticles dispersed on the surfaceof LaCoO3 after this catalyst is exposed to the reducing atmosphere.
In order to investigate the effect of pre-reduction duration onenhancing the catalytic activity of LaCo0.95Pd0.05O3 catalyst, thepre-reduction time was varied between 0 and 60 min at 180 8Cprior to each catalytic activity test. Temperatures of 50 and 90%conversion (T50 and T90) of CO and HC are extracted from thecatalytic activity test results. Fig. 5 shows the dependence of T50
and T90 of pollutants conversion on catalyst pre-reduction durationat 180 8C. To demonstrate the effect of Pd addition to LaCoO3 andits pre-reduction, dashed lines are also included to show the freshLaCoO3 levels of T50 and T90. The catalytic activity ofLaCo0.95Pd0.05O3 significantly enhances by the pre-reduction at180 8C, the duration of which has an optimum value of 30 min forboth CO and HC conversion. By pre-reduction, the T90’s of CO andHC reduce, respectively, by 56 and 41 8C at the optimum period of30 min reduction at 180 8C.
Fig. 5. Dependence of the temperature of 50% conversion (T50, ~) and 90%
conversion (T90, &) of (a) CO and (b) HC on LaCo0.95Pd0.05O3 catalyst reduction
duration at 180 8C. Dashed lines are T50 and T90 for the fresh LaCoO3.
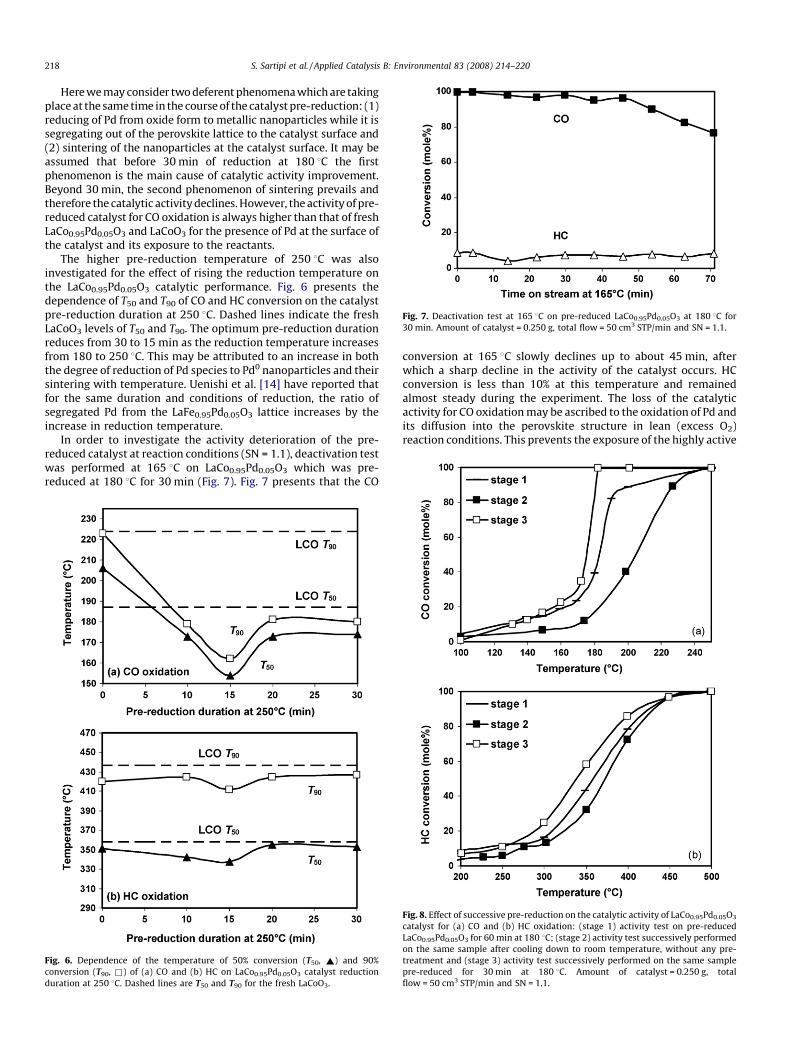
Fig. 7. Deactivation test at 165 8C on pre-reduced LaCo0.95Pd0.05O3 at 180 8C for
30 min. Amount of catalyst = 0.250 g, total flow = 50 cm3 STP/min and SN = 1.1.
