
Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1...
11
Cell Host & Microbe Article Nature and Duration of Growth Factor Signaling through Receptor Tyrosine Kinases Regulates HSV-1 Latency in Neurons Vladimir Camarena, 1,4 Mariko Kobayashi, 2 Ju Youn Kim, 2 Pamela Roehm, 3 Rosalia Perez, 1 James Gardner, 3 Angus C. Wilson, 2 Ian Mohr, 2 and Moses V. Chao 1,4,5,6,7, * 1 Molecular Neurobiology Program, Skirball Institute for Biomolecular Medicine 2 Department of Microbiology 3 Department of Otolaryngology 4 Department of Cell Biology 5 Department of Physiology and Neuroscience 6 Department of Psychiatry 7 Center for Neural Science New York University School of Medicine, 540 First Avenue, New York, NY 10016, USA *Correspondence: [email protected] DOI 10.1016/j.chom.2010.09.007 SUMMARY Herpes simplex virus-1 (HSV-1) establishes life-long latency in peripheral neurons where productive repli- cation is suppressed. While periodic reactivation results in virus production, the molecular basis of neuronal latency remains incompletely understood. Using a primary neuronal culture model of HSV-1 latency and reactivation, we show that continuous signaling through the phosphatidylinositol 3-kinase (PI3-K) pathway triggered by nerve growth factor (NGF)-binding to the TrkA receptor tyrosine kinase (RTK) is instrumental in maintaining latent HSV-1. The PI3-K p110a catalytic subunit, but not the b or d isoforms, is specifically required to activate 3-phos- phoinositide-dependent protein kinase-1 (PDK1) and sustain latency. Disrupting this pathway leads to virus reactivation. EGF and GDNF, two other growth factors capable of activating PI3-K and PDK1 but that differ from NGF in their ability to persistently acti- vate Akt, do not fully support HSV-1 latency. Thus, the nature of RTK signaling is a critical host parameter that regulates the HSV-1 latent-lytic switch. INTRODUCTION The ability of herpes simplex virus to establish and maintain a life-long latent infection in peripheral neurons is fundamental to its survival and function as a human pathogen. Classically, the latent state is defined as the absence of infectious virus production despite the presence of episomal viral genomes in neuronal nuclei. Expression of the more than 80 ORFs encoded by HSV-1 is highly restricted in latently infected neurons (Knipe and Cliffe, 2008). The exception is a latency-associated RNA transcript (LAT) that accumulates to high levels in the neuronal nucleus. Several functions have been proposed for LAT, including the ability to modulate the chromatin state of the viral episome, inhibit apoptosis, and produce microRNAs that suppress lytic gene expression (Bloom et al., 2010). Periodically, the virus changes its relationship with the neuronal host and reactivation from latency ensues, resulting in the coordinate expression of lytic genes and production of infec- tious virus that spreads back to the epithelium. A variety of conditions can promote reactivation, including exposure to UV light, stress, fever, anxiety, and nerve trauma (Cushing, 1905; Glaser and Kiecolt-Glaser, 2005; Warren et al., 1940; Wheeler, 1975). While herpes reactivation following surgery on the trigeminal ganglion was first reported over a century ago, the mechanisms underlying latency and reactivation remain largely unknown. Experiments using animal model systems have been instru- mental in understanding latency (Wagner and Bloom, 1997). In addition to defining viral genes required for reactivation, these systems have revealed important roles for components of both innate and acquired immunity in modulating viral reactivation (Knickelbein et al., 2008; Leib et al., 1989a, 1989b; Thompson et al., 2009). At its core, however, latency involves a precisely tuned interaction between the virus and host neuron. Conse- quently, the intricate details of this relationship are difficult to tease out in animal models due to the confounding influence of nonneuronal cell types and the actions of immune defenses. Instead, a detailed molecular understanding of HSV-1 latency in neurons requires a cell-culture model that utilizes a homoge- nous neuronal population that faithfully recapitulates the hall- marks of latency and reactivation. Sympathetic neurons can be cultured as a pure population of cells that depend upon trophic support from nerve growth factor (NGF) or glial-derived neurotrophic factor (GNDF) (Glebova and Ginty, 2005). Indeed, latency can be established in primary sympathetic neurons cultured in the presence of NGF (Wilcox and Johnson, 1987, 1988; Wilcox et al., 1990). This agrees with studies in latently infected rabbits showing that NGF withdrawal can induce HSV-1 reactivation in sensory and sympathetic neurons in vitro or after anti-NGF treatment in vivo (Hill et al., 1997). Importantly, NGF stimulates a range of physiological 320 Cell Host & Microbe 8, 320–330, October 21, 2010 ª2010 Elsevier Inc.
Transcript of Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1...

Cell Host & Microbe
Article
Nature and Duration of Growth Factor Signalingthrough Receptor Tyrosine KinasesRegulates HSV-1 Latency in NeuronsVladimir Camarena,1,4 Mariko Kobayashi,2 Ju Youn Kim,2 Pamela Roehm,3 Rosalia Perez,1 James Gardner,3
Angus C. Wilson,2 Ian Mohr,2 and Moses V. Chao1,4,5,6,7,*1Molecular Neurobiology Program, Skirball Institute for Biomolecular Medicine2Department of Microbiology3Department of Otolaryngology4Department of Cell Biology5Department of Physiology and Neuroscience6Department of Psychiatry7Center for Neural Science
New York University School of Medicine, 540 First Avenue, New York, NY 10016, USA
*Correspondence: [email protected]
DOI 10.1016/j.chom.2010.09.007
SUMMARY
Herpes simplex virus-1 (HSV-1) establishes life-longlatency in peripheral neurons where productive repli-cation is suppressed. While periodic reactivationresults in virus production, the molecular basis ofneuronal latency remains incompletely understood.Using a primary neuronal culture model of HSV-1latency and reactivation, we show that continuoussignaling through the phosphatidylinositol 3-kinase(PI3-K) pathway triggered by nerve growth factor(NGF)-binding to the TrkA receptor tyrosine kinase(RTK) is instrumental in maintaining latent HSV-1.The PI3-K p110a catalytic subunit, but not the b ord isoforms, is specifically required to activate 3-phos-phoinositide-dependent protein kinase-1 (PDK1) andsustain latency.Disrupting thispathway leads tovirusreactivation. EGF and GDNF, two other growthfactors capable of activating PI3-K and PDK1 butthat differ fromNGF in their ability to persistently acti-vateAkt, donot fully supportHSV-1 latency. Thus, thenature of RTK signaling is a critical host parameterthat regulates the HSV-1 latent-lytic switch.
INTRODUCTION
The ability of herpes simplex virus to establish and maintain
a life-long latent infection in peripheral neurons is fundamental
to its survival and function as a human pathogen. Classically,
the latent state is defined as the absence of infectious virus
production despite the presence of episomal viral genomes in
neuronal nuclei. Expression of the more than 80 ORFs encoded
by HSV-1 is highly restricted in latently infected neurons (Knipe
and Cliffe, 2008). The exception is a latency-associated RNA
transcript (LAT) that accumulates to high levels in the neuronal
nucleus. Several functions have been proposed for LAT,
320 Cell Host & Microbe 8, 320–330, October 21, 2010 ª2010 Elsevie
including the ability to modulate the chromatin state of the viral
episome, inhibit apoptosis, and produce microRNAs that
suppress lytic gene expression (Bloom et al., 2010).
Periodically, the virus changes its relationship with the
neuronal host and reactivation from latency ensues, resulting in
the coordinate expression of lytic genes and production of infec-
tious virus that spreads back to the epithelium. A variety of
conditions can promote reactivation, including exposure to UV
light, stress, fever, anxiety, and nerve trauma (Cushing, 1905;
Glaser and Kiecolt-Glaser, 2005; Warren et al., 1940; Wheeler,
1975). While herpes reactivation following surgery on the
trigeminal ganglion was first reported over a century ago, the
mechanisms underlying latency and reactivation remain largely
unknown.
Experiments using animal model systems have been instru-
mental in understanding latency (Wagner and Bloom, 1997). In
addition to defining viral genes required for reactivation, these
systems have revealed important roles for components of both
innate and acquired immunity in modulating viral reactivation
(Knickelbein et al., 2008; Leib et al., 1989a, 1989b; Thompson
et al., 2009). At its core, however, latency involves a precisely
tuned interaction between the virus and host neuron. Conse-
quently, the intricate details of this relationship are difficult to
tease out in animal models due to the confounding influence of
nonneuronal cell types and the actions of immune defenses.
Instead, a detailed molecular understanding of HSV-1 latency
in neurons requires a cell-culture model that utilizes a homoge-
nous neuronal population that faithfully recapitulates the hall-
marks of latency and reactivation.
Sympathetic neurons can be cultured as a pure population of
cells that depend upon trophic support from nerve growth factor
(NGF) or glial-derived neurotrophic factor (GNDF) (Glebova and
Ginty, 2005). Indeed, latency can be established in primary
sympathetic neurons cultured in the presence of NGF (Wilcox
and Johnson, 1987, 1988; Wilcox et al., 1990). This agrees with
studies in latently infected rabbits showing that NGF withdrawal
can induce HSV-1 reactivation in sensory and sympathetic
neurons in vitro or after anti-NGF treatment in vivo (Hill et al.,
1997). Importantly, NGF stimulates a range of physiological
r Inc.

Cell Host & Microbe
PI3-Kinase Signaling Regulates HSV-1 Latency
responses in neurons including but not limited to differentiation,
survival, inflammation, regeneration, cell-cycle arrest, and cell
death by interacting with multiple cell surface receptors and trig-
gering at least five independent signaling pathways. Surprisingly,
since publication of the initial reports describing NGF-dependent
latency, the specific NGF-responsive receptors and signal-
transduction pathways required to maintain latency and prevent
reactivation have not been deciphered.
Here we have developed a simple, real-time readout for reac-
tivation in living neurons and employed small-molecule chemical
inhibitors along with gene-silencing techniques to determine the
signaling components that control HSV-1 latency. Significantly,
we find that a continuous neuronal signaling program mediated
by NGF through the TrkA receptor, PI3-kinase (PI3-K) p110a iso-
form, PDK1, and Akt is required to suppress HSV productive
(lytic) growth and maintain latency. Disrupting this signaling
pathway, even transiently, using selective small molecule inhib-
itors or shRNA-mediated gene silencing resulted in efficient
reactivation. Moreover, these studies reveal that the duration
of growth factor signaling to Akt is a critical parameter regulating
latency in neurons. Specific growth factors therefore have
different abilities to support latency and suppress lytic HSV-1
replication.
RESULTS
To define the cellular requirements to sustain HSV-1 latency in
neurons, we modified a primary neuronal cell culture model for
establishing HSV-1 latency in vitro (Wilcox and Johnson, 1987,
1988), such that reactivation can be monitored in real time.
