
Mycobacterium tuberculosis Hsp16.3 Nonamers are Assembled and Re-assembled via Trimer and Hexamer...
11
Mycobacterium tuberculosis Hsp16.3 Nonamers are Assembled and Re-assembled via Trimer and Hexamer Intermediates Abuduaini Abulimiti 1,2,3 †, Xinmiao Fu 1,2,3 †, Liangcai Gu 1,2,3 Xiuguang Feng 1,2,3 and Zengyi Chang 1,2,3 * 1 Department of Biological Science and Biotechnology School of Life Science, Tsinghua University, Beijing 100084 People’s Republic of China 2 Department of Biochemistry and Molecular Biology, School of Medicine, Tsinghua University, Beijing 100084 People’s Republic of China 3 The Protein Science Laboratory of the Education Ministry Beijing 100084, People’s Republic of China Hsp16.3, a small heat shock protein from Mycobacterium tuberculosis pro- posed to form specific trimer-of-trimers structures, acts as a molecular chaperone in vitro. The assembly and re-assembly mechanisms of this oligomeric protein were studied and compared using in vitro transcrip- tion/translation and denaturization/renaturization systems. Analysis using a combination of non-denaturing pore gradient polyacrylamide gel electrophoresis, chemical cross-linking, and size-exclusion chromato- graphy demonstrate that the predominant form of Hsp16.3 produced in the in vitro transcription/translation system is the trimer, which can be further assembled into a nonameric structure via a hexamer intermediate in the presence of purified exogenous Hsp16.3 proteins. Meanwhile, an “inert” Hsp16.3 dimer, which does not seem to participate in nonamer assembly but may be involved in forming other forms of Hsp16.3, was also detected in the in vitro expression system. On the other hand, our current data clearly show that the re-assembly of Hsp16.3 nonamers also occurs via a very similar mechanism, with the formation of trimers and hexamers. The presence of high levels of macromolecular crowding pro- tein agent in the in vitro expression system promoted the formation of the nonamers to a very limited degree, indicating that the assembly of proteins like Hsp16.3 may depend mainly on its own concentration instead of those of the macromolecules in the environment. q 2003 Elsevier Science Ltd. All rights reserved Keywords: small heat shock protein16.3; in vitro transcription/translation assembly/re-assembly; subunit exchange; macromolecular crowding *Corresponding author Introduction A majority of proteins in modern cells exist as oligomers, among which many are homo- oligomers. 1–3 Such oligomerization of protein sub- units is generally believed to be needed for protein stability and/or regulation. 1,2 To assess the oligo- merization process, the re-assembly, instead of assembly, of such proteins has often been investi- gated by performing denaturization/renaturiza- tion experiments on purified proteins. 4,5 A literature search indicates that not even the re-assembly process of oligomeric proteins has been thoroughly studied, let alone the assembly process. 6 Small heat shock proteins (sHsps) are a family of proteins that form characteristic oligomeric struc- tures having various numbers of subunits. 7 Dimeric forms are often found to be the basic struc- tural unit of many small heat shock proteins. 8–10 Recent studies by Delmas and co-workers indi- cated that the trimeric species could be the sub- structures of the dodecamerric Lo18 protein (the sHsp from Oenococcus oeni). 11 Being molecular chaperones by themselves, sHsps have been found to be able to effectively refold and reassemble in vitro under optimal conditions after being denatured. 4,12 Hsp16.3, originally identified as an immuno- dominant antigen and later found to be a major 0022-2836/03/$ - see front matter q 2003 Elsevier Science Ltd. All rights reserved † These two authors contributed equally to this work. E-mail address of the corresponding author: [email protected] Abbreviations used: BSA, bovine serum albumin; sHsp, small heat shock protein; DTSSP, 3,3 0 -dithiobis (sulfosuccinimidyl) propionate; PBS, phosphate-buffered salinee. doi:10.1016/S0022-2836(03)00018-4 J. Mol. Biol. (2003) 326, 1013–1023
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Mycobacterium tuberculosis Hsp16.3 Nonamers are Assembled and Re-assembled via Trimer and Hexamer...

Mycobacterium tuberculosis Hsp16.3 Nonamers areAssembled and Re-assembled via Trimer andHexamer Intermediates
Abuduaini Abulimiti1,2,3†, Xinmiao Fu1,2,3†, Liangcai Gu1,2,3
Xiuguang Feng1,2,3 and Zengyi Chang1,2,3*
1Department of BiologicalScience and BiotechnologySchool of Life Science, TsinghuaUniversity, Beijing 100084People’s Republic of China
2Department of Biochemistryand Molecular Biology, Schoolof Medicine, TsinghuaUniversity, Beijing 100084People’s Republic of China
3The Protein Science Laboratoryof the Education MinistryBeijing 100084, People’sRepublic of China
Hsp16.3, a small heat shock protein from Mycobacterium tuberculosis pro-posed to form specific trimer-of-trimers structures, acts as a molecularchaperone in vitro. The assembly and re-assembly mechanisms of thisoligomeric protein were studied and compared using in vitro transcrip-tion/translation and denaturization/renaturization systems. Analysisusing a combination of non-denaturing pore gradient polyacrylamide gelelectrophoresis, chemical cross-linking, and size-exclusion chromato-graphy demonstrate that the predominant form of Hsp16.3 produced inthe in vitro transcription/translation system is the trimer, which can befurther assembled into a nonameric structure via a hexamer intermediatein the presence of purified exogenous Hsp16.3 proteins. Meanwhile, an“inert” Hsp16.3 dimer, which does not seem to participate in nonamerassembly but may be involved in forming other forms of Hsp16.3, wasalso detected in the in vitro expression system. On the other hand, ourcurrent data clearly show that the re-assembly of Hsp16.3 nonamers alsooccurs via a very similar mechanism, with the formation of trimers andhexamers. The presence of high levels of macromolecular crowding pro-tein agent in the in vitro expression system promoted the formation ofthe nonamers to a very limited degree, indicating that the assembly ofproteins like Hsp16.3 may depend mainly on its own concentrationinstead of those of the macromolecules in the environment.
q 2003 Elsevier Science Ltd. All rights reserved
Keywords: small heat shock protein16.3; in vitro transcription/translationassembly/re-assembly; subunit exchange; macromolecular crowding*Corresponding author
Introduction
A majority of proteins in modern cells exist asoligomers, among which many are homo-oligomers.1 – 3 Such oligomerization of protein sub-units is generally believed to be needed for proteinstability and/or regulation.1,2 To assess the oligo-merization process, the re-assembly, instead ofassembly, of such proteins has often been investi-gated by performing denaturization/renaturiza-tion experiments on purified proteins.4,5 A
literature search indicates that not even there-assembly process of oligomeric proteins hasbeen thoroughly studied, let alone the assemblyprocess.6
Small heat shock proteins (sHsps) are a family ofproteins that form characteristic oligomeric struc-tures having various numbers of subunits.7
Dimeric forms are often found to be the basic struc-tural unit of many small heat shock proteins.8 – 10
Recent studies by Delmas and co-workers indi-cated that the trimeric species could be the sub-structures of the dodecamerric Lo18 protein (thesHsp from Oenococcus oeni).11 Being molecularchaperones by themselves, sHsps have been foundto be able to effectively refold and reassemble invitro under optimal conditions after beingdenatured.4,12
Hsp16.3, originally identified as an immuno-dominant antigen and later found to be a major
0022-2836/03/$ - see front matter q 2003 Elsevier Science Ltd. All rights reserved
† These two authors contributed equally to this work.
E-mail address of the corresponding author:[email protected]
Abbreviations used: BSA, bovine serum albumin;sHsp, small heat shock protein; DTSSP, 3,30-dithiobis(sulfosuccinimidyl) propionate; PBS, phosphate-bufferedsalinee.
doi:10.1016/S0022-2836(03)00018-4 J. Mol. Biol. (2003) 326, 1013–1023
membrane protein,13,14 is the small heat shockprotein from Mycobacterium tuberculosis. Therecombinant Hsp16.3 protein was found to existas a nonamer, forming the specific trimer-of-trimers structure.15,16 This oligomeric protein wasrevealed to be able to suppress the aggregation ofdenaturing proteins, thus having chaperone-likeactivity in vitro.4,12,15,17 – 19 Our previous studiesdemonstrated that the Hsp16.3 protein possessesdynamic dissociation and re-association ability.20
A stepwise model has been proposed to accountfor the re-assembly process of this protein in vitro.4
Whether such a stepwise re-assembly mechanismcan be extended to the assembly process ofHsp16.3 (newly synthesized from the ribosomes)is examined in the studies reported here, using anin vitro transcription/translation system. There arevaluable advantages of using such in vitroexpression system to study protein folding andthe assembly mechanism of polypeptide chains,although it is often more difficult to perform.21,22
Recently, the effect of macromolecular crowdingagents on protein refolding and re-assembly hasbeen investigated on a few proteins.5,23 Neverthe-less, no studies have been reported on such acrowding effect on protein folding and assemblyof polypeptides newly synthesized from ribosomesas occurs in the in vitro transcription/translationsystem.
