
Mycobacterium tuberculosis decreases human macrophage IFN-γ responsiveness through miR-132 and...
12
of April 15, 2016. This information is current as through miR-132 and miR-26a Responsiveness γ Human Macrophage IFN- Decreases Mycobacterium tuberculosis Michelle B. Landes and Larry S. Schlesinger Bin Ni, Murugesan V. S. Rajaram, William P. Lafuse, http://www.jimmunol.org/content/193/9/4537 doi: 10.4049/jimmunol.1400124 September 2014; 2014; 193:4537-4547; Prepublished online 24 J Immunol Material Supplementary 4.DCSupplemental.html http://www.jimmunol.org/content/suppl/2014/09/24/jimmunol.140012 References http://www.jimmunol.org/content/193/9/4537.full#ref-list-1 , 13 of which you can access for free at: cites 37 articles This article Subscriptions http://jimmunol.org/subscriptions is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/ji/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/cgi/alerts/etoc Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved. Copyright © 2014 by The American Association of 9650 Rockville Pike, Bethesda, MD 20814-3994. The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on April 15, 2016 http://www.jimmunol.org/ Downloaded from by guest on April 15, 2016 http://www.jimmunol.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Mycobacterium tuberculosis decreases human macrophage IFN-γ responsiveness through miR-132 and...

of April 15, 2016.This information is current as
through miR-132 and miR-26a ResponsivenessγHuman Macrophage IFN- DecreasesMycobacterium tuberculosis
Michelle B. Landes and Larry S. SchlesingerBin Ni, Murugesan V. S. Rajaram, William P. Lafuse,
http://www.jimmunol.org/content/193/9/4537doi: 10.4049/jimmunol.1400124September 2014;
2014; 193:4537-4547; Prepublished online 24J Immunol
MaterialSupplementary
4.DCSupplemental.htmlhttp://www.jimmunol.org/content/suppl/2014/09/24/jimmunol.140012
Referenceshttp://www.jimmunol.org/content/193/9/4537.full#ref-list-1
, 13 of which you can access for free at: cites 37 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2014 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on April 15, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on April 15, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
Mycobacterium tuberculosis Decreases Human MacrophageIFN-g Responsiveness through miR-132 and miR-26a
Bin Ni,*,†,‡ Murugesan V. S. Rajaram,†,‡ William P. Lafuse,†,‡ Michelle B. Landes,‡ and
Larry S. Schlesinger*,†,‡
IFN-g–activated macrophages play an essential role in controlling intracellular pathogens; however, macrophages also serve as
the cellular home for the intracellular pathogen Mycobacterium tuberculosis. Based on previous evidence that M. tuberculosis can
modulate host microRNA (miRNA) expression, we examined the miRNA expression profile of M. tuberculosis–infected primary
human macrophages. We identified 31 differentially expressed miRNAs in primary human macrophages during M. tuberculosis
infection by NanoString and confirmed our findings by quantitative real-time RT-PCR. In addition, we determined a role for two
miRNAs upregulated upon M. tuberculosis infection, miR-132 and miR-26a, as negative regulators of transcriptional coactivator
p300, a component of the IFN-g signaling cascade. Knockdown expression of miR-132 and miR-26a increased p300 protein levels
and improved transcriptional, translational, and functional responses to IFN-g in human macrophages. Collectively, these data
validate p300 as a target of miR-132 and miR-26a, and demonstrate a mechanism by which M. tuberculosis can limit macrophage
responses to IFN-g by altering host miRNA expression. The Journal of Immunology, 2014, 193: 4537–4547.
Macrophages are among the frontline defenders againstinvading organisms (1). However, they can also act ascellular hosts for intracellular pathogens, including
Mycobacterium tuberculosis, the causative agent of tuberculosis(2). The lymphocyte secreted cytokine IFN-g is the predominantactivator of macrophage microbicidal activity, turning on a tran-scriptional program for the killing of intracellular pathogensthrough JAK-STAT signaling downstream of the type II IFNR (3).Phosphorylation of STAT1 and formation of homodimers upontype II IFNR engagement results in translocation to the nucleuswhere the STAT1 homodimers engage with transcriptional coac-tivators, including the coactivator p300, to initiate transcription ofIFN-g–induced genes (4). p300 interacts with a variety of tran-scription factors and has been identified as an important transcrip-tional mediator of immune defenses, including IFN-g responsegenes, against many intracellular pathogens (5–9).IFN-g activity is essential for control of M. tuberculosis in
murine models (10); however, the effects of IFN-g are not as clearin humans. Although lack of IFN-g signaling predisposes indi-viduals to infections with intracellular pathogens, including sev-eral mycobacteria species (11–13), high levels of IFN-g have beendetected in the pleural fluid of TB patients (14), and IFN-g therapyfor tuberculosis in humans has not demonstrated the same strikingresults as in murine models (15). These observations can beaccounted for by the discovery that M. tuberculosis blocks IFN-g
signaling and subsequent activation of human macrophage func-tions by interrupting the p300 association with STAT1 homodimerswithin the nucleus, causing reduced binding at the g-activation se-quences of IFN-g response genes (9).MicroRNAs (miRNAs) are endogenous, noncoding, small RNAs
that function as gene regulators, most commonly by mediatingtranslational repression or degradation of target mRNAs, which canhave widespread effects on downstream pathways (16). Manipu-lation of host miRNA expression may be another mechanism bywhich M. tuberculosis is able to subvert immune detection andpersist intracellularly within macrophages. Recent discoveriesfrom our laboratory demonstrate that virulent M. tuberculosis canmodify the human macrophage immune response by modulatingmiR-125b expression, which, in turn, decreases levels of TNF-a,another major cytokine for M. tuberculosis control (17). In thisstudy, we sought to identify additional miRNAs whose expressionis altered during M. tuberculosis infection of primary human mac-rophages and elucidate the functions of some of these miRNAs.We provide evidence that M. tuberculosis induces host miRNAsmiR-132 and miR-26a. Induction of these miRNAs reduces p300expression, thereby diminishing transcription of IFN-g–inducedgenes and macrophage responsiveness to this key lymphokine.
Materials and MethodsReagents and buffers
Dulbecco’s PBS without Ca2+ and Mg2+, RPMI medium 1640 withL-glutamine, DMEM medium, HEPES buffer, and TRIzol were purchasedfrom Invitrogen. 7H11 agar plates were prepared with Bacto Middlebrook7H11 from Difco Laboratories, oleic acid-albumin-dextrose-catalase en-richment medium, and glycerol. Human serum albumin was purchasedfrom CSL Behring, and recombinant human IFN-g was obtained fromR&D Systems.