Dissociated superior cervical ganglia (SCG) neurons from E21
rat embryos were cultured with 50 ng/ml NGF in the presence
of 5-fluorouracil and aphidicolin to remove nonneuronal cells.
SCG neurons isolated in this manner resulted in sufficiently
pure populations of neurons to enable a study of virus-neuron
interactions without interference from other cell types.
Once established, these neuronal cultures were subsequently
infected with HSV-1 (Figure 1A). An otherwise wild-type HSV-1
strain expressing GFP fused to the Us11 true-late (g2) protein
served as a reporter to follow the lytic phase of the viral life cycle
and allowed reactivation to be detected in living neurons
(Benboudjema et al., 2003). Replicate wells of virus-infected
neurons were treated with acyclovir (ACV) for up to 6 days to
suppress lytic HSV-1 replication. At this point, ACV can be
removed and the infected cultures maintained for weeks without
the production of infectious virus as detected by plaque assay
(Figure 1B). Likewise, there was no detectable expression of
mRNAencoding ICP27 (Figure 1C, lanes 2 and3), a critical imme-
diate-early (IE) regulator essential for productive replication,
indicating that the virus had entered a nonreplicating state. This
was reinforced by the accumulation of LAT transcripts, which
were readily detected by RT-PCR in SCG neurons (Figure 1C),
and reproducibly found in 20% of the neuronal nuclei (three
independent experiments, n = 1500) by in situ hybridization after
ACV removal (Figure 1D). Finally, accumulation of GFP-Us11,
a reporter gene expressed late in the productive growth cycle,
was also not detected (Figure 1E, top panels). The absence of
(1) detectable infectious virus production, (2) detectable produc-
tive lytic cycle gene expression, and (3) the concurrent accumu-
Cell Hos
lation of nuclear LATs are accepted hallmarks of latency in
neurons (Knipe and Cliffe, 2008; Bloom et al., 2010).
Depletion of NGF using an anti-NGF antibody resulted in
productive viral replication (reactivation), evident from the
production of infectious virus measured 6 days after adding
anti-NGF (Figure 1B), the selective accumulation of ICP27
mRNA in GFP-positive cultures (Figure 1C), and late GFP-Us11
reporter expression, which was readily detected after 1–2 days,
and steadily increased up until day 6 (Figures 1E and 1F). LATs
were detected in all cultures even during productive viral growth
(Figure 1C), consistent with studies showing that LAT expression
is not limited to latently infected cells (Devi-Rao et al., 1991) (data
not shown). Importantly, GFP-US11 accumulation was routinely
observed in approximately 10%–20% of wells in each experi-
ment, representing a baseline level of spontaneous reactivation.
Taken together, these results indicate that NGF depletion repro-
ducibly activated expression of viral productive cycle genes in
latently infected neurons and thereby verified the reported
requirement for NGF to suppress productive replication and
maintain latency in cultured sensory neurons (Smith et al.,
1994). Activation of productive cycle lytic genes in latently in-
fected neurons, culminating in the release of infectious virus, is
the hallmark of HSV-1 reactivation from latency.
NGF withdrawal results in apoptosis of SCG neurons (Deck-
werth and Johnson, 1993), and it is conceivable that HSV-1
reactivation occurs through activation of a cell-death pathway.
To address this, a pan-caspase inhibitor, Z-VAD-fmk, was
added to the cultures prior to NGFwithdrawal. While the inhibitor
effectively prevented cell death in response to NGF depletion
under these conditions (Figure 2A), latently infected SCGs
reactivated to equivalent levels (Figure 2B). In the absence of
Z-VAD-fmk, GFP-positive cells induced by NGF withdrawal
displayed intact nuclei by Hoechst staining (data not shown).
Thus, caspase-dependent apoptosis per se was not required
for viral reactivation induced by NGFdeprivation.
Latency Requires NGF-TrkA SignalingNext we began to explore the mechanism(s) by which NGF sup-
pressed lytic replication and maintained latency. NGF interacts
with two receptors, the TrkA receptor tyrosine kinase (RTK) and
the p75 neurotrophin receptor (Figure 2C). The earlier in vitro
studies were carried out prior to the identification of TrkA as an
NGF receptor (Kaplan et al., 1991; Wilcox and Johnson, 1988;
Wilcox et al., 1990) and before the multiple NGF signaling path-
ways were defined; consequently, little information is available
on the role of individual NGF receptors in controlling HSV-1
latency. A large body of work has established that NGF signaling
through the Trk and p75 receptors is remarkably complex and
capable of triggering at least five major signaling pathways that
orchestrate diverse physiological responses (Chao, 2003).
To address the receptor requirements for NGF-dependent
latency, infected SCG cultures were treated with the pharmaco-
logical agent K252a (Figure 2D), at a concentration (200 nM) that
selectively blocks Trk receptors (Figure S1), but not other RTKs
(Berg et al., 1992). Addition of K252a resulted in reactivation
levels and kinetics similar to those observed upon NGF depletion
using anti-NGF antibody. To test whether the p75 receptor
participates in HSV-1 reactivation, cells were treated with the
anti-p75 antibody (9651), which blocks NGF binding to the
t & Microbe 8, 320–330, October 21, 2010 ª2010 Elsevier Inc. 321
A
PhaseE F
Days post-treatment
6 days
Dissect & plate SCG neurons + NGF
Infect with GFP-HSV1 + ACV
ACV removal & GFP reporter screen
6 days
anti-NGF
NGF anti-NGF
control antibodies
Us11- GFP
D
LAT Hoechst Merge NF200
20
80
40
60
1 2 3 4 5 60
6 days
10
30
50
70
% G
FP+
wel
ls
C
GFP+
LAT
ICP27
GAPDH
HSV + + + +_
1 2 3 4 5
B
anti-NGF
PFU
/Wel
l
controlantibody
1
101
210
310
410
510
GFP-
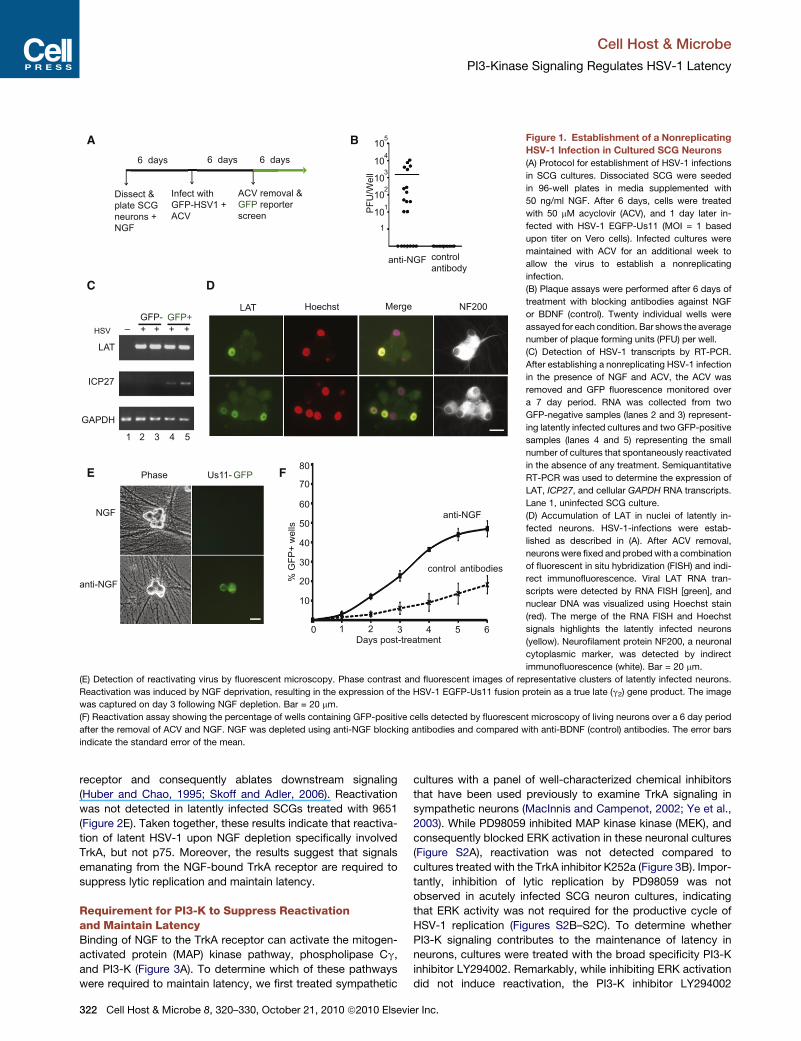
Figure 1. Establishment of a Nonreplicating
HSV-1 Infection in Cultured SCG Neurons
(A) Protocol for establishment of HSV-1 infections
in SCG cultures. Dissociated SCG were seeded
in 96-well plates in media supplemented with
50 ng/ml NGF. After 6 days, cells were treated
with 50 mM acyclovir (ACV), and 1 day later in-
fected with HSV-1 EGFP-Us11 (MOI = 1 based
upon titer on Vero cells). Infected cultures were
maintained with ACV for an additional week to
allow the virus to establish a nonreplicating
infection.
(B) Plaque assays were performed after 6 days of
treatment with blocking antibodies against NGF
or BDNF (control). Twenty individual wells were
assayed for each condition. Bar shows the average
number of plaque forming units (PFU) per well.
(C) Detection of HSV-1 transcripts by RT-PCR.
After establishing a nonreplicating HSV-1 infection
in the presence of NGF and ACV, the ACV was
removed and GFP fluorescence monitored over
a 7 day period. RNA was collected from two
GFP-negative samples (lanes 2 and 3) represent-
ing latently infected cultures and two GFP-positive
samples (lanes 4 and 5) representing the small
number of cultures that spontaneously reactivated
in the absence of any treatment. Semiquantitative
RT-PCR was used to determine the expression of
LAT, ICP27, and cellular GAPDH RNA transcripts.
Lane 1, uninfected SCG culture.
(D) Accumulation of LAT in nuclei of latently in-
fected neurons. HSV-1-infections were estab-
lished as described in (A). After ACV removal,
neurons were fixed and probedwith a combination
of fluorescent in situ hybridization (FISH) and indi-
rect immunofluorescence. Viral LAT RNA tran-
scripts were detected by RNA FISH [green], and
nuclear DNA was visualized using Hoechst stain
(red). The merge of the RNA FISH and Hoechst
signals highlights the latently infected neurons
(yellow). Neurofilament protein NF200, a neuronal
cytoplasmic marker, was detected by indirect
immunofluorescence (white). Bar = 20 mm.
(E) Detection of reactivating virus by fluorescent microscopy. Phase contrast and fluorescent images of representative clusters of latently infected neurons.
Reactivation was induced by NGF deprivation, resulting in the expression of the HSV-1 EGFP-Us11 fusion protein as a true late (g2) gene product. The image
was captured on day 3 following NGF depletion. Bar = 20 mm.