The data reported here strongly suggest that theHsp16.3 protein that is newly synthesized fromthe ribosomes assembles into the nonamer form
by forming trimer and hexamer intermediates.Based on this observation, the re-assembly processof this protein was re-examined using chemicalcross-linking techniques, and indeed trimers andhexamers and nonamers were found to be themost dominant form of cross-linked Hsp16.3proteins during the quick dilution/reassemblyprocess. The significance of the previous in vitrore-assembly results that led to the proposal of thestepwise re-assembly model is discussed. Inaddition to the trimers, hexamers, and nonamers,a relatively inert dimer form was also repeatedlyrevealed when the Hsp16.3 protein was expressedusing the in vitro transcription/translation system.The presence of high concentrations of BSA in thein vitro expression system acting as the crowdingagent was found to significantly enhance theconversion of trimers to nonamers.
Results
The Hsp16.3 protein produced in theEscherichia coli in vitro transcription/translation system exists mainly as trimers
The native size of Hsp16.3 protein produced inthe E. coli cell-free system was first analyzed usingnon-denaturing pore gradient PAGE and size-exclusion chromatography. The protein, appearingafter one minute of reaction (at 37 8C) apparentlyexisted mainly as trimers, with a small portion
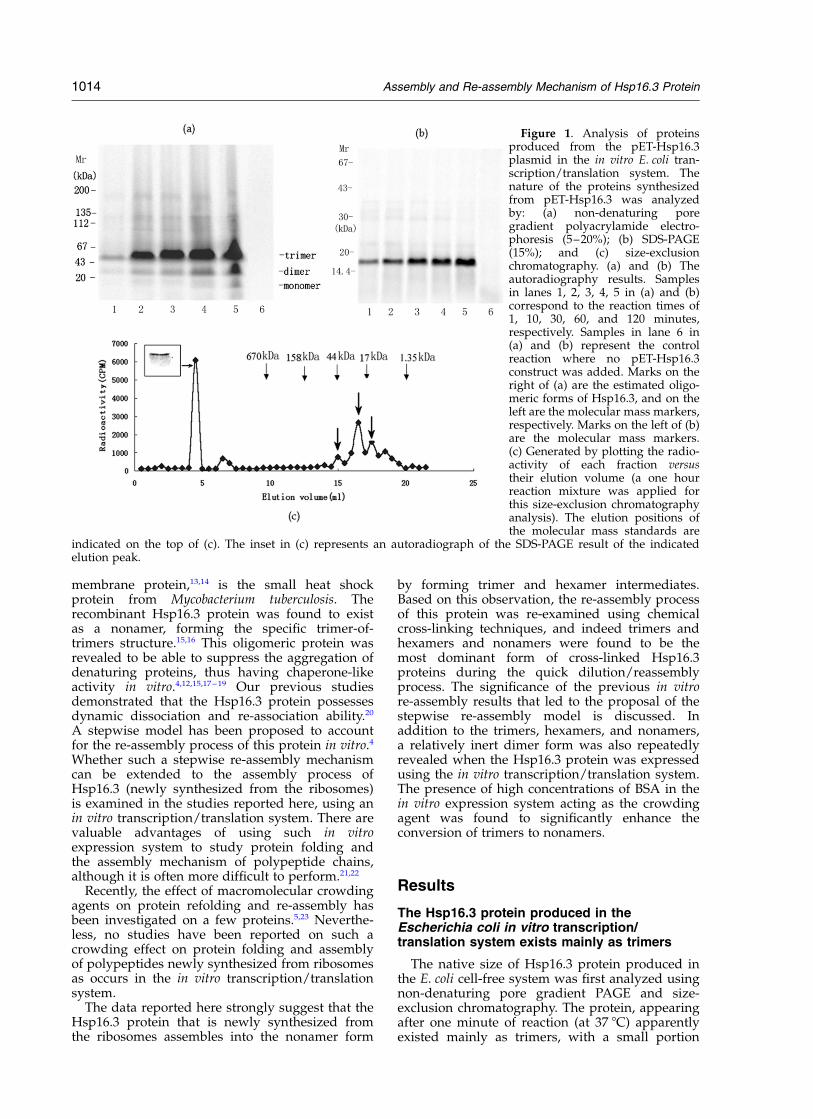
Figure 1. Analysis of proteinsproduced from the pET-Hsp16.3plasmid in the in vitro E. coli tran-scription/translation system. Thenature of the proteins synthesizedfrom pET-Hsp16.3 was analyzedby: (a) non-denaturing poregradient polyacrylamide electro-phoresis (5–20%); (b) SDS-PAGE(15%); and (c) size-exclusionchromatography. (a) and (b) Theautoradiography results. Samplesin lanes 1, 2, 3, 4, 5 in (a) and (b)correspond to the reaction times of1, 10, 30, 60, and 120 minutes,respectively. Samples in lane 6 in(a) and (b) represent the controlreaction where no pET-Hsp16.3construct was added. Marks on theright of (a) are the estimated oligo-meric forms of Hsp16.3, and on theleft are the molecular mass markers,respectively. Marks on the left of (b)are the molecular mass markers.(c) Generated by plotting the radio-activity of each fraction versustheir elution volume (a one hourreaction mixture was applied forthis size-exclusion chromatographyanalysis). The elution positions ofthe molecular mass standards are
indicated on the top of (c). The inset in (c) represents an autoradiograph of the SDS-PAGE result of the indicatedelution peak.
1014 Assembly and Re-assembly Mechanism of Hsp16.3 Protein
existing as dimers and monomers (Figure 1(a)), asdescribed. 20 The detection of only one single bandof about 16 kDa by SDS-PAGE (Figure 1(b))demonstrated unequivocally that all the proteinbands (representing the Hsp16.3 forms havingvarious sizes) detected by the non-denaturingpore gradient gel electrophoresis were indeedderived from Hsp16.3.
When the reaction mixture was analyzed by size-exclusion chromatography, three peaks of radio-activity, roughly corresponding to the sizes oftrimers, dimers and monomers were detected(marked by the three bold arrows in Figure 1(c)).In contrast to what was observed by the non-denaturing pore gradient gel electrophoresis, thetrimer forms detected by size-exclusion FPLC areapparently present at a level much lower than thatof the dimers (Figure 1(c)). It is likely that thedramatic dilution of a minute amount of Hsp16.3(here the loading amount is estimated to be 2.0 to2.5 ng/ml, where the total amount of protein pro-duced in the transcription/translation system isabout 50 ng to 200 ng/50 ml reaction mixtureaccording to the instruction of the kit provider) viasize-exclusion chromatography caused the dis-sociation of the trimer forms. Similarly, the non-americ form that was previously found to be themajor form of purified Hsp16.3,15 was not detectedunder these extremely diluted conditions. Strik-ingly, another major peak corresponding to anapparent molecular size of no less than,1000 kDa was repeatedly detected by size-exclusion FPLC analysis (Figure 1(c)). SDS-PAGEanalysis of samples from this peak clearly demon-strated the presence of Hsp16.3 (Figure 1(c), inset).
The small heat shock protein in M. bovis BCG(with an amino acid sequence identical to that ofHsp16.3 from M. tuberculosis) was found to beassociated with ribosomes.24 Judging from the sizealone, the Hsp16.3 found in this peak may rep-resent those that tightly bound to the ribosome inthe E. coli extracts. Consistent with this conjecture,the size of the peak did not change at all, evenwhen the total level of Hsp16.3 production wasincreased by adding purified exogenous Hsp16.3protein to the reaction mixture (Figure 4(c),below). The possibility that Hsp16.3 protein is ableto bind tightly to ribosomes and the biologicalimplications of this ability are worth furtherinvestigation.