Bacterial strains and preparation for infection
Lyophilized Mycobacterium tuberculosis H37Rv (ATCC #25618) was ob-tained from the American Tissue Culture Collection, reconstituted, andused as described previously (18). The concentration of bacteria and thedegree of clumping (#10%) were determined by Petroff-Hausser chambercounting; bacteria prepared in this fashion are $90% viable by CFU assay.For paraformaldehyde-killed M. tuberculosis, the single-cell suspensionwas pelleted and resuspended in 4% paraformaldehyde for 10 min, washed
*Biomedical Sciences Graduate Program, The Ohio State University, Columbus, OH43210; †Department of Microbial Infection and Immunity, The Ohio State University,Columbus, OH 43210; and ‡Center for Microbial Interface Biology, The Ohio StateUniversity, Columbus, OH 43210
Received for publication January 16, 2014. Accepted for publication August 27,2014.
Address correspondence and reprint requests to Prof. Larry Schlesinger, Department ofMicrobial Infection and Immunity, The Ohio State University, 460 West 12th Avenue,BRT 798, Columbus, OH 43210. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: MDM; monocyte-derived macrophage; miRNA,microRNA; qRT-PCR, quantitative real-time RT-PCR; UTR, untranslated region.
Copyright� 2014 by The American Association of Immunologists, Inc. 0022-1767/14/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1400124
by guest on April 15, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

once in PBS, and resuspended in RPMI 1640 medium with 10 mM HEPESand 0.4% human serum albumin for Petroff-Hausser counting.
Isolation of human monocyte-derived macrophages and cellculture of monocyte-derived macrophages and HEK293 cells
To obtain monocyte-derived macrophages (MDMs), we obtained bloodfrom healthy, purified protein derivative–negative human volunteers usinga protocol approved by The Ohio State University Institutional ReviewBoard and processed as described previously (19, 20). In brief, PBMCswere isolated from heparinized blood on a Ficoll-Paque cushion (GEHealthcare) and cultured in Teflon wells (Savillex) in RPMI 1640 mediumcontaining 20% autologous human serum for 5 d at 37˚C/5% CO2 fordifferentiation of monocytes to MDMs. On day 5, MDMs were harvestedfrom Teflon wells and used for transfection experiments or cultured for anadditional 7 d in RPMI 1640 with 20% autologous human serum for in-fection experiments. HEK293 cells were maintained in DMEM mediasupplemented with 10% (v/v) FBS.
M. tuberculosis infection of human macrophages
Six- or 12-d-old MDMs were incubated with M. tuberculosis at a multi-plicity of infection of 5:1 in RPMI 1640 medium with 10 mM HEPES and0.4% human serum albumin for 30 min at 37˚C in 5% CO2 on a platformshaker for equal dispersion of bacteria followed by an additional 90-minincubation without shaking. MDMs were washed three times with warmRPMI 1640 and repleted with RPMI 1640 containing 2% autologous hu-man serum for the remainder of the experiment.
NanoString nCounter miRNA expression profiling
The digital multiplexed NanoString nCounter miRNA assay (NanoStringTechnologies) was used for miRNA expression profiling of 24- and 72-hM. tuberculosis–infected MDMs (n = 4 individual donors). A total of 100 nghigh-quality total RNA, including small RNAs, isolated by phenol-chloroform extraction was used as input material. Small RNAs were pre-pared for detection by ligating a specific DNA tag (miR-tag) onto the 39end of each mature miRNAwith the aid of a bridge oligomer according tothe manufacturer’s instructions. miR-tags serve not only to normalize thewide range of miRNA melting temperatures, but also aid in identificationbetween homologous miRNA family members, enabling single-bp dis-crimination. Excess tags were removed by restriction digestion, and theresulting material was hybridized with miRNA:tag-specific nCounterCapture and barcoded Reporter probes. Hybridization reactions were in-cubated at 64˚C for 18 h according to the manufacturer’s instructionsbefore removal of excess probes by a magnetic bead-based purificationprocess; purified hybridized probe complexes were immobilized on astreptavidin-coated cartridge using the nCounter Prep Station. The nCounterDigital Analyzer was used to count individual fluorescent barcodes andquantify target miRNA molecules present in each sample. For each assay,a high-density scan (600 fields of view) was performed. Data normalizationand analysis were performed by the Ohio State University BioinformaticsCore Facility according to the manufacturer’s guidelines.
NanoString data analysis
Specific background correction factors were applied to certain miRNAsaccording to the manufacturer’s directions to account for nonhybridization-dependent interactions of some bridge oligomers, miR-tags, or capture andreporter probes. Technical normalization of the code counts was performedusing spiked mRNA+ controls according to the manufacturer’s instruc-tions, and background was determined by the included negative controls.Each sample was then normalized to the geometric mean of the top 50 mosthighly expressed miRNAs. miRNAs with normalized counts ,50 (averagebackground count) in all groups were removed, and Student t test was usedto calculate the statistical significances of pairwise comparisons of theremaining miRNAs with a cutoff of p , 0.05 considered significant. Cal-culations were performed using the R statistical computing environment.
Transfection
PBMCs were transfected with 100 nM each of mirVana miR-132 and miR-26a inhibitors or mimics, together with the appropriate negative controls(Applied Biosystems) by the Amaxa Nucleofector (Lonza Group) as de-scribed previously (21, 22). In brief, 5-d-old PBMCs (1 3 107) wereresuspended in 100 ml nucleofector solution, followed by the addition ofsiRNA or control, incubated at room temperature for 5 min, and nucleo-fected according to the manufacturer’s instructions. Plasmids and miRNAmimics were cotransfected into HEK293 cells using Lipofectamine 2000(Invitrogen) according to manufacturer’s instructions.
39 Untranslated region cloning and luciferase reporter assay
A fragment of the human p300 39 untranslated region (UTR) containingbinding sites for miR-132 and miR-26a was amplified using the primers59-GCTCTAGATTTCTCTGGGTGCAAAGATG-39 and 59-GCTCTAGA-TGTCTGTCTCACACAGTTTATT-39. The amplified product was subcl-oned into the XbaI site downstream of the luciferase gene on thepGL3_CMV vector (a modified form of Promega’s pGL3). The miR-132and miR-26a binding sites in the 39UTR of p300 were mutated using theQ5 Site-Directed Mutagenesis kit (New England BioLabs) using primers59-CACTGTATAGGAGTTTAAATTTGATTTCTTATTACCTATTGTTA-AATAAAC-39 and 59-ATCCAAAGTTCATATTTATAAGTAATC-39 formiR-132 and 59- TTTCTATAAAGACCGAAAATAGCAAAAACCCTCA-ACTG-39 and 59- GTGTGTGTGTGTGTGTGTG-39 for miR-26a. Se-quencing was performed to verify the correct alignment of all constructs. Forluciferase reporter assays, HEK293 cells were cotransfected with 5 ng p30039UTR constructs and 100 nM control miRNA or miR-132 and/or miR-26amimics (Applied Biosystems) by Lipofectamine 2000. Forty-eight hoursafter transfection, luciferase activity was measured using the PromegaLuciferase Assay System according to the manufacturer’s protocol.
