
Nonlinear Optical Properties of Ni(Me 6 pzS 2 )MX (M = Ni, Pd, Pt; X = Me 2 timdt, mnt)
21
Nonlinear optical properties of Ni(Me 6 pzS 2 )MX (M=Ni, Pd, Pt; X=Me 2 timdt, mnt) P. Romaniello 1,2 , M. C. D’Andria 3 , and F. Lelj 3 1 Laboratoire des Solides Irradi´ es UMR 7642, CNRS-CEA/DSM, ´ Ecole Polytechnique, F-91128 Palaiseau, France 2 European Theoretical Spectroscopy Facility (ETSF), F-91128 Palaiseau, France and 3 LaMI Dipartimento di Chimica and LaSCAMM, INSTM Sezione Basilicata, Universit` a della Basilicata, Via N. Sauro 85, Potenza 85100, Italy (Dated: November 30, 2009) Abstract In this work we investigate the second-order response of complexes of formula Ni(Me 6 pzS 2 )MX (M=Ni, Pd, Pt; X=Me 2 timdt, mnt=maleonitriledithiolate): by binding the porphyrazine to the metal-dithiolene the electron asymmetry and π-conjugation of the latter is increased, and its second- order response can result enhanced. By performing ab-initio calculations of the ground-state and response properties of these compounds, we predict the molecular first hyperpolarizability, we elu- cidate its electronic origin, and we illustrate its dependence on the metal and the dithiolene ligand. Our study indicates that these complexes show a very high second-order response, comparable to that of organic ’push-pull’ materials, and that the appropriate metal-dithiolene combination can significantly enhance it. 1
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Nonlinear Optical Properties of Ni(Me 6 pzS 2 )MX (M = Ni, Pd, Pt; X = Me 2 timdt, mnt)

Nonlinear optical properties of Ni(Me6pzS2)MX (M=Ni, Pd, Pt;
X=Me2timdt, mnt)
P. Romaniello1,2, M. C. D’Andria3, and F. Lelj3
1Laboratoire des Solides Irradies UMR 7642, CNRS-CEA/DSM,
Ecole Polytechnique, F-91128 Palaiseau, France
2European Theoretical Spectroscopy Facility (ETSF), F-91128 Palaiseau, France and
3LaMI Dipartimento di Chimica and LaSCAMM,
INSTM Sezione Basilicata, Universita della Basilicata,
Via N. Sauro 85, Potenza 85100, Italy
(Dated: November 30, 2009)
Abstract
In this work we investigate the second-order response of complexes of formula Ni(Me6pzS2)MX
(M=Ni, Pd, Pt; X=Me2timdt, mnt=maleonitriledithiolate): by binding the porphyrazine to the
metal-dithiolene the electron asymmetry and π-conjugation of the latter is increased, and its second-
order response can result enhanced. By performing ab-initio calculations of the ground-state and
response properties of these compounds, we predict the molecular first hyperpolarizability, we elu-
cidate its electronic origin, and we illustrate its dependence on the metal and the dithiolene ligand.
Our study indicates that these complexes show a very high second-order response, comparable to
that of organic ’push-pull’ materials, and that the appropriate metal-dithiolene combination can
significantly enhance it.
1

I. INTRODUCTION
With their wide range of possible applications, such as information processing, optical
switching, optical frequency conversion, and telecomunications [1, 2], nowadays nonlinear
optical materials play an important role in the technological development [3, 4]. The proper-
ties of these materials have been studied experimentally and computationally with a major
focus on organic compounds. Recently, there has been increasing interest in organometallic
and coordination complexes [2].
Metal 1,2-dithiolenes represent a very interesting class of coordination complexes due
to their unique properties, such as high thermal and photochemical stabilities, reversibly
connected oxidation states, and a very intense vis-IR absorption [5–8]. In particular the
dithiolenes derived from the dmit ligand (dmit = C3 S25, 1,3-dithiole-2-thione-4,5-dithiolate)
together with the d8 metal ions are the most known due to their low temperature elec-
trical superconducting properties [9–16]. In the last decade numerous dithiolenes of for-
mula M(R,R′timdt)2x− (M=Ni, Pd, Pt; R,R′timdt= monoanion of N, N ′-disubstituted
imidazolidine-2,4,5-trithione; x=0, 1, 2) complexes have been synthesized and fully char-
acterized both experimentally and theoretically. In particular, the neutral form shows a
very intense near-infrared (NIR) absorption, which has been attributed to a π → π∗ elec-
tronic transition between the HOMO and the LUMO, and occurs at energy values depending
on the nature of the R and R′ substituents, the metal, and the environment [6, 17]. The
wavelength and the high intensity of this NIR absorption band together with the high ther-
mal and photochemical stability candidate this class of dithiolenes for laser applications (see
Mitsubishi patent [18]) [19–21]. In particular, the presence of an intense low-energy charge-
transfer band, which involves a large difference between the dipole moment of the ground
state and the excited state, could lead to a strong second-order response, as predicted by the
two-level model [22]. In this model, only one excited state is responsible for the second-order
response, and the first hyperpolarizability tensor (second-harmonic generation, see later in
the text) can be expressed as
βiii ∝~ωegfeg∆µi
eg
[(~ω)2eg − (2~ω)2][(~ω)2
eg − (~ω)2], (1)
(in atomic units) where βiii represents the dominant tensorial component, ~ωeg and feg are
the excitation energy and oscillator strength, respectively, of the transition to the selected
2

excited state, ∆µeg is the difference between the excited-state and the ground-state dipole
moments, and ~ω is the frequency of the external laser source.
In Ref. [19] the second-order response of M(R,R′timdt)2 metal-dithiolenes has been in-
vestigated. This study shows that these compounds have a promising second-order re-
sponse, which can be enhanced by increasing the charge-tranfer character of the NIR ab-
sorption. This is, indeed, the case in the MXY mixed-ligand compounds (M=Ni, Pd, Pt;
X,Y=R2timdt, dmit, mnt (maleonitriledithiolate); X 6=Y) [23], which exhibit an enhanced
second-order response. This is in line with the well-established empirical observations that
the second-order response can be enhanced by increasing the electronic asymmetry (stronger
donator and acceptor moieties) and/or the π-conjugation between the donor and acceptor
groups.
These findings suggested us that one could obtain large second-order response by com-
bining metal-dithiolenes with porphyrazines. The latter are molecules with high delocalised
electronic structure in which the four pyrrole moieties are linked to each other by four aza
bridges. Recently unsymmetrical porphyrazines bearing a peripheral functionality capable
of binding metal ions have been investigated [24–29]. The peripheral functionalities can
significantly influence the electronic structure of the π-system in the porphyrazine, and con-
sequently the optical spectrum. In this paper we focus mainly on the porphyrazine of the
kind Ni[pz(A;B3)], where A e B are functional groups directly bound at the β-positions of
the pyrroles. The peripheral B moieties involve alkyl groups. The peripheral A moieties can
be an heteroatom of the kind of N or S (in our study it is a sulphur atom), which can bind a
metal ions (M= Ni, Pd, Pt) coordinated to a dithiolene ( mnt and Me2timdt). These com-
plexes have features in the UV-vis spectra that depend on the metal coordinating the ’core’
of the porphyrazine and on the peripheral substitution of the complex, hence allowing a fine
tuning of the electronic properties. Moreover, these complexes have two metals linked by
a polarizable bridge. Similar push-pull bimetallic complexes have already been investigated
in literature for their nonlinear optical properties [30, 31].
To fully characterise these complexes and their possible applications we perform ab-initio
calculations of their ground-state and response properties. The paper is organized as follows.
In Sec. II we give some theoretical and computational details. In Sec. III we discuss the
results calculated for the geometry, electronic structure, electronic excitations, and second-
order response of these complexes, and we focus, in particular, on the role played by the
3