Trimeric and dimeric forms were detected afterthe chemical cross-linking reaction of theHsp16.3 proteins produced in thetranscription/translation system
The nature of the oligomers present in the reac-tion mixture was further investigated by chemical
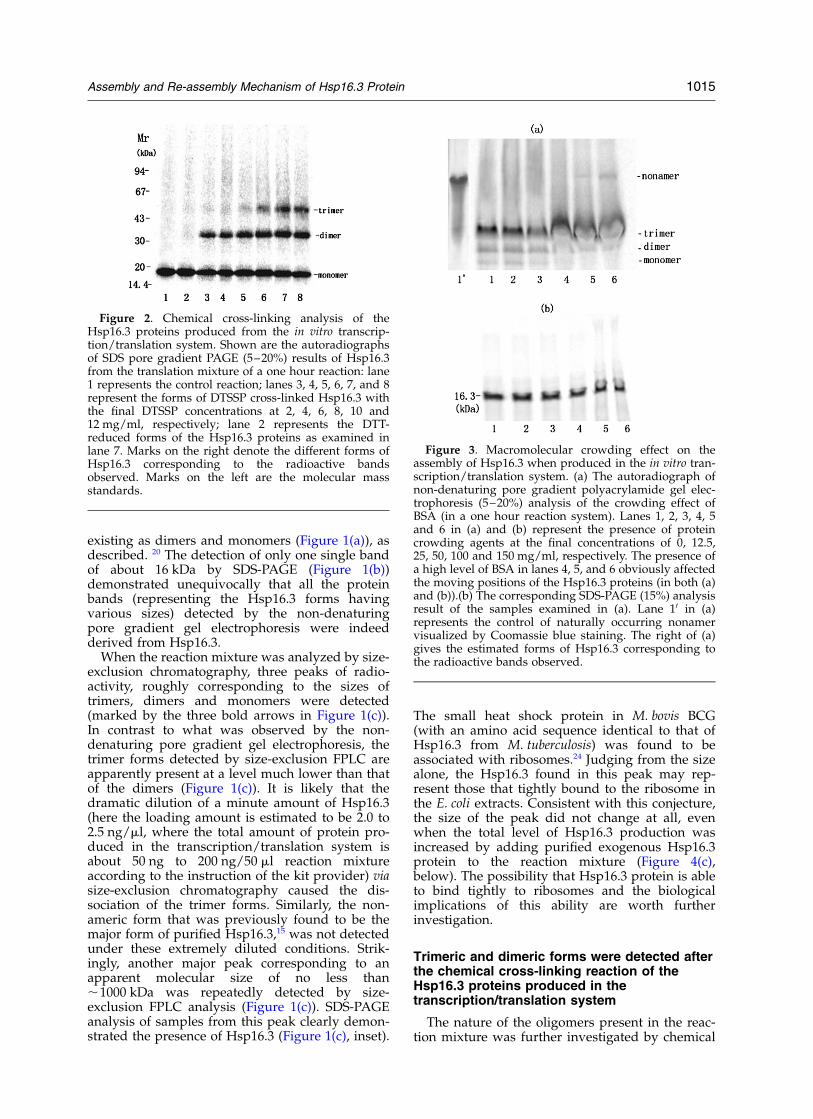
Figure 2. Chemical cross-linking analysis of theHsp16.3 proteins produced from the in vitro transcrip-tion/translation system. Shown are the autoradiographsof SDS pore gradient PAGE (5–20%) results of Hsp16.3from the translation mixture of a one hour reaction: lane1 represents the control reaction; lanes 3, 4, 5, 6, 7, and 8represent the forms of DTSSP cross-linked Hsp16.3 withthe final DTSSP concentrations at 2, 4, 6, 8, 10 and12 mg/ml, respectively; lane 2 represents the DTT-reduced forms of the Hsp16.3 proteins as examined inlane 7. Marks on the right denote the different forms ofHsp16.3 corresponding to the radioactive bandsobserved. Marks on the left are the molecular massstandards.
Figure 3. Macromolecular crowding effect on theassembly of Hsp16.3 when produced in the in vitro tran-scription/translation system. (a) The autoradiograph ofnon-denaturing pore gradient polyacrylamide gel elec-trophoresis (5–20%) analysis of the crowding effect ofBSA (in a one hour reaction system). Lanes 1, 2, 3, 4, 5and 6 in (a) and (b) represent the presence of proteincrowding agents at the final concentrations of 0, 12.5,25, 50, 100 and 150 mg/ml, respectively. The presence ofa high level of BSA in lanes 4, 5, and 6 obviously affectedthe moving positions of the Hsp16.3 proteins (in both (a)and (b)).(b) The corresponding SDS-PAGE (15%) analysisresult of the samples examined in (a). Lane 10 in (a)represents the control of naturally occurring nonamervisualized by Coomassie blue staining. The right of (a)gives the estimated forms of Hsp16.3 corresponding tothe radioactive bands observed.
Assembly and Re-assembly Mechanism of Hsp16.3 Protein 1015
cross-linking studies using 3,30-dithiobis (sulfo-succinimdyl) propionate (DTSSP). The SDS-PAGEanalysis of the forms of Hsp16.3 that resultedfrom the cross-linking reaction revealed thepresence of monomers, dimers and trimers(Figure 2, lanes 5, 6, 7 and 8) strongly suggestingthat trimers are indeed the major oligomeric formsof Hsp16.3 produced in the in vitro transcription/translation system (Figure 2). The failure to convertall the Hsp16.3 proteins to the trimer form bychemical cross-linking (Figure 2, lanes 3–8) mayindicate that a dynamic dissociation/re-associationexists for the trimeric Hsp16.3. This dynamicproperty would result in the irreversible dis-association of a cross-linked dimer from the trimer,producing a high level of dimer and monomer inthe reaction mixture. Such a dynamic dis-sociation/re-association phenomenon was indeedobserved for the nonameric Hsp16.3 proteins.20
The presence of a protein-crowding agent hasa significant but small effect onHsp16.3 assembly
The assembly environment in the bacterial cells,with the total concentration of proteins and RNAsbeing about 200–300 mg/ml,25,26 was mimickedhere by adding an increasing amount of BSA pro-tein in the in vitro transcription/translation system
(with the total protein concentration before addingthe crowding agent being about 3.6 mg/ml). Thedata presented in Figure 3(a) and (b) indicate thatonly a very small amount of nonameric Hsp16.3was formed when BSA was present at the final con-centrations of 100 mg/ml and 150 mg/ml (Figure3(a), lanes 5 and 6). However, no such nonamerswere observed when the BSA protein was addedafter the transcription/translation reaction wasstopped (data not shown). This suggests that thecrowding BSA molecules acted during (not after)the folding/assembly stage.
