
Oxidized LDL Upregulates Angiotensin II Type 1 Receptor Expression in Cultured Human Coronary Artery...
8
Dayuan Li, Tom Saldeen, Francesco Romeo and Jawahar L. Mehta B κ NF- Human Coronary Artery Endothelial Cells: The Potential Role of Transcription Factor Oxidized LDL Upregulates Angiotensin II Type 1 Receptor Expression in Cultured Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 2000 American Heart Association, Inc. All rights reserved. is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231 Circulation doi: 10.1161/01.CIR.102.16.1970 2000;102:1970-1976 Circulation. http://circ.ahajournals.org/content/102/16/1970 World Wide Web at: The online version of this article, along with updated information and services, is located on the http://circ.ahajournals.org//subscriptions/ is online at: Circulation Information about subscribing to Subscriptions: http://www.lww.com/reprints Information about reprints can be found online at: Reprints: document. Permissions and Rights Question and Answer this process is available in the click Request Permissions in the middle column of the Web page under Services. Further information about Office. Once the online version of the published article for which permission is being requested is located, can be obtained via RightsLink, a service of the Copyright Clearance Center, not the Editorial Circulation in Requests for permissions to reproduce figures, tables, or portions of articles originally published Permissions: by guest on September 25, 2014 http://circ.ahajournals.org/ Downloaded from by guest on September 25, 2014 http://circ.ahajournals.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Oxidized LDL Upregulates Angiotensin II Type 1 Receptor Expression in Cultured Human Coronary Artery...

Dayuan Li, Tom Saldeen, Francesco Romeo and Jawahar L. MehtaBκNF-
Human Coronary Artery Endothelial Cells: The Potential Role of Transcription Factor Oxidized LDL Upregulates Angiotensin II Type 1 Receptor Expression in Cultured
Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 2000 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation doi: 10.1161/01.CIR.102.16.1970
2000;102:1970-1976Circulation.
http://circ.ahajournals.org/content/102/16/1970World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circ.ahajournals.org//subscriptions/
is online at: Circulation Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialCirculationin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on September 25, 2014http://circ.ahajournals.org/Downloaded from by guest on September 25, 2014http://circ.ahajournals.org/Downloaded from

Oxidized LDL Upregulates Angiotensin II Type 1 ReceptorExpression in Cultured Human Coronary Artery
Endothelial CellsThe Potential Role of Transcription Factor NF-kB
Dayuan Li, MD, PhD; Tom Saldeen, MD, PhD; Francesco Romeo, MD; Jawahar L. Mehta, MD, PhD
Background—We demonstrated earlier that angiotensin II (Ang II), by AT1 receptor activation, upregulates oxidized LDL(ox-LDL) endothelial receptor LOX-1 gene expression and uptake of ox-LDL in human coronary artery endothelial cells(HCAECs). In this study, we investigated the regulation of Ang II receptors (AT1R and AT2R) by ox-LDL and the roleof the redox-sensitive transcription factor NF-kB in this process.
Methods and Results—HCAECs were incubated with ox-LDL for 24 hours. Ox-LDL (10 to 40mg protein/mL)upregulated AT1R but not AT2R, mRNA, or protein. Ox-LDL degraded IkBa in cytoplasm and activated transcriptionfactor NF-kB (P65) in HCAEC nuclear extract. Treatment of cells with the antioxidanta-tocopherol (10 to 50mmol/L)attenuated ox-LDL–mediated degradation of IkBa and activation of NF-kB (P65) and inhibited the upregulation ofAT1R mRNA and protein. The role of NF-kB signal transduction was further examined by use of an NF-kB inhibitor,caffeic acid phenethyl ester (CAPE). Pretreatment of cells with CAPE inhibited ox-LDL–mediated degradation of IkBaand NF-kB activation and inhibited ox-LDL–induced upregulation of AT1R expression. Incubation of cells with bothox-LDL and Ang II increased cell injury, measured as cell viability and LDH release, compared with either ox-LDL orAng II alone.a-Tocopherol as well as the specific AT1R blocker CV11974 (candesartan) attenuated the cell-injuriouseffects of ox-LDL.
Conclusions—These observations suggest an important role of ox-LDL–mediated AT1R upregulation in cell injury. In thisprocess, NF-kB activation seems to play a critical role in signal transduction. These findings provide a basis for the useof antioxidants and AT1R blockers in designing therapy of atherosclerosis.(Circulation. 2000;102:1970-1976.)
Key Words: angiotensinn receptorsn lipoproteinsn endotheliumn antioxidants
The renin-angiotensin system plays an important role inatherogenesis. Angiotensin II (Ang II) activates at least 2
distinct types of cell-surface receptors, the type 1 (AT1R) andthe type 2 (AT2R).1,2 Most studies suggest that AT1Ractivation mediates most known effects of Ang II in thecardiac tissues.2,3 However, some studies also show thatactivation of the AT2R receptor mediates myocardial ische-mia-reperfusion injury4 and exerts proapoptotic effect onmyocytes.5 Both AT1R and AT2R exist in rat coronary arteryendothelial cells.6 Recent work from our laboratory7 indicatesthat both AT1R and AT2R exist in human coronary arteryendothelial cells (HCAECs), and AT1R activation inducesapoptosis of HCAECs. Another recent study8 showed thatactivation of AT1R causes myocardial dysfunction duringmyocardial ischemia-reperfusion in isolated rat hearts.