peripheral metal and the dithiolene ligand in the studied properties. We show, in particular,
that increasing the electron asymmetry, passing from Me2timdt to mnt, and red-shifting
the dominant low-energy excitation (if there is a dominant excitation that determines the
second-order response), passing from Pd to Pt and Ni, the second-order response is very large
and comparable to that of extended π-organics molecules. Finally we draw our conclusions.
II. METHOD AND COMPUTATIONAL DETAILS
All results showed in this paper refer to ab-initio calculations based on density-functional
theory. In particular, for the response calculations we used time-dependent density func-
tional theory (TDDFT). Within this approach, after the first order equations have been
solved, one can calculate the first hyperpolarizability tensors, which govern the second-
order properties. In this work we will consider only the case in which the frequency of the
external perturbation is either 0 or ω, which gives rise to the following first hyperpolariz-
ability tensors: the static tensor β(0; 0, 0), and the tensors β(−2ω; ω, ω), β(−ω; 0, ω), and
β(0;−ω, ω), which govern the second-harmonic generation (SHG), the ElectroOptic Pockels
Effect (EOPE), and the Optical Rectification (OR), respectively.
Since d-metals are involved, scalar relativistic effects are taken into account in all our
calculations [17] by using the zeroth-order regular approximation (ZORA) [32]. We used a
valence double-ζ STO (Slater-type orbital) basis set with one polarization function (DZP) for
main element atoms and a triple-ζ STO basis set with one polarization function (TZP) for the
transition metals. The cores (C, N, O: 1s; S, Ni: 1s-2p; Pd: 1s-3d; Pt: 1s-4d) have been kept
frozen. All the results are converged with respect to the basis set size. Geometries have been
optimized by using the exchange-correlation potential proposed by Becke [33] and Perdew
[34] (BP). Excited states have been computed by using the asymptotically correct potential
proposed by van Leeuwen and Baerends (LB94) [35] and the SAOP (statistical average
of orbital potentials) [36, 37], and the hyperpolarizabilities by using the LB94 potential.
The choice of these functionals has been motivated by our previous calculations on similar
compounds [17, 19, 20, 23, 38, 39].
All the calculations have been performed using the ADF2003 computational package [40].
4

III. RESULTS
In this section we present the results obtained for the geometry, the electronic structure,
the excited states, and the second-order response of the Ni(pzMe6S2)MX (M=Ni, Pd, Pt;
X=mnt, Me2timdt) complexes (see Fig. 1). For simplicity, in the following we will indicate
with A and B the series with X=Me2timdt and X=mnt, respectively, and with AM and BM
(M= Ni, Pd, Pt) the complexes of each series.
A. Ground-state properties
1. Geometries
The geometries have been optimized assuming the system planar in the xz plane (see Fig.
1), with symmetry C2v and the singlet electronic ground state belonging to the A1 irreducible
representation. The dipole moments are along the z-axis, and they have opposite direction in
the two series. Although we have no experimental data for these complexes, the geometries
show that the bond lengths and bond angles are in agreement with the experimental data
found for similar complexes [24]. In Table I we compare the most representative bond
lengths and angles calculated for complex BPd with experimental data recorded for the
similar compound Ni[Pr6pz(NMe2)2]Pd(mnt) [24]: the agreement is in line with previous
studies on metal-dithiolenes [17, 19, 23, 38]. The geometry of the porphyrazine ring is not
influenced by the nature of the metal, as it is clear from the data reported in Table I, as
well as by the nature of the dithiolene ligand, since the bond lengths and angles of the
porphyrazine ring are similar in the two series. The M-S1 (S2) (and similarly the M-S3 (S4))
bond length is influenced both by the metal and by the dithiolene ligand. This bond length
increases passing from Ni to Pt, with a larger change from Ni to Pd than from Pd to Pt, as
already found in other metal-dithiolenes [17, 19, 23, 38]. The bond M-S3 is only marginally
affected by the nature of the dithiolene ligand: the variation between the two series are of
0.017 A , 0.008 A , and 0.007 A for Ni, Pd, and Pt, respectively, with series B showing the
largest values. The bond M-S1, instead, strongly feels the nature of the dithiolene ligand:
the variation between the two series are of 0.050 A, 0.035 A, and 0.037 A for Ni, Pd and Pt,
respectively, with series A showing the largest values. The remaining bond lengths as well
as the bond angles show in general small changes among the members of the two series.
5

2. Electonic structure
In order to better characterize these complexes we have analyzed the ground-state elec-
tronic structure in terms of the two ligand fragments Ni(Me6S2pz)x and Xy and the metal
M2+, with x =2-, y=0 for X=Me2timdt, and x =0, y =2- for X=mnt. This choice is based
on the Mulliken population analysis and on the calculated values of the ground-state dipole
moments (see later in the paper), which indicate as acceptor the porphyrazine ligand in
series A and the dithiolene ligand in series B. In Fig. 2 a schematic representation of the
energy levels of the two series is depicted. The analysis of this figure leads to the following
observations: the energy levels are similar within each series; in general, the energy levels of
series A are higher than those of series B, as it can be expected since each complex of the
former series has 8 electrons more than the corresponding complex of the latter series; the
LUMO is quite far from the LUMO+1 in both series; the HOMO tends to separate from the
other occupied MOs in series A, whereas it is quite close to the occupied MOs in series B;
the HOMO-LUMO energy gap of series A complexes is larger than that of the corresponding
series B complexes, with complexes APd and BPd showing the smallest gap (0.63 eV), and
complex ANi the largest gap (0.76 eV).
For both series the calculated MOs are, in general, mainly localized on the porphyrazine
ring. However, some of the orbitals that are involved in intense excitations of the systems, as
we will see in the next section, have large contributions from the peripheral metal and from
the dithiolene ligand as well. In Figs 3-4 we reported some selected orbitals (those which
will be mainly involved in the low-energy excitations) with the percentage compositions
in terms of the MOs of the fragments. In series A the HOMO (20b2) is delocalized both
on the porphyrazine ring and on the dithiolene ligand with a negligible contribution from
the peripheral metal. In series B, instead, the HOMO (13a2) is localized mainly on the
porphyrazine ring with a small contribution from the peripheral metal orbital ndxy, namely
9.8 %, 3.5 %, and 5.5 % for BNi, BPd, and BPt, respectively. The LUMO (21b2 for series
A and 19b1 for series B) is localized on the coordination sphere of peripheral metal, which
also contribute through the orbital ndyz with a percentage of 12.4 %, 8.9 %, and 10.6 %
for ANi, APd, and APt, respectively, and of 15.18 %, 10.5 %, and 13.4 % for BNi, BPd,
and BPt, respectively. The other selected orbitals in Figs 3-4 are mainly localized on the
porphyrazine ligand in both the series; the orbitals 17b1 and 18 b1 in series B have important
6

