
On the Motive Power of Chemical Transformations in Open Systems
20
J Elast (2011) 104:229–248 DOI 10.1007/s10659-011-9305-x On the Motive Power of Chemical Transformations in Open Systems Roger Fosdick · Gianni Royer-Carfagni Received: 29 November 2010 / Published online: 10 February 2011 © Springer Science+Business Media B.V. 2011 Abstract The classical basic concepts of cyclic processes and the efficiency of heat engines are used here to conjecture about the laws of thermodynamics for open systems that can exchange matter with a surrounding environment. An ideal chemomechanical elastic bar is envisioned that changes its stiffness while undergoing a chemical transformation which is, in turn, influenced by the axial strain of the bar. Stable equilibrium states are identified as minimizers of the total energy, which is assumed to be nonconvex in type. If the bar is loaded and then alternatively placed in environments at chemical potentials either μ i or μ s >μ i , a reversible cycle analogous to the classical Carnot cycle may be traced. In this case, the environmental “chemical potential” plays the role of the temperature and the “chemical work” the role of heat. For the system, the main form of interaction with the exterior, other than mechanical work, is the exchange of mass of a component at different environmental chemical potentials. It is then possible to obtain an elementary theory of chemical engines in which efficiency estimates (in terms of environmental chemical potentials) and related pertinent issues can be discussed. This model may serve as a basis for analyzing coupled chemo-mechanical processes occurring in materials such as ionized gels for possible appli- cations as actuators, and to interpret complex phenomena in biological systems, such as the chemical kinetics of smooth muscles. Keywords Chemical transformation · Phase transition · Chemomechanics · Chemomechanical engine · Efficiency · Non-convex minimization · Muscles Mathematics Subject Classification (2000) 74B20 · 74F20 · 74F25 · 82D60 Dedicated to the memory of Donald Carlson, a friend and special colleague. R. Fosdick ( ) Department of Aerospace Engineering and Mechanics, University of Minnesota, Minneapolis, MN 55455, USA e-mail: [email protected] G. Royer-Carfagni Department of Civil-Environmental Engineering and Architecture, University of Parma, Parco Area delle Scienze 181/A, 43100 Parma, Italy
Transcript of On the Motive Power of Chemical Transformations in Open Systems

J Elast (2011) 104:229–248DOI 10.1007/s10659-011-9305-x
On the Motive Power of Chemical Transformationsin Open Systems
Roger Fosdick · Gianni Royer-Carfagni
Received: 29 November 2010 / Published online: 10 February 2011© Springer Science+Business Media B.V. 2011
Abstract The classical basic concepts of cyclic processes and the efficiency of heat enginesare used here to conjecture about the laws of thermodynamics for open systems that canexchange matter with a surrounding environment. An ideal chemomechanical elastic bar isenvisioned that changes its stiffness while undergoing a chemical transformation which is,in turn, influenced by the axial strain of the bar. Stable equilibrium states are identified asminimizers of the total energy, which is assumed to be nonconvex in type. If the bar is loadedand then alternatively placed in environments at chemical potentials either μi or μs > μi ,a reversible cycle analogous to the classical Carnot cycle may be traced. In this case, theenvironmental “chemical potential” plays the role of the temperature and the “chemicalwork” the role of heat. For the system, the main form of interaction with the exterior, otherthan mechanical work, is the exchange of mass of a component at different environmentalchemical potentials. It is then possible to obtain an elementary theory of chemical enginesin which efficiency estimates (in terms of environmental chemical potentials) and relatedpertinent issues can be discussed. This model may serve as a basis for analyzing coupledchemo-mechanical processes occurring in materials such as ionized gels for possible appli-cations as actuators, and to interpret complex phenomena in biological systems, such as thechemical kinetics of smooth muscles.
Keywords Chemical transformation · Phase transition · Chemomechanics ·Chemomechanical engine · Efficiency · Non-convex minimization · Muscles
Mathematics Subject Classification (2000) 74B20 · 74F20 · 74F25 · 82D60
Dedicated to the memory of Donald Carlson, a friend and special colleague.
R. Fosdick (�)Department of Aerospace Engineering and Mechanics, University of Minnesota, Minneapolis,MN 55455, USAe-mail: [email protected]
G. Royer-CarfagniDepartment of Civil-Environmental Engineering and Architecture, University of Parma, Parco Areadelle Scienze 181/A, 43100 Parma, Italy