miRNA and gene expression studies by quantitative real-timeRT-PCR
MDM monolayers were treated with 40 ng/ml recombinant human IFN-gfor varying times. M. tuberculosis–infected or IFN-g–stimulated MDMs intriplicate wells were lysed in TRIzol reagent, and total RNA (includingsmall RNA) was isolated according to manufacturer’s instructions. RNAquantity and quality were evaluated by NanoDrop 2000 spectrophotometer.Total RNA (100 ng) was reversed transcribed to cDNA by SuperScript IIreverse transcriptase (Invitrogen), and expression of CD64, CYBB, IRF1,HLA-DRa, CXCL10, and CD86 were determined by quantitative real-timeRT-PCR (qRT-PCR) using TaqMan Gene Expression Assays (AppliedBiosystems) and normalized to b-actin as a housekeeping gene. miR-132and miR-26a cDNA were generated from RNA using the TaqManMicroRNA Reverse Transcription kit (Applied Biosystems). Expressionof miR-132 and miR-26a was determined by qRT-PCR using TaqManMicroRNA Assays with RNU44 as housekeeping control (Applied Bio-systems). Negative controls in all qRT-PCRs included no reverse tran-scriptase and no template (cDNA) groups in the reactions. Triplicatesamples were analyzed in duplicate wells in each experiment.
Western blotting
MDMmonolayers were washed once in PBS and lysed in TN-1 lysis buffer(50 mM Tris, 10 mM EDTA, 10 mM Na4PO7, 10 mM NaF, 1% TritonX-100, 125 mM NaCl, 10 mM Na3VO4, 10 mg/ml each aprotinin and leu-peptin) at 4˚C for 10 min and centrifuged at 10,000 3 g for 10 min at 4˚Cto remove cell debris (23). Protein concentrations of the cleared lysateswere measured by the BCA protein assay kit (Pierce). Protein-matchedtotal cell lysates were separated by SDS-PAGE and analyzed by Westernblot using the following Abs: p300 and b-actin (Santa Cruz), STAT1,JAK1, and JAK2 (Cell Signaling), HLA-DR (clone L243; BD Biosciences),and CD64 (clone 22.2), generously provided by Dr. Clark Anderson (TheOhio State University). The protein band intensities were measured usingthe online ImageJ software provided by the National Institutes of Health.Background intensity was subtracted from each sample and then normal-ized to the b-actin loading control.
Confocal microscopy
Transfected MDMs were adhered to glass coverslips in 24-well tissueculture plates for 2–3 h at 37˚C and washed to remove lymphocytes.MDMs were treated with IFN-g for 48 h, washed, and fixed in 2% para-formaldehyde, then blocked in blocking reagent (5% BSA, 10% FBS inPBS). Coverslips were incubated with primary Ab or the appropriateisotype control for 1 h at room temperature, washed with blocking reagent,and counterstained with an Alexa Fluor–conjugated secondary Ab (Mo-lecular Probes) for 1 h at room temperature. Nuclei were labeled with theDNA stain DAPI (Molecular Probes). Finally, coverslips were washed andmounted on glass slides and viewed using an Olympus Fluoview confocalmicroscope.
Phagocytosis assay
IgG-coated SRBCs were prepared as described previously (24) and thenincubated with transfected MDMs on coverslips at a ratio of 50:1 for 1 h inRPMI 1640 at 37˚C. Cells were subjected to brief hypotonic lysis for re-moval of extracellularly bound SRBCs and fixed in paraformaldehyde.Phagocytosis through the FcgRs was confirmed by the lack of ingested
4538 miR-132 AND miR-26a ALTER HUMAN MACROPHAGE IFN-g RESPONSES
by guest on April 15, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from
SRBCs by MDMs incubated with SRBCs not opsonized with IgG (datanot shown). Phagocytosis was measured by the total number of ingestedSRBCs in 100 MDMs (phagocytic index).
Statistical analysis
Most experiments were performed at least three independent times using threedifferent donors and yielded similar results. The magnitude of the responsefrom each independent experiment varied among the donors; however, thepattern of experimental results was the same from donor to donor. To accountfor this variability, we normalized the data to an internal control in eachexperiment. A ratio of experimental results to control was obtained, and themean ratio was then tested for a significant difference from one using tstatistics. An unpaired one-tailed Student t test was used to analyze differ-ences between groups. A p value , 0.05 was considered significant.
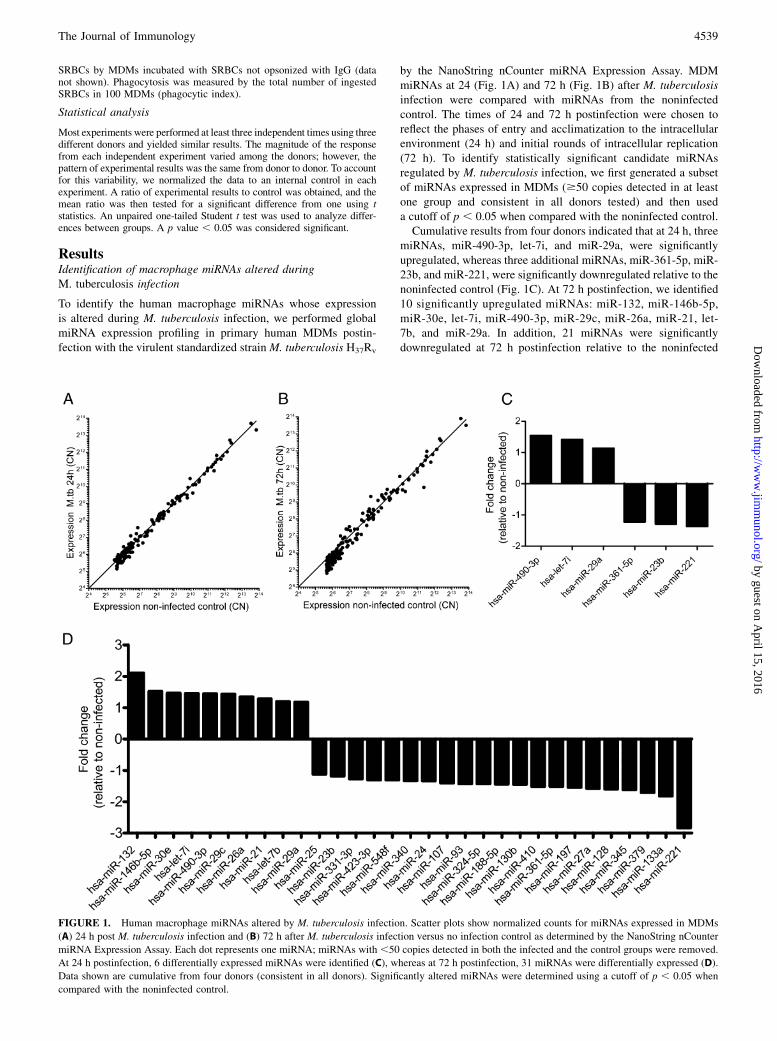
ResultsIdentification of macrophage miRNAs altered duringM. tuberculosis infection
To identify the human macrophage miRNAs whose expressionis altered during M. tuberculosis infection, we performed globalmiRNA expression profiling in primary human MDMs postin-fection with the virulent standardized strain M. tuberculosis H37Rv
by the NanoString nCounter miRNA Expression Assay. MDMmiRNAs at 24 (Fig. 1A) and 72 h (Fig. 1B) after M. tuberculosisinfection were compared with miRNAs from the noninfectedcontrol. The times of 24 and 72 h postinfection were chosen toreflect the phases of entry and acclimatization to the intracellularenvironment (24 h) and initial rounds of intracellular replication(72 h). To identify statistically significant candidate miRNAsregulated by M. tuberculosis infection, we first generated a subsetof miRNAs expressed in MDMs ($50 copies detected in at leastone group and consistent in all donors tested) and then useda cutoff of p , 0.05 when compared with the noninfected control.Cumulative results from four donors indicated that at 24 h, three
miRNAs, miR-490-3p, let-7i, and miR-29a, were significantlyupregulated, whereas three additional miRNAs, miR-361-5p, miR-23b, and miR-221, were significantly downregulated relative to thenoninfected control (Fig. 1C). At 72 h postinfection, we identified10 significantly upregulated miRNAs: miR-132, miR-146b-5p,miR-30e, let-7i, miR-490-3p, miR-29c, miR-26a, miR-21, let-7b, and miR-29a. In addition, 21 miRNAs were significantlydownregulated at 72 h postinfection relative to the noninfected
FIGURE 1. Human macrophage miRNAs altered by M. tuberculosis infection. Scatter plots show normalized counts for miRNAs expressed in MDMs
(A) 24 h post M. tuberculosis infection and (B) 72 h after M. tuberculosis infection versus no infection control as determined by the NanoString nCounter
miRNA Expression Assay. Each dot represents one miRNA; miRNAs with ,50 copies detected in both the infected and the control groups were removed.
