
Negative Feedback Regulation of Met-Dependent Invasive Growth by Notch
15
MOLECULAR AND CELLULAR BIOLOGY, May 2005, p. 3982–3996 Vol. 25, No. 10 0270-7306/05/$08.000 doi:10.1128/MCB.25.10.3982–3996.2005 Copyright © 2005, American Society for Microbiology. All Rights Reserved. Negative Feedback Regulation of Met-Dependent Invasive Growth by Notch M. Cristina Stella,* Livio Trusolino, Selma Pennacchietti, and Paolo M. Comoglio Division of Molecular Oncology, Institute for Cancer Research and Treatment (IRCC), University of Turin School of Medicine, Str. Prov. 142, Km. 3,95, 10060 Candiolo, Torino, Italy Received 15 July 2004/Returned for modification 8 October 2004/Accepted 11 February 2005 The hepatocyte growth factor (HGF) receptor encoded by the Met oncogene controls a genetic program— known as “invasive growth”—responsible for several developmental processes and involved in cancer invasion and metastasis. This program functions through several regulatory gene products, as yet largely unknown, both upstream and downstream of Met. Here we show that activation of the Notch receptor results in transcriptional down-regulation of Met, suppression of HGF-dependent Ras signaling, and impairment of HGF-dependent cellular responses. In turn, Met activation leads to transcriptional induction of the Notch ligand Delta and the Notch effector HES-1, indicating that Met is able to self-tune its own protein levels and the ensuing biochemical and biological outputs through stimulation of the Notch pathway. By using branching morphogenesis of the tracheal system in Drosophila as a readout of invasive growth, we also show that exogenous expression of a constitutively active form of human Met induces enhanced sprouting of the tracheal tree, a phenotype that is further increased in embryos lacking Notch function. These results unravel an in-built mechanism of negative feedback regulation in which Met activation leads to transcriptional induction of Notch function, which in turn limits HGF activity through repression of the Met oncogene. The ability of cells to arrange into three-dimensional struc- tures for shaping complex architectural patterns involves intri- cate genetic programs that are far from being elucidated. One of these programs is branching morphogenesis, the creation of a hierarchical organization of tubular networks starting from a relatively uniform group of cells. Successful implementation of such a process can be accomplished only through the coordi- nated and sequential execution of several events: first, cells dismantle lateral contacts and bud off from their primitive residency; then, they organize cord-like expansions that pene- trate into the surrounding tissues and become resistant to the proapoptotic cues that are normally exerted on cells dislodged from their natural habitat; finally, cells differentiate and polar- ize to turn cords into hollow cylinders and proliferate to ex- pand and ramify the nascent tubular plexus. From a conceptual point of view, branching morphogenesis represents the physi- ological paradigm of invasive growth, a multistep phenomenon that incorporates stimulation of cell multiplication, induction of a motile phenotype, and promotion of cell survival. Not surprisingly, the pathological counterpart of invasive growth is cancer progression and metastasis, which occur when neoplas- tic cells abandon the primary tumor mass and infiltrate the adjacent compartments and the vasculature as a prelude for colonization of distant organs (58). Spatial and chronological orchestration of the various steps of invasive growth is optimally accomplished by a family of growth factors known as scatter factors, whose prototypic member is hepatocyte growth factor (HGF), together with their tyrosine kinase receptors, in particular, the HGF receptor encoded by the Met protooncogene. HGF induces various mor- phogenetic responses in epithelial cells deriving from different tissues when these are grown in three-dimensional gels, includ- ing formation of branching tubules in kidney, breast, and pros- tate epithelial cells (for a review, see reference 43), and it is a potent angiogenic factor that promotes remodeling of capillary networks (5). In vivo, HGF stimulates tubulogenesis in the liver and kidneys during organ regeneration and promotes differentiation of the ductal tree in the mammary glands during early pregnancy (24, 29, 36, 52, 65). Although the role of HGF in the promotion of invasive growth is well documented and the intracellular signals trig- gered by Met activation and leading to execution of HGF- dependent responses have been extensively investigated (4, 18, 41, 42, 44, 45), fundamental questions remain unanswered. First, which are the nuclear factors that regulate Met transcrip- tion? This issue is particularly intriguing in light of the fre- quently observed neo- or overexpression of Met in human solid tumors (10) and of the recent finding that Met is transcription- ally induced by hypoxic stimuli (39). Second, which are, in turn, the transcriptional targets of Met? In spite of the huge amount of data gathered on the various signaling cascades that are primed upon Met stimulation, very little is known about how these signals affect the nuclear transcription machinery to in- duce or repress specific gene products. Answers to such ques- tions, together with the functional annotation of the molecules identified, are particularly important to categorize negative regulators of HGF-dependent invasive growth, which can be exploited to hamper cancer progression and metastasis. In this paper, we took advantage of an in silico analysis of the Met promoter region to identify the transmembrane re- ceptor Notch (and, specifically, its downstream effector HES-1) as a transcriptional repressor of Met, and thus as a physiological inhibitor of Met-dependent invasive growth, both * Institute for Cancer Research and Treatment (IRCC), University of Turin School of Medicine, Division of Molecular Oncology, IV Floor, Str. Prov. 142, Km. 3,95, 10060 Candiolo, Torino, Italy. Phone: (39) 011 9933232. Fax: (39) 011 9933225. E-mail: mariacristina [email protected]. 3982 on January 12, 2016 by guest http://mcb.asm.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Negative Feedback Regulation of Met-Dependent Invasive Growth by Notch

MOLECULAR AND CELLULAR BIOLOGY, May 2005, p. 3982–3996 Vol. 25, No. 100270-7306/05/$08.00�0 doi:10.1128/MCB.25.10.3982–3996.2005Copyright © 2005, American Society for Microbiology. All Rights Reserved.
Negative Feedback Regulation of Met-Dependent InvasiveGrowth by Notch
M. Cristina Stella,* Livio Trusolino, Selma Pennacchietti, and Paolo M. ComoglioDivision of Molecular Oncology, Institute for Cancer Research and Treatment (IRCC), University of Turin School
of Medicine, Str. Prov. 142, Km. 3,95, 10060 Candiolo, Torino, Italy
Received 15 July 2004/Returned for modification 8 October 2004/Accepted 11 February 2005
The hepatocyte growth factor (HGF) receptor encoded by the Met oncogene controls a genetic program—known as “invasive growth”—responsible for several developmental processes and involved in cancer invasionand metastasis. This program functions through several regulatory gene products, as yet largely unknown, bothupstream and downstream of Met. Here we show that activation of the Notch receptor results in transcriptionaldown-regulation of Met, suppression of HGF-dependent Ras signaling, and impairment of HGF-dependentcellular responses. In turn, Met activation leads to transcriptional induction of the Notch ligand Delta and theNotch effector HES-1, indicating that Met is able to self-tune its own protein levels and the ensuing biochemicaland biological outputs through stimulation of the Notch pathway. By using branching morphogenesis of thetracheal system in Drosophila as a readout of invasive growth, we also show that exogenous expression of aconstitutively active form of human Met induces enhanced sprouting of the tracheal tree, a phenotype that isfurther increased in embryos lacking Notch function. These results unravel an in-built mechanism of negativefeedback regulation in which Met activation leads to transcriptional induction of Notch function, which in turnlimits HGF activity through repression of the Met oncogene.
The ability of cells to arrange into three-dimensional struc-tures for shaping complex architectural patterns involves intri-cate genetic programs that are far from being elucidated. Oneof these programs is branching morphogenesis, the creation ofa hierarchical organization of tubular networks starting from arelatively uniform group of cells. Successful implementation ofsuch a process can be accomplished only through the coordi-nated and sequential execution of several events: first, cellsdismantle lateral contacts and bud off from their primitiveresidency; then, they organize cord-like expansions that pene-trate into the surrounding tissues and become resistant to theproapoptotic cues that are normally exerted on cells dislodgedfrom their natural habitat; finally, cells differentiate and polar-ize to turn cords into hollow cylinders and proliferate to ex-pand and ramify the nascent tubular plexus. From a conceptualpoint of view, branching morphogenesis represents the physi-ological paradigm of invasive growth, a multistep phenomenonthat incorporates stimulation of cell multiplication, inductionof a motile phenotype, and promotion of cell survival. Notsurprisingly, the pathological counterpart of invasive growth iscancer progression and metastasis, which occur when neoplas-tic cells abandon the primary tumor mass and infiltrate theadjacent compartments and the vasculature as a prelude forcolonization of distant organs (58).
Spatial and chronological orchestration of the various stepsof invasive growth is optimally accomplished by a family ofgrowth factors known as scatter factors, whose prototypicmember is hepatocyte growth factor (HGF), together withtheir tyrosine kinase receptors, in particular, the HGF receptor
encoded by the Met protooncogene. HGF induces various mor-phogenetic responses in epithelial cells deriving from differenttissues when these are grown in three-dimensional gels, includ-ing formation of branching tubules in kidney, breast, and pros-tate epithelial cells (for a review, see reference 43), and it is apotent angiogenic factor that promotes remodeling of capillarynetworks (5). In vivo, HGF stimulates tubulogenesis in theliver and kidneys during organ regeneration and promotesdifferentiation of the ductal tree in the mammary glands duringearly pregnancy (24, 29, 36, 52, 65).
Although the role of HGF in the promotion of invasivegrowth is well documented and the intracellular signals trig-gered by Met activation and leading to execution of HGF-dependent responses have been extensively investigated (4, 18,41, 42, 44, 45), fundamental questions remain unanswered.First, which are the nuclear factors that regulate Met transcrip-tion? This issue is particularly intriguing in light of the fre-quently observed neo- or overexpression of Met in human solidtumors (10) and of the recent finding that Met is transcription-ally induced by hypoxic stimuli (39). Second, which are, in turn,the transcriptional targets of Met? In spite of the huge amountof data gathered on the various signaling cascades that areprimed upon Met stimulation, very little is known about howthese signals affect the nuclear transcription machinery to in-duce or repress specific gene products. Answers to such ques-tions, together with the functional annotation of the moleculesidentified, are particularly important to categorize negativeregulators of HGF-dependent invasive growth, which can beexploited to hamper cancer progression and metastasis.
In this paper, we took advantage of an in silico analysis ofthe Met promoter region to identify the transmembrane re-ceptor Notch (and, specifically, its downstream effectorHES-1) as a transcriptional repressor of Met, and thus as aphysiological inhibitor of Met-dependent invasive growth, both
* Institute for Cancer Research and Treatment (IRCC), Universityof Turin School of Medicine, Division of Molecular Oncology, IVFloor, Str. Prov. 142, Km. 3,95, 10060 Candiolo, Torino, Italy. Phone:(39) 011 9933232. Fax: (39) 011 9933225. E-mail: [email protected].
3982
on January 12, 2016 by guesthttp://m
cb.asm.org/
Dow
nloaded from

in vitro and in vivo. Intriguingly, in an opposite but comple-mentary fashion, Met activation results in stimulation of theNotch pathway through transcriptional induction of the Notchligands Delta1 and Delta4, together with HES-1. Taken to-gether, these results unravel an in-built mechanism of negativefeedback regulation in which Met activation leads to transcrip-tional induction of Notch function, which in turn limits HGFactivity through repression of Met-triggered signals. Such aregulatory circuit can be exploited to prevent HGF-driven can-cer progression and metastasis.