contributions also from the dithiolene ligand (> 20%).
From the analysis of the bonding energy in terms of the fragments [42] it results that
these complexes are very stable (the total bonding energy being -3339.8, -3303.8, and -3463.9
kJ/mol for ANi, APd, and APt, respectively, and -3457.1, -3411.1, and -3577.2 kJ/mol for
BNi, BPd, and BPt, respectively), which is an essential requirement for laser applications.
B. Electronic spectra
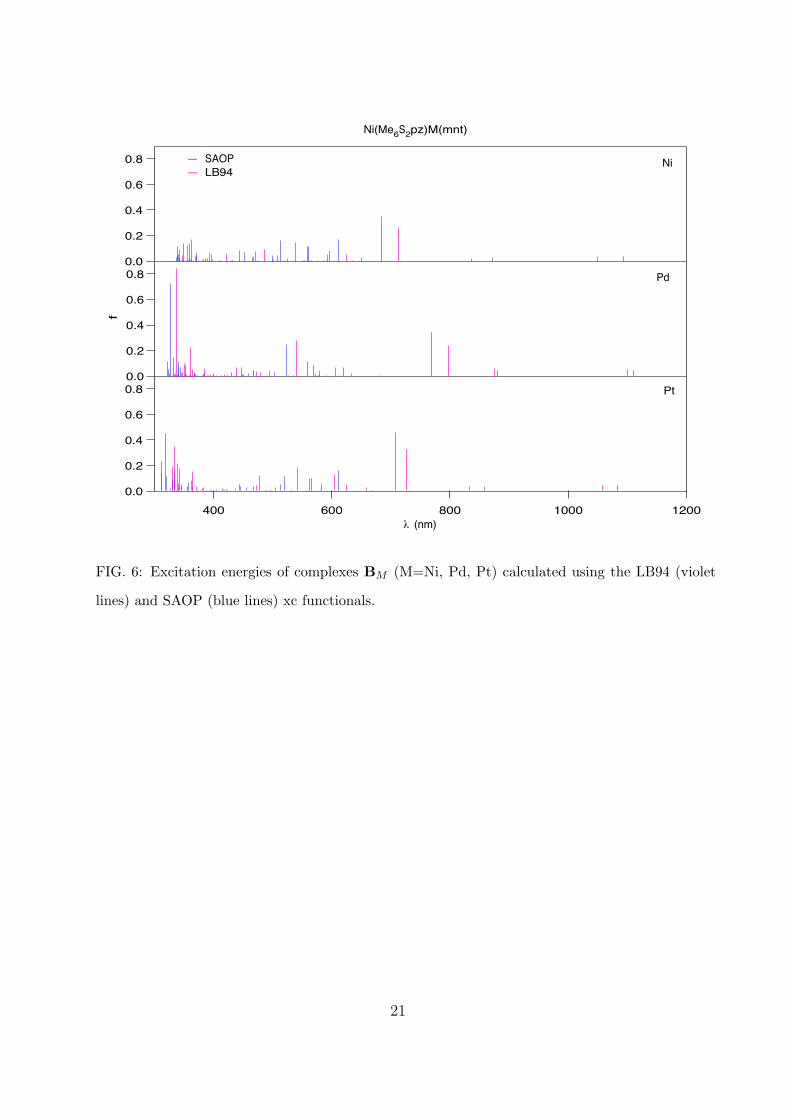
We have calculated the excitation energies for the Ni(Me6S2pz)MX ( M=Ni, Pd, Pt;
X=mnt, Me2timdt) complexes in the range around 300-1000 nm using both the LB94 and
SAOP xc functionals. This choice of the functionals is justified by previous studies on
metal-dithiolenes and porphyrazines [17, 38, 39, 41]: in that case the two functionals give
substantially the same overall picture of the spectra, with the LB94 better describing the
high wavelength range and the SAOP the lower wavelength range. The symmetry point
group of these complexes is the C2v, with the singlet ground-state belonging to the A1
irreducible representation. The spin- and symmetry-allowed excitations are those to the
singlet excited states belonging to the A1, B1, and B2 irriducible representations. In Figs 5-
6 we reported the calculated excitation energies. All the complexes show a similar spectrum.
Considering only the most dominant transitions (oscillator strength ≥ 0.1, mainly belonging
to the A1 representation), the spectra can be arbitrarily separated in three main regions:
the first two in the ranges 300-400 nm and 400-650 nm, which can be identified with the
N,B and Q bands, respectively, of the porphyrazine, and the third region at wavelengths
higher than 650 nm, which can be identified with the excitations of the metal-dithiolene.
Furthermore, for the near infrared (NIR) peaks we observe within each series the same
trend found for other metal-dithiolenes: a blue-shift of the peaks in the complex with Pd
with respect to the complexes with Ni and Pt. The analysis of the main excitations in
terms of monoelectronic transitions reveals a similar nature of the spectra with the two xc
functionals, although SAOP yields in general a stronger multiconfigurational character of
the spectra, specially for low wavelengths. In general the spectra of these complexes show
strong charge transfers (CT) from/to the metals to/from the ligands, and inter/intraligands
CT. Large charge transfers from one side of the molecule to another can result in a large
change in the dipole moment, which, in turn, can play an important role in the second-order
7