S. Sartipi et al. / Applied Catalysis B: Environmental 83 (2008) 214–220218
Here we may consider two deferent phenomena which are takingplace at the same time in the course of the catalyst pre-reduction: (1)reducing of Pd from oxide form to metallic nanoparticles while it issegregating out of the perovskite lattice to the catalyst surface and(2) sintering of the nanoparticles at the catalyst surface. It may beassumed that before 30 min of reduction at 180 8C the firstphenomenon is the main cause of catalytic activity improvement.Beyond 30 min, the second phenomenon of sintering prevails andtherefore the catalytic activity declines. However, the activity of pre-reduced catalyst for CO oxidation is always higher than that of freshLaCo0.95Pd0.05O3 and LaCoO3 for the presence of Pd at the surface ofthe catalyst and its exposure to the reactants.
The higher pre-reduction temperature of 250 8C was alsoinvestigated for the effect of rising the reduction temperature onthe LaCo0.95Pd0.05O3 catalytic performance. Fig. 6 presents thedependence of T50 and T90 of CO and HC conversion on the catalystpre-reduction duration at 250 8C. Dashed lines indicate the freshLaCoO3 levels of T50 and T90. The optimum pre-reduction durationreduces from 30 to 15 min as the reduction temperature increasesfrom 180 to 250 8C. This may be attributed to an increase in boththe degree of reduction of Pd species to Pd0 nanoparticles and theirsintering with temperature. Uenishi et al. [14] have reported thatfor the same duration and conditions of reduction, the ratio ofsegregated Pd from the LaFe0.95Pd0.05O3 lattice increases by theincrease in reduction temperature.
In order to investigate the activity deterioration of the pre-reduced catalyst at reaction conditions (SN = 1.1), deactivation testwas performed at 165 8C on LaCo0.95Pd0.05O3 which was pre-reduced at 180 8C for 30 min (Fig. 7). Fig. 7 presents that the CO
Fig. 6. Dependence of the temperature of 50% conversion (T50, ~) and 90%
conversion (T90, &) of (a) CO and (b) HC on LaCo0.95Pd0.05O3 catalyst reduction
duration at 250 8C. Dashed lines are T50 and T90 for the fresh LaCoO3.
conversion at 165 8C slowly declines up to about 45 min, afterwhich a sharp decline in the activity of the catalyst occurs. HCconversion is less than 10% at this temperature and remainedalmost steady during the experiment. The loss of the catalyticactivity for CO oxidation may be ascribed to the oxidation of Pd andits diffusion into the perovskite structure in lean (excess O2)reaction conditions. This prevents the exposure of the highly active
Fig. 8. Effect of successive pre-reduction on the catalytic activity of LaCo0.95Pd0.05O3
catalyst for (a) CO and (b) HC oxidation: (stage 1) activity test on pre-reduced
LaCo0.95Pd0.05O3 for 60 min at 180 8C; (stage 2) activity test successively performed
on the same sample after cooling down to room temperature, without any pre-
treatment and (stage 3) activity test successively performed on the same sample
pre-reduced for 30 min at 180 8C. Amount of catalyst = 0.250 g, total
flow = 50 cm3 STP/min and SN = 1.1.

Fig. 9. Catalytic activity of fresh LaCo0.95Pd0.05O3 (LCPO) and aged LaCo0.95Pd0.05O3
at 850 8C for 660 min in reaction conditions (aged LCPO) for (a) CO and (b) HC
oxidation. Amount of catalyst = 0.250 g, total flow = 50 cm3 STP/min and SN = 1.1.
Fig. 10. XRD pattern of aged LaCo0.95Pd0.05O3 at 850 8C for 660 min in reaction
conditions (aged LCPO).
S. Sartipi et al. / Applied Catalysis B: Environmental 83 (2008) 214–220 219
Pd nanoparticles to the CO and HC pollutant gases. Uenishi et al.[14] have proposed that by exposing the pre-reduced Pd contain-ing iron perovskite, on surface of which metallic Pd nanoparticlesare dispersed, to an oxidative atmosphere, Pd oxidizes again anddiffuses into the perovskite lattice to substitute the B site cation.
