
Mild Systemic Inflammation has a Neuroprotective Effect After Stroke in Rats

The FASEB Journal express article 10.1096/fj.03-1203fje. Published online May 7, 2004.
Neuroprotective effects of spermine following hypoxia-ischemia-induced brain damage: A mechanistic study Andrew N. Clarkson, Hanzhong Liu, Lachlan Pearson, Mohit Kapoor, Joanne C. Harrison, Ivan A. Sammut, David M. Jackson, and Ian Appleton Department of Pharmacology and Toxicology, University of Otago, PO Box 913, Dunedin, New Zealand
Corresponding author: Dr Ian Appleton Department of Pharmacology and Toxicology University of Otago, P.O. Box 913 Dunedin New Zealand; E-mail: [email protected]
ABSTRACT
The polyamines (spermine, putrescine, and spermidine) can have neurotoxic or neuroprotective properties in models of neurodegeneration. However, assessment in a model of hypoxia–ischemia (HI) has not been defined. Furthermore, the putative mechanisms of neuroprotection have not been elucidated. Therefore, the present study examined the effects of the polyamines in a rat pup model of HI and determined effects on key enzymes involved in inflammation, namely, nitric oxide synthase (NOS) and arginase. In addition, effects on mitochondrial function were investigated. The polyamines or saline were administered i.p. at 10mg/kg/day for 6 days post-HI. Histological assessment 7 days post-HI revealed that only spermine significantly (P<0.01) reduced infarct size from 46.14 ± 10.4mm3 (HI + saline) to 4.9 ± 2.7 mm3. NOS activity was significantly increased following spermine treatment in the left (ligated) hemisphere compared with nonintervention controls (P<0.01) and HI + saline (P<0.05). In contrast, spermine decreased arginase activity compared with HI + saline but was still significantly elevated in comparison to nonintervention controls (P<0.01). Assessment of mitochondrial function in the HI + saline group, revealed significant and extensive damage to complex-I (P<0.01) and IV (P<0.001) and loss of citrate synthase activity (P<0.05). No effect on complex II-III was observed. Spermine treatment significantly prevented all these effects. This study has therefore confirmed the neuroprotective effects of spermine in vivo. However, for the first time, we have shown that this effect may, in part, be due to increased NOS activity and preservation of mitochondrial function.
Key words: polyamines • arginase activity • nitric oxide synthase
cute tissue damage due to cerebral hypoxia–ischemia (HI) in neonates can result in functional morbidity or mortality. The resultant pathologies include mental retardation, cerebral palsy, and epilepsy. Cerebral HI has been shown to resemble many animal
models of stroke and acute ischemic stroke in humans (1). Ischemia triggers an excessive release of glutamate, which in turn, initiates a cascade of inflammatory and immune responses ultimately leading to neuronal damage (2). Although at present, no effective therapeutic interventions are available to treat neuronal outcomes following a stroke, numerous, largely unsuccessful, attempts have been made to retard stroke-induced brain damage.
A
Page 1 of 21(page number not for citation purposes)

The naturally occurring polyamines, putrescine, spermine, and spermidine, are important for cell growth, proliferation, regeneration, and differentiation (3). In addition, their involvement in cell homeostasis extends to regulatory mechanisms of programmed cell death (4), modulation of nitric oxide synthase activity (NOS; 5), regulation of mitochondrial Ca2+ transport (6, 7), inhibition of mitochondrial membrane permeability (8), and free radical scavenging (9, 10).
The polyamines are low molecular-weight aliphatic amines, present in moderately high concentrations in the brain. Their production is regulated by ornithine decarboxylase (ODC), which converts ornithine to putrescine (11). Previous studies have shown that the polyamines play an important role in the ischemic cascade (12, 13). Changes in the pathophysiological state of the body as a result of an ischemic insult result in significant changes to ODC activity and putrescine levels (14, 15). Most importantly, an increase in ODC activity and putrescine levels can occur with little or no change to spermidine or spermine levels (14, 15). It has also been demonstrated that elevated polyamine levels consequent to an ischemic event are implicated in neuronal damage (16–19). In addition, elevated levels of putrescine have been shown to result in blood–brain barrier dysfunction as well as vasogenic edema in ischemic events (15–17).
The involvement of polyamines in the pathophysiology of ischemic damage has not, as yet, been fully elucidated. Both global and focal models of brain ischemia have shown evidence of increased polyamine metabolism, characterized by an increase in ODC activity and putrescine levels. In contrast, the concentration of endogenous spermine in the brain has proven variable under different pathophysiological conditions. In a model of global ischemia, spermine levels are either slightly reduced (20), or somewhat elevated (21), whereas, in a model of focal ischemia, spermine levels are either considerably reduced (22), or unaltered (14).
To date, no study has ascertained the effects of exogenous administration of the polyamines in a model of HI-induced brain damage. Furthermore, the mechanisms of a putative polyamine-induced neuroprotection remain undetermined. Therefore, in this study, we have used a modified “Levine” rat-pup model (unilateral carotid artery ligation, coupled with global hypoxia), to evaluate the effects of the polyamines on HI-induced brain damage. We have also investigated the biochemical and molecular energetic implications associated with these effects.
METHODS
Surgery and treatment
All procedures were carried out in accordance with the guidelines on care and use of laboratory animals by the University of Otago Animal Ethics Committee. Male Wister rat pups, of age post-natal-day (PND) 21 were randomly sorted into 5 groups: untreated, no intervention controls, HI + saline controls, HI + spermidine, HI + spermine and HI + putrescine. A total of n = 6 animals per group were used for histological analysis of infarct size and n = 8 animals per group for biochemical and molecular analysis.
On PND 26, a small longitudinal incision was made along the neckline under halothane anesthesia, and the left common carotid artery was exposed and permanently double-ligated using 5-0 silk surgical sutures (Southern Medical Products, Dunedin, NZ). The incision was then closed with 4-0 sutures and animals were left to recover for a period of 2 hours. After recovery,
Page 2 of 21(page number not for citation purposes)