(F) Reactivation assay showing the percentage of wells containing GFP-positive cells detected by fluorescent microscopy of living neurons over a 6 day period
after the removal of ACV and NGF. NGF was depleted using anti-NGF blocking antibodies and compared with anti-BDNF (control) antibodies. The error bars
indicate the standard error of the mean.
Cell Host & Microbe
PI3-Kinase Signaling Regulates HSV-1 Latency
receptor and consequently ablates downstream signaling
(Huber and Chao, 1995; Skoff and Adler, 2006). Reactivation
was not detected in latently infected SCGs treated with 9651
(Figure 2E). Taken together, these results indicate that reactiva-
tion of latent HSV-1 upon NGF depletion specifically involved
TrkA, but not p75. Moreover, the results suggest that signals
emanating from the NGF-bound TrkA receptor are required to
suppress lytic replication and maintain latency.
Requirement for PI3-K to Suppress Reactivationand Maintain LatencyBinding of NGF to the TrkA receptor can activate the mitogen-
activated protein (MAP) kinase pathway, phospholipase Cg,
and PI3-K (Figure 3A). To determine which of these pathways
were required to maintain latency, we first treated sympathetic
322 Cell Host & Microbe 8, 320–330, October 21, 2010 ª2010 Elsevie
cultures with a panel of well-characterized chemical inhibitors
that have been used previously to examine TrkA signaling in
sympathetic neurons (MacInnis and Campenot, 2002; Ye et al.,
2003). While PD98059 inhibited MAP kinase kinase (MEK), and
consequently blocked ERK activation in these neuronal cultures
(Figure S2A), reactivation was not detected compared to
cultures treated with the TrkA inhibitor K252a (Figure 3B). Impor-
tantly, inhibition of lytic replication by PD98059 was not
observed in acutely infected SCG neuron cultures, indicating
that ERK activity was not required for the productive cycle of
HSV-1 replication (Figures S2B–S2C). To determine whether
PI3-K signaling contributes to the maintenance of latency in
neurons, cultures were treated with the broad specificity PI3-K
inhibitor LY294002. Remarkably, while inhibiting ERK activation
did not induce reactivation, the PI3-K inhibitor LY294002
r Inc.
D
DMSO
K252aanti-NGF
E
anti-NGF
IgG controlanti-p75
1 2 3 4 5 60 1 3 4 5 60
% re
activ
ated
wel
ls
% re
activ
ated
wel
ls
Days post-treatment Days post-treatment
20
80
40
60
10
30
50
70
20
80
40
60
10
30
50
70
anti-
NG
F
NG
F
anti-
NG
F+
ZVAD
0
200
50
150
100
BA
Dea
d ce
lls
anti-NGF
IgG controlIgG + ZVAD
anti-NGF + ZVAD
20
80
40
60
10
1 2 3 4 5 60%
reac
tivat
ed w
ells
Days post-treatment
30
50
70
p75
NGF
TrkA
cell cycle arrest
neurite outgrowth
C
2
γplasticity
differentiationsurvival
survival growth
κinflamation
survival
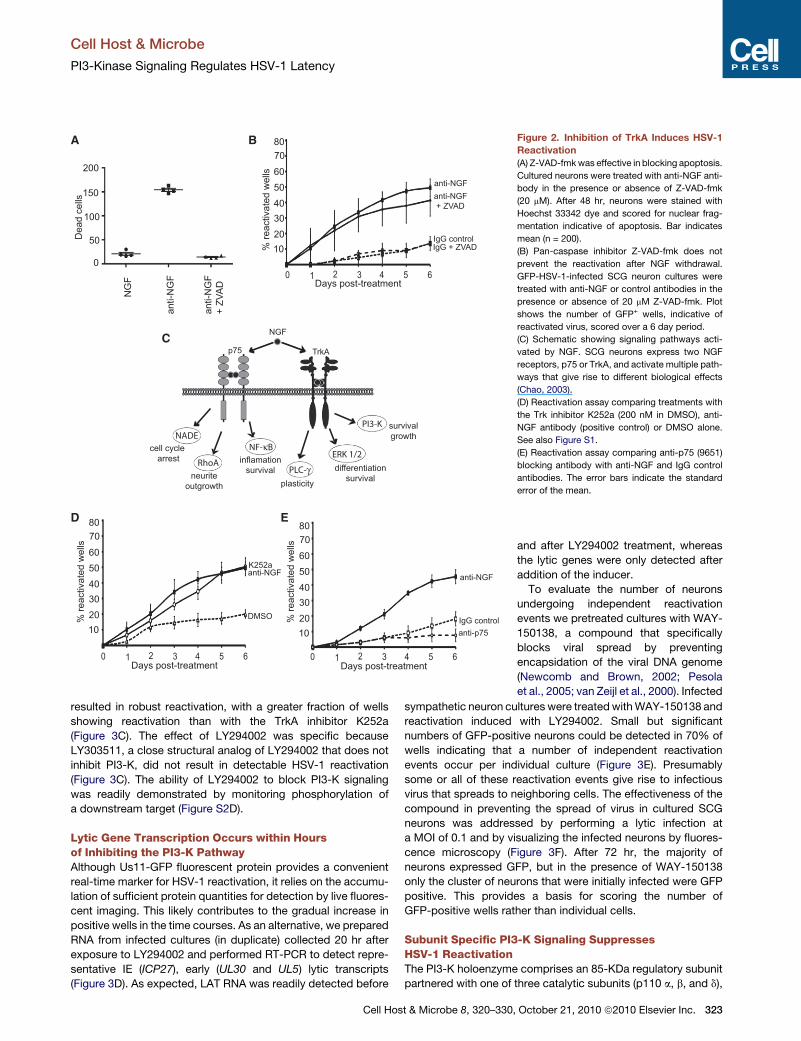
Figure 2. Inhibition of TrkA Induces HSV-1
Reactivation
(A) Z-VAD-fmkwas effective in blocking apoptosis.
Cultured neurons were treated with anti-NGF anti-
body in the presence or absence of Z-VAD-fmk
(20 mM). After 48 hr, neurons were stained with
Hoechst 33342 dye and scored for nuclear frag-
mentation indicative of apoptosis. Bar indicates
mean (n = 200).
(B) Pan-caspase inhibitor Z-VAD-fmk does not
prevent the reactivation after NGF withdrawal.
GFP-HSV-1-infected SCG neuron cultures were
treated with anti-NGF or control antibodies in the
presence or absence of 20 mM Z-VAD-fmk. Plot
shows the number of GFP+ wells, indicative of
reactivated virus, scored over a 6 day period.
(C) Schematic showing signaling pathways acti-
vated by NGF. SCG neurons express two NGF
receptors, p75 or TrkA, and activate multiple path-
ways that give rise to different biological effects
(Chao, 2003).
(D) Reactivation assay comparing treatments with
the Trk inhibitor K252a (200 nM in DMSO), anti-
NGF antibody (positive control) or DMSO alone.
See also Figure S1.
(E) Reactivation assay comparing anti-p75 (9651)
blocking antibody with anti-NGF and IgG control
antibodies. The error bars indicate the standard
error of the mean.
Cell Host & Microbe
PI3-Kinase Signaling Regulates HSV-1 Latency
resulted in robust reactivation, with a greater fraction of wells
showing reactivation than with the TrkA inhibitor K252a
(Figure 3C). The effect of LY294002 was specific because
LY303511, a close structural analog of LY294002 that does not
inhibit PI3-K, did not result in detectable HSV-1 reactivation
(Figure 3C). The ability of LY294002 to block PI3-K signaling
was readily demonstrated by monitoring phosphorylation of
a downstream target (Figure S2D).
Lytic Gene Transcription Occurs within Hoursof Inhibiting the PI3-K PathwayAlthough Us11-GFP fluorescent protein provides a convenient
real-time marker for HSV-1 reactivation, it relies on the accumu-
lation of sufficient protein quantities for detection by live fluores-
cent imaging. This likely contributes to the gradual increase in
positive wells in the time courses. As an alternative, we prepared
RNA from infected cultures (in duplicate) collected 20 hr after
exposure to LY294002 and performed RT-PCR to detect repre-
sentative IE (ICP27), early (UL30 and UL5) lytic transcripts
(Figure 3D). As expected, LAT RNA was readily detected before
Cell Host & Microbe 8, 320–330,
and after LY294002 treatment, whereas
the lytic genes were only detected after
addition of the inducer.
To evaluate the number of neurons
undergoing independent reactivation
events we pretreated cultures with WAY-
150138, a compound that specifically
blocks viral spread by preventing
encapsidation of the viral DNA genome
(Newcomb and Brown, 2002; Pesola
et al., 2005; van Zeijl et al., 2000). Infected
sympathetic neuron cultures were treatedwithWAY-150138 and
reactivation induced with LY294002. Small but significant
numbers of GFP-positive neurons could be detected in 70% of
wells indicating that a number of independent reactivation
events occur per individual culture (Figure 3E). Presumably
some or all of these reactivation events give rise to infectious
virus that spreads to neighboring cells. The effectiveness of the
compound in preventing the spread of virus in cultured SCG
neurons was addressed by performing a lytic infection at
a MOI of 0.1 and by visualizing the infected neurons by fluores-
cence microscopy (Figure 3F). After 72 hr, the majority of
neurons expressed GFP, but in the presence of WAY-150138
only the cluster of neurons that were initially infected were GFP
positive. This provides a basis for scoring the number of
GFP-positive wells rather than individual cells.
