
P3.70. Transforming growth factor beta-1 (TGF beta-1) polymorphisms are infrequent but exist in...
7
Indian J Dent Res, 21(3), 2010 413 Address for correspondence: Dr. Sukumaran Anil E-mail: [email protected] Received : 09-05-09 Review completed : 12-09-09 Accepted : 21-07-10 PubMed ID : *** DOI: 10.4103/0970-9290.70815 ORIGINAL RESEARCH Transforming growth factor-β-1 polymorphisms are infrequent but exist at selected loci in oral submucous fibrosis Rajendran R, Harish RK 1 , Sukumaran Anil, Ravi Vidyadharan 1 , Moinak Banerjee 1 Oral submucous fibrosis (OSF) is widely considered to be a potent precancerous condition whose predominant characteristics are the excessive and abnormal deposition of extracellular matrix (ECM) components that may affect adversely the routine oral functions. From a clinico- pathologic point of view, fibrosis may be considered as a somewhat irreversible state of tissue alteration, during which resolution of the healing process fails to occur. Increasingly, it has become appreciated that certain of College of Dentistry, King Saud University, Post Box: 60169, Riyadh 11545, Kingdom of Saudi Arabia, 1 Human Molecular Genetics Laboratory, Rajeev Gandhi Centre for Biotechnology (RGCB) Trivandrum 695 032, Kerala, India ABSTRACT Background: Oral submucous fibrosis (OSF) may be considered a collagen metabolic disorder resulting from areca-nut alkaloid exposure and individual variation in collagen metabolism. Due to the complexity of OSF pathogenesis, it is important to elucidate independent and interactive effects of polymorphisms of collagen-related genes on OSF risk. Materials and Methods: This study is focused on seven polymorphisms (SNPs) of transforming growth factor-beta-1 (TGF-beta-1) gene in patients with oral submucous fibrosis (OSF), belonging to south Indian ethnic extraction. The mean age at presentation was 43.9 years, range 23-72 years (n=50, M:F ratio, 2.6:1). DNA samples from 50 subjects of the same ethnic group and comparable demographic features who have had practiced the habit of areca-chewing of almost equal duration, but remained free of disease constituted the controls. All DNA samples were collected progressively and purified from peripheral blood employing standard protocols and tested for SNPs. They included two polymorphisms in the promoter region (C-509T and G-800A), three polymorphisms in exon-1 (Arg25Pro(G915C), Leu10Pro(T869C), Glu47Gly(A979G) and two in 5 ¢UTR regions (C→T(rs13306708) and G→A (rs9282871). The extracted DNA samples along with the primers underwent PCR amplification and the genotypic and allelic frequencies were calculated. All calculations were performed using the SPSS software. The PCR products were purified and subsequently sequenced using Flour S™ multi-imager system (Biorad). The sequenced data were analyzed using the BioEdit sequence analysis software. Results: Out of the seven polymorphisms analyzed, six such as two in the promoter region, three in exon-1 and one in 5¢UTR were found to have a “P” value above 0.05 and hence were not significant. The C→T transition (rs13306708) in the 5¢UTR region recorded a “P” value of 0.03 on comparison and hence was found to be significant. The allelic frequencies for this C→T transition in patients were 68.7% C and 31.2% T (27CC, 15CT, 8TT) and that in controls were 89.5% C and 10.4% T (42CC, 6CT, 2TT). Conclusions: The polymorphism in 5¢UTR C-T in TGF beta 1 gene has a significant association with OSF, being a prime determinant in the pro-angiogenic pathway which has got direct bearing with the pathophysiology of the disease. The proximity of this polymorphism to the transcription site and the associated risk involved is discussed. Key words: Cytokines, fibrogenesis, oral submucous fibrosis, TGF-β1, SNPs these actions of ECM derive from its ability to sequester and modulate the activity of specific growth factors. [1] Of all of the growth factors, none has been found to have the diversity of effects on ECM ascribed to transforming growth factor-β (TGF-β). This peptide plays a critical role not only in synthesis and degradation of ECM but also in response of cells to ECM mediated through integrin receptors; moreover, specific components of the ECM, in turn, can both deliver TGF-β and regulate its activity. [2,3] Isoform-1 of the TGF-β family has been extensively studied in fibrotic diseases such as sarcoidosis with radiographic stage IV [4,5] and idiopathic pulmonary fibrosis. [6,7] In contrast to www.ijdr.in
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of P3.70. Transforming growth factor beta-1 (TGF beta-1) polymorphisms are infrequent but exist in...