MATERIALS AND METHODS
Molecular biology. Standard molecular biology procedures, if not differentlyindicated, were done according to reference 46. PCRs were performed using PfuDNA polymerase (Promega). DNAs were sequenced with a BigDye Terminatorcycle sequencing kit (PE Applied Biosystems) and analyzed using 310 GeneticAnalyzer ABI Prism (Perkin Elmer). pB4021 and pP{W8}-SE-hsp70-torso4021cDNAs, described in references 53 and 11, respectively, were a gift of F.Sprenger. torso4021 was amplified using pB4021 as template and the oligonucle-otides 5� GGAGCACAGCATTAATATGCAGCATATTGG 3� and 5� TCTTTGATCTATTCAAAGGAGGTACGG 3� as primers. Met cDNA was amplifiedwith the primers 5� CCAATATGCTGCATATTAATGCTGTGCTCCCTGACGTTCTGCAAAAAGAGAAAGCAAA 3� and 5� CTGATCTTCTGGAAAAGTAGCTCGG 3�. The underlined residues represent conservative mutations inthe transmembrane domain of torso4021 that leads to generation of the restric-tion site specific for the nuclease AseI. Both torso4021- and Met-amplified frag-ments were treated with AseI and joined together (torso4021-MetTM). Then,torso4021-MetTM was treated with XhoI and XmnI. pB4021 was treated withXhoI and ClaI, Met cDNA with XmnI and ClaI. pB4021XhoI-ClaI, MetXmnI-ClaI,and torso4021-Met-TMXhoI-XmnI were joined together. This intermediate con-struct was validated by sequencing, and then it was treated with XhoI and BamHIand the resulting torso-Met fragment was inserted in pP{W8}-SE-hsp70-tor-so4021, previously digested with the two restriction enzymes XhoI and BamHI.N4-TM was generated starting from the following Human Genome MappingProject I.M.A.G.E. clones 5370162 (a), 317393 (b), 5391233 (c), 1224677 (d), and5372052 (e). Specific cDNA fragments were generated by PCRs using the fol-lowing primers: a, 5� ACGCGAATTCATGGCAGCAGTGGGAGCTCTGGAGCCC 3� and 5� CACTCCCCCACAGAAGACGGC 3�; b, 5� TTGGCCGCTCAACAGCGCGTG 3� and 5� GTCTTCTGCTGCCAATAGGAG 3�; c, 5� GCCAACCCCAATCAGCCAGACCG 3� and 5� TATTATCGAGCTCCAGCAACAGCTGC 3�; d, 5� TTTTCCTGGCAACGCGTGAAGGAG 3� and 5� TATTTATCATCCGTGATGCCCTAG 3�; e, 5� CTAGGGCATCACAGGATGAC 3�and 5� TAATCGCTCGAGTTACTTTCCAAGTCGGTTCATCTCTATGTCTGTATAGTTCAGATTTCTTACAACCGAGTTTAA 3�. The different PCRfragments were treated with the following nucleases: (a) EcoRI and Tsp45I, (b)Tsp45I and MslI, (c) MslI and BssHII, (d) AflIII and BfaI, and (e) BfaI andXhoI. The fragments were joined together in pBluescript SKII (Stratagene). Theresulting N4-TM cDNA was validated by sequencing. It was indistinguishablefrom the previously published sequence (GenBank accession no. NM-010929,residues 4347 to 6008), except for one conservative mutation (C 5429 T) that weinserted in order to generate a restriction site specific for AflIII for joining clonesc and d. N4-TM was transferred into the vector pBabe (35) treated with EcoRIand XhoI. For amplifying Delta mRNA in Drosophila embryos, we used thefollowing oligonucleotides: 5� CTGACCGACGCCCAGCGCTTC 3� and 5� ATCGTCGCGGGGCCGGCAGAA 3�. The primers for GPDH mRNA were 5�CCGCTTGCGAGCTTATCGCACCAC 3� and 5� ATTAATCAATTGTAATTGTACTGC 3�. The amplifications of HES-1, J1-2, Dl1-4, and actin in MDA-MB-435-�4 were done using the primers and the experimental conditions pre-viously described in reference 38. The PCR signals were visualized using aFluorimager apparatus (Molecular Dynamics) and quantified by densitometrywith the software ImageQant (Molecular Dynamics). HES-1 cDNA was obtainedby direct amplification of MDA-MB-435-�4 nucleic acids. Cells were treatedwith HGF in the presence of the proteasome inhibitor MG132 (Sigma; 50 �M)for 3 h, and then mRNAs were collected and retrotranscribed. The resultingcDNAs were amplified using the following primers: 5� TT AAT ATT CGC AGATCT ATG CCA GCT GAT ATA ATG GAG AAA AAT TCC 3� and 5� TATTAA CAA TTG TCA GTT CCG CCA CGG CCT CC 3�. The PCR-amplifiedfragment was validated by sequencing and then cloned into the eukaryotic ex-pression vector pcDNA3.1 (Invitrogen) by blunt-end ligation (pcDNA-HES-1).
Cell maintenance and retroviral infection. MDA-MB-435-�4 cells were grownin Dulbecco’s modified Eagle medium (DMEM; Sigma) supplemented with 10%fetal calf serum (FCS; Sigma) and 1 mg/ml G418 (Gibco); MDCK cells weregrown in DMEM (Sigma) supplemented with 10% FCS (HyClone). Viral hybridvectors were produced by transient transfection of 293T cells with an emptypBabe vector or with an N4-TM-pBabe construct, both containing a puromycinresistance cassette, a gag pol packaging construct derived from Moloney murineleukemia virus, and a third plasmid expressing the vesicular stomatitis virus(VSV) envelope. Pools of stably transfected cells were obtained by retroviralinfections. After infection, puromycin-resistant cells were isolated by growth inselective medium (DMEM, 10% FCS, 1 mg/ml G418 [Gibco], and 1 mg/mlpuromycin [Sigma] for MDA-MB-435-�4 cells, and DMEM, 10% FCS, and 5mg/ml puromycin [Sigma] for MDCK cells). Overexpressing HES-1 cells wereobtained by transfection of MDA-MB-435-�4 cells with the construct pcDNA-HES-1 using Lipofectamine (Invitrogen) together with Plus reagent (Invitrogen),according to the instructions of the manufacturer; transfection with an emptypcDNA3.1 vector was used to produce mock cells. The same transfection methodwas utilized for getting pBabe or N4-TM MDA-MB-435-�4 cells to expresseither control or HES-1 small interfering RNAs (siRNAs) (gift of S. Herzig;described in reference 20). All cells were maintained at 37°C in 5% CO2.
Electrophoretic mobility shift assays (EMSA). MDA-MB-435-�4 nuclear ex-tracts were obtained using a nuclear extraction kit (Panomics), following theinstructions of the manufacturer, and then they were dialyzed in a buffer con-taining 10 mM HEPES, pH 7.9, 25 mM KCl, 1 mM EDTA, 5 mM MgCl2, 1 mMdithiothreitol, 10% glycerol, and 0.05% NP-40 in the presence of protease andphosphatase inhibitors. 5� GCGAGGCAGACAGACACGTGCTGGGGCGGGCAGG 3� together with 5� CTCGCCTGCCCGCCCCAGCACGTGTCTGTCTGCC 3� and 5� GCGAGGCAGACAGACAAAAGCTGGGGCGGGCAGG3� together with 5� CTCGCCTGCCCGCCCCAGCTTTTGTCTGTCTGCC 3�were annealed to generate, respectively, HES-1-wt and HES-1-mut double-stranded Met-promoter oligonucleotides. Probes were obtained by a Klenowfill-in reaction of 100 ng of each of the two double-stranded oligonucleotides inthe presence of 100 �Ci of [�-32P]GTP, according to reference 9. The probe forGAA was obtained as described in reference 64. Labeled oligonucleotides (5 �104 cpm) were incubated with 10 �g of nuclear extracts and 1 mg/ml poly(dIdC)(Amersham-Pharmacia) for 45 min on ice. In some experiments, nuclear extractswere preincubated for 30 min on ice with 2 �g of either control immunoglobulinG (Sigma) or TransCruz Gel Supershift anti-HES-1 antibodies (Santa Cruz)(66). Complexes were separated using a 5% polyacrylamide–0.5% Tris-borate-EDTA gel and visualized using a Storm apparatus (Molecular Dynamics).
Transcriptional analysis. The wild-type Met-promoter construct used for thetranscriptional analysis has been previously described (15). The putative HESbinding sequence “CACGTG” (�20 from transcriptional start site) was mutatedby a recombinant PCR-based approach (34) to “CAAAAG.” Accuracy of themutagenesis procedure was verified by direct sequencing. Analysis of promoteractivity was performed in MDA-MB-435-�4 cells. By using Lipofectamine (In-vitrogen), 1.8 � 106 cells/100-mm-diameter plate were transfected with either 20�g of the appropriate promoter construct, with 3.5 �g of pcDNA-HES-1 orcontrol pcDNA3.1 vector together with 15 �g of the promoter constructs, or with2.8 �g of the siRNA constructs together with 10 �g of the appropriate promoterconstructs. For the rescuing experiments (see Fig. 4E and F), 8 � 106 of MDA-MB-435-�4 cells/100-mm-diameter plate were transfected with 10 �g of pcDNA-HES-1; after 3 days, these cells were divided and retransfected with increasingamounts of HES-1 siRNAs (8, 16, 24, and 48 �g) or control siRNAs (48 �g). Theresults showed that 16 and 24 �g of HES-1 siRNAs were sufficient for silencingexogenously induced overexpression of HES-1. After 4 days, these cells weretransfected with 25 �g of the appropriate promoter constructs. For the experi-ments presented in Fig. 5C and D, 8 � 106 N4-TM MDA-MB-435-�4 cells/100-mm-diameter plate were transfected with 20 �g of HES-1 siRNAs; after 3 days,these cells were divided and retransfected with increasing amounts of pcDNA-HES-1 (4, 8, 12, 24, and 36 �g). The down-regulation of HES-1 visible in HES-1siRNA-transfected cells is abrogated by 8 and 12 �g of HES-1. After 4 days,these cells were transfected with 25 �g of the appropriate promoter constructs.A TK-Renilla reporter plasmid (Promega) (0.2 �g) was added to each transfec-tion for efficiency standardization. After 48 h, cells were processed using adual-luciferase reporter assay system (Promega) according to the manufacturer’sinstructions. Ten microliters of cell lysate was used to determine reporter enzymeactivity by using a Lumat LB 9507 luminometer (Berthold). Each experimentalpoint was performed in quadruplicate.
Cell immunofluorescent staining. Cells were seeded and processed as previ-ously described (57). The staining was performed using anti-VSV monoclonalantibody (MAb) (clone P5D4, diluted 1:200; Sigma) followed by FITC-labeledanti-mouse antibodies (1:200; Molecular Probes), tetramethyl rhodamine isocya-
VOL. 25, 2005 Met NEGATIVE FEEDBACK REGULATION BY Notch 3983
on January 12, 2016 by guesthttp://m
cb.asm.org/
Dow
nloaded from

nate-conjugated phalloidin, to evidence actin cytoskeleton, and To-Pro-I3 (1:2,000; Molecular Probes), to label nuclei. The stained cells were mounted inMowiol, and then they were observed and photographed using a confocal mi-croscope (Molecular Dynamics).
Biological assays. In all the biological assays we used baculovirus recombinantHGF (37). Conditioned medium of uninfected insect cells, treated as the super-natant derived from HGF–baculovirus-infected cells, provided the negative con-trol. In the migration assays, 5 � 105 cells in 200 �l of DMEM supplementedwith 0.2% FCS were seeded in the upper compartment of Transwell chambers(6.5 �m, polycarbonate filter with 8-�m pore size; Costar Corporation). DMEM(500 �l) plus 0.2% FCS, containing either control medium or 40 ng/ml of HGF,was added to the lower chamber. The plates were incubated at 37°C for 12 h, andthen the cells on the upper side of the polycarbonate filter were mechanicallyremoved. Cells that had migrated to the lower side of the filter were stained withcrystal violet and counted using a Fast-Read 102 New Grid counting chamber(Roche Diagnostics) under a Dialux microscope (Leitz). The invasion assayswere performed as the migration assays described above, but here the uppersides of the filters were coated with 15 �g/cm2 of reconstituted Matrigel base-ment membrane (Collaborative Research). For the tubulogenesis assays, thetrypsinized cells were suspended to a final concentration of 1.5 � 105 cells/ml(MDA-MB-435-�4) or 1 � 105 cells/ml (MDCK) in gelling solution prepared asfollows: 800 �l of type I collagen S (3.4 mg/ml; Collaborative Biomed), 1�DMEM, 40 mM HEPES, 37 g/liter NaHCO3, all to a final volume of 1 ml. Eightymicroliters of this mixture was seeded into 96 microtiter plate wells and allowedto gel for 10 min at 37°C before overlying with 80 �l DMEM containing 20%FCS. After 2 h at 37°C, the upper aqueous phase was removed and substitutedwith fresh DMEM medium plus 10% FCS. After further 24 h, either controlmedium or 80 ng/ml of HGF was added in the upper aqueous phase. Tubuleswere scored every day for 2 weeks.
Biochemical methods. For immunoprecipitation, 1 � 106 cells or 100 embryoswere lysed for 15 min at 4°C with 1 ml of lysis buffer (50 mM HEPES, pH 7.4,5 mM EDTA, 2 mM EGTA, 150 mM NaCl, 10% glycerol, and 1% NP-40), in thepresence of protease and phosphatase inhibitors. Extracts were quantified with abicinchoninic acid protein assay reagent kit (Pierce), and an equal amount ofproteins was incubated with MAbs anti-VSV (clone P5D4; Sigma) or anti-hMet-DQ13 (produced in our laboratory). Immune complexes were collected withSepharose-protein A resin (Amersham-Pharmacia) supplemented with 1.2 �g ofrabbit anti-mouse immunoglobulin (RAMIG; Pierce) for 2 h at 4°C and thenwashed 4� in ice-cold lysis buffer and eluted in boiling Laemmli buffer. Westernblot analysis was performed using sodium dodecyl sulfate-polyacrylamide gelelectrophoresis gels. The gels were blotted onto a nylon membrane (polyvinyli-dene difluoride transfer membrane; Millipore Corporation); the membrane wasblocked using 10% bovine serum albumin in Tris-buffered saline–Tween for 2 hat 45°C; the primary antibodies (MAb anti-VSV, Sigma; polyclonal antibody[PAb] anti-Met-C12, Santa Cruz; MAb anti-phosphotyrosine, Upstate Biotech-nology; PAb anti-active-MAPK, Promega; PAb anti-MAPK, Cell Signaling; PAbanti-actin, Santa Cruz; PAb anti-active-AKT, Cell Signaling; PAb anti-AKT, CellSignaling; PAb anti-HES-1, kindly provided by T. Sudo; MAb anti-panRAS,Transduction Laboratories) were incubated in 5% bovine serum albumin inTris-buffered saline–Tween according to the dilutions suggested by the manu-facturers, for 12 h at 4°C. Specific signals were detected with peroxidase-conju-gated secondary antibodies (Amersham-Pharmacia) and an enhanced chemilu-minescence system (ECL; Amersham-Pharmacia). Pool-down experiments weredone according to reference 47; cells were maintained in serum-free medium for24 h, and then they were either left untreated or stimulated with 50 ng/ml ofHGF for the indicated times. The construct Raf-RBD-GST-pGEX (gift of E.Audero) was expressed in bacteria and purified as described in reference 47. Ineach experimental point, 1 mg of total protein extracts was incubated with 40 �gof Raf-RBD-GST fusion protein. For auto-kinase assays, immunoprecipitatedembryonic extracts were washed twice with ice-cold KB buffer (20 mM HEPES,pH 7.1, 5 mM MgCl2, 100 mM NaCl), and the phosphorylation reaction wasperformed in the presence of [�-32P]ATP and 10 mM unlabeled ATP for 15 minat 37°C. The reactions were stopped by adding boiling Laemmli buffer and thesamples were analyzed by sodium dodecyl sulfate-polyacrylamide gel electro-phoresis gels. The signals were visualized with a Storm apparatus (MolecularDynamics).
