
Non-syndromic vestibular disorder with otoconial agenesis in tilted/mergulhador mice caused by...
13
Non-syndromic vestibular disorder with otoconial agenesis in tilted/mergulhador mice caused by mutations in otopetrin 1 Belen Hurle 1 , Elena Ignatova 2 , Silvia M. Massironi 3 , Tomoji Mashimo 4 , Xavier Rios 1 , Isolde Thalmann 2 , Ruediger Thalmann 2 and David M. Ornitz 1, * 1 Department of Molecular Biology and Pharmacology, and 2 Department of Otolaryngology, Washington University Medical School, 660 South Euclid Ave, St Louis, MO 63110, USA, 3 Department of Immunology, Institute of Biomedical Sciences, University of Sa ˜ o Paulo, Av. Professor Lineu Prestes, 1730, Cidade Universita ´ ria, CEP 05508-900, Sa ˜o Paulo, SP, Brazil and 4 Unite de Genetique des Mammiferes, Institut Pasteur, 25 Rue du Docteur Roux, F-75724 Paris Cedex 15, France Received December 10, 2002; Revised and Accepted February 5, 2003 Otoconia are biominerals within the utricle and saccule of the inner ear that are critical for the perception of gravity and linear acceleration. The classical mouse mutant tilted (tlt) and a new allele, mergulhador (mlh), are recessive mutations that affect balance by impairing otoconial morphogenesis without causing collateral deafness. The mechanisms governing otoconial biosynthesis are not known. Here we show that tlt and mlh are mutant alleles of a novel gene (Otopetrin 1, Otop1), encoding a multi-transmembrane domain protein that is expressed in the macula of the developing otocyst. Both mutants carry single point mutations leading to non-conservative amino acid substitutions that affect two putative transmembrane (TM) domains (tlt, Ala 151 !Glu in TM3; mlh, Leu 408 !Gln in TM8). Otop1 and Otop1-like paralogues, Otop2 and Otop3, define a new gene family with homology to the C. elegans and D. melanoganster DUF270 genes. INTRODUCTION The vestibular system within the inner ear is responsible for the perception of gravity and motion, and for the maintenance of balance. The peripheral vestibular organs form a complex three-dimensional structure of interconnected chambers located within the inner ear that include three semicircular canals, the utricle and the saccule. Sensory transduction depends on the inertial mass of thousands of tiny biomineral particles, otoconia, embedded in a gelatinous membrane that overlies the sensory epithelium of the saccule and utricle (1,2). Forces due to gravity and linear acceleration act through otoconia to deflect the stereocilia of the sensory hair cells and create vestibular evoked potentials (VsEPs). Otoconia are critical for the correct processing of orientation and positional information. In humans, dislodging of otoconia due to ototoxic drugs, infection or trauma causes incapacitating chronic recurrent vertigo (3,4). Age-dependent degeneration of otoconia is a high-risk factor for loss of balance, contributing to hip fracture and accidental death in the elderly (5,6). Similarly, the absence of gravitational stimulation of otoconia under microgravity leads to space adaptation syndrome in astronauts, akin to terrestrial motion sickness (7). In mice, congenital absence of otoconia completely abolishes the VsEPs in response to linear acceleration (8). Almost nothing is known about the development, main- tenance and potential for regeneration of otoconia, in part because animal models with non-syndromic vestibular phenotypes are extremely rare (9,10). The original tilted mutant arose spontaneously in the C57BL/6J background and is one of the three classical mouse mutants presenting with a non-syndromic vestibular disorder [tilted (tlt; Chr5) (11); tilted head (thd; Chr1) (12–14); head tilted (het; Chr17) (15)]. Homozygous tlt mice lack a perception of gravity and spatial orientation, show head tilting behavior and an inability to swim. The non-swimming phenotype correlates with the total absence of otoconia in the saccule and utricule or, infrequently, with the presence of a few giant otoconia in the saccule (16). Unlike most available mutants and knockout mice with abnormal otoconia (www.jax.org/hmr/index.html/) (17), tlt is not accompanied by deafness, congenital or progressive degeneration of the cochlea, degeneration of the *To whom correspondence should be addressed. Email: [email protected] Human Molecular Genetics, 2003, Vol. 12, No. 7 777–789 DOI: 10.1093/hmg/ddg087 Human Molecular Genetics, Vol. 12, No. 7 # Oxford University Press 2003; all rights reserved by guest on July 23, 2016 http://hmg.oxfordjournals.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Non-syndromic vestibular disorder with otoconial agenesis in tilted/mergulhador mice caused by...

Non-syndromic vestibular disorder with otoconialagenesis in tilted/mergulhador mice caused bymutations in otopetrin 1
Belen Hurle1, Elena Ignatova2, Silvia M. Massironi3, Tomoji Mashimo4, Xavier Rios1,
Isolde Thalmann2, Ruediger Thalmann2 and David M. Ornitz1,*
1Department of Molecular Biology and Pharmacology, and 2Department of Otolaryngology, Washington University
Medical School, 660 South Euclid Ave, St Louis, MO 63110, USA, 3Department of Immunology, Institute of
Biomedical Sciences, University of Sao Paulo, Av. Professor Lineu Prestes, 1730, Cidade Universitaria,
CEP 05508-900, Sao Paulo, SP, Brazil and 4Unite de Genetique des Mammiferes, Institut Pasteur,
25 Rue du Docteur Roux, F-75724 Paris Cedex 15, France
Received December 10, 2002; Revised and Accepted February 5, 2003
Otoconia are biominerals within the utricle and saccule of the inner ear that are critical for the perception ofgravity and linear acceleration. The classical mouse mutant tilted (tlt) and a new allele, mergulhador (mlh), arerecessive mutations that affect balance by impairing otoconial morphogenesis without causing collateraldeafness. The mechanisms governing otoconial biosynthesis are not known. Here we show that tlt and mlhare mutant alleles of a novel gene (Otopetrin 1, Otop1), encoding a multi-transmembrane domain protein thatis expressed in the macula of the developing otocyst. Both mutants carry single point mutations leading tonon-conservative amino acid substitutions that affect two putative transmembrane (TM) domains (tlt,Ala151!Glu in TM3; mlh, Leu408!Gln in TM8). Otop1 and Otop1-like paralogues, Otop2 and Otop3, define anew gene family with homology to the C. elegans and D. melanoganster DUF270 genes.
INTRODUCTION
The vestibular system within the inner ear is responsible for theperception of gravity and motion, and for the maintenance ofbalance. The peripheral vestibular organs form a complexthree-dimensional structure of interconnected chambers locatedwithin the inner ear that include three semicircular canals, theutricle and the saccule. Sensory transduction depends on theinertial mass of thousands of tiny biomineral particles,otoconia, embedded in a gelatinous membrane that overliesthe sensory epithelium of the saccule and utricle (1,2). Forcesdue to gravity and linear acceleration act through otoconia todeflect the stereocilia of the sensory hair cells and createvestibular evoked potentials (VsEPs).
Otoconia are critical for the correct processing of orientationand positional information. In humans, dislodging of otoconiadue to ototoxic drugs, infection or trauma causes incapacitatingchronic recurrent vertigo (3,4). Age-dependent degeneration ofotoconia is a high-risk factor for loss of balance, contributing tohip fracture and accidental death in the elderly (5,6). Similarly,the absence of gravitational stimulation of otoconia under
microgravity leads to space adaptation syndrome in astronauts,akin to terrestrial motion sickness (7). In mice, congenitalabsence of otoconia completely abolishes the VsEPs inresponse to linear acceleration (8).
Almost nothing is known about the development, main-tenance and potential for regeneration of otoconia, in partbecause animal models with non-syndromic vestibularphenotypes are extremely rare (9,10). The original tiltedmutant arose spontaneously in the C57BL/6J background andis one of the three classical mouse mutants presenting with anon-syndromic vestibular disorder [tilted (tlt; Chr5) (11);tilted head (thd; Chr1) (12–14); head tilted (het; Chr17) (15)].Homozygous tlt mice lack a perception of gravity and spatialorientation, show head tilting behavior and an inability toswim. The non-swimming phenotype correlates with the totalabsence of otoconia in the saccule and utricule or,infrequently, with the presence of a few giant otoconia inthe saccule (16). Unlike most available mutants and knockoutmice with abnormal otoconia (www.jax.org/hmr/index.html/)(17), tlt is not accompanied by deafness, congenital orprogressive degeneration of the cochlea, degeneration of the
*To whom correspondence should be addressed. Email: [email protected]
Human Molecular Genetics, 2003, Vol. 12, No. 7 777–789DOI: 10.1093/hmg/ddg087
Human Molecular Genetics, Vol. 12, No. 7 # Oxford University Press 2003; all rights reserved
by guest on July 23, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

vestibular organs, abnormal central vestibular function and/ormulti-organ involvement (16). Thus, the identification of thegene responsible for the tlt phenotype could provide, for thefirst time, a tool to investigate the specific mechanisms thatregulate otoconia development and function.