response. Therefore, the analysis of the excitation energy spectrum and of the nature of
the predominant transitions can give a first insight in the second-order response of these
complexes. In particular, in Table II we reported the major monoelectronic transitions
contributing to the LB94 excitations for λ > 650 nm (SAOP compositions are essentially
similar).
1. Uv-vis-IR spectrum of Ni(Me6S2pz)M(Me2timdt])complexes
We first analyze the spectrum of complexes ANi, APd, and APt. In general, the spectrum
of complex ANi has a dominant ligand-to-ligand (both inter and intraligand) and metal-to-
ligand charge transfer character. However, due to the complexity of the molecules, also
metal-to-metal and ligand-to-metal charge transfers are present. In the low-energy region
(λ > 650 nm) there are three main excited states responsible for the absorption, namely 1
A1, 2 A1, and 3 A1. Intra/interligand and metal-to-ligand charge transfers are predominant,
as it becomes clear from Table II and from the MOs depicted in Fig. 3. In particular, we
observe that there are two opposite tendencies in the one-electron excitations involved in
the low-energy absorption: charge transfers from the porphyrazine ligand towards the metal
and the dithiolene ligand (namely 19b2 → 21b2), but also the viceversa (namely 20b2 →
22b2). This might result in a small dipole moment in the related excited state. Furthermore,
only the one-electron excitation 20b2 → 21b2 (HOMO→LUMO) involves MOs with large
overlap, which explains also why the excited state 2 A1 shows the largest oscillator strength.
Note also that, unlike the corresponding symmetric metal-dithiolenes, the NIR absorption is
not predominantly due to the HOMO→ LUMO transition. Complexes APd and APt show
similar spectral features, in particular the low-energy region has essentially a similar nature
as in complex ANi.
2. Uv-vis-IR spectrum of Ni(Me6S2pz)M(mnt) complexes
In general, ligand-to-ligand and metal-to-ligand charge transfers are predominant in the
spectra of series B, as in series A. However, the nature of the low-energy region is different
from series A. First, there exist a net charge transfer from the porphirazine ligand to the
peripheral metal and dithiolene ligand; this can yield a large dipole moment in the related
8

excited state. Second, no HOMO→LUMO transition is involved in the low-energy region.
In fact the lowest excitation energies for these complexes (related to the excited state 1B2)
is a pure HOMO→LUMO transition; it is, however, very weak (oscillator strength <10−3)
and hence not appearing in Fig. 6. We also notice that the lowest-energy absorption falls at
λ > 1000 nm in these complexes, whereas it falls at λ < 1000 nm in series A.
C. Second-order response
The analysis of the excitation energies in the previous section showed that combining
a metal-dithiolene with a porphyrazine one build complexes in which there can be intense
excitations that fall at low energies and for which the difference in the dipole moment
between the ground-state and the relative excited state (∆µeg) can be large. According to
the two-level model (see in the Introduction) this could lead to a large second-order response.
Therefore, we have calculated the off-resonance (0.35 eV) second-order response of these
complexes. In Table III the vector component of the β tensors along the dipole moment
direction (the z axes in the studied complexes) and the predominant tensorial component βzzz
are reported. We have also indicated the value of the dipole moment, in order to indicate the
electronic asymmetry in the ground-state. The studied complexes show a significant second-
order response, much larger than that calculated for the (quasi)symmetric- and mixed-ligand
dithiolenes previously investigated [19, 23]. Series B shows a larger second-order response
than series A, with complex BPd having the largest values of β (βSHGvec =355.5 · 10−30 esu).
The low-energy excitations are probably the main responsible for the second-order response
in these complexes, since they fall at very low energies and they can have strong oscillator
strengths. Moreover, these excitations are characterized by ∆µeg values that can be expected
larger in series B than in series A, in line with the larger β values calculated in the former.
The trend of the β values within series B seems to be explained by the two-level model,
assuming that the excited state 3 A1, which has the largest oscillator strength in the low-
energy region, gives the dominant contribution to the first hyperpolarizability. To show this,
we stress that the three main excitations in the low-energy region have similar nature (hence
one could assume a similar ∆µeg ) and similar oscillator strengths among the complexes of
the series. Furthermore, for each complex of the series the three excitations show a similar
nature, whereas the oscillator strengths are very different, with the excited state 3 A1 showing
9

a much larger oscillator strength than 1 A1 and 2 A1. If one assumed this excited state as
the main responsible for the second-order response in series B, then complex BPd, which
has the lowest-energy excitation, would have the largest β vector according to the two-level
model and in line with the values of β reported in Table III.
The two-level model, instead, does not explain the trend of the β values in series A. In
this case, assuming a predominant excitation in the low-energy region and using the same
arguments valid for series B, one would get the largest second-order response for complex
APd, whereas the results in Table III show the trend βAPt> βANi
> βAPd. The ”failure” of
the two-level model may be explained by the fact that the nature of the three main excited
states in the low-energy region is not as uniform as in series B and not coherent with the
trend of the oscillator strengths and excitation energies. To be more specific, the excited
states 1 A1 and 3 A1 show a stronger CT character than the excited state 2 A1, but a lower
oscillator strength. Therefore, there might not be a predominant excitation, but rather the
whole region at λ > 700 nm could be responsible of the second-order response.
IV. CONCLUSIONS
We investigated the ground state and response properties of Ni(Me6pzS2)MX (M=Ni,
Pd, Pt; X=Me2timdt, mnt) complexes within (time-dependent) density functional theory.
These compounds combine together a porphyrazine, which has a very delocalized π-system,
with a metal-dithiolene, which, with their intense π → π∗ NIR band and high thermal
and photochemical stabilities, are promising candidates for laser applications. From the
analysis of the ground state it results that, whereas in the complexes with X=Me2timdt
(series A in the article) the charge is quite uniformly distributed (small dipole moment) in
the molecules, in the complexes with X=mnt (series B in the article) there is a net tendency
of the porphirazine ligand to behave as donor and the dithiolene ligand as acceptor, which
resembles the structure of nonlineral push/pull organic materials. The calculation of the
excitation energies shows that also these complexes exhibit intense low-energy absorptions
with strong change transfers between the two ligands and between the ligands and the
peripheral metal. Series A shows charge transfers from the porphyrazine ligand to the metal
and dithiolene ligand and viceversa, which can result in a small dipole moment in the exited
states; series B, instead, shows a net CT from the porphyrazine to the metal and dithiolene
10