At 24 h postinfection, 6 differentially expressed miRNAs were identified (C), whereas at 72 h postinfection, 31 miRNAs were differentially expressed (D).
Data shown are cumulative from four donors (consistent in all donors). Significantly altered miRNAs were determined using a cutoff of p , 0.05 when
compared with the noninfected control.
The Journal of Immunology 4539
by guest on April 15, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

control: miR-25, miR-23b, miR-331-3p, miR-423-3p, miR-548f,miR-340, miR-24, miR-107, miR-93, miR-324-5p, miR-188-5p,miR-130b, miR-410, miR-361-5p, miR-197, miR-27a, miR-128,miR-345, miR-379, miR-133a, and miR-221 (Fig. 1D). Of note,all six of the significantly altered miRNAs at 24 h postinfectionwere also significantly altered with the same patterns at 72 hpostinfection. Therefore, we focused our efforts on further char-acterization of miRNA functions on the regulated miRNAs at 72 hpost M. tuberculosis infection.
Validation of miRNA expression patterns by qRT-PCR
To validate the miRNA expression profiling results, we used qRT-PCR to assay expression of several upregulated and downregulatedmiRNAs including miR-132 (the most upregulated miRNA at 72 hpostinfection; Fig. 2A, 2B) and miR-26a (Fig. 2C, 2D), and ad-ditional miRNAs of interest: let-7i, miR-221 (the most downreg-ulated miRNA at 72 h postinfection), and miR-128 (Supplemental
Fig. 1). When compared with the expression profiles as estab-lished by NanoString, the qRT-PCR validation results demonstrategreat congruence between the expression patterns determinedusing these two techniques for these miRNAs. Although wecannot conclude that the patterns of alteration for these miRNAsare specific responses to M. tuberculosis, our results confirm thatmiR-132, miR-26a, let-7i, miR-221, and miR-128 are distinctlyregulated in human macrophages after M. tuberculosis infection.
Paraformaldehyde-killedM. tuberculosis also induces miR-132and miR-26a upregulation
Because the observation of increased miR-132 and miR-26aexpression was observed at 72 h after M. tuberculosis infection,which is the time at which M. tuberculosis is undergoing earlyrounds of replication in our model, we investigated whether in-duction of miR-132 and miR-26a expression requires replicatingM. tuberculosis. We stimulated MDMs with paraformaldehyde-
FIGURE 2. Validation of macrophage miRNA expression patterns during live M. tuberculosis infection or infection with paraformaldehyde-killed
M. tuberculosis. Expression patterns of selected miRNAs of interest were validated by qRT-PCR; comparison of miR-132 expression in M. tuberculosis–
infected MDMs as measured by (A) NanoString and (B) qRT-PCR, and miR-26a expression as measured by (C) NanoString and (D) qRT-PCR. NanoString
data are normalized cumulative results from four donors (mean 6 SEM). MDM expression of (E) miR-132 and (F) miR-26a were assayed by qRT-PCR
after treatment with paraformaldehyde-killed M. tuberculosis. The qRT-PCR data were performed in triplicate, normalized to RNU44 endogenous control,
and plotted as a percentage of the noninfected control expression (black bars); shown are cumulative results from three donors for live M. tuberculosis and
two donors for paraformaldehyde-killed M. tuberculosis (mean 6 SEM). *p , 0.05, **p , 0.01, ***p , 0.001.
4540 miR-132 AND miR-26a ALTER HUMAN MACROPHAGE IFN-g RESPONSES
by guest on April 15, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

killed M. tuberculosis and assessed miR-132 and miR-26a expres-sion by qRT-PCR. Paraformaldehyde-fixed M. tuberculosis is nolonger viable and cannot secrete mycobacterial products, but itpossesses an intact cell-wall structure (25). The results in Fig. 2Eand 2F show that treatment of MDMs with paraformaldehyde-killedM. tuberculosis also induced miR-132 and miR-26a expression at72 h, indicating that replication is not required for the effect observed
and suggesting that an M. tuberculosis cell-wall component(s) issufficient to induce miR-132 and miR-26a expression in MDMs.
Identification of transcriptional coactivator p300 as a target ofM. tuberculosis upregulated miR-132 and miR-26a
We selected miR-132 for further analysis because it was the mostupregulated miRNA at 72 h postinfection and its expression pattern
FIGURE 3. p300 is a target of miR-132 and miR-26a in human macrophages. Knockdown (A and B) or overexpression (C and D) of miR-132 and miR-
26a was achieved by transfection of specific inhibitors or mimics and confirmed by qRT-PCR. p300 protein levels were analyzed by Western blot of protein-
matched total cell lysates from (E) double miRNA knockdown or (F) double miRNA overexpressing MDMs (left panels). Blots were probed with actin as
a loading control. Densitometry analysis of the immunoblot results show an increase in p300 levels with miR-132 and miR-26a knockdown, whereas p300
protein levels decreased with miR-132 and miR-26a overexpression (right panels). Shown are representative Western blots and cumulative densitometry
data representing data from five individual experiments for the knockdown and six individual experiments for the overexpressor groups (mean 6 SEM,
*p , 0.05, **p , 0.01, ***p , 0.001).
