
Capsular Transformation of a Multidrug‐Resistant Streptococcus pneumoniae In Vivo
Upload
niperraebareliCategory
view
4download
0
Biomedicine & Pharmacotherapy 65 (2011) 387–394
Original article
Overcoming multidrug resistance (MDR) in cancer in vitro and in vivo by aquinoline derivative
Avishek Ganguly a, Kaushik Banerjee a, Paramita Chakraborty a, Satyajit Das a, Avijit Sarkar b,Abhijit Hazra c, Maitrayee Banerjee c, Arindam Maity c, Mitali Chatterjee b, Nirup B. Mondal c,Soumitra Kumar Choudhuri a,*a Department of In Vitro Carcinogenesis and Cellular Chemotherapy, Chittaranjan National Cancer Institute, 37, S.P. Mukherjee Road, Calcutta700 026, Indiab Department of Pharmacology, Institute of Post Graduate Medical Education and Research, Calcutta, Indiac Steroid and Terpenoid chemistry division, Indian institute of Chemical Biology, Jadavpur, Calcutta, India
A R T I C L E I N F O
Article history:
Received 22 March 2011
Accepted 23 April 2011
Available online 12 June 2011
Keywords:
Multidrug resistance
Quinoline derivatives
Apoptosis
A B S T R A C T
Multidrug resistance (MDR) mediated by the over expression of drug efflux protein P-glycoprotein (P-gp)
is one of the major impediments to successful treatment of cancer. P-gp acts as an energy-dependent
drug efflux pump and reduces the intracellular concentration of structurally unrelated drugs inside the
cells. Therefore, there is an urgent need for development of new compound that are less toxic and
effective against drug resistance in cancer. Preclinical studies have shown that quinoline derivatives
possess anticancer activities. Here, we report the antitumor potential of quinoline derivative, 2-(2-
Methyl-quinolin-4ylamino)-N-phenyl acetamide (S4). To evaluate the cytotoxic potential of S4, we used
four different cell lines (Hela, HCT-116, CCRF-CEM, and CEM/ADR 5000) in vitro, and showed that S4 kills
doxorubicin resistant T lymphoblastic leukemia cell, CEM/ADR 5000 in a concentration dependent
manner while others remains unaffected. Moreover, S4 induces apoptosis in CEM/ADR 5000 cells through
generation reactive oxygen species (ROS). This is substantiated by the fact that the antioxidant N-acetyle-
cysteine (NAC) completely blocks ROS generation and, subsequently, abrogates S4 induced apoptosis.
Furthermore, in vivo treatment with S4 significantly increases the life span of swiss albino mice bearing
sensitive and doxorubicin resistant subline of Ehrlich ascites carcinoma. In addition, intraperitoneal
application of S4 in mice does not show any systemic toxicity at concentrations that in preliminary trials
in a mice Ehrlich ascites carcinoma model. Therefore, present report provides evidence that S4, a
quinoline derivative, may be a promising new therapeutic agent against drug resistant cancers.
� 2011 Elsevier Masson SAS. All rights reserved.
1. Introduction
Search for new chemopreventive and antitumor agents that aremore effective and less toxic has kindled great interest in medicinalchemistry [1]. Several authors have reported that numerousquinoline derivatives show strong antiproliferative activity andinduce apoptosis in various cancer cell lines [1,2]. All these studiesstrongly support the potential therapeutic applications of quinolinederivatives, making them attractive for their further evaluation asnovel therapeutic agents for cancer treatment. Conventional cancerchemotherapy is seriously limited by tumor cells exhibiting
Abbreviations: MDR, Multidrug resistance; ROS, Reactive oxygen species; S4, 2-(2-
Methyl-quinolin-4ylamino)-N-phenyl acetamide; MTT, (3-[4,5-dimethylthiazol- 2-
yl]-2, 5-diphenyltetrazolium bromide; NAC, N acetyl cystein; DCFDA, 20 , 70-
dihydrodichlorofluorescin diacetate.
* Corresponding author. Tel.: +91 33 2476 5101/02/04x332;
fax: +91 33 2475 7606.
E-mail address: [email protected] (S.K. Choudhuri).
0753-3322/$ – see front matter � 2011 Elsevier Masson SAS. All rights reserved.
doi:10.1016/j.biopha.2011.04.024
multidrug resistance (MDR), a phenotype of cross-resistance tocytostatic or cytotoxic actions of multiple, structurally dissimilarand functionally divergent drugs commonly used in cancerchemotherapy [3]. Although the underlying mechanisms arediverse, the role of ATP-dependent drug efflux-proteins on the cellmembrane is accepted as a major cause behind MDR. Theinvolvement of multiple mechanisms of MDR, particularly in tumorsthat comprise a heterogeneous population of cells, makes thedevelopment of effective therapies for multidrug resistant cancer aformidable task [4]. A number of tumor cells have been reported toundergo apoptotic cell death when treated with such chemothera-peutic agents as etoposide, camptothecin, cisplatin, 1-b-D-arabi-nofuranosyl cytosine (Ara-C), mitomycin C, adriamycin, andvincristine. The findings indicate that apoptosis in tumor cells playsa critical role in chemotherapy-induced tumor cell killing and alsosuggest that blockade of the apoptosis-inducing pathway could beanother mechanism for multidrug resistance (MDR) [5]. Although aplethora of agents have been developed that modify, modulate orreverse the P-gp-mediated MDR phenotype, unfortunately most of

A. Ganguly et al. / Biomedicine & Pharmacotherapy 65 (2011) 387–394388
these compounds are not useful to overcome the problem of drugresistance in clinical settings. In some cases, either there is a lack ofpotency or, alternatively, the MDR reversing agent may expose thepatient to unacceptable side-effects or toxicity at the doses requiredfor effectiveness. These limitations have impelled efforts to searchfor new compounds with low toxicity and high efficacy inmodulation of MDR [6].
Cancer cells, especially those in advanced disease stages, havebecome highly adapted to intrinsic oxidative stress with upregu-lated antioxidant capacity. This redox adaptation not only enablesthe cancer cells to survive under increased ROS stress, but alsoprovides a mechanism of resistance to many anticancer agents,owing to increased tolerance of exogenous stress, an upregulation ofsurvival molecules and increased capacity for drug inactivation [7].Redox regulation has been shown to be an important component ofmalignant cell survival. However, tipping the cellular redox balancethrough pharmacologic regulation in favour of increasing intracel-lular ROS and/or depleting protective reducing metabolites (such asglutathione [GSH] and nicotinamide adenine dinucleotide phos-phate) may lead to oxidative stress and resulting in induction ofapoptosis for the treatment of cancer [8]. Applying these notions, wehave reported earlier that iron chelate viz. iron N- (2-hydroxyacetophenone) glycinate (FeNG) overcome drug resistance byinvolving ROS for induction of apoptosis in CEM/ADR5000 cell line[9]. Therefore, agent that is capable to modulate intracellular redoxbalance may be a potential weapon to target MDR.