ligand, which can be a signal of large dipole moment in the excited states. All these features
can lead to a large second-order response. Indeed our values of the first hyperpolarizability
tensors, calcuclated off-resonance (0.35 eV) for the second-harmonic generation(SHG), the
ElectroOptic Pockels Effect (EOPE), and the Optical Rectification (OR), respectively, show
that all the complexes have a second-order response comparable to the typical push/pull
organic molecules. In particular, the response is strongly enhanced in series B (βvec >
100 · 10−30), with the complex of palladium showing the largest values of the β tensors
(βSHGvec =355.5 · 10−30). The trend of the β values in series B can be explained by the
two-level model, assuming one of the low-energy excitations as the main responsible for the
second-order response. In series A, instead, all the main low-energy excitations are probably
responsible for the second-order response.
[1] Marder, S. R.; Sohn, J. E.; Stucky, G. D.; Eds. ACS Symposium series 455; American Chemical
Society: Washington, DC, 1991.
[2] Benson, M. T.; Cundari, T. R.; Lim, S. J.; Nguyen, H. D.; Pierce-Beaver, K. J. Am. Chem.
Soc. , 1994, 116, 3955-3966.
[3] Wagner, R. W.; Lindsey, J. S.; Seth, J.; Palaniappan, V.; Bocian, D. F. J. Am. Chem. Soc.
1996, 118, 3996-3997.
[4] Reimers, J. R.; Lu, T. X.; Crossley, M. J.; Hush, N. S. Chem. Phys. Lett. 1996, 256, 353-359.
[5] Nakamura, T.; Underhill, A. E.; Coomber, A. T.; Friend, R. H.; Tajima, H.; Kobayashi, H.
Inorg. Chem. 1995, 34, 870-876.
[6] Aragoni, M. C.; Arca, M.; Demartin, F.; Devillanova, F. A.; Garau, A.; Isaia, F.; Lelj, F.;
Lippolis, V.; Verani, G. J. Am. Chem. Soc. 1999, 121, 7098-7107.
[7] Williams, R.; Billing, E.; Waters, J. H.; Gray, H. B. J. Am. Chem. Soc. 1966, 88, 43-50.
[8] Ferraro, J. R.; Williams, J. M. Introduction to synthetic electrical conductors; Academic: New
York, 1987.
[9] Cassoux, P.; Valade, L.; Kobayashi, H.; Kobayashi, A.; Clark, R. A.; Underhill, A. E. Coord.
Chem. Rev. 1991, 110, 115-160.
[10] Pomarede, B.; Garreau, B.; Malfante, I.; Valade, L.; Cassoux, P.; Legros, J. P.; Audouard, A.;
Brossard, L. Ulmet, J. P.; Doublet, M. L.; Canadell, E. Inorg. Chem. , 1994, 33, 3401-3414.
11

[11] Sun, S.; Wu, P.; Zhu, D.; Ma, Z.; Shi, N. Inorg. Chim. Acta 1998, 268, 103-107.
[12] Sato, A.; Kobayashi, H.; Naito, T.; Sakai, F.; Kobayashi, A. Inorg. Chem. 1997, 36, 5262-
5269.
[13] Kochurani; Singh, H. B.; Jasinski, J. P.; Paight, E. S.; Butcher, R. J. Polyhedron 1997, 16,
3505-3510.
[14] Matsubayashi, G.; Natsuaki, K.; Nakano, M.; Tamura, H.; Arakow, R. Inorg. Chim. Acta
1997, 262, 103-107.
[15] Veldhuizen, Y.; Veldman, N.; Lakin, M. T.; Spek, A. L.; Paulus, P. M.; Faulmann, C.;
Haasnoot, J. G.;, Maaskant, M. J. A.; Reedijk, J. Inorg. Chim. Acta 1996, 245, 27-34.
[16] Fujiwara, H.; Arai, E.; Kobayashi, H. Chem. Commun. 1997, 9, 837-838.
[17] Romaniello, P.; Aragoni, M. C.; Arca, M.; Cassano; T. Denotti, C.; Devillanova, F. A.; Isaia,
F.; Lelj, F.; Lippolis, V.; Tommasi, R. J. Phys. Chem. A 2003, 107, 9679-9687.
[18] Sato, I.; Saito, Y., Patent JP 2005099755, 2005.
[19] Romaniello, P.; Lelj, F. J. Mol. Struct. 2003, 636, 23-37.
[20] Cassano, T.; Tommasi, R.; Nitti, L.; Aragoni, M. C.; Arca, M.; Denotti, C.; Devillanova, F.
A.; Isaia, F.; Lippolis, V.; Lelj, F.; Romaniello, P. J. Chem. Phys. 2003, 118, 5995-6002.
[21] Curreli S.; Deplano P.; Faulmann C.; Ienco A.; Mealli C.; Mercuri M. L.;, Pilia L.; Pintus G.;
Serpe A.; Trogu E. F. Inorg. Chem. 2004, 43, 5069-5079.
[22] Oudar, J. L. J. Chem. Phys. 1977, 67, 446-457; Oudar, J. L; Chemla, D. S. J. Chem. Phys.
1977, 66, 2664-2668.
[23] Romaniello, P.; Lelj, F. Chem. Phys. Lett. 2003, 372, 51-58.
[24] Lange, S. J.; Nie, H.; Stern, C. L.; Barrett, A. G. M.; Hoffman, B. M. Inorg. Chem. 1998,
37, 6435-6443.
[25] Sibert, J. W.; Baumann, T. F.; Williams, D. J.; White, A. J. P.; Barrett, A. G. M.; Hoffman,
B. M. J. Am. Chem. Soc. 1996, 118, 10487-10493.
[26] Sakellariou, E. G.; Montalban, A. G.; Meunier, H. G.; Rumbles, G.; Phillips, D.; Ostler, R.
B.; Suhling, K.; Barrett, A. G. M.; Hoffman, B. M. Inorg. Chem. 2002, 41, 2182-2187.
[27] Lee, S. W.; White, A. J. P.; Williams, D. J.; Barrett, A. G. M.; Hoffman, B. M. J. Org. Chem.
2001, 66, 461-465.
[28] Baumann, T. F.; Nasir, M. S.; Sibert, J. W.; White, A. J. P.; Olmstead, M. M.; Williams, D.
J.; Barrett, A. G. M.; Hoffman, B. M. J. Am. Chem. Soc. 1996, 118, 10479-10486.
12