Indian J Dent Res, 21(3), 2010413
Address for correspondence: Dr. Sukumaran Anil E-mail: [email protected]
Received : 09-05-09Review completed : 12-09-09Accepted : 21-07-10PubMed ID : ***DOI: 10.4103/0970-9290.70815
oriGinal research
Transforming growth factor-β-1 polymorphisms are infrequent but exist at selected loci in oral
submucous fibrosis
Rajendran R, Harish RK1, Sukumaran Anil, Ravi Vidyadharan1, Moinak Banerjee1
Oral submucous fibrosis (OSF) is widely considered to be a potent precancerous condition whose predominant characteristics are the excessive and abnormal deposition of extracellular matrix (ECM) components that may affect adversely the routine oral functions. From a clinico-pathologic point of view, fibrosis may be considered as a somewhat irreversible state of tissue alteration, during which resolution of the healing process fails to occur. Increasingly, it has become appreciated that certain of
College of Dentistry, King Saud University, Post Box: 60169, Riyadh 11545, Kingdom of Saudi Arabia, 1Human Molecular Genetics Laboratory, Rajeev Gandhi Centre for Biotechnology (RGCB) Trivandrum 695 032, Kerala, India
ABSTRACTBackground: Oral submucous fibrosis (OSF) may be considered a collagen metabolic disorder resulting from areca-nut alkaloid exposure and individual variation in collagen metabolism. Due to the complexity of OSF pathogenesis, it is important to elucidate independent and interactive effects of polymorphisms of collagen-related genes on OSF risk. Materials and Methods: This study is focused on seven polymorphisms (SNPs) of transforming growth factor-beta-1 (TGF-beta-1) gene in patients with oral submucous fibrosis (OSF), belonging to south Indian ethnic extraction. The mean age at presentation was 43.9 years, range 23-72 years (n=50, M:F ratio, 2.6:1). DNA samples from 50 subjects of the same ethnic group and comparable demographic features who have had practiced the habit of areca-chewing of almost equal duration, but remained free of disease constituted the controls. All DNA samples were collected progressively and purified from peripheral blood employing standard protocols and tested for SNPs. They included two polymorphisms in the promoter region (C-509T and G-800A), three polymorphisms in exon-1 (Arg25Pro(G915C), Leu10Pro(T869C), Glu47Gly(A979G) and two in 5 ¢UTR regions (C→T(rs13306708) and G→A (rs9282871). The extracted DNA samples along with the primers underwent PCR amplification and the genotypic and allelic frequencies were calculated. All calculations were performed using the SPSS software. The PCR products were purified and subsequently sequenced using Flour S™ multi-imager system (Biorad). The sequenced data were analyzed using the BioEdit sequence analysis software. Results: Out of the seven polymorphisms analyzed, six such as two in the promoter region, three in exon-1 and one in 5¢UTR were found to have a “P” value above 0.05 and hence were not significant. The C→T transition (rs13306708) in the 5¢UTR region recorded a “P” value of 0.03 on comparison and hence was found to be significant. The allelic frequencies for this C→T transition in patients were 68.7% C and 31.2% T (27CC, 15CT, 8TT) and that in controls were 89.5% C and 10.4% T (42CC, 6CT, 2TT). Conclusions: The polymorphism in 5¢UTR C-T in TGF beta 1 gene has a significant association with OSF, being a prime determinant in the pro-angiogenic pathway which has got direct bearing with the pathophysiology of the disease. The proximity of this polymorphism to the transcription site and the associated risk involved is discussed.
Key words: Cytokines, fibrogenesis, oral submucous fibrosis, TGF-β1, SNPs
these actions of ECM derive from its ability to sequester and modulate the activity of specific growth factors.[1] Of all of the growth factors, none has been found to have the diversity of effects on ECM ascribed to transforming growth factor-β (TGF-β). This peptide plays a critical role not only in synthesis and degradation of ECM but also in response of cells to ECM mediated through integrin receptors; moreover, specific components of the ECM, in turn, can both deliver TGF-β and regulate its activity.[2,3]
Isoform-1 of the TGF-β family has been extensively studied in fibrotic diseases such as sarcoidosis with radiographic stage IV[4,5] and idiopathic pulmonary fibrosis.[6,7] In contrast to
www.ijdr.in
Avinash K
Rectangle

Indian J Dent Res, 21(3), 2010 414
their isoforms TGF-β2 and TGF-β3 which have yet to receive due attention as profibrogenic cytokines, polymorphisms in the genes encoding for all three TGF-β isoforms have been identified and linked to variations of protein expression or function. Single nucleotide polymorphisms (SNPs) in the TGF-β1 gene are found associated with interindividual variation in TGF-β1 production.[8] These, as well as other polymorphisms of TGF-β1, have been shown to confer an increased risk for pathologic fibrosis. The cytokine TGF-β1 is considered to have a central role in inducing myofibroblastic phenotype, and its expression is increased under numerous fibrotic conditions. Fibrogenic cytokines secreted by activated macrophages or “T” cells are very important in the development of fibrotic disorders.
The main histopathologic characteristic of OSF is the deposition of collagen in the oral submucosa.[9] It has been found that areca-nut alkaloid exposure of buccal mucosal fibroblasts may result in the accumulation of collagen[10] at least in a small percentage of habitués. This evidence implies that OSF may be considered a collagen-metabolic disorder resulting from alkaloid exposure and individual variation in collagen metabolism. Because of the complexity of OSF pathogenesis, it is important to elucidate independent and interactive effects of polymorphisms of collagen-related genes on OSF risk.
This study focused on seven polymorphisms in TGF-β1 gene. They include two polymorphisms in the promoter region (C-509 T and G-800 A), three polymorphisms in exon-1 (Arg 25 Pro(G 915 C), Leu 10 Pro (T869C), Glu 47 Gly (A979 G), and two in 5UTR region (C→T (rs 13306708) and G -A (rs 9282871).
MATERIALS AND METHODS
Case DNA was obtained from 50 patients with OSF of mean age 43.9 years, range 23-72 years (M:F ratio 2.6:1) diagnosed according to established criteria[9] and proven histopathologically, belonging to south Indian ethnic extraction. Control DNA samples from 50 subjects of the same ethnic group and demographic features who had practiced the habit of areca chewing of comparable duration and frequency but remained free of disease were collected. All DNA samples were purified from peripheral blood by standard techniques[11] after the subjects provided written informed consent and reviewed and approved by the ethical committee. Case subjects and controls were well matched genetically without any evidence of population stratification/mixing.
Sample collectionPeripheral blood samples of volume 10 ml each were collected by anti-cubital vein puncture from patients and control groups in plastic falcon tubes containing EDTA, who were free of any systemic illnesses or minor infections and kept at 4°C.