Through a positional cloning strategy, we have characterizedtlt as a mutant allele of a novel gene designated Otopetrin 1(Otop1). A second mutant allele, Mergulhador (mlh), or diverin Portuguese, arose in a medium-scale ENU (N-ethyl-N-nitrosourea) mutagenesis screen (18), and exhibits non-swimming behavior and lack of otoconia, similar to the tltmutant mouse. Both tlt and mlh carry single-point mutationsleading to non-conservative amino acid substitutions that affecttwo putative transmembrane domains of Otop1. This is the firstmolecular characterization of mutations that specifically impairthe morphogenesis of otoconia without affecting other organs,other inner ear structures or hearing function.
RESULTS
Tlt and mlh mutations are allelic
Mlh arose from a medium-scale ENU mutagenesis screen (18).Mlh/mlh mice are viable and fertile. Homozygotes hold theirhead tilted to one side, shake when suspended by the tail, andexhibit non-swimming behavior indicative of vestibulardysfunction. Mlh and tlt phenotypes are indistinguishable.Mlh was mapped using a standard interspecific backcross to
mouse Chr 5 (Fig. 1) and was found to co-segregate withmarkers D5Mit13, D5Mit75, D5Mit127, D5Mit149, D5Mit182,D5Mit351 and D5Mit354. This region overlapped with theregion where tlt had previously been mapped (19). To testwhether tlt and mlh could be alleles of the same gene,homozygous mice were mated. All of the offspring of tlt/tlt�mlh/mlh matings showed a non-swimming phenotype, indicat-ing that the two mutations do not complement each other invivo. Both the mapping and complementation data suggestedthat the tlt and mlh mutations are alleles of a single gene.
Evaluation of candidate genes within the tlt interval
To pinpoint the position of tlt within the available 900 kbphysical map spanning the tlt locus (20), new SSLP markerswere developed and mapped throughout the minimum tilingcontig. The minimum genetic interval for tlt was narrowed to a450 kb region delineated by D5Dmo9 and D5Dmo10, with onlyone crossover between tlt and each marker in 894 meioses(Fig. 2A, haplotype data not shown). Three known genes (Drd5,Wdr1 and Glut9) lie within this segment. BlastN (21) andGENSCAN (22) inspection of the remaining genomic sequencesuggested an additional new gene on BAC RPC23-426E16 thatpartially matched UniGene cluster Mm.204765. Expression ofWdr1 and Mm.204765 (but not Drd5 or Glut9) was observed inE18.5 mouse otocyst and P0 mouse inner ear RNA by RT–PCR(data not shown). Sequencing of the exons and splice junctionsof Drd5, Wdr1 and Glut9 in tlt/tlt mice, and the parental C57BL/6J strain, did not detect abnormalities that could be related to
Figure 1. Integrated genetic map of mlh and tlt on mouse Chr5. A (BALB/cJ-mlh�C57BL/6J)F1�BALB/cJ-mlh interspecific backcross was established for thehaplotype analysis of mlh. Genome-wide scanning with 54 SSLP markers showed linkage between mlh and D5Mit254 (LodScore 6.0, data not shown). For thehigh-resolution mapping of mlh, 18 additional D5Mit markers were tested. Markers are shown ordered from proximal to distal (left) according to the haplotypeanalysis results obtained (right). The black boxes represent the BALB/cJ allele and the white boxes represent the C57BL/6J allele. The number of mlh/mlh micewith each haplotype is shown at the top of each column. The distances between pairs of loci are expressed in cM as calculated by MapManager QTb28. Loci thatcould not be ordered are listed adjacent to each other. Mlh co-localizes with D5Mit13, D5Mit75, D5Mit127, D5Mit149, D5Mit182, D5Mit351 and D5Mit354(brackets). Boxed markers, D5Mit354 and D5Mit353 are the genetic boundaries for the 1.5 cM genetic interval spanning the tlt locus as determined by Yinget al. (19).
778 Human Molecular Genetics, 2003, Vol. 12, No. 7
by guest on July 23, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

Figure 2. Physical, transcription and mutation map of the tlt locus. (A) The tlt physical map from D5Dmo3 to D5Dmo5 (20) links five overlapping RPCI-23 BACclones (241E18, 426E16, 268D19, 115A12 and 456A210) that span �900 kb of genomic DNA. The asterisks indicate the T7 end of each clone, the open circlesrepresent relevant SSLP markers within the contig. D5Dmo9 and D5Mit10 delimit the tlt minimum region with only one crossover between tlt and each marker inthe F2 intercross panel (19). Candidate genes within this interval (Drd5, Wdr1 and Glut9 and UniGene Mm.204765) and their transcriptional orientation are shown(bold arrows). (B) Map of UniGene Mm.204765 showing coding exons (black boxes); untranslated regions (white boxes); CpG dinucleotide rich regions (orangeboxes). The positions of the tlt and mlh mutations (exons 3 and 6, respectively) are indicated by asterisks. (C) Sequence chromatographs spanning the tlt (top) andmlh (bottom) mutation sites compared with that of wild-type controls. Nucleotide changes and resulting non-conservative amino acid changes are highlighted inred. Amino acids are numbered according to the position of the first methionine in the most abundant transcript (Otop1-a). The tlt and mlh point mutations werereconfirmed by restriction analysis that detects a new TaqI restriction site in the Otop1tlt allele and loss of a Msc1 restriction site in the Otop1mlh allele. The sizes ofthe PCR products (PCR) and endonuclease restriction (RE) fragments are indicated.
Human Molecular Genetics, 2003, Vol. 12, No. 7 779
by guest on July 23, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

the tlt phenotype (data not shown). We therefore focused oncharacterization of the Mm.204765 cluster.
To identify the Mm.204765 full-length transcript, overlappingcDNA products were obtained from E18.5 mouse otocyst RNAby RT–PCR using primers positioned in predicted exons. Thestart of the gene was characterized by 50 RACE. A total of sevenexons were identified and assembled into two ORFs, differing intheir 50-ends (a and b, Figs 2B and 3A). The most abundanttranscript in mouse otocyst mRNA, (a), joined exons 2–7 andcontained a 1802 bp ORF and a 1313 bp 30 UTR with threeconsensus polyadenylation sites (positions 2416, 3113 and3129) all within exon 7. The putative translation initiation codonin Otop1-a is an in-frame methionine in exon 2 at position 25 bpin the cDNA. A rare alternative splice form, (b), in which exon 1is joined to an internal splice site at position þ130 within exon 2to produce an alternative ORF of 1846 bp with a different 50 endwas also detected. The first in-frame methionine for thistranscript is located in exon 1, within a favorable context forinitiation with a purine at position �3 and a guanine at þ4 (23).The existence of exon 1 is consistent with the Mm.204765 genestructure predicted by the Celera algorithm (www.celera.com).
Mutation screening of the Mm.204765 gene revealed that thetlt allele contained a C476!A change in exon 3, leading to anAla151!Glu substitution. The mlh allele carried a T1247!Achange in exon 6 leading to a Leu408!Gln substitution(Fig. 2C). To genotype these alleles, two independent PCR/restriction endonuclease assays were developed that specificallytarget the mutation sites in tlt and mlh. These assays wereconducted on genomic DNA amplified from strains SJL/J,C3H/H, BALB/CJ, 101/H and C57BL/6J, and mutations wereonly found in DNA from heterozygous or homozygous tlt andmlh mice (Fig. 2C). To investigate further whether the tlt andmlh substitutions could be naturally occurring polymorphisms,43 high-throughput genomic reads from the Celera mouse tracedatabase that partially span the Mm.204765 gene inthree additional backgrounds (DBA/2J, A/J and 129/SVJ) wereretrieved and analyzed. Overall, nine strain-specific poly-morphisms (seven silent and two non-silent) were found but inno case were the tlt or mlh sequence-variants detected. Wetherefore concluded that the observed non-conservative sub-stitutions in Mm.204765 (subsequently named Otopetrin 1,Otop1) are not neutral polymorphisms, and account for theobserved phenotype of tlt and mlh mice.