The effect of GroEL/GroES (a well-studiedmolecular chaperone) on the assembly of Hsp16.3was also examined. The data (not presented here)indicate that trimers were still the predominantform present; no higher oligomers were observed.However, the total amount of Hsp16.3 protein syn-thesized was significantly decreased by thepresence of GroEL/GroES. In addition, the pro-duction of Hsp16.3 protein in the transcription/translation system was almost completely inhibitedwhen both GroEL/GroES and ATP were present(data not shown). These results were somehowopposite to our expectation, considering thatGroEL/GroES is supposed to aid the protein fold-ing process. These results also ruled out thepossibility that the absence of oligomers higherthan trimers is due to the scarcity of GroEL/
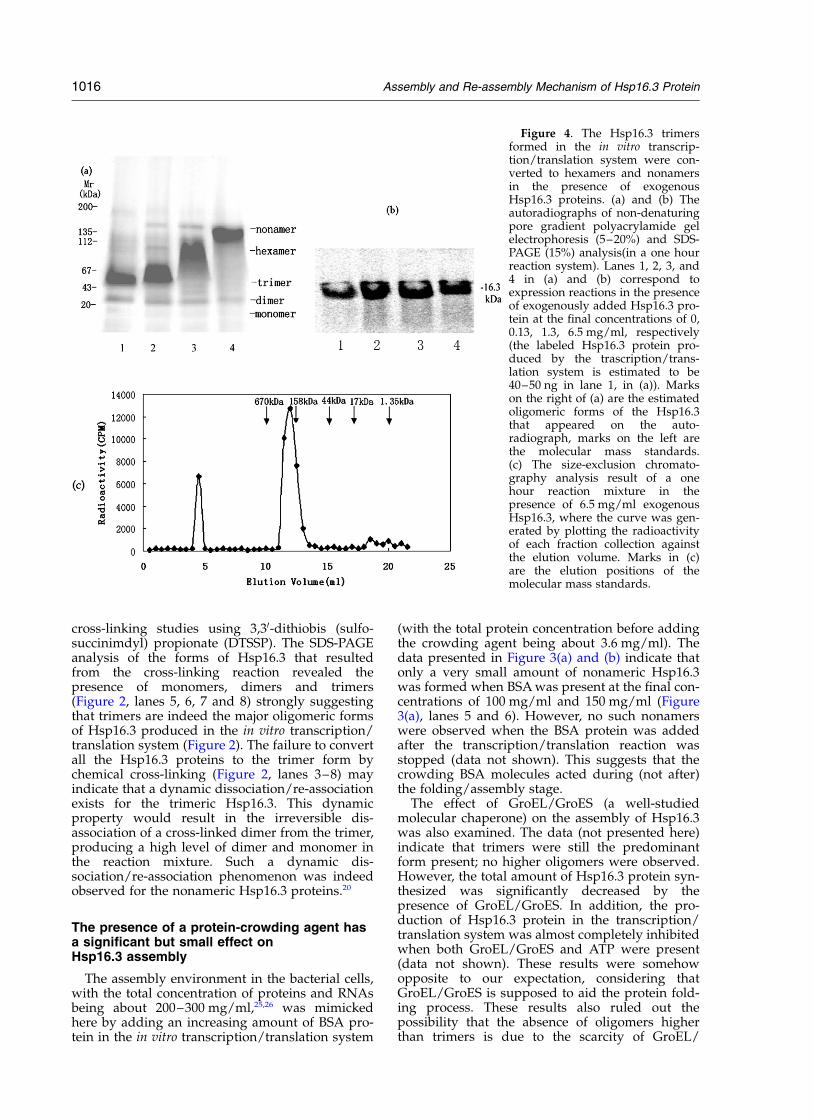
Figure 4. The Hsp16.3 trimersformed in the in vitro transcrip-tion/translation system were con-verted to hexamers and nonamersin the presence of exogenousHsp16.3 proteins. (a) and (b) Theautoradiographs of non-denaturingpore gradient polyacrylamide gelelectrophoresis (5–20%) and SDS-PAGE (15%) analysis(in a one hourreaction system). Lanes 1, 2, 3, and4 in (a) and (b) correspond toexpression reactions in the presenceof exogenously added Hsp16.3 pro-tein at the final concentrations of 0,0.13, 1.3, 6.5 mg/ml, respectively(the labeled Hsp16.3 protein pro-duced by the trascription/trans-lation system is estimated to be40–50 ng in lane 1, in (a)). Markson the right of (a) are the estimatedoligomeric forms of the Hsp16.3that appeared on the auto-radiograph, marks on the left arethe molecular mass standards.(c) The size-exclusion chromato-graphy analysis result of a onehour reaction mixture in thepresence of 6.5 mg/ml exogenousHsp16.3, where the curve was gen-erated by plotting the radioactivityof each fraction collection againstthe elution volume. Marks in (c)are the elution positions of themolecular mass standards.
1016 Assembly and Re-assembly Mechanism of Hsp16.3 Protein

GroES in the in vitro transcription/translationsystem.
The Hsp16.3 protein produced in the in vitrotranscription/translation system assemblesinto higher oligomeric forms in the presenceof exogenous Hsp16.3
Molecules similar in sizes to the target proteinsare believed to be the most effective crowdingagent for protein folding (or refolding) andassembly (or re-assembly).27 In consideration ofthis common belief, as well as the fact thatHsp16.3 is able to act as a molecular chaperone,the following question was asked: would thepurified recombinant Hsp16.3 protein be able tohelp the oligomeric forms (being predominantlytrimers) of Hsp16.3 produced in the in vitro systemto assemble into higher oligomeric forms? Datapresented in Figure 4(a) demonstrate that when anincreasing amount of exogenous Hsp16.3 proteinwas present in the in vitro protein synthesis system,hexamers and nonamers were indeed formed. Themajor oligomeric forms of newly synthesizedHsp16.3, as detected on the gel, depend stronglyon the concentration of the exogenously addedHsp16.3, with trimers, hexamers or nonamersbeing the most prominent when exogenousHsp16.3 was present at a low (0.13 mg/ml),medium (1.3 mg/ml) or high (6.5 mg/ml) levels,respectively (corresponding to lanes 2–4, respect-ively, in Figure 4(a)).
Interestingly, two minor bands corresponding tothe hexamer and nonamer sizes were also detectedwhen exogenous Hsp16.3 was present at a lowconcentration (Figure 4(a), lane 2). When exo-genous Hsp16.3 was further increased to amedium level, a minor band at the nonamer pos-ition was still detected, but no band was detected
at the trimer position anymore (Figure 4(a), lane3). At high exogenous Hsp16.3 concentrations, thenonamer was the only oligomeric form detected,with no trimers or hexamers at all, as shown byFigure 4(a), lane 4, as well as by the size-exclusionchromatography results shown in Figure 4(c).
Promotion of such assembly processes was notobserved when an increasing amount of eithera-crystallin or GroEL/GroES, both being molecularchaperones, were added in this system (data notshown), ruling out the likelyhood that the exo-genous Hsp16.3 added here acted as a molecularchaperone in promoting the further assembly ofthe newly synthesized trimeric forms. The datapresented in Figure 4(a) can thus be explained asfollows. The exogenously added Hsp16.3 non-amers underwent dynamic dissociation/re-association,20 meaning the trimer and hexamerforms also existed. By interacting with such non-radiolabeled exogenous trimers and hexamers, theradiolabeled newly produced Hsp16.3 trimersassemble into oligomers of a higher level, i.e.hexamers and nonamers. All these mean that theassembly process occurred via subunit exchangeas demonstrated.20 Here the assembly (of thenewly produced protein) and re-association (of theexogenously added protein) processes are occur-ring with the same nature. In other words, whatwe see in Figure 4(a) reflects the assembly processof newly produced radiolabeled Hsp16.3 protein.
Another interesting phenomenon is that thedimer form of the radiolabeled Hsp16.3 remainedall the time, although with its amount slightlydecreased when the exogenous Hsp16.3 waspresent at an increased concentration (Figure 4(a)).It should also be pointed out that bands corre-sponding to the monomer size (although margin-ally visible) could still be detected in all thesample lanes (Figure 4(a)). In addition, one or twominor bands were also marginally visible betweenthe trimer and dimer bands when the exogenousHsp16.3 was present at a medium level when thetrimers were being converted to hexamers (Figure4(a), lane 3; as well as Figure 6(a), lanes 2 and 3,below). In addition, the total amount of Hsp16.3synthesized in the in vitro system also seems toincrease significantly in the presence of exogenousHsp16.3 (Figure 4(b) and (c)).
Hsp16.3 proteins of higher oligomericstructure are more resistant to trypsindigestion trypsin digestion
The compactness of the assembled oligomericstructures of Hsp16.3 was examined by analyzingtheir sensitivity towards trypsin digestion. TheHsp16.3 proteins synthesized from the in vitro tran-scription/translation system (being predominantlytrimers) and those produced in the presence ofmedium to high levels of exogenous Hsp16.3(being predominantly hexamers or nonamers)were subject to trypsin digestion. The datapresented in Figure 5 demonstrate that the higher
Figure 5. Analysis of the resistance to trypsin digestionof the different oligomeric forms of Hsp16.3 produced inthe in vitro transcription/translation system in thepresence of exogenously added Hsp16.3 proteins.Shown is the autoradiograph of the SDS-PAGE (15%)result of the reaction mixture after a one hour trypsindigestion (with the final concentration of 50 mg/ml).Lanes 2–5 represent the digested samples of the reactionmixture (of one hour) in the presence of exogenousHsp16.3 protein at final concentrations of 0, 0.13, 1.3and 6.5 mg/ml, respectively. Lane 1 represents the con-trol reaction, where no trypsin was added.
Assembly and Re-assembly Mechanism of Hsp16.3 Protein 1017
the oligomer form, the more resistant the oligomersare to trypsin digestion. These observationssuggest that the nonamers and hexamers are morecompact. Interestingly, a minor band, slightlysmaller than that of the complete Hsp16.3 andwith a density unaffected by the forms of oligo-mers related to nonamer assembly (i.e. trimers,hexamers, or nonamers), was consistentlyobserved in all the digested samples (Figure 5,lanes 2–5). It is likely that this smaller bandresulted from the trypsin digestion of the inert
dimer in light of its similar level with the dimerform and its unrelatedness to the nonamerassembly process.