Quantification of DNAQuantification of DNA samples was done using a spectrophotometer (Bio Spec-1601, Shimadzu). The wavelengths of 260 nm and 280 nm were used and readings at 260 nm allowed calculation of concentration of nucleic acid in the samples. Readings at 280 nm gave the amount of proteins. The DNA preparations gave OD260/OD280 values of 1.7 to 1.8 which was rated as the spectrum for pure preparations of DNA without contamination, in which case accurate quantification will not be possible.
Sample preparation for spectrophotometer analysis and calculation of DNA concentrationDNA samples kept at –20°C were thawed by keeping samples in ice and later in water bath at 65°C for 10 min. In 0.6 ml tubes, 98 µl of sterile water was added along with 2 µl of DNA samples. The tubes were vortexed and spun for spectrophotometer analysis.
For calculating the concentration of DNA:• 1 OD at 260nm of DNA=50 ng/µl • Dilution factor=50 (the samples were diluted to 50 µl)• Concentration of DNA=Absorbance at 260 nm × dilution
factor × 10D at 260 nm of DNA
Polymerase chain reactions (PCR) were performed to amplify the DNA stocks and check for the allelic variants. The extracted DNA samples along with the primers for the gene underwent PCR amplification.
GenotypingGenomic DNA from whole blood was extracted and purified using the phenol-chloroform method recommended in the 11th International HLA Workshop.[12] Based on the published sequences of the TGF-beta-1 gene,[8,13] two fragments were amplified by PCR with two sets of primers. (TGF-beta-1(a)f–5¢–ggggACACCATCTACAgTg-3¢ and TGF-beta-1(a)r-5¢ggAggAgggggCAACAgg-3¢ for fragment 1 and TGF-beta-1(b)f- 5¢-TTCAAgACCACCCACCTTCT-3¢ and TGF-beta-1(b)r- 5¢-TcgCgggTgCTgTTgTACA-3¢ for fragment 2). Reactions were performed in a total volume of 50 µl containing 0.5 µg of genomic DNA, 10 pmol of each primer , 0.2 mM each of the dNTPs, 1 unit of Ampli Taq DNA polymerase (Perkin-Elmer) , 50 mM KCl, 2 mM MgCl2, 10% dimethyl sulfoxide (fragment 2 only), and 10 mM Tris-HCl (pH 8.3). The cycling reaction for fragment 1 consisted of initial denaturation at 95°C for 1 min; 40 cycles of denaturation at 94 °C for 15 sec; annealing at 61°C for 30 sec; extension at 72°C for 40 sec; and a final extension at 72°C for 5 min. The thermocycling procedure for fragment 2 consisted of initial denaturation at 95°C for 5 min; 5 cycles of 1 min at 94°C , 60°C, and 72°C; then 40 cycles of 30 sec at 94°C, 56°C, and 72°C; and a final extension at 72°C for 1 min. The PCR products were purified and subsequently sequenced in Flour S™ multi-imager system (Biorad). The sequenced data were analyzed using the BioEdit sequence
TGF β-1 polymorphism in oral submucous fibrosis Rajendran, et al.
Indian J Dent Res, 21(3), 2010415
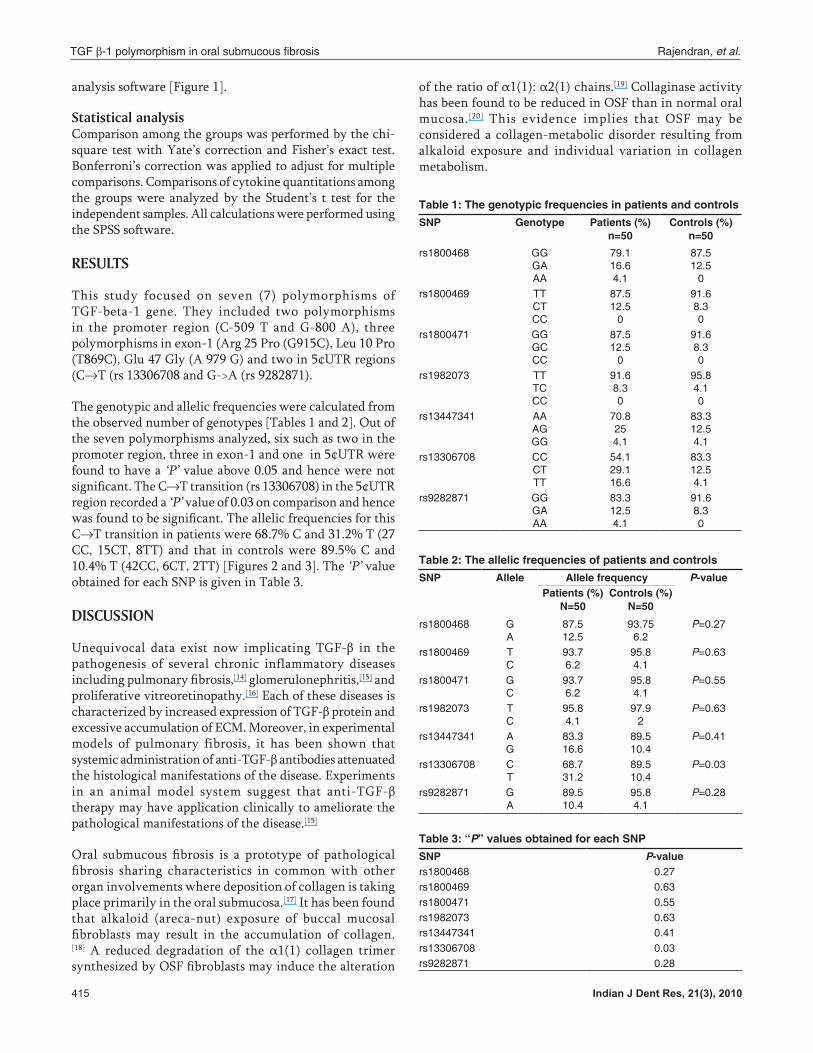
analysis software [Figure 1].