In atherosclerosis, oxidized LDL (ox-LDL) accumulates inthe vessel walls,9 decreases generation of nitric oxide (NO),10
and causes endothelial dysfunction.11 Ox-LDL is cytotoxic12
and acts as a chemotactic factor for monocytes,13 leading tothe accumulation of inflammatory cells and the generation ofoxygen-derived free radicals that can inactivate endothelium-derived NO.14 Ox-LDL induces apoptosis in vascular smoothmuscle cells,15 monocytes/macrophages,16 and endothelialcells.17 On the basis of these considerations, oxidative mod-ification of LDL is considered a key trigger in the initiationand progression of atherosclerosis.18 A recent study byMaziere et al19 demonstrated that ox-LDL induces activationof transcription factor NF-kB in endothelial cells and causescell injury. Activated NF-kB has indeed been detected inendothelial cells of atherosclerotic plaques.20
There is increasing evidence for an interaction betweenhyperlipidemia and the renin-angiotensin system in athero-genesis.21–23For example, AT1R expression is upregulated byLDL in vascular smooth muscle cells.21 Ang II facilitatesoxidation of LDL22 and its uptake by scavenger receptors onmonocytes/macrophages.23 We have recently demonstrated
Received March 30, 2000; revision received May 22, 2000; accepted May 22, 2000.From the Departments of Medicine and Physiology, University of Arkansas and VA Medical Center, Little Rock (D.L., J.L.M.); the University of
Rome, Tor Vergata, Rome, Italy (F.R.); and the Department of Forensic Medicine, University of Uppsala, Uppsala, Sweden (T.S.).Correspondence to J.L. Mehta, MD, PhD, University of Arkansas Medical Science, Mail Slot 532, 4301 West Markham, Little Rock, AR 72205-7199.© 2000 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org
1970
Basic Science Reports
by guest on September 25, 2014http://circ.ahajournals.org/Downloaded from

upregulation of specific lectin-like receptors for ox-LDL(LOX-1) in response to Ang II.24 In the present study, weprovide evidence that ox-LDL upregulates expression of AngII type AT1R in cultured HCAECs. We also demonstrate acritical role of transcription factor NF-kB activation in thisprocess.
MethodsCell CultureThe methodology for culture of HCAECs has been describedpreviously.17,24 In brief, the initial batch of HCAECs was purchasedfrom Clonetics Corp. The endothelial cells were pure on the basis ofmorphology and staining for factor VIII and acetylated LDL.HCAECs were cultured in microvascular endothelium growth me-dium (Clonetics) that consisted of 500 mL endothelial cell basalmedium, 5 ng human recombinant epidermal growth factor, 0.5 mghydrocortisone, 25 mg gentamycin, 50mg amphotericin B, 6 mgbovine brain extract, and 25 mL FBS. Fourth-generation HCAECswere used in this study.
HCAECs were incubated with ox-LDL (10 and 40mg protein/mL)for 24 hours, and expression of AT1R and AT2R was determined.Parallel groups of HCAECs were incubated with ox-LDL (40mgprotein/mL) alone or with the antioxidanta-tocopherol (10 and50 mmol/L) to study degradation of IkBa, NF-kB activation, andAT1R expression. Parallel groups of HCAECs also were pretreatedwith the antioxidanta-tocopherol, the AT1R blocker CV11974(candesartan, 1026 mol/L), or the AT2R blocker PD123319 (1026
mol/L) for 30 minutes, and HCAECs then were exposed to ox-LDL,Ang II, or both to study degradation of IkB, activation of NF-kB, andinduction of cell injury (cell viability and LDH release). To furtherdetermine the role of NF-kB activation, cells were incubated withTNF-a (40 ng/mL) and the NF-kB inhibitor caffeic acid phenethylester (CAPE), 20mg/mL, and degradation of IkBa and NF-kBactivation and AT1R expression was studied. TNF-a served as apositive control for NF-kB activation by ox-LDL. The concentra-tions of these reagents were based on previous studies.17,25,26
Preparation of LipoproteinsNative LDL and ox-LDL were prepared as described earlier.17 LDLwas oxidized by exposure to CuSO4 (5 mmol/L free Cu21) in PBS at37°C for 24 hours. The thiobarbituric acid–reactive substancescontent of ox-LDL was 17.561.6 versus 0.3860.11 nmol/100mgprotein in the native-LDL preparation (P,0.01). LDL and ox-LDLwere kept in 50 mmol/L Tris-HCl, 0.15 mol/L NaCl, and 2 mmol/LEDTA at pH 7.4 and were used within 10 days of preparation.Endotoxin concentration in the LDL was checked with the E-Toxatekit (Sigma) and found to be consistently,0.005 endotoxin units/mL(lowest detection limit).
RT-PCR for AT1R and AT2RThe detailed methodology for reverse transcription–polymerasechain reaction (RT-PCR) identification of AT1R and AT2R inHCAECs was published recently.24 Essentially, the reverse-transcribed material was amplified withTaq DNA polymerase(Promega) with a primer pair specific to human AT1R receptor(forward primer, 59-TCATTTACTTTTATATTGTAA-39; reverseprimer, 59-TGAATTTCATAAGCCTTCTT-39). PCR product was532 bp. For PCR, 35 cycles were used at 94°C for 1 minute, 50°C for1 minute, and 72°C for 2 minutes. The primer pair specific to humanAT2R receptor was (forward primer) 59-AATATGAAG-GGCAACTCCAC-39 and (reverse primer) 59-TTAAGAC-ACAAAGGTCTCCAT-39. The PCR product was 1100 bp. ForPCR, 35 cycles were used at 94°C for 1 minute, 58°C for 1 minute,and 72°C for 2 minute.