In the present communication, we report the apoptotic potentialof quinoline derivatives (S4) against drug resistant T lymphoblasticleukemia (CEM/ADR5000) in vitro and involvement of oxidativestress in S4 induced apoptosis. We have also described the antitumorproperty of S4 on in vivo Ehrlich ascite carcinoma cell and inaddition, we conducted a study on the systemic toxicity of S4 whenapplied in mice as a model system. The present report suggests thatthis compound displays properties as a potential therapeutic drug inthe treatment of drug resistance in cancer.
2. Materials and methods
2.1. Reagents
2-(2-Methyl-quinolin-4ylamino)-N-phenyl acetamide (S4),MTT dye (3-[4,5-dimethylthiazol- 2-yl]-2, 5-diphenyltetrazoliumbromide, N acetyl cystein (NAC) (Sigma Chemical Chompany, St.Louis, MO), propidiam iodide, 20, 70-dihydrodichlorofluorescindiacetate (H2-DCFDA, Molecular Probes), FITC-labeled Annexin V(BD Bioscience), DTNB (SRL India).
2.2. Synthesis of compound
The compound 2-(2-methylquinolin- 4-ylamino)-N-phenyla-cetamide (S4) was prepared as described earlier [10]. Briefly 2-Chloro-N-phenylacetamide, synthesized by reacting chloroacetylchloride with aniline, was condensed with 4-aminoquinaldine inthe presence of sodium hydride and dimethyl sulfoxide, giving riseto four products. The compounds were separated by columnchromatography over silica gel to furnish 2-(2-methylquinolin- 4-ylamino)-N-phenylacetamide.
2.3. Cell culture
The human T-cell acute lymphoblastic CEM/ADR5000 leukemiacell lines [11,12] were maintained in RPMI medium (GIBCOInvitrogen Corp., Carlsbad, California, USA) supplemented with10% fetal bovine serum (FBS), additional glutamine (0.15%), HEPES(25 mM) 66.67 mg/L penicillin and 100 mg/L streptomycin. Cells
were grown in plastic tissue culture flasks in a 5% CO2 atmosphere at37 8C. Cells were passages twice weekly. The doxorubicin resistantCEM/ADR5000 cell line was generated by treating CCRF-CEM cellswith doxorubicin doses up to a final concentration of 5000 ng/mldoxorubicin [13]. These cell lines were kindly provided by Prof TEfferth, University of Mainz, Germany. The CEM/ADR5000 specifi-cally overexpress P glycoprotein without concomitant over-expression of MRP1 or BCRP [14,15]. The human colorectalcarcinoma HCT 116 (wild-type p53 [wt-p53]) and human cervicalcarcinoma Hela cell lines were obtained from the American TypeCulture Collection. Cells were cultured in DMEM supplemented with10% FCS (invitrogen/life technologies), 2 mmol/L L-glutamine,66.67 mg/L penicillin, and 100 mg/L streptomycin (invitrogen/lifetechnologies). Cells from exponentially growing cultures were usedfor all experiments. All experiments were repeated three times.
2.4. Cytotoxicity assay (MTT assay)
The data generated were from three separate experiments, eachperformed in duplicate. Cell viability was determined using theMTT assay, which was carried out as described previously [16]with slight modification, briefly, cells were seeded in 96-wellplates at a density 4 � 104 of cells per well. For single-agentstudies, cells were seeded and allowed to settle for 24 hours beforetreatment with increasing concentrations of drug and incubate itfor further 72 hours with 5% CO2 at 37 8C. After completion ofincubation cells were incubated with 5 mg per ml of MTT dye (3-[4,5-dimethylthiazol- 2-yl]-2, 5-diphenyltetrazolium bromide;Sigma, France) for 4 hours at 37 8C. The monolayer was suspendedin 0.1 ml of DMSO and the absorbance at 540 nm was read by ELISAreader (Tecan 200). The control value corresponding to untreatedcells was taken as 100% and the viability of treated samples wereexpressed as a percentage of the control. The IC50 values weredetermined as the concentration that reduced cell viability by 50%.
2.5. Cell cycle analysis
Cell cycle analysis was studied by flow cytometry. In brief,5 � 105cells were seeded in 60 mm tissue culture plate and treatedwith drugs. At various time points, cells were recovered, washedtwice in PBS, fixed in 70% ethanol, and stored at 4 8C until analyzed.Immediately before analysis, cells were washed twice in PBS, andincubated with 250 mg/ml RNAse A for 30 minutes at roomtemperature, followed by staining with propidium iodide (PI,20 mg/ml final concentration) for 20 minutes incubation in dark.The cell cycle distribution and percentage of apoptotic cells weredetermined using a FACS calibur flow cytometer (Becton Dick-inson, USA). Ten thousand events were analyzed for each sample.Appropriate gating was used to select the single-cell population.The same gate was used on all samples, ensuring that themeasurements were made on a standardized cell population.
2.6. Annexin V binding assay
Staining the cells with Annexin V-FITC and propidium iodide(PI) can be used in a bivariate analysis to distinguish between cellsundergoing apoptosis (PI negative) and those that are necrotic ordead (PI positive). Cells (5 � 105) were incubated with FITC labeledAnnexin V and propidium iodide (PI) at room temperature for15 minutes in the dark and analyzed using a FACS Calibur(Becktone Dickinson).
2.7. Intracellular ROS accumulation study
Levels of ROS generation in cells were assessed fluorometricallyusing 20, 70-dihydrodichlorofluorescin diacetate (H2-DCFDA, Mo-

A. Ganguly et al. / Biomedicine & Pharmacotherapy 65 (2011) 387–394 389
lecular Probes). H2-DCFDA is a nonfluorescent, cell-permeantcompound. Endogenous esterases within the cell cleave the acetategroups, thus trapping the reduced form of the probe (DCHF)intracellularly. It is known that the probe can be readily oxidized toDCF by H2O2 or OH. Cells were treated with drug or left untreatedfor 1 to 6 hours. The cells were then washed with PBS and furtherincubated with H2-DCFDA for 30 minutes at 37 8C in dark. Afterincubation, cells were washed twice in PBS at room temperaturefor 5 minutes each time. The fluorescence was measured atexcitation and emission wavelengths of the oxidized form were504 and 529 nm respectively [9].
2.8. Caspase 3 activation assay
Cells washed in PBS and resuspended in 25 mM Hepes (pH 7.5),5 mM MgCl2, 5 mM EDTA, 5 mM dithiothreitol (DTT), 2 mMphenylmethylsulfonyl fluoride (PMSF), 10 mg/ml pepstatin A,and 10 mg/ml leupeptin after treatment. The kit (Caspasefluorometric assay system) was used to investigate caspaseactivity. Cells were lysed and clarified using centrifugation at 1,2000 g for 5 minutes. The clear lysates containing 50 mg of proteinwere incubated with 50 mM substrate Ac-DEVD-AMC at 30 8C for1 hour. Levels of released AMC were measured using a spectroflu-orometer (Varion) with excitation at 360 nm and emission at460 nm (Caspase Assay System, BD bioscience, USA).