animals were placed in a chamber maintained at 34°C, constantly perfused with a humidified hypoxic atmosphere of 8% O2/92% N2 for a period of 60 min. Animals were then returned to normal housing conditions until 7 days post-HI, at which time assessment of neuroprotection was carried out.
Drug (putrescine, spermine, and spermidine) administration was carried out with a single i.p. injection of 10 mg/kg, 1hour post-HI, and then subsequently once daily for the next 6 days. This dose is based on the finding that 10 mg/kg of putrescine, spermine, or spermidine resulted in a significant neuronal protection of CA1 and striatal neurons in a gerbil model of forebrain ischemia (23).
Histological assessment
Histological assessment at 7 days post-HI (PND 33) was carried out using hematoxylin and eosin (H and E) staining. Animals were deeply anaesthetized with 3% halothane. Briefly, the chest cavity was opened and the heart perfused with heparinized saline followed by 10% phosphate-buffered formalin. The brains were then removed and immediately stored in the same fixative until embedded in paraffin wax. 10 µm serial sections were cut at 1 mm intervals throughout the brain and stained with H and E. Computer image analyses (Zeiss Axiovision, Thornwood, NY, USA) of the histological sections were used to quantify the extent of neuronal infarct. The area of infarct (mm2) within the ipsilateral (left) hemisphere was normalized against the total noninfarcted area in the right hemisphere. The total volume (mm3) of infarction was then determined by calculating the area for each point throughout the brain (every 1mm) and an area under the curve formula used (GraphPad Prism, version 3.0a, San Diego, CA, USA).
Tissue preparation for biochemical and mitochondrial analysis
Animals were rapidly killed by decapitation, brains were removed, and the left and right hemispheres were dissected. Tissues were immediately frozen on dry ice and stored at –80°C until use. The samples were then homogenized on ice (4°C) in a mixture of protease inhibitors (10 µg/ml leupeptin, 10 µg/ml pepstatin A and 100 µg/ml phenylmethylsulphonyl fluoride) in 50 mM Tris-HCl, pH 7.5. Samples were centrifuged for 15 min at 2,000 g (4°C) and supernatants aliquoted for each assay.
For mitochondrial enzyme assays, samples of tissue homogenates were freeze-thawed three times to ensure complete mitochondrial lysis. Samples were then diluted to 1 mg/ml in 10 mM Tris-HCl, pH 7.4 containing 320 mM sucrose and 1 mM K+EDTA and stored at –80°C.
Protein concentrations for all samples were assayed using the Bradford assay (24) against bovine serum albumin (BSA) standards.
Nitric oxide synthase activity
Homogenates were incubated for 30 min at 37°C in 50 mM Tris (pH 7.4), containing NADPH (1mM), calmodulin (30nM), tetrahydrobiopterin (5µM), CaCl2 (2mM), L-Valine (50mM) and the substrate (a mixture of unlabeled and 10µM [3H] L-arginine). To assess the contribution of iNOS (calcium-independent) from total NOS (calcium-dependent), the reaction was then
Page 3 of 21(page number not for citation purposes)

repeated in the absence of CaCl2 and presence of 1 mM EGTA. Following incubation, the reaction was stopped, by the addition of ice-cold HEPES buffer (20mM, pH 5.5) containing 1 mM EDTA and 1 mM EGTA. The newly formed [3H] L-citrulline was separated from [3H] L-arginine by passing the mix over a Dowex (mesh size 50×) chromatographic column. Radioactivity was then assessed using liquid scintillation and results expressed as pmol [3H] L-citrulline/30 min/mg protein.
Arginase activity
50 µl of 10 mM MnCl2 in 50 mM Tris-HCl (pH 7.5) was incubated with 50 µl of tissue homogenate at 55°C for 10 min. 25 µl was then removed and incubated in the presence of 25 µl of 0.5 M L-arginine, pH 9.7 at 37°C for 1 hour. An acid mixture containing H2SO4, H3PO4, and H2O (1:3:7) was added to stop the reaction. 9% isonitrosopropiopheanone (in ethanol) was added and incubated at 100°C for 45 min. The amount of urea formed was determined spectrophotometrically at 540 nm. The results are expressed as µg urea/mg protein.
Mitochondrial complex I assay
The reaction mixture contained 25 mM phosphate buffer pH 7.2, 0.2 mM NADH, 10 mM MgCl2, 1 mM KCN, 2.5 mg BSA and 12 µg of tissue homogenate protein in a final volume of 260 µl. The reaction was initiated by the addition of ubiquinone (CoQ1; to give a final concentration of 50 µM) and read at 340 nm for 5 min. The inhibited rate was then measured in the presence of 0.5 mM rotenone for a further 5 min. Results are expressed as nmol/min/mg protein.
Mitochondrial complex II-III assay
To a reaction mixture containing 100 mM phosphate buffer pH 7.4, 0.3 mM K+EDTA, 1 mM KCN and 100 µM cytochrome c, 12 µg of tissue homogenate protein was added to give a final volume of 260 µl. The reaction was initiated by the addition of 20 µl of 1M succinate, and read at 550 nm at 30°C for 5 min. After this time, 10 µl of 2 mg/ml antimycin A was added, and the inhibited rate measured for a further 5 min. Results are expressed as nmol/min/mg protein.
Mitochondrial complex IV assay
The reaction mixture contained 10 mM phosphate buffer pH 7.0, 50 µM reduced cytochrome c and 1 mM K3FeCN6 in a final volume of 260 µl. The reaction was initiated by the addition of 12 µg tissue homogenate protein. The first-order rate constant (k) was calculated as the difference between the natural logarithms of the absorbance at t = 0 and at three time points—1, 2, and 3-min following addition of mitochondrial protein. The mean of these values (k) was taken, and the activity was expressed as k/min/mg protein.
Citrate synthase assay
The reaction mixture contained 100 mM Tris-HCl pH 8.0, 0.1 mM acetyl-CoA, 0.2 mM 5,5′ dithio-bis 2-nitrobenzoic acid, 0.1% (v/v) Triton X-100, and 12 µg of tissue homogenate protein in a final volume of 260 µl. The reaction was initiated by the addition of 10 µl of 20 mM oxaloacetate and read at 412 nm at 30°C. Results are expressed as nmol/min/mg protein.
Page 4 of 21(page number not for citation purposes)