Statistical analysisComparison among the groups was performed by the chi-square test with Yate’s correction and Fisher’s exact test. Bonferroni’s correction was applied to adjust for multiple comparisons. Comparisons of cytokine quantitations among the groups were analyzed by the Student’s t test for the independent samples. All calculations were performed using the SPSS software.
RESULTS
This study focused on seven (7) polymorphisms of TGF-beta-1 gene. They included two polymorphisms in the promoter region (C-509 T and G-800 A), three polymorphisms in exon-1 (Arg 25 Pro (G915C), Leu 10 Pro (T869C), Glu 47 Gly (A 979 G) and two in 5¢UTR regions (C→T (rs 13306708 and G->A (rs 9282871).
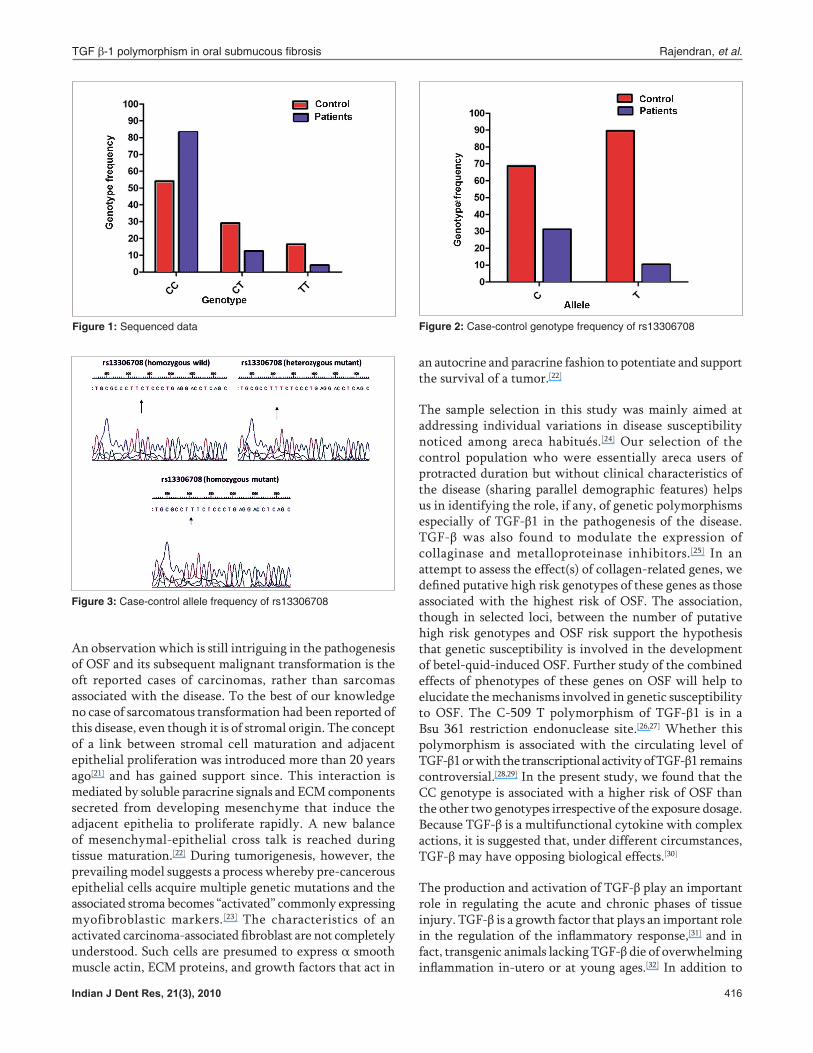
The genotypic and allelic frequencies were calculated from the observed number of genotypes [Tables 1 and 2]. Out of the seven polymorphisms analyzed, six such as two in the promoter region, three in exon-1 and one in 5¢UTR were found to have a ‘P’ value above 0.05 and hence were not significant. The C→T transition (rs 13306708) in the 5¢UTR region recorded a ‘P’ value of 0.03 on comparison and hence was found to be significant. The allelic frequencies for this C→T transition in patients were 68.7% C and 31.2% T (27 CC, 15CT, 8TT) and that in controls were 89.5% C and 10.4% T (42CC, 6CT, 2TT) [Figures 2 and 3]. The ‘P’ value obtained for each SNP is given in Table 3.