Subunit Specific PI3-K Signaling SuppressesHSV-1 ReactivationThe PI3-K holoenzyme comprises an 85-KDa regulatory subunit
partnered with one of three catalytic subunits (p110 a, b, and d),
October 21, 2010 ª2010 Elsevier Inc. 323
C
DMSO
K252a
LY294002
DMSO
K252a
PD98059
B
LY294002
DMSO
PIK-75
TGX115
IC87114
D
LY303511
LY303511
1 2 3 4 5 60
1 2 3 4 5 60
1 2 3 4 5 60
sllew detavitcaer
%
sllew detavitcaer
%
sllew detavitcaer
%
Days post-treatment
Days post-treatment
Days post-treatment
20
80
40
60
10
30
50
70
20
80
40
60
10
30
50
70
20
80
40
60
10
30
50
70
NGFTrkA
MEK1/2
ERK1/2
PLCγ PDK1
PI3-K
plasticitydifferentiation
survival
Latent HSV-1 genome
Cytoplasm
Nucleus
A
survival growth
-
LAT
ICP27
GAPDH
- + +LY294002:
UL30
UL5
DMSO LY294002(20 uM)
sllec + PFG fo .o
N
50
+ WAY-150138
100
150
200
E
DMSO
WAY-150138
>
phase GFPF G
••••••••••• •••• •••
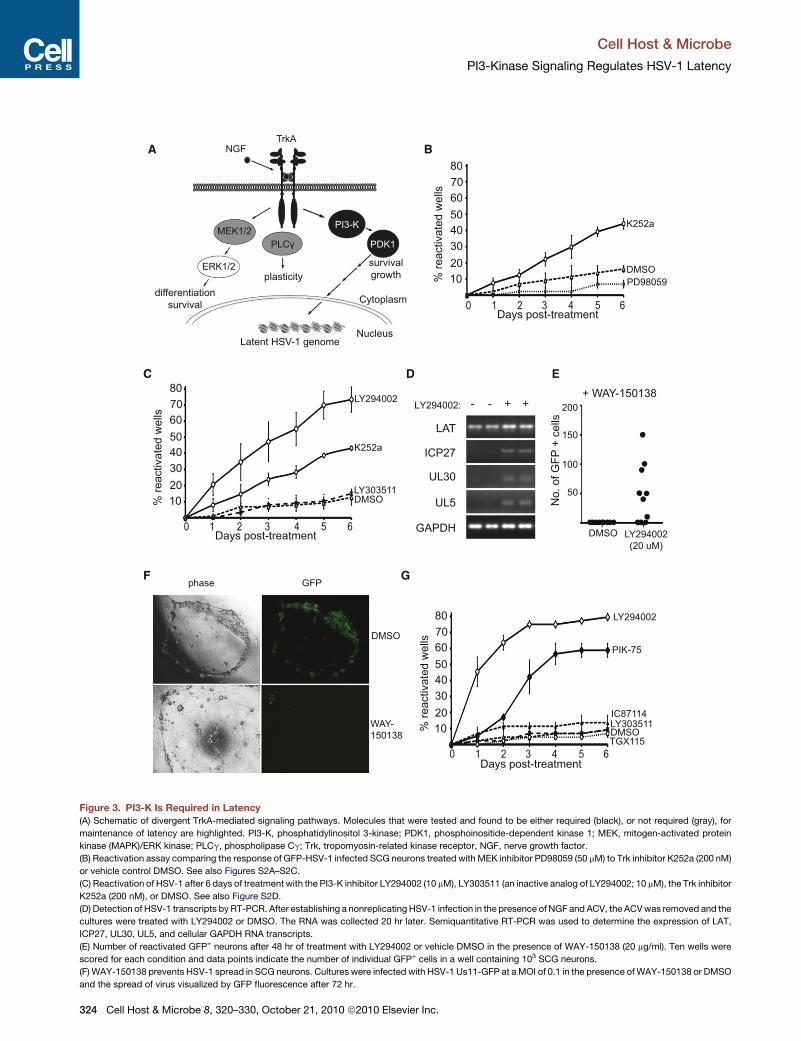
Figure 3. PI3-K Is Required in Latency
(A) Schematic of divergent TrkA-mediated signaling pathways. Molecules that were tested and found to be either required (black), or not required (gray), for
maintenance of latency are highlighted. PI3-K, phosphatidylinositol 3-kinase; PDK1, phosphoinositide-dependent kinase 1; MEK, mitogen-activated protein
kinase (MAPK)/ERK kinase; PLCg, phospholipase Cg; Trk, tropomyosin-related kinase receptor, NGF, nerve growth factor.
(B) Reactivation assay comparing the response of GFP-HSV-1 infected SCG neurons treated with MEK inhibitor PD98059 (50 mM) to Trk inhibitor K252a (200 nM)
or vehicle control DMSO. See also Figures S2A–S2C.
(C) Reactivation of HSV-1 after 6 days of treatment with the PI3-K inhibitor LY294002 (10 mM), LY303511 (an inactive analog of LY294002; 10 mM), the Trk inhibitor
K252a (200 nM), or DMSO. See also Figure S2D.
(D) Detection of HSV-1 transcripts by RT-PCR. After establishing a nonreplicating HSV-1 infection in the presence of NGF and ACV, the ACVwas removed and the
cultures were treated with LY294002 or DMSO. The RNA was collected 20 hr later. Semiquantitative RT-PCR was used to determine the expression of LAT,
ICP27, UL30, UL5, and cellular GAPDH RNA transcripts.
(E) Number of reactivated GFP+ neurons after 48 hr of treatment with LY294002 or vehicle DMSO in the presence of WAY-150138 (20 mg/ml). Ten wells were
scored for each condition and data points indicate the number of individual GFP+ cells in a well containing 103 SCG neurons.
(F) WAY-150138 prevents HSV-1 spread in SCG neurons. Cultures were infected with HSV-1 Us11-GFP at aMOI of 0.1 in the presence ofWAY-150138 or DMSO
and the spread of virus visualized by GFP fluorescence after 72 hr.
Cell Host & Microbe
PI3-Kinase Signaling Regulates HSV-1 Latency
324 Cell Host & Microbe 8, 320–330, October 21, 2010 ª2010 Elsevier Inc.

Cell Host & Microbe
PI3-Kinase Signaling Regulates HSV-1 Latency
each of which is expressed in sympathetic neurons (Bartlett
et al., 1999). LY294002 is a broad-spectrum inhibitor capable
of antagonizing all PI3-K p110 isoforms, but small molecule
inhibitors selective for each isoform have also been character-
ized (Feldman et al., 2005; Knight et al., 2006). Latently infected
cultures were treated with three of these inhibitors: TGX115,
a selective inhibitor of p110b and p110d; IC87114, selective for
p110d; and PIK75, an inhibitor of p110a. Surprisingly, treatment
with p110a-selective inhibitor PIK75 resulted in substantial reac-
tivation that was nearly as efficient as LY294002 (Figure 3G). In
contrast, treatment with the p110b and p110d inhibitors
TGX115 and IC87114 did not result in reactivation (Figure 3G).
Thus, the catalytic activity of the PI3-K p110a subunit is most
critical for maintaining latent HSV-1 in cultured sympathetic
neurons.
Depletion of PDK1 with shRNAs Resultsin HSV-1 ReactivationActivation of PI3-K stimulates phosphatidylinositol phosphoryla-
tion and leads to the recruitment of 3-phosphoinositide-depen-
dent protein kinase-1 (PDK1) to the plasma membrane. We
examined the involvement of PDK1 in maintaining latency, using
BX-795, a pyrimidine derivative that inhibits PDK1 by competing
for the ATP-binding pocket of the catalytic site (Feldman et al.,
2005). BX-795 treatment resulted in levels of reactivation similar
to those induced by LY294002 (Figure 4A). Again, inhibition
could be readily demonstrated by monitoring phosphorylation
of a downstream substrate (Figure S3).
Next, the requirement for PDK1 was confirmed using RNA
interference, an independent approach that does not rely
upon chemical inhibitors. PDK1 was depleted using shRNAs
expressed from a pLVTHM lentiviral vector (Figure 4B) that
had been modified to express mCherry, thereby allowing lentivi-
ral infection and HSV-1 reactivation to be monitored simulta-
neously in live cells. Infection with two different PDK1 shRNA
lentiviruses successfully depleted endogenous PDK1 protein
levels and significantly resulted in reactivation at levels compa-
rable to LY294002 (Figure 4C). Parallel infections with a control
lentivirus did not induce reactivation unless neurons were
treated with LY294002, confirming that coinfection with a lenti-
virus does not have a detectable effect on HSV-1 latency or
reactivation.
We also tested a lentivirus expressing shRNA to phospholi-
pase Cg (PLCg), an independent arm of TrkA signaling (Fig-
ure 3A). While PLCg levels were reduced significantly by the
shRNA (Figure 4D), no increase in HSV-1 reactivation was
detected (Figure 4E). Cultures treated with PLCg shRNAs were
still capable of reactivation in response to LY294002 (Figure 4E),
demonstrating that PLCg was not required for productive repli-
cation. Thus, loss of the PLCg from NGF-TrkA signaling is not
sufficient to reactivate latent HSV-1. This result also strengthens
the observations made with the PDK1 shRNAs by showing that
the methodology does not necessarily give rise to reactivation.
Taken together, these findings show that specifically interrupting
the PI3-K signaling pathway either by inhibiting PDK1 activity or
(G) Selective inhibition of the PI3-K catalytic subunit p110a leads to reactivation
PIK75 (PI3-K p110a inhibitor, 0.116 mM), TGX115 (PI3-K p110bd inhibitor, 2.6 mM
the standard error of the mean.
Cell Hos
by selectively depleting PDK1 protein using shRNA resulted in
efficient reactivation. Moreover, these experiments clearly
demonstrate that shRNAs can provide an effective tool to study
HSV-1 latency.
Differential Ability of Growth Factorsto Support HSV-1 LatencyNGF is not alone in its ability to bind its receptor and trigger
PI3-K-mediated signaling. Indeed, it is surprising that a relatively
ubiquitous RTK-linked signal pathway component such as PI3-K
would be involved in suppressing HSV-1 lytic replication and
maintaining latency. This raises the intriguing possibility that
other growth factors that act through the PI3-K pathway and
are expressed in SCG neurons, such as EGF and GDNF, might
also regulate HSV-1 latency.