2.9. Determination of intracellular GSH contents
Determination of cellular GSH content was performed by amodification of the method of [37]. Drug treated and untreated cells(5 � 105 cells) were washed three times with cold washing buffer(0.1 M sodium phosphate, 5 mM ethylenediaminetetraacetate[EDTA], pH 7.5) and immediately lysed in 100 ml lysis buffer(0.1% Triton-X, 0.1 M sodium phosphate, 5 mM EDTA, pH 7.5). Fiveminutes later, the lysates were acidified with 15 ml of 0.1 N HCl, andprotein was precipitated with 15 ml of 50% sulfosalicylic acid. Aftercentrifugation at 12,000 g for, supernatants were collected for GSHmeasurement. The supernatants were assayed for GSH content byspectrophotometric determination of the reduction of DTNB to 05-thio-2-nitrobenzonic acid at a wavelength of 412 nm [17].
2.10. Animals
Swiss albino mice, originally obtained from National Institute ofNutrition, Hyderabad, India and reared in the institute animalfacilities, were used for all in vivo experiments with prior approvalof the institutional animal ethics committee (IAEC). The experi-mental protocols described herein were approved by the IAEC
Table 1Effect of S4 on survivality of EAC/S and EAC/DOX bearing male Swiss albino mice (10
Group Drug
concentration
No. of
animals
Tot
(tum
flui
EAC/S Untreated control – 12 9.3
EAC/S treated 15 mg/kg of body weight 12 6.4
EAC/S treated 20 mg/kg of body weight 12 5.9
EAC/DOX untreated control – 12 10.2
EAC/DOX treated 15 mg/kg of body weight 12 8.8
EAC/DOX treated 20 mg/kg of body weight 12 8.3
S4 (at 15 and 20 mg/kg of body weight) increases the mean survival time (MST) of EAC/S
by dividing the MST value for mice receiving treatment by MST value for the control group
The total ascitic fluid (TAF) as well as the packed cell volume (PCV) also decreased as
mean � standard deviation (S.D.) for 12 mice in each group.
(Registration No.: 175/99/CPCSEA, dated 28.01.2000) in accor-dance with the ethical guidelines laid down by the Committee forthe purpose of control and supervision of experiments on animals(CPCSEA) by the Ministry of Social Justice and Empowerment,Government of India. Adult male Swiss albino mice weighing 18–20 g were kept for a quarantine period of 1 week at a temperatureof 25 � 2 8C, relative humidity of 55 � 2% and with photo cycle of 12-hour light/12-hour dark. Water and food pellets were provided ad
libitum.
2.11. In vivo chronic toxicity testing for S4
To define the subacute toxicity of S4, three experimental groupsof Swiss albino mice were treated i.p. with 0.75 mg/kg of bodyweight, 1.0 mg/kg body weight S4 or vehicle treated (controlgroup). There were six mice for each group. Treatment was done on5 days/week for 4 consecutive weeks. During the study period, themice were weighed twice a week. Clinical appearance of the micewas evaluated daily. After 4 weeks of continuous treatment, bloodwas collected from untreated as well as from treated mice. Bloodwas obtained via closed cardiac puncture with the help of a 22-guage hypodermic needle by subxiphoid approach. Blood fromeach group (0.75 mg/kg of body weight S4, 1.0 mg/kg body weightS4 treated vehicle treated) was pooled into separate glass tubesand treated with anticoagulant (heparin) or left untreated forserum collection. After that hematological parameters wereanalyzed by automated haematology analyzer (Sysmex, KX-21)and hematologic biochemical analyses (blood urea, creatinin,alkaline phosphatase, ALT, AST) was determined by automatedclinical chemistry analyzer (Olympus, AU400).
2.12. Cell line, tumor implantation and experimental protocol
EAC cell was maintained as an ascitic tumor in male Swissalbino mice. A Dox-resistant subline was developed by sequentialtransfer of Dox treated EAC cells to the subsequent generation ofhost mice with continuous Dox treatment [18,19]. Briefly, thetreatment regime consisted of 2 mg/kg per week Dox intraperito-neally (i.p.). The daily treatment dose was 0.4 mg/kg for 5 days.Dox was started 24 hours after inoculation of 106 EAC cells i.p. intomice. Six groups (each group containing six mice) of animals weretaken for animal survival studies. Each mouse was inoculated witheither 106 EAC/S cells (three groups) or 106 EAC/Dox cells (threegroups) i.p. 10 days later of i.p. inoculum 1.5 mg/kg of body weightor 2 mg/kg of body weight of S4 were injected (i.p.) daily forconsecutive 10 days to EAC/S or EAC/Dox bearing mice ofappropriate groups. Animals were checked daily for assessmentof ascitic growth and body weights were measured.
days after S4 injection).
al volume
or cells + ascite
d), mL
Total packed
cell volume, mL
Mean survival
time (MST)
T/C value (%)
� 2.1 5.5 � 1.4 35 � 3.3 100
� 1.6 3.9 � 0.98 52 � 3.9 148.6
� 1.08 3.5 � 0.81 53 � 4.1 151.4
� 1.9 6.2 � 1.5 30 � 2.8 100
� 1.6 5.3 � 1.1 41 � 3.1 136.7
� 0.9 5.0 � 0.78 39 � 2.9 130.0
and EAC/Dox cells bearing mice. T/C value (expressed in percentage) was calculated
, which comprised of EAC/S and EAC/Dox cells bearing mice receiving no treatment.