Otop1 belongs to a novel gene family
tBlastN searches, using murine Otop1 amino-acid sequence asa query, revealed multiple Otop1 orthologous and Otop1-likeparalogous sequences in the EST and genomic databases.Comparison of Otop1 homologous sequences was carried outthrough EST clustering and the alignment of EST assemblies tocorresponding genomic contigs, combined with GENSCANpredictions and RT–PCR amplification, to resolve ambiguitiesin gene structures.
Orthologous sequences were identified for mouse Otop1 inrat, human, F. rubripes (fugu) and D. renio (zebrafish), showing95, 77, 44 and 41% amino acid identity, respectively (Table 1).Complementary mapping data from the Ensembl projects(www.ensembl.org/), the WUZGR integrated zebrafish map(http://zfish.wustl.edu/) and the Ratmap database (http://
ratmap.gen.gu.se/) confirmed that these genes map to syntenicregions in mouse Chr5, human Chr4p16.2, rat Chr14p21-14q21and zebrafish LG14. No orthologous mapping information wasavailable for Fugu scaffold_3725, where Otop1 was electroni-cally identified. Additionally, tandem assemblies representingtwo Otop1-like genes (hereafter named Otop2 and Otop3), werefound on mouse Chr11q21 and its syntenic location on humanChr17q25.3. Mouse Otop2 and Otop3 showed 34 and 30%amino acid identity with Otop1, respectively.
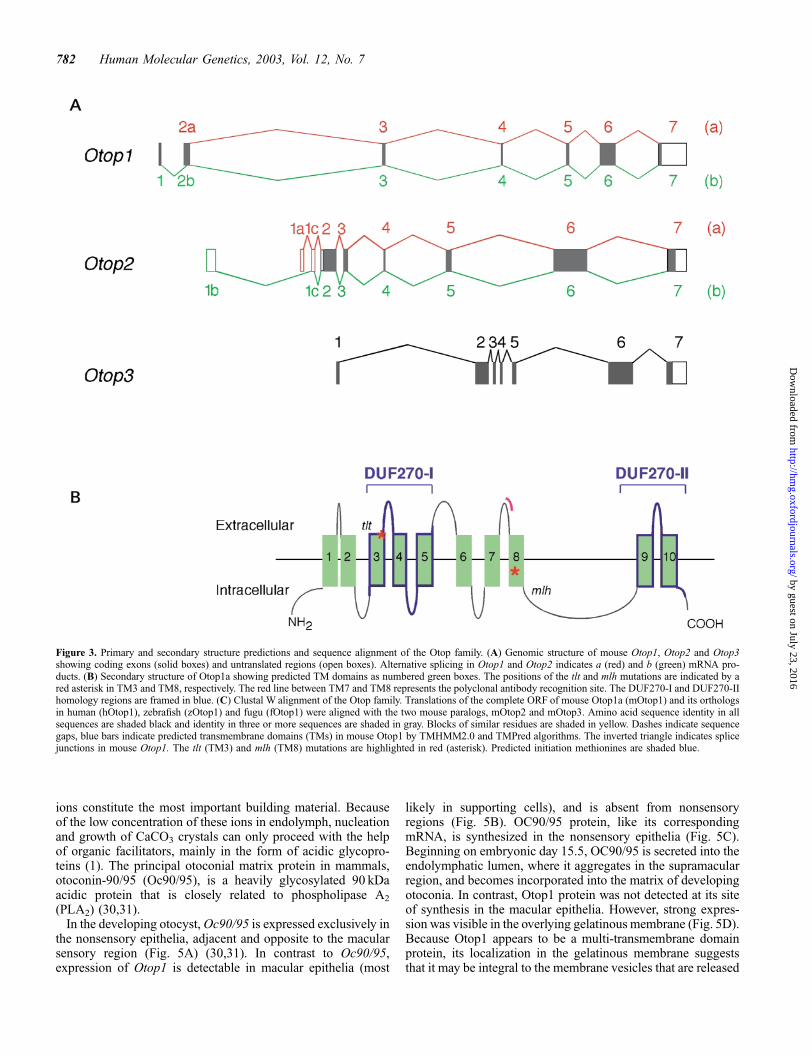
Comparison of the cDNA assemblies and genomic sequenceindicated that all Otop family members share a common genestructure, with conserved splice-site positions. Some variabilitywas observed in the structure of the 50 UTR and N-terminalcoding exons, and in the length of exon 6 (Fig. 3A). Among theOtop1 and Otop1-like proteins that have been identified, onlymouse Otop1-b showed a recognizable N-terminal signalpeptide. Pattern recognition algorithms TMHMM2.0 (24) andTMPred (25), and the protein localization program PSORT II(26), predicted a variable number of transmembrane (TM)domains. TM domain annotations were accepted according tothe number of algorithms predicting them, and their occurrencein an orthologous gene. A conserved pattern of seven TMdomains (namely TM1, TM2, TM3, TM6, TM7, TM9 andTM10) was strongly predicted in all family members by allalgorithms used. Three additional TMs, in conserved positions,were predicted by either TMHMM2.0 (TM5) or TMPred (TM8)or both (TM4). Based on these annotations we suggest that theOtop family members are 10-span transmembrane domainproteins, with membrane topology type 3a, and a cytoplasmicorientation of the N- and C-termini. The tlt and mlh mutationsoccur in TM3 and TM8 of mouse Otop1, respectively, and couldtherefore affect the transmembrane domain structure, and/ortertiary structure of the protein (Fig. 3B).
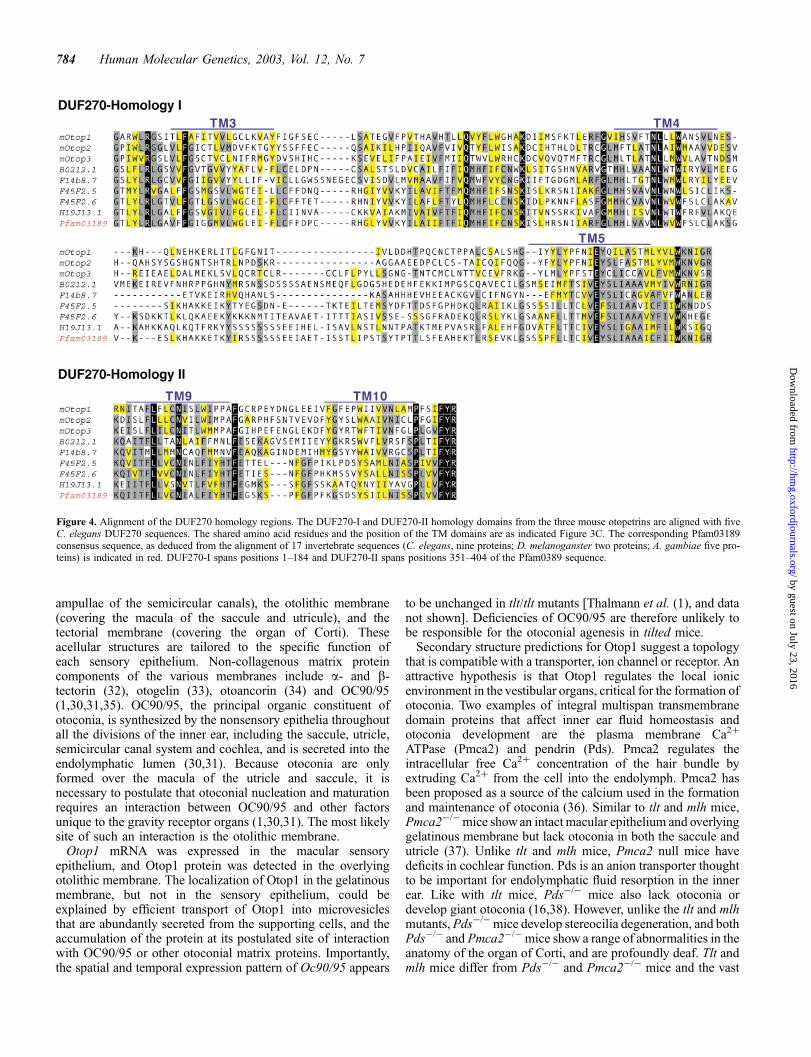
The extent of the sequence conservation in the Otop familywas examined by CLUSTALWalignment (27) of the amino acidsequences followed by manual adjustment (Fig. 3C). Aminoacid sequence divergence within the family occurred primarilyin the N-terminus and in the loop between TM8 and TM9. Tworecognizable regions showed higher levels of amino acidsequence conservation. These regions spanned TM3–TM5 inthe middle of the protein, and TM9 and TM10 in the C-terminus(Fig. 3B). Searches of the NCBI Conserved Domain Database(28) with these two regions identified significant homology withthe Pfam03189 motif. Pfam03189 is a 404 amino acid consensussequence domain of unknown function that defines the DUF270family, with members in C. elegans and D. melanoganster(Table 1). The two regions of maximum homology with the Otopfamily (hereafter designated DUF270-I and DUF270-II), matchpositions 1–184 and 351–404 of the Pfam03189 consensussequence, respectively (Figs 3B and 4). PSORT II also identifieda leucine zipper pattern (PS00029) in several Otop sequences,although its functionality is unclear because of its partial overlapwith either TM4 or TM9 (data not shown). No other identifiablefunctional motifs were detected.