Lactate dehydrogenase assay
Lactate dehydrogenase (LDH) activity in tissue samples was assessed using a commercially available kit (Sigma, Castle Hill, New South Wales, AU). The reaction mixture contained 4 parts of reagent B (16.2 mM pyruvate in Tris-base pH 7.5) to 10 parts of reagent A (0.194 mM NADH in 54 mM phosphate buffer pH 7.5) in a final volume of 290 µl. The reaction was initiated by the addition of 40 µg of tissue homogenate protein. LDH activity was calculated as the difference between the natural logarithms of the absorbance at 340 nm at 25°C for three time points; 1-, 2-, and 3-min following addition of tissue homogenate protein and is based on the conversion of NADH to NAD+. Results are expressed as U/L.
Statistics
Results are expressed as the mean ± SEM for n = 6–8 separate observations. Statistical analysis was performed using two-way ANOVA. The Mann-Whitney U-test was used to determine significance at individual points throughout the brain for each group. Student Newman-Keuls test was used for enzymatic post-hoc comparisons between control and treatment groups. A P value of <0.05 was considered significant.
RESULTS
Histological effects of polyamines on HI-induced brain damage
During the time course of HI and drug administration, body weight, temperature and blood glucose levels were monitored and shown not to vary between treatment groups (data not shown).
H and E staining 7 days post-HI revealed extensive neuronal damage ipsilateral to the carotid ligation, with no apparent anatomical damage contralaterally in the HI + saline-treated animals (Fig. 1). The extent of neuronal damage following HI + saline extends through all cortical layers (Fig. 1A), likewise all areas of the hippocampus (Fig. 1C). Treatment with spermine preserved anatomical regions in both the cortex and hippocampus (Fig. 1B and D, respectively), against HI-induced damage. Of the polyamines, only spermine significantly (P<0.01) decreased the infarct volume (4.9±2.7mm3) compared with HI + saline (46.14±10.4mm3). Quantification of the effects of the polyamines throughout the whole brain is shown in Fig. 2.
Effect of spermine on NOS activity
Spermine was the only polyamine that resulted in a significant neuroprotection (Fig. 2). Therefore, all subsequent biochemical and molecular analysis were only performed on spermine-treated animal samples. NOS activity 7 days post-HI revealed a significant increase in iNOS activity (absence of calcium) in the left (ligated) hemisphere from HI + saline-treated animals (P<0.05; Fig. 3B) compared with nonintervention controls. No change was seen in total NOS activity (addition of calcium) in the left hemisphere from HI + saline-treated animals (Fig. 3B). Treatment with spermine resulted in a significant increase in total NOS activity in comparison to nonintervention controls (P<0.01) and HI + saline (P<0.05), with no change in iNOS activity (Fig. 3B), compared with HI + saline controls. Assessment of iNOS and total NOS activity in the
Page 5 of 21(page number not for citation purposes)

right (nonligated) hemisphere, failed to reveal any significant differences between treatment groups (Fig. 3A).
Effect of spermine on arginase activity
Assessment of arginase activity in the left hemisphere of HI + saline-treated animals showed a significant increase in activity from 33.6±1.6 (nonintervention controls) to 58.9 ± 8.1 µg urea/mg protein (P<0.01; Fig. 4B). Treatment with spermine decreased arginase activity (45.7±2.0µg urea/mg protein) in comparison to HI + saline controls. However, the level of activity was still significantly up-regulated compared with untreated controls (P<0.01; Fig. 4B). Assessment of arginase activity in the right hemisphere, failed to reveal any significance between any groups (Fig. 4A).
Effect of spermine on Complex I activity
Complex I activity was significantly impaired in the left hemisphere following HI + saline (P<0.01; Fig. 5A), compared with nonintervention controls. Treatment with spermine reversed the HI-induced impairment of complex I to near normal nonintervention control levels of activity (P<0.01). Assessment of complex I activity in the right hemisphere failed to show any significant differences between any groups.
Effect of spermine on Complex II-III activity
Mitochondrial complex II-III activity for both hemispheres elicited no change in activity between any groups (Fig. 5B).
Effect of spermine on Complex IV activity
Hypoxia–ischemia resulted in a decrease in complex IV kinetics from 6.44 to 2.49 k/min/mg protein in the left hemisphere (P<0.001; Fig. 5C), compared with nonintervention controls. Spermine treatment provided significant protection to mitochondrial complex IV kinetics (P<0.001) compared with HI + saline, but was still down-regulated compared with nonintervention controls. No change in mitochondrial complex IV kinetics was seen in the right hemisphere between any groups.
Effect of spermine on citrate synthase activity
The mitochondrial membrane matrix marker, citrate synthase was significantly impaired in the left hemisphere following HI + saline (P<0.05; Fig. 5D), compared with nonintervention controls. Treatment with spermine-preserved citrate synthase activity to nonintervention control levels of activity (P<0.05). Assessment of citrate synthase activity in the right hemisphere failed to show any significant differences between any groups.
Effect of spermine on LDH activity
Assessment of LDH activity showed a significant decrease in the left hemisphere following HI + saline (P<0.001; Fig. 6), compared with nonintervention controls. Treatment with spermine reversed the HI-induced decrease in LDH activity back to nonintervention control levels
Page 6 of 21(page number not for citation purposes)