Immunoprecipitation and Western Blot for AT1Rand AT2RCell lysates (120mg) were subjected to immunoprecipitation andthen Western analysis. In brief, the cell lysates containing equal
amounts of soluble proteins were immunoprecipitated. Precipitateswere washed and then resuspended in SDS-PAGE sample buffer andboiled for 5 minutes. Samples were separated by 12% SDS-PAGE,and then transferred to nitrocellulose membranes. After incubation inblocking solution (4% nonfat milk, Sigma), membranes were incu-bated with primary antibody (polyclonal antibody to AT1R orpolyclonal antibody to AT2R, Santa Cruz Laboratory) for 2 hours atroom temperature. Membranes were washed and then incubated with1:3000 dilution secondary antibody (Amersham) for 1 hour, detectedwith the ECL system, and relative intensities of protein bands wereanalyzed by MSF-300G Scanner (Microtek Laboratory).27
Preparation of Nuclear Extracts and Western Blotfor NF-kBThe detailed methodology for preparation of nuclear extracts andWestern blot for NF-kB in HCAECs has been published recently.28
We used a monoclonal antibody to the P65 subunit of NF-kB frommouse to mouse hybrid cells (Boehringer Mannheim). The antibodyrecognizes an epitope overlapping the nuclear location signal of theP65 subunit and therefore selectively binds the activated form ofNF-kB.
Western Blot for I kBThe detailed methodology for Western blot for IkB in HCAECs waspublished recently.28 The antibody used was a rabbit polyclonalanti-IkBa (Santa Cruz Biotechnology).
Measurement of LDHHCAEC supernatants were collected for determination of LDH. Anenzyme activity method based on oxidation of lactate with measure-ment of rate of increase in absorbance at 340 nm was used. Theactivity of LDH was expressed as units per milligram protein.24
Figure 1. Upregulation of AT1R mRNA by ox-LDL in HCAECs.Incubation of HCAECs with ox-LDL (10 and 40 mg protein/mL)induced progressive upregulation of AT1R mRNA expression.Incubation of cells with ox-LDL had no significant effect onAT2R mRNA expression. Top, Result of a representative experi-ment; bottom, summary of data from 6 independently per-formed experiments (mean6SD). AU indicates arbitrary units.
Li et al Oxidized LDL and Ang II Receptors 1971
by guest on September 25, 2014http://circ.ahajournals.org/Downloaded from
Cell ViabilityA small aliquot of cells was incubated in 0.1% trypan blue for a fewminutes, and the cells were viewed under a light microscope. Deadcells are permeable to trypan blue and thus become colored, whereasviable cells do not take up the dye. By counting 100 cells, thepercentage of viable cells was calculated.24
Data AnalysisAll data represent the mean of duplicate samples from 6 indepen-dently performed experiments. Data are presented as mean6SD.Statistical significance was determined in multiple comparisonsamong independent groups of data in which ANOVA and the F testindicated the presence of significant differences. A value ofP,0.05was considered significant.
ResultsOx-LDL and Regulation of AT1R and AT2Rin HCAECsAs described earlier,24 AT1R and AT2R mRNA and proteinwere identified by RT-PCR and Western blot analyses,respectively, in all HCAEC aliquots. Incubation of HCAECswith ox-LDL (10 and 40mg protein/mL) induced progressiveupregulation of AT1R mRNA. Incubation of cells withox-LDL had no significant effect on AT2R mRNA (Figure 1).Incubation with 80mg/mL of ox-LDL had a smaller effect onAT1R expression than the 40mg/mL concentration (data notshown), perhaps a result of the cell-injurious effect of highconcentrations of ox-LDL.
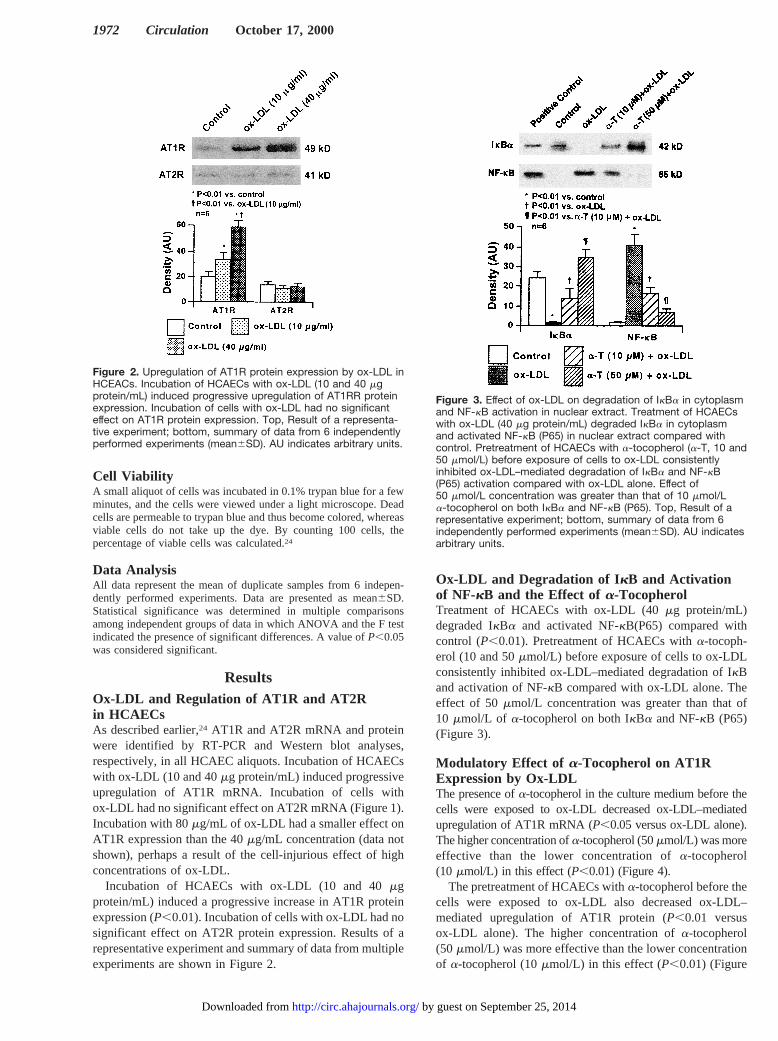
Incubation of HCAECs with ox-LDL (10 and 40mgprotein/mL) induced a progressive increase in AT1R proteinexpression (P,0.01). Incubation of cells with ox-LDL had nosignificant effect on AT2R protein expression. Results of arepresentative experiment and summary of data from multipleexperiments are shown in Figure 2.