230 R. Fosdick, G. Royer-Carfagni
1 Introduction
The concept of a cyclic process has played a central role in the origins of thermodynam-ics, especially in the formulation of its fundamental laws as restriction on what an engineworking in a cycle can or cannot do. In classical work, reference is made to the Gedanken-experiment of a cylinder containing an ideal gas with a movable piston at one of its ends.The piston is alternatively placed in contact with an insulator or heat reservoirs at two dif-ferent temperatures Tmin and Tmax > Tmin, and the gas is allowed to expand and contract“reversibly”, either adiabatically or isothermally, following the Carnot cycle. From the prin-ciples of thermodynamics, the maximum efficiency obtainable from any heat engine work-ing between the two temperatures Tmin and Tmax is well-known to be equal to 1 −Tmin/Tmax.The result is universal: no other device employing thermal energy and heat reservoirs attemperatures lying between Tmin and Tmax could ever do better, because of the second lawfor cyclic processes.
Here, we use the idea of cycles to discuss and make some conjectures about the laws ofthermodynamics for open systems, i.e., systems that can exchange matter with the surround-ing environment. No formal theory as a modification or alternative to the classical theoriesis presented: we limit ourselves mostly to conjecture on what the concepts needed for a pos-sible extension of the fundamental principles of thermodynamics to open system might be.As a Gedankenexperiment, we draw upon our work in [1] and consider an elastic bar whichundergoes a chemical transformation due to the mass transport of some substance S1 froma surrounding environment. The substance S1 reacts with the substance S0 of which the baris made. The reaction is supposed to follow a prescribed stoichiometric relationship and thesystem is “ideal”, in the sense that the chemical transformation is reversible. The volume ofthe bar, as well as its elastic modulus, varies with the degree of chemical reaction and if thebar is loaded axially, this load will affect not only the strain in the bar, but also its chemicalaffinity and, consequently, the degree of reaction.
In nature there are materials for which the ideal chemomechanical system describedabove represents a fair approximation. An example is given by filaments of an ionic gel[16] that are bathed in an acid solution: the gel can swell or contract due to variations of thechemical composition of the surrounding environment. Moreover, the kinetics of biologicalsystems such as muscles is also chemically driven by receptors and changes in ion concentra-tion. Understanding the maximum efficiency obtainable by a chemomechanical open systemmay serve as a criterion to conceive optimal artificial actuators and, possibly, to understandthe underlying processes in biological systems such as muscles, for the optimization of anathletic performance.
The plan of the paper is as follows. In Sect. 2, ionized gels are briefly reviewed as par-adigmatic examples of chemomechanical materials. In Sect. 3 a model is proposed basedupon the minimization of a non-convex energy, conceived to be a function of the strain anda chemical state-variable. In Sect. 4 the counterpart of the Carnot cycle for a chemomechan-ical open system is discussed, deriving an upper threshold for its efficiency. Conclusionsabout possible abstract formulations of the second law of thermodynamics for isothermalchemomechanical cycles are discussed in the last Sect. 5.
2 Chemomechanical Materials
The aim of this section is to present examples of chemomechanical materials to which themodel hereafter presented may apply. To this respect, one of the most interesting class of
On the Motive Power of Chemical Transformations in Open Systems 231
materials is that of polymer gels, i.e., cross-linked networks of polymers swollen with a liq-uid. Polymer gels usually exist in two distinct phases, swollen or collapsed, and the volumetransition occurs between the phases either continuously or discontinuously in response tochemical and physical stimuli such as temperature, solvent composition and PH variation,among others [17].
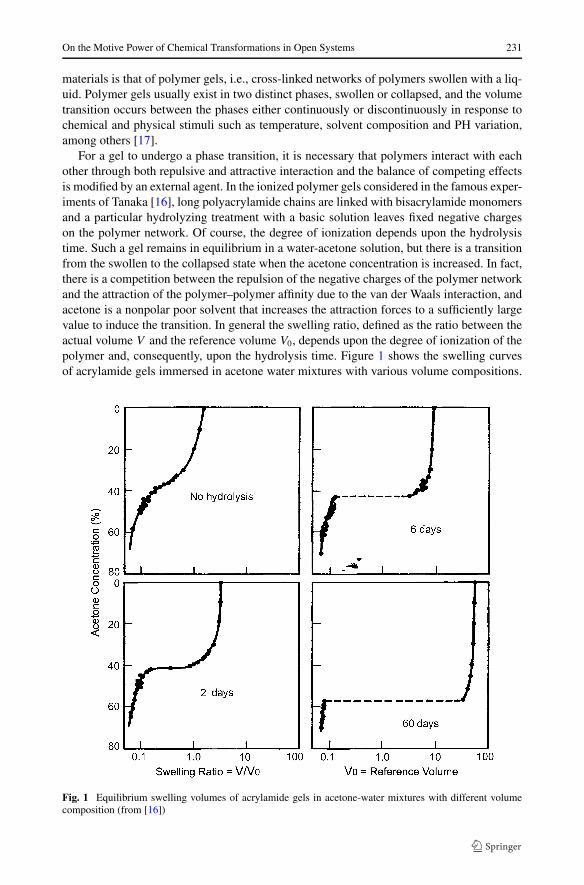
For a gel to undergo a phase transition, it is necessary that polymers interact with eachother through both repulsive and attractive interaction and the balance of competing effectsis modified by an external agent. In the ionized polymer gels considered in the famous exper-iments of Tanaka [16], long polyacrylamide chains are linked with bisacrylamide monomersand a particular hydrolyzing treatment with a basic solution leaves fixed negative chargeson the polymer network. Of course, the degree of ionization depends upon the hydrolysistime. Such a gel remains in equilibrium in a water-acetone solution, but there is a transitionfrom the swollen to the collapsed state when the acetone concentration is increased. In fact,there is a competition between the repulsion of the negative charges of the polymer networkand the attraction of the polymer–polymer affinity due to the van der Waals interaction, andacetone is a nonpolar poor solvent that increases the attraction forces to a sufficiently largevalue to induce the transition. In general the swelling ratio, defined as the ratio between theactual volume V and the reference volume V0, depends upon the degree of ionization of thepolymer and, consequently, upon the hydrolysis time. Figure 1 shows the swelling curvesof acrylamide gels immersed in acetone water mixtures with various volume compositions.
Fig. 1 Equilibrium swelling volumes of acrylamide gels in acetone-water mixtures with different volumecomposition (from [16])
232 R. Fosdick, G. Royer-Carfagni
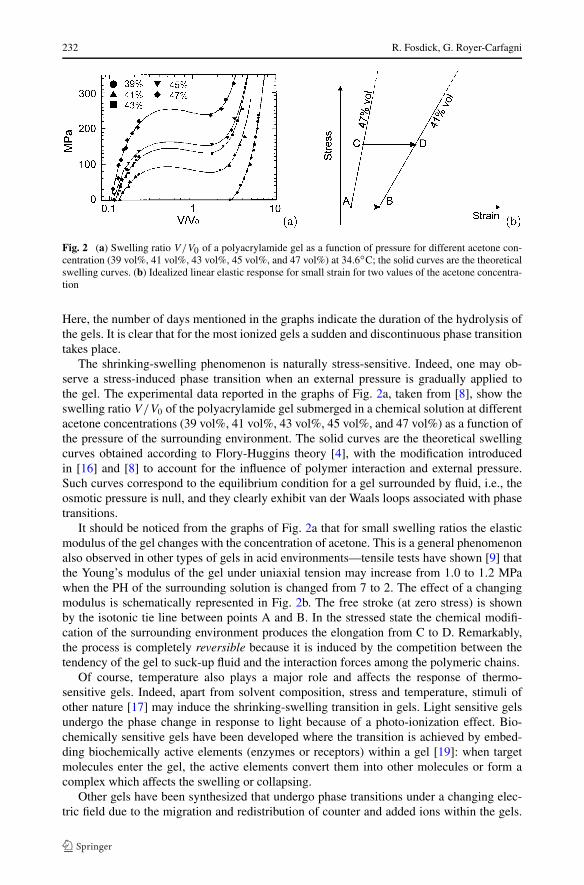
Fig. 2 (a) Swelling ratio V/V0 of a polyacrylamide gel as a function of pressure for different acetone con-centration (39 vol%, 41 vol%, 43 vol%, 45 vol%, and 47 vol%) at 34.6◦C; the solid curves are the theoreticalswelling curves. (b) Idealized linear elastic response for small strain for two values of the acetone concentra-tion
Here, the number of days mentioned in the graphs indicate the duration of the hydrolysis ofthe gels. It is clear that for the most ionized gels a sudden and discontinuous phase transitiontakes place.
The shrinking-swelling phenomenon is naturally stress-sensitive. Indeed, one may ob-serve a stress-induced phase transition when an external pressure is gradually applied tothe gel. The experimental data reported in the graphs of Fig. 2a, taken from [8], show theswelling ratio V/V0 of the polyacrylamide gel submerged in a chemical solution at differentacetone concentrations (39 vol%, 41 vol%, 43 vol%, 45 vol%, and 47 vol%) as a function ofthe pressure of the surrounding environment. The solid curves are the theoretical swellingcurves obtained according to Flory-Huggins theory [4], with the modification introducedin [16] and [8] to account for the influence of polymer interaction and external pressure.Such curves correspond to the equilibrium condition for a gel surrounded by fluid, i.e., theosmotic pressure is null, and they clearly exhibit van der Waals loops associated with phasetransitions.
It should be noticed from the graphs of Fig. 2a that for small swelling ratios the elasticmodulus of the gel changes with the concentration of acetone. This is a general phenomenonalso observed in other types of gels in acid environments—tensile tests have shown [9] thatthe Young’s modulus of the gel under uniaxial tension may increase from 1.0 to 1.2 MPawhen the PH of the surrounding solution is changed from 7 to 2. The effect of a changingmodulus is schematically represented in Fig. 2b. The free stroke (at zero stress) is shownby the isotonic tie line between points A and B. In the stressed state the chemical modifi-cation of the surrounding environment produces the elongation from C to D. Remarkably,the process is completely reversible because it is induced by the competition between thetendency of the gel to suck-up fluid and the interaction forces among the polymeric chains.
Of course, temperature also plays a major role and affects the response of thermo-sensitive gels. Indeed, apart from solvent composition, stress and temperature, stimuli ofother nature [17] may induce the shrinking-swelling transition in gels. Light sensitive gelsundergo the phase change in response to light because of a photo-ionization effect. Bio-chemically sensitive gels have been developed where the transition is achieved by embed-ding biochemically active elements (enzymes or receptors) within a gel [19]: when targetmolecules enter the gel, the active elements convert them into other molecules or form acomplex which affects the swelling or collapsing.
Other gels have been synthesized that undergo phase transitions under a changing elec-tric field due to the migration and redistribution of counter and added ions within the gels.