In vitro reassembly of Hsp16.3 protein occurvia trimer and hexamer intermediates
Only trimers and hexamers were observed asassembly intermediates for the Hsp16.3 proteinsthat are newly synthesized from the ribosomes, asrevealed by the results described above. Since thisstudy proposed a stepwise model to explain there-assembly process of Hsp16.3,4 it is important tore-examine the reassembly process of Hsp16.3using new approaches.
In the first approach, the purified (unlabeled)Hsp16.3 was denatured in 8 M urea and concen-trated, then various amounts of denatured proteinswere quickly diluted into the in vitro transcription/translation system in which (radiolabeled) Hsp16.3was already synthesized. The oligomeric formshaving radiolabeled Hsp16.3 were examined vianon-denaturing pore gradient PAGE and auto-radiography. Results presented Figure 6(a) clearlyshow a similar pattern as observed when nativeexogenous Hsp16.3 was added before protein syn-thesis was initiated (as shown in Figure 4), withhexamer intermediates observed on the way ofconverting the radiolabeled trimers to nonamers.The data presented in Figure 6(a) represent theresult of two events: one is the re-assembly of thedenatured Hsp16.3 in the in vitro expression sys-tem; the other is the exchange of subunits of theradiolabeled Hsp16.3 and non-labeled Hsp16.3 asdemonstrated by us.20 These results stronglysuggest that trimers and hexamers are indeedintermediates for the re-assembly of Hsp16.3 pro-teins in the in vitro transcription/translation sys-tem. Similar to that observed in the data presentedin Figure 4(a), the “inertness” of the dimer form,as well as the appearance of a minor band betweenthe dimer and trimer forms were also observedhere (Figure 6(a), lanes 2 and 3).
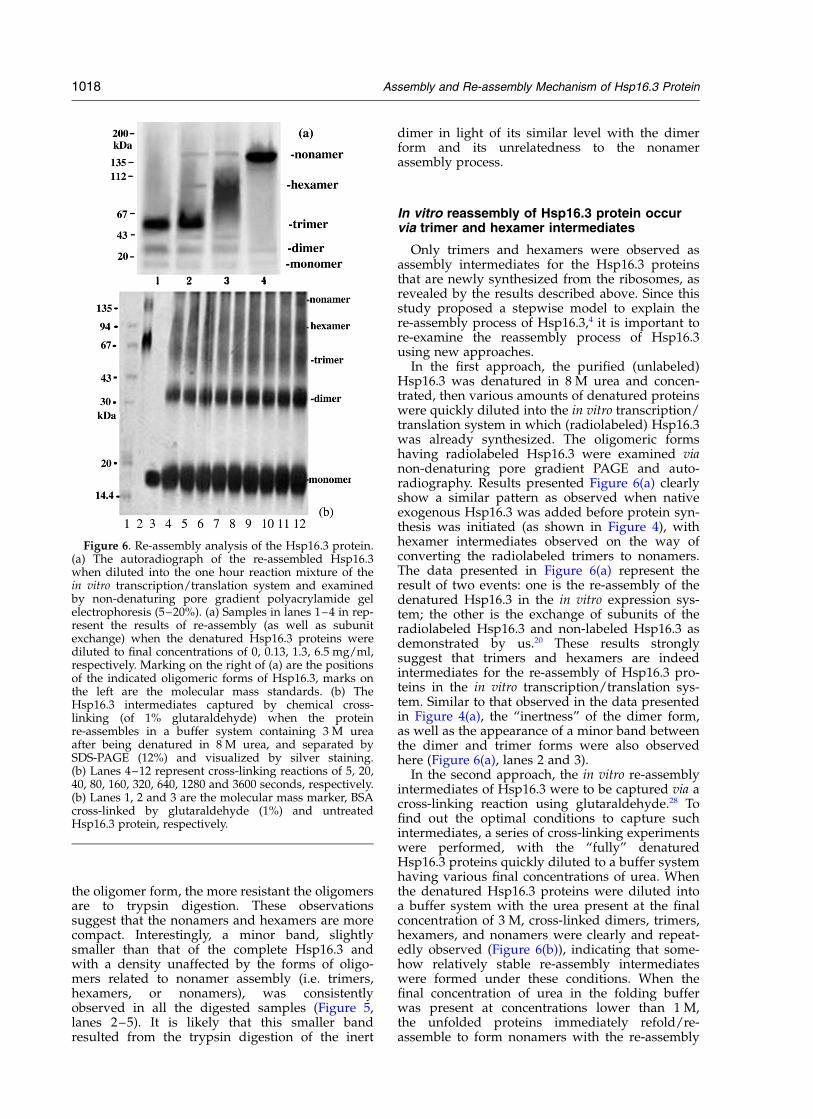
In the second approach, the in vitro re-assemblyintermediates of Hsp16.3 were to be captured via across-linking reaction using glutaraldehyde.28 Tofind out the optimal conditions to capture suchintermediates, a series of cross-linking experimentswere performed, with the “fully” denaturedHsp16.3 proteins quickly diluted to a buffer systemhaving various final concentrations of urea. Whenthe denatured Hsp16.3 proteins were diluted intoa buffer system with the urea present at the finalconcentration of 3 M, cross-linked dimers, trimers,hexamers, and nonamers were clearly and repeat-edly observed (Figure 6(b)), indicating that some-how relatively stable re-assembly intermediateswere formed under these conditions. When thefinal concentration of urea in the folding bufferwas present at concentrations lower than 1 M,the unfolded proteins immediately refold/re-assemble to form nonamers with the re-assembly
Figure 6. Re-assembly analysis of the Hsp16.3 protein.(a) The autoradiograph of the re-assembled Hsp16.3when diluted into the one hour reaction mixture of thein vitro transcription/translation system and examinedby non-denaturing pore gradient polyacrylamide gelelectrophoresis (5–20%). (a) Samples in lanes 1–4 in rep-resent the results of re-assembly (as well as subunitexchange) when the denatured Hsp16.3 proteins werediluted to final concentrations of 0, 0.13, 1.3, 6.5 mg/ml,respectively. Marking on the right of (a) are the positionsof the indicated oligomeric forms of Hsp16.3, marks onthe left are the molecular mass standards. (b) TheHsp16.3 intermediates captured by chemical cross-linking (of 1% glutaraldehyde) when the proteinre-assembles in a buffer system containing 3 M ureaafter being denatured in 8 M urea, and separated bySDS-PAGE (12%) and visualized by silver staining.(b) Lanes 4–12 represent cross-linking reactions of 5, 20,40, 80, 160, 320, 640, 1280 and 3600 seconds, respectively.(b) Lanes 1, 2 and 3 are the molecular mass marker, BSAcross-linked by glutaraldehyde (1%) and untreatedHsp16.3 protein, respectively.
1018 Assembly and Re-assembly Mechanism of Hsp16.3 Protein

intermediates difficult to capture via cross-linking(data not shown).
Non-denaturing pore gradient polyacrylamidegel detects a dramatically decreased level oftrimers of L122A mutant Hsp16.3
The purified recombinant L122A mutantHsp16.3 protein was previously shown to be lessstable and to migrate at a significantly slower rateon a non-denaturing gel than the wild-typeprotein.18,19 The assembly property of this mutantprotein was also examined using a similarapproach as described above. Although the overall
level of protein production was significantlydecreased in the in vitro transcription/translationsystem, the non-denaturing pore gradient gel elec-trophoresis results show that trimers still appearto be the major oligomeric forms generated for theL122A mutant protein (Figure 7(a), lane 2). Thedata presented in Figure 7(a) also clearly showthat all the oligomeric forms of the L122A mutantHsp16.3 protein migrates at a slower rate thantheir corresponding forms of the wild-type protein,suggesting a less compact structure for the mutantoligomers. In contrast to the wild-type protein, theaddition of purified mutant Hsp16.3 in the reactionmixture did not seem to have any obvious effect onthe oligomeric forms of L122A mutant proteindetected on the gel, but lead to the formation of amore compact trimeric form (Figure 7(a), lanes3–6). The addition of purified mutant Hsp16.3 inthe reaction mixture seems to somehow increasethe translation rate of the Hsp16.3 protein (Figure7, lanes 3–6 in both panels).
