
Old and new biomarkers of oxidative stress in heart failure
31
Elsevier Editorial System(tm) for Drug Discovery Today: Therapeutic Strategies Manuscript Draft Manuscript Number: Title: Biomarkers of oxidative stress in heart failure and identification of novel carbonylated proteins in plasma of failing rabbit hearts Article Type: Heart failure 2013 Corresponding Author: Prof. Rainer Schulz, PhD MD Corresponding Author's Institution: Justus-Liebig University First Author: Sara Menazza, PhD Order of Authors: Sara Menazza, PhD; Marcella Canton, PhD; Elisa Sorato, PhD; Kerstin Boengler, PhD; Rainer Schulz, PhD MD; Fabio Di Lisa, MD
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Old and new biomarkers of oxidative stress in heart failure

Elsevier Editorial System(tm) for Drug Discovery Today: Therapeutic Strategies Manuscript Draft Manuscript Number: Title: Biomarkers of oxidative stress in heart failure and identification of novel carbonylated proteins in plasma of failing rabbit hearts Article Type: Heart failure 2013 Corresponding Author: Prof. Rainer Schulz, PhD MD Corresponding Author's Institution: Justus-Liebig University First Author: Sara Menazza, PhD Order of Authors: Sara Menazza, PhD; Marcella Canton, PhD; Elisa Sorato, PhD; Kerstin Boengler, PhD; Rainer Schulz, PhD MD; Fabio Di Lisa, MD

t
DIPARTIMENTO DI SCIENZE BIOMEDICHE
Viale Giuseppe Colombo, 3 - 35131 Padova - Italy
Direzione tel.: +39 049 827.6047 – 6061 Amministrazione tel.: +39 049 827.6045 – 6046 – 6042-5317
Fax: +39 049 827 6040 – 6049
C.F. 80006480281 – P.IVA 00742430283
Dr. Raymond Baker and Eliot Ohlstein Editors-in-Chief Drug Discovery Today: Therapeutic Strategies Padova, August 30, 2013 Dear Editors, Please find attached our manuscript entitled "Old and new biomarkers of oxidative stress in heart failure" that we are submitting upon your kind invitation to contribute to Drug Discovery Today: Therapeutic Strategies. The submitted manuscript has not been published previously and is not under consideration for publication elsewhere. All Authors have approved the manuscript in its current form. If accepted, this work will not be published elsewhere including electronically in the same form, in English or in any other language, without the written consent of the copyright-holder. Please note that as specified in the title page we propose to have both Prof. Rainer Schulz and Prof. Fabio Di Lisa as corresponding Authors. We hope that you will find our manuscript suitable for publication in Drug Discovery Today: Therapeutic Strategies. Best regards,
Prof. Fabio Di Lisa Department of Biomedical Sciences University of Padova Viale G. Colombo, 3 35131 Padova Italy
Cover Letter

Old and new biomarkers of oxidative stress in heart failure
Sara Menazzaa, Marcella Cantona, Elisa Soratoa, Kerstin Boenglerb, Rainer Schulzb*,
Fabio Di Lisaac*
a Department of Biomedical Sciences, University of Padova, Viale G. Colombo 3, 35131
Padova, Italy
b Institute of Physiology, Faculty of Medicine, Justus Liebig University, Aulweg 129, 35392
Giessen, Germany
c Institute of Neurosciences, CNR, University of Padova, Viale G. Colombo 3, 35131
Padova, Italy
* Corresponding Authors: Rainer Schulz and Fabio Di Lisa
Prof. Rainer Schulz
Institute of Physiology, Faculty of Medicine, Justus Liebig University, Aulweg 129,
35392 Giessen, Germany
Tel.: +496419947240
Email: [email protected]
Prof. Fabio Di Lisa
Department of Biomedical Sciences, University of Padova, Viale G. Colombo 3,
35131 Padova, Italy
Tel.: +390498276132
Fax: +390498276140
Email: [email protected]
ManuscriptClick here to view linked References

2
Highlights
Several molecular pathways and organs are involved in heart failure suggesting the
need for more objective measures for this disease.
Many oxidative stress biomarkers have been identified in plasma.
The relationship between oxidative stress within the failing heart and oxidative
event occurring in the plasma needs to be established.
Two novel targets of oxidative stress are identified in plasma and related to
contractile dysfunction in a heart failure model.
Abstract
Many cardiovascular diseases have been related to increased oxidative stress and
subsequent alterations in cardiomyocyte function and/or viability. As increased oxidative
stress might also modify non-cardiac proteins, quantitative relationships between plasma
proteins modified by reactive oxygen species and contractile abnormalities might be of
interest and become a diagnostic tool but have hardly been established yet. In the past
few decades several urine and serum biomarkers have been identified but the diagnostic
reproducibility of these tools as well as the scarcity of data evaluating their potential role in
heart failure development/progression is currently limited. Therefore, the different
biomarkers of oxidative stress and their relation to cardiac disease, especially heart failure,
are discussed and two novel plasma protein targets of oxidation – derived from
experimental studies - are identified.

3
Introduction
The utilization of oxygen as the terminal acceptor of electrons in the mitochondrial
respiratory chain gives a very large advantage in the conversion of nutrients into energy
that can be utilized by cells, especially in the form of ATP synthesis. For instance, the
complete oxidation of glucose to CO2 and H2O yields 19 times more ATP than the
anaerobic conversion to lactate. For this tremendous increase in energy efficiency, living
organisms have to pay the price of coping with oxygen that, besides its transformation into
water, easily generates reactive and potentially toxic molecules. In fact, since oxygen
accepts only one electron at a time, its complete reduction into water requiring 4 electrons
inevitably goes through three steps that generate partially reduced forms of oxygen,
namely superoxide anion (O2·-), hydrogen peroxide and hydroxyl radical (OH·)(as covered
by countless reviews; for instance see [1-4]). These molecules are generally termed as
reactive oxygen species (ROS) that include also singlet oxygen (1O2), hypochlorous acid
(HOCl), as well as alkoxyl (RO·), peroxyl (ROO·) and hydroperoxyl (HOO·) radicals. In
addition, oxygen is utilized in the formation of nitric oxide and nitrogen-centered reactive
species (RNS) that can further react with ROS, as in the case of peroxynitrite (ONOO-)
resulting from the interaction between NO and superoxide.
Cytochrome oxidase, the final reaction of the mitochondrial chain utilizing more than 90%
of oxygen supplied to cells, catalyzes the reduction into water proceeding through the
formation of potentially toxic intermediates that fortunately remain bound to the enzyme, so
that only the final product is released. Nevertheless, other physiological processes or
undesired reactions release ROS in every cell at any given time. A vast repertoire of
antioxidant defenses, both in the form of enzymatic activities and chemical scavengers,
abolishes ROS accumulation under physiological conditions. However, under pathological
conditions this delicate balance between ROS generation and removal is altered favoring
the increase in ROS levels as well as their deleterious attack to essential cellular
macromolecules, such as lipids, carbohydrates, proteins and nucleic acids. This imbalance
has been termed as oxidative stress that was originally introduced by H. Sies to describe
“a disturbance in the pro-oxidant–antioxidant balance in favour of the former, leading to
potential damage” [5].
Although ROS are involved in various physiological processes, the clinical interest related
to the development of diagnostic biomarkers is mostly, if not exclusively focused on the