Otop1 and Oc90/95 exhibit complementary mRNAexpression patterns
The inorganic phase of otoconia consists of CaCO3 crystalsarranged in a calcite lattice (29). Accordingly, Ca2þ and CO3
2�
780 Human Molecular Genetics, 2003, Vol. 12, No. 7
by guest on July 23, 2016http://hm
g.oxfordjournals.org/D
ownloaded from
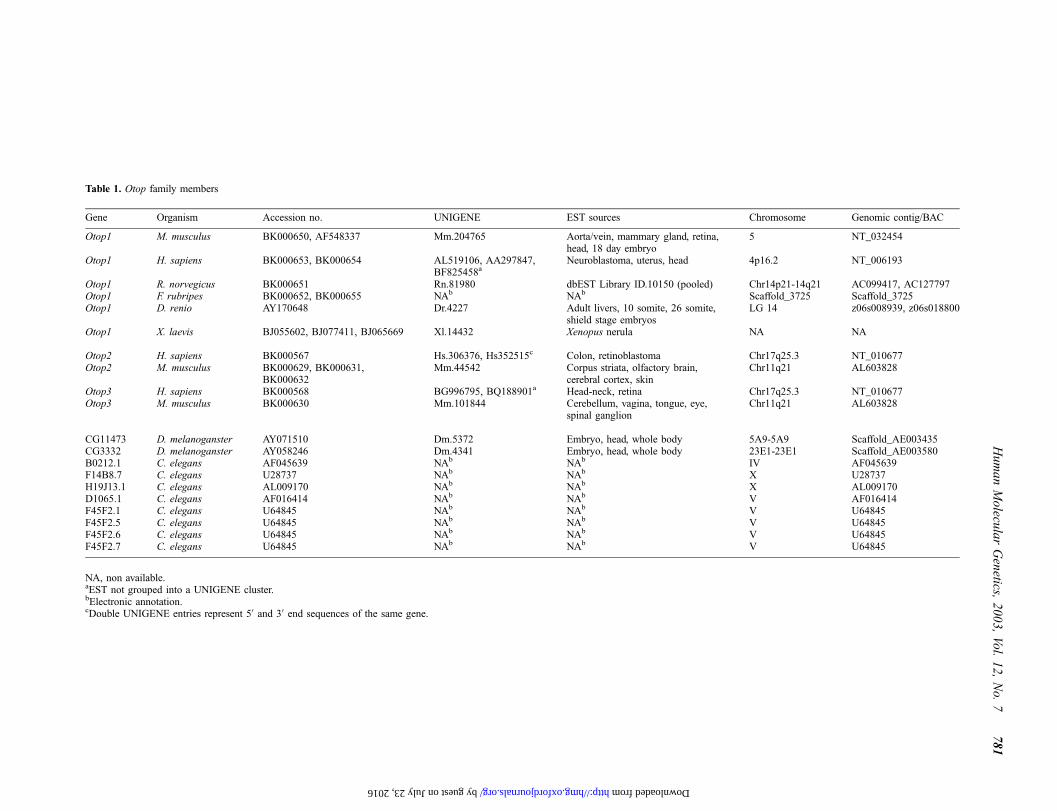
Table 1. Otop family members
Gene Organism Accession no. UNIGENE EST sources Chromosome Genomic contig/BAC
Otop1 M. musculus BK000650, AF548337 Mm.204765 Aorta/vein, mammary gland, retina,head, 18 day embryo
5 NT_032454
Otop1 H. sapiens BK000653, BK000654 AL519106, AA297847,BF825458a
Neuroblastoma, uterus, head 4p16.2 NT_006193
Otop1 R. norvegicus BK000651 Rn.81980 dbEST Library ID.10150 (pooled) Chr14p21-14q21 AC099417, AC127797Otop1 F. rubripes BK000652, BK000655 NAb NAb Scaffold_3725 Scaffold_3725Otop1 D. renio AY170648 Dr.4227 Adult livers, 10 somite, 26 somite,
shield stage embryosLG 14 z06s008939, z06s018800
Otop1 X. laevis BJ055602, BJ077411, BJ065669 Xl.14432 Xenopus nerula NA NA
Otop2 H. sapiens BK000567 Hs.306376, Hs352515c Colon, retinoblastoma Chr17q25.3 NT_010677Otop2 M. musculus BK000629, BK000631,
BK000632Mm.44542 Corpus striata, olfactory brain,
cerebral cortex, skinChr11q21 AL603828
Otop3 H. sapiens BK000568 BG996795, BQ188901a Head-neck, retina Chr17q25.3 NT_010677Otop3 M. musculus BK000630 Mm.101844 Cerebellum, vagina, tongue, eye,
spinal ganglionChr11q21 AL603828
CG11473 D. melanoganster AY071510 Dm.5372 Embryo, head, whole body 5A9-5A9 Scaffold_AE003435CG3332 D. melanoganster AY058246 Dm.4341 Embryo, head, whole body 23E1-23E1 Scaffold_AE003580B0212.1 C. elegans AF045639 NAb NAb IV AF045639F14B8.7 C. elegans U28737 NAb NAb X U28737H19J13.1 C. elegans AL009170 NAb NAb X AL009170D1065.1 C. elegans AF016414 NAb NAb V AF016414F45F2.1 C. elegans U64845 NAb NAb V U64845F45F2.5 C. elegans U64845 NAb NAb V U64845F45F2.6 C. elegans U64845 NAb NAb V U64845F45F2.7 C. elegans U64845 NAb NAb V U64845
NA, non available.aEST not grouped into a UNIGENE cluster.bElectronic annotation.cDouble UNIGENE entries represent 50 and 30 end sequences of the same gene.
Hu
ma
nM
olecu
lar
Gen
etics,2
00
3,
Vo
l.1
2,
No.
77
81
by guest on July 23, 2016 http://hmg.oxfordjournals.org/ Downloaded from
ions constitute the most important building material. Becauseof the low concentration of these ions in endolymph, nucleationand growth of CaCO3 crystals can only proceed with the helpof organic facilitators, mainly in the form of acidic glycopro-teins (1). The principal otoconial matrix protein in mammals,otoconin-90/95 (Oc90/95), is a heavily glycosylated 90 kDaacidic protein that is closely related to phospholipase A2
(PLA2) (30,31).In the developing otocyst, Oc90/95 is expressed exclusively in
the nonsensory epithelia, adjacent and opposite to the macularsensory region (Fig. 5A) (30,31). In contrast to Oc90/95,expression of Otop1 is detectable in macular epithelia (most
likely in supporting cells), and is absent from nonsensoryregions (Fig. 5B). OC90/95 protein, like its correspondingmRNA, is synthesized in the nonsensory epithelia (Fig. 5C).Beginning on embryonic day 15.5, OC90/95 is secreted into theendolymphatic lumen, where it aggregates in the supramacularregion, and becomes incorporated into the matrix of developingotoconia. In contrast, Otop1 protein was not detected at its siteof synthesis in the macular epithelia. However, strong expres-sion was visible in the overlying gelatinous membrane (Fig. 5D).Because Otop1 appears to be a multi-transmembrane domainprotein, its localization in the gelatinous membrane suggeststhat it may be integral to the membrane vesicles that are released
Figure 3. Primary and secondary structure predictions and sequence alignment of the Otop family. (A) Genomic structure of mouse Otop1, Otop2 and Otop3showing coding exons (solid boxes) and untranslated regions (open boxes). Alternative splicing in Otop1 and Otop2 indicates a (red) and b (green) mRNA pro-ducts. (B) Secondary structure of Otop1a showing predicted TM domains as numbered green boxes. The positions of the tlt and mlh mutations are indicated by ared asterisk in TM3 and TM8, respectively. The red line between TM7 and TM8 represents the polyclonal antibody recognition site. The DUF270-I and DUF270-IIhomology regions are framed in blue. (C) Clustal W alignment of the Otop family. Translations of the complete ORF of mouse Otop1a (mOtop1) and its orthologsin human (hOtop1), zebrafish (zOtop1) and fugu (fOtop1) were aligned with the two mouse paralogs, mOtop2 and mOtop3. Amino acid sequence identity in allsequences are shaded black and identity in three or more sequences are shaded in gray. Blocks of similar residues are shaded in yellow. Dashes indicate sequencegaps, blue bars indicate predicted transmembrane domains (TMs) in mouse Otop1 by TMHMM2.0 and TMPred algorithms. The inverted triangle indicates splicejunctions in mouse Otop1. The tlt (TM3) and mlh (TM8) mutations are highlighted in red (asterisk). Predicted initiation methionines are shaded blue.