(P<0.001). In contrast, LDH activity in the right hemisphere was increased following HI + saline, but this was not significant compared with the right hemisphere of the nonintervention controls. No change in LDH activity compared with nonintervention controls was seen in the right hemisphere following HI + spermine treatment.
DISCUSSION
The present study illustrates that HI induces pronounced lesions across cortical and subcortical regions ipsilateral to a carotid occlusion. In addition, modulation of critical biochemical enzymes, namely NOS and arginase and substantial impairment in mitochondrial energetics occurs following HI. Acute (6 days) administration of the polyamines showed that only spermine provided anatomical neuroprotection and virtually full preservation of mitochondrial energetics.
Our findings that HI produces substantial CNS damage ipsilateral to the occlusion, is consistent with those of other groups in this field. H and E staining, 7 days post-HI, revealed widespread regions of damage. Spermine provided significant neuroprotection. This is in keeping with previous work that has shown spermine to be neuroprotective in a model of forebrain ischemia (23). In this study, i.p. administration of spermine decreased the number of both hippocampal and striatal cell loss. A number of studies have also shown that the polyamines are neuroprotective, both in vivo (25, 26), and in vitro (27, 28), though spermine appears to provide the greatest amount of protection. However, in this study neither putrescine nor spermidine showed any neuroprotective effects. We have therefore confirmed the neuroprotective effects of spermine but have extended these observations by determining the biochemical and molecular events associated with these observations.
Nitric oxide (NO) is a key component in the brain’s response to HI-induced damage. NOS, of which three isoforms exist (iNOS, nNOS, and ecNOS), converts L-arginine to NO and L-citrulline. The present study showed an increase in iNOS activity in response to HI. However, this increase in iNOS activity only accounted for a minority of the total NOS activity, as shown in Fig. 3. Previous work has demonstrated that iNOS-derived NO is deleterious (29) and that iNOS knockout mice are more resistant to ischemic episodes (30). Furthermore, mice lacking the nNOS gene are more resistant to ischemic damage (31). In contrast, NO from ecNOS is neuroprotective, as ecNOS knockout mice have increased infarctions in comparison to their wild-type controls (32). Therefore, NO can be both neurotoxic if it is from iNOS or nNOS, but neuroprotective if derived from ecNOS. We have shown that treatment with spermine resulted in a further increase in total NOS activity in comparison to HI. This increase in total NOS activity is most likely to be attributed to an increase in either ecNOS or nNOS activity. Our findings therefore further support the evidence that an elevation of total NOS activity, in the absence of an increase in iNOS activity, is neuroprotective.
To date, little, if anything, is known about the pathoneurological role of the other L-arginine-metabolizing enzyme arginase. Arginase metabolizes L-arginine to urea and L-ornithine, which is the first step in the production of the polyamines. Because both NOS and arginase use L-arginine as a common substrate, modulating the activity of arginase may subsequently regulate the activity of NOS (33). The present study resulted in an elevated level of arginase activity following HI, which was reduced with spermine treatment. Thus, spermine increased NOS and decreased arginase activity with resultant neuroprotection. Therefore, we hypothesize that
Page 7 of 21(page number not for citation purposes)

exogenous administration of spermine indirectly suppresses arginase activity, with a resultant utilization of L-arginine by the NOS pathway. This leads to an increase in NO levels and thus results in neuroprotection.
Investigations over the past decade have substantiated mitochondria as being important sub-cellular targets for ischemic damage. It has been observed that brain mitochondrial respiratory chain complexes undergo significant changes in activity following an ischemic event, both in vivo and in vitro (34–38). As evidenced in the present study, HI-induced substantial damage to mitochondrial respiratory energetics. This decrease in mitochondrial energetics is consistent with other models of ischemic brain damage (39). A key finding in the present study was that spermine provided virtually complete preservation of mitochondrial respiratory chain complex activity.
Elevation of reactive oxygen species (ROS) and the resulting oxidative stress plays a pivotal role in neuronal damage. Accumulation of ROS and cytosolic Ca2+ may impair metabolic enzymes required for substrate production or promote disruption of mitochondrial membranes. ROS production has been shown to inhibit pyruvate dehydrogenase through oxidation, a process that is potentiated by elevated Ca2+ (40). These changes could explain the deficits seen at the level of complex I observed in the present study following HI. This decrease in activity has previously been seen in brain mitochondria following an ischemic episode, in which a decrease in state 3 NAD-linked respiration rates were observed (41). Inhibition of complex I activity, unlike the other complexes, leads to a rapid decrease in oxygen consumption initiating oxidative stress (42). This oxidative stress then triggers a sudden rise in Ca2+, as a consequence of inhibition of oxidative phosphorylation, preventing the energy-dependent pumping of Na+ and Ca2+ out of the cell, while continuously leaking Ca2+ into the cell at a normal rate (43). Spermine can restore phosphorylation in mitochondria isolated from liver that have been subjected to heat stress (44). The results of this study, therefore, confirm and extend pervious observations.
In addition to the effects of HI on complex I activity, other parts of the mitochondrial respiratory chain have been shown to be sensitive to stress-induced ROS. The formation of large quantities of oxygen free radicals has been proposed to initiate both lipid and protein peroxidation (45, 46). Complex IV is particularly susceptible to damage resulting from lipid peroxidation products (47). In the present study, HI induced a significant decrease in complex IV activity, which was reversed by spermine treatment. This finding is in support of previous studies that have shown that the polyamines can inhibit lipid peroxidation (48), suggesting that the damage seen to complex IV in our study is, in part, due to lipid peroxidation.
Citrate synthase activity was also significantly decreased following HI, indicating that mitochondrial structures were not intact, and that they were leaking important mitochondrial matrix components. Spermine provided full preservation of citrate synthase activity, protecting against the HI-induced mitochondrial membrane permeability. It has been demonstrated that spermine protects against mitochondrial membrane permeability transition, as measured by Ca2+ release, mitochondrial swelling and pyridine nucleotide (8).
Nitric oxide produced as a consequence of ischemic damage may react with other free radicals such as superoxide to form peroxynitrite (ONOO–), which is an extremely potent oxidizing agent (49, 50). Peroxynitrite has been shown to directly inhibit complexes II-III and IV activities in
Page 8 of 21(page number not for citation purposes)