4
relationships between ROS and diseases. This article is not meant at covering the current
debate on how and to what extent oxidative stress contributes to a wide array of
pathologies. Here, we aim at reviewing the association that has been established between
oxidation of plasma components and cardiac pathologies, especially heart failure. In
addition, two novel plasma biomarkers are described that we have shown to be linked with
intracellular oxidative stress and contractile dysfunction in an experimental model of heart
failure.
Biomarkers of oxidative stress in body fluids
Although the participation of ROS in functional and structural derangements is commonly
accepted, discrepancies exist on causal links whereby pathological states would depend
on oxidative stress. Besides negative results in clinical trials evaluating the efficacy of
antioxidant interventions [6], the lack of conclusive evidence is mostly due to uncertainties
on the methodologies used to detect oxidative stress in clinical settings.
Techniques available for assessing directly ROS formation in tissues can hardly apply to
studies in humans that are mostly based upon the detection of metabolites in plasma or
urine. Since ROS are compounds with an extremely short half-life, their assessment in
extracellular fluids withdrawn from patients is not feasible. This limitation is circumvented
by measuring more stable results of ROS-induced changes of molecular targets that are
considered as biomarkers of oxidative stress. However and unfortunately, the reliability of
all of them is far from being optimal (as reviewed in [7-10]). The most relevant
shortcomings are that the available markers of oxidative stress are not specific for any
disease and do not allow the prediction of the evolution of any given pathology. Therefore,
data can be collected just as associations between a disease state and the increase of a
given biomarker without providing conclusive information on the severity of the disease, its
prognosis, the mechanisms causing the increase in modified plasma components and the
causal links between the disease and oxidative stress. These limitations obviously apply to
the validation and monitoring of antioxidant treatment as well. Notably, while oxidative
stress can be reflected reliably by an increased oxidized state within cells, this might not
be the case in the extracellular space. In fact, while the intracellular ratio between thiols
(reduced) and disulfides is > 100:1, as shown by the redox state of glutathione, in plasma
the cysteine/cystine ratio is 1:40 [10]. Therefore, not only it is unlikely that antioxidants can

5
increase the reduced state in plasma, but also this might not represent a desired goal. In
addition, biomarkers adopted for oxidative stress hardly satisfy several other criteria
required reliability. Most of them are not chemically stable, their levels are frequently
influenced by diet or pathological states independently of oxidative stress and standard
levels are difficult to be determined since large variations are present among not only
individuals, but also assay procedures. Furthermore, some assays are technically
demanding and require expensive instruments limiting their use in routinely clinical
evaluations.
Despite all these limitations, the wide and strong interest to ROS involvement in
pathological states prompted the development of a large number of biomarkers. A detailed
review listed 71 different indicators in 2009 [10], a number that is likely to be higher at
present. Products of lipid and protein oxidation represent the majority of circulating
biomarkers of oxidative stress that include also DNA oxidation, as reflected by 8-hydroxy-
2'-deoxyguanosine [8,10].
Markers of lipid oxidation
The oxidative degradation of polyunsaturated lipids, commonly refereed as
lipoperoxidation, generates a large number of end-products the detection of which allows
the assessment of oxidative stress in tissues and in extracellular fluids. Among them the
most utilized for plasma assays are malondialdehyde (MDA) and isoprostanes, especially
in cardiovascular studies.
The assessment of MDA is based upon its reaction with thiobarbituric acid (TBA)
generating a stable chromophore that can be detected either spectrophotometrically or by
means of HPLC [11]. The first approach is the easiest and thus the most utilized, yet it is
flawed by the TBA reaction with other aldehydes that are not related to lipoperoxidation.
The specificity, though also the procedure complexity, is increased by separating and then
detecting the MDA-TBA adduct by HPLC. In any case sources of errors should be
considered, such as MDA derived from dietary sources, the artifactual generation of MDA
during sample preparation, and the sensitivity to metals, as well as to metal chelating
agents used to prevent blood clotting [8,10].
Notably, MDA is not a specific marker of lipoperoxidation, since it can result also from the
ROS attack on sialic acid and deoxyribose. An absolute specificity for lipoperoxidation is
attributed to isoprostanes (isoPs) formed by non-enzymatic free-radical-induced

6
peroxidation of arachidonic acid. These compounds have been termed F2-isoprostanes
(F2-IsoPs), since they are isomeric to prostaglandin (PG) F2 [8]. Although up to 64
isomers of F2-IsoPs can be formed, due to its abundance 15-F2-IsoP or 8-iso-PGF2 is the
product that is mostly used as a marker for oxidative stress [12].
IsoPs in plasma have a short half time, approximately 16 minutes [13], and are excreted
rapidly, which means that they must be formed constantly to maintain a steady-state
concentration. However, since IsoPs are structurally stable end-products, their excretion in
urine allows a cumulative detection of oxidative stress.
IsoPs can be assessed by either tandem mass-spectrometry (MS/MS) techniques (i.e.,
gas chromatograpy-MS/MS or liquid chromatography-MS/MS) or immunoassays, such as
EIA (enzyme immunoassay) or RIA (radioimmunoassay) [8,10,12]. MS-based assays are
more reliable and accurate, although they require a labor-intensive preparation of samples
along with expensive instruments.
IsoPs are likely to represent the best biomarker for lipid peroxidation. Nevertheless,
shortcomings have to be taken into account, such as elevation after fatty meal, plasma
concentrations close to detection limits, and direct excretion into urine of IsoPs generated
in the kidney.
Besides the lack of prognostic value, common concerns related to products of
lipoperoxidation are that they do not provide information on both the site of production and
the mechanisms linking oxidative stress in tissues with their elevation in body fluids. This
latter issue is complicated by the biological effects elicited by many of the molecules
generated by lipoperoxidation. Aldehydes are extremely reactive species that modify
covalently structures and functions of proteins and nucleic acids. In addition, by reacting
with glutathione aldehydes reduce its cellular content potentiating oxidative stress through
a decrease in antioxidant defenses [14,15]. IsoPs trigger signaling pathways upon their
binding to plasma membrane receptors [12,16]. Besides exerting vasoconstrictor and pro-
coagulant effects by means of effects on platelets and the endothelium [17,18], in liver
IsoPs have been shown to play a pro-fibrotic role by stimulating hepatic stellate cell
proliferation and collagen hyperproduction [16]. Therefore, the products of lipoperoxidation
generate a vicious cycle that is likely to amplify and exacerbate the initial injury. However,
causal relationships are ill defined, since it remains difficult to understand whether
molecules, such as MDA and IsoPs, are just biomarkers or contribute directly to causing
and maintaining pathological conditions.