DISCUSSION
Unequivocal data exist now implicating TGF-β in the pathogenesis of several chronic inflammatory diseases including pulmonary fibrosis,[14] glomerulonephritis,[15] and proliferative vitreoretinopathy.[16] Each of these diseases is characterized by increased expression of TGF-β protein and excessive accumulation of ECM. Moreover, in experimental models of pulmonary fibrosis, it has been shown that systemic administration of anti-TGF-β antibodies attenuated the histological manifestations of the disease. Experiments in an animal model system suggest that anti-TGF-β therapy may have application clinically to ameliorate the pathological manifestations of the disease.[15]
Oral submucous fibrosis is a prototype of pathological fibrosis sharing characteristics in common with other organ involvements where deposition of collagen is taking place primarily in the oral submucosa.[17] It has been found that alkaloid (areca-nut) exposure of buccal mucosal fibroblasts may result in the accumulation of collagen.[18] A reduced degradation of the α1(1) collagen trimer synthesized by OSF fibroblasts may induce the alteration
Table 1: The genotypic frequencies in patients and controlsSNP Genotype Patients (%)
n=50Controls (%)
n=50rs1800468 GG
GAAA
79.116.64.1
87.512.5
0rs1800469 TT
CTCC
87.512.5
0
91.68.30
rs1800471 GGGCCC
87.512.5
0
91.68.30
rs1982073 TTTCCC
91.68.30
95.84.10
rs13447341 AAAGGG
70.8254.1
83.312.54.1
rs13306708 CCCTTT
54.129.116.6
83.312.54.1
rs9282871 GGGAAA
83.312.54.1
91.68.30
Table 2: The allelic frequencies of patients and controlsSNP Allele Allele frequency P-value
Patients (%) N=50
Controls (%) N=50
rs1800468 GA
87.512.5
93.756.2
P=0.27
rs1800469 TC
93.76.2
95.84.1
P=0.63
rs1800471 GC
93.76.2
95.84.1
P=0.55
rs1982073 TC
95.84.1
97.92
P=0.63
rs13447341 AG
83.316.6
89.510.4
P=0.41
rs13306708 CT
68.731.2
89.510.4
P=0.03
rs9282871 GA
89.510.4
95.84.1
P=0.28
Table 3: “P” values obtained for each SNPSNP P-valuers1800468 0.27rs1800469 0.63rs1800471 0.55rs1982073 0.63rs13447341 0.41rs13306708 0.03rs9282871 0.28
of the ratio of α1(1): α2(1) chains.[19] Collaginase activity has been found to be reduced in OSF than in normal oral mucosa.[20] This evidence implies that OSF may be considered a collagen-metabolic disorder resulting from alkaloid exposure and individual variation in collagen metabolism.
TGF β-1 polymorphism in oral submucous fibrosis Rajendran, et al.
Indian J Dent Res, 21(3), 2010 416
An observation which is still intriguing in the pathogenesis of OSF and its subsequent malignant transformation is the oft reported cases of carcinomas, rather than sarcomas associated with the disease. To the best of our knowledge no case of sarcomatous transformation had been reported of this disease, even though it is of stromal origin. The concept of a link between stromal cell maturation and adjacent epithelial proliferation was introduced more than 20 years ago[21] and has gained support since. This interaction is mediated by soluble paracrine signals and ECM components secreted from developing mesenchyme that induce the adjacent epithelia to proliferate rapidly. A new balance of mesenchymal-epithelial cross talk is reached during tissue maturation.[22] During tumorigenesis, however, the prevailing model suggests a process whereby pre-cancerous epithelial cells acquire multiple genetic mutations and the associated stroma becomes “activated” commonly expressing myofibroblastic markers.[23] The characteristics of an activated carcinoma-associated fibroblast are not completely understood. Such cells are presumed to express α smooth muscle actin, ECM proteins, and growth factors that act in
an autocrine and paracrine fashion to potentiate and support the survival of a tumor.[22]
The sample selection in this study was mainly aimed at addressing individual variations in disease susceptibility noticed among areca habitués.[24] Our selection of the control population who were essentially areca users of protracted duration but without clinical characteristics of the disease (sharing parallel demographic features) helps us in identifying the role, if any, of genetic polymorphisms especially of TGF-β1 in the pathogenesis of the disease. TGF-β was also found to modulate the expression of collaginase and metalloproteinase inhibitors.[25] In an attempt to assess the effect(s) of collagen-related genes, we defined putative high risk genotypes of these genes as those associated with the highest risk of OSF. The association, though in selected loci, between the number of putative high risk genotypes and OSF risk support the hypothesis that genetic susceptibility is involved in the development of betel-quid-induced OSF. Further study of the combined effects of phenotypes of these genes on OSF will help to elucidate the mechanisms involved in genetic susceptibility to OSF. The C-509 T polymorphism of TGF-β1 is in a Bsu 361 restriction endonuclease site.[26,27] Whether this polymorphism is associated with the circulating level of TGF-β1 or with the transcriptional activity of TGF-β1 remains controversial.[28,29] In the present study, we found that the CC genotype is associated with a higher risk of OSF than the other two genotypes irrespective of the exposure dosage. Because TGF-β is a multifunctional cytokine with complex actions, it is suggested that, under different circumstances, TGF-β may have opposing biological effects.[30]
The production and activation of TGF-β play an important role in regulating the acute and chronic phases of tissue injury. TGF-β is a growth factor that plays an important role in the regulation of the inflammatory response,[31] and in fact, transgenic animals lacking TGF-β die of overwhelming inflammation in-utero or at young ages.[32] In addition to
Figure 2: Case-control genotype frequency of rs13306708
Figure 3: Case-control allele frequency of rs13306708
TGF β-1 polymorphism in oral submucous fibrosis Rajendran, et al.
Figure 1: Sequenced data

Indian J Dent Res, 21(3), 2010417
anti-inflammatory properties, TGF-β promotes fibrosis as part of altered tissue repair.[33,34] For example, in pulmonary fibrosis, TGF-β is found in the lung and the elevation in levels of TGF-β correlates with the extent of fibrosis. In animal models of pulmonary fibrosis, administering antibodies to TGF-β or blocking signal transduction modulated by active TGF-β by over expressing the inhibitory Smad7 blocks fibrosis.[35] Thus, defining the specific mechanisms regulating the production and activation of TGF-β may have therapeutic opportunities to help patients with fibrotic diseases. The role of persistent and chronic inflammation probably the result of a hypersensitivity response to oral irritants especially areca nut alkaloids and the resultant juxta-epithelial inflammatory response noted in OSF have already been reported.[36] These authors held that the hypersensitivity to local irritants is resulting in persistent mucosal inflammation which acts as the initiating factor for a protracted and defective inflammatory-reparative response, culminating in fibrotic healing. We believe that understanding the molecular regulation of TGF-β activation and recognition may provide opportunity to intercede on this process, being a significant mediator of tissue repair.