The Journal of Immunology 4541
by guest on April 15, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

was validated by qRT-PCR. To identify the gene targets of miR-132, we sought previously validated targets in literature, as wellas predicted gene targets by miRWalk, an online software formiRNA target prediction that aggregates miRNA target predictionsfrom 10 computational algorithms (DIANAmT, miRanda, miRDB,miRWalk, RNAhybrid, PICTAR4, PICTAR5, PITA, RNA22, andTargetscan). Among the putative target genes of interest wastranscriptional coactivator p300, previously identified as an miR-132 target in endothelial and monocytic models of viral infec-tion (5), and predicted by 9 of 10 algorithms as a target of miR-132 based on sequence. In addition, miR-26a, another miRNAsignificantly upregulated at 72 h after M. tuberculosis infection,was also predicted to target p300 by 7 of 10 algorithms, althoughno such biological evidence yet exists in the literature. Therefore,there is solid support for some of these differentially regulatedmiRNAs to have a functional role during M. tuberculosis infec-tion by targeting p300, a component of an important macrophageimmune response pathway.
Changes in p300 protein levels correlate with alterations inmiR-132 and miR-26a expression
To establish p300 as a target of miR-132 and miR-26a in humanmacrophages, we used transient knockdown or overexpression ofthese two miRNAs by transfection of miR-132– and miR-26a–specific inhibitors or mimics and assayed for p300 protein levelsby Western blot analysis of transfected total cell lysates. Confir-mation of miR-132 and miR-26a knockdown and overexpressionby qRT-PCR demonstrated significant decrease (Fig. 3A, 3B) orincrease (Fig. 3C, 3D) of miRNA expression when compared withthe appropriate transfection control. Knockdown of miR-132 andmiR-26a in MDMs by miRNA inhibitors resulted in an increasein p300 protein levels when compared with the inhibitor negativecontrol (Fig. 3E). Conversely, overexpression of miR-132 andmiR-26a by miRNA mimics led to reduced p300 protein levelswhen compared with the corresponding mimic negative control(Fig. 3F). Individual knockdown or overexpression of miR-132 ormiR-26a alone also produced similar results on p300 levels, butthe effects were not as pronounced as the combined knockdown oroverexpressing findings (Supplemental Fig. 2). Based on thesedata, we conclude that p300 is also a target of miR-132 in humanmacrophages; further, we demonstrate the novel finding that miR-26a also targets p300.
miR-132 and miR-26a target p300 by directly binding to thep300 39UTR
To demonstrate that miR-132 and miR-26a directly regulate p300protein levels by targeting p300 mRNA, we cloned the p300 39UTRfragment containing the binding sites for both miR-132 and miR-26a into a luciferase reporter plasmid (pGL3_CMV) downstreamof the luciferase gene. Using HEK293 cells for the target valida-tion assays, we cotransfected the reporter plasmid containing thep300 39UTR fragment along with either miR-132 or miR-26amimic alone, both mimics combined, or scrambled control byLipofectamine 2000, and after 48 h we assessed luciferase activity.Our results showed a significant decrease in light production withaddition of both miR-132 and miR-26a mimics as compared withthe scrambled control (Fig. 4). The combined effect of both miR-132 and miR-26a on quenching luciferase activity was strongerthan addition of the individual miRNAs. To further demonstratethe specificity of miR-132 and miR-26a for the p300 39UTR, wemutated four nucleotides in the seed sequence of the 39UTRbinding site for each miRNA (indicated in bold). Luciferase ex-pression from the plasmids with mutated p300 39UTR seedsequences remained at similar levels after cotransfection with both
miR-132 and miR-26a mimics, and transfection with the scram-bled control. These data provide evidence that miR-132 and miR-26a alter p300 protein levels by directly targeting p300 mRNAthrough binding sites in the p300 39UTR.
miR-132 and miR-26a regulate transcriptional andtranslational macrophage responses to IFN-g
Because p300 is an important transcriptional mediator of IFN-gresponses, we hypothesized that knockdown of miR-132 and miR-26a could improve IFN-g responses in human macrophages viaincreased p300 levels. We assessed the expression of six knownIFN-g–responsive genes encompassing a variety of macrophagefunctions, including FcgRI/CD64, CYBB, IRF1, HLA-DRa,CXCL10, and B.7.2/CD86, by qRT-PCR in miR-132 and miR-26aknockdown MDMs (24-h transfection) after treatment with rIFN-gfor 6 h or 24 h. Our results demonstrate increased transcriptionalexpression of all six genes upon IFN-g stimulation in miR-132and miR-26a knockdown cells when compared with the trans-fection control cells (also treated with IFN-g; Fig. 5A–F). Inaddition to increased mRNA levels, we observed increased cell-surface protein expression of FcgRI/CD64 (Fig. 5G) and HLA-DR (Fig. 5H) in knockdown MDMs by confocal microscopy at48 h after IFN-g treatment, indicating that there was also in-creased translational expression. Western blot analysis of FcgRIand HLA-DR expression in MDMs after miR-132 and miR-26aknockdown without IFN-g stimulation demonstrated no change inFcgRI expression and a small but significant increase in HLA-DR(data not shown). Therefore, we demonstrate that knockdown ofmiR-132 and miR-26a in human macrophages can improve tran-
FIGURE 4. miR-132 and miR-26a directly target the p300 39UTR.
Luciferase reporter plasmids were constructed with either the wild-type
p300 39UTR segment containing the miR-132 and miR-26a binding sites
(p300 UTR) or the p300 39UTR with seed sequences mutated as indicated
(p300 mut). Mutated sequences are indicated in bold. Luciferase activity
of HEK293 cells transfected with either the p300 UTR or the p300 mut
plasmid along with miR-132 and/or miR-26a mimics or scrambled control
by Lipofectamine 2000 was determined. Luciferase activity was normal-
ized to total protein in each group. Data shown are the mean6 SD of three
replicates of one representative experiment (n = 2). *p , 0.05.