7
Markers of protein oxidation
Proteins undergo various forms of oxidation [19-22] the most abundant of which is protein
carbonylation. This covalent modification can result from the following types of reactions:
(i) metals- or H2O2-catalyzed formation of semialdehydes at the level of lysine, arginine
and proline residues; (ii) in a process termed as Michael addition, the side chain of lysine,
histidine or cysteine can react via nucleophilic attack on C3 of , unsaturated aldehydes
generated by lipoperoxidation, such as 4-hydroxynonenal or the above mentioned MDA.
This second type is the most abundant; (iii) Schiff base formation between a lipid aldehyde
and the amino group of lysine residues. Notably, this type of lipid-protein modification, do
not generate a detectable free carbonyl group. Lysine residues can also react with
carbonyl derivatives formed by carbohydrate oxidation [20]. These glycation reactions
result in the formation of the so called advanced glycation end-products (AGEs) that are
suggested to play a relevant role in mechanisms of injury related to diabetes, whereas the
products of lipid-protein reactions, termed as advanced lipoxidation end products (ALEs),
are especially involved in atherosclerosis [10]. For instance, circulating MDA is not free,
but mostly bound to oxidized LDL (oxLDL) generated by macrophages within
atherosclerotic lesions and then released into the blood stream. Conceivably, the
assessment of MDA-oxLDL represents a valuable tool for investigating pathologies related
to atherosclerosis, including coronary artery disease [8,10,23].
Among the various amino acids in proteins, cysteinyl residues in the highly nucleophilic
thiolate form represent the major site for ROS attack. The initial oxidation to sulfenic acids
(R-SOH) can proceed to potentially reversible steps forming disulfides (R-SS-R),
sulfonamides and sulfinic acids that can further be oxidized irreversibly into sulfonic acids.
Additional reversible modifications of sulfenic acid are given by reacting with thiol
compounds, such as glutathione or cysteine, or with reactive nitrogen species generating
nitrosothiols in a process termed as S-nitrosation or S-nitrosylation. These reactions add to
cysteine carbonylation described above. The other sulfur-containing amino acid,
methionine, also is prone to oxidation generating methionine sulfoxide which can be
catalytically reduced by methionine sulfoxidases and methionine sulfoxide reductases
repairing oxidative damage to methionine in native proteins or further oxidized irreversibly
to methionine sulfone [22].

8
The general outcome of protein oxidation is inactivation. However, notable exceptions,
especially in signaling pathways, have been described in which carbonyl formation as well
as cysteine and methionine oxidation result in gain of function [20,22].
Products of protein oxidation can be detected by means of dedicated proteomic analyses,
or in some cases by immunoblotting [24]. Of note, protein carbonyls are assessed by
means of their reaction with 2,4 dinitrophenylhydrazine (DPNH) [25]. Western blot stained
with anti-DPNH antibodies (also termed as OxyBlot) is used to separate and identify the
oxidized proteins as also performed in the present study for the identification of novel
biomarkers.
Being the most abundant protein in plasma albumin is relevant as a circulating biomarker
of oxidative stress [26]. Albumin ability to bind and transport metals favors carbonylation.
On the other hand the redox properties of human serum albumin (HSA) are mostly related
to Cys34 whose low pKa (<6.7) favors the thiolate form. Cys34 in HSA represents the great
majority ( 80%) of all free thiols in plasma. Therefore, data on circulating free thiols by
which a decrease is a marker of oxidative stress can be considered a rough estimate of
HSA redox state that can be considered a significant fraction of plasma antioxidant
capacity. Various oxidized forms of albumin have been demonstrated in humans,
especially in kidney failure, but the relationships among them have not been investigated
yet.
Albumin exemplifies unsolved issues and open questions in the field of biomarkers for
oxidative stress and more in general in the clinical evaluation of ROS contribution to
pathophysiology, as well as in the diagnosis and therapy of a wide array of diseases.
Major questions appear to be as follows:
(i) Sensitivity and specificity. Is albumin oxidation more or less specific and sensitive than
that of other plasma proteins? Due to its exchange with the extravascular/interstitial space,
albumin could sense tissue oxidative stress before proteins confined to the intravascular
space. Its abundance could make it as a general probe of oxidative stress with little
specificity for a subset of diseases. A high sensitivity with a narrower broad of specificity
could pertain to proteins that are more directly involved in metal transport. By the way,
albumin oxidation affects its metal binding ability resulting in increases in free metal levels
that are likely to exacerbate oxidative stress;
(ii) Kinetics. How much oxidation affects plasma protein turnover/removal? And in the case
of albumin how much is HSA exchange among compartments impaired? For instance,

9
nitroalbumin crosses the blood-brain barrier 4 times faster than albumin, yet no information
is available for other modified forms;
(iii) Causal relationships. Do specific links exist between the various oxidized forms and
diseases?
Biomarkers of oxidative stress and heart failure
Oxidative stress is associated with most, if not all cardiovascular disorders and contributes
largely to their development and worsening. This applies to large ROS formation, since a
mild oxidative stress elicits protective mechanisms and appear to be necessary for
physiological responses to various forms of stress. The link between cardiovascular
diseases and abnormal ROS formation has been established by countless experimental
and clinical studies and covered by numerous excellent reviews (for instance, see [6,27-
33]). Initial evidence has been obtained in ischemia/reperfusion (I/R) injury during the
eighties [29]. Then, besides its role in inflammation and atherosclerosis, the contribution of
oxidative stress to HF, even independently of I/R, became evident. Indeed, ROS are
involved in every step leading to HF, such as development of hypertrophy, contractile
dysfunction, interstitial cardiac fibrosis, adverse remodelling after myocardial infarction,
loss of viability and endothelial dysfunction [32]. It is worth pointing out that, although ROS
contribution has been suggested for many cardiac diseases, including diabetic
cardiomyopathy [34] and aging [35], a non-ambiguous causal relationship can be
described only in a limited number of cases, such as Keshan syndrome due to selenium
deficiency [36] and anthracycline cardiotoxicity [37,38].
In HF, oxidative stress appears to be triggered and/or exacerbated by hormones and
cytokines released from the diseased heart creating a vicious cycle that amplifies the initial
injury. In this respect, catecholamines, angiotensin II, endothelin 1, aldosterone and TNF
appear to be the main agonists [39]. Their contribution is likely to be reinforced by
lipotoxicity in the case of diabetes and obesity [40-43]. All these factors trigger signaling
pathways that cause an increase in ROS formation within cardiomyocytes and vascular
cells. Among the many sources the most relevant are NADPH oxidase [44] and
mitochondrial dysfunction [45-48] that can be contributed by the increased activity of
specific enzymes, such as MAO [49,50] or p66Shc [51]. Eventually, the oxidative attack on
cellular components makes contractile derangements hardly surprising. In fact, ROS