cultured neurons (51) without affecting complex I activity. Here, we have shown that post HI, NOS activity is increased with concomitant damage to mitochondrial function. Thus, the formation of NO and or peroxynitrite could account for the damage to mitochondrial complex activity. However, spermine, which protected mitochondrial respiratory chain activity, further increased NOS activity. Furthermore, it has been shown in vitro, using cytosolic preparations of normal rat cerebellum, that large concentrations of spermine inhibit NOS activity (5). These results would initially seem contradictory. However, it now appears that the action of these endogenous compounds may be intrinsically interlinked and dependent on the relative concentration of each agent. In addition, the region of the brain studied and the state of activation may complicate interpretation of results.
The controversial range of actions reported is further confounded by the level of oxidative stress and other determining factors involved in the pathophysiological state of the tissue. Although pathophysiologically high levels of NO have been indicated to be detrimental to mitochondrial cytochrome redox state, targeting, in particular, the cytochrome oxidase super complex (complex IV) (52), low physiologically relevant NO levels have been shown to abrogate cardiac LDH release in association with a mitochondrial KATP channel-dependent pathway of protection against ischemic reperfusion injury. Up-regulation of tissue LDH activity has been indicated to be dependent on an increased transcription of a hypoxia-inducing factor (53), stabilized intriguingly, by high levels of NO in a hypoxic (3% O2) model (54). Our model did show that decreased complex IV activity did in fact coincide with elevated iNOS activity within the infarcted left hemisphere. Both mitochondrial integrity, as assessed by the matrix marker, citrate synthase (55), and LDH levels, were also severely compromised in this region. Spermine has also been shown to have a double-edged effect in that it can both inhibit (8, 56) and stimulate (57–59) cellular apoptosis through mitochondrial-linked pathways. This is through a modulatory effect of the N-methyl-D-aspartate (NMDA) receptor (60). However, this is a concentration-dependent effect, whereby low doses (<50 nmol) attenuate NMDA-induced damage, whereas larger doses potentiate excitotoxicity (61). Much of the evidence relating to spermine-mediated induction of mitochondrial cytochrome c release and caspase (3 and 9) activation is based on in vitro studies in cancer cell lines. Paradoxically, studies on neuronal cells, thymocytes, and in vivo cerebral tissue indicate a protective role for spermine, possibly mediated through a mitochondrial-related anti-apoptotic action. Ha and co-workers (9) also provided evidence to suggest that part of this protection may relate to a direct antioxidant capability of spermine. Spermine’s neuroprotective role may also be contingent on an increased mitochondrial Ca2+ buffering capacity. In our present study, spermine failed to provoke evidence of cellular or mitochondrial damage in either ipsilateral or contralateral regions. However, the results of this study clearly demonstrated that a daily regime of 10 mg/kg spermine, significantly restored left hemisphere indices of integrity and mitochondrial energetics damaged by HI. These effects coincided with a reduction in iNOS activity but not total NOS.
Lactate dehydrogenase is the enzyme catalyzed in the final step of the anaerobic metabolism, glycolysis, and is a key cytosolic marker. The present study resulted in a marked decrease in LDH activity following HI. In addition to the results of citrate synthase activity, this reinforces the hypothesis that the induction of HI results in a significant leak in the cytosolic membrane, which is preserved by treatment with spermine.
Page 9 of 21(page number not for citation purposes)

CONCLUSIONS
The present study demonstrates that following an HI insult, the lesioned (left) hemisphere undergoes extensive changes in both biochemical and mitochondrial energetics, which could account for a large proportion of the neuronal cell damage. Exogenous treatment with the naturally occurring polyamine spermine resulted in significant decrease in infarction size. This may be due to an up-regulation of NOS activity (more specifically ecNOS) and or a decrease in arginase activity. In addition, we have shown that HI results in severe impairment of mitochondrial function. Furthermore, spermine provided full preservation of mitochondrial respiratory chain energetics. This confirms and extends, our and other, previous studies, which have shown that NO is neuroprotective. More importantly, we speculate, for the first time, that modulation of the enzyme arginase may be a future, novel target for drug intervention in neurodegenerative pathologies.
ACKNOWLEDGMENTS
This work was supported by a New Zealand Neurological Foundation grant number 0121SPG and a New Zealand Lottery grant in aid, number AP97549.
REFERENCES
1. Rice, J. E., Vannucci, R. C., and Brierley, J. B. (1981) The influence of immaturity on hypoxic–ischemic brain damage in the rat. Ann. Neurol. 9, 131–141
2. Silverstein, F. S., Buchanan, K., and Johnston, M. V. (1986) Perinatal hypoxia–ischemia disrupts striatal high-affinity [3H] glutamate uptake into synaptosomes. J. Neurochem. 47, 1614–1619
3. Tabor, C. W., and Tabor, H. (1984) Polyamines. Annu. Rev. Biochem. 53, 749–790
4. Ha, H. C., Woster, P. M., Yager, J. D., and Casero, R. A., Jr. (1997) The role of polyamine catabolism in polyamine analogue-induced programmed cell death. Proc. Natl. Acad. Sci. U.S.A. 94, 11,557–11,562
5. Hu, J., Mahmoud, I. M., and El-Fakahany, E. E. (1994) Polyamines inhibit nitric oxide synthase in rat cerebellum. Neurosci. Lett. 175, 41–45
6. Jensen, J. R., Lynch, G., and Baudry, M. (1987) Polyamines stimulate mitochondrial calcium transport in rat brain. J. Neurochem. 48, 765–772
7. Lenzen, S., Münster, W., and Runstenbeck, I. (1992) Dual effect of spermine on mitochondrial Ca2+ transport. Biochem. J. 286, 597–602
8. Lapidus, R. G., and Sokolove, P. M. (1992) Inhibition by spermine of the inner permeability transition of isolated rat heart mitochondria. FEBS Lett. 313, 314–318
Page 10 of 21(page number not for citation purposes)