782 Human Molecular Genetics, 2003, Vol. 12, No. 7
by guest on July 23, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

into the gelatinous membrane. Although Otop1 is expressed intlt/tlt mice, the protein no longer appears localized to thegelatinous membrane (data not shown).
In addition to expression in the inner ear, Otop1 appears to beexpressed in a number of other tissues. An RT–PCR expressionscreen of a panel of mouse tissues identified expression ofOtop1 in thymus, heart, kidney, skin, stomach, adrenal glandand lactating mammary gland (data not shown). Furthermore,Otop1 and related sequences were represented in a wide rangeof EST libraries (Table 1).
DISCUSSION
The embryonic development, anatomy and physiology of theauditory and vestibular organs are intricately related, making itdifficult to study vestibular function apart from hearing. Manygenes affecting auditory function have been identified due tothe abundance of well characterized human and mousemutations (17). In contrast, the study of genes affectingvestibular function has lagged behind, in part because of thepaucity of animal models with non-syndromic vestibular
dysfunction. Nevertheless, otoconial pathology is a significantmedical problem that can lead to mild to severe vestibularimpairment, benign paroxysmal positional vertigo, dizziness,ataxia and, in the elderly, loss of balance and resulting injury oraccidental death (1). Chronic recurrent vertigo, unassociatedwith auditory or neurological symptoms, is one of the mostcommon reasons for referral to otolaryngology services (4).Despite the prevalence of vestibular dysfunction in humans, ofthe 150 murine loci linked to deafness and/or vestibulardysfunction, only three (tlt, thd and het) specifically affectotoconial development without causing collateral deafness,disrupted inner ear morphogenesis or stereocilia degeneration(www.jax.org/hmr/index.html/) (17). The identification ofOtop1 as the gene underlying the tlt and mlh mutationsprovides, for the first time, a tool to investigate the specificmechanisms that regulate otoconia development and function.
Morphogenesis of otoconia depends on a complex interactionbetween organic and inorganic processes that are not wellunderstood (1,30). Sensory epithelia within the inner ear organsare covered by an extracellular matrix which transmits forces tothe underlying hair cells. Three types of extracellular structureshave been described, the cupula (covering the crista within the
Figure 3. continued.
Human Molecular Genetics, 2003, Vol. 12, No. 7 783
by guest on July 23, 2016http://hm
g.oxfordjournals.org/D
ownloaded from
ampullae of the semicircular canals), the otolithic membrane(covering the macula of the saccule and utricule), and thetectorial membrane (covering the organ of Corti). Theseacellular structures are tailored to the specific function ofeach sensory epithelium. Non-collagenous matrix proteincomponents of the various membranes include a- and b-tectorin (32), otogelin (33), otoancorin (34) and OC90/95(1,30,31,35). OC90/95, the principal organic constituent ofotoconia, is synthesized by the nonsensory epithelia throughoutall the divisions of the inner ear, including the saccule, utricle,semicircular canal system and cochlea, and is secreted into theendolymphatic lumen (30,31). Because otoconia are onlyformed over the macula of the utricle and saccule, it isnecessary to postulate that otoconial nucleation and maturationrequires an interaction between OC90/95 and other factorsunique to the gravity receptor organs (1,30,31). The most likelysite of such an interaction is the otolithic membrane.
Otop1 mRNA was expressed in the macular sensoryepithelium, and Otop1 protein was detected in the overlyingotolithic membrane. The localization of Otop1 in the gelatinousmembrane, but not in the sensory epithelium, could beexplained by efficient transport of Otop1 into microvesiclesthat are abundantly secreted from the supporting cells, and theaccumulation of the protein at its postulated site of interactionwith OC90/95 or other otoconial matrix proteins. Importantly,the spatial and temporal expression pattern of Oc90/95 appears
to be unchanged in tlt/tlt mutants [Thalmann et al. (1), and datanot shown]. Deficiencies of OC90/95 are therefore unlikely tobe responsible for the otoconial agenesis in tilted mice.
Secondary structure predictions for Otop1 suggest a topologythat is compatible with a transporter, ion channel or receptor. Anattractive hypothesis is that Otop1 regulates the local ionicenvironment in the vestibular organs, critical for the formation ofotoconia. Two examples of integral multispan transmembranedomain proteins that affect inner ear fluid homeostasis andotoconia development are the plasma membrane Ca2þ
ATPase (Pmca2) and pendrin (Pds). Pmca2 regulates theintracellular free Ca2þ concentration of the hair bundle byextruding Ca2þ from the cell into the endolymph. Pmca2 hasbeen proposed as a source of the calcium used in the formationand maintenance of otoconia (36). Similar to tlt and mlh mice,Pmca2�/� mice show an intact macular epithelium and overlyinggelatinous membrane but lack otoconia in both the saccule andutricle (37). Unlike tlt and mlh mice, Pmca2 null mice havedeficits in cochlear function. Pds is an anion transporter thoughtto be important for endolymphatic fluid resorption in the innerear. Like with tlt mice, Pds�/� mice also lack otoconia ordevelop giant otoconia (16,38). However, unlike the tlt and mlhmutants, Pds�/� mice develop stereocilia degeneration, and bothPds�/� and Pmca2�/� mice show a range of abnormalities in theanatomy of the organ of Corti, and are profoundly deaf. Tlt andmlh mice differ from Pds�/� and Pmca2�/� mice and the vast
Figure 4. Alignment of the DUF270 homology regions. The DUF270-I and DUF270-II homology domains from the three mouse otopetrins are aligned with fiveC. elegans DUF270 sequences. The shared amino acid residues and the position of the TM domains are as indicated Figure 3C. The corresponding Pfam03189consensus sequence, as deduced from the alignment of 17 invertebrate sequences (C. elegans, nine proteins; D. melanoganster two proteins; A. gambiae five pro-teins) is indicated in red. DUF270-I spans positions 1–184 and DUF270-II spans positions 351–404 of the Pfam0389 sequence.
784 Human Molecular Genetics, 2003, Vol. 12, No. 7
by guest on July 23, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

majority of the other inner ear mouse mutants in that theirphenotype only affects otoconia morphogenesis. This may bedue to the very restricted expression of Otop1 in the macularsensory epithelium, to expression of Otop1 in a restricteddevelopmental stage, or to the possible hypomorphic nature ofthe missense mutations in Otop1.
The vestibular system is one of the phylogenetically oldestsensory systems in vertebrates (39,40). In zebrafish, the abilityto initiate otolith formation is limited to a critical period from18.5 to 24 h post fertilization (41,42). In contrast to the scarcityof mouse otoconia mutants, mutations that cause loss orabnormal development of otoliths, without affecting other earstructures, are well represented in zebrafish (43). A variety ofphenotypes have been identified, including complete bilateralloss of otoliths, only one otolith and supernumerary ormisplaced otoliths (44,45). Bilateral loss of utricular otolithsseverely impairs both balance and motor coordination, and isinvariably lethal. The presence of one utricular otolith in at leastone inner ear is necessary and sufficient for vestibular functionand survival (46). Overall, 12 different complementation groupsthat only have defective otolith development have beendescribed, although the genes underlying these mutationsremain to be identified (44,45). We have cloned the Otop1cDNA in zebrafish, reported its linkage to LG14, and partiallycharacterized its genomic structure. However, Otop1 ESTs werenot identified in the zebrafish embryonic inner ear EST databaseusing accession number queries (www.genoscope.cns.fr/zie)(47). Unfortunately, this database cannot currently be queried by
a direct BlastN search. Further studies are in progress todetermine the expression pattern of Otop1 in zebrafish. A direct-candidate gene screen would clarify the potential involvement ofzebrafish Otop1 in any of the available otolith mutants.Alternatively, morpholino antisense strategies, already provensuccessful in dissecting early otic placode induction pathways(48,49), will be tested.