As it was mentioned in Section 1, a regenerative catalyst keepsa high activity through redox fluctuations of the exhaust gas byadapting its structure to the reductive and oxidative atmospheresin a reversible manner. In order to investigate the regenerativefunction of LaCo0.95Pd0.05O3, effect of successive pre-reduction onthe catalytic activity performance of this catalyst was tested forCO and HC oxidation in three stages: (1) the catalytic activity testwas performed on a sample which was pre-reduced for 60 min at180 8C; (2) after cooling down the same sample to roomtemperature, it was tested again without any pre-treatmentand (3) finally the same sample was pre-reduced again for 30 minat 180 8C and then subjected to the catalytic activity test for thethird time. The catalytic activity test results are presented in Fig. 8.CO light-off temperature of LaCo0.95Pd0.05O3 pre-reduced for60 min at 180 8C is 23 8C lower than that of the fresh catalyst (seeFig. 5a). Comparing the test results of the first and second stagesreveals that the catalytic activity of the pre-reduced catalystdecreases after the first catalytic activity test, due to the oxidationof Pd at lean reaction conditions (SN = 1.1). However, pre-reduction of the same catalyst sample at the third stageregenerates the high catalytic activity. As it can be observed inFig. 8, the catalytic activity of the sample which is pre-reduced for30 min at the third stage (i.e., for the optimum reduction duration
in Fig. 5), is even higher than that of the catalyst which is pre-reduced for 60 min at the first stage. From this result it can beconcluded that even though the excessive reduction ofthe catalyst to 60 min in the first stage is not favorable for thepurposes of catalytic activity, due to sintering of Pd,LaCo0.95Pd0.05O3 is able to rearrange its structure in order tomaintain a high activity at the third stage and therefore is aregenerative catalyst. By in situ energy-dispersive X-ray absorp-tion fine-structure (DXAFS) analysis, it is reported that the changein structure of Pd in LaFe1�xPdxO3 catalyst is sufficiently fast torespond to the control frequency (1�4 Hz) of an actual gasolineengine [15,18,21].
Finally the catalyst was aged to see if the perovskite structure isstable enough and does not decompose under the reactionconditions. LaCo0.95Pd0.05O3 catalyst was aged at 850 8C for660 min in reaction conditions (total flow = 50 cm3 STP/min andSN = 1.1) and then subjected to catalytic activity test and XRDanalysis. Fig. 9 presents the catalytic activity of the aged catalyst aswell as the fresh one for oxidation of CO and HC. In Fig. 9 no activityloss is observed for LaCo0.95Pd0.05O3 catalyst after the agingtreatment. Moreover, XRD confirms the perovskite structure forthe aged catalyst (Fig. 10). Tanaka et al. [13,15,18] have reportedthat LaFe0.57Co0.38Pd0.05O3 and LaFe0.95Pd0.05O3 catalysts main-tained their high TWC activity during and after aging at 900 8C for100 h by the exhaust of a real engine.
4. Conclusions
The effect of Pd partial substitution in LaCoO3 perovskite and itsregenerative reduction is studied in a simulated exhaust gas foroxidation of CO and C3H8 present in air. Pd addition even lowersthe CO oxidation activity. However, partial reduction of the Pdcontaining catalyst for a proper duration of time significantlyenhances the oxidation activity of the catalyst, due to segregationof Pd from the perovskite lattice and its dispersion on the catalystsurface as metallic nanoparticles. Excessive reduction of the Pdcontaining catalyst for longer durations has an unfavorable effecton the oxidation activity of the catalyst, due probably to sinteringof the Pd nanoparticles. The high activity declines in lean reactionconditions after a period of time since Pd re-oxidizes and diffusesback into the perovskite lattice. However, successive reductionregenerates the high activity of the catalyst by re-dispersing Pdnanoparticles on the catalyst surface.
References
[1] R.M. Heck, R.J. Farrauto, Catalytic Air Pollution Control: Commercial Technology,Van Nostrand Reinhold, New York, 1995, pp. 95–102.

S. Sartipi et al. / Applied Catalysis B: Environmental 83 (2008) 214–220220
[2] Y. Nishihata, J. Mizuki, H. Tanaka, M. Uenishi, M. Kimura, J. Phys. Chem. Solids 66(2005) 274–282.