4542 miR-132 AND miR-26a ALTER HUMAN MACROPHAGE IFN-g RESPONSES
by guest on April 15, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

scriptional responses to IFN-g (via increased expression of p300),and these increased mRNA levels correspond to increases inprotein expression of IFN-g–induced genes.To demonstrate that the previously observed increases in tran-
scriptional and translational responses to IFN-g are a result ofmiR-132 and miR-26a targeting of p300 and not off-target effectsof these miRNAs on other IFN-g pathway proteins, we assessedeffects of miR-132 and miR-26a knockdown on integral downstreamIFN-g signaling proteins. Binding of IFN-g to its receptor resultsin recruitment of JAK1 and JAK2, which phosphorylates STAT1leading to its homodimerization and translocation to the nucleus toassociate with p300. Western blot analyses of STAT1, JAK1, andJAK2 (Fig. 6A–C) after miR-132 and miR-26a knockdown inMDMs show no significant changes in expression of these proteins
relative to the negative control group, indicating that miR-132 andmiR-26a do not affect expression of these IFN-g pathway proteins.In addition, to further demonstrate that the increased tran-
scriptional responses after miR-132 and miR-26a knockdown aremediated by increased p300 protein levels and not a result ofwidespread increases in transcription due to miRNA knockdown,we assessed transcriptional responses of IFN-g–induced JAK-STAT–independent genes. These genes include CCL3/MIP-1a,CCL4/MIP-1b, and CEBPb, which are known to be induced byIFN-g but whose signaling after IFN-g stimulation is not mediatedthrough the JAK-STAT signaling pathway, and thus do not requireassociation of STAT1 homodimers with p300 to initiate theirtranscription (26, 27). After miR-132 and miR-26a knockdown inMDMs and treatment with IFN-g, transcriptional levels of CCL3/
FIGURE 5. miR-132 and miR-26a knockdown improves macrophage responses to IFN-g. miR-132 and miR-26a were knocked down in MDMs by
transfection with miR-132 and miR-26a inhibitors, and then stimulated with 40 ng/ml recombinant human IFN-g. Cells were lysed at 6 or 24 h to assay for
transcription of known IFN-g–responsive genes (A) CD64/FcgRI (n = 5), (B) CYBB/p91-PHOX (n = 5), (C) IRF-1 (n = 4), (D) HLA-DRa (n = 4), (E)
CXCL10 (n = 3), and (F) CD86/B7.2 (n = 4) by qRT-PCR. Shown are cumulative data from multiple donors, normalized to b-actin, each performed in
triplicate (mean 6 SEM). Transfected MDMs on coverslips were stimulated with IFN-g for 48 h and stained with Ab for FcgRI (G) or HLA-DR (H) and
examined by confocal microscopy (original magnification 3400). Shown are representative sections from one of three independent experiments performed
on triplicate coverslips. Mean fluorescence intensity was calculated from at least 10 cross-sectional images and normalized to the number of nuclei, then
plotted as fold increase relative to the transfection control group (n = 3, mean 6 SEM). *p , 0.05, **p , 0.01, ***p , 0.001.
The Journal of Immunology 4543
by guest on April 15, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from
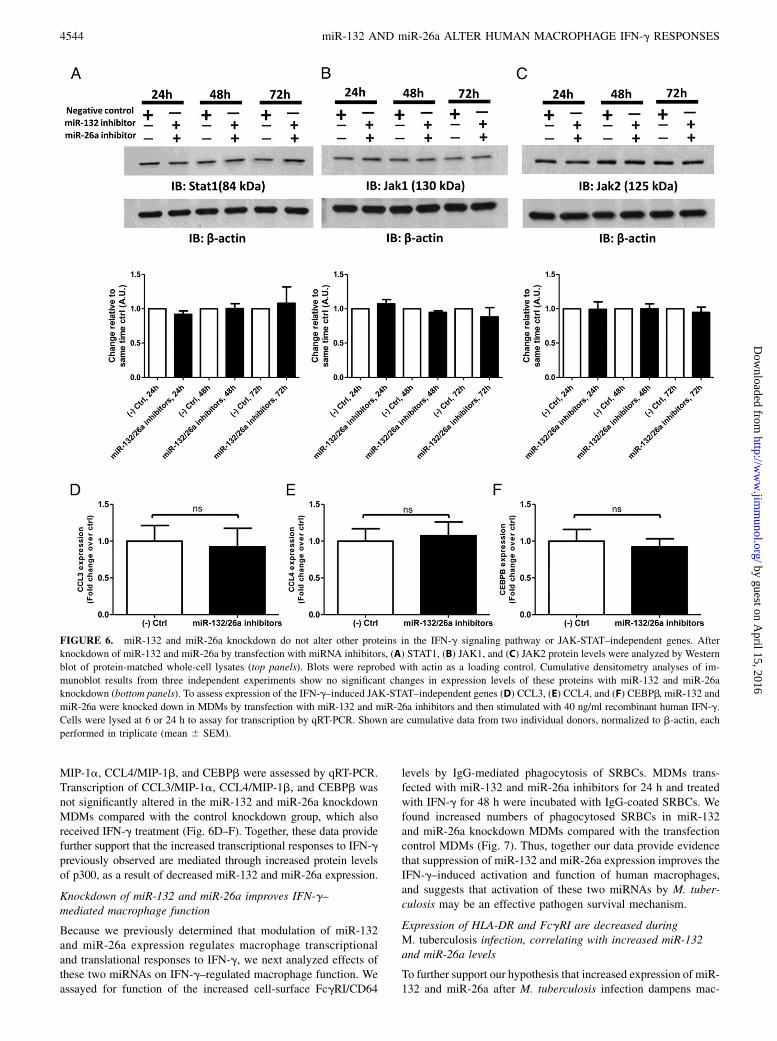
MIP-1a, CCL4/MIP-1b, and CEBPb were assessed by qRT-PCR.Transcription of CCL3/MIP-1a, CCL4/MIP-1b, and CEBPb wasnot significantly altered in the miR-132 and miR-26a knockdownMDMs compared with the control knockdown group, which alsoreceived IFN-g treatment (Fig. 6D–F). Together, these data providefurther support that the increased transcriptional responses to IFN-gpreviously observed are mediated through increased protein levelsof p300, as a result of decreased miR-132 and miR-26a expression.
Knockdown of miR-132 and miR-26a improves IFN-g–mediated macrophage function
Because we previously determined that modulation of miR-132and miR-26a expression regulates macrophage transcriptionaland translational responses to IFN-g, we next analyzed effects ofthese two miRNAs on IFN-g–regulated macrophage function. Weassayed for function of the increased cell-surface FcgRI/CD64
levels by IgG-mediated phagocytosis of SRBCs. MDMs trans-fected with miR-132 and miR-26a inhibitors for 24 h and treatedwith IFN-g for 48 h were incubated with IgG-coated SRBCs. Wefound increased numbers of phagocytosed SRBCs in miR-132and miR-26a knockdown MDMs compared with the transfectioncontrol MDMs (Fig. 7). Thus, together our data provide evidencethat suppression of miR-132 and miR-26a expression improves theIFN-g–induced activation and function of human macrophages,and suggests that activation of these two miRNAs by M. tuber-culosis may be an effective pathogen survival mechanism.
Expression of HLA-DR and FcgRI are decreased duringM. tuberculosis infection, correlating with increased miR-132and miR-26a levels
To further support our hypothesis that increased expression of miR-132 and miR-26a after M. tuberculosis infection dampens mac-
FIGURE 6. miR-132 and miR-26a knockdown do not alter other proteins in the IFN-g signaling pathway or JAK-STAT–independent genes. After
knockdown of miR-132 and miR-26a by transfection with miRNA inhibitors, (A) STAT1, (B) JAK1, and (C) JAK2 protein levels were analyzed by Western
blot of protein-matched whole-cell lysates (top panels). Blots were reprobed with actin as a loading control. Cumulative densitometry analyses of im-
munoblot results from three independent experiments show no significant changes in expression levels of these proteins with miR-132 and miR-26a
knockdown (bottom panels). To assess expression of the IFN-g–induced JAK-STAT–independent genes (D) CCL3, (E) CCL4, and (F) CEBPb, miR-132 and
miR-26a were knocked down in MDMs by transfection with miR-132 and miR-26a inhibitors and then stimulated with 40 ng/ml recombinant human IFN-g.