Ox-LDL and Degradation of I kB and Activationof NF-kB and the Effect of a-TocopherolTreatment of HCAECs with ox-LDL (40mg protein/mL)degraded IkBa and activated NF-kB(P65) compared withcontrol (P,0.01). Pretreatment of HCAECs witha-tocoph-erol (10 and 50mmol/L) before exposure of cells to ox-LDLconsistently inhibited ox-LDL–mediated degradation of IkBand activation of NF-kB compared with ox-LDL alone. Theeffect of 50mmol/L concentration was greater than that of10 mmol/L of a-tocopherol on both IkBa and NF-kB (P65)(Figure 3).
Modulatory Effect of a-Tocopherol on AT1RExpression by Ox-LDLThe presence ofa-tocopherol in the culture medium before thecells were exposed to ox-LDL decreased ox-LDL–mediatedupregulation of AT1R mRNA (P,0.05 versus ox-LDL alone).The higher concentration ofa-tocopherol (50mmol/L) was moreeffective than the lower concentration ofa-tocopherol(10 mmol/L) in this effect (P,0.01) (Figure 4).
The pretreatment of HCAECs witha-tocopherol before thecells were exposed to ox-LDL also decreased ox-LDL–mediated upregulation of AT1R protein (P,0.01 versusox-LDL alone). The higher concentration ofa-tocopherol(50 mmol/L) was more effective than the lower concentrationof a-tocopherol (10mmol/L) in this effect (P,0.01) (Figure
Figure 2. Upregulation of AT1R protein expression by ox-LDL inHCEACs. Incubation of HCAECs with ox-LDL (10 and 40 mgprotein/mL) induced progressive upregulation of AT1RR proteinexpression. Incubation of cells with ox-LDL had no significanteffect on AT1R protein expression. Top, Result of a representa-tive experiment; bottom, summary of data from 6 independentlyperformed experiments (mean6SD). AU indicates arbitrary units.
Figure 3. Effect of ox-LDL on degradation of IkBa in cytoplasmand NF-kB activation in nuclear extract. Treatment of HCAECswith ox-LDL (40 mg protein/mL) degraded IkBa in cytoplasmand activated NF-kB (P65) in nuclear extract compared withcontrol. Pretreatment of HCAECs with a-tocopherol (a-T, 10 and50 mmol/L) before exposure of cells to ox-LDL consistentlyinhibited ox-LDL–mediated degradation of IkBa and NF-kB(P65) activation compared with ox-LDL alone. Effect of50 mmol/L concentration was greater than that of 10 mmol/La-tocopherol on both IkBa and NF-kB (P65). Top, Result of arepresentative experiment; bottom, summary of data from 6independently performed experiments (mean6SD). AU indicatesarbitrary units.
1972 Circulation October 17, 2000
by guest on September 25, 2014http://circ.ahajournals.org/Downloaded from
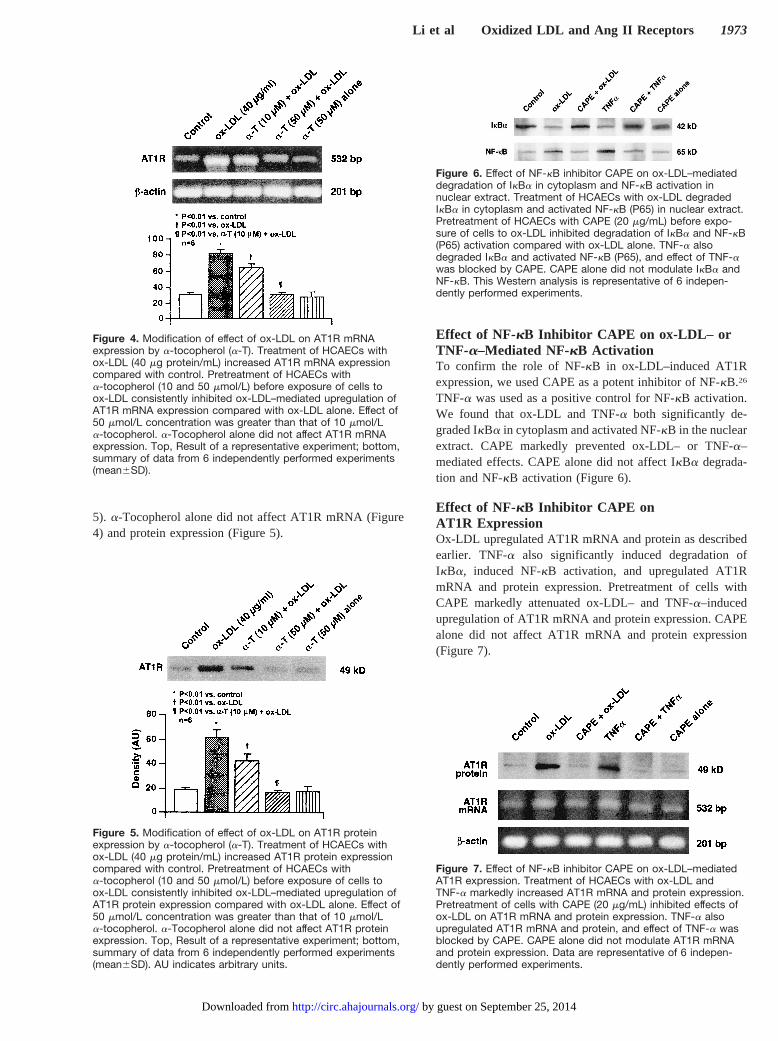
5). a-Tocopherol alone did not affect AT1R mRNA (Figure4) and protein expression (Figure 5).