On the Motive Power of Chemical Transformations in Open Systems 233
A material of this kind seems to be particularly interesting for the construction of electro-mechanical actuators. Importantly, the electrical stimulus influences the elastic modulus ofthe gel, which significantly influences the actuator strain. This allows for the possibilityof working against a dead weight and producing mechanical work from a cyclic processwherein the electric field is alternatively varied. The maximum work-per-cycle has beencalculated in [13] for various cases.
Some researchers consider that the phase transition of gels plays a vital role in muscularcontraction and relaxation, and also in neural excitation and signal transmission. Indeed,chemomechanical energy conversion by polymer gels has been noted to produce artificialmuscular action. Porous gel films made from poly(vinyl alcohol), poly(acrylic acid) andpoly(allylamine) mixed solutions have been used [15] to generate a mechanical power den-sity close to skeletal muscle by changing the solvent from water to acetone. These polymergels swell and shrink reversibly by chemical or thermal stimulations.
As a matter of fact, the chemical kinetics of muscle contraction has been interpreted interms of a mechanochemical model similar to that of gels. In the approach proposed in [14]for example, a free energy function is defined that depends upon the stretches that representcell stiffness and sarcomere contractile capability, four additional variables which representthe various biochemical components, and a chemical state variable which is driven by tem-perature and by the Ca2+-concentration in the surrounding environment. Mechanochemicalmodels of this kind are valuable for furnishing a tool to analyze coupled complex processes,such as the dependency of tissue properties on the chemical kinetics of smooth muscles.
3 The Model
For the forthcoming deductions, reference is made to the ideal chemomechanical systemalready proposed in [1]. Here we recall only the main results, referring to [1] for the details.
3.1 The Chemomechanical System
Consider a system composed of two distinct compounds, here referred to as C ′ and C ′′, andthree substances, say S0, S1 and S2. In particular, C ′ is a solid compound of S0 and S1,whereas C ′′ is a fluid (or a vapor) mixture of S1 and S2 only. Symbolically,
S0 + S1 ⇔ C ′, S1 + S2 ⇔ C ′′, (3.1)
where the first corresponds to a chemical reaction, whereas the second, in general, denotesa simple solution of S1 dispersed in S2. All transformations are supposed to be reversible.Substance S0 and compound C ′ may coexist in the solid body B′ ⊂ R
3, which is surroundedby the fluid environment C ′′ occupying the configuration B′′ ⊂ R
3. The reference config-urations are those in which B′ is composed of pure S0 in a natural and undistorted state,whereas B′′ is stress free and has an initial assigned concentration of S1. Let ρ ′
0 (ρ ′′1,0) repre-
sent the referential mass density of S0 (S1) in B′ (B′′). Due to the occurrence of the chemicalreaction (3.1)1, a particle x ∈ B′ may vary its chemical composition because some S1 can beabsorbed from the surrounding environment, supposed to remain homogeneous throughoutthe process. Let ρ ′
1(x) denote the actual mass density of S1 at x ∈ B′ and ρ ′′1 denote the
(uniform) actual mass density of S1 in B′′. Clearly, 0 ≤ ρ ′1(x) ≤ ρ̄ ′
1, where the lower (upper)bound is attained when the particle x is composed of pure S0 (C ′). Since the stoichiome-try of reaction (3.1)1 requires that the mass of C ′ contains masses of S0 and S1 in definite

234 R. Fosdick, G. Royer-Carfagni
proportions, then
ρ̄ ′1 = ρ ′ s1
s ′ t′1, (3.2)
where ρ ′ is the referential mass density of C ′, s1 is the molar mass of S1, s ′ is the molarmass of C ′ and t ′1 is a stoichiometric coefficient representing the number of moles of S1 inone mole of C ′. It is convenient in the following to introduce the mass fraction
c(x) = ρ ′1(x)
ρ̄ ′1
(3.3)
as a measure of the degree of reaction. When c(x) = 0 (c(x) = 1), then x is composed ofpure S0 (pure C ′).
3.2 The Minimization Problem
In the one-dimensional version, the present model characterizes the chemomechanical equi-librium of a bar in a chemically reacting environment. The bar B′, composed of a mixtureof S0 and C ′, can be uniaxially stressed by an external loading machine while remaining incontact with the surrounding solution C ′′ of S1 and S2. We recognize that the stress statein the bar can modify the chemical affinity relative to the surrounding environment and,consequently, influence the degree of the reaction (3.1)1, i.e., c(x) in B′. Suppose that roomtemperature and pressure are kept constant. Initially the system is in its reference state,i.e., the bar is undistorted with length L and cross sectional area A0, while V ′′ denotes thereference volume of B ′′, the environmental bath. Supposing that the cross sections remainflat and at right angle to the bar axis, the reference configuration is defined by the interval(0,L) ⊂ R and the deformation is measured by the axial strain e : (0,L) → (−1,+∞),where e = u′(x), with u(x) : (0,L) → R being the axial displacement. Supposing the extentof reaction to be homogeneous across each transversal section, then c(x) of (3.3) is restrictedto the domain (0,L), i.e., c : (0,L) → (0,1).
We suppose that the chemomechanical transformation is reversible and that configura-tions of chemomechanical equilibrium correspond to minimizers of the total energy
�[e(x), c(x), ρ ′′1 ] =
∫ L
0A0ψ
′(e(x), c(x))dx + V ′′ψ ′′(ρ ′′
1 ) + PE, (3.4)
where ψ ′(e, c) represents the specific free energy of the bar at constant room temperaturemeasured per unit reference volume of B′, ψ ′′(ρ ′′
1 ) denotes the free energy per unit referencevolume V ′′ of B ′′ at constant room temperature and fixed room pressure, and PE denotesthe potential energy of the bar B′ due to the external dead loads that are applied to the bar,possibly at its extremities (soft-device). When the loading device is hard, then the averagestrain is constrained and the contribution due to PE is omitted in the total energy expression(3.4). In the formulation of the aforementioned minimization problem it has been implic-itly assumed that the pressure of the environment B′′ does not vary in the process underconsiderations and, in any case, that it has a negligible effect on the reaction (3.1)1.
3.3 The Variational Characterization
The mass fraction c(x) in (3.4) is strictly correlated, through (3.3), with the mass densityρ ′
1(x) of S1 in B′. Moreover, since the mass of S1 is conserved, what is lost in B′′ must

On the Motive Power of Chemical Transformations in Open Systems 235
appear in B′ , i.e.,
V ′′(ρ ′′1,0 − ρ ′′
1 ) =∫ L
0A0ρ
′1(x) dx ⇒ ρ ′′
1 = ρ ′′1,0 − A0
V ′′ ρ̄′1
∫ L
0c(x) dx. (3.5)
A necessary condition for the chemomechanical equilibrium of the system is that the firstvariation of (3.4) must be non-negative. Consequently, if [e∗(x), c∗(x), ρ ′′
1∗] represents a
minimizer of (3.4), then it is necessary that
∫ L
0A0
∂ψ ′
∂c
(e∗(x), c∗(x)
)δc(x) dx + V ′′ dψ ′′
dρ ′′1
(ρ ′′
1∗)
δρ ′′1 ≥ 0, (3.6)
where the variations δc(x) and δρ ′′1 are correlated through (3.5).
Borrowing the terms from the work of Gibbs [7], we say that S1 is an “actual component”of B′ (or B′′) if S1 is present in that part. From a variational point of view, if S1 is an actualcomponent then the quantity of S1 in that part could be either increased or diminished. Onthe other hand, we say that S1 is a “possible component” of B′ (or B′′) if S1 is not presentin that part. In this case, only one-sided variations may be supported since S1 can be added,but not subtracted, and equilibrium is governed by variational inequalities which have beendiscussed at length in [1].
For the following, it is relevant to consider two particular reaction environments for thebar in either a hard or soft loading device.
– Environment 1. Chemical Bath.If B′′ is so large (A0L/V ′′ 1) that any allowed transport of S1 to B′ does not produce
an appreciable change in the initial mass density of S1 in B′′ so that ρ ′′1 = ρ ′′
1,0, then setting
μ̄ = ρ̄ ′1
dψ ′′
dρ ′′1
(ρ ′′
1,0
)> 0, (3.7)
which represents the (normalized) chemical potential of the environment, and using (3.5),we may approximate the second term on the right hand side of (3.4) by (see [1])
V ′′ψ ′′(ρ ′′1 ) ∼= −μ̄
∫ L
0A0c(x) dx + const. (3.8)
Therefore, when the bar B′ is elongated in a hard-device, it is reasonable to write theminimization problem characterized by the functional (3.4) in the form
PHCB : min
(e,c)∈AHCB
(∫ L
0A0ψ
′(e(x), c(x))dx − A0μ̄
∫ L
0c(x) dx
), (3.9)
where, denoting with β the average bar elongation (i.e., nominal strain), we define theadmissible class AH
CB as
AHCB ≡
{(e, c) :
∫ L
0e(x) dx = βL and 0 ≤ c(x) ≤ 1 ∀x ∈ (0,L)
}. (3.10)
Clearly, (3.6) and the bounds on c(x) in (3.10) produce the necessary conditions
∂cψ′(e∗(x), c∗(x)
) = μ̄ ∀x with 0 < c∗(x) < 1, (3.11)