Cells were lysed at 6 or 24 h to assay for transcription by qRT-PCR. Shown are cumulative data from two individual donors, normalized to b-actin, each
performed in triplicate (mean 6 SEM).
4544 miR-132 AND miR-26a ALTER HUMAN MACROPHAGE IFN-g RESPONSES
by guest on April 15, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

rophage immune functions, we assayed for expression of FcgRIand HLA-DR inM. tuberculosis–infected MDMs by Western blot.We observed decreased expression of both FcgRI (Fig. 8A) andHLA-DR (Fig. 8B) 72 h postinfection as compared with the time-matched noninfected lysates. These decreases in protein levelscorrelate with the increase in miR-132 and miR-26a expressionobserved during our initial screen of miRNAs altered duringM. tuberculosis infection. When increased expression of miR-132and miR-26a after M. tuberculosis infection was prevented by trans-fection of miR-132 and miR-26a inhibitors, we observed small butreproducible increases in FcgRI (Fig. 8C) and HLA-DR (Fig. 8D)expression as compared with the scrambled control at 72 h after
M. tuberculosis infection. Together, our data indicate that in-creased host miRNA expression of miR-132 and miR-26a down-regulate key macrophage host defense functions and provideanother mechanism used by M. tuberculosis to ensure its survival.
DiscussionRecent years have witnessed a spate of investigation on the dys-regulation and roles of miRNAs during bacterial infections (28),including M. tuberculosis (29). However, with some exceptions(17, 30–35), the majority of these studies have failed to identifydownstream effects on the host immune response for the miRNAsdetected with altered expression. A major contributing factor tothe lack of concrete targets is the use of mixed cell populations orsputum for the identification of differentially expressed miRNAsduring M. tuberculosis infection. M. tuberculosis is an ancientorganism that has coevolved with its human host; therefore, it isuniquely adapted for survival within its intracellular host: themacrophage (36). To date, little data exist on how miRNAs alterhuman macrophage immune responses during infection, which arethe first phagocyte immune responders to M. tuberculosis in thelung microenvironment (37). This study emphasizes the effects ofprimary M. tuberculosis infection, which determines the outcomeof active or latent infection, on macrophage miRNAs using a pureprimary human macrophage model of tuberculosis. We haveidentified 31 miRNAs differentially expressed in primary humanmacrophages during infection with virulent M. tuberculosis anddemonstrate an immune effect for two of these miRNAs: miR-132and miR-26a.By studying a pure population of human macrophages, we were
able to pinpoint a direct effect of two miRNAs altered duringM. tuberculosis infection on specific macrophage functions,which has broad implications for both M. tuberculosis and othermacrophage-mediated diseases. Our data demonstrate that througha single miRNA, or multiple miRNAs acting in concert, an in-
FIGURE 7. miR-132 and miR-26a knockdown improve FcgR-mediated
phagocytosis. miR-132 and miR-26a inhibitor or control transfected
MDMs were incubated with IgG-coated SRBCs. Phagocytosis was mea-
sured by counting the total number of SRBCs ingested by 100 MDMs
(phagocytic index). Three independent experiments were performed and
scored (mean 6 SEM, *p , 0.05).
FIGURE 8. miR-132 and miR-26a regulate macrophage responses to M. tuberculosis. Protein-matched total cell lysates from MDMs with and without
M. tuberculosis infection (72 h) were probed for FcgRI (A) and HLA-DR (B) by Western blot and then reprobed for actin as a loading control. miR-132 and
miR-26a inhibitor or scrambled control transfected MDMs were probed for FcgRI (C) and HLA-DR (D) at 72 h after M. tuberculosis infection and then
reprobed for actin as a loading control. Shown are representative Western blots and cumulative densitometry data representing data from two individual
experiments (mean 6 SEM, *p , 0.05, **p , 0.01, ***p , 0.001).
The Journal of Immunology 4545
by guest on April 15, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

trinsic macrophage activation pathway can be tuned up or down,in this case, to the benefit of the invading pathogen. In identifyingp300 as a target of upregulated miRNAs miR-132 and miR-26aduring M. tuberculosis infection, we have uncovered another layerto the conundrum of IFN-g function against human tuberculosis.Building on the work of Ting et al. (9) that M. tuberculosis in-fection results in decreased association of p300 at g-activationsequence elements after IFN-g treatment, our work supports therole of miRNAs in mediating this effect, providing a mechanismfor their observation. In total, this work supports the concept thatM. tuberculosis modulates miR-132 and miR-26a, in addition toother host miRNAs, which can attenuate host immune responsesto promote its own survival.It stands to reason that miR-132 and miR-26a may normally
function as negative regulators of macrophage activation by IFN-g.In the case of M. tuberculosis pulmonary infection, induction ofthese two miRNAs in alveolar macrophages serves to limit theimmune response and damage to the delicate alveolar tissue space.However, whether the induction of miR-132 and miR-26a is ini-tiated by M. tuberculosis or through internal host pathways wasnot addressed in this study and will be the focus of future work. Inaddition, whether this phenomenon is specific to virulent myco-bacterial strains also requires further study. Regardless of whetherit is the host or the pathogen that drives upregulation of miR-132and miR-26a expression, we have assigned a novel role for miR-132 and miR-26a in human macrophages as regulators of the IFN-g–induced macrophage activation pathway. This knowledge canlead the way to therapeutic regulation of macrophage activationvia miRNAs for diseases of overt Th1-type IFN-g–mediated mac-rophage activation, or as adjunct therapy with IFN-g for treatmentof tuberculosis.
AcknowledgmentsWe thank Drs. Susheela Tridandapani and Jon Butchar for providing IgG-
coated SRBCs, and Drs. Clark Anderson and Latha Prabha Ganesan for
providing the FcgRI Ab. The NanoString nCounter miRNA assay was
performed at The Ohio State University Comprehensive Cancer Center
Nucleic Acid Facility.
DisclosuresThe authors have no financial conflicts of interest.
References1. Pl€uddemann, A., S. Mukhopadhyay, and S. Gordon. 2011. Innate immunity to
intracellular pathogens: macrophage receptors and responses to microbial entry.Immunol. Rev. 240: 11–24.
2. Fenton, M. J. 1998. Macrophages and tuberculosis. Curr. Opin. Hematol. 5: 72–78.3. Schroder, K., P. J. Hertzog, T. Ravasi, and D. A. Hume. 2004. Interferon-gamma:
an overview of signals, mechanisms and functions. J. Leukoc. Biol. 75: 163–189.4. Platanias, L. C. 2005. Mechanisms of type-I- and type-II-interferon-mediated
signalling. Nat. Rev. Immunol. 5: 375–386.5. Lagos, D., G. Pollara, S. Henderson, F. Gratrix, M. Fabani, R. S. Milne, F. Gotch,
and C. Boshoff. 2010. miR-132 regulates antiviral innate immunity throughsuppression of the p300 transcriptional co-activator. Nat. Cell Biol. 12: 513–519.