compared to untreated EAC/S and EAC/Dox cells bearing mice. Values represent
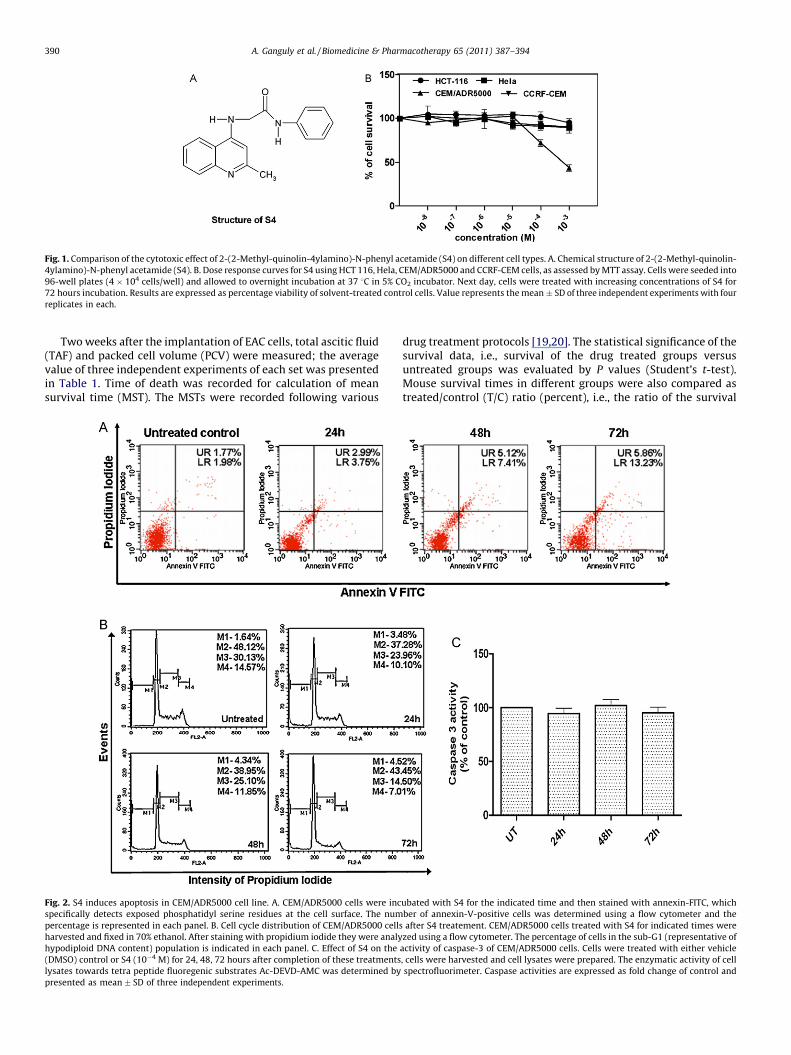
Fig. 1. Comparison of the cytotoxic effect of 2-(2-Methyl-quinolin-4ylamino)-N-phenyl acetamide (S4) on different cell types. A. Chemical structure of 2-(2-Methyl-quinolin-
4ylamino)-N-phenyl acetamide (S4). B. Dose response curves for S4 using HCT 116, Hela, CEM/ADR5000 and CCRF-CEM cells, as assessed by MTT assay. Cells were seeded into
96-well plates (4 � 104 cells/well) and allowed to overnight incubation at 37 8C in 5% CO2 incubator. Next day, cells were treated with increasing concentrations of S4 for
72 hours incubation. Results are expressed as percentage viability of solvent-treated control cells. Value represents the mean � SD of three independent experiments with four
replicates in each.
A. Ganguly et al. / Biomedicine & Pharmacotherapy 65 (2011) 387–394390
Two weeks after the implantation of EAC cells, total ascitic fluid(TAF) and packed cell volume (PCV) were measured; the averagevalue of three independent experiments of each set was presentedin Table 1. Time of death was recorded for calculation of meansurvival time (MST). The MSTs were recorded following various
Fig. 2. S4 induces apoptosis in CEM/ADR5000 cell line. A. CEM/ADR5000 cells were inc
specifically detects exposed phosphatidyl serine residues at the cell surface. The num
percentage is represented in each panel. B. Cell cycle distribution of CEM/ADR5000 cells
harvested and fixed in 70% ethanol. After staining with propidium iodide they were analy
hypodiploid DNA content) population is indicated in each panel. C. Effect of S4 on the
(DMSO) control or S4 (10�4 M) for 24, 48, 72 hours after completion of these treatments
lysates towards tetra peptide fluoregenic substrates Ac-DEVD-AMC was determined by
presented as mean � SD of three independent experiments.
drug treatment protocols [19,20]. The statistical significance of thesurvival data, i.e., survival of the drug treated groups versusuntreated groups was evaluated by P values (Student’s t-test).Mouse survival times in different groups were also compared astreated/control (T/C) ratio (percent), i.e., the ratio of the survival
ubated with S4 for the indicated time and then stained with annexin-FITC, which
ber of annexin-V-positive cells was determined using a flow cytometer and the
after S4 treatement. CEM/ADR5000 cells treated with S4 for indicated times were
zed using a flow cytometer. The percentage of cells in the sub-G1 (representative of
activity of caspase-3 of CEM/ADR5000 cells. Cells were treated with either vehicle
, cells were harvested and cell lysates were prepared. The enzymatic activity of cell
spectrofluorimeter. Caspase activities are expressed as fold change of control and

A. Ganguly et al. / Biomedicine & Pharmacotherapy 65 (2011) 387–394 391
time (in days) for treated mice to untreated control mice. As instandard National Cancer Institute protocols for screening newanticancer drugs, it was considered that the increase in survivaltime corresponding to T/C ratios around 120% to ‘‘marginal’’, T/Cratios between 120 and 150% to be ‘‘clear’’ and T/C ratios equal orsuperior to 150% to be ‘‘marked’’.
2.13. Statistical analysis
All data reported are the arithmetic mean from threeindependent experiments performed in triplicate �SD unlessstated otherwise. The unpaired Student’s t-test was used to evaluatethe significance differences between groups, accepting P < 0.05 as alevel of significance. Data analyses were performed using the Prismsoftware (GraphPad, San Diego, CA).
3. Result
3.1. S4 induces cytotoxicity in CEM/ADR 5000 cell line
We tested the cytotoxic effects of S4 (Fig. 1A) on four differenthuman carcinoma cell lines (CCRF-CEM, HCT-116, CEM/ADR 5000,Hela). Cell lines were treated with a range of concentrations (10�8
to 10�3 M) of S4 for 72 hours, or vehicle and then cell viability wasdetermined by MTT reduction assay (Fig. 1B). S4 induced growthinhibitory effect occurred in dose dependent manner in CEM/ADR5000 cell line with IC50 value 8.4 � 10�4 M (at 72 hourstreatment). However, under the same condition, S4 didn’t displaycytotoxic effect on any of other cell lines at given experimentalconcentration range. The result presented in the Fig. 1B showedthat S4 induces approx. Fifty-six percent reduction in cell viability
Fig. 3. S4 induces oxidative stress. (A) CEM/ADR5000 and (B) CCRF-CEM cells were eithe
measured (in terms of peroxide using dichlorofluorescein diacetate [DCF-DA]) as descr
percent of control and are presented as mean � SD of three independent experiments.
***P < 0.001, by unpaired Student’s t test. (C) S4 depletes intracellular glutathion (GSH) cont
treated with S4 (10�4 M) for indicated time points and intracellular GSH was measured as
independent experiments. Differences between untreated control and S4 treated cells are
after 72 hours treatment in CEM/ADR5000 cell line as compared tovehicle treated control cell. Thus Collateral sensitivities of CEM/ADR5000 cells were observed towards S4 as parental sensitive cellsCCRF-CEM remains unaffected after S4 treatment. ‘‘Collateralsensitivity’’ (CS) is a phenomenon where a resistant cancer cellshows hypersensitivity of to other drugs.
3.2. S4 induces apoptosis in CEM/ADR5000 cell lines without involving
caspase 3
Early cellular changes in apoptosis are characterized by thetranslocation of phosphatidylserine (PS) to the external surface ofthe plasma membrane where it can be detected by binding toannexin V-FITC. As cell membrane is further compromised and celldeath occurs, cellular DNA becomes accessible for staining with PI[21]. The flow cytometric analysis showed that (Fig. 2A), the CEM/ADR5000 cells that had been incubated with S4 for 24, 48 and72 hours and dual stained with annexin V-FITC and PI, there was aprogressive increase in the annexin V-FITC positive population ofcells (3.75, 7.41, 13.23% for 24, 48, 72 hours respectively) in atemporal manner as compared to untreated control (1.98%).