Fly stocks. The following fly strains were used in this study: the w1118 strain(gift of S. Roth) was used for P-element-mediated germ line transformation;trachealess enhancer trap line 1-eve-1 (Perrimon, 1991, no. 2014) was a gift of M.Affolter; and the NCo, CyO, and CyO-ftz-LacZ strains were provided from theBloomington Stock Centre.
Embryo fixation and staining. Embryos were staged according to reference 6.They were collected on apple juice agar plates following standard protocols (63),
and then they were processed as previously described (55). For the staining weused the following primary antibodies: MAb 2A12 (developed by C. Goodmanand N. Patel; diluted 1:10) and MAb C17.9C6 (anti-Notch, intracellular domain,diluted 1:5; developed by S. Artavanis-Tsakonas), both obtained from the De-velopmental Studies Hybridoma Bank. Anti-�-galactosidase (�- gal) antibodies,purchased from Cappel, were diluted 1:1,000; anti-human Met antibody, cloneC12, purchased from SantaCruz, after preabsorption using wild-type embryos,was used 1:100. The primary antibodies were diluted in 1% FCS. The incubationswith the primary antibodies were performed at 4°C for 12 h. The secondaryantibodies were as follows: biotinylated anti-rabbit and anti-mouse (diluted1:5,000; Vector Laboratories) and FITC-conjugated anti-rabbit and tetramethylrhodamine isocyanate-conjugated anti-mouse (diluted 1:500; Molecular Probes).All the secondary antibodies were incubated for 2 h at room temperature. Thestainings performed with the biotinylated antibodies were developed using aVectastain Elite ABC kit (Vector Laboratories) with 3�-3�-diaminobenzidine(Sigma) as chromogen. Subsequently, the embryos were dehydrated, rinsed inacetone, and transferred to a 1:1 mixture of acetone and Durcupan (Fluka)overnight. After acetone evaporation, embryos were observed and photographedby using a Dialux microscope (Leitz). For genotyping, embryos were first stainedwith biotinylated antibodies to label the tracheas and then with anti-�-gal andMAb C17.9C6 followed by fluorochrome-conjugated secondary antibodies. NCo
torso-Met mutants were identified for the absence of both �-gal and MAbC17.9C6 staining. They were collected under an Axioplan microscope (Zeiss),mounted in Durcupan (Fluka), and photographed as previously described.
RESULTS
HES protein binds to Met promoter. In silico analysis of thehuman Met gene reveals that the Met promoter contains oneputative binding site for HES proteins, a basic helix-loop-helixtype of transcriptional repressor known to be a downstreamtarget of Notch activity (Fig. 1A) (23). We therefore decided toinvestigate whether HES-1, the prototype of this family oftranscription factors, was able to bind to the Met promoter.This was achieved by EMSA analysis in MDA-MB-435-�4 cells(57) overexpressing HES-1 (Fig. 1B; see Materials and Meth-ods for details). For comparison, the binding of HES-1 to thepromoter of acid �-glucosidase (GAA), a known target gene ofthe HES-Notch pathway (64), was monitored. As expected,incubation of nuclear extracts with a probe containing the HESbinding site on the GAA promoter determined the formationof a complex (Fig. 1C, first lane). The same was true when thenuclear extracts were incubated with a probe containing thewild-type HES putative binding site on the Met promoter,whereas incubation with a probe mutated in this binding sitedid not lead to complex formation (Fig. 1C, compare the thirdand second lanes). The specificity of the complex was furtherexamined by competition experiments using either unlabeledHES wild-type or HES mutated Met promoter probes. By add-ing the mutated competitor, the complex was only slightlyreduced in a dose-independent fashion, suggesting an unspe-cific quenching effect of the mutated competitor over HES-1/Met complex formation (Fig. 1C, lanes six and seven). When a10-fold molar excess of wild-type competitor was added to thereaction, complex formation was reduced, and it was almosttotally abolished in the presence of a 50-fold molar excess (Fig.1C, lanes eight and nine). The addition of anti-HES-1 antibod-ies reduced the intensity of the HES-1/Met complex comparedto control unspecific immunoglobulins (Fig. 1C, lanes ten andeleven), thus indicating a specific interference of anti-HES-1antibodies in the complex formation. Altogether, these dataconfirm the in silico observation by demonstrating that indeedHES-1 binds to Met promoter.
3984 STELLA ET AL. MOL. CELL. BIOL.
on January 12, 2016 by guesthttp://m
cb.asm.org/
Dow
nloaded from

Activation of Notch down-regulates Met transcription andimpairs Met-dependent Ras activation and biological re-sponses. The observation that the Met promoter can bind theHES-1 transcription factor suggests that Met could be a tran-scriptional target of Notch. To study the potential effect ofNotch on Met transcription, we generated stable transfectantsexpressing constitutive active forms of Notch. Among the fourdifferent vertebrate homologues of Notch, we focused our at-tention on Notch4, which, similar to Met, has been demon-strated to affect the development of different tubular organssuch as the mammary gland (51, 59) and the vascular system(27, 30, 49). As cellular recipients, we chose two mammaliancell lines that have been thoroughly characterized in terms ofbiological responses to the Met ligand HGF, MDA-MB-435-�4 and MDCK (42, 67). Retroviral-mediated infection ofMDA-MB-435-�4 and MDCK cells with a constitutively activeform of Notch tagged with a VSV C-terminal epitope (N4-TM)resulted in expression and correct processing of the transgeneproduct. Western blot analysis demonstrated the presence of adoublet corresponding to the unprocessed transmembrane
protein and the cleaved intracellular portion of N4-TM (Fig.2A). Moreover, N4-TM properly localized in the nucleus, asshown by confocal immunofluorescence analysis of MDA-MB-435-�4 cells simultaneously stained for F-actin (red), a nuclearmarker (blue), and N4-TM (green; Fig. 2B).
Subsequently, we subcloned the human Met promoter (15),either of the wild type or mutated in the HES binding site, intoa reporter plasmid upstream of a luciferase gene and trans-fected it into mock and N4-TM MDA-MB-435-�4 cells. Lucif-erase activity of transfected cells was analyzed to determinepromoter activity. Constitutively active Notch depressed tran-scription from the Met promoter by 1.5-fold (Fig. 2C). Accord-ingly, mutagenesis of the HES binding site restored Met tran-scription in N4-TM transfectants. In line with these findings,reverse transcription (RT)-PCR analysis of Met mRNA inN4-TM and mock cells indicated that the Met transcript isdiminished in N4-TM cells compared to mock cells (Fig. 2D),which leads to reduced levels of Met protein (Fig. 2E). Takentogether, these data demonstrate that Notch can negativelymodulate Met synthesis at the transcriptional level.
FIG. 1. HES-1 binds to the Met promoter. (A) Sequence of the Met promoter. Bold residues represent the putative HES binding site. Shownin capital letters are the residues that were mutated in the EMSA and promoter analysis (CGT towards AAA). (B) Western blot of MDA-MB-435-�4 cell extracts either mock or HES-1 transfected. The same blot was stained with antibodies anti-HES-1 and anti-actin as loading control.(C) EMSA showing HES-1 binding activity to the GAA and to the Met promoter. Nuclear extracts from cells overexpressing HES-1 were incubatedwith the HES-1 DNA binding domain of the GAA gene (lane 1) and with mutated (lane 2) or wild-type (wt) (lanes 3 and 5 to 11) HES-1 DNAbinding domain of Met promoter. A complex is formed exclusively in the presence of both GAA and Met wild-type promoters. This complex isunaltered by adding an excess of mutated probe (lanes 6 and 7) but is affected by the addiction of wild-type probe (lanes 8 and 9). Addiction ofanti-HES-1 antibodies determines a reduction in the complex (lane 10) that is absent when control immunoglobulins (Ig) are added (lane 11).
VOL. 25, 2005 Met NEGATIVE FEEDBACK REGULATION BY Notch 3985
on January 12, 2016 by guesthttp://m
cb.asm.org/
Dow
nloaded from
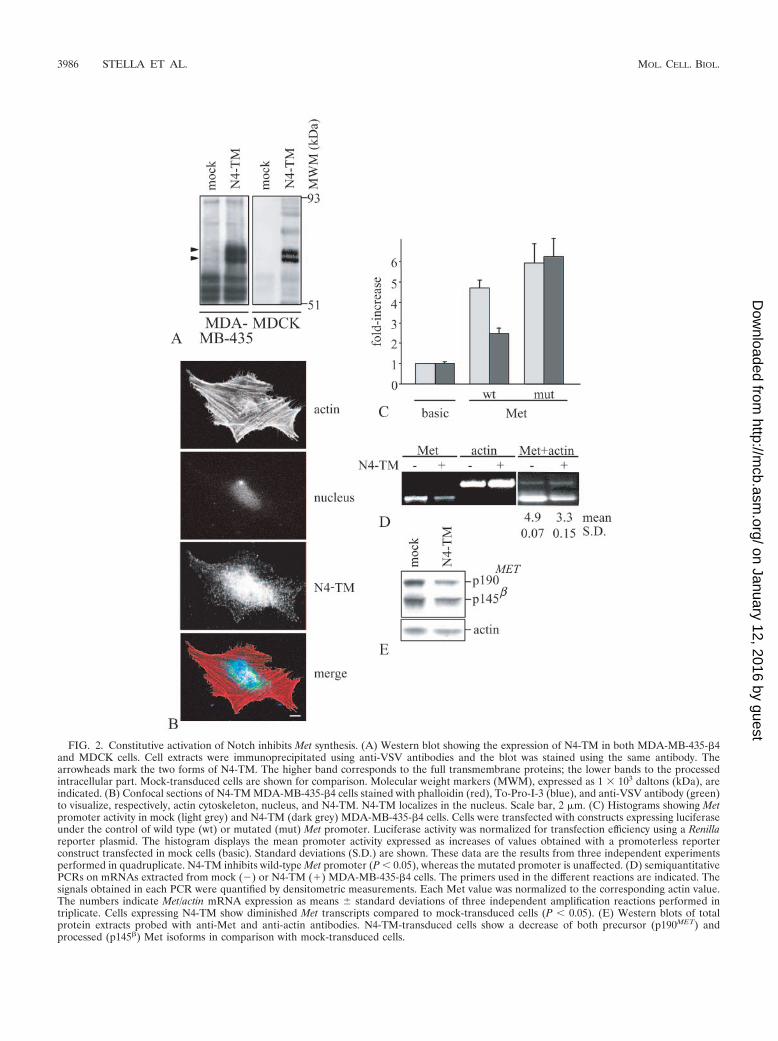
FIG. 2. Constitutive activation of Notch inhibits Met synthesis. (A) Western blot showing the expression of N4-TM in both MDA-MB-435-�4and MDCK cells. Cell extracts were immunoprecipitated using anti-VSV antibodies and the blot was stained using the same antibody. Thearrowheads mark the two forms of N4-TM. The higher band corresponds to the full transmembrane proteins; the lower bands to the processedintracellular part. Mock-transduced cells are shown for comparison. Molecular weight markers (MWM), expressed as 1 � 103 daltons (kDa), areindicated. (B) Confocal sections of N4-TM MDA-MB-435-�4 cells stained with phalloidin (red), To-Pro-I-3 (blue), and anti-VSV antibody (green)to visualize, respectively, actin cytoskeleton, nucleus, and N4-TM. N4-TM localizes in the nucleus. Scale bar, 2 �m. (C) Histograms showing Metpromoter activity in mock (light grey) and N4-TM (dark grey) MDA-MB-435-�4 cells. Cells were transfected with constructs expressing luciferaseunder the control of wild type (wt) or mutated (mut) Met promoter. Luciferase activity was normalized for transfection efficiency using a Renillareporter plasmid. The histogram displays the mean promoter activity expressed as increases of values obtained with a promoterless reporterconstruct transfected in mock cells (basic). Standard deviations (S.D.) are shown. These data are the results from three independent experimentsperformed in quadruplicate. N4-TM inhibits wild-type Met promoter (P � 0.05), whereas the mutated promoter is unaffected. (D) semiquantitativePCRs on mRNAs extracted from mock () or N4-TM (�) MDA-MB-435-�4 cells. The primers used in the different reactions are indicated. Thesignals obtained in each PCR were quantified by densitometric measurements. Each Met value was normalized to the corresponding actin value.The numbers indicate Met/actin mRNA expression as means standard deviations of three independent amplification reactions performed intriplicate. Cells expressing N4-TM show diminished Met transcripts compared to mock-transduced cells (P � 0.05). (E) Western blots of totalprotein extracts probed with anti-Met and anti-actin antibodies. N4-TM-transduced cells show a decrease of both precursor (p190MET) andprocessed (p145�) Met isoforms in comparison with mock-transduced cells.