In humans, three disease loci causing hearing impairment andvestibular dysfunction have been assigned to Chr17q25 (Fig. 6).The recessive disease, USHG1, is characterized by profoundcongenital hearing loss, vestibular dysfunction and prepubertalonset of retinitis pigmentosa (50). The USH1G genomic locusoverlaps with the candidate region for DFNA26 (www.uia.ac.be/dnalab/hhh/), and on its distal side with the DFNA20locus (51), two forms of dominant non-syndromic late-onsetbilateral and progressive sensorineural hearing loss (52). It isnot yet clear whether these disorders are attributable tomutations in the same gene. Two Otop1-like genes, OTOP2and OTOP3, map within the DFNA26 and USH1G candidateregions, and lie in close proximity to the DFNA20 candidateregion (Fig. 6). Interestingly, OTOP2 and OTOP3 representa-tion was found in human retinoblastoma and retina ESTdatabases (ESTs BM466860 and BQ188901, respectively).Based on available information, we suggest OTOP2 andOTOP3 should be screened as positional candidates forthese disease loci. Inner ear mutants mapped to theorthologous region of mouse Chr11, include shaker-2 (sh2)and jackson-shaker ( js). Sh2 is caused by a deletion in the
Figure 5. Expression of Otop1 and Otoconin-90/95 in the utricle of E16.5 mouse embryos. (A) Expression of Otoconin-90/95 (Oc90/95) in the nonsensory epithe-lium (arrowheads) (non-radioactive in situ). (B) Expression of Otop1 in the sensory epithelium (se) (radioactive in situ). (C) Immunolocalization of OC90/95 in thegelatinous membrane/otoconia (arrow) and nonsensory epithelium (arrowhead). (D) Immunolocalization of Otop1 in the gelatinous membrane/otoconia (arrow).Similar results were obtained in the saccule (data not shown).
Human Molecular Genetics, 2003, Vol. 12, No. 7 785
by guest on July 23, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

myosin XV gene (53). Js is characterized by deafness, head-bobbing and circling behavior, indicative of vestibular dys-function (54), and has been proposed as a model for DFNA26/20 and USH1G (50). Although mutation screening of exon andsplice junction sequences of Otop2 and Otop3 in js/js mice didnot identify abnormalities that could account for the observedphenotype (data not shown), it is possible that other ‘regu-latory’ mutations could affect the expression of these genes.
MATERIALS AND METHODS
Primers
SSLP markers delineating the minimal non-recombinantinterval for the tlt mutation are as follows: D5Dmo9 [50-GTTGAC CTC TGA CCT ACA TGC (forward), 50-GAT AGA GGT
CAA TAG TGT GC (reverse)] and D5Dmo10 [50-TGT ACTTAA TTT ACT ATT TGC (forward), 50-CTC TAG TGT CTGCCC AAG TGT CTA G (reverse)]. Primers to amplify acrossthe Otop1tltC476>A mutation are 50-CAC TGT TTG GTC TTGGTA CC (forward) and 50-CAG CTC ATTATT CCT GAC AAG(reverse). Primers to amplify across the Otop1mlh T1247>Amutation are 50-TGA GAA GTC TCT GGA TGA GTC(forward) and 50-GAA TAA CAA CAG CTT GAT GAA G(reverse).
Generation and haplotype analysis of the mlh mice
The mlh mutation was induced using ENU as describedpreviously (18). Briefly, adult BALB/cJ males were injected i.p.with a single dose of ENU (250 mg/kg) and mated to wt DBA/2adult females after 13 weeks. Fertile F1 males were mated with
Figure 6. Colocalization of OTOP2 and OTOP3 and the DFNA20/26 and USHG1 hearing impairment loci on human Ch17. Ideogram of human Chr17 indicatingthe cytogenetic region where DFNA20/26 and USHG1 are mapped (top). Genomic supercontigs spanning bands Chr17q24.3–Chr17q25.3 derived from the goldenpath assembly (http://genome.ucsc.edu/, June 2002 freeze). Accession numbers for several supercontigs are indicated. Candidate regions for DFNA20, DFNA26and USHG1 and their flanking D17S markers are indicated (bottom) (50,51) (www.uia.ac.be/dnalab/hhh/). Human OTOP2 and OTOP3 map to BAC AC024725 insupercontig NT_010677.
786 Human Molecular Genetics, 2003, Vol. 12, No. 7
by guest on July 23, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

wt BALB/cJ females and F2 females were back-crossed to theF1 parents. Homozygous mlh mice were identified in the F3
generation by their inability to swim and head tilting and havebeen subsequently maintained in a BALB/cJ background formore that 20 generations.
For the genetic mapping of the mlh mutation, a (BALB/cJ-mlh�C57BL/6J)F1�BALB/cJ-mlh interspecific back-crosswas established. Twenty-seven back-cross mice were typedwith 54 SSLP markers evenly distributed over the wholegenome (data not shown). Genotype data were analyzed withMapManager QTb28 software. When evidence of linkage withmarker D5Mit254 became obvious (LodScore 6.0), a high-resolution genetic scan of Chr5 was carried out with 18additional D5Mit markers (Fig. 1).
Mutation screening
The genomic structures of mouse Wdr1 (NM_011715), Drd5(S77992), Glut9 (NM_145559) and Otop1 (AF548337) weredetermined by alignment of their full-length cDNAs andavailable ESTs with the genomic sequence of RPC23 clones426E16 (AC084071), 268D19, (AC084070) and 376E18(AC084322). A total of 37 exons (Otop1, 7 exons; Wdr1, 15exons; Drd5, intronless; Glut9, 14 exons) were identified andsubsequently amplified from tlt genomic DNA and a controlsample of parental strain C57BL/6J using intronic primersflanking the splice junction sequences. In the mlh mutant and itsparental strain BALB/cJ, only Otop1 was screened. The tlt andmlh point mutations were reconfirmed by a restrictionendonuclease assay which detected an introduced TaqI restric-tion site in the Otop1tlt allele and the loss of a Msc1 restrictionsite in the Otop1mlh allele (Fig. 2). The restriction endonucleaseassay was conducted on the SJL/J, C3H/H, 101/H and DBA/2Jinbread strains with parental-like results in all cases.
Characterization of the mouse Otop1 cDNA andgene structure
Mouse Otop1 oligonucleotide primers spanning multiple exonsof the ORF were designed from UNIGENE cluster Mm.204765(19 ESTs from aorta-vein, retina and mammary glandlibraries). RT–PCR and RACE fragments were prepared fromtotal RNA obtained from E18.5 mouse otocyst. PCR productswere cloned into the pGEM-T Easy vector (Promega) andsequenced using ABI Big Dye terminator chemistry (PEApplied Biosystems, Foster City, CA, USA). The Otop1full-length cDNA was obtained by assembling overlapping 50
and 30 RACE fragments generated with the SMARTTM RACEcDNA amplification kit (Clonetech Inc.) following themanufacturer’s instructions. The gene structure of Otop1 wasinitially predicted by running GENSCAN on the sequence ofBAC RP23CI-426E16 and later curated by alignment of thefull-length cDNA with the genomic sequence.
Characterization of Otop family members
The clustering of ESTs was seeded by an initial tBlastN searchusing the mouse Otop1 amino-acid sequence masked for lowcomplexity regions as a query against the mouse, human andzebrafish subsets of the EMBL EST databases. All the EST
matches with a tBlastN bit-score of 45 or greater were searchedagainst the EST database of the appropriate species using BlastNto identify all the overlapping EST sequences. OverlappingESTs were assembled into contigs, masked for interspersedrepeat elements using RepeatMasker and searched against theEST databases until no new ESTs were obtained.
This analysis identified three human and three mouse ESTassemblies matching different UniGene clusters that corres-pond to the Otop1, Otop2 and Otop3 genes (Table 1). Thesesequences were electronically aligned to the Ensembl (www.ensembl.org/) and Golden Path (http://genome.ucsc.edu/)genome drafts to determine their respective chromosomallocations and the complete gene structures. Conserved splicejunctions positions across the gene family and GENSCANpredictions were used to resolve point ambiguities.