In addition, S4 induced apoptosis was also determined by cellcycle analysis of PI stained CEM/ADR5000 cell line by flowcyto-metry after S4 treatment. It was found that (Fig. 2B), the increase inthe counts of sub diploidal (sub G1/G0) cells in a time dependentway as compared to untreated control. Moreover, no significantchanges in cell cycle distribution were observed in CEM/ADR5000cells exposed to S4. Taken together, these results indicated that celldeath induced by S4 was mainly caused by apoptosis.
Activation of caspase 3 plays the central role in the initiation ofapoptosis and is a hallmark of apoptosis in response to death
r kept untreated or treated with S4 (10�4 M) and intracellular ROS generation was
ibed under Materials and Methods at different time points. Data are expressed as
Differences between control and S4 treated cells are significant *P < 0.05, **P < 0.01,
ents of CEM/ADR5000 cells and (D) CCRF-CEM cells. Cells were either kept untreated or
described under Materials and Methods. Results are presented as mean � SD of three
significant **P < 0.01 by unpaired Student’s t test.
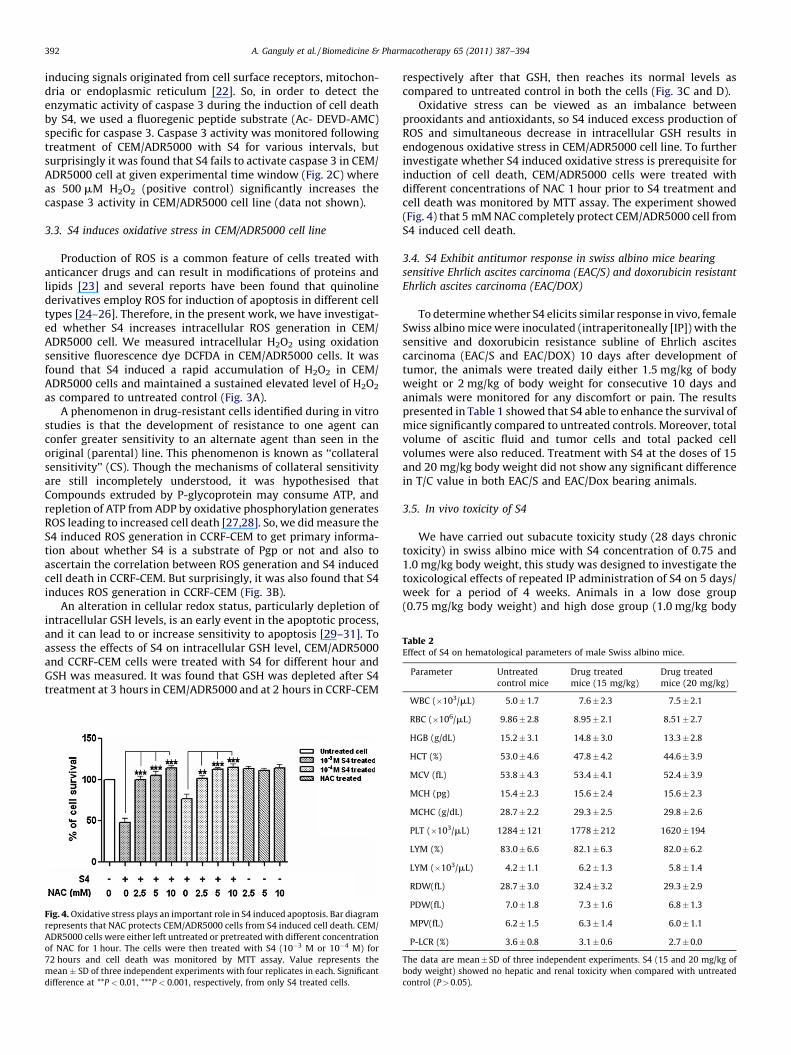
Table 2Effect of S4 on hematological parameters of male Swiss albino mice.
Parameter Untreated
control mice
Drug treated
mice (15 mg/kg)
Drug treated
mice (20 mg/kg)
3
A. Ganguly et al. / Biomedicine & Pharmacotherapy 65 (2011) 387–394392
inducing signals originated from cell surface receptors, mitochon-dria or endoplasmic reticulum [22]. So, in order to detect theenzymatic activity of caspase 3 during the induction of cell deathby S4, we used a fluoregenic peptide substrate (Ac- DEVD-AMC)specific for caspase 3. Caspase 3 activity was monitored followingtreatment of CEM/ADR5000 with S4 for various intervals, butsurprisingly it was found that S4 fails to activate caspase 3 in CEM/ADR5000 cell at given experimental time window (Fig. 2C) whereas 500 mM H2O2 (positive control) significantly increases thecaspase 3 activity in CEM/ADR5000 cell line (data not shown).
3.3. S4 induces oxidative stress in CEM/ADR5000 cell line
Production of ROS is a common feature of cells treated withanticancer drugs and can result in modifications of proteins andlipids [23] and several reports have been found that quinolinederivatives employ ROS for induction of apoptosis in different celltypes [24–26]. Therefore, in the present work, we have investigat-ed whether S4 increases intracellular ROS generation in CEM/ADR5000 cell. We measured intracellular H2O2 using oxidationsensitive fluorescence dye DCFDA in CEM/ADR5000 cells. It wasfound that S4 induced a rapid accumulation of H2O2 in CEM/ADR5000 cells and maintained a sustained elevated level of H2O2
as compared to untreated control (Fig. 3A).A phenomenon in drug-resistant cells identified during in vitro
studies is that the development of resistance to one agent canconfer greater sensitivity to an alternate agent than seen in theoriginal (parental) line. This phenomenon is known as ‘‘collateralsensitivity’’ (CS). Though the mechanisms of collateral sensitivityare still incompletely understood, it was hypothesised thatCompounds extruded by P-glycoprotein may consume ATP, andrepletion of ATP from ADP by oxidative phosphorylation generatesROS leading to increased cell death [27,28]. So, we did measure theS4 induced ROS generation in CCRF-CEM to get primary informa-tion about whether S4 is a substrate of Pgp or not and also toascertain the correlation between ROS generation and S4 inducedcell death in CCRF-CEM. But surprisingly, it was also found that S4induces ROS generation in CCRF-CEM (Fig. 3B).
An alteration in cellular redox status, particularly depletion ofintracellular GSH levels, is an early event in the apoptotic process,and it can lead to or increase sensitivity to apoptosis [29–31]. Toassess the effects of S4 on intracellular GSH level, CEM/ADR5000and CCRF-CEM cells were treated with S4 for different hour andGSH was measured. It was found that GSH was depleted after S4treatment at 3 hours in CEM/ADR5000 and at 2 hours in CCRF-CEM
Fig. 4. Oxidative stress plays an important role in S4 induced apoptosis. Bar diagram
represents that NAC protects CEM/ADR5000 cells from S4 induced cell death. CEM/
ADR5000 cells were either left untreated or pretreated with different concentration
of NAC for 1 hour. The cells were then treated with S4 (10�3 M or 10�4 M) for
72 hours and cell death was monitored by MTT assay. Value represents the
mean � SD of three independent experiments with four replicates in each. Significant
difference at **P < 0.01, ***P < 0.001, respectively, from only S4 treated cells.
respectively after that GSH, then reaches its normal levels ascompared to untreated control in both the cells (Fig. 3C and D).