3986 STELLA ET AL. MOL. CELL. BIOL.
on January 12, 2016 by guesthttp://m
cb.asm.org/
Dow
nloaded from
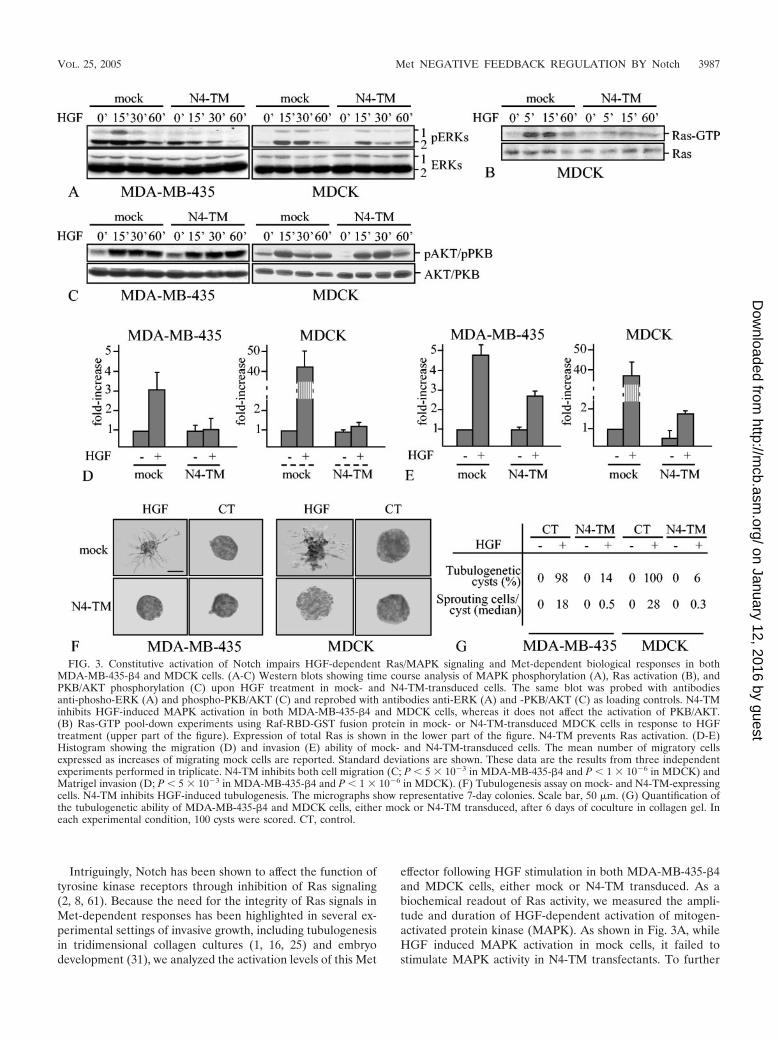
Intriguingly, Notch has been shown to affect the function oftyrosine kinase receptors through inhibition of Ras signaling(2, 8, 61). Because the need for the integrity of Ras signals inMet-dependent responses has been highlighted in several ex-perimental settings of invasive growth, including tubulogenesisin tridimensional collagen cultures (1, 16, 25) and embryodevelopment (31), we analyzed the activation levels of this Met
effector following HGF stimulation in both MDA-MB-435-�4and MDCK cells, either mock or N4-TM transduced. As abiochemical readout of Ras activity, we measured the ampli-tude and duration of HGF-dependent activation of mitogen-activated protein kinase (MAPK). As shown in Fig. 3A, whileHGF induced MAPK activation in mock cells, it failed tostimulate MAPK activity in N4-TM transfectants. To further
FIG. 3. Constitutive activation of Notch impairs HGF-dependent Ras/MAPK signaling and Met-dependent biological responses in bothMDA-MB-435-�4 and MDCK cells. (A-C) Western blots showing time course analysis of MAPK phosphorylation (A), Ras activation (B), andPKB/AKT phosphorylation (C) upon HGF treatment in mock- and N4-TM-transduced cells. The same blot was probed with antibodiesanti-phosho-ERK (A) and phospho-PKB/AKT (C) and reprobed with antibodies anti-ERK (A) and -PKB/AKT (C) as loading controls. N4-TMinhibits HGF-induced MAPK activation in both MDA-MB-435-�4 and MDCK cells, whereas it does not affect the activation of PKB/AKT.(B) Ras-GTP pool-down experiments using Raf-RBD-GST fusion protein in mock- or N4-TM-transduced MDCK cells in response to HGFtreatment (upper part of the figure). Expression of total Ras is shown in the lower part of the figure. N4-TM prevents Ras activation. (D-E)Histogram showing the migration (D) and invasion (E) ability of mock- and N4-TM-transduced cells. The mean number of migratory cellsexpressed as increases of migrating mock cells are reported. Standard deviations are shown. These data are the results from three independentexperiments performed in triplicate. N4-TM inhibits both cell migration (C; P � 5 � 103 in MDA-MB-435-�4 and P � 1 � 106 in MDCK) andMatrigel invasion (D; P � 5 � 103 in MDA-MB-435-�4 and P � 1 � 106 in MDCK). (F) Tubulogenesis assay on mock- and N4-TM-expressingcells. N4-TM inhibits HGF-induced tubulogenesis. The micrographs show representative 7-day colonies. Scale bar, 50 �m. (G) Quantification ofthe tubulogenetic ability of MDA-MB-435-�4 and MDCK cells, either mock or N4-TM transduced, after 6 days of coculture in collagen gel. Ineach experimental condition, 100 cysts were scored. CT, control.
VOL. 25, 2005 Met NEGATIVE FEEDBACK REGULATION BY Notch 3987
on January 12, 2016 by guesthttp://m
cb.asm.org/
Dow
nloaded from

validate this result, we monitored HGF-dependent Ras activa-tion in mock and N4-TM MDCK cells by Ras-GTP pull-downsusing the Ras-binding domain of Raf-1 fused to glutathioneS-transferase (GST). HGF treatment determined an increaseof Ras-GTP in mock cells but not in N4-TM cells, thus con-firming a Notch-based repression of HGF-triggered Ras sig-naling (Fig. 3B). Ras-GTP pull-downs in MDA-MB-435-�4cells were hampered by low expression levels of Ras. Differ-ently from Ras, HGF-triggered activation of AKT/PKB, adownstream target of PI3K crucially involved in Met-depen-dent invasive growth, was unaffected in N4-TM versus mockcells (Fig. 3C). Combined, these data suggest that Notch can,on one hand, decrease Met transcription and, on the other,selectively impinge on specific interruption of the Ras signalingcascade.
To explore whether Notch-dependent transcriptional down-regulation of Met and disturbance of Met-triggered Ras sig-naling can modify HGF-dependent biological responses, wecompared the ability of mock versus N4-TM transfectants torespond to HGF in migration, invasion, and tubulogenesisassays. Interestingly, in both MDA-MB-435-�4 and MDCKcells, N4-TM inhibited HGF-stimulated motility across cell-permeable filters (Fig. 3D), decreased cell invasion through athree-dimensional matrix of Matrigel (Fig. 3E), and abolishedformation of branching tubules in collagen gels (Fig. 3F; forquantification, see Fig. 3G). The striking abrogation of HGF-dependent branching morphogenesis in cells expressingN4-TM is likely due to the combined action of reduced Metexpression and Notch-based selective attenuation of Ras sig-naling; indeed, HGF-dependent branching morphogenesis inthree-dimensional collagen gels crucially relies on a fine equi-librium between Ras and PI3K activity, and any imbalancedisrupting this equilibrium severely hampers tubule formation(1, 16, 25). Together, all these findings indicate that activationof Notch strongly impairs Met-dependent biological responses,thus supporting a role of Notch as a repressor of Met activity.
Met activation induces Notch signaling, which throughHES-1 represses Met mRNA synthesis. It is now becomingincreasingly clear that subtle regulation of tyrosine kinase ac-tivity may rely on negative checkpoints that counter-feedbackpositive inputs. One of the possible mechanisms whereby thiscan be achieved is through transcriptional induction of regu-latory molecules that, in turn, can affect the activity of tran-scription factors and coactivators (3, 48). Based on this infor-mation, we hypothesized that the inhibitory function of Notchon Met could be induced by Met itself, within an intrinsiccircuit of transcriptional cross talk.
Notch activity is first primed by elevation of the cellularlevels of its ligands (in vertebrates, two homologues of Serrate[Jagged1 and -2] [26, 60] and four Delta-like genes [Delta 1 to-4] [17, 23, 50]) and further proceeds through nuclear translo-cation, association with the transcription factor Suppressor ofHairless/CBF-1, and transactivation of several target genescoding for basic helix-loop-helix proteins, namely the genesbelonging to the hairy/Enhancer of Split that include HES-1[E(spl) family] (23).
To validate the prediction that Met stimulates the activity ofits own repressor, we performed a time course RT-PCR anal-ysis to track HGF-dependent transcriptional induction of thevarious Notch ligands and of HES-1. Indeed, HGF stimulation
of MDA-MB-435-�4 cells induced the transient transcriptionof HES-1 and of two Delta mRNAs, Dl1 and Dl4 (Fig. 4A).Among the other ligands, J1 was expressed constitutively (Fig.4A), whereas J2 and Dl3 were not expressed nor were theyinduced by HGF (data not shown).
Given that HGF induces HES-1 mRNA and that activationof Notch, the HES-1 upstream regulator, leads to Met inhibi-tion, we tested if altered expression of HES-1 protein results inmodifications of Met mRNA transcription. This was done bymonitoring the luciferase activity of Met-promoter reporterconstructs (see above) in MDA-MB-435-�4 cells in whichHES-1 was overexpressed by transient transfection. In agree-ment with the inhibitory function of the Notch pathway on Metsynthesis, overexpression of HES-1 led to a 30% decrease inMet-promoter activity (Fig. 4B). As expected, luciferase activ-ity of the Met-promoter construct in which the HES-1 bindingsite was mutated was insensitive to dose alterations of HES-1(Fig. 4B). Accordingly, overexpression of HES-1 resulted indown-regulation of the Met protein (Fig. 4C), which leads toreduced biological responses to HGF treatment, measured asgrowth in tridimensional collagen gel (Fig. 4D). To furthersupport the correlation between HES-1 activity and Met ex-pression, we used the RNA interference technology to pro-gressively down-modulate HES-1 protein levels in MDA-MB-435-�4 cells exogenously overexpressing HES-1. Addition of aHES-1-specific siRNA resulted in a dose-response increase inMet protein (Fig. 4E, lanes three and four), whereas an unre-lated siRNA had no effect (see Fig. 4E, lane five). Accordingly,luciferase activity analysis of the Met-promoter construct inthese cells showed that siRNA-mediated inhibition of HES-1expression abrogated the transcriptional down-regulation ofthe Met gene (Fig. 4F).
By using an opposite but complementary approach, we mon-itored luciferase activity of the Met-promoter constructs usingMDA-MB-435-�4 cells in which HES-1 function was knockeddown by RNA interference technology. Ablation of HES-1 incells expressing N4-TM restored a promoter activity similar tothat observed in mock cells; as previously shown, mutations inthe HES-1 binding domain of the Met promoter region abol-ished Notch-dependent down-regulation of Met synthesis irre-spective of HES-1 expression (Fig. 5A). Consistent with thesefindings, HES-1 inactivation increased expression of the Metprotein in N4-TM-expressing cells (Fig. 5B). Again, gradualincrease of HES-1 expression in these HES-1 cells led torecovery of the Notch inhibitory activity, with consequent pro-gressive reduction of Met protein levels (Fig. 5, panels C andD).