9. Ha, H. C., Sirisoma, N. S., Kuppusamy, P., Zweier, J. L., Woster, P. M., and Casero, R. A., Jr. (1998) The natural polyamine spermine functions directly as a free radical scavenger. Proc. Natl. Acad. Sci. U.S.A. 95, 11,140–11,145
10. Tadolini, B. (1988) Polyamine inhibition of lipoperoxidation: The influence of polyamines on iron oxidation in the presence of compounds mimicking phospholipid polar heads. Biochem. J. 249, 33–36
11. Pegg, A. E., and McCann, P. P. (1982) Polyamine metabolism and function. Am. J. Physiol. 243, 212–221
12. Kleihaus, P., Hossmann, K. A., Pegg, A. E., Kobayashi, K., and Zimmermann, V. (1975) Resuscitation of the monkey brain after one hour ischemia, III: Indications of metabolic recovery. Brain Res. 95, 61–73
13. Paschen, W., Csiba, L., Rőhn, G., and Bereczki, D. (1991) Disturbances in polyamine metabolism in reversible focal cerebral ischemia of rat. J. Cereb. Blood Flow Metab. 11, suppl 2, 59 (abstract)
14. Başkaya, M. F., Rao, A. M., Doğan, A., Donaldson, D., Gellin, G., and Dempsey, R. J. (1997) Regional brain polyamine levels in permanent focal cerebral ischemia. Brain Res. 744, 302–308
15. Rao, A. M., Başkaya, M. K., Maley, M. E., Prasad, M. R., and Dempsey, R. J. (1995) Ornithine decarboxylase activity and edema formation in cerebral ischemia of conscious gerbils. J. Neurochem. 65, 2639–2643
16. Rao, A. M., Hatcher, J. F., Doğan, A., and Dempsey, R. J. (2000) Elevated N1-Acetylspermidine levels in gerbil and rat brains after CNS injury. J. Neurochem. 74, 1106–1111
17. Rao, V. L. R., Başkaya, M. K., Rao, A. M., Doğan, A., and Dempsey, R. J. (1998) Increased ornithine decarboxylase activity and protein level in the cortex following traumatic brain injury in rats. Brian Res. 783, 163–166
18. Doğan, A., Rao, A. M., Hatcher, J., Rao, V. L. R., Başkaya, M. K., and Dempsey, R. J. (1999) Effects of MDL 72527, a specific inhibitor of polyamine oxidase, on brain edema, ischemic injury volume, and tissue polyamine levels in rats after temporary middle cerebral artery occlusion. J. Neurochem. 72, 765–770
19. Henley, C. M., Wey, K., Takashima, A., Mills, C., Granmayeh, E., Krishnppe, I., and Robertson, C. S. (1997) S-Adenosylmethionine decarboxylase activity is decreased in the rat cortex after traumatic brain injury. J. Neurochem. 69, 259–265
20. Paschen, W., Csiba, L., Rőhn, G., and Bereczki, D. (1991) Polyamine metabolism in transient focal ischemia of rat brain. Brain Res. 566, 354–357
Page 11 of 21(page number not for citation purposes)

21. Koenig, H., Goldstone, A. D., Lu, C. Y., and Trout, J. J. (1992) Brain polyamines are controlled by N-methyl-D-aspartate receptors during ischemia recirculation. Stroke 21, 98–102
22. Paschen, W., Widman, R., and Weber, C. (1992) Changes in regional polyamine profiles in rat brains after transient cerebral ischemia (single versus repetitive ischemia): Evidence for release of polyamines from injured neurons. Neurosci. Lett. 135, 121–124
23. Gilad, G. M., and Gilad, V. H. (1991) Polyamines can protect against ischemia-induced nerve cell death in gerbil forebrain. Exp. Neurol. 111, 349–355
24. Bradford, M. M. (1979) A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Annals Biochem. 72, 248–254
25. Coert, B. A., Anderson, R. E., and Meyer, F. B. (2000) Exogenous spermine reduces ischemic damage in a model of focal cerebral ischemia in the rat. Neurosci. Lett. 282, 5–8
26. Farbiszewski, R., Bielawski, K., Bielawski, A., and Sobaniec, W. (1995) Spermine protects in vivo the antioxidant enzymes in transiently hypoperfused rat brain. Acta Neurobiol. Exp. 55, 254–258
27. Abe, K., Chida, N., Nishiyama, N., and Saito, H. (1993) Spermine promotes the survival of primary cultured brain neurons. Brain Res. 605, 322–326
28. Ferchmin, P. A., Perez, D., and Biello, M. (2000) Spermine is neuroprotective against anoxia and N-methyl-D-aspartate in hippocampal slices. Brain Res. 859, 273–279
29. Escott, K. J., Beech, J. S., Haga, K. K., Williams, S. C., Meldrm, B. S., and Bath, P. M. (1998) Cerebroprotective effects of the nitric oxide synthase inhibitors, 1-(2-trifluoromethylphenyl) imidazole and 7-nitro indazole, after transient focal cerebral ischemia in the rat. J. Cereb. Blood Flow Metab. 18, 281–287
30. Iadecola, C., Zhang, F., Casey, R., Nagayama, M., and Ross, M. E. (1997) Delayed reductions in ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J. Neurosci. 17, 9157–9164
31. Ferriero, D. M., Holtzman, D. M., Black, S. M., and Shelon, R. A. (1996) Neonatal mice lacking neuronal nitric oxide synthase are less vulnerable to hypoxic–ischemic injury. Neurobiol. Dis. 3, 64–71
32. Huang, Z., Huang, P. L., Ma, J., Meng, G., Ayata, C., Fishman, M. C., and Moskowitz, M. A. (1996) Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J. Cereb. Blood Flow Metab. 15, 378–384
33. Wang, W. W., Jenkinson, C. P., Griscavage, J. M., Kern, R. M., Arabolos, N. S., Byns, R. E., Cederbaum, S. D., and Ignarro, L. J. (1995) Co-induction of arginase and nitric oxide
Page 12 of 21(page number not for citation purposes)

synthase in murine macrophages activated by lipopolysaccharide. Biochem. Biophys. Res. Commun. 210, 1009–1016
34. Pérez-Pinzón, M. A., Mumford, P. L., Rosenthal, M., and Sick, T. J. (1997) Antioxidants, mitochondrial hyperoxidation and electrical activity recovery after anoxia in hippocampal slices. Brain Res. 754, 163–170
35. Pérez-Pinzón, M. A., Mumford, P. L., and Sick, T. J. (1998) Prolonged anoxia depolarization exacerbates NADH hyperoxidation and promotes poor electrical recovery after anoxia in hippocampal slices. Brain Res. 786, 165–170
36. Rosenthal, M., Feng, Z. C., Raffin, C. N., Harrison, M., and Sick, T. J. (1995) Mitochondrial hyperoxidation signals residual intracellular dysfunction after global ischemia in rat neocortex. J. Cereb. Blood Flow Metab. 15, 655–665
37. Welsh, F. A., Vannucci, R. C., and Brierley, J. B. (1982) Columnar alterations of NADH fluorescence during hypoxia-ischemia in immature rat brain. J. Cereb. Blood Flow Metab. 2, 221–228
38. Welsh, F. A., Marcy, V. R., and Sims, R. E. (1991) NADH fluorescence and regional energy metabolites during focal ischemia and reperfusion of rat brain. J. Cereb. Blood Flow Metab. 11, 459–465
39. Almeida, A., Allen, K. L., Bates, T. E., and Clark, J. B. (1995) Effect of reperfusion following cerebral ischemia on the activity of the mitochondrial respiratory chain in the gerbil brain. J. Neurochem. 65, 1698–1703
40. Bogaert, Y. E., Rosenthal, R. E., and Fiskum, G. (1994) Postischemic inhibition of cerebral cortex pyruvate dehydrogenase. Free Rad Biol Med. 16, 811–820
41. Sciamanna, M. A., Zinkel, J., Fabi, A. Y., and Lee, C. P. (1992) Ischemic injury to rat forebrain mitochondria and cellular calcium homeostasis. Biochim. Biophys. Acta 1134, 223–232
42. Davey, R. M., Peuchen, S., and Clark, J. B. (1998) Energy thresholds in brain mitochondria: Potential involvement in neurodegeneration. J. Biol. Chem. 273, 12,753–12,757
43. Lee, J. A., and Allen, D. G. (1992) Changes in intracellular free Ca2+ concentrations during long exposures to stimulated ischemia in isolated mammalian ventricular muscle. Circ. Res. 71, 58–59
44. Phillips, J. E., and Chaffee, R. R. J. (1982) Restorative effects of spermine on oxidative phosphorylation and respiration in heat-aged mitochondria. Biochem. Biophys. Res. Commun. 108, 174–181
45. Braughler, J. M., and Hall, E. D. (1989) Central nervous system trauma and stroke. I. Biochemical considerations for free radical formatio ad lipid peroxidation. Free Rad. Biol. Med. 6, 289–301
Page 13 of 21(page number not for citation purposes)