Oxidative stress can be viewed as an imbalance betweenprooxidants and antioxidants, so S4 induced excess production ofROS and simultaneous decrease in intracellular GSH results inendogenous oxidative stress in CEM/ADR5000 cell line. To furtherinvestigate whether S4 induced oxidative stress is prerequisite forinduction of cell death, CEM/ADR5000 cells were treated withdifferent concentrations of NAC 1 hour prior to S4 treatment andcell death was monitored by MTT assay. The experiment showed(Fig. 4) that 5 mM NAC completely protect CEM/ADR5000 cell fromS4 induced cell death.
3.4. S4 Exhibit antitumor response in swiss albino mice bearing
sensitive Ehrlich ascites carcinoma (EAC/S) and doxorubicin resistant
Ehrlich ascites carcinoma (EAC/DOX)
To determine whether S4 elicits similar response in vivo, femaleSwiss albino mice were inoculated (intraperitoneally [IP]) with thesensitive and doxorubicin resistance subline of Ehrlich ascitescarcinoma (EAC/S and EAC/DOX) 10 days after development oftumor, the animals were treated daily either 1.5 mg/kg of bodyweight or 2 mg/kg of body weight for consecutive 10 days andanimals were monitored for any discomfort or pain. The resultspresented in Table 1 showed that S4 able to enhance the survival ofmice significantly compared to untreated controls. Moreover, totalvolume of ascitic fluid and tumor cells and total packed cellvolumes were also reduced. Treatment with S4 at the doses of 15and 20 mg/kg body weight did not show any significant differencein T/C value in both EAC/S and EAC/Dox bearing animals.
3.5. In vivo toxicity of S4
We have carried out subacute toxicity study (28 days chronictoxicity) in swiss albino mice with S4 concentration of 0.75 and1.0 mg/kg body weight, this study was designed to investigate thetoxicological effects of repeated IP administration of S4 on 5 days/week for a period of 4 weeks. Animals in a low dose group(0.75 mg/kg body weight) and high dose group (1.0 mg/kg body
WBC (�10 /mL) 5.0 � 1.7 7.6 � 2.3 7.5 � 2.1
RBC (�106/mL) 9.86 � 2.8 8.95 � 2.1 8.51 � 2.7
HGB (g/dL) 15.2 � 3.1 14.8 � 3.0 13.3 � 2.8
HCT (%) 53.0 � 4.6 47.8 � 4.2 44.6 � 3.9
MCV (fL) 53.8 � 4.3 53.4 � 4.1 52.4 � 3.9
MCH (pg) 15.4 � 2.3 15.6 � 2.4 15.6 � 2.3
MCHC (g/dL) 28.7 � 2.2 29.3 � 2.5 29.8 � 2.6
PLT (�103/mL) 1284 � 121 1778 � 212 1620 � 194
LYM (%) 83.0 � 6.6 82.1 � 6.3 82.0 � 6.2
LYM (�103/mL) 4.2 � 1.1 6.2 � 1.3 5.8 � 1.4
RDW(fL) 28.7 � 3.0 32.4 � 3.2 29.3 � 2.9
PDW(fL) 7.0 � 1.8 7.3 � 1.6 6.8 � 1.3
MPV(fL) 6.2 � 1.5 6.3 � 1.4 6.0 � 1.1
P-LCR (%) 3.6 � 0.8 3.1 � 0.6 2.7 � 0.0
The data are mean � SD of three independent experiments. S4 (15 and 20 mg/kg of
body weight) showed no hepatic and renal toxicity when compared with untreated
control (P > 0.05).

Table 3Effect of S4 on hepatic and renal functions of male Swiss albino mice.
Group Alkaline phosphatase (U/L) SGOT (U/L) SGPT (U/L) Urea (mg/dL) Creatinin (mg/dL)
Untreated control mice 60 � 3.6 216 � 7.3 59 � 2.8 51 � 2.3 0.3 � 0.009
Drug treated mice (15 mg/kg) 119 � 5.1 234 � 7.7 73 � 3.1 63 � 2.6 0.4 � 0.02
Drug treated mice (20 mg/kg) 107 � 5.8 212 � 6.9 60 � 2.3 80 � 3.1 0.4 � 0.023
The data are mean � SD of three independent experiments. S4 (15 and 20 mg/kg of body weight) showed no hematological toxicity when compared with untreated control
(P > 0.05).
A. Ganguly et al. / Biomedicine & Pharmacotherapy 65 (2011) 387–394 393
weight) did not reveal any pathological changes as compared to thecontrol group of animals. No mortality or toxic symptoms wereobserved in test and control groups of animals. No significantdifferences were observed in the body weight gain/loss pattern,organ weight, hematological or biochemical parameters (seeMaterial and Methods) of all the test groups when compared tothe control group (Tables 2 and 3).
4. Discussion
In this present communication, we report the anticancerproperty of a quinoline derivative, 2-(2-Methyl-quinolin-4yla-mino)-N-phenyl acetamide (S4). Although several biologicalproperties of S4 have been documented [10,32,33], no study hasbeen performed regarding cytotoxic potential of this compound. Totest the cytotoxic potential of S4, we choose four different cell linesincluding one drug resistant lymphoblastic leukemia cell line CEM/ADR5000 but surprisingly, all of the cell lines were foundunresponsive towards S4 except CEM/ADR5000. The compoundshowed concentration dependent reduction of cell viability inCEM/ADR5000 cell line and, on the contrary, the parental sensitiveline CCRF/CEM was totally unaffected within stipulated experi-mental concentration and time frame. As multidrug-resistant cellswere not cross-resistant to the compound S4 the phenomenon‘‘collateral sensitivity’’ might not be ruled out, although themechanistic basis of collateral sensitivity towards this compoundis unknown. There is a growing realization that chemotherapeuticagents act primarily by inducing cancer cell death throughinvolvement of apoptosis [34]. Apoptotic cell death is character-ized by several hallmarks such as phosphatidyl serine exposure toplasma membrane surface, cell shrinkage and DNA fragmentation,which distinguish it from necrosis, another type of cell death. Toinvestigate the mode of cell death involved in S4 inducedcytotoxicity, we carried out annexin V/PI binding assay; we havefound that S4 increases the percentage of annexin V positivepopulation in time dependent manner whereas the percentage ofboth annexin V and PI positive population remains negligible astime progressed. These results clearly indicate that S4 kills CEM/ADR5000 cells through induction of apoptosis. Further studies oncell cycle analysis of CEM/ADR5000 cells after S4 treatment revealsan increase in the sub-diploidal population which represent cellswith significant DNA damage indicating a late apoptotic stage withrespect to cycling cells. However, S4 does not affect cell cyclecheckpoints in CEM/ADR5000 cell line within experimental timeframe. The caspase protease family plays a central role in apoptoticcell death especially caspase-3, known to be critically involved inthe execution phase of apoptosis, [35,36]. But we fail to determinethe activation of caspase 3 in S4 induced apoptosis in CEM/ADR5000 cells. Although activation of caspase-3 is generallyregarded as essential for apoptosis to proceed, realization of theapoptotic program has also been observed in the essential absenceof caspase-3 or in the presence of its inhibitors [37]. Several reportshas been documented that quinoline derivatives itself are capableto inhibit caspase 3 in different mechanistic ways [25,38,39],
therefore, inhibitory role of S4 on activation of caspase 3 cannot beprecluded.