[29] Ghosh, A.; Gassman, P. G.; Almlof, J. J. Am. Chem. Soc. 1994, 116, 1932-1940.
[30] Pizzotti, M.; Ugo, R.; Roberto, D.; Bruni, S.; Fantucci, P. C.; Rovizzi, C. Organometallics
2002, 21, 5830-5840.
[31] Bruschi, M.; Fantucci, P. C.; Pizzotti, M. J. Phys. Chem. A 2005, 109, 9637-9645.
[32] van Lenthe, E.; Baerends, E. J.; Snijders, J. G. J. Chem. Phys. 1993, 99, 4597-4610; J.
Chem. Phys. 1994, 101, 9783-9792.
[33] Becke, A. Phys. Rev. A 1988, 38, 3098-3100.
[34] Perdew, J. P. Phys. Rev. B 1986, 33, 8822-8824.
[35] van Leeuwen, R.; Baerends, E. J. Phys. Rev. A 1994, 49, 2421-2431.
[36] Gritsenko, O. V.; Schipper, P. R. T.; Baerends, E. J. Chem. Phys. Lett. 1999, 302, 199-207.
[37] Schipper, P. R. T.; Gritsenko, O. V.; van Gisbergen, S. J. A.; Baerends, E. J. J. Chem. Phys.
2000, 112, 1344-1352.
[38] Romaniello P.; Lelj, F.; Arca, M.; Devillanova, F. A. Theor. Chem. Acc. 2007, 117, 621-635.
[39] Infante, I.; Lelj, F. Chem. Phys. Lett. 2003, 367, 308-318.
[40] Fonseca Guerra, C.; Visser, O.; Snijders, J. G.; te Velde, G.; Baerends, E. J. In Methods and
Techniques of Computational Chemistry ; Clementi, E. , Corongiu, G. (Eds.): STEF, Cagliari,
1995; pp. 303-395.
[41] Baerends, E. J.; Ricciardi, G.; Rosa, A.; van Gisbergen, S. J. A. Coord. Chem. Rev. 2002,
230, 5-27.
[42] Ziegler, T.; Rauk, A. Theor. Chim. Acta 1977, 46, 1-10.
13
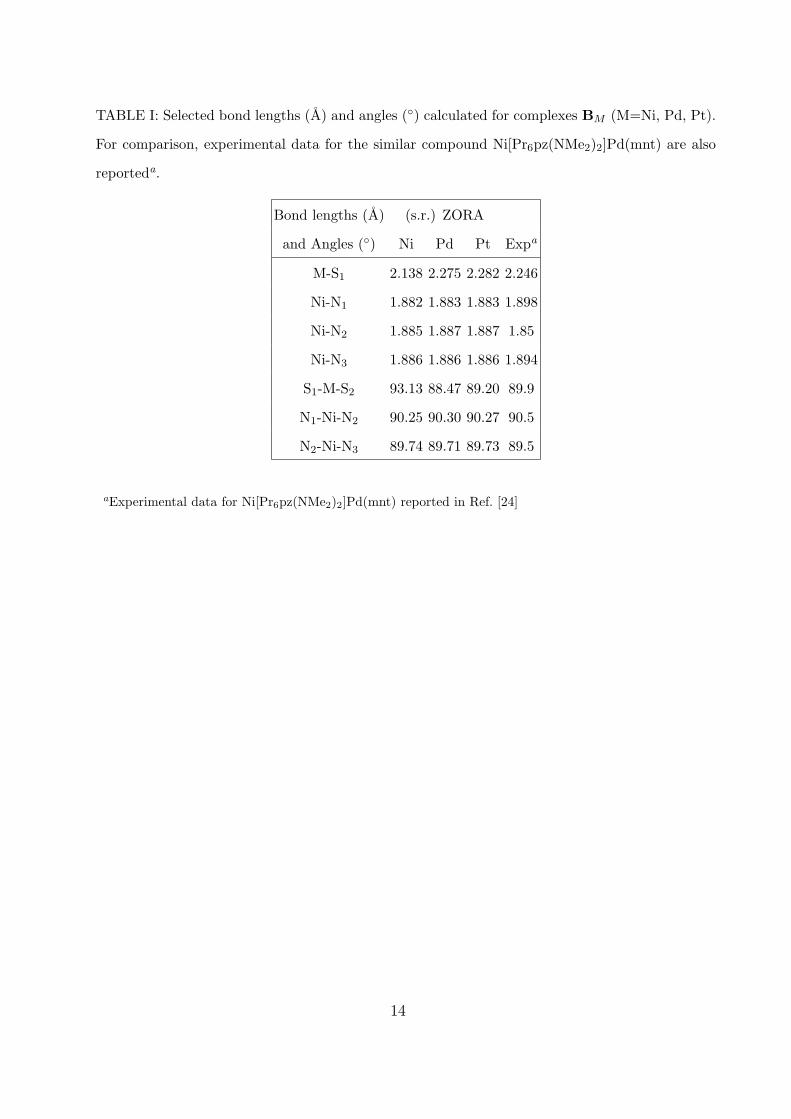
TABLE I: Selected bond lengths (A) and angles (◦) calculated for complexes BM (M=Ni, Pd, Pt).
For comparison, experimental data for the similar compound Ni[Pr6pz(NMe2)2]Pd(mnt) are also
reporteda.
Bond lengths (A) (s.r.) ZORA
and Angles (◦) Ni Pd Pt Expa
M-S1 2.138 2.275 2.282 2.246
Ni-N1 1.882 1.883 1.883 1.898
Ni-N2 1.885 1.887 1.887 1.85
Ni-N3 1.886 1.886 1.886 1.894
S1-M-S2 93.13 88.47 89.20 89.9
N1-Ni-N2 90.25 90.30 90.27 90.5
N2-Ni-N3 89.74 89.71 89.73 89.5
aExperimental data for Ni[Pr6pz(NMe2)2]Pd(mnt) reported in Ref. [24]
14