10
hamper mitochondrial function and energy metabolism [52], channel activities and ion
homeostasis [53,54], and structure-function relationships in myofibrillar proteins [55-58].
These concepts have been established by experimental studies allowing to obtain direct
evidence of ROS formation and causal relationships with biochemical and functional
alterations occurring within cardiomyocytes. Nevertheless, many of the above mentioned
biomarkers have been utilized to provide clinical support to the association between
oxidative stress and HF [23,59-75] (reviewed in [6] and [76]). Most of the markers utilized
belongs to products of lipoperoxidation. Initial evidence was obtained by assessing MDA,
especially in patients suffering from ischemic heart disease [59-61]. In this respect, the
assessment of MDA-oxLDL has been suggested to provide reliable information of the
severity of vascular lesions along with having a significant prognostic value [23]. As
discussed above, MDA and oxLDL have an intrinsic link with atherosclerotic lesions and
coronary artery disease. Evidence of oxidative stress in HF independent of myocardial
ischemia has been obtained by measuring IsoPs in various body fluids and especially in
urine of HF patients [65,67,69,70,72-74]. IsoPs were found elevated also in patients
suffering for coronary artery disease [59,76], indicating that independently of
etiopathogenesis HF is associated with an abnormal generation of ROS that is reflected by
plasma and urinary biomarkers of oxidative stress. Indeed, MDA and IsoPS were
significantly higher in severe heart failure (NYHA class III and IV) and their levels
correlated with the severity of contractile dysfunction and/or left ventricle dilation
[64,65,67,69,70,72,73].
Recently, the involvement of oxidative stress in human I/R injury has been argued by
showing the absence of changes in arterio-venous differences of several circulating
biomarkers of oxidative stress in patients undergoing I/R protocols due to surgical
interventions on heart or kidney [77]. The argument is based upon measurements during
only the first hour of reperfusion. Interestingly, at least in kidney evidence of tissue
oxidative stress was documented by an increased expression of Nrf2, a transcription factor
stimulating the expression of antioxidant defenses. Therefore, this study does not appear
to challenge the involvement of oxidative stress in human I/R injury, as also demonstrated
previously by other means [78]. Rather, evidence is provided indicating that a prolonged
and/or more severe oxidative stress in tissues is required for detecting changes in
circulating biomarkers, and also that in acute phases oxidation events follows ROS-
induced changes within tissues. This might not be the case in chronic diseases, especially
when inflammatory responses involving circulating cells become relevant.

11
The increase in MDA and IsoPs was also associated with a decrease of circulating
antioxidants [61,63,75] along with an increase in SOD [61,75] and a decrease in
glutathione peroxidase [64,75]. However, since these enzymes are released by tissues,
the interpretation of their changes in plasma is not straightforward, also because the
activity assessment was not followed by structural studies documenting their oxidation.
This is a general caveat in the available studies that monitored lipoperoxidation products
with scarce attention to oxidation-induced changes in plasma proteins. For instance, it is
rather surprising that, at least to our knowledge, albumin oxidation was not assessed
directly, so that the evidence of its occurrence is left to indirect measurements. Few
studies reported the decrease in plasma free thiols [61,73] that is likely to be mostly
related to the oxidation of Cys34 in HSA. In addition, the increase in plasma carbonyls was
documented without identifying the proteins involved [75].
Even if direct evidence of albumin oxidation is obtained, due to its exchange with the
interstitial space it would be difficult to understand whether the observed changes are
generated by cardiac cells (and/or other tissues) or occur directly within the blood. This is
a relevant issue when effects of antioxidant or protective interventions are considered. In
fact, the important goal is to reduce oxidative stress in the injured organ. A decrease in the
levels of oxidation biomarkers in plasma is important only if it reflects reliably the reduction
of oxidative stress in the failing heart. In addition, components present in only plasma (i.e.,
not released from failing cardiomyocytes or diseased vessels) should be characterized to
understand whether and how much plasma oxidation follows oxidative stress generated
within failing hearts.
In this respect, also experimental studies fell short of relating oxidative events in plasma
with oxidation of intracellular components that are relevant for ensuing contractile
dysfunction. Our findings described below are aimed at filling these blanks. After
identifying novel targets of oxidative stress in plasma, we related their changes with both
the occurrence of oxidation in myofibrillar proteins and contractile dysfunction in a model of
heart failure, such as rapid atrial pacing in rabbit, devoid of ischemia.
Identification of novel proteins as biomarkers of heart failure independent of
coronary heart disease

12
Evidence of oxidative stress in pacing-induced HF model
The aim of our study was to find new specific biomarkers for HF, easily monitored in a
clinical setting. HF was induced in a rabbit model by rapid left ventricular (LV) pacing for 3
weeks. A total of 8 sham-operated rabbits and 7 HF rabbits were enrolled in this study.
Heart morphology and function are significantly modified after 3 weeks of LV pacing. HF
was evident from clinical signs, such as a strong decrease of LV fractional shortening (FS)
and a significant increase of hypertrophy and apoptosis. In an additional group, rabbit
received vitamins C and E for 3 weeks without (Sham-Vit, n=3) or with LV pacing (HF-Vit,
n=6). Administration of antioxidant vitamins during the 3 weeks of pacing significantly
decreased LVFS and reduced cardiomyocyte cross-sectional area and the number of
TUNEL-cardiomyocyte.
In our previous studies we developed quantitative assessment of oxidation of myofibrillar
proteins, in particular actin and tropomyosin, to detect the progression of HF in several
animal models as well as in human HF [55,58,79]. In this rabbit model the degree of
oxidative modifications of myofibrillar proteins is assessed using the oxyblot technique, a
method to measure protein carbonylation. Figure 1 shows that actin carbonylation is
significantly increased in hearts subjected to pacing induced HF compared to sham
operated rabbits, confirming our previous finding in other cardiac disease models. The
degree of actin carbonylation, measured by densitometric analysis, is approximately 50%
higher in HF compared to sham-operated rabbits (p=0.0043) (Fig.1 panel B). This result
confirms both the occurence of HF and the presence of oxidative stress in the myocardium
of this rabbit model.
Two novel markers of oxidative stress in plasma
To assess whether plasma proteins could be subjected to carbonylation, plasma samples
were collected from HF and sham-operated rabbits. To detect protein carbonylation the
plasma proteins were subjected to DNP-derivatization and then analyzed by immunoblot
probed with anti-DNP antibody. The representative 2D gel probed with anti-DNP antibody
in Figure 2 (panel A) shows a strong increase in plasma protein carbonylation in HF
compared to sham-operated rabbits, especially at the level of two proteins with a
molecular weight around 80 kDa. The identity of two major carbonylated spots was
confirmed using proteomic analysis as histidine-rich glycoprotein (HRG) and

13
serotransferrin (TRF), the upper and the lower spot respectively in the figure 2, panel A. In
order to quantify the amount of HRG and TRF carbonylation in plasma HF we used
western blot analysis: after incubation with anti-DNP antibody the membranes are stained
with anti-HRG and anti-TRF antibodies (Fig. 2 panel B). Densitometric analysis illustrated
in Figure 2 (panel C) shows that the amount of carbonylation in both the plasma proteins
was significantly increased in HF rabbits (a raise of 2.9 fold and 1.5 fold in HF vs sham-
operated group, HRG and TRF respectively, p<0.05). Administration of antioxidant
vitamins C and E throughout the three weeks of LV pacing completely and significantly
abolished the increase in plasma HRG and TRF carbonylation (Figure 2, panel C).
These results show, for the first time, the occurrence of plasma protein carbonylation in a
HF model, identifying two new major targets of oxidative stress in the plasma. Our finding
demonstrates that antioxidant treatment completely rescues protein plasma carbonylation,
keeping the degree of HRG and TRF carbonylation to physiological values. Moreover, this
evidence is entirely novel in the case of HRG carbonylation, demonstrating a new post-
modification at the level of this protein. Unlike HRG, TRF has been demonstrated to be
carbonylated in several stress-related diseases, including cancer, abdominal aortic
aneurysm, and Alzheimer disease and in aging mice [80-82]. Moreover, Guidi et al.
demonstrated a decrease in TRF carbonylation after regular physical exercise, a condition
that leads to cardiovascular protection reducing risk of obesity and of diabetes [83]. TRF is
an iron (Fe3+)-binding and -transporting protein. Thanan et al. suggested that the
accumulation of iron into a carcinoma tissue model is a result of the accumulation and
dysfunction of the iron transporter TRF due to carbonylation [84].
HRG and TRF carbonylation correlated with actin carbonylation and contractile
dysfunction in rabbit HF
Next, we aimed to demonstrate that the increased oxidative stress inside the heart is significantly
correlated with the occurrence of oxidative stress in plasma proteins. In our previous work we
correlated myofibrillar protein carbonylation with contractile dysfunction in human HF. However,
the process occurring within cardiomyocytes has not been related to oxidative stress in plasma
that can be easily monitored in both experimental and clinical settings. Here, we correlated HRG
and TRF carbonylation levels with actin carbonylation that, as we previously demonstrated, plays
a relevant role in contractile failure. Our findings demonstrated a positive linear correlation
between HRG and TRF to actin carbonylation (R2=0.7188 and R2= 0.4539, respectively) (Fig. 3).