Effect of NF-kB Inhibitor CAPE on ox-LDL– orTNF-a–Mediated NF-kB ActivationTo confirm the role of NF-kB in ox-LDL–induced AT1Rexpression, we used CAPE as a potent inhibitor of NF-kB.26
TNF-a was used as a positive control for NF-kB activation.We found that ox-LDL and TNF-a both significantly de-graded IkBa in cytoplasm and activated NF-kB in the nuclearextract. CAPE markedly prevented ox-LDL– or TNF-a–mediated effects. CAPE alone did not affect IkBa degrada-tion and NF-kB activation (Figure 6).
Effect of NF-kB Inhibitor CAPE onAT1R ExpressionOx-LDL upregulated AT1R mRNA and protein as describedearlier. TNF-a also significantly induced degradation ofIkBa, induced NF-kB activation, and upregulated AT1RmRNA and protein expression. Pretreatment of cells withCAPE markedly attenuated ox-LDL– and TNF-a–inducedupregulation of AT1R mRNA and protein expression. CAPEalone did not affect AT1R mRNA and protein expression(Figure 7).
Figure 4. Modification of effect of ox-LDL on AT1R mRNAexpression by a-tocopherol (a-T). Treatment of HCAECs withox-LDL (40 mg protein/mL) increased AT1R mRNA expressioncompared with control. Pretreatment of HCAECs witha-tocopherol (10 and 50 mmol/L) before exposure of cells toox-LDL consistently inhibited ox-LDL–mediated upregulation ofAT1R mRNA expression compared with ox-LDL alone. Effect of50 mmol/L concentration was greater than that of 10 mmol/La-tocopherol. a-Tocopherol alone did not affect AT1R mRNAexpression. Top, Result of a representative experiment; bottom,summary of data from 6 independently performed experiments(mean6SD).
Figure 5. Modification of effect of ox-LDL on AT1R proteinexpression by a-tocopherol (a-T). Treatment of HCAECs withox-LDL (40 mg protein/mL) increased AT1R protein expressioncompared with control. Pretreatment of HCAECs witha-tocopherol (10 and 50 mmol/L) before exposure of cells toox-LDL consistently inhibited ox-LDL–mediated upregulation ofAT1R protein expression compared with ox-LDL alone. Effect of50 mmol/L concentration was greater than that of 10 mmol/La-tocopherol. a-Tocopherol alone did not affect AT1R proteinexpression. Top, Result of a representative experiment; bottom,summary of data from 6 independently performed experiments(mean6SD). AU indicates arbitrary units.
Figure 6. Effect of NF-kB inhibitor CAPE on ox-LDL–mediateddegradation of IkBa in cytoplasm and NF-kB activation innuclear extract. Treatment of HCAECs with ox-LDL degradedIkBa in cytoplasm and activated NF-kB (P65) in nuclear extract.Pretreatment of HCAECs with CAPE (20 mg/mL) before expo-sure of cells to ox-LDL inhibited degradation of IkBa and NF-kB(P65) activation compared with ox-LDL alone. TNF-a alsodegraded IkBa and activated NF-kB (P65), and effect of TNF-awas blocked by CAPE. CAPE alone did not modulate IkBa andNF-kB. This Western analysis is representative of 6 indepen-dently performed experiments.
Figure 7. Effect of NF-kB inhibitor CAPE on ox-LDL–mediatedAT1R expression. Treatment of HCAECs with ox-LDL andTNF-a markedly increased AT1R mRNA and protein expression.Pretreatment of cells with CAPE (20 mg/mL) inhibited effects ofox-LDL on AT1R mRNA and protein expression. TNF-a alsoupregulated AT1R mRNA and protein, and effect of TNF-a wasblocked by CAPE. CAPE alone did not modulate AT1R mRNAand protein expression. Data are representative of 6 indepen-dently performed experiments.
Li et al Oxidized LDL and Ang II Receptors 1973
by guest on September 25, 2014http://circ.ahajournals.org/Downloaded from
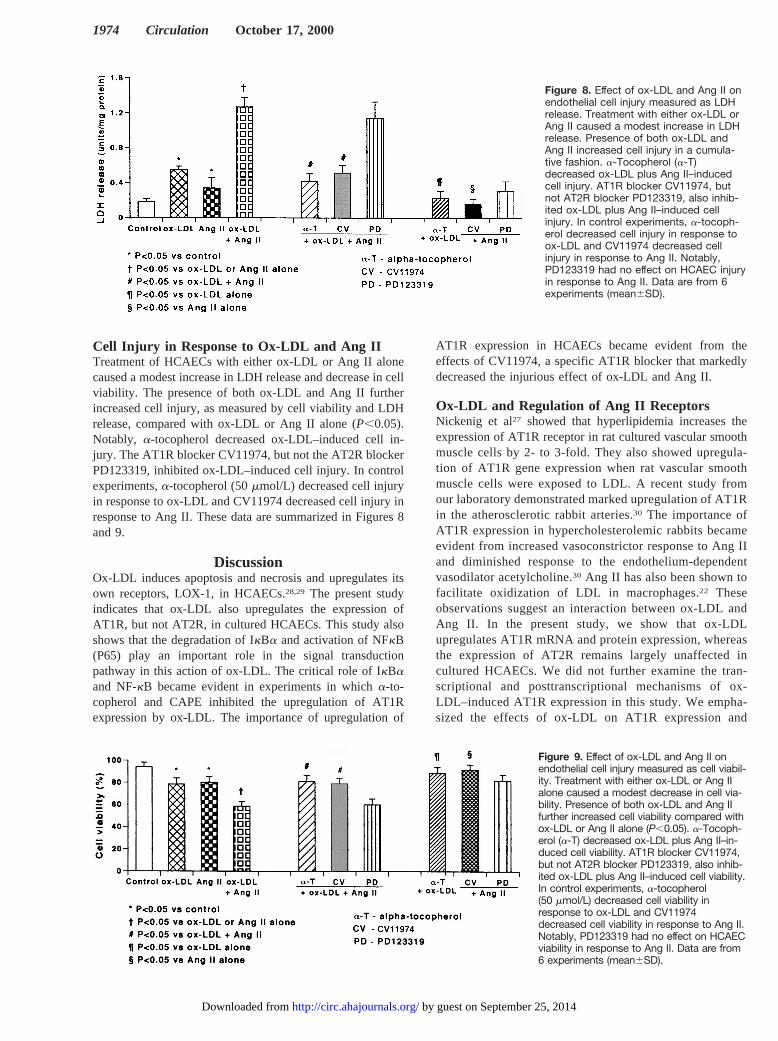
Cell Injury in Response to Ox-LDL and Ang IITreatment of HCAECs with either ox-LDL or Ang II alonecaused a modest increase in LDH release and decrease in cellviability. The presence of both ox-LDL and Ang II furtherincreased cell injury, as measured by cell viability and LDHrelease, compared with ox-LDL or Ang II alone (P,0.05).Notably, a-tocopherol decreased ox-LDL–induced cell in-jury. The AT1R blocker CV11974, but not the AT2R blockerPD123319, inhibited ox-LDL–induced cell injury. In controlexperiments,a-tocopherol (50mmol/L) decreased cell injuryin response to ox-LDL and CV11974 decreased cell injury inresponse to Ang II. These data are summarized in Figures 8and 9.