46. Gutteridge, J. M. C., and Halliwell, B. (1990) Reoxygenation injury and antioxidant protection: A tale of two paradoxes. Arch. Biochem. Biophys. 283, 223–226
47. Soussi, B., Idstrőm, J.-P., Scherstén, T., and Bylund-Fellenius, A.-C. (1990) Cytochrome c oxidase and cardiolipin alterations in response to skeletal muscle ischemia and reperfusion. Acta Physiol. Scand. 138, 107–114
48. Ohmori, S., Misaizu, T., Kitada, M., Kitagama, H., Igarashi, K., Hirose, S., and Kanakubo, Y. (1988) Polyamines lowered the hepatic lipid peroxide level in rat. Res. Commun. Chem. Pathol. Pharmacol. 62, 235–249
49. Beckman, J. S., Beckman, T. W., Chen, J., Marshall, P. A., and Freeman, B. A. (1990) Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 87, 1620–1624
50. Radi, R., Beckman, J. S., Bush, K. M., Freeman, B. A. (1991) Peroxynitrite oxidation of sulphydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 266, 4244–4250
51. Bolaños, J. P., Heales, S. J. R., Land, J. M., and Clark, J. B. (1994) Effect of peroxynitrite on the mitochondrial respiratory chain: Differential susceptibility of neurons and astrocytes in primary culture. J. Neurochem. 64, 1965–1972
52. Brown, G. C., and Borutaite, V. (2002) Nitric oxide inhibition of mitochondrial respiration and its role in cell death. Free Rad. Biol. Med. 33, 1440–1450
53. Rossignol, F., Solares, M., Balanza, E., Coudert, J., and Clottes, E. (2003) Expression of lactate dehydrogenase A and B genes in different tissues of rats adapted to chronic hypobaric hypoxia. J. Cell. Biochem. 89, 67–79
54. Mateo, J., Garcia-Lecea, M., Cadenas, S., Hernandez, C., and Moncada, S. (2003) Regulation of hypoxic-inducible factor-1alpha by nitric oxide through mitochondria-dependent and -independent pathways. Biochem. J. 376, 537–544
55. Sammut, I. A., Jayakumar, J., Latif, N., Rothery, S., Severs, N. J., Smolenski, R. T., Bates, T. E., and Yacoub, M. H. (2001) Heat stress contributes to the enhancement of cardiac mitochondrial complex activity. Am. J. Path. 158, 1821–1831
56. Hegardt, C., Andersson, G., and Oredsson, S. M. (2003) Spermine prevents cytocrome c release in glucocorticoid-induced apoptosis in mouse thymocytes. Cell Biol. Int. 27, 115–121
57. Stefanelli, C., Bonavita, F., Stanic’, I., Mignani, M., Facchini, A., Pignatti, C., Flamigni, F., Caldarera, C. M. (1998) Spermine causes caspase activation in leukaemia cells. FEBS Lett. 437, 233–236
Page 14 of 21(page number not for citation purposes)

58. Stefanelli, C., Bonavita, F., Stanic’, I., Pignatti, C., Flamigni, F., Guarnieri, C., Caldarera, C. M. (1999) Spermine triggers the activation of caspase-3 in a cell-free model of apoptosis. FEBS Lett. 451, 95–98
59. Maccarrone, M., Bari, M., Battista, N., Di Rinzo, M., Falciglia, K., and Finazi Agrò, A. (2001) Oxidation products of polyamines induce mitochondrial uncoupling and cytochrome c release. FEBS Lett. 507, 30–34
60. Simon, R. P., Swan, J. H., Griffiths, T., and Meldrum, B. S. (1984) Blockade of N-methyl-D-aspartate receptors may protect against ischemic damage in the brain. Science 266, 850–852
61. Munir, M., Subramamiam, S., and McGonigle, P. (1993) Polyamines modulate the neurotoxic effects of NMDA in vivo. Brain Res. 616, 163–170
Received January 15, 2004; accepted April 2, 2004.
Page 15 of 21(page number not for citation purposes)