14
HF is well established in causing contractile impairment, as shown by a strong reduction of FS
after three weeks of LV-pacing (table 1). In this work, we provided evidence of a correlation
between HRG and TRF plasma carbonylation and contractile dysfunction. Figure 4 (panel A)
shows a negative correlation between both plasma protein carbonylation and the decrease in FS
that occurs in hearts subjected to pacing induced HF model (R2=0.518 and R2= 0.517, for HRG
and TRF respectively). Of interest, figure 4 (panel B) shows that the loss in HRG and TRF
carbonylation after antioxidant treatment correlates with the higher values of LVFS that we
detected in HF-vitamin treated rabbits.
Taken together, these findings propose a strong rationale to exploiting protein plasma oxidation
as a marker to evaluate both HF diagnosis and, more importantly, to improve the efficacy of the
therapeutic interventions. We found two possible biomarkers associated with rabbit HF model,
focusing the attention on plasma-associated markers rather than on tissue-associated markers
because the application of circulating biomarkers in diagnostic laboratories would be relatively
simple. Further study is required to confirm the potential and the clinical role of HRG and TRF
carbonylation in plasma HF and to further investigate whether and how these proteins participate
in this disease in other HF animal models as well as in human HF.
Methods
The present study was approved by the bioethical committee of the district of Düsseldorf,
Germany, and all animals were treated and cared for in accordance with the Guide for the
Care and Use of Laboratory Animals published by the US National Institutes of Health
(NIH publication No. 85-23, revised 1996).
Experimental model
Male Chinchilla bastard rabbits (Charles River, Kisslegg, Germany) weighing 3-4 kg were
anesthetized, and a pacing lead was sutured onto the apical region of the left ventricle.
The pacing lead was connected to a pacemaker (Medtronic, Düsseldorf, Germany), which
was implanted subcutaneously as previously described [79]. After euthanasia of the
rabbits the hearts were quickly removed and stored in liquid nitrogen until use. Plasma
samples were collected by puncture of the LV at the end of the study and stored at -80oC.
Development of heart failure

15
HF was induced by rapid LV pacing (400 bpm) for 3 weeks (HF, n=7). Eight sham-
operated rabbits served as controls (sham). In two additional groups rabbits received
either placebo or vitamins C (100mg/kg/day per os) and E (200mg/day per os) without
(sham-vit, n=3) or with rapid LV pacing (HF-vit, n=6).
Clinical parameters
Heart rate and LV function were measured as previously described [85]. LV fractional
shortening (LVFS) was calculated using this equation: ((LV end-diastolic diameter –end-
systolic diameter)/ LV end-diastolic diameter X 100).
Apoptosis was assessed by means the TdT-mediated dUTP nick end labeling (TUNEL)
technique (In Situ Death Detection Kit, La Roche Diagnostic, Mannheim, Germany) and
the TUNEL-positive nuclei were counted using fluorescence microscopy (Leica DMLB,
Bensheim, Germany). Cardiomyocyte cross-sectional area was measured staining the
tissue sections with haematoxylin and eosin. Fibrosis was measured in random fields by
planimetry and the degree of tissue damage was expressed as percentage of the entire
field of view.
Protein extraction
Heart protein extraction was performed as previously reported [55]. Briefly, heart samples
were homogenized in ice-cold PBS, pH 7.2 containing 5 mM EDTA and then were
centrifuged at 12 000 x g for 10 min at 4°C. The resulting pellet was resuspended in
sample buffer (2% SDS, 5% glycerol, 125 mM Tris–HCl, and 10% -mercaptoethanol pH
6.8). Plasma samples were stored in liquid nitrogen and then were diluted (1:10) in ice-
cold PBS, pH 7.2. Protein concentration was determined by Bradford assay (Bio-Rad) both
for the heart and the plasma samples.
Oxyblot technique (protein carbonylation)
Total myocardial protein and plasma proteins carbonylation was measured using the
Oxyblot Protein Oxidation Detection Kit (Chemicon, USA) according to the manufacturer’s
protocol. Briefly, after protein quantification, carbonyl groups were derivatized by reaction
with 2,4-dinitrophenylhydrazine for 15 minutes at room temperature. Dinitrophenyl-(DNP-)
derivatized proteins were then separated using SDS-PAGE and transferred to a
nitrocellulose membrane. Membranes were incubated overnight at 4°C with anti-DNP
antibody (1:150 dilution), washed three times with TBS-Tween and incubated with

16
secondary goat anti-rabbit/horseradish peroxidase antibody (1:300) for 1 hour at room
temperature. Membranes were incubated with ECL reagent. Bands were visualized using
a Kodak X-Omat film processor. Successively, the membranes were stripped and probed
with anti--sarcomeric actin 5C5 clone (Sigma, USA), anti-HRG (Abcam) and anti- TRF
(GenWay Biotech, Inc.) antibodies. Quantification of actin, TRF and HRG carbonyl
modification was made by the ratio between the densitometric values of the corresponding
band in the oxyblot or of the bands in the Red Ponceau and those of the bands stained
with the corresponding antibody, analyzed using the ImageJ software (NIH, USA).
2 Dimension Electrophoresis
Plasma proteins (150 μg) were solubilized in a lysis buffer containing 8 M urea, 4%3-[(3-
Cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS), 20 mM dithiothreitol
(DTT) and 2.0% 4–7 nonlinear immobilized pH gradient (IPG) buffer.
The proteins were resolved by isoelectric focusing on 11-cm immobilized pH gradient
strips (pH 3-10). The IPG-strips were rehydrated at 30v for 10 hr and were focused
according to the following conditions:
Step 1:250V for 250vhr
Step 2: 500V for 500vhr
Step 3: 1000V for 1000vhr
Step 4: 8000V “Gradient” for 1hr
Step 5: 8000V for 66,667vhr
Step 6: 8000V for 1hr
After focusing, the IPG-stips were equilibrated for 10 min in 6 M urea, 30% glycerol, 2%
Sodium Dodecyl Sulfate (SDS), 0.05 M Tris–HCl, pH 6.8, 2% DTT, and subsequently for
10 min in the same urea/SDS/Tris buffer solution but substituting the 2% DTT with 2.5%
iodoacetamide. After IEF, IPG-strips were subjected to in-strip DNP derivatization
(derivatization of protein carbonyls) for 10 min.
The strips were loaded onto a gel and the proteins separated by electrophoresis on SDS-
PAGE. The membranes transferred to a nitrocellulose membrane and were incubated with
anti-DNP antibody as described in the paragraph above.
Mass spectrometry analysis
Plasma proteins were subjected to 2-dimensional electrophoresis followed by DNP
derivatization and a separation by SDS-PAGE. The gels were stained with colloidal

17
Coomassie Blue G-250 (Sigma) and the two interested bands were cut and sent to the
proteomic core facilities VIMM, University of Padua (Italy) for protein identification.
Statistics
Data are expressed as mean values±SEM. Cardiomyocyte cross-sectional area, TUNEL-
positive cardiomyocite and plasma protein carbonylaiton were compared between
untreated and treated sham and HF rabbits using one-way ANOVA. Actin carbonylation
between sham and HF rabbits was compared by two-tailed unpaired Student’s t-test.
Linear regression was performed to determine correlation between plasma protein
carbonylation and LVFS parameter. All statistical analysis was performed using Prism
program and values of P < 0.05 were considered significant.