Free radicals, particularly reactive oxygen species (ROS), havebeen proposed as common mediators for apoptosis. Many agentsthat induce apoptosis are either oxidants or stimulators of cellularoxidative metabolism. Oxidative stress may cause a shift in cellularredox state that ultimately causes apoptosis [40]. Quinolinederivatives are such compounds that are known to associate withROS generation in induction of apoptosis in many cell lines [24–26]. Moreover, substances that are causing collateral sensitivitywhen extruded by P-glycoprotein may consume ATP, and repletionof ATP from ADP by oxidative phosphorylation generates ROSleading to increased cell death [41]. These observations promptedus to enquire whether ROS is involved in the mechanism of actionof S4 induced apoptosis. Our data discloses that S4 induces celldeath in CEM/ADR5000 although it generates ROS and depletesGSH in both CCRF-CEM and CEM/ADR5000. Moreover, NAC blocksthe ROS production and abrogates at least in part, S4 inducedapoptosis in CEM/ADR5000 cells. So this result clearly indicatesthat the generation of ROS and intracellular GSH depletion mayexert an additive effect to cause cellular redox imbalance oroxidative stress that may play a pivotal role in S4 inducedapoptosis in CEM/ADR5000 whereas S4 fails to induce cell death inCCRF-CEM as this cell well tolerated such amount of oxidativeinsult generated by S4. In addition, as S4 generates greater amountof ROS in CCRF-CEM than CEM/ADR5000, we may infer that S4 maynot be a substrate for P-glycoprotein and it supports the indirect P-glycoprotein mediated collateral sensitivity where the drug is notbe a substrate or inhibitor of P-glycoprotein but functional P-glycoprotein is needed for their activity. Therefore, our in vitro dataindicates that S4 causes apoptosis in drug resistance CEM/ADR5000 cells, but couldn’t affect the parental CCRF-CEM and,thus, support the phenomenon of collateral sensitivity.
In addition with this, we can also conclude in agreement withour in vitro data, the proof of concept study carried out in apreclinical model of Ehrlich ascites carcinoma shows that S4 iscapable of favourably influencing the course of the disease evenwhen administered under a ‘‘therapeutic’’ regime to mice that havealready developed tumor. S4 significantly increases the life spanand showing almost equal effects on T/C values of EAC/S and EAC/DOX bearing mice.
Moreover, we conducted a standardized study (28-day chronictoxicity) on the systemic toxicity of S4 when applied in mice as amodel system. Intraperitoneal administration of S4 did not showany systemic toxicity at the concentration (15 and 20 mg/kg bodyweight) that in our preliminary treatment trials of S4 in a miceEhrlich ascites carcinoma cell model. This observation proves thenon-toxic nature of S4, which is the primary criteria and importantstep for anticancer drugs development.
In conclusion, our in vitro data discloses that S4 preferentiallykills drug resistance CEM/ADR5000 cells through induction ofapoptosis but left parental CCRF-CEM unaffected and, thus,support the phenomenon of collateral sensitivity. Moreover, S4induced oxidative stress plays a pivotal role for cell death of drug

A. Ganguly et al. / Biomedicine & Pharmacotherapy 65 (2011) 387–394394
resistance cells. Furthermore, in vivo treatment with S4 signifi-cantly increases the life span of Swiss albino mice bearing sensitiveand doxorubicin resistant subline of Ehrlich ascites carcinoma. Inaddition, S4 does not show any systemic toxicity at concentrationsthat in preliminary trials in a mice Ehrlich ascites carcinomamodel. Therefore, present report provides evidence that a non-toxic quinoline derivative S4 is a growth-suppressing agent in vitrofor drug-resistant cell and may be a promising new therapeuticagent against drug resistant cancers.
Disclosure of interest
The authors declare that they have no conflicts of interestconcerning this article.
Acknowledgments
This investigation received financial support from Indian Councilof Medical Research (ICMR), New Delhi, no. 3/2/2/200/2009NCD-III.The funders had no role in study design, data collection and analysis,decision to publish, or preparation of the manuscript.
References
[1] Via LD, Gia O, Gasparotto V, et al. Discovery of a new anilino-3H-pyrrolo[3,2-f]quinoline derivative as potential anti-cancer agent. Eur J Med Chem2008;43:429–34.
[2] Hawtin RE, Stockett DE, Byl JAW, et al. Voreloxin is an anticancer quinolonederivative that intercalates DNA and poisons topoisomerase II. PLoS ONE2010;5(4):e10186.
[3] Ye W, Chang HL, Wang LS, et al. Modulation of multidrug-resistance geneexpression in human breast cancer cells by (–)-gossypolenriched cottonseedoil. Anticancer Res 2007;27:107–16.
[4] Liscovitch M, Lavie Y. Cancer multidrug resistance: a review of recent drugdiscovery research. I Drugs 2002;5:1–7.
[5] Tsuruo T, Naito M, Tomida A, et al. Molecular targeting therapy of cancer: drugresistance, apoptosis and survival signal. Cancer Sci 2003;94:15–21.
[6] Chou TC, Depew KM, Zheng YH, et al. Reversal of anticancer multidrugresistance by the ardeemins. Proc Natl Acad Sci 1998;95:8369–74.
[7] Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS mediatedmechanisms: a radical therapeutic approach? Nat Rev Drug Discov2009;8:579–91.
[8] Engel RH, Evens AM. Oxidative stress and apoptosis: a new treatment para-digm in cancer. Frontiers Biosci 2006;11:300–12.
[9] Ganguly A, Basu S, Chakraborty P, et al. Targeting mitochondrial cell deathpathway to overcome drug resistance with a newly developed iron chelate.PLoS ONE 2010;5(6):e11253.
[10] Sahu NP, Pal C, Mandal NB, et al. Synthesis of a novel quinoline derivative, 2-(2-Methylquinolin-4-ylamino)-N-phenylacetamide – a potential antileishmanialagent. Bioorg Med Chem 2002;10:1687–93.
[11] Mookerjee A, Basu JM, Dutta P, et al. Overcoming drug-resistant in cancer by anewly developed copper chelate through host protective cytokine mediatedapoptosis. Clin Cancer Res 2006;12:4339–49.