236 R. Fosdick, G. Royer-Carfagni
∂cψ′(e∗(x), c∗(x)
) ≥ μ̄ ∀x with c∗(x) ≡ 0, (3.12)
∂cψ′(e∗(x), c∗(x)
) ≤ μ̄ ∀x with c∗(x) ≡ 1, (3.13)
where here, and in the following, we write ∂cψ′ := ∂ψ ′/∂c, for convenience. For a soft
loading device, if σ0 is the applied engineering stress on the ends of the bar, the analogousproblem to consider is
PSCB : min
(e,c)∈ASCB
(∫ L
0A0ψ
′(e(x), c(x))dx − A0μ̄
∫ L
0c(x) dx − A0σ0
∫ L
0e(x) dx
),
(3.14)where the admissible class AS
CB associated with (3.14) is defined as
ASCB ≡ {
(e, c) : 0 ≤ c(x) ≤ 1 ∀x ∈ (0,L)}. (3.15)
– Environment 2. Ideal.Suppose that conditions are such that either no chemical reaction occurs, so that
c(x) = 0 ∀x ∈ (0,L), or the maximal reaction takes place, i.e., c(x) = 1 and ρ ′1(x) = ρ̄ ′
1∀x ∈ (0,L). In the first case, according to (3.5) a minimizer of (3.4) will be of the form[e∗(x),0, ρ ′′
1,0] and, from (3.6) and the definition (3.7), together with the correlation of thevariations δc(x) and δρ ′′
1 through (3.5), we see that it is necessary that
∂cψ′(e∗(x),0
) ≥ μ̄, ∀x ∈ (0,L). (3.16)
If this condition is not satisfied, the chemical reaction will take place at some point in B′.In the case that c(x) ≡ 1, the chemical reaction (3.1) is maximal in B′ and, conse-
quently, the bar contains S1 at full strength mass density ρ̄ ′1. Then, according to (3.5) a
minimizer of (3.4) will be of the form [e∗(x),1, ρ ′′1
∗], where ρ ′′1
∗ = ρ ′′1,0 − ρ̄ ′
1A0L/V ′′.Moreover, according to (3.6) the following necessary condition holds:
∂cψ′(e∗(x),1
) ≤ ρ̄ ′1∂ρ′′
1ψ ′′ (ρ ′′
1∗)
, ∀x ∈ (0,L). (3.17)
Of course, for an ideal environment in a chemical bath it is clear that ρ ′′1
∗ = ρ ′′1,0 and,
according to (3.7), in this case we may simplify (3.17) to
∂cψ′(e∗(x),1
) ≤ μ̄, ∀x ∈ (0,L). (3.18)
4 The Chemomechanical Cycle
4.1 Ideal Chemomechanical Bar: An Example
An ideal chemomechanical bar is amenable of reversible quasi-static chemomechanicaltransformations and its response in either the soft or hard loading device is determined byan energy functional of the form (3.4). While the specific Helmholtz free energy ψ ′(e, c)can be evaluated experimentally, as discussed in [1], here, to render the calculations elemen-tary, only a particular form will be considered; our considerations apply to more complexfunctions. Thus, we assume that
ψ ′(e, c) = 1
2E(c)e2, E(c) = (
(1 − c)E0 + cE1
), (4.1)
On the Motive Power of Chemical Transformations in Open Systems 237
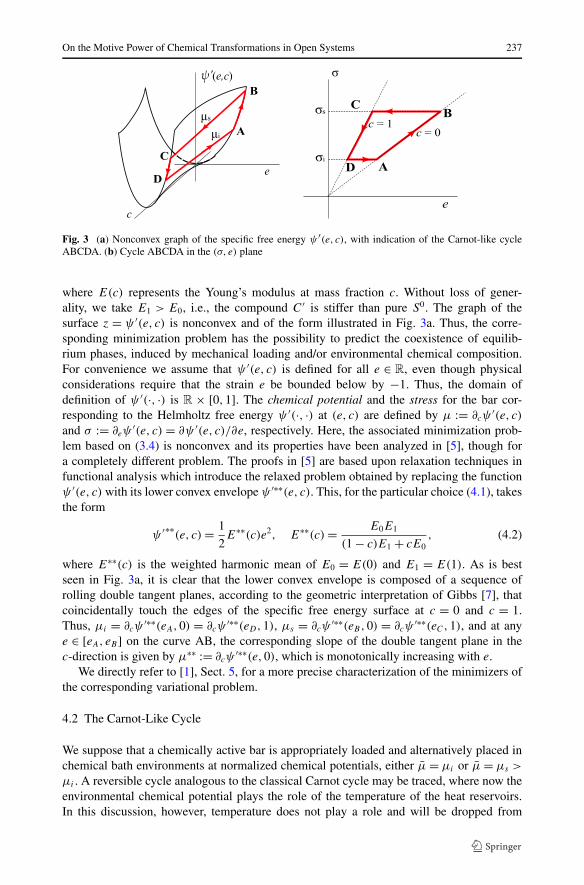
Fig. 3 (a) Nonconvex graph of the specific free energy ψ ′(e, c), with indication of the Carnot-like cycleABCDA. (b) Cycle ABCDA in the (σ, e) plane
where E(c) represents the Young’s modulus at mass fraction c. Without loss of gener-ality, we take E1 > E0, i.e., the compound C ′ is stiffer than pure S0. The graph of thesurface z = ψ ′(e, c) is nonconvex and of the form illustrated in Fig. 3a. Thus, the corre-sponding minimization problem has the possibility to predict the coexistence of equilib-rium phases, induced by mechanical loading and/or environmental chemical composition.For convenience we assume that ψ ′(e, c) is defined for all e ∈ R, even though physicalconsiderations require that the strain e be bounded below by −1. Thus, the domain ofdefinition of ψ ′(·, ·) is R × [0,1]. The chemical potential and the stress for the bar cor-responding to the Helmholtz free energy ψ ′(·, ·) at (e, c) are defined by μ := ∂cψ
′(e, c)and σ := ∂eψ
′(e, c) = ∂ψ ′(e, c)/∂e, respectively. Here, the associated minimization prob-lem based on (3.4) is nonconvex and its properties have been analyzed in [5], though fora completely different problem. The proofs in [5] are based upon relaxation techniques infunctional analysis which introduce the relaxed problem obtained by replacing the functionψ ′(e, c) with its lower convex envelope ψ ′∗∗(e, c). This, for the particular choice (4.1), takesthe form
ψ ′∗∗(e, c) = 1
2E∗∗(c)e2, E∗∗(c) = E0E1
(1 − c)E1 + cE0, (4.2)
where E∗∗(c) is the weighted harmonic mean of E0 = E(0) and E1 = E(1). As is bestseen in Fig. 3a, it is clear that the lower convex envelope is composed of a sequence ofrolling double tangent planes, according to the geometric interpretation of Gibbs [7], thatcoincidentally touch the edges of the specific free energy surface at c = 0 and c = 1.Thus, μi = ∂cψ
′∗∗(eA,0) = ∂cψ′∗∗(eD,1), μs = ∂cψ
′∗∗(eB,0) = ∂cψ′∗∗(eC,1), and at any
e ∈ [eA, eB ] on the curve AB, the corresponding slope of the double tangent plane in thec-direction is given by μ∗∗ := ∂cψ
′∗∗(e,0), which is monotonically increasing with e.We directly refer to [1], Sect. 5, for a more precise characterization of the minimizers of
the corresponding variational problem.
4.2 The Carnot-Like Cycle
We suppose that a chemically active bar is appropriately loaded and alternatively placed inchemical bath environments at normalized chemical potentials, either μ̄ = μi or μ̄ = μs >
μi . A reversible cycle analogous to the classical Carnot cycle may be traced, where now theenvironmental chemical potential plays the role of the temperature of the heat reservoirs.In this discussion, however, temperature does not play a role and will be dropped from