Discussion
The assembly pathway of Hsp16.3 produced byin vitro transcription/translation was studied. Ourprinciple discoveries include the following. (1)Non-denaturing pore gradient polyacrylamide gelelectrophoresis and chemical cross-linking studies(data presented in Figures 1 and 2) all suggest thatthe trimer is the predominant form of Hsp16.3 forthe Hsp16.3 proteins synthesized in the in vitrotranscription/translation system. (2) Results fromdisassociation/re-association experiments usingradiolabeled and non-radiolabeled Hsp16.3 indi-cate that the trimeric form is able to furtherassemble directly into hexamers and nonamers(Figures 4 and 6). (3) A remarkably stable dimerform (Figures 3, 4 and 6) of Hsp16.3, which doesnot seem to participate in the formation of thenonamers, was consistently detected from the invitro transcription/translation products. (4) Are-examination of the re-assembly process ofHsp16.3 strongly suggests that trimers and hex-amers are indeed also intermediates for thedenatured Hsp16.3 proteins to refold/re-assemble.
In light of all these observations, a model is pro-posed (Figure 8) to explain the assembly processof the newly synthesized Hsp16.3 proteins to form
Figure 7. Analysis of L122A mutant Hsp16.3 proteinsproduced from the in vitro E. coli transcription/trans-lation system in the presence of exogenously addedL122A mutant proteins. This Figure shows the auto-radiographs of (a) the non-denaturing pore gradientpolyacrylamide gel electrophoresis (5–20%) and (b)SDS-PAGE (15%) results of the L122A mutant Hsp16.3from a one hour reaction in the absence (lane 2 in bothpanels) or in the presence of exogenously added L122Amutant Hsp16.3 proteins. Lanes 3–6 in both (a) and (b)represent the results having the exogenously addedmutant Hsp16.3 proteins at the final concentrations of0.13, 1.3, 6.5 and 13 mg/ml, respectively. Added in lane1 was the in vitro reaction mixture making the wild-typeHsp16.3. Marks on the left and right of (a) are the esti-mated oligomeric forms of Hsp16.3 and L122A mutantHsp16.3, respectively.
Figure 8. The assembly model of Hsp16.3 protein.
Assembly and Re-assembly Mechanism of Hsp16.3 Protein 1019

the nonameric trimer-of-trimers structure,15,16 inwhich the nonameric form of Hsp16.3 protein isassembled via the trimer and hexamer inter-mediates. In other words, the newly synthesizedHsp16.3 polypeptide chains somehow first collideto form the trimer intermediates, which willfurther assemble to form the hexamer inter-mediates and eventually the most stable nonamersin a concentration-dependent manner. Koteicheet al. also recently demonstrated via truncationstudies that trimers are a basic structural unit forthe Hsp16.3 protein.29
Formation of an inert dimer is also proposed toaccount for the consistent appearance of the dimerband and its inertness towards Hsp16.3 assemblyunder all our experimental conditions (Figures 3, 4and 6). The slight decrease on the band density ofthe dimeric form when higher concentrations ofeither BSA or exogenous Hsp16.3 was present(Figures 3, 4 and 6, respectively) may reflect theco-existence of a “competent” dimer. The com-petent dimer may be further assembled to higheroligomers, apparently via a certain conformationchange before being converted to the trimer form,as indicated by the detection of one or two minorbands between the dimer and trimer bands inFigures 4(a) and 6(a). A dimer form of Hsp16.3was also observed when the protein was examinedby cryo-electron microscopy in the presence ofcertain types of lipid bilayers (Yong Chen et al.,Tsinghua University, unpublished data). This inertform of dimer may participate in the formation ofa layer of Hsp16.3 proteins that is formed on theoutside surface of the M. tuberculosis cells or thefiber structure formed in the cytoplasm underoxidative stress conditions.30 Lee et al.14 revealedthat the Hsp16.3 protein is a “major membraneprotein”, whether the inert dimer has anything todo with this membrane formation of Hsp16.3 alsoneeds further investigation.
The protein folding/assembly problem is oftenapproached by studying the in vitro refolding/re-assembly pathways of denatured proteins. Thevalidity of utilizing such results to explain the fold-ing and assembly mechanism of newly synthesizedpolypeptide chains has often been questioned.31 – 33
Our previous studies on the refolding/re-assemblyprocess of denatured Hsp16.3 proteins, examinedmainly via urea-gradient polyacrylamide gel elec-trophoresis and urea-containing pore-gradientpolyacrylamide gel electrophoresis, suggested astepwise process for the re-assembly of Hsp16.3.4
However, the data reported here strongly suggestthat trimers and hexamers are always inter-mediates formed no matter whether the nonamerHsp16.3 is assembled from the newly synthesizedpolypeptide chains or re-assembled from thedenatured proteins. The data provided here clearlydemonstrate that the assembly and re-assembly ofthe Hsp16.3 nonamers may use a similar mechan-ism. Given that Hsp16.3 is a molecular chaperonethat was found to be able to effectively re-assemblein a spontaneous way,4 it cannot be stated that both
the assembly and re-assembly of any oligomericprotein occur in a similar mechanism until similarcomparison studies are performed for more pro-teins. The multiple inter-supportive data presentedhere forced us to re-appraise our previous dataobtained and the stepwise model proposed for there-assembly process of the Hsp16.3 nonamer.4
Looking back, one possible explanation for thenine bands observed on the urea-gradient gel afterelectrophoretic separation of the denaturedHsp16.3 protein may be that they actuallyrepresent nine intermediates of the protein dis-assembly, instead of the re-assembly.
In light of the similar mechanisms used by theassembly of Hsp16.3 polypeptide chains newlysynthesized from the ribosomes and the re-assembly of the unfolded intact Hsp16.3 proteins,it is conceivable that the assembly of the Hsp16.3nonamers could have occurred post-translationallyin vivo. In other words, for homo-oligomericproteins that assemble after the synthesis of thewhole polypeptide chain ends (i.e. post-trans-lationally) and that assemble spontaneously, themechanism revealed for the re-assembly processmay be extended to explain the process for proteinassembly in vivo. Of course, one can argue thatsuch an assembly mechanism revealed by usingthe in vitro transcription/translation system stilldoes not represent what actually happens insidethe living cells.
The macromolecular crowding effect on proteinrefolding and re-assembly has been examined inthe in vitro denaturization/renaturization system.5
However, we have seen no report in which such acrowding effect is examined for the actual proteinfolding or assembly processes. The data presentedhere represent the first report where the crowdingeffect for the assembly of proteins that are beingsynthesized from the ribosomes is studied. Thepresence of a high concentration (close to that incells) of crowding proteins (BSA) in our in vitrotranscription/translation system indeed promoted,however, to a limited degree, the conversion of theHsp16.3 trimers to the nonamers. The failure toobserve the Hsp16.3 hexamer intermediates in thistype of crowding experiment may indicate thatthe nonamers are indeed more stable than the hex-amers (Figure 3(a)). The limited crowding effect weobserved seems to suggest that the assembly pro-cess is more affected by the total concentration ofthe assembly protein (here it is Hsp16.3), insteadof the level of the macromolecules present in theenvironment.
The difference in the behavior of the L122Amutant and wild-type Hsp16.3 genes/proteins inthe in vitro transcription/translation system isremarkable. Consistent with our previous obser-vations obtained by examining the purified recom-binant proteins,18,19 the mutant protein wasproduced at a much lower level than that of thewild-type protein (obvious by comparing theresults in Figures 1 and 7). In contrast to what wasobserved for the wild-type protein, the assembly
1020 Assembly and Re-assembly Mechanism of Hsp16.3 Protein

process of the mutant protein was hardly affectedby the exogenous addition of high levels of eitherthe purified L122A mutant (Figure 7) or the wild-type Hsp16.3 proteins to the transcription/translation system expressing the mutant Hsp16.3proteins. Given the fact that proteases are absentfrom the in vitro expression system, it is surprisingthat the mutant protein level is so much lowerthan that of the wild-type protein. It appears thatthe lower efficiency of protein folding andassembly affected the protein expression leveldramatically for the mutant Hsp16.3 protein.Whether there exists a close relation betweenprotein folding/assembly and polypeptide syn-thesis on the ribosome merits further studies.These results also suggest that the leucine residueat position 122 (being the only conserved residuefor all the sHsps so far analyzed and beingreplaced by an alanine residue in the mutantHsp16.3 protein) plays an important role in form-ing the Hsp16.3 trimers.