If HES-1 is the Notch effector responsible for attenuation ofMet synthesis, then overexpression of HES-1 should mimicNotch activity in impairing Met-dependent signals. To test thishypothesis, we compared Met expression, PI3-kinase stimula-tion (in terms of activated PKB/AKT), and Ras stimulation (interms of MAPK activity) upon HGF treatment in MDA-MB-435-�4 cells, expressing either N4-TM or HES-1. As previouslyshown, both N4-TM- and HES-1-expressing cells displayedreduced Met expression (Fig. 5E, first panel), which correlateswith the expression level of HES-1 (Fig. 5E, sixth panel).Moreover, cells expressing either N4-TM or HES-1 exhibitedreduced MAPK activation (Fig. 5E, second panel) in responseto HGF stimulation, whereas PKB/AKT activity was unaf-
3988 STELLA ET AL. MOL. CELL. BIOL.
on January 12, 2016 by guesthttp://m
cb.asm.org/
Dow
nloaded from
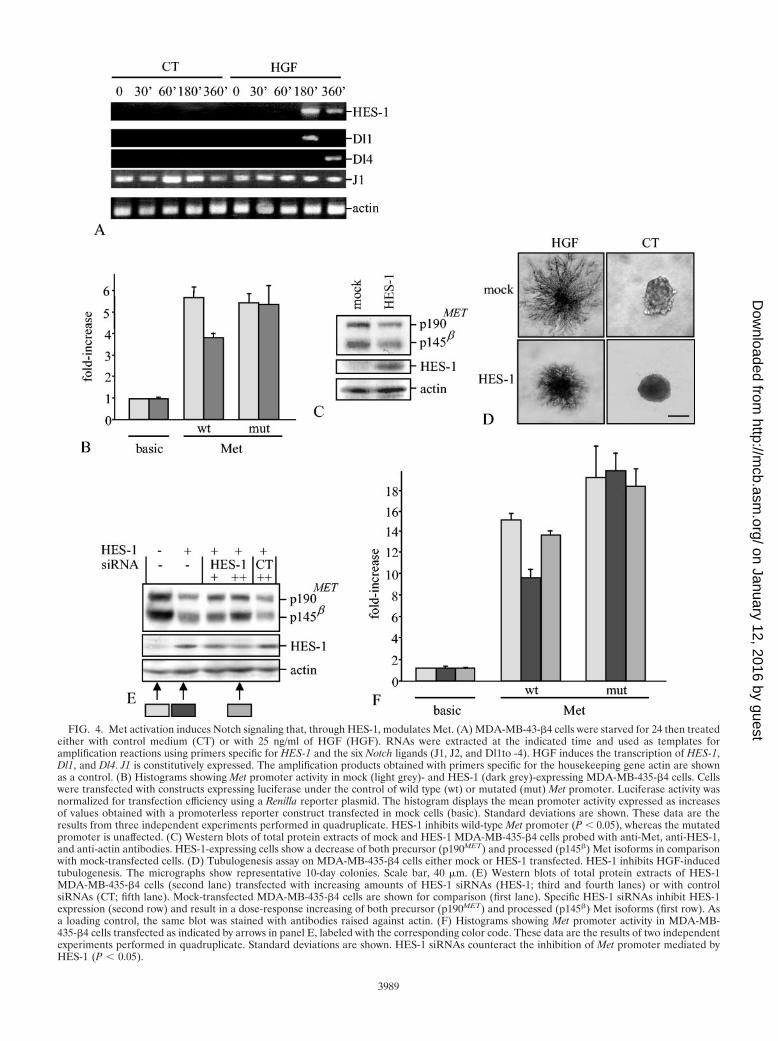
FIG. 4. Met activation induces Notch signaling that, through HES-1, modulates Met. (A) MDA-MB-43-�4 cells were starved for 24 then treatedeither with control medium (CT) or with 25 ng/ml of HGF (HGF). RNAs were extracted at the indicated time and used as templates foramplification reactions using primers specific for HES-1 and the six Notch ligands (J1, J2, and Dl1to -4). HGF induces the transcription of HES-1,Dl1, and Dl4. J1 is constitutively expressed. The amplification products obtained with primers specific for the housekeeping gene actin are shownas a control. (B) Histograms showing Met promoter activity in mock (light grey)- and HES-1 (dark grey)-expressing MDA-MB-435-�4 cells. Cellswere transfected with constructs expressing luciferase under the control of wild type (wt) or mutated (mut) Met promoter. Luciferase activity wasnormalized for transfection efficiency using a Renilla reporter plasmid. The histogram displays the mean promoter activity expressed as increasesof values obtained with a promoterless reporter construct transfected in mock cells (basic). Standard deviations are shown. These data are theresults from three independent experiments performed in quadruplicate. HES-1 inhibits wild-type Met promoter (P � 0.05), whereas the mutatedpromoter is unaffected. (C) Western blots of total protein extracts of mock and HES-1 MDA-MB-435-�4 cells probed with anti-Met, anti-HES-1,and anti-actin antibodies. HES-1-expressing cells show a decrease of both precursor (p190MET) and processed (p145�) Met isoforms in comparisonwith mock-transfected cells. (D) Tubulogenesis assay on MDA-MB-435-�4 cells either mock or HES-1 transfected. HES-1 inhibits HGF-inducedtubulogenesis. The micrographs show representative 10-day colonies. Scale bar, 40 �m. (E) Western blots of total protein extracts of HES-1MDA-MB-435-�4 cells (second lane) transfected with increasing amounts of HES-1 siRNAs (HES-1; third and fourth lanes) or with controlsiRNAs (CT; fifth lane). Mock-transfected MDA-MB-435-�4 cells are shown for comparison (first lane). Specific HES-1 siRNAs inhibit HES-1expression (second row) and result in a dose-response increasing of both precursor (p190MET) and processed (p145�) Met isoforms (first row). Asa loading control, the same blot was stained with antibodies raised against actin. (F) Histograms showing Met promoter activity in MDA-MB-435-�4 cells transfected as indicated by arrows in panel E, labeled with the corresponding color code. These data are the results of two independentexperiments performed in quadruplicate. Standard deviations are shown. HES-1 siRNAs counteract the inhibition of Met promoter mediated byHES-1 (P � 0.05).
3989
on January 12, 2016 by guesthttp://m
cb.asm.org/
Dow
nloaded from
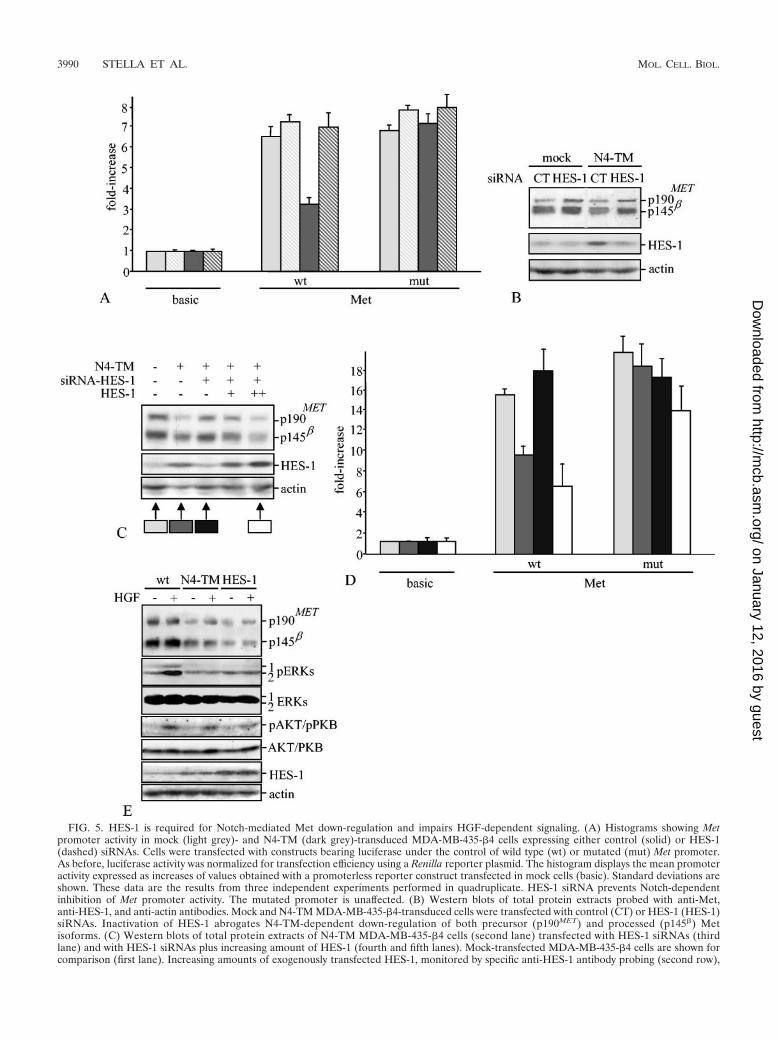
FIG. 5. HES-1 is required for Notch-mediated Met down-regulation and impairs HGF-dependent signaling. (A) Histograms showing Metpromoter activity in mock (light grey)- and N4-TM (dark grey)-transduced MDA-MB-435-�4 cells expressing either control (solid) or HES-1(dashed) siRNAs. Cells were transfected with constructs bearing luciferase under the control of wild type (wt) or mutated (mut) Met promoter.As before, luciferase activity was normalized for transfection efficiency using a Renilla reporter plasmid. The histogram displays the mean promoteractivity expressed as increases of values obtained with a promoterless reporter construct transfected in mock cells (basic). Standard deviations areshown. These data are the results from three independent experiments performed in quadruplicate. HES-1 siRNA prevents Notch-dependentinhibition of Met promoter activity. The mutated promoter is unaffected. (B) Western blots of total protein extracts probed with anti-Met,anti-HES-1, and anti-actin antibodies. Mock and N4-TM MDA-MB-435-�4-transduced cells were transfected with control (CT) or HES-1 (HES-1)siRNAs. Inactivation of HES-1 abrogates N4-TM-dependent down-regulation of both precursor (p190MET) and processed (p145�) Metisoforms. (C) Western blots of total protein extracts of N4-TM MDA-MB-435-�4 cells (second lane) transfected with HES-1 siRNAs (thirdlane) and with HES-1 siRNAs plus increasing amount of HES-1 (fourth and fifth lanes). Mock-transfected MDA-MB-435-�4 cells are shown forcomparison (first lane). Increasing amounts of exogenously transfected HES-1, monitored by specific anti-HES-1 antibody probing (second row),
3990 STELLA ET AL. MOL. CELL. BIOL.
on January 12, 2016 by guesthttp://m
cb.asm.org/
Dow
nloaded from

fected (Fig. 5E, fourth panel). The amounts of extracellularsignal-regulated kinases (ERKs), AKT/PKB, and actin con-firmed equal loading in all the experimental conditions (Fig.5E, third, fifth, and sixth panels). This indicates that HES-1overexpression recapitulates the repression of Met signalingobserved in N4-TM transfectants.
In summary, these data define a novel epistatic relationshipin which Met activation results in induction of Notch signalingthat in turn represses Met activity through HES-1.
Cross talk between Met and Notch signaling in Drosophila.To date, four vertebrate Notch genes have been identified(Notch1 to -4) (23) together with six Notch ligands (17, 23, 26,50, 60). The complexity of Notch and Notch ligand familymembers, as well as the evidence that loss of either HGF orMet in the mouse results in early embryonic lethality, points tothe necessity of exploiting alternative animal models to explorein vivo the interaction between Met and Notch. This modelshould not require Met function during development and inadult life, and in the meantime it should be easily amenable togenetic manipulation. We thus decided to scrutinize the effectsof Met-Notch interactions during development of the trachealtree in Drosophila melanogaster. The advantages of such anexperimental approach are twofold: Met is not expressed inDrosophila (54), so this organism represents a naive recipientfor clear-cut dissection of Met-dependent responses withoutany kind of endogenous developmental interference; more-over, formation of the tracheal tree in Drosophila is a strikingexample of branching morphogenesis in vivo, in which the roleof Notch pathway has been extensively documented (21, 28).
The Drosophila tracheal system is an elaborate network ofepithelial tubes that ramifies in some 10,000 branches. Thenetwork arises from segmentally repeated clusters of ectoder-mal cells, the placodes, which invaginate to form sac-like struc-tures called tracheal pits. Each tracheal pit sprouts successivelyfiner branches, some of which ultimately fuse to form an ar-borescent pattern throughout the embryo. Unlike sprouting ofthe major tracheal branches, which is simple and stereotyped,ramification of the terminal branches is complex and variableand is affected by several morphogenetic regulators. Amongthese, the Notch signaling pathway is needed in the trachealcells for the fusion and terminal branching programs (21, 28).In particular, Notch is first required to single out the cells thatwill control branch fusion from a group of competent trachealcells and later to select the correct number of terminalbranches arising from some primary branches.
To confirm in this in vivo setting our assumption that Metactivation results in induction of Notch function, which in turnrepresses Met activity, we followed three subsequent experi-mental lines: first, we analyzed whether expression of a con-stitutively active Met in Drosophila leads to Notch stimulation
(through increased expression of its physiological ligands);then, as a basal read-out of Met function, we examined thetracheal phenotype of Met-expressing flies; finally, we analyzedwhether this phenotype is affected in a Notch-null background.
Construction and expression of the chimeric torso-Met re-ceptor in Drosophila. To generate transgenic flies in which Metsignaling is constitutively activated, we produced a chimericfusion protein in which the extracellular and transmembranedomains of human Met were substituted with the correspond-ing domains of torso4021 (torso-Met). torso4021 is a Drosophilatyrosine kinase mutant receptor bearing a single amino acidsubstitution (Y 327 C) that leads to spontaneous dimerizationand consequent constitutive activation of the receptor (53).The expression of this hybrid receptor was driven by the pro-moter hsp70, which allows a ubiquitous and heat-inducibleexpression of the protein.