18
Acknowledgements
This work was supported by the COST Action "EU-ROS" BM1203 (to R.S and F.D.L.) and
grants from the University of Padova, Fondazione Cariparo, and CNR (to F.D.L.).

19
References
1 Halliwell, B. (1991) Reactive oxygen species in living systems: source,
biochemistry, and role in human disease. Am J Med 91 (3C), 14S-22S
2 Finkel, T. and Holbrook, N.J. (2000) Oxidants, oxidative stress and the biology of
ageing. Nature 408 (6809), 239-247
3 Droge, W. (2002) Free radicals in the physiological control of cell function. Physiol
Rev. 82 (1), 47-95
4 Murphy, M.P. (2009) How mitochondria produce reactive oxygen species.
Biochem.J. 417, 1-13
5 Sies, H. (1985) Oxidative stress: introductory remarks. In Oxidative Stress (Sies, H.,
ed.), pp. 1-8, Academic Press
6 Sugamura, K. and Keaney, J.F., Jr. (2011) Reactive oxygen species in
cardiovascular disease. Free Radic Biol Med 51 (5), 978-992
7 Griffiths, H.R. et al. (2002) Biomarkers. Mol Aspects Med 23 (1-3), 101-208
8 Halliwell, B. and Whiteman, M. (2004) Measuring reactive species and oxidative
damage in vivo and in cell culture: how should you do it and what do the results
mean? Br J Pharmacol 142 (2), 231-255
9 Dalle-Donne, I. et al. (2006) Biomarkers of oxidative damage in human disease.
Clin Chem 52 (4), 601-623
10 Giustarini, D. et al. (2009) Oxidative stress and human diseases: Origin, link,
measurement, mechanisms, and biomarkers. Crit Rev Clin Lab Sci 46 (5-6), 241-
281
11 Pompella, A. et al. (1987) Measurement of lipid peroxidation in vivo: a comparison
of different procedures. Lipids 22 (3), 206-211
12 Roberts, L.J., 2nd and Morrow, J.D. (2002) Products of the isoprostane pathway:
unique bioactive compounds and markers of lipid peroxidation. Cell Mol Life Sci 59
(5), 808-820
13 Proudfoot, J. et al. (1999) Measurement of urinary F(2)-isoprostanes as markers of
in vivo lipid peroxidation-A comparison of enzyme immunoassay with gas
chromatography/mass spectrometry. Anal Biochem 272 (2), 209-215
14 Del Rio, D. et al. (2005) A review of recent studies on malondialdehyde as toxic
molecule and biological marker of oxidative stress. Nutr Metab Cardiovasc Dis 15
(4), 316-328

20
15 Roede, J.R. et al. (2010) 9.26 - Hepatotoxicity of Reactive Aldehydes. In
Comprehensive Toxicology (Second Edition) (Editor-in-Chief: Charlene, A.M., ed.),
pp. 581-594, Elsevier
16 Comporti, M. et al. (2008) F2-isoprostanes are not just markers of oxidative stress.
Free Radic Biol Med 44 (3), 247-256
17 Minuz, P. et al. (1998) The F2-isoprostane 8-epiprostaglandin F2alpha increases
platelet adhesion and reduces the antiadhesive and antiaggregatory effects of NO.
Arterioscler Thromb Vasc Biol 18 (8), 1248-1256
18 Hoffman, S.W. et al. (1997) Isoprostanes: free radical-generated prostaglandins
with constrictor effects on cerebral arterioles. Stroke 28 (4), 844-849
19 Berlett, B.S. and Stadtman, E.R. (1997) Protein oxidation in aging, disease, and
oxidative stress. J.Biol.Chem. 272 (33), 20313-20316
20 Grimsrud, P.A. et al. (2008) Oxidative stress and covalent modification of protein
with bioactive aldehydes. J Biol Chem 283 (32), 21837-21841
21 Winterbourn, C.C. and Hampton, M.B. (2008) Thiol chemistry and specificity in
redox signaling. Free Radic Biol Med 45 (5), 549-561
22 Burgoyne, J.R. et al. (2013) Hydrogen peroxide sensing and signaling by protein
kinases in the cardiovascular system. Antioxid Redox Signal 18 (9), 1042-1052
23 Tsutsui, T. et al. (2002) Plasma oxidized low-density lipoprotein as a prognostic
predictor in patients with chronic congestive heart failure. J Am Coll Cardiol 39 (6),
957-962
24 Eaton, P. (2006) Protein thiol oxidation in health and disease: techniques for
measuring disulfides and related modifications in complex protein mixtures. Free
Radic.Biol.Med. 40 (11), 1889-1899
25 Levine, R.L. et al. (2000) Determination of carbonyl groups in oxidized proteins.
Methods Mol.Biol. 99, 15-24
26 Colombo, G. et al. (2012) Redox albuminomics: oxidized albumin in human
diseases. Antioxid Redox Signal 17 (11), 1515-1527
27 Dhalla, A.K. et al. (1996) Role of oxidative stress in transition of hypertrophy to
heart failure. J Am Coll Cardiol 28 (2), 506-514
28 Singal, P.K. et al. (1998) The role of oxidative stress in the genesis of heart
disease. Cardiovasc Res 40 (3), 426-432
29 Bolli, R. and Marban, E. (1999) Molecular and cellular mechanisms of myocardial
stunning. Physiol.Rev. 79 (2), 609-634