238 R. Fosdick, G. Royer-Carfagni
consideration. To be somewhat more descriptive, consider an axially uniformly extendedbar B′ , composed of a pure substance S0, that is placed in a chemical bath composed ofa mixture S1 + S2 ⇔ C ′′ having the chemical potential μi . While under certain conditionssubstances S0 and S1 could react to form the solid compound S0 + S1 ⇔ C ′, under thepresent conditions the bath and the bar are considered to be in chemical equilibrium withno reaction taking place. Thus, the bar is considered to be in a minimal energy state with auniform nominal strain and a mass fraction c = 0, and the chemical potential of the bath isμi , as is exhibited by point A in Fig. 3a. We now suppose that the bar undergoes a sequenceof controlled axial extensions of increasing nominal strain e while the chemical potentialof the bath μ̄ is assigned such that μi ≤ μ̄ ≤ ∂cψ
′∗∗(e,0). This means that in this sequencethe chemical potential of the bath, μ̄, may be gradually modified to equal any value greaterthan or equal to its starting value μi and less than or equal to the value ∂cψ
′∗∗(e,0) which,according to the lower convex envelope (4.2), determines the current slope of the doubletangent plane in the c-direction. In particular, the chemical potential of the bath could befixed at μ̄ = μi in this sequence. In this case, the minimal energy equilibrium path requiresthat the substances S0 and S1 do not react and, therefore, the mass fraction is maintainedat c = 0. In fact, the environmental chemical potential μ̄ is too weak [1] for any reactionof S0 and S1 to occur and the bar remains composed of the pure substance S0 during thistransformation.
At a certain value of the controlled axial nominal strain, the chemical potential of the bathis fixed1 at the value μ̄ = μs > μi , which corresponds to the equilibrium minimal energycondition for a quasi-static chemical reaction of S0 and S1 to initiate and to form the purecompound S0 + S1 ⇔ C ′. Coincidentally, the axial strain control is released and the axialload is fixed at its current value. Thus, a transformation, with an associated volume change,takes place and we envision a sequence of axial deformations of the bar wherein the axialnominal strain is reduced at fixed load, provoked by the aforementioned chemical reaction.We suppose that the whole mass of substance S0 contained in B′ takes part in this processand that the nominal mass fraction changes from c = 0 to c = 1, so that at the end of thetransformation the whole bar is composed of compound C ′ and the mass fraction is at c = 1.
At this point, μ̄ = μs and the bar is in a state of uniform axial strain with c = 1. Wenow suppose that the bar undergoes a sequence of controlled axial uniform contractions ofdecreasing nominal strain while the chemical potential of the bath, μ̄, is such that μs ≥ μ̄
≥ ∂cψ′∗∗(e,1). This means that in this sequence the chemical potential of the bath, μ̄, may
be gradually modified to equal any value less than or equal to its starting value μs and greaterthan or equal to the value ∂cψ
′∗∗(e,1) which, according to the lower convex envelope (4.2),determines the current slope of the double tangent plane in the c-direction. In particular, thechemical potential of the bath could be fixed at μ̄ = μs in this sequence. Then, the minimalenergy equilibrium path requires that no reaction takes place, the mass fraction remainsfixed at c = 1, and the pure compound C ′ for the composition of the bar is maintainedwhile, correspondingly, the bar shortens. In this case, the environmental chemical potential,μ̄, is too strong to initiate the reverse chemical reaction.
Finally, at a certain value of the axial nominal strain, the chemical potential of the bathis fixed2 at the original value μ̄ = μi < μs , which corresponds to the equilibrium minimal
1We can imagine this to take place either by adding some substance S1 to the same bath in order to changeits chemical potential from μi to μs , or by putting the bar into contact with a second chemical reservoir atμ̄ = μs .2Also for this, we may imagine either that some of the substance S1 contained in the bath is re-moved/annihilated through an auxiliary chemical process, or that the bar is placed into contact with anotherbath at μ̄ = μi .

On the Motive Power of Chemical Transformations in Open Systems 239
energy condition for the reverse reaction to initiate and for the compound C ′ of the barto return to the pure substance S0. Of course, the axial strain control is released and theaxial load is fixed at its current value. The resulting transformation is associated with avolume change, and we envision a sequence of axial deformations of the bar wherein theaxial nominal strain is increased at fixed end load and the nominal mass fraction changesfrom c = 1 to c = 0. At the end of this transformation the whole bar is again composed ofthe pure substance S0, it is uniformly strained at its original length, and the mass fraction isat c = 0.
This describes the elements of an ideal, reversible, chemomechanical engine that oper-ates between two chemical baths at different chemical potentials. In particular, such a cycleABCDA, drawn on the surface ψ = ψ ′(e, c) of Fig. 3a, is defined by the states A ≡ (eA, cA),B ≡ (eB, cB), C ≡ (eC, cC) and D ≡ (eD, cD) which must satisfy the following “commontangency conditions”:
ψ ′(eC,1) − ψ ′(eB,0) = σs(eC − eB) + μs, (4.3)
ψ ′(eA,0) − ψ ′(eD,1) = σi(eA − eD) − μi. (4.4)
Here, (σs,μs) and (σi,μi) correspond, respectively, to the components of the slope of twoplanes with respect to the e-axis and the c-axis. According to a well-known geometric inter-pretation of Gibbs [7], the slope (σs,μs) of the first plane is such that, when appropriatelytranslated, it coincidently touches (and supports from below) the specific energy surface atthe two points B and C, corresponding to the states (eB,0) and (eC,1), and (4.3) expressesthis common touching condition. From (4.1), we see that σs = E0eB = E1eC , so that σs rep-resents the (constant) axial stress at which the chemical transformation associated with thepath B → C at μ̄ = μs takes place. Similarly, the slope (σi,μi) of the second plane allowsthis plane to coincidently touch (and support from below) the specific energy surface at thepoints A and D corresponding to the states (eA,0) and (eD,1), and (4.4) expresses this com-mon touching condition. From (4.1), we see that σi = E1eD = E0eA, with σi denoting the(constant) axial stress at which the reverse chemical transformation associated with the pathD → A at μ̄ = μi occurs. States of stable equilibrium correspond to the lower support pointsof the stored energy surface and, because of the assumed form of the specific free energy(4.1), coexistent states (i.e., phases) are possible only if they correspond to points on the twoparabolas, defined by the intersection of the surface z = ψ ′(e, c) with the planes c = 0 andc = 1, that lie in a common touching lower support plane, as in the case of (4.3) and (4.4).Notice that these parabolas are also the locus of points where the surface z = ψ ′(e, c) andits lower convex envelope z = ψ ′∗∗(e, c) coincide [5]. In fact, the surface defined by the setof all lower support planes is the lower convex envelope and in this case it is constructed bythe corresponding sequence of rolling double-touching support planes.
More precisely, the cycle described above consists of the following four transformationpaths:
1: Path A → B.The bar B′ of pure substance S0 has an initial strain eA and it is emersed in a chemical
bath solution B′′ at chemical potential μ̄ ∈ [μi, ∂cψ′∗∗(e,0)]. In particular, μ̄ could be
held fixed at μ̄ = μi . While in contact with the solution, the bar undergoes an infinitelyslow reversible dilatation under a nominal strain driven path with nominal strain e varyingfrom eA in state A to eB in the state B. During the process, the work done by the bar is
WAB = −A0L
∫ eB
eA
σde = −A0L
∫ eB
eA
E0ede = −1
2A0LE0(e
2B − e2
A). (4.5)

240 R. Fosdick, G. Royer-Carfagni
The corresponding minimization problem is given by (3.9) and (3.10), wherein β is nowidentified with e. In this case, the minimizing mass fraction is c(x) ≡ 0. Note that becausec(x) ≡ 0 it follows that no mass of substance S1 enters B′ from B′′. That is, no chemi-cal reaction S0 + S1 in B′ takes place because, in particular, μ̄ ∈ [μi, ∂cψ
′∗∗(e,0)] ⇒μ̄ < ∂cψ
′(e,0), which means that the chemical potential of B′ is adverse to the influxof S1 from B′′ between A and B in the sense that the mass of a substance can only fluxfrom an environment of higher to lower chemical potential.3 This transformation can beconsidered the counterpart of one of the two adiabatic transformations in a Carnot cy-cle. However, in this case, there is no need of a “chemical insulation” analogous to thethermal insulation for the adiabatic transformations in the Carnot cycle, because it is theprescribed composition (i.e., sufficiently low chemical potential) of the chemical bath andthe nominal strain path that prevents the occurrence of the chemical reaction.
2: Path B → C.Now, the axial strain control is released and the axial load is maintained constant and
equal to A0E0eB = A0σs . In addition, the chemical potential of the chemical bath is set atthe value μ̄ = μs , so that the axial nominal strain in the bar undergoes a decrease from eB
to eC following the line BC due to the occurrence of a chemical reaction. The minimiza-tion problem is now given by (3.14) and (3.15), wherein, in particular, the nominal strainis not controlled. Of course, two coexistent phases of changing relative amounts appear inthe bar, corresponding to the points B and C of Fig. 3a. In other words, a chemical phasetransformation S0 + S1 ⇔ C ′ occurs and, as discussed in [1], S1 is adsorbed by the barfrom the solution until the whole bar is composed of pure compound C ′. The chemicalreaction is promoted because μs = ∂cψ
′∗∗(eB,0) = ∂cψ′∗∗(eC,1) > ∂cψ
′(eC,1), whichimplies that the chemical potential of the bar B′ in the state (eC,1) is now favorable forthe influx of S1 from B′′. At the end of this transformation, the nominal strain of the bar ise = eC , i.e., the bar is uniformly strained, and the minimizing mass fraction is c(x) ≡ 1.The work done by the bar in this transformation is equal to
WBC = −A0L
∫ eC
eB
σsde = A0LE0eB(eB − eC) = A0LE1eC(eB − eC)
= A0Lσs(eB − eC). (4.6)
During this process the bar B′ will adsorb from the surrounding environment B′′ a mass
M1 = ρ̄ ′1A0L, (4.7)
of substance S1, and it will transform to the pure compound C ′ and become shorter. If oneidentifies the chemical potential of the bath and the mass absorbed in a chemomechan-ical engine with the temperature and entropy change in a thermal engine, respectively,this transformation is the counterpart of the isothermal absorption of heat at the higherenvironmental temperature Tmax in the classical Carnot cycle.
3: Path C → D.Here, while the bar B′ is maintained in contact with the chemical bath solution B′′ at
chemical potential μ̄ ∈ [∂cψ′∗∗(e,1),μs], its axial nominal strain e is controlled and is
driven from eC in state C to eD in state D. Note, in particular, that μ̄ could be held fixedat μ̄ = μs . As in the path A → B, the minimization problem is that of (3.9) and (3.10),
3Recall the necessary condition (3.12).