The torso-Met construct was checked for correct expressionby in vitro translation experiments (data not shown) and thenintroduced into Drosophila embryos by P-element-mediatedgene transfer. Six independent P-element insertions weremapped by in situ hybridization onto polytene chromosomes,and different lines were generated using specific marked bal-ancer chromosomes. All the transformant lines were main-tained at 18°C to prevent torso-Met activation. To test theexpression and the biochemical properties of the torso-Metrecombinant protein in vivo, the transgene was induced by heatshock treatment (37°C, 20 min) in the progeny of the transfor-mant flies, and then the embryos were transferred at 18°C fora further 3 h. Immunoprecipitation of lysates from heat-shocked and untreated embryos with specific monoclonal an-tibodies raised against human Met followed by Western blotanalysis with anti-Met and anti-phosphotyrosine antibodiesshows that the recombinant protein was expressed exclusivelyin the embryos exposed to heat shock treatment and had theexpected molecular weight of 95,000 (Fig. 6A). Moreover, asfor activated human Met receptor, torso-Met was tyrosinephosphorylated (Fig. 6B) and underwent autophosphorylationin autokinase assays (Fig. 6C). This indicates that the recom-binant torso-Met protein expressed in Drosophila retains thecatalytic properties of human wild-type Met.
Active Met stimulates Notch function in Drosophila. To an-alyze whether Met activation results in induction of Notchfunction also in Drosophila, we employed semiquantitative RT-PCR to measure the overall expression of the Notch ligandDelta, which is responsible for Notch activation in the trachealsystem (21, 28), in wild-type and torso-Met embryos. Consis-tent with the findings obtained in mammalian cells, embryosexpressing torso-Met displayed an increase in Delta transcripts,which peaked 2 h after heat shock treatment (Fig. 6D).
result in a dose-response down-regulation of both precursor (p190MET) and processed (p145�) Met isoforms (first row, lanes four and five). As aloading control, the same blot was stained with antibodies raised against actin. (D) Histograms showing Met promoter activity in MDA-MB-435-�4cells transfected as indicated by arrows in panel C are labeled with the corresponding color code. These data are the results of two independentexperiments performed in quadruplicate. Standard deviations are shown. Exogenous transfection of HES-1 rescues the Notch-mediated inhibitionof Met mRNA expression in N4-TM MDA-MB-435-�4 cells transfected with HES-1 siRNAs. (E) Western blots of total protein extracts showinga comparison of HGF-dependent signaling pathways in N4-TM and HES-1 MDA-MB-435-�4 cells. The same blot was stained with antibodiesraised against Met, pERKs, pAKT/PKB, and HES-1. Similarly to N4-TM, HES-1 overexpression determines down-regulation of Met (first panel);ERK phosphorylation in response to HGF is impaired (second panel), whereas AKT/PKB phosphorylation is unaffected (third panel). Again, theblot was reprobed with antibodies anti-actin, anti-ERK, and anti-AKT/PKB (third, fifth, and sixth panels, respectively) as loading controls.
VOL. 25, 2005 Met NEGATIVE FEEDBACK REGULATION BY Notch 3991
on January 12, 2016 by guesthttp://m
cb.asm.org/
Dow
nloaded from
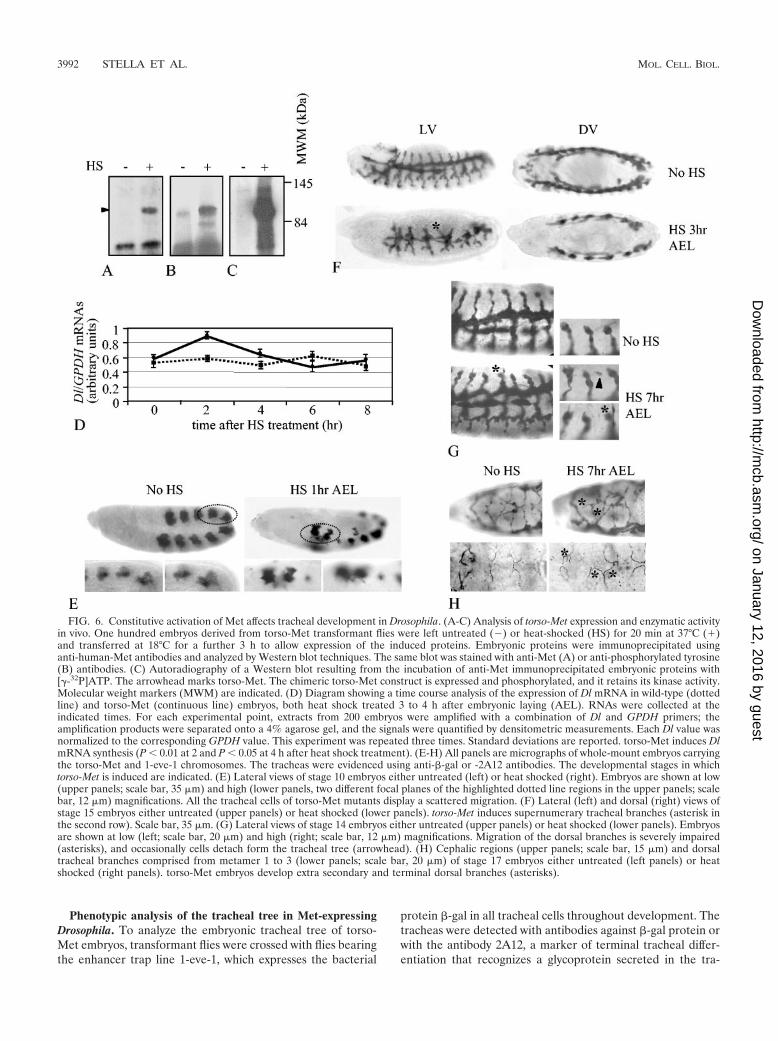
Phenotypic analysis of the tracheal tree in Met-expressingDrosophila. To analyze the embryonic tracheal tree of torso-Met embryos, transformant flies were crossed with flies bearingthe enhancer trap line 1-eve-1, which expresses the bacterial
protein �-gal in all tracheal cells throughout development. Thetracheas were detected with antibodies against �-gal protein orwith the antibody 2A12, a marker of terminal tracheal differ-entiation that recognizes a glycoprotein secreted in the tra-
FIG. 6. Constitutive activation of Met affects tracheal development in Drosophila. (A-C) Analysis of torso-Met expression and enzymatic activityin vivo. One hundred embryos derived from torso-Met transformant flies were left untreated () or heat-shocked (HS) for 20 min at 37°C (�)and transferred at 18°C for a further 3 h to allow expression of the induced proteins. Embryonic proteins were immunoprecipitated usinganti-human-Met antibodies and analyzed by Western blot techniques. The same blot was stained with anti-Met (A) or anti-phosphorylated tyrosine(B) antibodies. (C) Autoradiography of a Western blot resulting from the incubation of anti-Met immunoprecipitated embryonic proteins with[�-32P]ATP. The arrowhead marks torso-Met. The chimeric torso-Met construct is expressed and phosphorylated, and it retains its kinase activity.Molecular weight markers (MWM) are indicated. (D) Diagram showing a time course analysis of the expression of Dl mRNA in wild-type (dottedline) and torso-Met (continuous line) embryos, both heat shock treated 3 to 4 h after embryonic laying (AEL). RNAs were collected at theindicated times. For each experimental point, extracts from 200 embryos were amplified with a combination of Dl and GPDH primers; theamplification products were separated onto a 4% agarose gel, and the signals were quantified by densitometric measurements. Each Dl value wasnormalized to the corresponding GPDH value. This experiment was repeated three times. Standard deviations are reported. torso-Met induces DlmRNA synthesis (P � 0.01 at 2 and P � 0.05 at 4 h after heat shock treatment). (E-H) All panels are micrographs of whole-mount embryos carryingthe torso-Met and 1-eve-1 chromosomes. The tracheas were evidenced using anti-�-gal or -2A12 antibodies. The developmental stages in whichtorso-Met is induced are indicated. (E) Lateral views of stage 10 embryos either untreated (left) or heat shocked (right). Embryos are shown at low(upper panels; scale bar, 35 �m) and high (lower panels, two different focal planes of the highlighted dotted line regions in the upper panels; scalebar, 12 �m) magnifications. All the tracheal cells of torso-Met mutants display a scattered migration. (F) Lateral (left) and dorsal (right) views ofstage 15 embryos either untreated (upper panels) or heat shocked (lower panels). torso-Met induces supernumerary tracheal branches (asterisk inthe second row). Scale bar, 35 �m. (G) Lateral views of stage 14 embryos either untreated (upper panels) or heat shocked (lower panels). Embryosare shown at low (left; scale bar, 20 �m) and high (right; scale bar, 12 �m) magnifications. Migration of the dorsal branches is severely impaired(asterisks), and occasionally cells detach form the tracheal tree (arrowhead). (H) Cephalic regions (upper panels; scale bar, 15 �m) and dorsaltracheal branches comprised from metamer 1 to 3 (lower panels; scale bar, 20 �m) of stage 17 embryos either untreated (left panels) or heatshocked (right panels). torso-Met embryos develop extra secondary and terminal dorsal branches (asterisks).
3992 STELLA ET AL. MOL. CELL. BIOL.
on January 12, 2016 by guesthttp://m
cb.asm.org/
Dow
nloaded from

cheal lumen. The heat shock treatment used for transgeneexpression did not alter the tracheal trees of embryos resultingfrom the cross of wild-type and 1-eve-1 flies (data not shown).Since the tracheas of these embryos were indistinguishablefrom those of uninduced embryos carrying the 1-eve-1 andtorso-Met chromosomes, we elected uninduced 1-eve-1; torso-MET embryos as control organisms. About 50% of the heat-shocked embryos resulting from the cross between torso-Metand 1-eve-1 flies that expressed �-gal protein displayed tra-cheal alterations.
In the first stages of tracheal development, when the tra-cheal placodes start to invaginate to form the tracheal pits,expression of torso-Met resulted in scattering of the cells thatcompose the tracheal pits, which appeared as starred aggre-gates of loosely associated cells (Fig. 6E), whereas in the im-mediately subsequent developmental stage, torso-Met led tothe formation of supernumerary tracheal branches (Fig. 6F,second row). At later stages of development, the cells that formthe primary dorsal branches start a highly coordinated migra-tory program towards the dorsal side of the embryo that endsup in the formation of the secondary and terminal dorsaltracheal branches. When expressed in this stage, torso-Metimpaired this stereotyped migratory behavior (Fig. 6G). Thecells that compose the stalk were much more disorganized and,in some cases, one of the uppermost two-paired cells acquiredan unusual migratory route, eventually breaking its connectionwith the neighboring cells. As a consequence, the formation ofdorsal secondary and terminal tracheal branches was severelyaffected (Fig. 6H), resulting in the presence of unfused dorsalbranches and an excess of terminal branches.
In summary, these results suggest that exogenous expressionof active Met amplifies the overall genetic program of Dro-sophila tracheal morphogenesis, with enhanced sprouting,primitive hyperbranching, and increased terminal ramification.
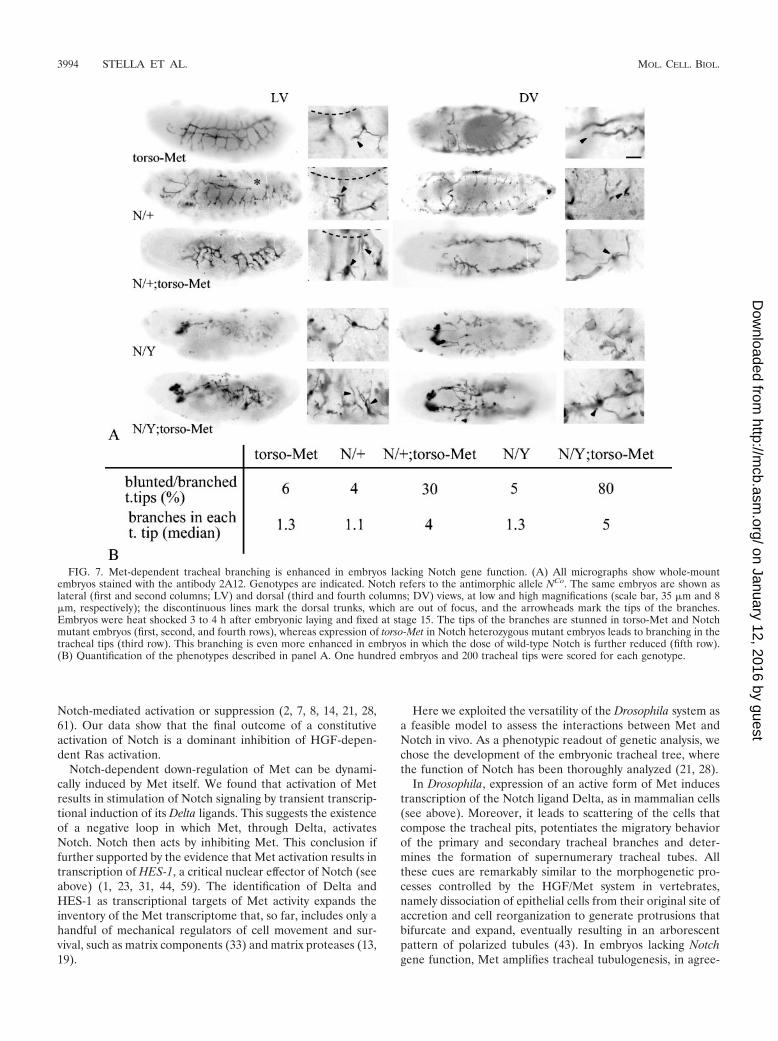
Absence of Notch enhances Met-dependent tracheal pheno-type in Drosophila. If in invertebrates, as in vertebrates, Notchelicits an inhibitory effect on Met-dependent branching mor-phogenesis, then abrogation of Notch gene function in Dro-sophila embryos should enhance torso-Met-controlled trachealbranching.