21
30 Sawyer, D.B. et al. (2002) Role of oxidative stress in myocardial hypertrophy and
failure. J.Mol.Cell.Cardiol. 34 (4), 379-388
31 Giordano, F.J. (2005) Oxygen, oxidative stress, hypoxia and heart failure.
J.Clin.Invest 115, 500-508
32 Seddon, M. et al. (2007) Oxidative stress and redox signalling in cardiac
hypertrophy and heart failure. Heart 93 (8), 903-907
33 Sawyer, D.B. (2011) Oxidative stress in heart failure: what are we missing? Am J
Med Sci 342 (2), 120-124
34 Boudina, S. and Abel, E.D. (2007) Diabetic cardiomyopathy revisited. Circulation
115 (25), 3213-3223
35 Dai, D.F. et al. (2012) Cardiac aging: from molecular mechanisms to significance in
human health and disease. Antioxid Redox Signal 16 (12), 1492-1526
36 Fairweather-Tait, S.J. et al. (2011) Selenium in human health and disease. Antioxid
Redox Signal 14 (7), 1337-1383
37 Chen, B. et al. (2007) Molecular and cellular mechanisms of anthracycline
cardiotoxicity. Cardiovasc Toxicol 7 (2), 114-121
38 Eschenhagen, T. et al. (2011) Cardiovascular side effects of cancer therapies: a
position statement from the Heart Failure Association of the European Society of
Cardiology. Eur J Heart Fail 13 (1), 1-10
39 Al Ghouleh, I. et al. (2011) Oxidases and peroxidases in cardiovascular and lung
disease: new concepts in reactive oxygen species signaling. Free Radic Biol Med
51 (7), 1271-1288
40 Opie, L.H. and Knuuti, J. (2009) The adrenergic-fatty acid load in heart failure. J Am
Coll Cardiol 54 (18), 1637-1646
41 Wende, A.R. and Abel, E.D. (2010) Lipotoxicity in the heart. Biochim Biophys Acta
1801 (3), 311-319
42 Goldberg, I.J. et al. (2012) Lipid metabolism and toxicity in the heart. Cell Metab 15
(6), 805-812
43 Stanley, W.C. et al. (2012) Dietary fat and heart failure: moving from lipotoxicity to
lipoprotection. Circ Res 110 (5), 764-776
44 Nabeebaccus, A. et al. (2011) NADPH oxidases and cardiac remodelling. Heart Fail
Rev 16 (1), 5-12
45 Murray, A.J. et al. (2007) Mitochondria and heart failure. Curr Opin Clin Nutr Metab
Care 10 (6), 704-711

22
46 Marin-Garcia, J. and Goldenthal, M.J. (2008) Mitochondrial centrality in heart
failure. Heart Fail Rev 13 (2), 137-150
47 Rosca, M.G. and Hoppel, C.L. (2010) Mitochondria in heart failure. Cardiovasc.Res.
88 (1), 40-50
48 Osterholt, M. et al. (2012) Alterations in mitochondrial function in cardiac
hypertrophy and heart failure. Heart Fail Rev
49 Kaludercic, N. et al. (2011) Monoamine oxidases (MAO) in the pathogenesis of
heart failure and ischemia/reperfusion injury. Biochim.Biophys.Acta 1813 (7), 1323-
1332
50 Villeneuve, C. et al. (2013) p53-PGC-1alpha pathway mediates oxidative
mitochondrial damage and cardiomyocyte necrosis induced by monoamine
oxidase-A upregulation: role in chronic left ventricular dysfunction in mice. Antioxid
Redox Signal 18 (1), 5-18
51 Di Lisa, F. et al. (2009) Mitochondrial pathways for ROS formation and myocardial
injury: the relevance of p66(Shc) and monoamine oxidase. Basic Res.Cardiol. 104
(2), 131-139
52 Di Lisa, F. et al. (2011) Mitochondrial injury and protection in ischemic pre- and
postconditioning. Antioxid.Redox.Signal. 14 (5), 881-891
53 Zima, A.V. and Blatter, L.A. (2006) Redox regulation of cardiac calcium channels
and transporters. Cardiovasc Res 71 (2), 310-321
54 Csordas, G. and Hajnoczky, G. (2009) SR/ER-mitochondrial local communication:
calcium and ROS. Biochim Biophys Acta 1787 (11), 1352-1362
55 Canton, M. et al. (2006) Oxidative modification of tropomyosin and myocardial
dysfunction following coronary microembolization. Eur.Heart J. 27 (7), 875-881
56 Chung, H.S. et al. (2013) Cysteine oxidative posttranslational modifications:
emerging regulation in the cardiovascular system. Circ Res 112 (2), 382-392
57 Steinberg, S.F. (2013) Oxidative stress and sarcomeric proteins. Circ Res 112 (2),
393-405
58 Canton, M. et al. (2011) Oxidation of myofibrillar proteins in human heart failure.
J.Am.Coll.Cardiol. 57 (3), 300-309
59 Chopra, M. et al. (1990) Oxidative damage in chronic heart failure: protection by
captopril through free radical scavenging? Adv Exp Med Biol 264, 251-255
60 Belch, J.J. et al. (1991) Oxygen free radicals and congestive heart failure. Br Heart
J 65 (5), 245-248

23
61 McMurray, J. et al. (1993) Evidence of oxidative stress in chronic heart failure in
humans. Eur.Heart J. 14 (11), 1493-1498
62 Diaz-Velez, C.R. et al. (1996) Increased malondialdehyde in peripheral blood of
patients with congestive heart failure. Am.Heart J. 131 (1), 146-152
63 Reilly, M.P. et al. (1997) Increased formation of the isoprostanes IPF2alpha-I and 8-
epi-prostaglandin F2alpha in acute coronary angioplasty: evidence for oxidant
stress during coronary reperfusion in humans. Circulation 96 (10), 3314-3320
64 Keith, M. et al. (1998) Increased oxidative stress in patients with congestive heart
failure. J Am Coll Cardiol 31 (6), 1352-1356
65 Mallat, Z. et al. (1998) Elevated levels of 8-iso-prostaglandin F2alpha in pericardial
fluid of patients with heart failure: a potential role for in vivo oxidant stress in
ventricular dilatation and progression to heart failure. Circulation 97 (16), 1536-1539
66 Nishiyama, Y. et al. (1998) Oxidative stress is related to exercise intolerance in
patients with heart failure. Am Heart J 135 (1), 115-120
67 Cracowski, J.L. et al. (2000) Increased formation of F(2)-isoprostanes in patients
with severe heart failure. Heart 84 (4), 439-440
68 Tsutamoto, T. et al. (2001) Relationship between tumor necrosis factor-alpha
production and oxidative stress in the failing hearts of patients with dilated
cardiomyopathy. J Am Coll Cardiol 37 (8), 2086-2092
69 Nonaka-Sarukawa, M. et al. (2003) Increased urinary 15-F2t-isoprostane
concentrations in patients with non-ischaemic congestive heart failure: a marker of
oxidative stress. Heart 89 (8), 871-874
70 Polidori, M.C. et al. (2004) Increased F2 isoprostane plasma levels in patients with
congestive heart failure are correlated with antioxidant status and disease severity.
J Card Fail 10 (4), 334-338
71 Tingberg, E. et al. (2006) Lipid peroxidation is not increased in heart failure patients
on modern pharmacological therapy. Int J Cardiol 112 (3), 275-281
72 Wolfram, R. et al. (2005) Enhanced oxidative stress in coronary heart disease and
chronic heart failure as indicated by an increased 8-epi-PGF(2alpha). Eur J Heart
Fail 7 (2), 167-172
73 Radovanovic, S. et al. (2008) Markers of oxidative damage in chronic heart failure:
role in disease progression. Redox Rep 13 (3), 109-116