To address this, SCG neuron cultures were established and
maintained in media containing either NGF and EGF, or NGF
and GDNF (each at 50 ng/ml). Latent HSV-1 infections were
then established in each culture and assayed for reactivation
using blocking antibodies to individual growth factors. Removal
of NGF resulted in reactivation regardless of the presence
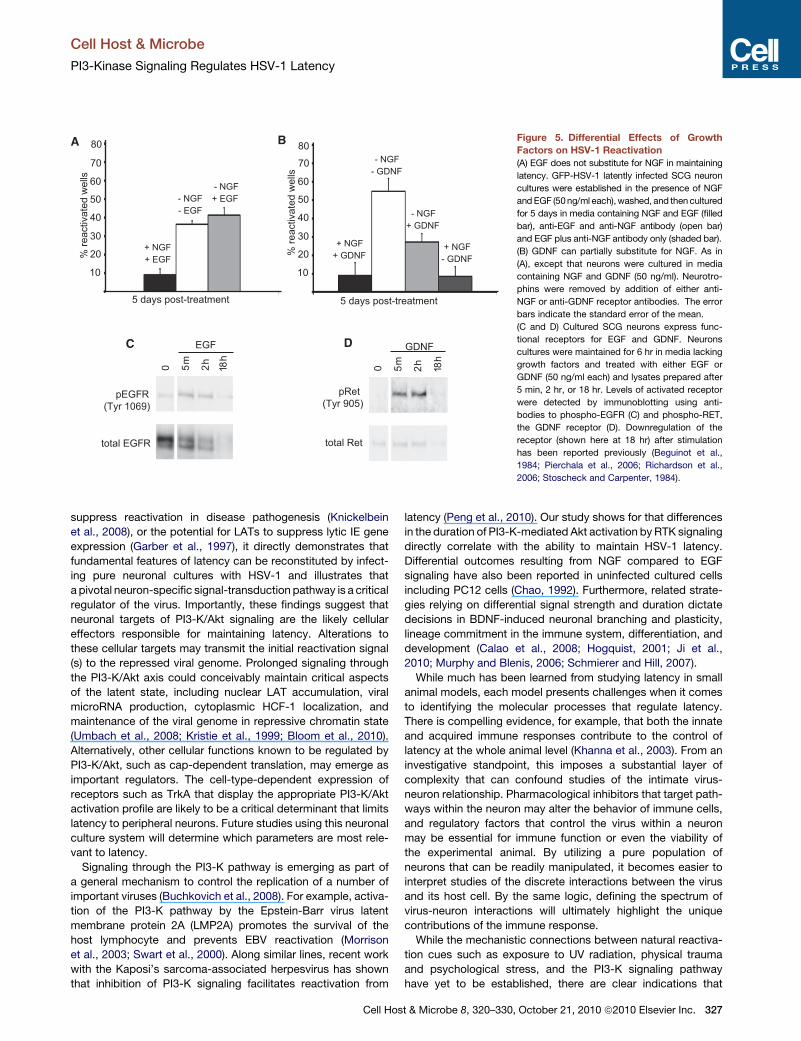
or absence of EGF (Figure 5A). In contrast, inclusion of GDNF
resulted in smaller numbers of GFP+ wells suggesting that
GDNF has some capacity to maintain latency after NGF deple-
tion (Figure 5B). Removal of both NGF and GDNF was required
to achieve maximal reactivation in cultures established and
maintained in the presence of both factors. The differential ability
of EGF and GDNF to maintain HSV-1 latency was not due to lack
of RTK activity, since both factors stimulated their respective
receptors, EGFR and c-RET (Figures 5C and 5D). Thus, despite
their ability to bind ligand and stimulate RTK signaling via a PI3K-
dependent pathway, NGF, EGF, and GDNF differed in their
ability to suppress lytic replication and maintain HSV-1 latency
in neurons.
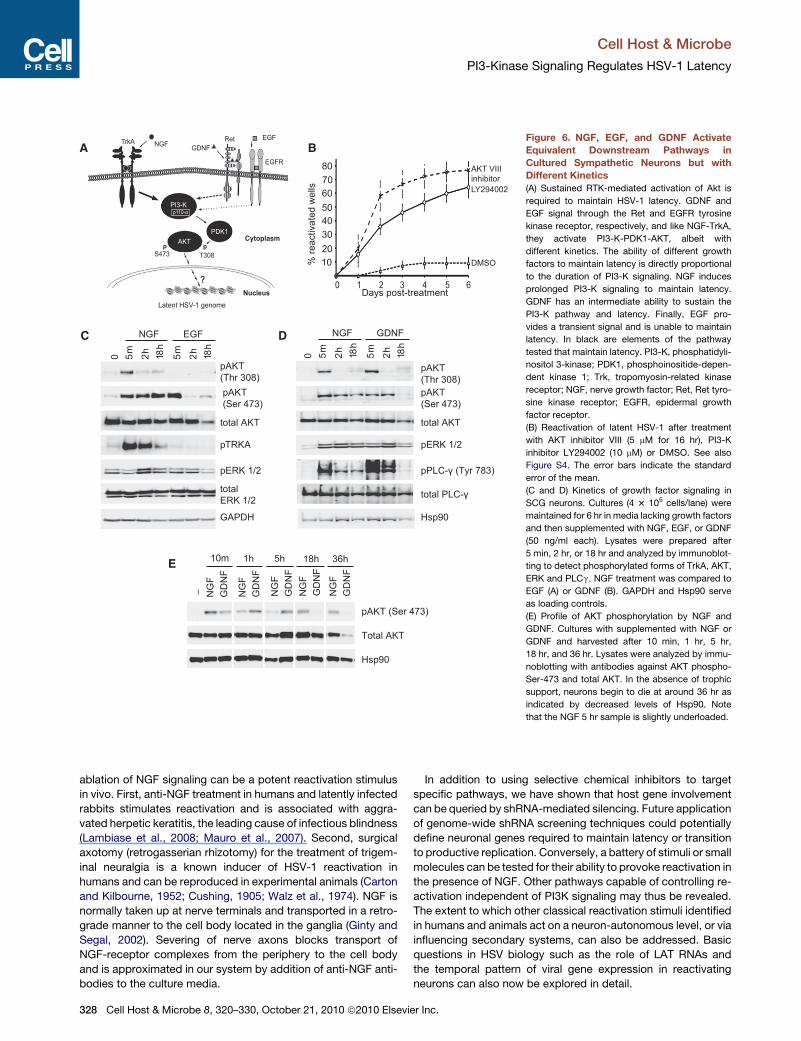
Duration of Akt Activation Is Criticalto Maintain Latency in NeuronsThe serine/threonine kinase Akt represents a key component of
the PI3-K pathway and regulates fundamental cellular processes
such as apoptosis and protein synthesis. Because Akt is a prom-
inent substrate for PDK1-mediated phosphorylation, we treated
latently infected neurons with AKT inhibitor VIII, a cell permeable
allosteric inhibitor of Akt (Calleja et al., 2009), in the presence of
NGF (Figure 6B). Treatment with the inhibitor resulted in robust
activation with 60% of wells scoring positive for GFP in 2 days.
The ability of this compound to prevent activation of Akt as
measured by phosphorylation at serine 473 was confirmed by
immunoblotting (Figure S4). This result demonstrates that activa-
tion of Akt is necessary to maintain latent HSV-1 in sympathetic
neuron cultures.
The differential ability of NGF, EGF, and GDNF to maintain
latency cannot be explained by a simple lack of receptor expres-
sion or PI3-K activity and suggests that the duration of signaling
might be more important. Therefore, the kinetics of growth-
factor signaling in sympathetic neurons was examined. We
. Infected cultures were treated with the PI3-K inhibitor LY294002, LY303511,
), IC87114 (PI3-K p110d inhibitor, 2.6 mM), or DMSO. The error bars indicate
t & Microbe 8, 320–330, October 21, 2010 ª2010 Elsevier Inc. 325
DMSO
LY294002 BX-795A
C
Control lentivirus
ShRNA PDK1(3)
Control + LY294002
ShRNA PDK1(2)
B
Con
trol
ShR
NA
PDK1
(3)
ShR
NA
PDK1
(2)
PDK1
GAPDH
% re
activ
ated
wel
ls
% re
activ
ated
wel
ls
6 days post-treatment
6 days post-ACV removal
20
80
40
60
10
30
50
70
20
80
40
60
10
30
50
70
D
ShR
NA
PLC
-gam
ma
cont
rol
PLC-γ
GAPDH
EshRNA
PLC-γ + LY294002
Control% re
activ
ated
wel
ls
5 days post ACV removal
20
80
40
60
10
30
50
70
90100
shRNA PLC-γ
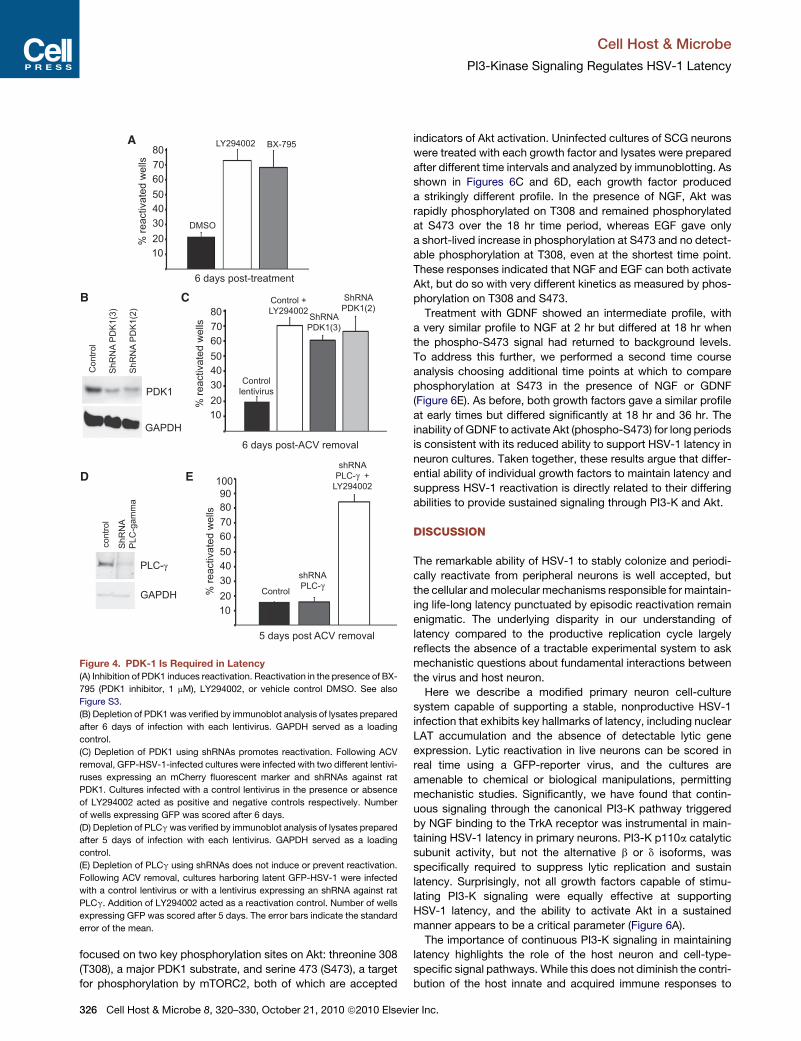
Figure 4. PDK-1 Is Required in Latency
(A) Inhibition of PDK1 induces reactivation. Reactivation in the presence of BX-
795 (PDK1 inhibitor, 1 mM), LY294002, or vehicle control DMSO. See also
Figure S3.
(B) Depletion of PDK1 was verified by immunoblot analysis of lysates prepared
after 6 days of infection with each lentivirus. GAPDH served as a loading
control.
(C) Depletion of PDK1 using shRNAs promotes reactivation. Following ACV
removal, GFP-HSV-1-infected cultures were infected with two different lentivi-
ruses expressing an mCherry fluorescent marker and shRNAs against rat
PDK1. Cultures infected with a control lentivirus in the presence or absence
of LY294002 acted as positive and negative controls respectively. Number
of wells expressing GFP was scored after 6 days.
(D) Depletion of PLCgwas verified by immunoblot analysis of lysates prepared
after 5 days of infection with each lentivirus. GAPDH served as a loading
control.
(E) Depletion of PLCg using shRNAs does not induce or prevent reactivation.
Following ACV removal, cultures harboring latent GFP-HSV-1 were infected
with a control lentivirus or with a lentivirus expressing an shRNA against rat
PLCg. Addition of LY294002 acted as a reactivation control. Number of wells
expressing GFP was scored after 5 days. The error bars indicate the standard
error of the mean.