Accordingly, we expressed torso-Met in flies bearing themutation encoded by the NCo allele, which lacks the intracel-lular portion of the receptor and behaves as a dominant neg-ative (62). Moreover, since NCo is an antimorphic allele, theseverity of the phenotype may be modulated by the dose ofwild-type allele present in the embryo. Males bearing the NCo
allele were crossed with torso-Met females carrying a balancerchromosome labeled with �-gal. In the progeny of this cross,the tracheas were evidenced using 2A12 antibody and theembryos were genotyped using anti-�-gal antibodies and anti-bodies raised against the C-terminal portion of Notch receptor;embryos lacking �-gal staining and expressing barely detect-able full-length Notch protein were considered having the ge-notype NCo/�; torso-Met/�.
The NCo mutants presented the typical alteration of Notchloss-of-function mutants (Fig. 7, second row): the dorsal trunkshowed conspicuous interruptions; some dorsal branches wereirregularly fused, and some others were missing. Interestingly,when analyzing the tracheal trees following activation of torso-Met in NCo mutants, we found that lack of Notch function
results in enhanced tracheal branching. In particular, the ter-minal portions of the tracheas, instead of being blunt-endtubes, as in torso-Met and NCo mutants (Fig. 7, first and secondrows), developed additional ramifications with reiterated seg-mentations of the distal tips (Fig. 7, third row).
To confirm the finding that inhibition of Notch activity re-sults in enhanced branching effects of torso-Met on terminaltracheation, we took advantage of the antimorphic propertiesof NCo to increase the dose of this allele versus wild-typeNotch, thus enforcing the dominant negative activity inducedby NCo. To this aim, females harboring the genotype NCo/�;torso-Met/� were mated with males bearing torso-Met and a�-gal-labeled second balancer chromosome. The resultingprogeny (NCo/Y; torso-Met/�) displayed gross developmentalalterations, most likely as a consequence of the high dominantnegative activity of NCo that suppresses the maternal contribu-tion of wild-type Notch (for comparison, see Fig. 7, fourthrow). However, although rudimentary, the tracheas still devel-oped and displayed many terminal branches (Fig. 7, fifth row),which appeared more ramified and elongated than those ob-served in NCo/�; torso-Met/� embryos (Fig. 7, compare thethird and fifth rows). Therefore, further reduction of Notchfunction results in potentiation of torso-Met-dependent extra-branching. This overbranching phenotype is likely due to thepossibility for exogenous Met to hyperactivate the Ras pathwayin the absence of Notch, which in turn results in potentiation ofterminal tracheogenesis. Indeed, the crucial role of Ras inDrosophila tracheogenesis has been repeatedly documented(21, 28). In contrast, we could not explore direct transcrip-tional repression of Met by Notch in Drosophila because, inthis system, Met is under the control of an artificial promoter.
DISCUSSION
In this paper we demonstrate that Notch is an inhibitor ofMet-dependent branching morphogenesis and invasive growth.This inhibitory activity resides in two major mechanisms. First,Notch suppresses Met transcription. So far, the only otherexample of vertebrate tyrosine kinase receptor that can betranscriptionally down-regulated by Notch is VEGF receptor-2/KDR, which is crucially involved in the control of anotherparadigmatic example of branching morphogenesis, the capil-lary sprouting of endothelial cells (56). The transcriptionalsuppression elicited by Notch requires induction of genes be-longing to the HES family. These genes encode class C helix-loop-helix proteins that act mainly as transcriptional suppres-sors by binding to specific consensus sequences in thepromoters of the target genes (22, 32). Here we show thatHES-1 binds in vitro to the class C consensus site containedwithin the Met promoter and that mutagenesis of such a re-sponsive element results in increased Met transcription. More-over, we demonstrate that HES-1 is sufficient to induce Metdown-regulation and that inactivation of this transcription fac-tor abrogates Notch-dependent inhibition of Met synthesis.
A second level of negative regulation relies on the ability ofNotch to suppress Met-dependent activation of the Ras-MAPK cascade. The interactions between Ras and Notch arecomplex. Much evidence, both in invertebrates and in verte-brates, suggests that the Ras pathway has a role in Notchactivation, but at the same time Ras can be a target of either
VOL. 25, 2005 Met NEGATIVE FEEDBACK REGULATION BY Notch 3993
on January 12, 2016 by guesthttp://m
cb.asm.org/
Dow
nloaded from
Notch-mediated activation or suppression (2, 7, 8, 14, 21, 28,61). Our data show that the final outcome of a constitutiveactivation of Notch is a dominant inhibition of HGF-depen-dent Ras activation.
Notch-dependent down-regulation of Met can be dynami-cally induced by Met itself. We found that activation of Metresults in stimulation of Notch signaling by transient transcrip-tional induction of its Delta ligands. This suggests the existenceof a negative loop in which Met, through Delta, activatesNotch. Notch then acts by inhibiting Met. This conclusion iffurther supported by the evidence that Met activation results intranscription of HES-1, a critical nuclear effector of Notch (seeabove) (1, 23, 31, 44, 59). The identification of Delta andHES-1 as transcriptional targets of Met activity expands theinventory of the Met transcriptome that, so far, includes only ahandful of mechanical regulators of cell movement and sur-vival, such as matrix components (33) and matrix proteases (13,19).
Here we exploited the versatility of the Drosophila system asa feasible model to assess the interactions between Met andNotch in vivo. As a phenotypic readout of genetic analysis, wechose the development of the embryonic tracheal tree, wherethe function of Notch has been thoroughly analyzed (21, 28).
In Drosophila, expression of an active form of Met inducestranscription of the Notch ligand Delta, as in mammalian cells(see above). Moreover, it leads to scattering of the cells thatcompose the tracheal pits, potentiates the migratory behaviorof the primary and secondary tracheal branches and deter-mines the formation of supernumerary tracheal tubes. Allthese cues are remarkably similar to the morphogenetic pro-cesses controlled by the HGF/Met system in vertebrates,namely dissociation of epithelial cells from their original site ofaccretion and cell reorganization to generate protrusions thatbifurcate and expand, eventually resulting in an arborescentpattern of polarized tubules (43). In embryos lacking Notchgene function, Met amplifies tracheal tubulogenesis, in agree-
FIG. 7. Met-dependent tracheal branching is enhanced in embryos lacking Notch gene function. (A) All micrographs show whole-mountembryos stained with the antibody 2A12. Genotypes are indicated. Notch refers to the antimorphic allele NCo. The same embryos are shown aslateral (first and second columns; LV) and dorsal (third and fourth columns; DV) views, at low and high magnifications (scale bar, 35 �m and 8�m, respectively); the discontinuous lines mark the dorsal trunks, which are out of focus, and the arrowheads mark the tips of the branches.Embryos were heat shocked 3 to 4 h after embryonic laying and fixed at stage 15. The tips of the branches are stunned in torso-Met and Notchmutant embryos (first, second, and fourth rows), whereas expression of torso-Met in Notch heterozygous mutant embryos leads to branching in thetracheal tips (third row). This branching is even more enhanced in embryos in which the dose of wild-type Notch is further reduced (fifth row).(B) Quantification of the phenotypes described in panel A. One hundred embryos and 200 tracheal tips were scored for each genotype.
3994 STELLA ET AL. MOL. CELL. BIOL.
on January 12, 2016 by guesthttp://m
cb.asm.org/
Dow
nloaded from

ment with the inhibitory role of Notch over Met-dependentmorphogenesis postulated in this paper.
In conclusion, our findings suggest that Met can self-limitthe strength and duration of its positive signals by a twofoldmechanism: on the one hand, Met-dependent induction ofNotch function leads to a direct repression of Met levels; onthe other, Notch can further subsidize this negative tuning bydecreasing Ras signaling, with consequent lessening of Met-dependent transduction pathways. This unconventional mech-anism of Met negative modulation goes along with the well-established roles of receptor endocytosis and ubiquitin-mediated degradation for attenuation of receptor signaling(12, 40). A delicate balance between positive and negativesignals is critical for normal cell homeostasis, and a prevalenceof positive signals leading to excessive cell stimulation is com-monly found in cancers. Not surprisingly, aberrant execution ofHGF-dependent invasive growth in neoplastic cells leads tocancer invasion and metastasis. The finding that Notch canefficiently harness Met-dependent transcription and Met sig-naling discloses a novel inhibitory pathway that might be ex-ploited to hamper Met-based cancer progression.
ACKNOWLEDGMENTS
P. Larghero and E. Laguzzi were of invaluable help during the earlystages of this work. We thank T. Sudo, S. Herzig, S. Roth, M. Affolter,and F. Sprenger for sharing reagents, all the colleagues and especiallythe “Lilla’s group,” and E. Audero and L. Primo for helpful discussionsand comments. We acknowledge N. Azpiazu for critical reading of themanuscript. We are grateful to A. Cignetto for secretarial assistance.The excellent technical assistance of L. Palmas, R. Albano, F. Grasso,and S. Tyenga is acknowledged.
This work was supported by research grants of AIRC, CNR-MIUR,FIRB-MIUR, MIUR-PRIN, and the Foundation Compagnia di SanPaolo to P.M.C.
REFERENCES
1. Bardelli, A., M. L. Basile, E. Audero, S. Giordano, S. Wennstrom, S. Men-ard, P. M. Comoglio, and C. Ponzetto. 1999. Concomitant activation ofpathways downstream of Grb2 and PI 3-kinase is required for MET-medi-ated metastasis. Oncogene 18:1139–1146.
2. Berset, T., E. F. Hoier, G. Battu, S. Canevascini, and A. Hajnal. 2001. Notchinhibition of RAS signaling through MAP kinase phosphatase LIP-1 duringC. elegans vulval development. Science 291:1055–1058.
3. Blume-Jensen, P., and T. Hunter. 2001. Oncogenic kinase signalling. Nature411:355–365.
4. Boccaccio, C., M. Ando, L. Tamagnone, A. Bardelli, P. Michieli, C. Battis-tini, and P. M. Comoglio. 1998. Induction of epithelial tubules by growthfactor HGF depends on the STAT pathway. Nature 391:285–288.
5. Bussolino, F., M. F. Di Renzo, M. Ziche, E. Bocchietto, M. Olivero, L.Naldini, G. Gaudino, L. Tamagnone, A. Coffer, and P. M. Comoglio. 1992.Hepatocyte growth factor is a potent angiogenic factor which stimulatesendothelial cell motility and growth. J. Cell Biol. 119:629–641.
6. Campos-Ortega, A., and V. Hartenstein. 1997. The embryonic developmentof Drosophila melanogaster, 2nd ed. Springer-Verlag KG, Berlin, Germany.
7. Carmena, A., E. Buff, M. S. Halfon, S. Gisselbrecht, F. Jimenez, M. K.Baylies, and A. M. Michelson. 2002. Reciprocal regulatory interactions be-tween the Notch and Ras signaling pathways in the Drosophila embryonicmesoderm. Dev. Biol. 244:226–242.
8. Chihara, T., and S. Hayashi. 2000. Control of tracheal tubulogenesis byWingless signaling. Development 127:4433–4442.
9. Coligan, S. E., B. M. Dunn, H. L. Ploegh, D. W. Speicher, and P. Wingfield.1998. Current protocols in protein science, vol. 1. J. Wiley & Sons, Inc., NewYork, N.Y.
10. Danilkovitch-Miagkova, A., and B. Zbar. 2002. Dysregulation of Met recep-tor tyrosine kinase activity in invasive tumors. J. Clin. Investig. 109:863–867.
11. Dickson, B., F. Sprenger, and E. Hafen. 1992. Prepattern in the developingDrosophila eye revealed by an activated torso-sevenless chimeric receptor.Genes Dev. 6:2327–2339.
12. Dikic, I., and S. Giordano. 2003. Negative receptor signalling. Curr. Opin.Cell Biol. 15:128–135.
13. Dunsmore, S., J. Rubin, S. Kovacs, M. Chedid, W. Parks, and H. Welgus.
1996. Mechanisms of hepatocyte growth factor stimulation of keratinocytemetalloproteinase production. J. Biol. Chem. 271:24576–24582.
14. Fitzgerald, K., A. Harrington, and P. Leder. 2000. Ras pathway signals arerequired for notch-mediated oncogenesis. Oncogene 19:4191–4198.
15. Gambarotta, G., S. Pistoi, S. Giordano, P. M. Comoglio, and C. Santoro.1994. Structure and inducible regulation of the human MET promoter.J. Biol. Chem. 269:12852–12857.