On the Motive Power of Chemical Transformations in Open Systems 241
wherein β is identified with e, and this implies that the minimizing mass fraction mustremain fixed at c(x) ≡ 1. In fact, because μ̄ ∈ [∂cψ
′∗∗(e,1),μs] ⇒ μ̄ > ∂cψ′(e,1), the
chemical potential of the bath B′′ is now adverse to the efflux of S1 from the bar B′ to B′′
between C and D and stoichiometry prevents further influx of S1 to the bar B′ from B′′.Consequently, the bar remains composed of the pure compound C ′. The work done bythe bar during this path is
WCD = −A0L
∫ eD
eC
σde = −A0L
∫ eD
eC
E1ede = 1
2A0LE1(e
2C − e2
D). (4.8)
This is the second counterpart of the two adiabatic transformations in a Carnot cycle.4
4: Path D → A.In the final transformation of the cycle, the strain control is released and the axial load
in the bar is kept constant and equal to A0E1eD = A0σi . In addition, the chemical potentialof the chemical bath is set at the equilibrium value μ̄ = μi . Again, the minimizationproblem is now given by (3.14) and (3.15), wherein the nominal strain is not controlled.Then, due to the occurrence of the reverse chemical reaction during which the compoundC ′ of the bar transforms back to the pure substance S0, the axial nominal strain in the barundergoes an increase from eD to eA following the line DA. Coexistent phases of changingrelative amounts, whose states correspond to points D and A, now occur. A chemicalphase transformation S0 + S1 ⇔ C ′, opposite to the one that occurred in the path B → C,takes place, and the substance S1 is expelled by the bar and dispersed in the surroundingsolution until the bar B′ is again composed of pure substance S0. This chemical reaction ispromoted because μi = ∂cψ
′∗∗(eD,1) = ∂cψ′∗∗(eA,0) < ∂cψ
′(eA,0), which implies thatthe chemical potential of the bar B′ in the state (eA,0) is now favorable for the efflux ofS1 to B′′. At the end of the chemical transformation the state of the bar is the same as atthe beginning of the cycle, characterized by the uniform strain e = eA and the minimizingmass fraction c(x) ≡ 0. The work done by the bar in this transformation is equal to
WDA = −A0L
∫ eA
eD
σide = −A0LE1eD(eA − eD)
= −A0LE0eA(eA − eD) = −A0Lσi(eA − eD). (4.9)
During this transformation the bar B′ expels the same mass M1 of substance S1 to thechemical bath B′′ that it had adsorbed during the path B → C, as indicated by (4.7). Thistransformation is analogous to the isothermal emission of heat at the lower environmentaltemperature Tmin in the classical Carnot cycle.
The points that define the four transformations of the cyclic process are represented inFig. 3a. To clarify the cyclic process, the various steps are followed in Fig. 4 on the specificenergy surface with a schematic indication of the deformation of the chemomechanical bar.
In synthesis, the bar adsorbs the mass M1 of substance S1 from a chemical bath at thenormalized chemical potential μ̄ = μs on the path BC and returns the same amount of massto the chemical bath at the chemical potential μ̄ = μi on path DA. Thus, mass exchangeplays the same role as entropy exchange in a classical thermal engine.
4Also for this transformation, there is no need of a “chemical insulation”.

242 R. Fosdick, G. Royer-Carfagni
Fig. 4 Schematic representation of the four transformations of the cyclic process on the energy surface
4.3 Efficiency of the Ideal, Reversible Chemomechanical Engine
Figure 3b represents the cycle ABCDA in the σ–e plane where, without risk of confusion,the variable e here stands for the axial average, i.e., nominal, strain of the bar. It is clearfrom (4.5), (4.6), (4.8) and (4.9) that, for each cycle, a net positive amount of work
W = WAB + WBC + WCD + WDA, (4.10)
proportional to the area enclosed inside of ABCD, is done by the bar.In a heat engine, the efficiency is defined as the ratio of the mechanical work done by
the engine to the heat adsorbed from the heat reservoir during the cycle. A transformationtakes place between the two forms of energy represented by heat and mechanical work. Fora Carnot cycle the efficiency turns out to be given by eff = 1 − Tmin/Tmax, where Tmin andTmax are the minimum and maximum operating environmental temperatures, and this repre-sents an upper threshold for the efficiency of any device operating in a cycle and employingthermal energy and heat reservoirs at temperatures lying between Tmin and Tmax.
Here, thermal issues, such as the absorption and emission of heat for the operation ofan engine, are not of major interest, but the possibility of chemical reaction and the conse-quential change of composition through the absorption and emission of mass is our pointof concern. According to traditional ideas of energy conservation, in order for a system toperform a net positive amount of work during a cycle a certain amount of energy must be