[12] Majumder S, Dutta P, Mukherjee P, et al. Reversal of drug resistance in P-glycoprotein-expressing T-cell acute lymphoblastic CEM leukemia cells bycopper N-(2-hydroxy acetophenone) glycinate and oxalyl bis (N-phenyl)hydroxamic acid. Cancer Lett 2006;244:16–23.
[13] Kimmig A, Gekeler V, Neumann M, et al. Susceptibility of multidrug-resistanthuman leukemia cell lines to human interleukin 2-activated killer cells. CancerRes 1990;50:6793–9.
[14] Gillet JP, Efferth T, Steinbach D, et al. Microarray-based detection of multidrugresistance in human tumor cells by expression profiling of ATP-bindingcassette transporter genes. Cancer Res 2004;64:8987–93.
[15] Efferth T, Sauerbrey A, Olbrich A, et al. Molecular modes of action of artesunatein tumor cell lines. Mol Pharmacol 2003;64:382–94.
[16] Muscella A, Greco S, Elia MG, et al. Angiotensin II stimulation of Na/KATPaseactivity and cell growth by calcium-independent pathway in MCF-7 breastcancer cells. J Endocrinol 2002;173:315–23.
[17] Mookerjee A, Basu JM, Majumder S, et al. A novel copper complex induces ROSgeneration in doxorubicin resistant Ehrlich ascitis carcinoma cells andincreases activity of antioxidant enzymes in vital organs in vivo. BMC Cancer2006;6:267–77.
[18] Friche E, Danks MK, Beck WT. Characterization of tumor cell resistance to 4-deoxy-4-iododoxorubicin developed in Ehrlich Ascites cells in vitro. CancerRes 1992;52:5701–6.
[19] Choudhuri SK, Chatterjee A. Reversal of resistance against doxorubicin by anewly developed compound, oxalyl bis(N-phenyl) hydroxamic acid in vitro.Anticancer Drugs 1998;9:825–32.
[20] Tsuoro T, Iisida H, Tsukagoshi S, et al. Overcoming vincristine resistance in P-388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine,vinblastin by verapamil. Cancer Res 1981;41:1967–72.
[21] Vermes I, Haanen C, Steens-Nakken H, et al. A novel assay for apoptosis, flowcytometric detection of hosphatidylserine expression on early apoptotic cellsusing fluorescein labelled Annexin V. J Immunol Methods 1995;184:39–51.
[22] Yang L, Wu S, Zhang Q, et al. 23,24-Dihydrocucurbitacin B induces G2/M cell-cycle arrest and mitochondria-dependent apoptosis in human breast cancercells (Bcap37). Cancer Lett 2007;256:267–78.
[23] Chandra J. Oxidative stress by targeted agents promotes cytotoxicity inhematologic malignancies. Antioxid Redox Signal 2009;11:1123–37.
[24] Mizutani H, Tada-Oikawa S, Hiraku Y, et al. Mechanism of apoptosis inducedby a new topoisomerase inhibitor through the generation of hydrogen perox-ide. J Biol Chem 2002;277:30684–9.
[25] Du JQ, Wu J, Zhang HJ, et al. Isoquinoline-1,3,4-trione derivatives inactivateCaspase-3 by generation of reactive oxygen species. J Biol Chem2008;283:30205–1.
[26] Soha Y, Shin MH, Lee JS, et al. Oxidative DNA damage and glioma cell deathinduced by tetrahydropapaveroline. Mutat Res 2003;544:129–42.
[27] Hall MD, Handley MD, Gottesman MM. Is resistance useless? Multidrugresistance and collateral sensitivity. Trends Pharmacol Sci 2009;30:546–56.
[28] Horwedel C, Tsogoeva SB, Wei S, et al. Cytotoxicity of artesunic acid homo- andheterodimer molecules toward sensitive and multidrug-resistant CCRF-CEMleukemia cells. J Med Chem 2010;53:4842–8.
[29] Ikeda T, Sporn M, Honda T, et al. The novel triterpenoid CDDO and itsderivatives induce apoptosis by disruption of intracellular redox balance.Cancer Res 2003;63:5551–8.
[30] Lizard G, Gueldry S, Sordet O, et al. Glutathione is implied in the control of 7-ketocholesterol-induced apoptosis, which is associated with radical oxygenspecies production. FASEB J 1998;12:1651–63.
[31] Liu W, Kato M, Akhand AA, et al. 4-hydroxynonenal induces a cellular redoxstatus-related activation of the caspase cascade for apoptotic cell death. J CellSci 2000;113:635–41.
[32] Deb I, Paira P, Hazra A, et al. Synthesis and characterizations of novel quinolinederivatives having mixed ligand activities at the k and m receptors: potentialtherapeutic efficacy against morphine dependence. Bioorg Med Chem2009;17:5782–90.
[33] Mondal SK, Mondal NB, Banerjee S, et al. Determination of drug like propertiesof a novel antileismanial compound: in vitro absorption, distribution, metab-olism, and excretion studies. Indian J Pharmacol 2009;41:176–81.
[34] Clarke MJ. Ruthenium metalo pharmaceuticals. Coord Chem Rev2003;236:209–33.
[35] Ghavami S, Hashemi M, Ande SR, et al. Apoptosis and cancer: mutations withincaspase genes. J Med Genet 2009;46:497–510.
[36] Slee EA, Adrain C, Martin SJ. Executioner Caspase-3, -6, and -7 perform distinct,non-redundant roles during the demolition phase of apoptosis. J Biol Chem2001;276:7320–6.
[37] Lavoie JN, Nguyen M, Marcellus RC, et al. E4orf4, a novel adenovirus deathfactor that induces p53-independent apoptosis by a pathway that is notinhibited by zVAD-fmk. J Cell Biol 1998;140:637–45.
[38] Kravchenko DV, Kuzovkova YA, Kysil VM, et al. Synthesis and structure –activity relationship of 4-substituted 2-(2-Acetyloxyethyl)-8-(morpholine- 4-sulfonyl)pyrrolo[3,4-c]quinoline- 1,3-diones as potent Caspase-3 inhibitors. JMed Chem 2005;48:3680–3.
[39] Lee D, Long SA, Adams JL, et al. Potent and selective nonpeptide inhibitors ofCaspases 3 and 7 inhibit apoptosis and maintain cell functionality. J Biol Chem2000;275:16007–14.
[40] Hadad J. Redox and oxidant-mediated regulation of apoptosis signaling path-ways: immuno-pharmaco-redox conception of oxidative siege versus celldeath commitment. Int Immunopharmacol 2004;4:475–93.
[41] Trompier D, Chang XB, Barattin R, et al. Verapamil and its derivative triggerapoptosis through glutathione extrusion by multidrug resistance proteinMRP1. Cancer Res 2004;64:4950–6.
Copyright © 2022 FDOKUMEN












![Understanding the solvatochromism of 10-hydroxybenzo[h]quinoline. An appraisal of a polarity calibrator](https://static.fdokumen.com/doc/165x107/63174024bc8291e22e0e2a62/understanding-the-solvatochromism-of-10-hydroxybenzohquinoline-an-appraisal-of.jpg)