16. Giordano, S., A. Bardelli, Z. Zhen, S. Menard, C. Ponzetto, and P. M.Comoglio. 1997. A point mutation in the MET oncogene abrogates metas-tasis without affecting transformation. Proc. Natl. Acad. Sci. USA 94:13868–13872.
17. Gray, G., R. Mann, R. Mitsiadis, D. Henrique, M. Carcangiu, A. Banks, J.Leiman, D. Ward, D. Ish-Horowitz, and S. Artavanis-Tsakonas. 1999. Hu-man ligands of the Notch receptor. Am. J. Pathol. 154:785–794.
18. Gual, P., S. Giordano, T. Williams, S. Rocchi, E. Van Obberghen, and P.Comoglio. 2000. Sustained recruitment of phospholipase C-gamma to Gab1is required for HGF-induced branching tubulogenesis. Oncogene 19:1509–1518.
19. Harvey, P., I. Clark, M. Jaurand, R. Warn, and D. Edwards. 2000. Hepato-cyte growth factor/scatter factor enhances the invasion of mesothelioma celllines and the expression of matrix metalloproteinases. Br. J. Cancer 89:1147–1153.
20. Herzig, S., S. Hedrick, I. Morantte, S. H. Koo, F. Galimi, and M. Montminy.2003. CREB controls hepatic lipid metabolism through nuclear hormonereceptor PPAR-gamma. Nature 426:190–193.
21. Ikeya, T., and S. Hayashi. 1999. Interplay of Notch and FGF signalingrestricts cell fate and MAPK activation in the Drosophila trachea. Develop-ment 126:4455–4463.
22. Iso, T., L. Kedes, and Y. Hamamori. 2003. HES and HERP families: multipleeffectors of the Notch signaling pathway. J. Cell. Physiol. 194:237–255.
23. Joutel, A., and E. Tournier-Lasserve. 1998. Notch signaling pathway andhuman diseases. Semin. Cell Dev. Biol. 9:619–625.
24. Kawaida, K., K. Matsumoto, H. Shimazu, and T. Nakamura. 1994. Hepa-tocyte growth factor prevents acute renal failure and accelerates renal re-generation in mice. Proc. Natl. Acad. Sci. USA 91:4357–4361.
25. Khwaja, A., K. Lehmann, B. M. Marte, and J. Downward. 1998. Phospho-inositide 3-kinase induces scattering and tubulogenesis in epithelial cellsthrough a novel pathway. J. Biol. Chem. 273:18793–18801.
26. Lindsell, C. E., C. J. Shawber, J. Boulter, and G. Weinmaster. 1995. Jagged:a mammalian ligand that activates Notch1. Cell 80:909–917.
27. Liu, Z. J., T. Shirakawa, Y. Li, A. Soma, M. Oka, G. P. Dotto, R. M. Fairman,O. C. Velazquez, and M. Herlyn. 2003. Regulation of Notch1 and Dll4 byvascular endothelial growth factor in arterial endothelial cells: implicationsfor modulating arteriogenesis and angiogenesis. Mol. Cell. Biol. 23:14–25.
28. Llimargas, M. 1999. The Notch pathway helps to pattern the tips of theDrosophila tracheal branches by selecting cell fates. Development 126:2355–2364.
29. Maher, J. J. 1993. Cell-specific expression of hepatocyte growth factor inliver. Upregulation in sinusoidal endothelial cells after carbon tetrachloride.J. Clin. Investig. 91:2244–2252.
30. Mailhos, C., U. Modlich, J. Lewis, A. Harris, R. Bicknell, and D. Ish-Horowicz. 2001. Delta4, an endothelial specific notch ligand expressed atsites of physiological and tumor angiogenesis. Differentiation 69:135–144.
31. Maina, F., G. Pante, F. Helmbacher, R. Andres, A. Porthin, A. M. Davies, C.Ponzetto, and R. Klein. 2001. Coupling Met to specific pathways results indistinct developmental outcomes. Mol. Cell 7:1293–1306.
32. Massari, M. E., and C. Murre. 2000. Helix-loop-helix proteins: regulators oftranscription in eucaryotic organisms. Mol. Cell. Biol. 20:429–440.
33. Medico, E., G. Gambarotta, A. Gentile, P. M. Comoglio, and P. Soriano.2001. A gene trap vector system for identifying transcriptionally responsivegenes. Nat. Biotechnol. 19:579–582.
34. Michieli, P., C. Basilico, S. Pennacchietti, A. Maffe, L. Tamagnone, S. Gior-dano, A. Bardelli, and P. M. Comoglio. 1999. Mutant Met-mediated trans-formation is ligand-dependent and can be inhibited by HGF antagonists.Oncogene 18:5221–5231.
35. Morgenstern, J. P., and H. Land. 1990. A series of mammalian expressionvectors and characterisation of their expression of a reporter gene in stablyand transiently transfected cells. Nucleic Acids Res. 18:1068–1078.
36. Nagaike, M., S. Hirao, H. Tajima, S. Noji, S. Taniguchi, K. Matsumoto, andT. Nakamura. 1991. Renotropic functions of hepatocyte growth factor inrenal regeneration after unilateral nephrectomy. J. Biol. Chem. 266:22781–22784.
37. Naldini, L., E. Vigna, A. Bardelli, A. Follenzi, F. Galimi, and P. M. Comoglio.1995. Biological activation of pro-HGF (hepatocyte growth factor) by uroki-nase is controlled by a stoichiometric reaction. J. Biol. Chem. 270:603–611.
38. Nijjar, S. S., L. Wallace, H. A. Crosby, S. G. Hubscher, and A. J. Strain.2002. Altered Notch ligand expression in human liver disease: further evi-dence for a role of the Notch signaling pathway in hepatic neovascularizationand biliary ductular defects. Am. J. Pathol. 160:1695–1703.
39. Pennacchietti, S., P. Michieli, M. Galluzzo, M. Mazzone, S. Giordano, andP. M. Comoglio. 2003. Hypoxia promotes invasive growth by transcriptionalactivation of the met protooncogene. Cancer Cell 3:347–361.
VOL. 25, 2005 Met NEGATIVE FEEDBACK REGULATION BY Notch 3995
on January 12, 2016 by guesthttp://m
cb.asm.org/
Dow
nloaded from

40. Petrelli, A., G. F. Gilestro, S. Lanzardo, P. M. Comoglio, N. Migone, and S.Giordano. 2002. The endophilin-CIN85-Cbl complex mediates ligand-de-pendent downregulation of c-Met. Nature 416:187–190.
41. Ponzetto, C., A. Bardelli, Z. Zhen, F. Maina, P. dalla Zonca, S. Giordano, A.Graziani, G. Panayotou, and P. M. Comoglio. 1994. A multifunctional dock-ing site mediates signaling and transformation by the hepatocyte growthfactor/scatter factor receptor family. Cell 77:261–271.
42. Ponzetto, C., Z. Zhen, E. Audero, F. Maina, A. Bardelli, M. L. Basile, S.Giordano, R. Narsimhan, and P. Comoglio. 1996. Specific uncoupling ofGRB2 from the Met receptor. Differential effects on transformation andmotility. J. Biol. Chem. 271:14119–14123.
43. Rosario, M., and W. Birchmeier. 2003. How to make tubes: signaling by theMet receptor tyrosine kinase. Trends Cell Biol. 13:328–335.
44. Royal, I., N. Lamarche-Vane, L. Lamorte, K. Kaibuchi, and M. Park. 2000.Activation of cdc42, rac, PAK, and rho-kinase in response to hepatocytegrowth factor differentially regulates epithelial cell colony spreading anddissociation. Mol. Biol. Cell 11:1709–1725.
45. Royal, I., and M. Park. 1995. Hepatocyte growth factor-induced scatter ofMadin-Darby canine kidney cells requires phosphatidylinositol 3-kinase.J. Biol. Chem. 270:27780–27787.
46. Sambrock, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular Cloning, ColdSpring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
47. Sander, E. E., S. van Delft, J. P. ten Klooster, T. Reid, R. A. van derKammen, F. Michiels, and J. G. Collard. 1998. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cellmigration and is regulated by phosphatidylinositol 3-kinase. J. Cell Biol.143:1385–1398.
48. Schlessinger, J. 2000. Cell signaling by receptor tyrosine kinases. Cell 103:211–225.
49. Shirayoshi, Y., Y. Yuasa, T. Suzuki, K. Sugaya, E. Kawase, T. Ikemura, andN. Nakatsuji. 1997. Proto-oncogene of int-3, a mouse Notch homologue, isexpressed in endothelial cells during early embryogenesis. Genes Cells2:213–224.
50. Shutter, J., S. Scully, W. Fan, W. Richards, J. Kitajewski, G. Deblandre, C.Kintner, and K. Stark. 2000. Dll4, a novel Notch ligand expressed in arterialendothelium. Genes Dev. 14:1313–1318.
51. Smith, G. H., D. Gallahan, F. Diella, C. Jhappan, G. Merlino, and R.Callahan. 1995. Constitutive expression of a truncated INT3 gene in mousemammary epithelium impairs differentiation and functional development.Cell Growth Differ. 6:563–577.
52. Soriano, J. V., M. S. Pepper, T. Nakamura, L. Orci, and R. Montesano. 1995.Hepatocyte growth factor stimulates extensive development of branchingduct-like structures by cloned mammary gland epithelial cells. J. Cell Sci.108:413–430.
53. Sprenger, F., and C. Nuesslein-Volhard. 1992. Torso receptor activity isregulated by a diffusible ligand produced at the extracellular terminal regionsof the Drosophila egg. Cell 71:987–1001.
54. Tamagnone, L., S. Artigiani, H. Chen, Z. He, G. I. Ming, H. Song, A.Chedotal, M. L. Winberg, C. S. Goodman, M. Poo, M. Tessier-Lavigne, andP. M. Comoglio. 1999. Plexins are a large family of receptors for transmem-brane, secreted, and GPI-anchored semaphorins in vertebrates. Cell 99:71–80.
55. Tautz, D., and C. Pfeifle. 1989. A non-radioactive in situ hybridizationmethod for the localization of specific RNAs in Drosophila embryos revealstranslational control of the segmentation gene hunchback. Chromosome98:81–85.
56. Taylor, K. L., A. M. Henderson, and C. C. Hughes. 2002. Notch activationduring endothelial cell network formation in vitro targets the basic HLHtranscription factor HESR-1 and downregulates VEGFR-2/KDR expres-sion. Microvasc. Res. 64:372–383.
57. Trusolino, L., A. Bertotti, and P. M. Comoglio. 2001. A signaling adapterfunction for alpha6beta4 integrin in the control of HGF-dependent invasivegrowth. Cell 107:643–654.
58. Trusolino, L., and P. M. Comoglio. 2002. Scatter-factor and semaphorinreceptors: cell signalling for invasive growth. Nat. Rev. Cancer 2:289–300.
59. Uyttendaele, H., J. V. Soriano, R. Montesano, and J. Kitajewski. 1998.Notch4 and Wnt-1 proteins function to regulate branching morphogenesis ofmammary epithelial cells in an opposing fashion. Dev. Biol. 196:204–217.
60. Valsecchi, V., C. Ghezzi, A. Ballabio, and E. I. Rugarli. 1997. JAGGED2: aputative Notch ligand expressed in the apical ectodermal ridge and in sites ofepithelial-mesenchymal interactions. Mech. Dev. 69:203–207.
61. Weijzen, S., P. Rizzo, M. Braid, R. Vaishnav, S. M. Jonkheer, A. Zlobin, B. A.Osborne, S. Gottipati, J. C. Aster, W. C. Hahn, M. Rudolf, K. Siziopikou,W. M. Kast, and L. Miele. 2002. Activation of Notch-1 signaling maintainsthe neoplastic phenotype in human Ras-transformed cells. Nat. Med. 8:979–986.
62. Wesley, C. S., and L. Saez. 2000. Analysis of Notch lacking the carboxylterminus identified in Drosophila embryos. J. Cell Biol. 149:683–696.
63. Wieschaus, E., and C. Nuesslein-Volhard. 1998. Looking at embryos, p.179–214. In D. B. Roberts (ed.), Drosophila, a practical approach. IRL Press,Oxford, England.
64. Yan, B., N. Rabe, and H. Plotz. 2002. HES-1, a known transcriptional re-pressor, acts as a transcriptional activator for the human acid �-glucosidasegene in human fibroblast cells. Biochem. Biophys. Res. Commun. 291:582–587.
65. Yang, Y., E. Spitzer, D. Meyer, M. Sachs, C. Niemann, G. Hartmann, K. M.Weidner, C. Birchmeier, and W. Birchmeier. 1995. Sequential requirementof hepatocyte growth factor and neuregulin in the morphogenesis and dif-ferentiation of the mammary gland. J. Cell Biol. 131:215–226.
66. Zayzafoon, M., S. A. Abdulkadir, and J. M. McDonald. 2004. Notch signalingand ERK activation are important for the osteomimetic properties of pros-tate cancer bone metastatic cell lines. J. Biol. Chem. 279:3662–3670.
3996 STELLA ET AL. MOL. CELL. BIOL.
on January 12, 2016 by guesthttp://m
cb.asm.org/
Dow
nloaded from




