Cell Host & Microbe
PI3-Kinase Signaling Regulates HSV-1 Latency
focused on two key phosphorylation sites on Akt: threonine 308
(T308), a major PDK1 substrate, and serine 473 (S473), a target
for phosphorylation by mTORC2, both of which are accepted
326 Cell Host & Microbe 8, 320–330, October 21, 2010 ª2010 Elsevie
indicators of Akt activation. Uninfected cultures of SCG neurons
were treated with each growth factor and lysates were prepared
after different time intervals and analyzed by immunoblotting. As
shown in Figures 6C and 6D, each growth factor produced
a strikingly different profile. In the presence of NGF, Akt was
rapidly phosphorylated on T308 and remained phosphorylated
at S473 over the 18 hr time period, whereas EGF gave only
a short-lived increase in phosphorylation at S473 and no detect-
able phosphorylation at T308, even at the shortest time point.
These responses indicated that NGF and EGF can both activate
Akt, but do so with very different kinetics as measured by phos-
phorylation on T308 and S473.
Treatment with GDNF showed an intermediate profile, with
a very similar profile to NGF at 2 hr but differed at 18 hr when
the phospho-S473 signal had returned to background levels.
To address this further, we performed a second time course
analysis choosing additional time points at which to compare
phosphorylation at S473 in the presence of NGF or GDNF
(Figure 6E). As before, both growth factors gave a similar profile
at early times but differed significantly at 18 hr and 36 hr. The
inability of GDNF to activate Akt (phospho-S473) for long periods
is consistent with its reduced ability to support HSV-1 latency in
neuron cultures. Taken together, these results argue that differ-
ential ability of individual growth factors to maintain latency and
suppress HSV-1 reactivation is directly related to their differing
abilities to provide sustained signaling through PI3-K and Akt.
DISCUSSION
The remarkable ability of HSV-1 to stably colonize and periodi-
cally reactivate from peripheral neurons is well accepted, but
the cellular andmolecular mechanisms responsible for maintain-
ing life-long latency punctuated by episodic reactivation remain
enigmatic. The underlying disparity in our understanding of
latency compared to the productive replication cycle largely
reflects the absence of a tractable experimental system to ask
mechanistic questions about fundamental interactions between
the virus and host neuron.
Here we describe a modified primary neuron cell-culture
system capable of supporting a stable, nonproductive HSV-1
infection that exhibits key hallmarks of latency, including nuclear
LAT accumulation and the absence of detectable lytic gene
expression. Lytic reactivation in live neurons can be scored in
real time using a GFP-reporter virus, and the cultures are
amenable to chemical or biological manipulations, permitting
mechanistic studies. Significantly, we have found that contin-
uous signaling through the canonical PI3-K pathway triggered
by NGF binding to the TrkA receptor was instrumental in main-
taining HSV-1 latency in primary neurons. PI3-K p110a catalytic
subunit activity, but not the alternative b or d isoforms, was
specifically required to suppress lytic replication and sustain
latency. Surprisingly, not all growth factors capable of stimu-
lating PI3-K signaling were equally effective at supporting
HSV-1 latency, and the ability to activate Akt in a sustained
manner appears to be a critical parameter (Figure 6A).
The importance of continuous PI3-K signaling in maintaining
latency highlights the role of the host neuron and cell-type-
specific signal pathways. While this does not diminish the contri-
bution of the host innate and acquired immune responses to
r Inc.
A
+ NGF+ EGF
- NGF- EGF
- NGF+ EGF
5 days post-treatment
B
+ NGF+ GDNF
- NGF+ GDNF
+ NGF- GDNF
- NGF- GDNF
pEGFR (Tyr 1069)
total EGFR
0 m5 h2
EGFC D
0 m5 h2
h81
GDNF
pRet (Tyr 905)
total Ret
5 days post-treatment
20
80
40
60
10
30
50
70
20
80
40
60
10
30
50
70
% re
activ
ated
wel
ls
% re
activ
ated
wel
ls
h81
Figure 5. Differential Effects of Growth
Factors on HSV-1 Reactivation
(A) EGF does not substitute for NGF in maintaining
latency. GFP-HSV-1 latently infected SCG neuron
cultures were established in the presence of NGF
andEGF (50ng/ml each),washed, and then cultured
for 5 days in media containing NGF and EGF (filled
bar), anti-EGF and anti-NGF antibody (open bar)
and EGF plus anti-NGF antibody only (shaded bar).
(B) GDNF can partially substitute for NGF. As in
(A), except that neurons were cultured in media
containing NGF and GDNF (50 ng/ml). Neurotro-
phins were removed by addition of either anti-
NGF or anti-GDNF receptor antibodies. The error
bars indicate the standard error of the mean.
(C and D) Cultured SCG neurons express func-
tional receptors for EGF and GDNF. Neurons
cultures were maintained for 6 hr in media lacking
growth factors and treated with either EGF or
GDNF (50 ng/ml each) and lysates prepared after
5 min, 2 hr, or 18 hr. Levels of activated receptor
were detected by immunoblotting using anti-
bodies to phospho-EGFR (C) and phospho-RET,
the GDNF receptor (D). Downregulation of the
receptor (shown here at 18 hr) after stimulation
has been reported previously (Beguinot et al.,
1984; Pierchala et al., 2006; Richardson et al.,
2006; Stoscheck and Carpenter, 1984).
Cell Host & Microbe
PI3-Kinase Signaling Regulates HSV-1 Latency
suppress reactivation in disease pathogenesis (Knickelbein
et al., 2008), or the potential for LATs to suppress lytic IE gene
expression (Garber et al., 1997), it directly demonstrates that
fundamental features of latency can be reconstituted by infect-
ing pure neuronal cultures with HSV-1 and illustrates that
a pivotal neuron-specific signal-transduction pathway is a critical
regulator of the virus. Importantly, these findings suggest that
neuronal targets of PI3-K/Akt signaling are the likely cellular
effectors responsible for maintaining latency. Alterations to
these cellular targets may transmit the initial reactivation signal
(s) to the repressed viral genome. Prolonged signaling through
the PI3-K/Akt axis could conceivably maintain critical aspects
of the latent state, including nuclear LAT accumulation, viral
microRNA production, cytoplasmic HCF-1 localization, and
maintenance of the viral genome in repressive chromatin state
(Umbach et al., 2008; Kristie et al., 1999; Bloom et al., 2010).
Alternatively, other cellular functions known to be regulated by
PI3-K/Akt, such as cap-dependent translation, may emerge as
important regulators. The cell-type-dependent expression of
receptors such as TrkA that display the appropriate PI3-K/Akt
activation profile are likely to be a critical determinant that limits
latency to peripheral neurons. Future studies using this neuronal
culture system will determine which parameters are most rele-
vant to latency.
Signaling through the PI3-K pathway is emerging as part of
a general mechanism to control the replication of a number of
important viruses (Buchkovich et al., 2008). For example, activa-
tion of the PI3-K pathway by the Epstein-Barr virus latent
membrane protein 2A (LMP2A) promotes the survival of the
host lymphocyte and prevents EBV reactivation (Morrison
et al., 2003; Swart et al., 2000). Along similar lines, recent work
with the Kaposi’s sarcoma-associated herpesvirus has shown
that inhibition of PI3-K signaling facilitates reactivation from
Cell Hos
latency (Peng et al., 2010). Our study shows for that differences
in the duration of PI3-K-mediated Akt activation by RTK signaling
directly correlate with the ability to maintain HSV-1 latency.
Differential outcomes resulting from NGF compared to EGF
signaling have also been reported in uninfected cultured cells
including PC12 cells (Chao, 1992). Furthermore, related strate-
gies relying on differential signal strength and duration dictate
decisions in BDNF-induced neuronal branching and plasticity,
lineage commitment in the immune system, differentiation, and
development (Calao et al., 2008; Hogquist, 2001; Ji et al.,
2010; Murphy and Blenis, 2006; Schmierer and Hill, 2007).
While much has been learned from studying latency in small
animal models, each model presents challenges when it comes
to identifying the molecular processes that regulate latency.
There is compelling evidence, for example, that both the innate
and acquired immune responses contribute to the control of
latency at the whole animal level (Khanna et al., 2003). From an
investigative standpoint, this imposes a substantial layer of
complexity that can confound studies of the intimate virus-
neuron relationship. Pharmacological inhibitors that target path-
ways within the neuron may alter the behavior of immune cells,
and regulatory factors that control the virus within a neuron
may be essential for immune function or even the viability of
the experimental animal. By utilizing a pure population of
neurons that can be readily manipulated, it becomes easier to
interpret studies of the discrete interactions between the virus
and its host cell. By the same logic, defining the spectrum of
virus-neuron interactions will ultimately highlight the unique
contributions of the immune response.
While the mechanistic connections between natural reactiva-
tion cues such as exposure to UV radiation, physical trauma
and psychological stress, and the PI3-K signaling pathway
have yet to be established, there are clear indications that
t & Microbe 8, 320–330, October 21, 2010 ª2010 Elsevier Inc. 327
D
0 m 5 h 2h 81 m 5 h 2h 81
NGF EGFC
pAKT (Thr 308) pAKT (Ser 473)
total AKT
pERK 1/2
total ERK 1/2
GAPDH
pTRKA pERK 1/2
total AKT
pAKT (Ser 473)
pAKT(Thr 308)
total PLC-γ
Hsp90
0 m 5 h 2h 81 m 5 h 2h 81
NGF GDNF
pPLC-γ (Tyr 783)
A NGFTrkA
Latent HSV-1 genome
Cytoplasm
Nucleus
PDK1
PI3-K
AKT
GDNFRet EGF
EGFR
S473 T308p p
p110-α
B
1 2 3 4 5 60
% re
activ
ated
wel
ls
Days post-treatment
20
80
40
60
10
30
50
70
DMSO
AKT VIII inhibitor LY294002
pAKT (Ser 473)
Hsp90
_ GD
NF
NG
F
GD
NF
NG
F
GD
NF
NG
F
GD
NF
NG
F
GD
NF
NG
F
10m 1h 5h 18h 36h
Total AKT
E
Figure 6. NGF, EGF, and GDNF Activate
Equivalent Downstream Pathways in
Cultured Sympathetic Neurons but with
Different Kinetics
(A) Sustained RTK-mediated activation of Akt is
required to maintain HSV-1 latency. GDNF and
EGF signal through the Ret and EGFR tyrosine
kinase receptor, respectively, and like NGF-TrkA,
they activate PI3-K-PDK1-AKT, albeit with
different kinetics. The ability of different growth
factors to maintain latency is directly proportional
to the duration of PI3-K signaling. NGF induces
prolonged PI3-K signaling to maintain latency.