DiscussionOx-LDL induces apoptosis and necrosis and upregulates itsown receptors, LOX-1, in HCAECs.28,29 The present studyindicates that ox-LDL also upregulates the expression ofAT1R, but not AT2R, in cultured HCAECs. This study alsoshows that the degradation of IkBa and activation of NFkB(P65) play an important role in the signal transductionpathway in this action of ox-LDL. The critical role of IkBaand NF-kB became evident in experiments in whicha-to-copherol and CAPE inhibited the upregulation of AT1Rexpression by ox-LDL. The importance of upregulation of
AT1R expression in HCAECs became evident from theeffects of CV11974, a specific AT1R blocker that markedlydecreased the injurious effect of ox-LDL and Ang II.
Ox-LDL and Regulation of Ang II ReceptorsNickenig et al27 showed that hyperlipidemia increases theexpression of AT1R receptor in rat cultured vascular smoothmuscle cells by 2- to 3-fold. They also showed upregula-tion of AT1R gene expression when rat vascular smoothmuscle cells were exposed to LDL. A recent study fromour laboratory demonstrated marked upregulation of AT1Rin the atherosclerotic rabbit arteries.30 The importance ofAT1R expression in hypercholesterolemic rabbits becameevident from increased vasoconstrictor response to Ang IIand diminished response to the endothelium-dependentvasodilator acetylcholine.30 Ang II has also been shown tofacilitate oxidization of LDL in macrophages.22 Theseobservations suggest an interaction between ox-LDL andAng II. In the present study, we show that ox-LDLupregulates AT1R mRNA and protein expression, whereasthe expression of AT2R remains largely unaffected incultured HCAECs. We did not further examine the tran-scriptional and posttranscriptional mechanisms of ox-LDL–induced AT1R expression in this study. We empha-sized the effects of ox-LDL on AT1R expression and
Figure 8. Effect of ox-LDL and Ang II onendothelial cell injury measured as LDHrelease. Treatment with either ox-LDL orAng II caused a modest increase in LDHrelease. Presence of both ox-LDL andAng II increased cell injury in a cumula-tive fashion. a-Tocopherol (a-T)decreased ox-LDL plus Ang II–inducedcell injury. AT1R blocker CV11974, butnot AT2R blocker PD123319, also inhib-ited ox-LDL plus Ang II–induced cellinjury. In control experiments, a-tocoph-erol decreased cell injury in response toox-LDL and CV11974 decreased cellinjury in response to Ang II. Notably,PD123319 had no effect on HCAEC injuryin response to Ang II. Data are from 6experiments (mean6SD).
Figure 9. Effect of ox-LDL and Ang II onendothelial cell injury measured as cell viabil-ity. Treatment with either ox-LDL or Ang IIalone caused a modest decrease in cell via-bility. Presence of both ox-LDL and Ang IIfurther increased cell viability compared withox-LDL or Ang II alone (P,0.05). a-Tocoph-erol (a-T) decreased ox-LDL plus Ang II–in-duced cell viability. AT1R blocker CV11974,but not AT2R blocker PD123319, also inhib-ited ox-LDL plus Ang II–induced cell viability.In control experiments, a-tocopherol(50 mmol/L) decreased cell viability inresponse to ox-LDL and CV11974decreased cell viability in response to Ang II.Notably, PD123319 had no effect on HCAECviability in response to Ang II. Data are from6 experiments (mean6SD).
1974 Circulation October 17, 2000
by guest on September 25, 2014http://circ.ahajournals.org/Downloaded from

subsequent functional significance of AT1R expression.We recently provided evidence that Ang II upregulatesspecific LOX-1 on HCAECs and facilitates the uptake ofI125-labeled ox-LDL into these cells; this effect of Ang IIcan be blocked with specific blockade of AT1R.24 Collec-tively, these studies provide strong evidence of cross talkbetween ox-LDL and Ang II in the regulation of celldysfunction and injury.