In summary, our studies of the assembly andre-assembly processes of the nonameric Hsp16.3protein using,respectively, the in vitro transcrip-tion/translation and denaturization/renaturizationsystems lead to the following conclusions: the non-amer of Hsp16.3 assembles/re-assembles via com-mon trimer and hexamer intermediates; an inertHsp16.3 dimer which does not seem to participatenonamer formation but may participate in formingother forms of Hsp16.3 structure was also observedin the in vitro transcription/translation system;macromolecular crowding (using bovine serumalbumin) affects, to a limited degree, the assemblyprocess of Hsp16.3 that is newly synthesized fromthe ribosomes, suggesting that the assembly ofoligomeric proteins like Hsp16.3 is not affectedmuch by the level of macromolecules in theenvironment, but more dependent on the concen-tration of the proteins being assembled.
Materials and Methods
Materials
BSA and trypsin were all purchased from Sigma;-[35S]methionine (1000 Ci/mmol) was purchased fromAmersham Corp. The GroEL/GroES proteins werekindly provided by Dr Hai-Meng Zhou at the Depart-ment of Biological Science and Biotechnology, TsinghuaUniversity. All other chemical reagents were of analyticalpurity.
Plasmid construction, protein expressionand purification
The construction of the expression plasmid vector forHsp16.3 (pET-Hsp16.3), the over-expression and purifi-cation of the recombinant protein from E. coli cells wereall as described.15 The generation of the L122A mutantHsp16.3 gene and protein was also as described.18
In vitro transcription and translation of Hsp16.3
Transcription and translation was performed using thein vitro E. coli T7 S30 Extract System for Circular DNASystem (Promega) according to the provider’s instruc-tion. Template DNA (being pET-Hsp16.3) of 1.8 mg wasadded per 50 ml of reaction volume. Each proteinexpression reaction, having 10 mCi of 35S-labeledmethionine, was performed at 37 8C and was stoppedby cooling with ice. The mixture was either examinedimmediately or stored at 220 8C before usage. The totalprotein concentration in the reaction mixture was deter-mined using the Bradford assay (with BSA being usedas the standard protein). The crowding agent wasadded to the reaction mixture before the in vitrotranscription/translation reaction was initiated.
Electrophoresis and autoradiography
Cell-free translation products were analyzed usingSDS-PAGE and non-denaturing pore gradient polyacryl-amide gel electrophoresis. SDS-PAGE (15%, w/v) wasperformed as described with the protein components inthe reaction mixture precipitated via acetone treatment,thus removing the free radioactive methionine residuesand other waste.34,35 Non-denaturing pore gradient poly-acrylamide gel electrophoresis (5–20%) was prepared ina 125 mm £ 100 mm £ 1 mm mold and performedaccording to methods described,20 with the followingproteins used as molecular mass standards: soybeantrypsin inhibitor (20 kDa), chicken egg ovalbumin(43 kDa), BSA monomer (67 kDa), b-lactamase tetramer(112 kDa, expressed from a PinPointe control vector pro-vided with the in vitro expression kit of Promega), BSAdimer (135 kDa) and BSA trimer (200 kDa). Proteinbands were visualized either by Coomassie blue R-250staining or by autoradiography (after the gel was dried).The radioactive protein bands were quantified byPhosphorimager analysis (Molecular Dynamics).
TCA protein precipitation assay for aminoacid incorporation
Trichloroacetic acid (TCA) precipitable radioactivity,representing the part that has been incorporated intonewly synthesized proteins, was determined by spottingthe sample aliquots onto Whatman GF/A glass fiberfilters after NaOH hydrolysis. Radioactivity wasmeasured using a 1209 RackBeta liquid scintillationcounter.
Detection of the compactness of proteinconformation via proteolysis
An in vitro transcription/translation reaction mixturethat expresses the Hsp16.3 protein in the absence orpresence of purified exogenous Hsp16.3 was acetoneprecipitated and the pellet was then subject to trypsin(with a final concentration of 50 mg/ml) digestion at37 8C for one hour. The treated reaction mixture wasthen applied for SDS-PAGE and autoradiographyanalysis.
Size-exclusion FPLC
Size exclusion FPLC was performed using an AKTAExplorer with the Surperdex 200 column (Pharmacia,Uppsala, Sweden). The translation mixture containing
Assembly and Re-assembly Mechanism of Hsp16.3 Protein 1021

the newly synthesized Hsp16.3 proteins (50 ml) wasapplied onto the column equilibrated with 50 mM PBS,200 mM NaCl (pH 7.0). Fractions of 0.5 ml were collectedat 0.5 ml/minute, with the radioactivity of each collec-tion counted after being precipitated with trichloroaceticacid (TCA). The column was calibrated with the follow-ing high molecular mass standards (Bio-Rad): thyro-globulin, 670 kDa; bovine gamma globulin, 158 kDa;chicken ovalbumin, 44 kDa; equine myoglobin, 17 kDa;vitamin B-12, 1.35 kDa.
Protein cross-linking
A water-soluble and thiol-cleavable cross-linkerDTSSP (3,30-dithiobis (sulfosuccinimdyl) propionate),purchased from Soltec Ventures, which reacts rapidlyand specifically with primary amines (up to ,12 Aapart), was used. Hsp16.3 (present in the in vitro trans-lation mixture of a one hour reaction) was mixed withfreshly prepared DTSSP in 50 mM PBS (pH 7.5) and thereaction was allowed to occur at 25 8C for three hours.The reaction was stopped via quenching by adding anexcessive amount of 1 M Tris–HCl (pH 7.5), incubatedat 25 8C for 15 minutes. After the reaction the samplewas then divided into two aliquots, the free radioactivemethionine and other waste were removed by acetoneprecipitation, with one being treated with 100 mM DTTat 37 8C for 60 minutes (which will break the cross-link-ing bonds by reduction), and the other being untreated.The samples were then applied for SDS pore gradientPAGE (5–20%) and autoradiography analysis. The sizesof the autoradiography bands were estimated by usingthe Low Molecular Weight Calibration Kit (Pharmacia,Uppsala, Sweden) which contains the following: phos-phorylase b (94 kDa), bovine serum albumin (67 kDa),ovalbumin (43 kDa), carbonic anhydrase (30 kDa), tryp-sin inhibitor (20 kDa) and a-lactalbumin(14.4 kDa).
Refolding and reassembly reaction
Two approaches were used to investigate the refoldingand reassembly mechanism of the Hsp16.3 protein. Inthe first approach, Hsp16.3 protein samples were pre-treated with 8 M urea at 25 8C for 24 hours and thenwere concentrated in urea. The samples were thendiluted rapidly by adding them to the in vitro E. coli tran-scription/translation extracts with radiolabeled Hsp16.3protein (acting as the refolding buffer) to final concen-trations of 0.13 mg/ml, 1.3 mg/ml and 6.5 mg/ml,respectively. The samples were then analyzed by usingnon-denaturing pore gradient PAGE (5–20%) and auto-radiography. In the second approach, the purifiedHsp16.3 protein was denatured in 8 M urea at 25 8Covernight, and then the protein sample was dilutedquickly with the refolding buffer (50 mM PBS (pH7.0))to the final concentrations of 3 M urea and 2 mg/mlHsp16.3. Meanwhile, intermediates of Hsp16.3 formedduring the refolding/re-assembling processes werecaptured by time-course cross-linking with 1% (v/v)glutaraldehyde for 5, 20, 40, 80, 160, 320, 640, 1280, and3600 seconds. The cross-linking reaction was thenstopped by adding an equivalent volume of 1 M Tris–HCl (pH 7.0). The cross-linking results were examinedthrough SDS-PAGE (12%) and protein bands werevisualized through silver staining.
Acknowledgements
We thank Dr Duanqing Pei at the Department ofPharmacology, School of Medicine, University ofMinnesota, USA and Mr Wangwang Jiao at theDepartment of Biological Science and Biotechnol-ogy, Tsinghua University for helpful discussions.This work was supported by grants from theNational Key Basic Research Foundation of China(number G1999075607), National Science Foun-dation for Outstanding Young Scientists in China(number G39725008) (to Z.C.) and sponsored bythe Scientific Foundation for Returned OverseasChinese Scholars, Ministry of Education (to A.A.).
References
1. Goodsell, D. S. & Olson, A. J. (2000). Structuralsymmetry and protein function. Annu. Rev. Biophys.Biomol. Struct. 29, 105–153.