GDNF has an intermediate ability to sustain the
PI3-K pathway and latency. Finally, EGF pro-
vides a transient signal and is unable to maintain
latency. In black are elements of the pathway
tested that maintain latency. PI3-K, phosphatidyli-
nositol 3-kinase; PDK1, phosphoinositide-depen-
dent kinase 1; Trk, tropomyosin-related kinase
receptor; NGF, nerve growth factor; Ret, Ret tyro-
sine kinase receptor; EGFR, epidermal growth
factor receptor.
(B) Reactivation of latent HSV-1 after treatment
with AKT inhibitor VIII (5 mM for 16 hr), PI3-K
inhibitor LY294002 (10 mM) or DMSO. See also
Figure S4. The error bars indicate the standard
error of the mean.
(C and D) Kinetics of growth factor signaling in
SCG neurons. Cultures (4 3 105 cells/lane) were
maintained for 6 hr in media lacking growth factors
and then supplemented with NGF, EGF, or GDNF
(50 ng/ml each). Lysates were prepared after
5 min, 2 hr, or 18 hr and analyzed by immunoblot-
ting to detect phosphorylated forms of TrkA, AKT,
ERK and PLCg. NGF treatment was compared to
EGF (A) or GDNF (B). GAPDH and Hsp90 serve
as loading controls.
(E) Profile of AKT phosphorylation by NGF and
GDNF. Cultures with supplemented with NGF or
GDNF and harvested after 10 min, 1 hr, 5 hr,
18 hr, and 36 hr. Lysates were analyzed by immu-
noblotting with antibodies against AKT phospho-
Ser-473 and total AKT. In the absence of trophic
support, neurons begin to die at around 36 hr as
indicated by decreased levels of Hsp90. Note
that the NGF 5 hr sample is slightly underloaded.
Cell Host & Microbe
PI3-Kinase Signaling Regulates HSV-1 Latency
ablation of NGF signaling can be a potent reactivation stimulus
in vivo. First, anti-NGF treatment in humans and latently infected
rabbits stimulates reactivation and is associated with aggra-
vated herpetic keratitis, the leading cause of infectious blindness
(Lambiase et al., 2008; Mauro et al., 2007). Second, surgical
axotomy (retrogasserian rhizotomy) for the treatment of trigem-
inal neuralgia is a known inducer of HSV-1 reactivation in
humans and can be reproduced in experimental animals (Carton
and Kilbourne, 1952; Cushing, 1905; Walz et al., 1974). NGF is
normally taken up at nerve terminals and transported in a retro-
grade manner to the cell body located in the ganglia (Ginty and
Segal, 2002). Severing of nerve axons blocks transport of
NGF-receptor complexes from the periphery to the cell body
and is approximated in our system by addition of anti-NGF anti-
bodies to the culture media.
328 Cell Host & Microbe 8, 320–330, October 21, 2010 ª2010 Elsevie
In addition to using selective chemical inhibitors to target
specific pathways, we have shown that host gene involvement
can be queried by shRNA-mediated silencing. Future application
of genome-wide shRNA screening techniques could potentially
define neuronal genes required to maintain latency or transition
to productive replication. Conversely, a battery of stimuli or small
molecules can be tested for their ability to provoke reactivation in
the presence of NGF. Other pathways capable of controlling re-
activation independent of PI3K signaling may thus be revealed.
The extent to which other classical reactivation stimuli identified
in humans and animals act on a neuron-autonomous level, or via
influencing secondary systems, can also be addressed. Basic
questions in HSV biology such as the role of LAT RNAs and
the temporal pattern of viral gene expression in reactivating
neurons can also now be explored in detail.
r Inc.

Cell Host & Microbe
PI3-Kinase Signaling Regulates HSV-1 Latency
EXPERIMENTAL PROCEDURES
Cell Culture and HSV-1 Infection
Superior cervical ganglia (SCG) neurons from E21 rat embryos were dissoci-
ated in trypsin (0.1%) at 37�C for 30 min. Approximately 5000 neurons per
well were plated in a 96-well plate coated with rat tail collagen (0.66 mg/ml,
08-115, Millipore). SCG neurons isolated in this manner provide a relatively
pure population of neurons expressing the TrkA receptor (Glebova and Ginty,
2005) andcontain fewnonneuronal cells. The cellsweremaintainedwith neuro-
basal media, glucose (0.4%), B27 supplement, NGF (50 ng/ml), and glutamine
(2 mM) and treated with 5-fluorouracil and aphidicolin to eliminate any dividing
cells that contaminate the cultures. After 6 days, the cells were pretreated with
acyclovir, (ACV, 50mM) for 20 hr, and subsequently infectedwithHSV-1 (Patton
strain GFP-Us11; multiplicity of infection (MOI) = 1 based upon titer on Vero
cells) for 2 hr in the presence of ACV to block productive HSV-1 replication.
Neurons were maintained in ACV for at least 6 days. After ACV removal,
infected neuronal cultures were exposed to different reactivation stimuli. In
an experiment, 22 independently infected wells were analyzed per individual
stimulus. Graphs summarize a minimum of three separate experiments and
error bars indicate the standard error of the mean.
RT-PCR
RNA was isolated from approximately 30,000 latently infected neurons and
analyzed by standard methodologies. The primer sequences are posted in
the supplementary section.
Combined Fluorescent In Situ Hybridization
and Indirect Immunofluorescence
Cells were cultured and infected with HSV-1 (GFP-Us11) as described above
but plated onto 8-well chamber slides at a density of �2 3 104 neurons/
chamber. In situ hybridization was performed by adding a mix containing
four LAT probes (200 nM each) for 5 hr at 42�C. LAT-specific oligonucleotides
were designed against the�2 kb intron region of HSV-1 strain 17 (Farrell et al.,
1991), and were synthesized with a fluorescein tag on the 50 end. All subse-quent incubations for immunofluorescence (IF) were done at RT. Additional
details can be found in the supplement.
shRNA Lentivirus Infection
Lentiviruses expressing shRNAs against rat PDK1 and rat PLCg were gener-
ated using a pLVTHM vector that included an mCherry expression cassette.
SCG cultures were infected with lentivirus (MOI = 5) for 12 hr prior infection
with HSV-1. The efficiency of lentiviral infection as judged by mCherry expres-
sion was approximately 90%. The shRNA sequences are posted in the supple-
mentary section.
Plaque Assay
Cell lysates were prepared from neuron cultures by freeze thawing, and the
amount of infectious virus determined by plaque assay using Vero cells and
serial dilutions of each lysate.
Viability Assay by Hoechst Staining
One week after plating, SCG cultures were treated with anti-NGF blocking
antibodies in the presence or absence of 20 mM ZVAD-fmk (627610, Calbio-
chem). After 48 hr, cells were stained with Hoechst 33342 (Molecular Probes)
and visualized by light microscopy. The number of apoptotic nuclei was deter-
mined by counting 200 cells.
Additional materials and methods can be found in the Supplemental
Information.
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures, Supplemental Experimental
Procedures, and references and can be found with this article online at
doi:10.1016/j.chom.2010.09.007.
Cell Hos
ACKNOWLEDGMENTS
We thank Kevan Shokat for the specific PI3K inhibitors, Josh Bloom (Wyeth)
for WAY-150138 and Eugene Johnson for advice. This work was supported by
grants to MVC (NS21072, HD23315), ACW (GM61139, S10RR017970) and IM
(AI073898, GM056927) from the NIH.
Received: March 9, 2010
Revised: July 10, 2010
Accepted: August 20, 2010
Published: October 20, 2010
REFERENCES
Bartlett, S.E., Reynolds, A.J., Tan, T., Heydon, K., and Hendry, I.A. (1999).
Differential mRNA expression and subcellular locations of PI3-kinase isoforms
in sympathetic and sensory neurons. J. Neurosci. Res. 56, 44–53.
Beguinot, L., Lyall, R.M., Willingham, M.C., and Pastan, I. (1984). Down-regu-
lation of the epidermal growth factor receptor in KB cells is due to receptor
internalization and subsequent degradation in lysosomes. Proc. Natl. Acad.
Sci. USA 81, 2384–2388.
Benboudjema, L., Mulvey, M., Gao, Y., Pimplikar, S.W., and Mohr, I. (2003).
Association of the herpes simplex virus type 1 Us11 gene product with the
cellular kinesin light-chain-related protein PAT1 results in the redistribution
of both polypeptides. J. Virol. 77, 9192–9203.
Berg, M.M., Sternberg, D.W., Parada, L.F., and Chao, M.V. (1992). K-252a
inhibits nerve growth factor-induced trk proto-oncogene tyrosine phosphory-
lation and kinase activity. J. Biol. Chem. 267, 13–16.
Bloom, D.C., Giordani, N.V., and Kwiatkowski, D.L. (2010). Epigenetic regula-
tion of latent HSV-1 gene expression. Biochim. Biophys. Acta 1799, 246–256.
Buchkovich, N.J., Yu, Y., Zampieri, C.A., and Alwine, J.C. (2008). The TORrid
affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR
signalling pathway. Nat. Rev. Microbiol. 6, 266–275.
Calao, M., Burny, A., Quivy, V., Dekoninck, A., and Van Lint, C. (2008). A perva-
sive role of histone acetyltransferases and deacetylases in an NF-kappaB-
signaling code. Trends Biochem. Sci. 33, 339–349.
Calleja, V., Laguerre, M., Parker, P.J., and Larijani, B. (2009). Role of a novel
PH-kinase domain interface in PKB/Akt regulation: structural mechanism for
allosteric inhibition. PLoS Biol. 7, e17.
Carton, C.A., and Kilbourne, E.D. (1952). Activation of latent herpes simplex by
trigeminal sensory-root section. N. Engl. J. Med. 246, 172–176.
Chao, M.V. (1992). Growth factor signaling: where is the specificity? Cell 68,
995–997.
Chao, M.V. (2003). Neurotrophins and their receptors: a convergence point for
many signalling pathways. Nat. Rev. Neurosci. 4, 299–309.
Cushing, H. (1905). The surgical aspects of major neuralgia of the trigeminal
nerve. J. Am. Med. Assoc. XLIV, 773–779.
Deckwerth, T.L., and Johnson, E.M., Jr. (1993). Temporal analysis of events
associated with programmed cell death (apoptosis) of sympathetic neurons
deprived of nerve growth factor. J. Cell Biol. 123, 1207–1222.
Devi-Rao, G.B., Goodart, S.A., Hecht, L.M., Rochford, R., Rice, M.K., and
Wagner, E.K. (1991). Relationship between polyadenylated and nonpolyade-
nylated herpes simplex virus type 1 latency-associated transcripts. J. Virol.
65, 2179–2190.
Farrell, M.J., Dobson, A.T., and Feldman, L.T. (1991). Herpes simplex virus
latency-associated transcript is a stable intron. Proc. Natl. Acad. Sci. USA
88, 790–794.