Ox-LDL and the Transcription Factor NF- kBNF-kB is an oncogenic protein that regulates transcription ofa variety of cellular genes, including immune and inflamma-tory response and growth control.31 NF-kB is present in thecytosol as a heterodimer composed of NF-kB1 (P50) and Rel(P65) subunits bound to an inhibitor protein, IkB. Degrada-tion of IkBa protein seems to be necessary for the activationof NF-kB.32 After activation, NF-kB translocates from thecytosol to the nucleus of the cell, binds to specific DNAsequences, and initiates transcription. Maziere et al19 showedthat ox-LDL activates NF-kB in fibroblasts, smooth musclecells, and endothelial cells. Collins33 suggested oxidativeactivation of endothelial cell transcription factors, especiallyNF-kB, as a mechanism for changing endothelial cell pheno-type and for initiating atherosclerotic lesions. Hernan dez-Presa and colleagues20 provided direct evidence for NF-kBactivation in early atherosclerotic lesions. Other studies haveshown a critical role of NF-kB activation in apoptosis inmyocytes34 and endothelial cells.35 In the present study, wedemonstrate that ox-LDL degrades IkBa protein and acti-vates NF-kB in HCAECs. An obvious question relates to thebasis of degradation of IkBa and activation of NF-kB byox-LDL. ox-LDL–induced free radical release may play acritical role in this process. The redox-sensitive nature ofNF-kB activation became clear from the observation that afree radical scavenger,a-tocopherol, and the NF-kB inhibitorCAPE not only inhibited ox-LDL–mediated degradation ofIkBa and activation of NF-kB but also inhibited the upregu-lation of AT1R in HCAECs. A recent study26 reported thatCAPE inhibits transcription factor NF-kB activation withoutany effect on other transcription factors, such as AP-1, Oct-1,and TFIID. This study26 also showed that CAPE exerts itseffects by inhibiting reactive oxygen intermediates. Theseobservations collectively indicate that activation of NF-kBmay be an important signal transduction pathway in theeffects of ox-LDL on HCAECs.
Interaction Between ox-LDL and Ang II inCell InjuryExperimental studies21,24,27,30suggest that cross talk betweenhypercholesterolemia and Ang II may have pathophysiolog-ical significance. For example, upregulation of AT1R medi-ated by hypercholesterolemia leads to enhanced Ang II–induced vasoconstriction.27,30 In contrast, Ang II increasesoxidation of LDL22 and upregulates ox-LDL endothelialreceptor (LOX-1).24
Both Ang II and ox-LDL are important factors in inducingendothelial dysfunction and injury. Work from our laboratoryhas shown that Ang II7 and ox-LDL17 decrease NO genera-tion and increase lipid peroxidation and LDH release in
cultured HCAECs. Both Ang II and ox-LDL enhance anoxia-reoxygenation–mediated HCAEC injury. Work from otherlaboratories36,37 also suggests that ox-LDL and Ang II causeinjury to endothelial cells. In the present study, we demon-strate that ox-LDL and Ang II induce cell injury in acumulative fashion. The mechanism of ox-LDL–mediatedcell injury may be related, at least in part, to the upregulationof AT1R expression. NF-kB activation may play an impor-tant role in this process. This became evident in experimentsin which a-tocopherol significantly inhibited the actions ofox-LDL in conjunction with inhibition of upregulation ofAT1R by ox-LDL. The AT1R blocker CV11974 (candesar-tan) also attenuated the cell-injurious effects of ox-LDL.These observations suggest thata-tocopherol and AT1Rblockers may reduce the adverse effect of cross talk betweenox-LDL and Ang II.
In summary, this study shows that ox-LDL upregulates theexpression of AT1R, but not AT2R, in cultured humancoronary endothelial cells. The cross talk between ox-LDLand Ang II serves to promote injury to coronary endothelialcells. Degradation of IkBa and activation of NF-kB appear tobe important signal transduction pathways involved in theaction of ox-LDL.
AcknowledgmentsThis study was supported by a Merit Review Award from the VACentral Office, a contract with the Department of Defense, and fundsfrom the Swedish Medical Research Council.
References1. Feolde E, Vigne P, Frelin C. Angiotensin II receptor subtypes and bio-
logical responses in the rat heart.J Mol Cell Cardiol. 1993;25:1359–1367.
2. Yang BC, Phillips MI, Ambuehl PEJ, et al. Increase in angiotensin II type1 receptor expression immediately following ischemia-reperfusion inisolated rat hearts.Circulation. 1997;96:922–926.
3. Timmermans PB, Smith RD. Angiotensin II receptor subtypes: selectiveantagonists and functional correlates.Eur Heart J. 1994;15:79–87.
4. Ford WR, Clanachan AS, Jugdutt BI. Opposite effects of angiotensin AT1and AT2 receptor antagonists on recovery of mechanical function afterischemia-reperfusion in isolated working rat hearts.Circulation. 1996;94:3087–3089.
5. Yamada T, Horiuchi M, Dzau VJ. Angiotensin II type 2 receptor mediatesprogrammed cell death.Proc Natl Acad Sci U S A. 1996;93:156–160.
6. Stoll M, Steckelings M, Paul M, et al. The angiotensin AT2R-receptormediates inhibition of cell proliferation in coronary endothelial cells.J Clin Invest. 1995;95:651–657.
7. Li DY, Yang BC, Philips MI, et al. Pro-apoptotic effects of angiotensinII in human coronary artery endothelial cells: role of AT1R receptor andPKC activation.Am J Physiol. 1999;276:H786–H792.
8. Yang BC, Philips MI, Zhang YC, et al. Critical role of AT1 receptorexpression after ischemia-reperfusion in isolated rat hearts: beneficialeffect of antisense oligodeoxynucleotides directed at AT1R receptormRNA. Circ Res. 1998;83:552–559.
9. Yla-Herttuala S, Palinski W, Rosenfeld ME, et al. Evidence for thepresence of oxidatively modified low density lipoprotein in athero-sclerotic lesions of rabbit and man.J Clin Invest. 1989;84:1086–1095.
10. Chin JH, Azhar S, Hoffman BB. Inactivation of endothelium-derivedrelaxing factor by oxidized lipoproteins.J Clin Invest. 1992;89:10–18.
11. Kugiyama K, Kerns SA, Morrisett JD, et al. Impairment of endothelium-dependent arterial relaxation by lysolecithin in modified low-densitylipoproteins.Nature. 1990;344:160–162.