The mechanism of action of TGF-β in promoting angiogenesis is unknown and will involve an entire cascade of cellular and molecular events.[37,38] including the possibilities that TGF-β may be chemotactic for cells involved in angiogenesis, or even less directly that TGF-β may induce cells to secrete other peptides with angiogenic activity. It has not yet been demonstrated that TGF-β itself has any direct effect on proliferation of capillary endothelial cells. Mucosal vasculature aimed at segmentation, characterization, and quantification of sub-epithelial microvasculature in OSF has been reported.[39] The mean vascular percentage area and luminal diameter were found to increase with the progress of the disease (P<0.001).[39] The vascularity of mucosa was assessed by estimating the mean vascular density, the mean vascular area percentage, and the mean vascular luminal diameter. The vascular alteration noted was assumed to be an adaptive response to compensate for the transient ischemia/hypoxia likely to have occurred during the course of the disease process when collagenization of the submucosa predominates. The failure to notice an increase in the mean vascular density in OSF mucosa irrespective of the disease stage and the possible role of TGF-β1 along with other pro-angiogenic cytokines (e.g. VEGF and NO) in this regard is worthy of further investigation.
The mechanism of action of TGF-β in promoting collagen formation is similarly unknown. Certainly the increased number of fibroblasts at the site of injection of TGF-β in experimental animals might be sufficient to explain the increased collagen deposition.[40] However, in-vivo experiments suggest a direct effect of TGF-β on collagen synthesis. It follows from the above that sustained, aberrant expression of TGF-β would result in pathological
accumulation of matrix as is the case in OSF. The events in collagen metabolism modulated by TGF-β can be grouped in to two main pathways; collagen production and degradation pathways. The events modulated by TGF-β in the production pathway include (a) activation of pro-collagen genes (COL1A2, COL3A1, COL6A1, COL6A3, and COL7A1), (b) elevation of pro-collagen proteinase levels, and (c) upregulation of lysyl oxidase activity. The two main events that are modulated by TGF-β in the collagen degradation pathway are: (a) activation of tissue inhibitor of matrix metalloproteinase (TIMPs) genes and (b) activation of plasminogen activator inhibitor (PAI) genes.[41] The identification of alternate signaling pathways for TGF-β remains critically important. For example, the role of Smad2 downstream of TGF-β is rather poorly understood. Identification of Smad2 target genes will likely shed some light on alternate mechanisms by which TGf-β may affect connective tissue remodeling. Although in this report we have emphasized the action of TGF-β1 by itself, it is clear that in-vivo TGF-β acts in combination with other peptide growth regulators, such as EGF or its homolog, TGF-α,[42] PDGF,[43] and basic fibroblast growth factor,[44] and that better understanding of its role will require a further study of these interactions.
Although the precise mechanism of TGF-β activation in-vivo remains unknown, several candidate molecules, including plasmin, thrombospondin-1, and αVβ6 integrin, have been found to activate latent TGF-β,[45-47] either by proteolysis of the latent complex in the case of the plasmin or by induction of conformational changes as a result of direct physical interaction. A statistically significant increase in the levels of stromal expression of MMP-1, MMP-2, and MMP-9 and TIMP-1 and TIMP-2 using monospecific antibodies reacting against tissue antigens had been reported in OSF.[48] The simultaneous increase in reactivity of MMPs and TIMPs posed difficulty in interpreting the result of this study. Interestingly, MMP-9 and MMP-2 produce different TGF-β proteolytic cleavage products, suggesting different recognition/cleavage sites. The basis for the difference in sensitivity to MMP-9-mediated activation among the three latent TGF-β isoforms is unclear at present. One possible explanation may be that structural variations among the isoforms influence the accessibility of the cleavage site.
TGF-β i s a pleotropic cytokine with mult iple immunosuppressive properties.[49] These properties include inhibition of T-cell proliferation, inhibition of T-cell differentiation in to cytotoxic T lymphocytes (CTLs) and helper T cells, and inhibition of the T-cell stimulatory functions of antigen-presenting cells (APCs). Suppression of these functions by TGF-β could be an important mechanism whereby it inhibits immune response. Importantly, TGF-β production has been associated with tumor growth of a vast variety of tumors.[50] In addition to the TGF-β production by cancerous cells, TGF-β can be produced by
TGF β-1 polymorphism in oral submucous fibrosis Rajendran, et al.

Indian J Dent Res, 21(3), 2010 418
a wide variety of non-cancerous cells present at the tumor sites. Specifically, tumor-associated T cells, natural killer (NK) cells, macrophages, epithelial cells, and stromal cells have all been shown to produce TGF-β in various tumor models.[51] Therefore, regardless of its source, TGF-β production by tumors can have an important role in the suppression of anti-tumor immune responses. Extrapolating these data in the context of OSF, certain parallels could be drawn in that the presence of large numbers of lymphocytes and fibroblasts, as well as plasma cells in moderate number, suggest the importance of a sustained lymphocytic infiltration in the maintenance of the tissue reaction in OSF.[24] A significant reduction in the cell-mediated immune response reflected by a decrease in high affinity rosette-forming cells (HARFC) and an increase in serum levels of IgA, IgD, and IgE have been reported in OSF.[52] TGF-β signaling pathway has been considered both as a tumor suppressor pathway and a promoter of tumor progression and invasion,[53] and at present the role of this cytokine in modifying the patho-physiology of OSF remains speculative. The consistent immune suppression noticed in OSF needs redressal against this backdrop.