On the Motive Power of Chemical Transformations in Open Systems 243
supplied in some form. In a chemomechanical engine, one looks for the production of me-chanical work as a result of the transformation of another type of energy rather than heat,a transformation which is produced by the exchange of mass in an environment of chang-ing chemical potential. We call the energy supply corresponding to this mass exchange the“chemical work”, thinking of it as a form of energy interaction similar to heat and mechani-cal work.
To explore the efficiency of a chemomechanical engine for the cycle ABCDA of Fig. 3a,it is convenient to first introduce (e(α), c(α)) as the parametric representation of the graphof the projection of this cycle onto the (e, c)-plane of the “state space” in the sense that:
(i) Path A → B: (e(α), c(α)) = (eBα + eA(1 − α),0), α ∈ [0,1];(ii) Path B → C: (e(α), c(α)) = (eCα + eB(1 − α),α), α ∈ [0,1];
(iii) Path C → D: (e(α), c(α)) = (eDα + eC(1 − α),1), α ∈ [0,1];(iv) Path D → A: (e(α), c(α)) = (eAα + eD(1 − α),1 − α), α ∈ [0,1].
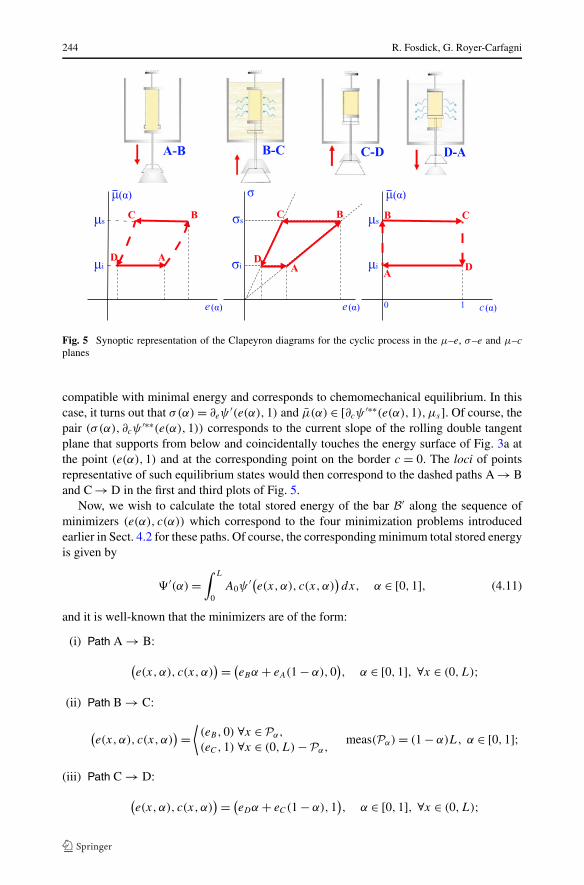
A synoptic representation of the various processes that correlate with the minimizingstates of a cycle may be interpreted by considering the counterparts, in the chemomechani-cal system, of the Clapeyron diagrams for the Carnot cycle. These are represented in Fig. 5.Here, e(α) and c(α) are interpreted as the average, i.e., nominal, values of the axial strainand mass fraction (equivalently, extent of reaction), respectively, σ(α) is the correspondingaxial stress, and μ̄ denotes, as usual, the chemical potential of the environmental bath withwhich the bar is in contact. Notice that paths A → B and C → D in the first and third graphsare represented by dashed arrows because, in the cycle upon which these graphs are basedthe environmental chemical potential for A → B need only satisfy μ̄ ∈ [μi, ∂cψ
′∗∗(e,0)]and for C → D need only satisfy μ̄ ∈ [∂cψ
′∗∗(e,1),μs]. Thus, for A → B, when the barB′ consists of the pure substance S0, the environmental chemical potential may be fixed atμ̄ = μi or it may undergo a strain-controlled increase from μi to μs , with incrementallysmall enough increase to prevent a reaction and consequential mass transfer from the en-vironmental bath to the bar. Similarly, for C → D, when the bar B′ consists of the purecompound C ′, the environmental chemical potential may be fixed at μ̄ = μs or it may un-dergo a strain-controlled decrease from μs to μi , with incrementally small enough decreaseto prevent a reaction and associated mass transfer from the bar to the environmental bath.Also, in the case studied here, during this path stoichiometry plays a part and prevents thebar from reacting any further with the environment to combine the substances S0 and S1 andto form more of the pure compound C ′.
To emphasize, one may envision a cycle wherein bath reservoirs are employed at interme-diate chemical potentials comprised between μi and μs . In this case, during the process cor-responding to the path A → B the nominal strain of the bar is increased from its value eA at Aand the bath reservoir slowly changes its chemical potential in a controlled manner, little bylittle, from μ̄ = μi to μ̄ = μs > μi by the addition of substance S1 while the bar undergoesan infinitely slow reversible dilatation in such a way that, at each instant, the current valueof the reservoir chemical potential μ̄(α) and the bar nominal strain e(α) and stress σ(α)
are those compatible with minimal energy and corresponding to chemomechanical equilib-rium. In this case, it turns out that σ(α) = ∂eψ
′(e(α),0) and μ̄(α) ∈ [μi, ∂cψ′∗∗(e(α),0)].
According to the geometric interpretation of Gibbs [7], in the surface plot of Fig. 3a thepair (σ (α), ∂cψ
′∗∗(e(α),0)) would correspond to the current slope of the “rolling doubletangent plane” that supports from below and coincidentally touches the energy surface atthe point (e(α),0) and at the corresponding point on the border c = 1. Analogously, duringthe process that corresponds to the path C → D the nominal strain of the bar is decreasedfrom its value eC at C and the chemical potential of the bath may continuously vary in a con-trolled manner, little by little, from μ̄ = μs to μ̄ = μi < μs so that the pair (σ (α), μ̄(α)) is
244 R. Fosdick, G. Royer-Carfagni
Fig. 5 Synoptic representation of the Clapeyron diagrams for the cyclic process in the μ–e, σ–e and μ–c
planes
compatible with minimal energy and corresponds to chemomechanical equilibrium. In thiscase, it turns out that σ(α) = ∂eψ
′(e(α),1) and μ̄(α) ∈ [∂cψ′∗∗(e(α),1),μs]. Of course, the
pair (σ (α), ∂cψ′∗∗(e(α),1)) corresponds to the current slope of the rolling double tangent
plane that supports from below and coincidentally touches the energy surface of Fig. 3a atthe point (e(α),1) and at the corresponding point on the border c = 0. The loci of pointsrepresentative of such equilibrium states would then correspond to the dashed paths A → Band C → D in the first and third plots of Fig. 5.
Now, we wish to calculate the total stored energy of the bar B′ along the sequence ofminimizers (e(α), c(α)) which correspond to the four minimization problems introducedearlier in Sect. 4.2 for these paths. Of course, the corresponding minimum total stored energyis given by
′(α) =∫ L
0A0ψ
′(e(x,α), c(x,α))dx, α ∈ [0,1], (4.11)
and it is well-known that the minimizers are of the form:
(i) Path A → B:
(e(x,α), c(x,α)
) = (eBα + eA(1 − α),0
), α ∈ [0,1], ∀x ∈ (0,L);
(ii) Path B → C:
(e(x,α), c(x,α)
) =⟨(eB,0) ∀x ∈ Pα,
(eC,1) ∀x ∈ (0,L) − Pα,meas(Pα) = (1 − α)L, α ∈ [0,1];
(iii) Path C → D:
(e(x,α), c(x,α)
) = (eDα + eC(1 − α),1
), α ∈ [0,1], ∀x ∈ (0,L);

On the Motive Power of Chemical Transformations in Open Systems 245
(iv) Path D → A:
(e(x,α), c(x,α)
) =⟨(eD,1) ∀x ∈ Pα,
(eA,0) ∀x ∈ (0,L) − Pα,meas(Pα) = (1 − α)L, α ∈ [0,1].
Thus, using (4.11), it readily follows that:
(i) Path A → B: ′(α) = A0Lψ ′(eBα + eA(1 − α),0), α ∈ [0,1];(ii) Path B → C: ′(α) = A0L(1 − α)ψ ′(eB,0) + A0Lαψ ′(eC,1), α ∈ [0,1];
(iii) Path C → D: ′(α) = A0Lψ ′(eDα + eC(1 − α),1), α ∈ [0,1];(iv) Path D → A: ′(α) = A0L(1 − α)ψ ′(eD,1) + A0Lαψ ′(eA,0), α ∈ [0,1];and, noting (4.5), (4.6), (4.8) and (4.9), the change in the minimum total stored energy ofthe bar B′ for each of the four paths is given by:
(i) Path A → B:
� ′A→B = ′(1) − ′(0) = A0L
(ψ ′(eB,0) − ψ ′(eA,0)
)
= 1
2A0LE0(e
2B − e2
A)
= −WAB. (4.12)
(ii) Path B → C:
� ′B→C = ′(1) − ′(0) = A0L
(ψ ′(eC,1) − ψ ′(eB,0)
)= A0L
(σs(eC − eB) + μs
)= −WBC + A0Lμs. (4.13)
(iii) Path C → D:
� ′C→D = ′(1) − ′(0) = A0L
(ψ ′(eD,1) − ψ ′(eC,1)
)
= 1
2A0LE1(e
2D − e2
C)
= −WCD. (4.14)
(iv) Path D → A:
� ′D→A = ′(1) − ′(0) = A0L
(ψ ′(eA,0) − ψ ′(eD,1)
)= A0L
(σi(eA − eD) − μi
)= −WDA − A0Lμi. (4.15)
In (4.12) and (4.14) we have used (4.1) and in (4.13) and (4.15) we have used the commontangency conditions (4.3) and (4.4). Clearly, for the cycle ABCDA we have � ′
A→A = 0,and, thus, the total work done by the bar is
W = WAB + WBC + WCD + WDA = A0L(μs − μi). (4.16)
Also, it is clear from (4.13) and (4.15) that the total chemical work supplied to the barduring the cycle is A0Lμs > 0 and this is supplied only on the path B → C when the