Feldman, R.I., Wu, J.M., Polokoff, M.A., Kochanny, M.J., Dinter, H., Zhu, D.,
Biroc, S.L., Alicke, B., Bryant, J., Yuan, S., et al. (2005). Novel small molecule
inhibitors of 3-phosphoinositide-dependent kinase-1. J. Biol. Chem. 280,
19867–19874.
Garber, D.A., Schaffer, P.A., and Knipe, D.M. (1997). A LAT-associated
function reduces productive-cycle gene expression during acute infection
t & Microbe 8, 320–330, October 21, 2010 ª2010 Elsevier Inc. 329

Cell Host & Microbe
PI3-Kinase Signaling Regulates HSV-1 Latency
of murine sensory neurons with herpes simplex virus type 1. J. Virol. 71, 5885–
5893.
Ginty, D.D., and Segal, R.A. (2002). Retrograde neurotrophin signaling: Trk-ing
along the axon. Curr. Opin. Neurobiol. 12, 268–274.
Glaser, R., and Kiecolt-Glaser, J.K. (2005). Stress-induced immune dysfunc-
tion: implications for health. Nat. Rev. Immunol. 5, 243–251.
Glebova, N.O., and Ginty, D.D. (2005). Growth and survival signals controlling
sympathetic nervous system development. Annu. Rev. Neurosci. 28, 191–222.
Hill, J.M., Garza, H.H., Jr., Helmy, M.F., Cook, S.D., Osborne, P.A., Johnson,
E.M., Jr., Thompson, H.W., Green, L.C., O’Callaghan, R.J., and Gebhardt,
B.M. (1997). Nerve growth factor antibody stimulates reactivation of ocular
herpes simplex virus type 1 in latently infected rabbits. J. Neurovirol. 3,
206–211.
Hogquist, K.A. (2001). Signal strength in thymic selection and lineage commit-
ment. Curr. Opin. Immunol. 13, 225–231.
Huber, L.J., and Chao, M.V. (1995). A potential interaction of p75 and trkA NGF
receptors revealed by affinity crosslinking and immunoprecipitation. J. Neuro-
sci. Res. 40, 557–563.
Ji, Y., Lu, Y., Yang, F., Shen, W., Tang, T.T., Feng, L., Duan, S., and Lu, B.
(2010). Acute and gradual increases in BDNF concentration elicit distinct
signaling and functions in neurons. Nat. Neurosci. 13, 302–309.
Kaplan, D.R., Hempstead, B.L., Martin-Zanca, D., Chao, M.V., and Parada,
L.F. (1991). The trk proto-oncogene product: a signal transducing receptor
for nerve growth factor. Science 252, 554–558.
Khanna, K.M., Bonneau, R.H., Kinchington, P.R., and Hendricks, R.L. (2003).
Herpes simplex virus-specific memory CD8+ T cells are selectively activated
and retained in latently infected sensory ganglia. Immunity 18, 593–603.
Knickelbein, J.E., Khanna, K.M., Yee, M.B., Baty, C.J., Kinchington, P.R., and
Hendricks, R.L. (2008). Noncytotoxic lytic granule-mediated CD8+ T cell inhi-
bition of HSV-1 reactivation from neuronal latency. Science 322, 268–271.
Knight, Z.A., Gonzalez, B., Feldman, M.E., Zunder, E.R., Goldenberg, D.D.,
Williams, O., Loewith, R., Stokoe, D., Balla, A., Toth, B., et al. (2006). A phar-
macological map of the PI3-K family defines a role for p110alpha in insulin
signaling. Cell 125, 733–747.
Knipe, D.M., and Cliffe, A. (2008). Chromatin control of herpes simplex virus
lytic and latent infection. Nat. Rev. Microbiol. 6, 211–221.
Kristie, T.M., Vogel, J.L., and Sears, A.E. (1999). Nuclear localization of the C1
factor (host cell factor) in sensory neurons correlates with reactivation of
herpes simplex virus from latency. Proc. Natl. Acad. Sci. USA 96, 1229–1233.
Lambiase, A., Coassin, M., Costa, N., Lauretti, P., Micera, A., Ghinelli, E., Aloe,
L., Rama, P., and Bonini, S. (2008). Topical treatment with nerve growth factor
in an animal model of herpetic keratitis. Graefes Arch. Clin. Exp. Ophthalmol.
246, 121–127.
Leib, D.A., Bogard, C.L., Kosz-Vnenchak, M., Hicks, K.A., Coen, D.M., Knipe,
D.M., and Schaffer, P.A. (1989a). A deletion mutant of the latency-associated
transcript of herpes simplex virus type 1 reactivates from the latent state with
reduced frequency. J. Virol. 63, 2893–2900.
Leib, D.A., Coen, D.M., Bogard, C.L., Hicks, K.A., Yager, D.R., Knipe, D.M.,
Tyler, K.L., and Schaffer, P.A. (1989b). Immediate-early regulatory gene
mutants define different stages in the establishment and reactivation of herpes
simplex virus latency. J. Virol. 63, 759–768.
MacInnis, B.L., and Campenot, R.B. (2002). Retrograde support of neuronal
survival without retrograde transport of nerve growth factor. Science 295,
1536–1539.
Mauro, C., Pietro, L., and Emilio, C.C. (2007). The use of nerve growth factor in
herpetic keratitis: a case report. J. Med. Case Reports 1, 124.
Morrison, J.A., Klingelhutz, A.J., and Raab-Traub, N. (2003). Epstein-Barr virus
latent membrane protein 2A activates beta-catenin signaling in epithelial cells.
J. Virol. 77, 12276–12284.
Murphy, L.O., and Blenis, J. (2006). MAPK signal specificity: the right place at
the right time. Trends Biochem. Sci. 31, 268–275.
330 Cell Host & Microbe 8, 320–330, October 21, 2010 ª2010 Elsevie
Newcomb, W.W., and Brown, J.C. (2002). Inhibition of herpes simplex virus
replication by WAY-150138: assembly of capsids depleted of the portal and
terminase proteins involved in DNA encapsidation. J. Virol. 76, 10084–10088.
Peng, L., Wu, T.T., Tchieu, J.H., Feng, J., Brown, H.J., Feng, J., Li, X., Qi, J.,
Deng, H., Vivanco, I., et al. (2010). Inhibition of the phosphatidylinositol
3-kinase-Akt pathway enhances gamma-2 herpesvirus lytic replication and
facilitates reactivation from latency. J. Gen. Virol. 91, 463–469.
Pesola, J.M., Zhu, J., Knipe, D.M., and Coen, D.M. (2005). Herpes simplex
virus 1 immediate-early and early gene expression during reactivation from
latency under conditions that prevent infectious virus production. J. Virol.
79, 14516–14525.
Pierchala, B.A., Milbrandt, J., and Johnson, E.M., Jr. (2006). Glial cell line-
derived neurotrophic factor-dependent recruitment of Ret into lipid rafts
enhances signaling by partitioning Ret from proteasome-dependent degrada-
tion. J. Neurosci. 26, 2777–2787.
Richardson, D.S., Lai, A.Z., and Mulligan, L.M. (2006). RET ligand-induced
internalization and its consequences for downstream signaling. Oncogene
25, 3206–3211.
Schmierer, B., andHill, C.S. (2007). TGFbeta-SMADsignal transduction:molec-
ular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 8, 970–982.
Skoff, A.M., and Adler, J.E. (2006). Nerve growth factor regulates substance P
in adult sensory neurons through both TrkA and p75 receptors. Exp. Neurol.
197, 430–436.
Smith, R.L., Escudero, J.M., and Wilcox, C.L. (1994). Regulation of the herpes
simplex virus latency-associated transcripts during establishment of latency in
sensory neurons in vitro. Virology 202, 49–60.
Stoscheck, C.M., and Carpenter, G. (1984). Down regulation of epidermal
growth factor receptors: direct demonstration of receptor degradation in
human fibroblasts. J. Cell Biol. 98, 1048–1053.
Swart, R., Ruf, I.K., Sample, J., and Longnecker, R. (2000). Latent membrane
protein 2A-mediated effects on the phosphatidylinositol 3-Kinase/Akt
pathway. J. Virol. 74, 10838–10845.
Thompson, R.L., Preston, C.M., and Sawtell, N.M. (2009). De novo synthesis of
VP16 coordinates the exit fromHSV latency in vivo. PLoSPathog. 5, e1000352.
Umbach, J.L., Kramer, M.F., Jurak, I., Karnowski, H.W., Coen, D.M., and
Cullen, B.R. (2008). MicroRNAs expressed by herpes simplex virus 1 during
latent infection regulate viral mRNAs. Nature 454, 780–783.
van Zeijl, M., Fairhurst, J., Jones, T.R., Vernon, S.K., Morin, J., LaRocque, J.,
Feld, B., O’Hara, B., Bloom, J.D., and Johann, S.V. (2000). Novel class of thio-
urea compounds that inhibit herpes simplex virus type 1 DNA cleavage and
encapsidation: resistance maps to the UL6 gene. J. Virol. 74, 9054–9061.
Wagner, E.K., and Bloom, D.C. (1997). Experimental investigation of herpes
simplex virus latency. Clin. Microbiol. Rev. 10, 419–443.
Walz, M.A., Price, R.W., and Notkins, A.L. (1974). Latent ganglionic infection
with herpes simplex virus types 1 and 2: viral reactivation in vivo after neurec-
tomy. Science 184, 1185–1187.
Warren, S.L., Carpenter, C.M., and Boak, R.A. (1940). Symptomatic herpes,
a sequela of artificially induced fever: incidence and C aspects; recovery of
a virus from herpetic vesicles, and comparison with a K strain of herpes virus.
J. Exp. Med. 71, 155–168.
Wheeler, C.E., Jr. (1975). Pathogenesis of recurrent herpes simplex infections.
J. Invest. Dermatol. 65, 341–346.
Wilcox, C.L.J., and Johnson, E.M., Jr. (1987). Nerve growth factor deprivation
results in the reactivation of latent herpes simplex virus in vitro. J. Virol. 61,
2311–2315.
Wilcox, C.L., and Johnson, E.M., Jr. (1988). Characterization of nerve growth
factor-dependent herpes simplex virus latency in neurons in vitro. J. Virol.
62, 393–399.
Wilcox, C.L., Smith, R.L., Freed, C.R., and Johnson, E.M., Jr. (1990). Nerve
growth factor-dependence of herpes simplex virus latency in peripheral
sympathetic and sensory neurons in vitro. J. Neurosci. 10, 1268–1275.
Ye, H., Kuruvilla, R., Zweifel, L.S., and Ginty, D.D. (2003). Evidence in support
of signalling endosome-based retrograde survival of sympathetic neurons.
Neuron 39, 57–68.
r Inc.




