The role of TGF-β in epithelial malignancy is complex, but it is becoming clear that, in the early stages of carcinogenesis, the protein acts as a potent tumor suppressor, while later, TGF-β can function to advance tumor progression.[54] In the setting of a precancerous condition like in OSF, the role of this cytokine in inducing carcinogenesis is currently left for speculation. The truncated C→T polymorphism (rs 13306708) in 5¢UTR region of TGF-β1 isomer gene noted in this study is, in any way, contribute to disease progression is debatable at this time. The proximity of this polymorphism and its sequelae to the transcriptional regulator sites and the associated “hot spot “ locales of the TGF-β1 gene have an area poised for future research.
REFERENCES
1. Nathan C, Sporn M. Cytokines in context. J Cell Biol 1991;113:981-6.2. Roberts AB, Flanders KC, Kondaiah P, Thompson NL, Van Obberghen-
Schilling E, Wakefield L, et al. Transforming growth factor beta: Biochemistry and roles in embryogenesis, tissue repair and remodeling, and carcinogenesis. Recent Prog Horm Res 1988;44:157-97.
3. Roberts AB, Heine UI, Flanders KC, Sporn MB. Transforming growth factor-beta. Major role in regulation of extracellular matrix. Ann N Y Acad Sci 1990;580:225-32.
4. Salez F, Gosset P, Copin MC, Lamblin Degros C, Tonnel AB, Wallaert B. Transforming growth factor-beta1 in sarcoidosis. Eur Respir J 1998;12:913-9.
5. Zissel G, Homolka J, Schlaak J, Schlaak M, Muller-Quernheim J. Anti-inflammatory cytokine release by alveolar macrophages in pulmonary sarcoidosis. Am J Respir Crit Care Med 1996;154:713-9.
6. Baughman RP, Lower EE, Miller MA, Bejarano PA, Heffelfinger SC. Overexpression of transforming growth factor-alpha and epidermal growth factor-receptor in idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 1999;16:57-61.
7. Bergeron A, Soler P, Kambouchner M, Loiseau P, Milleron B, Valeyre D, et al. Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for TGF-beta and IL-10. Eur Respir J 2003;22:69-76.
8. Awad MR, El-Gamel A, Hasleton P, Turner DM, Sinnott PJ, Hutchinson IV. Genotypic variation in the transforming growth factor-beta1 gene: Association with transforming growth factor-beta1 production, fibrotic lung disease, and graft fibrosis after lung transplantation. Transplant 1998;27:1014-20.
9. Pindborg JJ. Oral submucous fibrosis: A review. Ann Acad Med Singapore 1989;18:603-7.
10. Meghji S, Scutt A, Harvey W, Canniff JP. An in-vitro comparison of human fibroblasts from normal and oral submucous fibrosis tissue. Arch Oral Biol 1987;32:213-5.
11. Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning. A Laboratory Manual. 2nd ed, Vol. 2. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989.
12. Kimura A, Dong RP, Harada H, Sasazuki T. DNA typing of HLA class II genes in B-lymphoblastoid cell lines homozygous for HLA. Tissue Antigens 1992;40:5-12.
13. Syrris P, Carter ND, Metcalfe JC, Kemp PR, Grainger DJ, Kaski JC, et al. Transforming growth factor-beta1 gene polymorphisms and coronary artery disease. Clin Sci (Lond) 1998;95:659-67.
14. Khalil N, Bereznay O, Sporn M, Greenberg AH. Macrophage production of transforming growth factor beta and fibroblast collagen synthesis in chronic pulmonary inflammation. J Exp Med 1989;170:727-37.
15. Border WA, Okuda S, Languino LR, Sporn MB, Ruoslahti E. Suppression of experimental glomerulonephritis by antiserum against transforming growth factor beta 1. Nature 1990;346:371-4.
16. Connor TB Jr, Roberts AB, Sporn MB, Danielpour D, Dart LL, Michels RG, et al. Correlation of fibrosis and transforming growth factor-beta type 2 levels in the eye. J Clin Invest 1989;83:1661-6.
17. Shiau YY, Kwan HW. Submucous fibrosis in Taiwan. Oral Surg Oral Med Oral Pathol 1979;47:453-7.
18. Harvey W, Scutt A, Meghji S, Canniff JP. Stimulation of human buccal mucosa fibroblasts in vitro by betel-nut alkaloids. Arch Oral Biol 1986;31:45-9.
19. Kuo MY, Chen HM, Hahn LJ, Hsieh CC, Chiang CP. Collagen biosynthesis in human oral submucous fibrosis fibroblast cultures. J Dent Res 1995;74:1783-8.
20. Shieh TY, Yang JF. Collagenase activity in oral submucous fibrosis. Proc Natl Sci Counc Repub China B 1992;16:106-10.
21. Cunha GR, Reese BA, Sekkingstad M. Induction of nuclear androgen-binding sites in epithelium of the embryonic urinary bladder by mesenchyme of the urogenital sinus of embryonic mice. Endocrinology 1980;107:1767-70.
22. Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. science 2004;303:848-51.
23. Ronnov-Jessen L, Petersen OW, Bissell MJ. Cellular changes involved in conversion of normal to malignant breast: Importance of the stromal reaction. Physiol Rev 1996;76:69-125.
24. Rajendran R. Oral submucous fibrosis: Etiology, pathogenesis, and future research. Bull World Health Organ 1994;72:985-96.
25. Edwards DR, Murphy G, Reynolds JJ, Whitham SE, Docherty AJ, Angel P, et al. Transforming growth factor beta modulates the expression of collagenase and metalloproteinase inhibitor. Embo J 1987;6:1899-904.
26. Wang XL, Sim AS, Wilcken DE. A common polymorphism of the transforming growth factor-beta1 gene and coronary artery disease. Clin Sci (Lond) 1998;95:745-6.