24
74 Caruso, R. et al. (2012) Severity of oxidative stress and inflammatory activation in
end-stage heart failure patients are unaltered after 1 month of left ventricular
mechanical assistance. Cytokine 59 (1), 138-144
75 Radovanovic, S. et al. (2012) Markers of oxidative damage and antioxidant enzyme
activities as predictors of morbidity and mortality in patients with chronic heart
failure. J Card Fail 18 (6), 493-501
76 Davies, S.S. and Roberts, L.J., 2nd. (2011) F2-isoprostanes as an indicator and risk
factor for coronary heart disease. Free Radic Biol Med 50 (5), 559-566
77 de Vries, D.K. et al. (2013) Oxidative damage in clinical ischemia/reperfusion injury:
a reappraisal. Antioxid.Redox.Signal. 19 (6), 535-545
78 Grech, E.D. et al. (1996) Evidence for free radical generation after primary
percutaneous transluminal coronary angioplasty recanalization in acute myocardial
infarction. Am.J.Cardiol. 77 (2), 122-127
79 Heusch, P. et al. (2010) The contribution of reactive oxygen species and p38
mitogen-activated protein kinase to myofilament oxidation and progression of heart
failure in rabbits. Br J Pharmacol 160 (6), 1408-1416
80 Gamberi, T. et al. (2011) A proteomic approach to identify plasma proteins in
patients with abdominal aortic aneurysm. Mol Biosyst 7 (10), 2855-2862
81 Yu, H.L. et al. (2003) Aberrant profiles of native and oxidized glycoproteins in
Alzheimer plasma. Proteomics 3 (11), 2240-2248
82 Jana, C.K. et al. (2002) Specificity of age-related carbonylation of plasma proteins
in the mouse and rat. Arch Biochem Biophys 397 (2), 433-439
83 Guidi, F. et al. (2011) Plasma protein carbonylation and physical exercise. Mol
Biosyst 7 (3), 640-650
84 Thanan, R. et al. (2012) Inflammation-induced protein carbonylation contributes to
poor prognosis for cholangiocarcinoma. Free Radic Biol Med 52 (8), 1465-1472
85 Schulz, R. et al. (2003) Stress kinase phosphorylation is increased in pacing-
induced heart failure in rabbits. Am J Physiol Heart Circ Physiol 285 (5), H2084-
2090

Figure legends
Fig.1. Actin carbonylation is increased in HF.
Septum proteins extract from biopsies of HF and Sham operated rabbits were subjected to
DNP-derivatization to detect protein carbonylation and then analyzed by immunoblot probed
with anti-DNP antibodies. (A) Red Poceau staining was performed for protein loading. Actin was
identified by immunoblot. (B) The Oxidation Index is given by the ratio between the
densitometric values of the actin band in the oxyblot and those of the corresponding bands
stained with Red Ponceau. * p<0.05 vs Sham. Sham: sham-operated rabbits (n=8); HF: heart
failure rabbits 3 weeks (n=7); Mean ± SEM.
Fig.2. Plasma protein carbonylation is increased in HF rabbits.
(A) Plasma proteins were extracted and subjected to 2D gel electrophoresis followed by oxyblot
analyses. (B) Plasma proteins from HF, Sham-operated, and HF and Sham-operated pretreated
with vitamins C and E rabbits were subjected to DNP-derivatization to detect protein
carbonylation and then analyzed by immunoblot probed with anti-DNP antibodies. Red Poceau
staining was performed for protein loading. (C) The densitometric analysis is given by the ratio
between the densitometric values of the STF or HRG bands in the oxyblot and the albumin band
stained with Red Ponceau. * p<0.05 vs. Sham, # p<0.05 vs HF. Sham: sham-operated rabbits
(n=8); Sham-Vit: sham-operated rabbits receiving vitamins C and E (n=3); HF: heart failure
rabbits (n=7); HF: heart failure rabbits receiving vitamins C and E (n=6); Mean ± SEM.
Fig.3. Relationship between HRG and STF carbonylation with Actin carbonylation in
plasma HF samples.
Sham: sham-operated rabbits (n=6); HF: heart failure rabbits, 3 weeks (n=7);
Fig.4. Carbonylation of HRG and STF correlates with contractile dysfunction.
(A) The degree of HRG and STF carbonylation correlates with the decrease in fractional
shortening during HF. (B) The degree of HRG and STF carbonylation in HF rabbits receiving
vitamins C and E treatment shows a linear trend line with the improvement in fractional
shortening. Sham: sham-operated rabbits (n=8); HF: heart failure rabbits (n=7); HF: heart failure
rabbits receiving vitamins C and E (n=6)
Figure legends

Figure 1Click here to download high resolution image

Figure 2Click here to download high resolution image

Figure 3Click here to download high resolution image
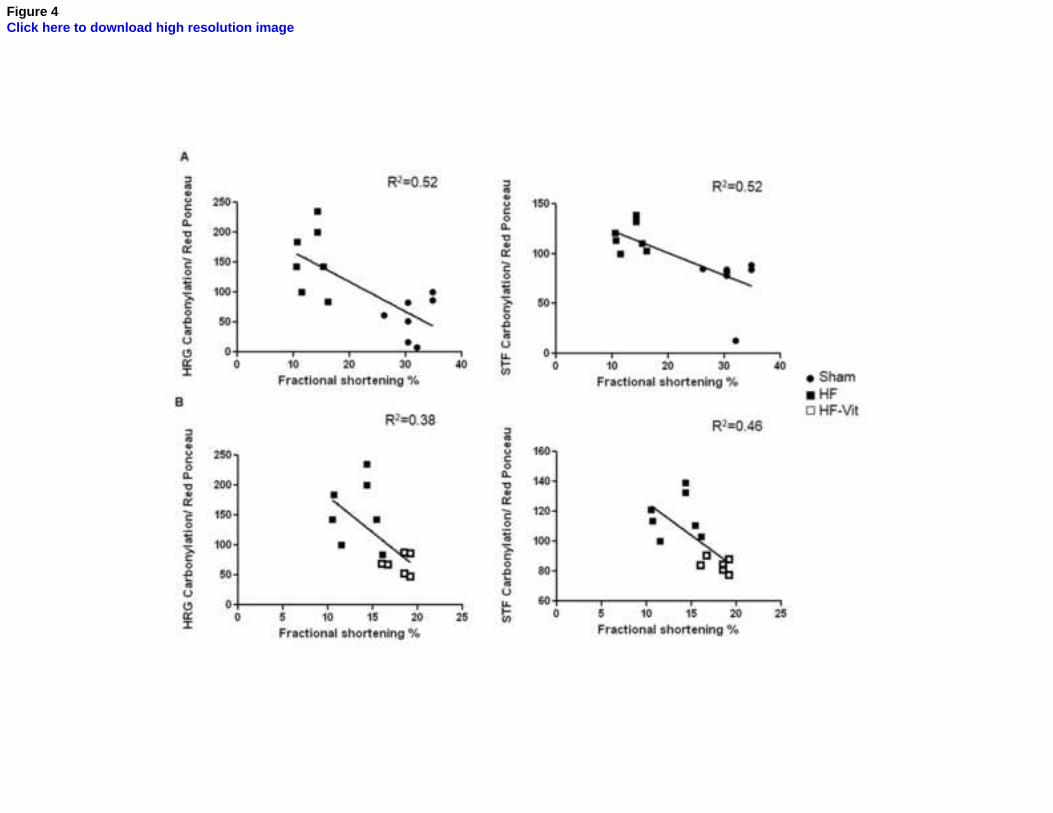
Figure 4Click here to download high resolution image




