12. Morel DW, Hessler GM, Chisolm GM. Low density lipoprotein cyto-toxicity induced by free radical peroxidation of lipid. J Lipid Res. 1983;24:1070–1076.
13. Quinn MT, Parthasarathy S, Fong LG, et al. Oxidatively modified lowdensity lipoprotein: a potential role in recruitment and retention of mono-
Li et al Oxidized LDL and Ang II Receptors 1975
by guest on September 25, 2014http://circ.ahajournals.org/Downloaded from

cyte/macrophages during atherogenesis. Proc Natl Acad Sci U S A. 1987;84:2995–2998.
14. Gryglewski RJ, Palmer RM, Moncada S. Superoxide anion is involved inthe breakdown of endothelium-derived vascular relaxing factor.Nature.1986;320:454–456.
15. Nishio E, Watanabe Y. Oxysterols induced apoptosis in cultured smoothmuscle cells through CPP32 protease activation and bcl-2 protein down-regulation. Biochem Biophys Res Commun. 1996;226:928–934.
16. Yang X, Galeano NF, Szaboles M, et al. Oxidized low densitylipoproteins alter macrophage lipid uptake, apoptosis, viability and nitricoxide synthesis.J Nutr. 1996;126:10272s-10275s.
17. Li DY, Yang BC, Mehta JL. Oxidized low density lipoprotein inducesapoptosis in cultured human coronary artery endothelial cells: role ofPKC, PTK, bcl-2, and Fas.Am J Physiol. 1998;275:H568–H576.
18. Jialal I, Devaraj S. The role of oxidized low density lipoprotein inatherogenesis. J Nutr. 1996;126:1053S–1057S.
19. Maziere C, Auclair M, Djavaheri-Mergny M, et al. Oxidized low densitylipoprotein induces activation of the transcription factor NF-kB in fibro-blasts, endothelial and smooth muscle cells.Biochem Mol Biol Int.1996;39:1201–1207.
20. Hernan dez-Presa M, Bustos C, Ortego M, et al. Angiotensin-convertingenzyme inhibition prevents arterial nuclear factor-kB activation,monocyte chemoattractant protein-1 expression, and macrophage infil-tration in a rabbit model of early accelerated atherosclerosis.Circulation.1997;95:1532–1541.
21. Nickenig G, Bohm M. Regulation of the angiotensin AT1R receptorexpression by hypercholesterolemia.Eur J Med Res. 1997;2:285–289.
22. Keidar S, Kaplan M, Hoffman A, et al. Angiotensin II stimulates mac-rophage-mediated oxidation of low density lipoproteins.Atherosclerosis.1995;115:201–215.
23. Keidar S, Attias J. Angiotensin II injection into mice increases the uptakeof oxidized LDL by their macrophages via a proteoglycan-mediatedpathway.Biochem Biophys Res Commun. 1997;239:63–67.
24. Li DY, Zhang YC, Philips MI, et al. Upregulation of endothelial receptorfor oxidized low-density lipoprotein (LOX-1) in cultured human coronaryartery endothelial cells by angiotensin II type 1 receptor activation.CircRes. 1999;84:1043–1049.
25. Li DY, Yang BC, Mehta JL. Tumor necrosis factor-a enhances hypoxia-reox-ygenation-mediated apoptosis in cultured human coronary artery endothelialcells: critical role of protein kinase C.Cardiovasc Res. 1999;42:805–813.
26. Natarajan K, Singh S, Burke T Jr, et al. Caffeic acid phenethyl ester is apotent and specific inhibitor of activation of nuclear transcription factorNF-kB. Proc Natl Acad Sci U S A. 1996;93:9090–9095.
27. Nickenig G, Jung O, Strehlow K, et al. Hypercholesterolemia is asso-ciated with enhanced angiotensin AT1R-receptor expression.Am JPhysiol.1997;272:H2701–H2707.
28. Li D, Saldeen T, Mehta JL. Gamma-tocopherol decreases ox-LDL-mediated activation of nuclear factor-kappaB and apoptosis in humancoronary artery endothelial cells.Biochem Biophys Res Commun. 1999;259:157–161.
29. Mehta JL, Li DY. Identification and autoregulation of receptor forox-LDL in cultured human coronary artery endothelial cells.BiochemBiophys Res Commun. 1998;298:511–514.
30. Yang BC, Phillips MI, Mohuczy D, et al. Increased angiotensin II type 1receptor expression in hypercholesterolemic atherosclerosis in rabbits.Arterioscler Thromb Vasc Biol. 1998;18:1433–1439.
31. Baeuerle PA. The inducible transcription activator NF-kB: regulation bydistinct protein subunits.Biochim Biophys Acta. 1991;1072:63–80.
32. Baeuerle PA, Baltimore D. IkB: a specific inhibitor of the NF-kB tran-scription factor.Science. 1998;242:540–546.
33. Collins T. Endothelial nuclear factor-kB and the initiation of the athero-sclerotic lesion.Lab Invest. 1993;68:499–508.
34. Pelzer T, Schumann M, Neumann M, et al. 17 Beta-estradiol preventsprogrammed cell death in cardiac myocytes.Biochem Biophys ResCommun.2000;268:192–200.
35. DeMeester SL, Buchman TG, Qiu Y, et al. Heat shock induces I kappaB-alpha and prevents stress-induced endothelial cell apoptosis. ArchSurg. 1997;132:1283–1287.
36. Islam KN, Devaraj S, Jialal I.a-Tocopherol enrichment of monocytesdecreases agonist-induced adhesion to human endothelial cells.Circu-lation. 1998;98:2255–2261.
37. Dimmeler S, Rippmann V, Weiland U, et al. Angiotensin II inducesapoptosis of human endothelial cells: protective effect of nitric oxide.Circ Res. 1997;81:970–976.
1976 Circulation October 17, 2000
by guest on September 25, 2014http://circ.ahajournals.org/Downloaded from




