In zebrafish, 5 ESTs (BI888217, BI890046, AI544512,AI545839 and BG985802) were retrieved and joined withoverlapping RT–PCR sequences amplified from RNA from 26somite embryos into a transcript of 2150 bp encoding the ORF ofOtop1 (1760 bp). The genomic structure was partially deducedfrom the alignment of the cDNA with contigs z06s008939 andz06s018800 from zebrafish assembly06. The mapping informa-tion was obtained through EST AI544512 that maps to LG14,centiRay Position 57 on the radiation hybrid panel LN54 orcentiRay Position 13 in the GF radiation hybrid panel [theWUZGR integrated zebrafish map (http://zfish.wustl.edu/)]. Thezebrafish Otop1 amino acid sequence was used in a tBlastNquery against the F. rubripes genomic assembly. A 1781 bp ORFspliced from six exons in fugu scaffold_3725 that encodes aprotein 60% identical to zebrafish Otop1 was electronicallyannotated. Otop1 related sequences were also observed in fuguScaffold_299 and Scaffold_3932 (data not shown).
Other partial Otop1 orthologs found in the databasesincluded one rat EST (BM387360, UniGene Rn.81980), twogenomic draft sequences spanning five exons of the rat Otop1gene (AC099417 and AC127797) and three Xenopus ESTs(BJ055602, BJ077411, BJ065669, UniGene Xl.14432).
In situ hybridization
Sagittal hemi-heads from E16.5 mice where fixed in PBS/4%paraformaldehyde overnight, paraffin-embedded and sectioned(10 mm). Sections were deparaffinzed in xylene and rehydratedin a series of graded ethanols prior to use. A 1800 bp fragmentspanning the 30 UTR of mouse Otop1 was used as a probe forin situ hybridization. [33P]labeled Otop1 probes (both senseand antisense) were hybridized following the proceduredescribed by Naski et al. (55) with minor modification.Sections were autoradiographed for up to 4 weeks prior todevelopment and counter staining with eosin–hematoxylin. Thesense and antisense Oc90/95 probes spanning the full-lengthcDNA were labeled with DIG, hybridized, and developedaccording to the manufacturer’s instructions (RocheDiagnostics Corp., Indianapolis, IN, USA).
Antibody generation
A 14 amino acid peptide (LDESKNPARKLDVD) from themouse Otop1 protein sequence was synthesized and conjugatedto keyhole limpet hemocyanin (Sigma-Genosys, Woodlands,
Human Molecular Genetics, 2003, Vol. 12, No. 7 787
by guest on July 23, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

TX, USA). Two rabbits were immunized with the peptide-conjugated carrier according to an established schedule(Sigma-Genosys, Woodlands, TX, USA). The antiserum wastested for the presence of the desired antibodies and the titerestablished by ELISA.
Immunohistochemistry
Paraffin-embedded tissue sections were deparaffinized inxylene and rehydrated in graded ethanol solutions. Residualendogenous peroxidase activity was blocked by incubating theslides in 7% H2O2/H2O for 15 min. The non-specific binding ofthe primary antibody was blocked by 5% normal goat serumfor 30 min. The primary antibody was diluted to 14 mg/ml in theblocking buffer and incubated overnight. The biotinylatedsecondary antibody and the color development were performedaccording to the instructions of the ABC Vectastain kit (Vectorlaboratories, Burlingame, CA, USA). Control experimentswere carried out with antiserum that had been preabsorbedovernight with the antigenic peptide.
ACKNOWLEDGEMENTS
We thank K. Steel and A. Erven for examination of mlh innerear structures and S. Johnson for providing zebrafish cloneFb76b02. We are grateful to J.L. Guenet, I. Hughes andJ. Gibson-Brown for critical reading of the manuscript andinsightful discussion. This work was supported by NIH grantDC02236 and the NIH mouse BAC sequencing project.
REFERENCES
1. Thalmann, R., Ignatova, E., Kachar, B., Ornitz, D.M. and Thalmann, I.(2001) Development and maintenance of otoconia: biochemical con-siderations. In Goebel, J. and Highstein, S.M. (eds), The VestibularLabyrinth in Health and Disease. Annals of the New York Academy ofSciences, New York, Vol. 942, pp. 162–178.
2. Lins, U., Farina, M., Kurc, M., Riordan, G., Thalmann, R., Thalmann, I.and Kachar, B. (2000) The otoconia of the guinea pig utricle: internalstructure, surface exposure, and interactions with the filament matrix.J. Struct. Biol., 131, 67–78.
3. El-Kashlan, H.K. and Telian, S.A. (2000) Diagnosis and initiating treatmentfor peripheral system disorders: imbalance and dizziness with normalhearing. Otolaryngol. Clin. N. Am., 33, 563–578.
4. Oas, J.G. (2001) Benign paroxysmal positional vertigo: a clinician’sperspective. Ann. NY Acad. Sci., 942, 201–209.
5. Ross, M.D., Johnsson, L.-G., Peacor, D. and Allard, L.F. (1976)Observations on normal and degenerating human otoconia. Ann. Otol.Rhinol. Laryngol., 85, 1–17.
6. Melton, L.J., III (1996) Epidemiology of hip fractures: implications of theexponential increase with age. Bone, 18, 121S–125S.
7. Parker, D.E. (1998) The relative roles of the otolith organs and semicircularcanals in producing space motion sickness. J. Vestib. Res., 8, 57–59.
8. Jones, S.M., Erway, L.C., Bergstrom, R.A., Schimenti, J.C. and Jones, T.A.(1999) Vestibular responses to linear acceleration are absent in otoconia-deficient C57BL/6JEi-het mice. Hear. Res., 135, 56–60.
9. Lyon, M.F. (1955) The developmental origin of hereditary absence ofotoliths in mice. J. Embryol. Exp. Morphol., 3, 230–241.
10. Lyon, M.F. (1951) Hereditary absence of otoliths in the house mouse.J. Physiol., 114, 410–418.
11. Lane, P.W. (1987) New mutants and linkages: Tilted Mouse News Lett.,77, 129.
12. Lim, D.J. and Erway, L.C. (1974) Influence of manganese on geneticallydefective otolith: a behavioral and morphological study. Ann. Otol. Rhinol.Laryngol., 83, 565–581.
13. Lim, D.J., Erway, L.C. and Clark, D.L. (1978) Tilted-head mice withgenetic otoconial anomaly. Behavioural and morphological correlates. InHood, J.D. (ed.), Vestibular Mechanisms in Health and Disease. AcademicPress, London, pp. 195–206.
14. Erway, L.C., Fraser, A.S. and Hurley, L.S. (1971) Prevention of congenitalotolith defect in pallid mutant mice by manganese supplementation.Genetics, 67, 97–108.
15. Bergstrom, R.A., You, Y., Erway, L.C., Lyon, M.F. and Schimenti, J.C.(1998) Deletion mapping of the head tilt (het) gene in mice: a vestibularmutation causing specific absence of otoliths. Genetics, 150, 815–822.
16. Ornitz, D.M., Bohne, B.A., Thalmann, I., Harding, G.W. and Thalmann, R.(1998) Otoconial agenesis in tilted mutant mice. Hear. Res., 122, 60–70.
17. Kiernan, A.E. and Steel, K.P. (2000) Mouse homologues for humandeafness. Adv. Otorhinolaryngol., 56, 233–243.
18. Massironi, S.M., Dagli, M.L., Lima, M.R., Alvarez, J.M. and Kipnis, T.L.(1994) A new mutant hairless mouse with lymph node hyperplasia and lateonset of autoimmune pathology. Braz. J. Med. Biol. Res., 27, 2401–2405.
19. Ying, H.C., Hurle, B., Wang, Y., Bohne, B.A., Wuerffel, M.K. andOrnitz, D.M. (1999) High resolution mapping of tlt, a mouse mutantlacking otoconia. Mam. Genome, 10, 544–548.
20. Hurle, B., Lane, K., Kenney, J., Tarantino, L.M., Bucan, M.,Brownstein, B.H. and Ornitz, D.M. (2001) Physical mapping of the mousetilted locus identifies an association between human deafness loci DFNA6/14 and vestibular system development. Genomics, 77, 189–199.
21. Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W.and Lipman, D.J. (1997) Gapped BLAST and PSI-BLAST: a new generationof protein database search programs. Nucl. Acids Res., 25, 3389–3402.
22. Burge, C. and Karlin, S. (1997) Prediction of complete gene structures inhuman genomic DNA. J. Mol. Biol., 268, 78–94.
23. Kozak, M. (1987) An analysis of 50-noncoding sequences from 699vertebrate messenger RNAs. Nucl. Acids Res., 15, 8125–8148.
24. Moller, S., Croning, M.D. and Apweiler, R. (2001) Evaluation of methods forthe prediction of membrane spanning regions. Bioinformatics, 17, 646–653.
25. Krogh, A., Larsson, B., von Heijne, G. and Sonnhammer, E.L. (2001)Predicting transmembrane protein topology with a hidden Markov model:application to complete genomes. J. Mol. Biol., 305, 567–580.