TABLE II: Low-energy excitations (λ > 650 nm) and major one-electron transition contributions
calculated for complexes AM and BM (M=Ni, Pd, Pt) using the LB94 xc functional. In brackets
the oscillator strengths are reported as well.
State ANi APd APt BNi BPd BPt
1 A1 926 (0.12) 941 (0.14) 945 (0.07) 1049 (0.04) 1100 (0.05) 1057 (0.05)
76% 20b2 → 22b2 79% 20b2 → 22b2 88% 20b2 → 22b2 61% 17b1 → 19b1 70% 17b1 → 19b1 60% 17b1 → 19b1
21% 20b2 → 21b2 19% 20b2 → 21b2 10% 20b2 → 21b2 35% 18b1 → 19b1 26% 18b1 → 19b1 37% 18b1 → 19b1
2 A1 885 (0.23) 923 (0.24) 890 (0.29) 836 (0.02) 875 (0.06) 833 (0.03)
49% 20b2 → 21b2 51% 20b2 → 21b2 57% 20b2 → 21b2 88% 16b1 → 19b1 80% 16b1 → 19b1 86% 16b1 → 19b1
30% 19b2 → 21b2 34% 19b2 → 21b2 34% 19b2 → 21b2 15% 18b1 → 19b1 8% 18b1 → 19b1
15% 20b2 → 22b2 11% 20b2 → 22b2
3 A1 776 (0.15) 808 (0.13) 780 (0.24) 713 (0.26) 798 (0.24) 726 (0.32)
58% 19b2 → 21b2 58% 19b2 → 21b2 56% 19b2 → 21b2 47% 18b1 → 19b1 52% 18b1 → 19b1 47% 18b1 → 19b1
20% 20b2 → 21b2 23% 20b2 → 21b2 28% 20b2 → 21b2 28% 17b1 → 19b1 21% 17b1 → 19b1 29% 17b1 → 19b1
8% 16b1 → 19b1 18% 16b1 → 19b1 12% 16b1 → 19b1
TABLE III: βvec(10−30esu)a values calculated for complexes AM and BM (M=Ni, Pd, Pt) at 0.35
eV (3.50 µm) using the LB94 xc functional. The calculated dipole moments along the z-axis, µz
(D), are also reported.
ANi APd APt BNi BPd BPt
βstaticvec 68.8 (69.8)b 39.4 (40.6) 82.5 (83.3) 136.4 (-134.7) 166.4 (-163.7) 140.7 (-138.9)
βSHGvec 78.2 (80.2) 33.5 (36.0) 94.3 (95.9) 270.1 (-257.5) 355.5 (-333.6) 280.1 (-268.8)
βEOPE/ORvec 71.7 (72.9) 38.4 (39.8) 86.1 (87.1) 165.7 (-162.8) 205.2 (-201.0) 171.6 (-168.6)
µz 1.95 1.93 1.73 -14.28 -14.84 -14.49
aHere βvec = µz·βz
|µ| , with βz = 13
∑i(βzii + βizi + βiiz), (i=x,y,z), and z the axis of the dipole moment.
bIn brackets the dominant component of the hyperpolarizability tensor, i.e. βzzz, is reported.
15
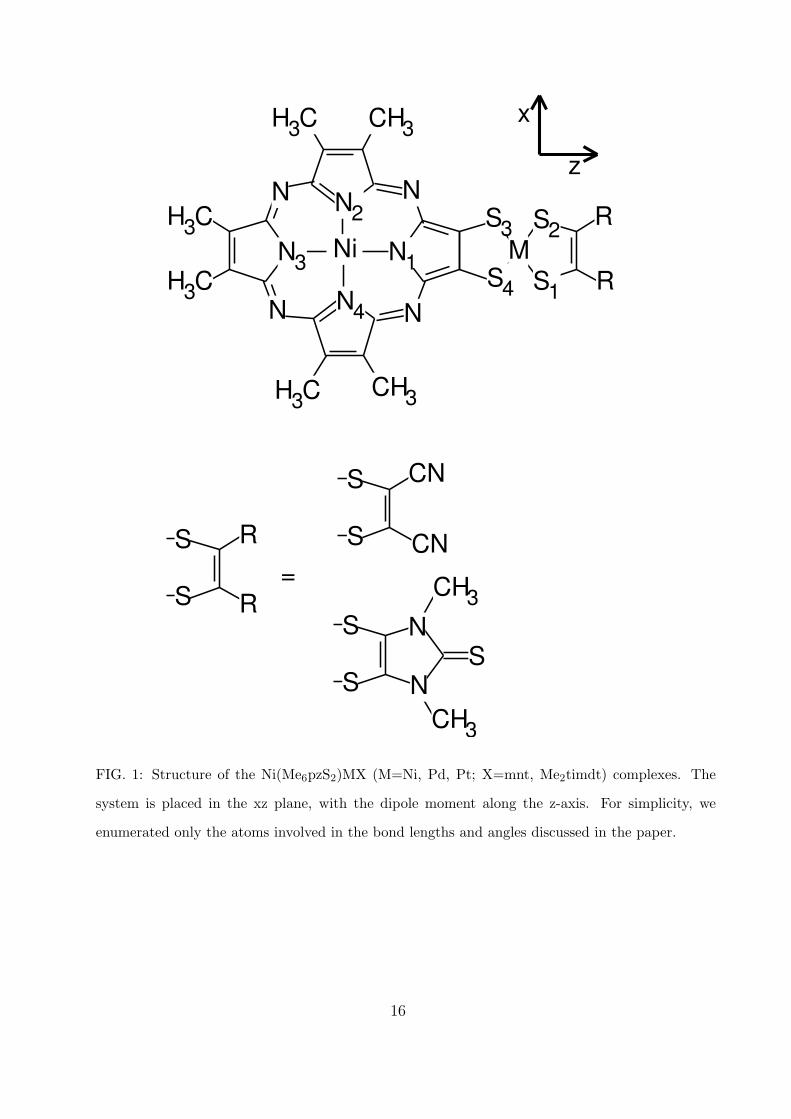
Ni
N2
N4
N3 N1 M
NN
N N
S3 S2
S4 S1
CH3
CH3H3C
H3C
H3C
H3C
R
R
R
R
S
S=
S
S
CN
CN
S
SS
CH3N
CH3
N
x
z
FIG. 1: Structure of the Ni(Me6pzS2)MX (M=Ni, Pd, Pt; X=mnt, Me2timdt) complexes. The
system is placed in the xz plane, with the dipole moment along the z-axis. For simplicity, we
enumerated only the atoms involved in the bond lengths and angles discussed in the paper.
16