246 R. Fosdick, G. Royer-Carfagni
chemical potential of the chemical bath is at its maximum value over the whole cycle. Thus,the efficiency of the chemomechanical engine for the cycle, being defined as the ratio of thetotal work done by the bar to the total chemical work that is supplied to the bar, is given by
eff = W
A0Lμs
= 1 − μi
μs
(4.17)
which has an evident similarity with the efficiency of a heat engine operating in a Carnotcycle between heat reservoirs at temperatures Tmin and Tmax.
The importance of (4.17) is highlighted by the following observation. If a chemomechan-ical engine operating in chemical baths at normalized chemical potentials lying between μi
and μs having an efficiency greater than (4.17) was possible, one could construct a device ca-pable of producing mechanical work without consumption of chemical work. Equivalently,for such a device, it would be possible to transfer some mass of substance S1 from a chem-ical bath at a certain normalized chemical potential to a chemical bath at a higher chemicalpotential with no consumption of mechanical work. That this is not possible is the counter-part in chemomechanics to Maxwell’s classical statement in thermodynamics that “heat, inthe form of heat,5 never passes out of a body except when it flows by conduction or radiationinto a colder body” ([10], p. 154). Paraphrasing a similar statement of Planck [11], p. 85, andexpressly stated by Clausius [3], p. 11, we may assert that for chemomechanics, chemicalwork cannot pass (i.e., mass exchange cannot take place) from an environment of a lowerchemical potential to an environment of a higher chemical potential without compensation.
An intuitive proof of the aforementioned statements follows from the most classical ideasof Carnot [2]. Recalling the initial assumption about the reversibility of the transformations,then by following the reverse cycle ADCBA, it would be possible to let the mass M1 ofsubstance S1 flow from the chemical bath with μ̄ = μi to a chemical bath with μ̄ = μs atthe expenses of mechanical work. If there existed any method whatever to make the chem-ical engine produce a quantity of motive power greater than that implied by (4.17), then itwould be sufficient to divert a portion of this power, by the method just indicated, so as tomake the substance S1 move from the reservoir at lower chemical potential to that at higherpotential in order to restore the initial conditions and, thus, to be ready to commence againan operation precisely similar to the former. Equivalently, if a method existed to produce anamount of work greater than that predicted by (4.17) at the expenses of the passage of themass M1 of substance S1 from the reservoir at μ̄ = μs to the reservoir at μ̄ = μi , then onecould use this work by performing a certain number of reversed cycles ADCBA with theeffect of letting a mass M∗
1 > M1 of S1 pass from the reservoir at μi to the reservoir at μs .The final result would be the passage of the mass M∗
1 − M1 from the reservoir at μ̄ = μi
to the reservoir at μ̄ = μs at the expenses of no other chemomechanical transformation ofany kind. One should then conclude that the maximum of mechanical work resulting fromthe employment of chemical power according to (4.17) is also the maximum of mechanicalwork realizable by any means whatever.
5 Discussion and Conclusions
In the example described above, the main form of interaction of the body with the exteriorbath other than mechanical work is the exchange of mass of a component of the bath and the
5Although strongly tautological, this is the original sentence of Maxwell as written in [10], p.154. As aninterpretation, one might replace the first “heat” in this sentence with “energy”.

On the Motive Power of Chemical Transformations in Open Systems 247
body at different environmental chemical potentials, an energy source that has been definedhere as the “chemical work”. If one believes in the laws of thermodynamics as universallaws governing the direction of all natural processes and the way in which different forms ofenergy may be interconverted, for which the different known expressions of the “secondlaw” in terms of thermal cycles are not more than particular instances, then one wouldexpect there to be “laws” such as those of Clausius, Kelvin and Planck that characterize thepossibilities or non-possibilities of certain isothermal chemomechanical cycles. One suchlaw might be: It is impossible for a thermally isolated body to produce positive mechanicalwork during a chemomechanical cycle if no mass is emitted.
In order to propose, albeit tentatively, an abstract formulation for isothermal chemome-chanical cycles in which no heat is exchanged, we draw upon the fundamental work of Serrin[12]. Let C(P ) and W(P ) denote the chemical work supplied to and the mechanical workdone by a body during a cyclic process P . Then, as an abstract first law one might take:
I. If P is a cyclic process of a body then W(P ) > 0 ⇒ C(P ) > 0.
Moreover, as was considered in [12] for classical thermodynamics, one may postulate theexistence of an accumulation function C(P,μ) which gives the chemical work transferredto the body during the chemomechanical process P at chemical potential less than or equalto μ and take an ad hoc version of the second law to be:
II. If P is a cyclic process of a body, then C(P ) > 0 ⇒ C(P,μ∗) < 0 for some μ∗ > 0.
Of course, for general processes in which chemical work can be converted into heat andvice-versa, a more comprehensive form of the general laws should be anticipated.
In closing, we emphasize that for a chemomechanical engine the chemical work playsthe same role as heat plays for a heat engine; it is the form of energy from which mechanicalwork is produced. In time-dependent chemomechanical processes, the primitive notion ofchemical working as a source of chemical power is of fundamental importance. For futurework, we wish to develop an elementary temporal theory for chemomechanical engines. Ef-ficiency estimates and the generalization to continuum chemomechanical processes will beof prime concern. Our inquiry will draw motivation from the elementary work of Truesdelland Bharatha [18] and it is expected to follow the earlier work of Fosdick and Serrin [6].
Acknowledgements Gianni Royer-Carfagni acknowledges the Italian MURST for its partial support underthe PRIN2008 program. This work also has been partially supported by Progetto Strategico Regione Puglia“S.I.S.M.A. Strutture Innovative e Sperimentazione di Materiali Avanzati”. Roger Fosdick acknowledges thePolitecnico di Bari, Dipartimento di Ingegneria Civile e Ambientale, for its hospitality and support during his2009-2010 sabbatical leave visit. We are in debt to an anonymous referee for keen comments that led to asubtle revision of a former version of this work. We also gratefully acknowledge the special help of MarcoRossi in the preparation of the graphs and figures.
References
1. Buonsanti, M., Fosdick, R., Royer-Carfagni, G.: Chemomechanical equilibrium of bars. J. Elast. 84,167–188 (2006)
2. Carnot, S.: Réflections sur la Puissance Motrice du Feu et sur le Machines propres a developer cettePuissance. Bachelier, Paris (1824). Reflections on the Motive Power of Fire, and on Machines Fitted toDevelop that Power. Translated by R.H. Thurston. Dover, New York (1960)
3. Clausius, R.: On the Motive Power of Heat, and on the Laws which can be Deduced from It for theTheory of Heat. Dover, New York (1960). Translated by W.F. Magie
4. Flory, P.J.: Principles of Polymer Chemistry. Cornell University Press, Ithaca (1957)5. Fosdick, R., Royer-Carfagni, G.: The static state of a two-phase solid mixture in a stressed elastic bar.
Int. J. Solids Struct. 33, 2267–2281 (1996)

248 R. Fosdick, G. Royer-Carfagni
6. Fosdick, R., Serrin, J.: Global properties of continuum thermodynamic processes. Arch. Ration. Mech.Anal. xx, 97–109 (1975)
7. Gibbs, J.W.: On the equilibrium of heterogeneous substances. Trans. Conn. Acad. III, 108–248 (1875–1878)
8. Kato, E.: Pressure-induced volume phase transition of polyacrylamide gels in acetone-water mixtures. J.Chem. Phys. 113, 1310–1314 (2000)
9. Kim, S.J., Spinks, G.M., Prosser, S., Whitten, P.G., Wallace, G.G., Kim, S.I.: Surprising shrinkage ofexpanding gels under an external load. Nat. Mater. 5, 48–51 (2005)
10. Maxwell, J.C.: Theory of Heat. Longmans, Green, London (1872)11. Planck, M.: Treatise on Thermodynamics, 3rd edn. Dover, New York (1945). Translated from the seventh
German edition by Alexander Ogg12. Serrin, J.: An outline of thermodynamical structure. In: Serrin, J. (ed.) New Perspective in Thermody-
namics, pp. 3–31. Springer, Heidelberg (1986)13. Spinks, G., Truong, V.T.: Work-per-cycle analysis for electromechanical actuators. Sens. Actuators A
119, 455–461 (2005)14. Stalhand, J., Klarbring, A., Holzapfel, G.A.: Smooth muscle contraction: Mechanichemical formulation
for homogeneous finite strains. Biophys. Mol. Biol. 96, 465–481 (2008)15. Suzuki, M., Hirasa, O.: An approach to artificial muscle using polymer gels formed by micro-phase
separation. Adv. Polym. Sci. 110, 241–261 (1993)16. Tanaka, T.: Collapse of gels at critical endpoint. Phys. Rev. Lett. 40, 820–823 (1978)17. Tanaka, T.: Phase transitions of gels. In: Harland, R., Prud’homme, R. (eds.) Polyelectrolyte Gels. ACS
Symposium Series, pp. 1–20. American Chemical Society, Washington DC (1992)18. Truesdell, C., Bharatha, S.: Classical Thermodynamics as a Theory of Heat Engines. Springer, New York
(1977)19. Zhang, Y.Q., Tanaka, T., Shibayama, M.: Super-absorbency and phase transition of gels in physiological
salt solutions. Nature 360, 142–144 (1992)




