26. Nakai, K. and Kanehisa, M. (1992) A knowledge base for predictingprotein localization sites in eukaryotic cells. Genomics, 14, 897–911.
27. Thompson, J.D., Higgins, D.G. and Gibson, T.J. (1994) CLUSTAL W:improving the sensitivity of progressive multiple sequence alignmentthrough sequence weighting, position-specific gap penalties and weightmatrix choice. Nucl. Acids Res., 22, 4673–4680.
28. Marchler-Bauer, A., Panchenko, A.R., Shoemaker, B.A., Thiessen, P.A.,Geer, L.Y. and Bryant, S.H. (2002) CDD: a database of conserved domainalignments with links to domain three-dimensional structure. Nucl. AcidsRes., 30, 281–283.
29. Pote, K.G. and Ross, M.D. (1991) Each otoconia polymorph has a proteinunique to that polymorph. Comp. Biochem. Physiol., 98B, 287–295.
30. Verpy, E., Leibovici, M. and Petit, C. (1999) Characterization of otoconin-95,the major protein of murine otoconia, provides insights into the formation ofthese inner ear biominerals. Proc. Natl Acad. Sci. USA, 96, 529–534.
31. Wang, Y., Kowalski, P.E., Thalmann, I., Ornitz, D.M., Mager, D.L. andThalmann, R. (1998) Otoconin-90, the mammalian otoconial matrix proteincontains two domains of homology to secretory phospholipase A2. Proc.Natl Acad. Sci. USA, 95, 15345–15350.
32. Legan, P.K., Rau, A., Keen, J.N. and Richardson, G.P. (1997) The mousetectorins. Modular matrix proteins of the inner ear homologous to compo-nents of the sperm-egg adhesion system. J. Biol. Chem., 272, 8791–8801.
33. Cohen-Salmon, M., El-Amraoui, A., Leibovici, M. and Petit, C. (1997)Otogelin: a glycoprotein specific to the acellular membranes of the innerear. Proc. Natl Acad. Sci. USA, 94, 14450–14455.
34. Zwaenepoel, I., Mustapha, M., Leibovici, M., Verpy, E., Goodyear, R.,Liu, X.Z., Nouaille, S., Nance, W.E., Kanaan, M., Avraham, K.B. et al.(2002) Otoancorin, an inner ear protein restricted to the interface betweenthe apical surface of sensory epithelia and their overlying acellular gels,is defective in autosomal recessive deafness DFNB22. Proc. Natl Acad.Sci. USA, 99, 6240–6245.
35. Pote, K.G. and Ross, M.D. (1986) Ultrastructural morphology and proteincontent of the internal organic material of rat otoconia. J. Ultrastruct. Mol.Struct. Res., 95, 61–70.
788 Human Molecular Genetics, 2003, Vol. 12, No. 7
by guest on July 23, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

36. Yamoah, E.N., Lumpkin, E.A., Dumont, R.A., Smith, P.J., Hudspeth, A.J.and Gillespie, P.G. (1998) Plasma membrane Ca2þ-ATPase extrudes Ca2þ
from hair cell stereocilia. J. Neurosci., 18, 610–624.37. Kozel, P.J., Friedman, R.A., Erway, L.C., Yamoah, E.N., Liu, L.H.,
Riddle, T., Duffy, J.J., Doetschman, T., Miller, M.L., Cardell, E.L. et al.(1998) Balance and hearing deficits in mice with a null mutation in thegene encoding plasma membrane Ca2þ-ATPase isoform 2. J. Biol.Chem., 273, 18693–18696.
38. Everett, L.A., Belyantseva, I.A., Noben-Trauth, K., Cantos, R., Chen, A.,Thakkar, S.I., Hoogstraten-Miller, S.L., Kachar, B., Wu, D.K. andGreen, E.D. (2001) Targeted disruption of mouse Pds provides insightabout the inner-ear defects encountered in Pendred syndrome. Hum. Mol.Genet., 10, 153–161.
39. Haddon, C. and Lewis, J. (1996) Early ear development in the embryo ofthe zebrafish, Danio Rerio. J. Comp. Neurol., 365, 113–128.
40. Moens, C.B., Cordes, S.P., Giorgianni, M.W., Barsh, G.S. and Kimmel, C.B.(1998) Equivalence in the genetic control of hindbrain segmentation in fishand mouse. Development, 125, 381–391.
41. Riley, B.B., Zhu, C., Janetopoulos, C. and Aufderheide, K.J. (1997) Acritical period of ear development controlled by distinct populations ofciliated cells in the zebrafish. Dev. Biol., 191, 191–201.
42. Peterson, R.T., Link, B.A., Dowling, J.E. and Schreiber, S.L. (2000) Smallmolecule developmental screens reveal the logic and timing of vertebratedevelopment. Proc. Natl Acad. Sci. USA, 97, 12965–12969.
43. Whitfield, T.T., Riley, B.B., Chiang, M.Y. and Phillips, B. (2002)Development of the zebrafish inner ear. Dev. Dyn., 223, 427–458.
44. Malicki, J., Schier, A.F., Solnica-Krezel, L., Stemple, D.L., Neuhauss, S.C.,Stainier, D.Y., Abdelilah, S., Rangini, Z., Zwartkruis, F. and Driever, W.(1996) Mutations affecting development of the zebrafish ear. Development,123, 275–283.
45. Whitfield, T.T., Granato, M., Vaneeden, F.J.M., Schach, U., Brand, M.,Furutaniseiki, M., Haffter, P., Hammerschmidt, M., Heisenberg, C.P.,Jiang, Y.J. et al. (1996) Mutations affecting development of the zebrafishinner ear and lateral line. Development, 123, 241–254.
46. Riley, B.B. and Moorman, S.J. (2000) Development of utricular otoliths,but not saccular otoliths, is necessary for vestibular function and survival inzebrafish. J. Neurobiol., 43, 329–337.
47. Coimbra, R.S., Weil, D., Brottier, P., Blanchard, S., Levi, M., Hardelin, J.P.,Weissenbach, J. and Petit, C. (2002) A subtracted cDNA library from thezebrafish (Danio rerio) embryonic inner ear. Genome Res., 12, 1007–1011.
48. Solomon, K.S. and Fritz, A. (2002) Concerted action of two dlx paralogs insensory placode formation. Development, 129, 3127–3136.
49. Maroon, H., Walshe, J., Mahmood, R., Kiefer, P., Dickson, C. and Mason, I.(2002) Fgf3 and Fgf8 are required together for formation of the otic placodeand vesicle. Development, 129, 2099–2108.
50. Mustapha, M., Chouery, E., Torchard-Pagnez, D., Nouaille, S., Khrais, A.,Sayegh, F.N., Megarbane, A., Loiselet, J., Lathrop, M., Petit, C. et al.(2002) A novel locus for Usher syndrome type I, USH1G, maps tochromosome 17q24–25. Hum. Genet., 110, 348–350.
51. Morell, R.J., Friderici, K.H., Wei, S., Elfenbein, J.L., Friedman, T.B. andFisher, R.A. (2000) A new locus for late-onset, progressive, hereditaryhearing loss DFNA20 maps to 17q25. Genomics, 63, 1–6.
52. Elfenbein, J.L., Fisher, R.A., Wei, S., Morell, R.J., Stewart, C.,Friedman, T.B. and Friderici, K. (2001) Audiologic aspects of the searchfor DFNA20: a gene causing late-onset, progressive, sensorineural hearingloss. Ear Hear., 22, 279–288.
53. Anderson, D.W., Probst, F.J., Belyantseva, I.A., Fridell, R.A., Beyer, L.,Martin, D.M., Wu, D., Kachar, B., Friedman, T.B., Raphael, Y. et al. (2000)The motor and tail regions of myosin XV are critical for normal structureand function of auditory and vestibular hair cells. Hum. Mol. Genet., 9,1729–1738.
54. Kitamura, K., Kakoi, H., Yoshikawa, Y. and Ochikubo, F. (1992)Ultrastructural findings in the inner ear of Jackson shaker mice. ActaOtolaryngol., 112, 622–627.
55. Naski, M.C., Colvin, J.S., Coffin, J.D. and Ornitz, D.M. (1998)Repression of hedgehog signaling and BMP4 expression in growthplate cartilage by fibroblast growth factor receptor 3. Development, 125,4977–4988.
Human Molecular Genetics, 2003, Vol. 12, No. 7 789
by guest on July 23, 2016http://hm
g.oxfordjournals.org/D
ownloaded from




