2. Traut, T. W. (1994). Dissociation of enzyme oligo-mers: a mechanism for allosteric regulation. Crit.Rev. Biochem. Mol. Biol. 29, 125–163.
3. Bennett, M. J., Schlunegger, M. P. & Eisenberg, D.(1995). 3D domain swapping: a mechanism for oligo-mer assembly. Protein Sci. 4, 2455–2468.
4. Feng, X., Huang, S., Fu, X., Abulimiti, A. & Chang, Z.(2002). The reassembling process of the nonamericMycobacterium tuberculosis small heat-shock proteinHsp16.3 occurs via a stepwise mechanism. Biochem.J. 363, 329–334.
5. Galan, A., Sot, B., Llorca, O., Carrascosa, J. L.,Valpuesta, J. M. & Muga, A. (2001). Excluded volumeeffects on the refolding and assembly of an oligo-meric protein. GroEL, a case study. J. Biol. Chem.276, 957–964.
6. Reddy, P. S. & Corley, R. B. (1998). Assembly, sorting,and exit of oligomeric proteins from the endoplasmicreticulum. Bioessays, 20, 546–554.
7. de Jong, W. W., Caspers, G. & Leunissen, J. A. (1998).Genealogy of the alpha-crystallin small heat-shockprotein superfamily. Int. J. Biol. Macromol. 22,151–162.
8. Van Montfort, R. L. M., Basha, E., Friedrich, K. L.,Slingsby, C. & Vierling, E. (2001). Crystal structureand assembly of a eukaryotic small heat shockprotein. Nature Struct. Biol. 8, 1025–1030.
9. Kim, K. K., Kim, R. & Kim, S. H. (1998). Crystalstructure of a small heat-shock protein. Nature, 394,595–599.
10. Dudich, I. V., Zav’yalov, V. P., Pfeil, W., Gaestel, M.,Zav’yalova, G. A., Denesyuk, A. & Korpela, T.(1995). Dimer structure as a minimum cooperativesubunit of small heat-shock proteins. Biochim.Biophys. Acta, 1253, 163–168.
11. Delmas, F., Pierre, F., Coucheney, F., Divies, C. &Guzzo, J. (2001). Biochemical and physiologicalstudies of the small heat shock protein Lo18 fromthe lactic acid bacterium Oenococcus oeni. J. Mol.Microbiol. Biotechnol. 3, 601–610.
12. Mao, Q., Ke, D., Feng, X. & Chang, Z. (2001). Preheattreatment for Mycobacterium tuberculosis Hsp16.3: cor-relation between a structural phase change at 60 8Cand a dramatic increase in chaperone-like activity.Biochem. Biophys. Res. Commun. 284, 942–947.
13. Kingston, A. E., Salgame, P. R., Mitchison, N. A. &Colston, M. J. (1987). Immunological activity of a
1022 Assembly and Re-assembly Mechanism of Hsp16.3 Protein

14-kilodalton recombinant protein of Mycobacteriumtuberculosis H37Rv. Infect. Immun. 3149, 3154.
14. Lee, B. Y., Hefta, S. A. & Brennan, P. J. (1992). Majormembrane protein of virulent Mycobacteriumtuberculosis. Infect. Immun. 60, 2066–2074.
15. Chang, Z., Primm, T. P., Jakana, J., Lee, I. H., Chiu,W., Gilbert, H. F. & Quicho, F. A. (1996).Mycobacterium tuberculosis 16-kDa antigen (Hsp16.3)functions as an oligomeric structure in vitro tosuppress thermal aggregation. J. Biol. Chem. 271,7218–7223.
16. Berengian, A. R., Parfenova, M. & Mchaourab, H. S.(1999). Site-directed spin labeling study of subunitinteractions in the alpha-crystallin domain of smallheat-shock proteins. Comparison of the oligomersymmetry in alpha-crystallin, HSP 27, and HSP 16.3.J. Biol. Chem. 274, 6305–6314.
17. Yang, H., Huang, S., Dai, H., Gong, Y., Zheng, C. &Chang, Z. (1999). The Mycobacterium tuberculosissmall heat shock protein Hsp16.3 exposes hydro-phobic surfaces at mild conditions: conformationalflexibility and molecular chaperone activity. ProteinSci. 8, 174–179.
18. Dai, H., Mao, Q., Yang, H., Huang, S. & Chang, Z.(2000). Probing the roles of the only universally con-served leucine residue (Leu122) in the oligomeriza-tion and chaperone-like activity of Mycobacteriumtuberculosis small heat shock protein Hsp16.3.J. Protein Chem. 19, 319–326.
19. Mao, Q. & Chang, Z. (2001). Site-directed mutationon the only universally conserved residue Leu122 ofsmall heat shock protein Hsp16.3. Biochem. Biophys.Res. Commun. 289, 1257–1261.
20. Gu, L. C., Abulimiti, A., Li, W. & Zengyi, C. (2002).Monodisperse Hsp16.3 nonamer exhibits dynamicdissociation and reassociation, with the nonamer dis-sociation prerequisite for chaperone-like activity.J. Mol. Biol. 319, 517–526.
21. Federov, A. N. & Baldwin, T. O. (1998). Protein fold-ing and assembly in a cell-free expression system.Methods. Enzymol. 290, 1–17.
22. Jermutus, L. A., Ryabova, L. & Pluckthun, A. (1998).Recent advances in producing and selecting func-tional proteins by using cell-free translation. Curr.Opin. Biotechnol. 9, 534–548.
23. Rivas, G., Fermandez, J. A. & Minton, A. P. (2001).Direct observation of the enhancement of non-cooperative protein self-assembly by macromolecu-
lar crowding: indefinite linear self-association ofbacterial cell division protein FtsZ. Proc. Natl Acad.Sci, 98, 3150–3155.
24. Tabira, T., Ohara, N., Ohara, N., Kitaura, H.,Matsumoto, S., Naito, M. & Yamada, T. (1998). The16 kDa a-crystallin-like protein of Mycobacteriumbovic BCG is produced under conditions of oxygendeficiency and is associated with ribosomes. Res.Microbiol. 149, 255–264.
25. Zimmerman, S. B. & Trach, S. O. (1991). Estimation ofmacromolecule concentrations and excluded volumeeffects for the cytoplasm of Escherichia coli. J. Mol.Biol. 222, 599–620.
26. Ellis, R. J. (2001). Macromolecular crowding: animportant but neglected aspect of the intracellularenvironment. Curr. Opin. Struct. Biol. 11, 114–119.
27. Minton, A. P. (2001). The influence of macromolecu-lar crowding and macromolecular confinement onbiochemical reactions in physiological media. J. Biol.Chem. 276, 10577–10580.
28. Jaenicke, R. & Rudolph, R. (1986). Refolding andassociation of oligomeric protein. Methods Enzymol.131, 218–250.
29. Koteiche, H. A. & Mchaourab, H. S. (2002). Thedeterminants of the oligomeric structure in Hsp16.5are encoded in the alpha-crystallin domain. FEBSLetters, 519, 16–22.
30. Cunningham, A. F. & Spreadbury, C. L. (1998). Myco-bacterial stationary phase induced by low oxygentension: cell wall thickening and localization of the16-kilodalton alpha-crystallin homolog. J. Bacteriol.180, 801–808.
31. Tsou, C. L. (1988). Folding of the nascent peptidechain into a biologically active protein. Biochemistry,27, 1809–1812).
32. Basharov, M. A. (2000). The posttranslational conceptof protein folding: how valid is it? Biochemistry(Moscow), 65, 1184–1191.
33. Finkelstein, A. V. (1997). Can protein unfolding simu-late protein folding? Protein. Eng. 10, 843–845.
34. Ausubel, F. M., Brent, R., Kingston, R. E., Moore,D. D., Seidman, J. G., Smith, J. G. & Struhl, K.(1995). Current Protocols in Molecular Biology, 3rdedit., Wiley, New York.
35. Lesley, S. A. (1995). In vitro transcription and trans-lation protocols. Methods in Molecular Biology, vol. 37,pp. 265–278, Humana Press Inc, Totowa, NJ.
Edited by M. Moody
(Received 24 June 2002; received in revised form 7 October 2002; accepted 18 December 2002)
Assembly and Re-assembly Mechanism of Hsp16.3 Protein 1023




