27. Cambien F, Ricard S, Troesch A, Mallet C, Generenaz L, Evans A, et al. Polymorphisms of the transforming growth factor-beta 1 gene in relation to myocardial infarction and blood pressure. The Etude Cas-Temoin de l'Infarctus du Myocarde (ECTIM) Study. Hypertension 1996;28:881-7.
28. Grainger DJ, Heathcote K, Chiano M, Snieder H, Kemp PR, Metcalfe JC, et al. Genetic control of the circulating concentration of transforming growth factor type beta1. Hum Mol Genet 1999;8:93-7.
29. Luedecking EK, DeKosky ST, Mehdi H, Ganguli M, Kamboh MI. Analysis of genetic polymorphisms in the transforming growth factor-beta1 gene and the risk of Alzheimer's disease. Hum Genet 2000;106:565-9.
30. Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med 1994;331:1286-92.
31. Chen W, Wahl SM. Manipulation of TGF-beta to control autoimmune
TGF β-1 polymorphism in oral submucous fibrosis Rajendran, et al.

Indian J Dent Res, 21(3), 2010419
and chronic inflammatory diseases. Microbes Infect 1999;1:1367-80.32. Kulkarni AB, Karlsson S. Transforming growth factor-beta 1 knockout
mice. A mutation in one cytokine gene causes a dramatic inflammatory disease. Am J Pathol 1993;143:3-9.
33. Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, et al. Transforming growth factor type beta: Rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U S A 1986;83:4167-71.
34. Raghow R. Role of transforming growth factor-beta in repair and fibrosis. Chest 1991;99:61S-5.
35. Giri SN, Hyde DM, Hollinger MA. Effect of antibody to transforming growth factor beta on bleomycin induced accumulation of lung collagen in mice. Thorax 1993;48:959-66.
36. Rajendran R, Vijayakumar T, Vasudevan DM. An alternative pathogenetic pathway for oral submucous fibrosis (OSMF). Med Hypotheses 1989;30:35-7.
37. Folkman J, Cotran R. Relation of vascular proliferation to tumor growth. Int Rev Exp Pathol 1976;16:207-48.
38. Lobb RR, Alderman EM, Fett JW. Induction of angiogenesis by bovine brain derived class 1 heparin-binding growth factor. Biochemistry 1985;24:4969-73.
39. Rajendran R, Paul S, Mathews PP, Raghul J, Mohanty M. Characterisation and quantification of mucosal vasculature in oral submucous fibrosis. Indian J Dent Res 2005;16:83-91.
40. Amento EP, Beck LS. TGF-beta and wound healing. Ciba Found Symp 1991;157:115-23.
41. Verrecchia F, Mauviel A. Transforming growth factor-beta and fibrosis. World J Gastroenterol 2007;13:3056-62.
42. Assoian RK, Grotendorst GR, Miller DM, Sporn MB. Cellular transformation by coordinated action of three peptide growth factors from human platelets. Nature 1984;309:804-6.
43. Shimokado K, Raines EW, Madtes DK, Barrett TB, Benditt EP, Ross R. A significant part of macrophage-derived growth factor consists of at least two forms of PDGF. Cell 1985;43:277-86.
44. Esch F, Ueno N, Baird A, Hill F, Denoroy L, Ling N, et al. Primary structure of bovine brain acidic fibroblast growth factor (FGF). Biochem Biophys Res Commun 1985;133:554-62.
45. Lyons RM, Gentry LE, Purchio AF, Moses HL. Mechanism of activation of latent recombinant transforming growth factor beta 1 by plasmin. J Cell Biol 1990;110:1361-7.
46. Sato Y, Tsuboi R, Lyons R, Moses H, Rifkin DB. Characterization of the activation of latent TGF-beta by co-cultures of endothelial cells and pericytes or smooth muscle cells: A self-regulating system. J Cell Biol 1990;111:757-63.
47. Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, et al. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell 1998;93:1159-70.
48. Rajendran R, Rajeesh MP, Shaikh S, Shanthi, Pillai MR. Expression of matrix metalloproteinases (MMP-1, MMP-2 and MMP-9) and their inhibitors (TIMP-1 and TIMP-2) in oral submucous fibrosis. Indian J Dent Res 2006;17:161-6.
49. Letterio JJ, Bottinger EP. TGF-beta knockout and dominant-negative receptor transgenic mice. Miner Electrolyte Metab 1998;24:161-7.
50. Pasche B. Role of transforming growth factor beta in cancer. J Cell Physiol 2001;186:153-68.
51. Chakravarthy D, Green AR, Green VL, Kerin MJ, Speirs V. Expression and secretion of TGF-beta isoforms and expression of TGF-beta-receptors I, II and III in normal and neoplastic human breast. Int J Oncol 1999;15:187-94.
52. Rajendran R, Sugathan CK, Remani P, Ankathil R, Vijayakumar T. Cell mediated and humoral immune responses in oral submucous fibrosis. Cancer 1986;58:2628-31.
53. Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet 2001;29:117-29.
54. Prime SS, Davies M, Pring M, Paterson IC. The role of TGF-beta in epithelial malignancy and its relevance to the pathogenesis of oral cancer (part II). Crit Rev Oral Biol Med 2004;15:337-47.
How to cite this article: Rajendran R, Harish RK, Anil S, Vidyadharan R, Banerjee M. Transforming growth factor-β-1 polymorphisms are infrequent but exist at selected loci in oral submucous fibrosis. Indian J Dent Res 2010;21:413-9Source of Support: Nil, Conflict of Interest: None declared.
TGF β-1 polymorphism in oral submucous fibrosis Rajendran, et al.




