-7.0
-6.5
-6.0
-5.5
-5.0
-4.5
-4.0
MO
s en
ergy
(eV)
ANi APd APt BNi BPd BPt
20b2
21b2
22b241b1
14a2
13a2
50a1
19b2
20b2
21b2
22b2
14a2
15a2
13a2
50a119b2
20b2
21b2
22b2
15a2
14a2
13a2
50a119b2
13a2 13a213a2
19b1
39b2
14a2
15a2
12a2
18b1
47a1
17b1
12a218b1
47a1
17b1
19b1
14a2
20b1
15a2
12a2
18b1
47a1
17b1
19b1
14a2
20b1
15a2
11a2 16b2 16b2
FIG. 2: Energy level scheme for complexes AM and BM (M=Ni, Pd, Pt). Only some selected
molecular orbitals are depicted. For the sake of homogeneity here and in the following the orbital
numbering of complexes APt and BPt does not include the 4f valence orbitals of Pt.
17
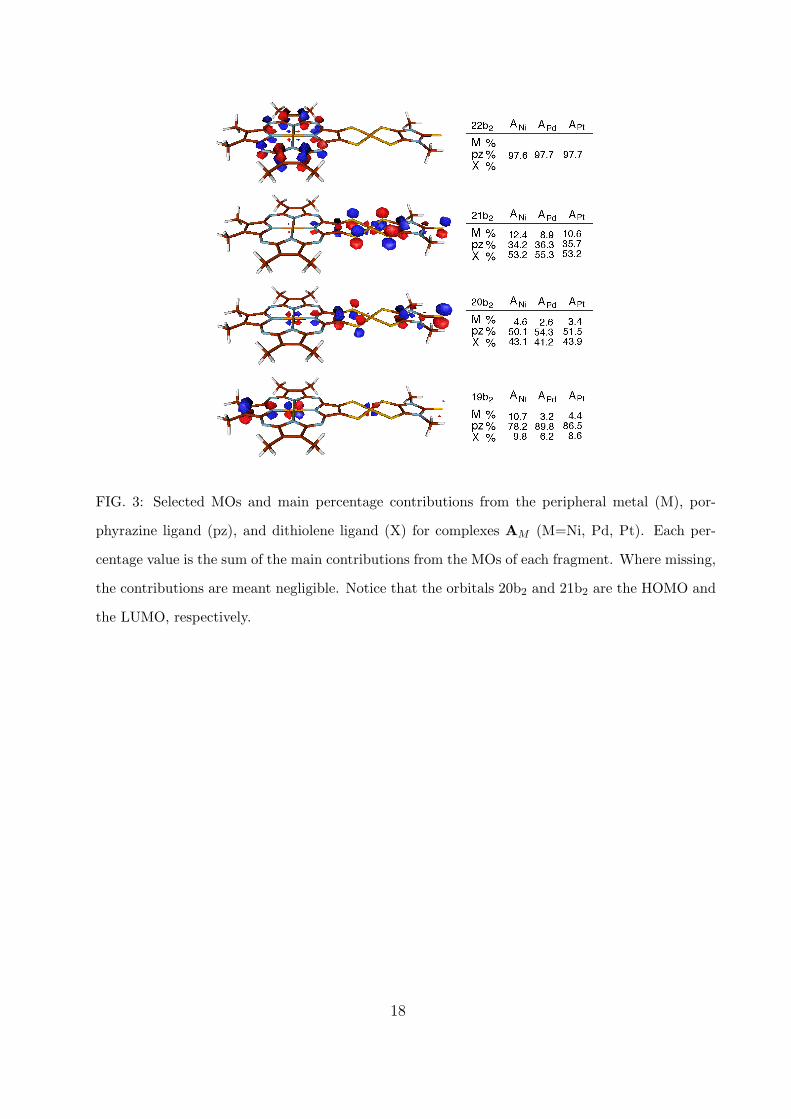
FIG. 3: Selected MOs and main percentage contributions from the peripheral metal (M), por-
phyrazine ligand (pz), and dithiolene ligand (X) for complexes AM (M=Ni, Pd, Pt). Each per-
centage value is the sum of the main contributions from the MOs of each fragment. Where missing,
the contributions are meant negligible. Notice that the orbitals 20b2 and 21b2 are the HOMO and
the LUMO, respectively.
18

FIG. 4: Selected MOs and main percentage contributions from the peripheral metal (M), por-
phyrazine ligand (pz), and dithiolene ligand for complexes BM (M=Ni, Pd, Pt). Each percentage
value is the sum of the main contributions from the MOs of each fragment. Where missing, the
contributions are meant negligible. Notice that the orbitals 13a2 and 19b1 are the HOMO and the
LUMO, respectively.
19
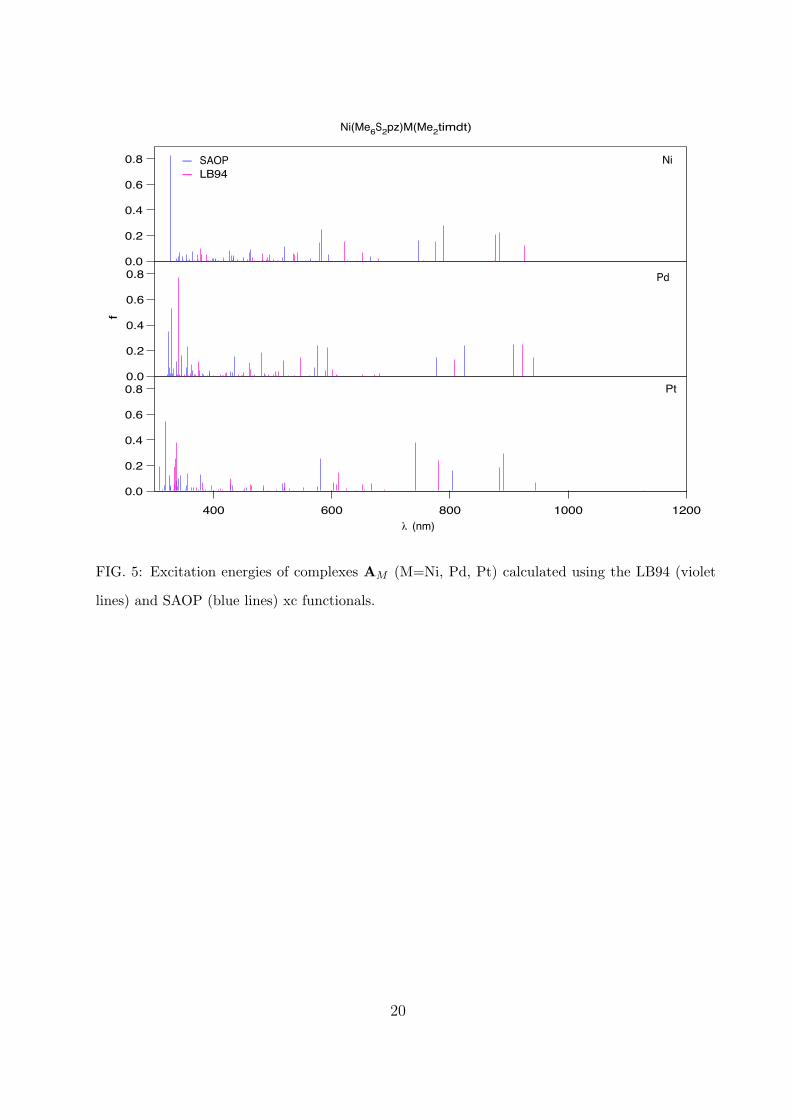
0.8
0.6
0.4
0.2
0.0
12001000800600400! (nm)
0.8
0.6
0.4
0.2
0.0
f
0.8
0.6
0.4
0.2
0.0
Ni
Pd
Pt
— SAOP— LB94
Ni(Me6S2pz)M(Me2timdt)
FIG. 5: Excitation energies of complexes AM (M=Ni, Pd, Pt) calculated using the LB94 (violet
lines) and SAOP (blue lines) xc functionals.
20
0.8
0.6
0.4
0.2
0.0
12001000800600400! (nm)
0.8
0.6
0.4
0.2
0.0
f
0.8
0.6
0.4
0.2
0.0
Ni
Pd
Pt
— SAOP— LB94
Ni(Me6S2pz)M(mnt)
FIG. 6: Excitation energies of complexes BM (M=Ni, Pd, Pt) calculated using the LB94 (violet
lines) and SAOP (blue lines) xc functionals.
21




![Synthesis, characterization and X-ray crystal structures of [Ni(Me-sal) 2dpt] and [Ni(Me-sal)dpt]Cl](https://static.fdokumen.com/doc/165x107/63171c928ebcb731770b81b7/synthesis-characterization-and-x-ray-crystal-structures-of-nime-sal-2dpt-and.jpg)