Fig. 1
Figure 1. Extent of neuronal infarct following HI + saline (A and C) and HI + spermine (B and D). Sample H and E stained sections of the left hemisphere 7 days post-HI + saline revealed extensive damage to cortical layers and hippocampal regions (A and C). The area of infarction is indicated by arrows. Treatment with spermine provided significant protection as evidenced by a decrease in the extent of neuronal damage for both the cortex and hippocampus (B and D). Magnification × 3.15.
Page 16 of 21(page number not for citation purposes)
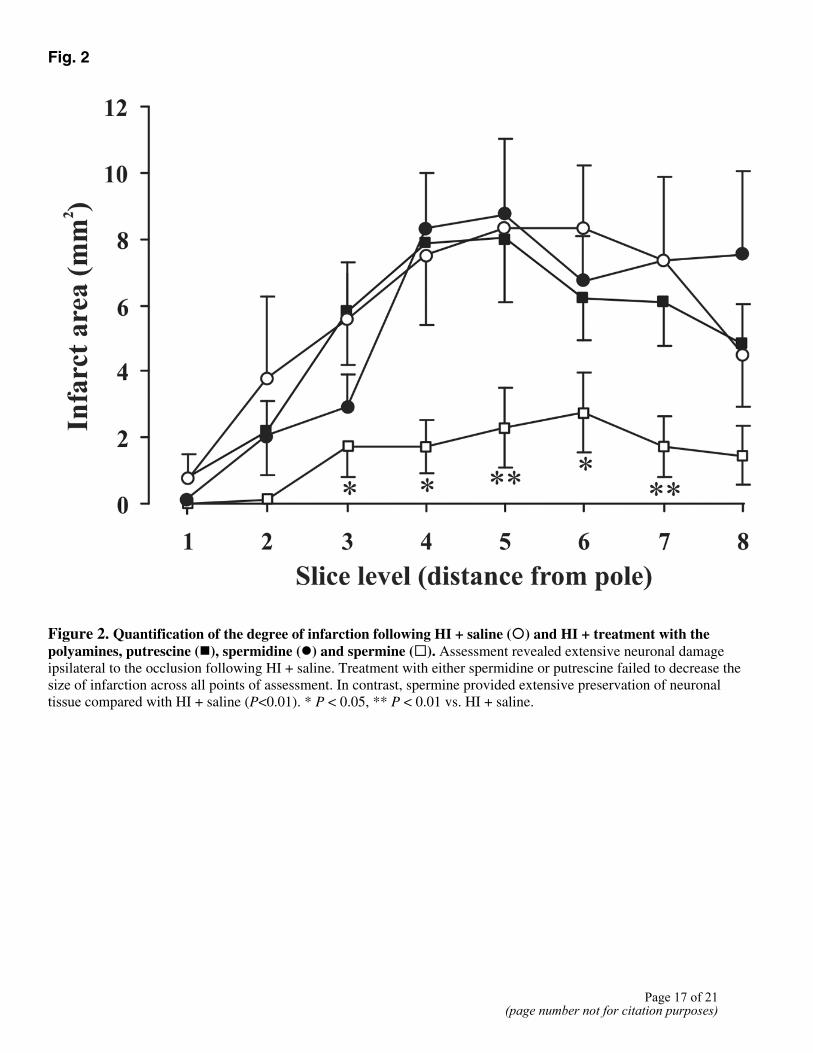
Fig. 2
Figure 2. Quantification of the degree of infarction following HI + saline (�) and HI + treatment with the polyamines, putrescine (�), spermidine (�) and spermine (�). Assessment revealed extensive neuronal damage ipsilateral to the occlusion following HI + saline. Treatment with either spermidine or putrescine failed to decrease the size of infarction across all points of assessment. In contrast, spermine provided extensive preservation of neuronaltissue compared with HI + saline (P<0.01). * P < 0.05, ** P < 0.01 vs. HI + saline.
Page 17 of 21(page number not for citation purposes)

Fig. 3
Figure 3. The effects of control, HI + saline and HI + spermine on total NOS activity (open bars) and iNOS activity(solid bars) in the right (A) and left (B) hemispheres. HI alone resulted in an increase in iNOS activity (P<0.05) in the left hemisphere, compared with nonintervention controls. Spermine treatment negated this increase in iNOS activity. However, this was not significantly different to either HI + saline nor nonintervention controls. Also, spermine treatment resulted in a significant increase in total NOS activity (P<0.05) in the left hemisphere, compared with HI + saline: + P < 0.05, ++ P < 0.01 vs. nonintervention control; *P < 0.05 vs. HI + saline.
Page 18 of 21(page number not for citation purposes)

Fig. 4
Figure 4. The effects of control (open bars), HI + saline (solid bars) and HI + treatment with spermine (hatchedbars) on arginase activity in the right (A) and left (B) hemispheres. HI alone resulted in a significant increase in arginase activity (P<0.01) in the left hemisphere, compared with nonintervention controls. Spermine treatment decreased the level of activity, but was still significantly elevated compared with nonintervention controls (P<0.01): ++ P < 0.01 vs. nonintervention control.
Page 19 of 21(page number not for citation purposes)

Fig. 5
Figure 5. Effects of control (open bars), HI + saline (solid bars) and HI + treatment with spermine (hatched bars) on mitochondrial respiratory chain complexes and the membrane permeability marker citrate synthase. Complexes I (A), II-III (B), IV (C) and citrate synthase (D) activities were assessed in tissue homogenates from both left and right hemispheres (data not shown). In the left hemisphere, HI alone resulted in a decrease in complex I, IV, and citrate synthase activities (P<0.01, P<0.001, and P<0.05, respectively), compared with nonintervention controls. In the left hemisphere, HI + spermine provided significant preservation of mitochondrial energetics at complex I, IV, and citrate synthase (P<0.01, P<0.001, and P<0.05, respectively), compared with HI + saline. No change in any complex or citrate synthase activity in the right hemisphere were observed (data not shown): + P < 0.05, ++ P < 0.01, +++ P < 0.001 vs. nonintervention control; * P < 0.05, ** P < 0.01, *** P < 0.001 vs. HI + saline.
Page 20 of 21(page number not for citation purposes)

Fig. 6
Figure 6. Effects of control (open bars), HI + saline (solid bars) and HI + spermine (hatched bars) on LDH activity in the right (A) and left (B) hemispheres. LDH activity was significantly impaired following HI + saline in the left hemisphere (P<0.001), compared with nonintervention controls. In the HI + saline group, LDH activity was elevated inthe right hemisphere, though LDH activity was not significantly different to nonintervention controls (P=0.071). Spermine treatment resulted in a significant preservation of LDH activity (P<0.001), compared with HI + saline. Assessment followingHI + spermine in the right hemisphere showed no change in activity compared with nonintervention controls and HI + saline. +++ P < 0.001 vs. nonintervention control; *** P < 0.001 vs. HI + saline.
Page 21 of 21(page number not for citation purposes)
Copyright © 2022 FDOKUMEN




