[3] M.A. Pena, J.L.G. Fierro, Chem. Rev. 101 (2001) 1981–2017.[4] H. Tanaka, H. Fujikawa, I. Takahashi, SAE Paper 930251, 1993.[5] N. Guilhaume, S.D. Peter, M. Primet, Appl. Catal. B 10 (1996) 325–344.[6] N. Guilhaume, M. Primet, J. Catal. 165 (1997) 197–204.[7] D.R. Rainer, M. Koranne, S.M. Vesecky, D.W. Goodman, J. Phys. Chem. B 101 (1997)
10769–10774.[8] J.C. Summers, W.B. Williamson, M.G. Henk, SAE Paper 880281, 1989.[9] D.D. Beck, J.W. Sommers, C.L. DiMaggio, Appl. Catal. B 3 (1994) 205–227.
[10] R. Van Yperen, D. Linder, L. Mussmann, E.S. Lox, T. Kreuzer, Stud. Surf. Sci. Catal.116 (1998) 51–60.
[11] H. Tanaka, M. Misono, Curr. Opin. Solid State Mater. Sci. 5 (2001) 381–387.[12] H. Tanaka, I. Tan, M. Uenishi, M. Kimura, K. Dohmae, Top. Catal. 16/17 (2001) 63–
70.[13] H. Tanaka, M. Taniguchi, N. Kajita, M. Uenishi, I. Tan, N. Sato, K. Narita, M. Kimura,
Top. Catal. 30/31 (2004) 389–396.[14] M. Uenishi, M. Taniguchi, H. Tanaka, M. Kimura, Y. Nishihata, J. Mizuki, T.
Kobayashi, Appl. Catal. B 57 (2005) 267–273.[15] H. Tanaka, Catal. Surv. Asia 9 (2005) 63–74.[16] H. Tanaka, I. Tan, M. Uenishi, M. Taniguchi, M. Kimura, Y. Nishihata, J. Mizuki, J.
Alloys Compd. 408/412 (2006) 1071–1077.
[17] M. Uenishi, H. Tanaka, M. Taniguchi, I. Tan, Y. Sakamoto, Sh. Matsunaga, K. Yokota,T. Kobayashi, Appl. Catal. A 296 (2005) 114–119.
[18] H. Tanaka, M. Uenishi, M. Taniguchi, I. Tan, K. Narita, M. Kimura, K. Kaneko, Y.Nishihata, J. Mizuki, Catal. Today 117 (2006) 321–328.
[19] H. Tanaka, M. Taniguchi, M. Uenishi, N. Kajita, I. Tan, Y. Nishihata, J. Mizuki, K.Narita, M. Kimura, K. Kaneko, Angew. Chem. Int. Ed. 45 (2006) 5998–6002.
[20] M. Taniguchi, H. Tanaka, M. Uenishi, I. Tan, Y. Nishihata, J. Mizuki, H. Suzuki, K.Narita, A. Hirai, M. Kimura, Top. Catal. 42/43 (2007) 367–371.
[21] M. Uenishi, H. Tanaka, M. Taniguchi, I. Tan, Y. Nishihata, J. Mizuki, T. Kobayashi,Catal. Commun. 9 (2008) 311–314.
[22] L. Abadian, A. Malekzadeh, Y. Mortazavi, A. Khodadadi, in: Proceedings of the 5thWorld Congress on Oxidation Catalysis (WCOC 2005), Sapporo, Japan, September25–30, 2005.
[23] R. Zhang, H. Alamdari, S. Kaliaguine, J. Catal. 242 (2006) 241–253.[24] S. Royer, F. Berube, S. Kaliaguine, Appl. Catal. A 282 (2005) 273–284.[25] M. Engelmann-Pirez, P. Granger, G. Leclercq, Catal. Today 107/108 (2005) 315–322.[26] K. Zhou, H. Chen, Q. Tian, Zh. Hao, D. Shen, X. Xu, J. Mol. Catal. A 189 (2002) 225–
232.[27] J.M. Giraudon, A. Elhachimi, F. Wyrwalski, S. Siffert, A. Aboukaıs, J.F. Lamonier, G.
Leclercq, Appl. Catal. B 75 (2007) 147–156.[28] W.J. Shen, M. Okumura, Y. Matsumura, M. Haruta, Appl. Catal. A 213 (2001) 225–
232.




