6. Bouhet, S., V. Lafont, E. Billard, A. Gross, and J. Dornand. 2009. TheIFNgamma-induced STAT1-CBP/P300 association, required for a normal re-sponse to the cytokine, is disrupted in Brucella-infected macrophages. Microb.Pathog. 46: 88–97.
7. Darieva, Z., E. B. Lasunskaia, M. N. Campos, T. L. Kipnis, and W. D. Da Silva.2004. Activation of phosphatidylinositol 3-kinase and c-Jun-N-terminal kinasecascades enhances NF-kappaB-dependent gene transcription in BCG-stimulatedmacrophages through promotion of p65/p300 binding. J. Leukoc. Biol. 75: 689–697.
8. Sen, N., M. R. Hara, M. D. Kornberg, M. B. Cascio, B. I. Bae, N. Shahani,B. Thomas, T. M. Dawson, V. L. Dawson, S. H. Snyder, and A. Sawa. 2008.Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apo-ptosis. Nat. Cell Biol. 10: 866–873.
9. Ting, L.-M., A. C. Kim, A. Cattamanchi, and J. D. Ernst. 1999. Mycobacteriumtuberculosis inhibits IFN-g transcriptional responses without inhibiting activa-tion of STAT1. J. Immunol. 163: 3898–3906.
10. Flynn, J. L., J. Chan, K. J. Triebold, D. K. Dalton, T. A. Stewart, andB. R. Bloom. 1993. An essential role for interferon gamma in resistance toMycobacterium tuberculosis infection. J. Exp. Med. 178: 2249–2254.
11. Jouanguy, E., F. Altare, S. Lamhamedi-Cherradi, and J. L. Casanova. 1997.Infections in IFNGR-1-deficient children. J. Interferon Cytokine Res. 17: 583–587.
12. Newport, M. J., C. M. Huxley, S. Huston, C. M. Hawrylowicz, B. A. Oostra,R. Williamson, and M. Levin. 1996. A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N. Engl. J. Med.335: 1941–1949.
13. Dorman, S. E., and S. M. Holland. 1998. Mutation in the signal-transducingchain of the interferon-gamma receptor and susceptibility to mycobacterial in-fection. J. Clin. Invest. 101: 2364–2369.
14. Ribera, E., I. Ocana, J. M. Martinez-Vazquez, M. Rossell, T. Espanol, andA. Ruibal. 1988. High level of interferon gamma in tuberculous pleural effusion.Chest 93: 308–311.
15. Rook, G. A., J. Steele, M. Ainsworth, and B. R. Champion. 1986. Activation ofmacrophages to inhibit proliferation ofMycobacterium tuberculosis: comparisonof the effects of recombinant gamma-interferon on human monocytes and mu-rine peritoneal macrophages. Immunology 59: 333–338.
16. Bartel, D. P. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function.Cell 116: 281–297.
17. Rajaram, M. V., B. Ni, J. D. Morris, M. N. Brooks, T. K. Carlson,B. Bakthavachalu, D. R. Schoenberg, J. B. Torrelles, and L. S. Schlesinger. 2011.Mycobacterium tuberculosis lipomannan blocks TNF biosynthesis by regulatingmacrophage MAPK-activated protein kinase 2 (MK2) and microRNA miR-125b.Proc. Natl. Acad. Sci. USA 108: 17408–17413.
18. Schlesinger, L. S., C. G. Bellinger-Kawahara, N. R. Payne, and M. A. Horwitz.1990. Phagocytosis of Mycobacterium tuberculosis is mediated by humanmonocyte complement receptors and complement component C3. J. Immunol.144: 2771–2780.
19. Horwitz, M. A., and S. C. Silverstein. 1980. Influence of the Escherichia colicapsule on complement fixation and on phagocytosis and killing by humanphagocytes. J. Clin. Invest. 65: 82–94.
20. Schlesinger, L. S. 1993. Macrophage phagocytosis of virulent but not attenuatedstrains of Mycobacterium tuberculosis is mediated by mannose receptors inaddition to complement receptors. J. Immunol. 150: 2920–2930.
21. Rajaram, M. V., M. N. Brooks, J. D. Morris, J. B. Torrelles, A. K. Azad, andL. S. Schlesinger. 2010. Mycobacterium tuberculosis activates human macro-phage peroxisome proliferator-activated receptor gamma linking mannose re-ceptor recognition to regulation of immune responses. J. Immunol. 185: 929–942.
22. Brooks, M. N., M. V. Rajaram, A. K. Azad, A. O. Amer, M. A. Valdivia-Arenas, J. H. Park, G. Nunez, and L. S. Schlesinger. 2011. NOD2 controls thenature of the inflammatory response and subsequent fate of Mycobacteriumtuberculosis and M. bovis BCG in human macrophages. Cell. Microbiol. 13:402–418.
23. Henning, L. N., A. K. Azad, K. V. Parsa, J. E. Crowther, S. Tridandapani,and L. S. Schlesinger. 2008. Pulmonary surfactant protein A regulatesTLR expression and activity in human macrophages. J. Immunol. 180:7847–7858.
24. Ganesan, L. P., G. Wei, R. A. Pengal, L. Moldovan, N. Moldovan,M. C. Ostrowski, and S. Tridandapani. 2004. The serine/threonine kinase AktPromotes Fc gamma receptor-mediated phagocytosis in murine macrophagesthrough the activation of p70S6 kinase. J. Biol. Chem. 279: 54416–54425.
25. Kang, P. B., A. K. Azad, J. B. Torrelles, T. M. Kaufman, A. Beharka, E. Tibesar,L. E. DesJardin, and L. S. Schlesinger. 2005. The human macrophage mannosereceptor directs Mycobacterium tuberculosis lipoarabinomannan-mediatedphagosome biogenesis. J. Exp. Med. 202: 987–999.
26. Ramana, C. V., M. P. Gil, R. D. Schreiber, and G. R. Stark. 2002. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling.Trends Immunol. 23: 96–101.
27. Hu, J., S. K. Roy, P. S. Shapiro, S. R. Rodig, S. P. Reddy, L. C. Platanias,R. D. Schreiber, and D. V. Kalvakolanu. 2001. ERK1 and ERK2 activateCCAAAT/enhancer-binding protein-beta-dependent gene transcription in re-sponse to interferon-gamma. J. Biol. Chem. 276: 287–297.
28. Eulalio, A., L. Schulte, and J. Vogel. 2012. The mammalian microRNA responseto bacterial infections. RNA Biol. 9: 742–750.
29. Singh, P. K., A. V. Singh, and D. S. Chauhan. 2013. Current understanding onmicro RNAs and its regulation in response to Mycobacterial infections. J.Biomed. Sci. 20: 14.
30. Dorhoi, A., M. Iannaccone, M. Farinacci, K. C. Fae, J. Schreiber, P. Moura-Alves, G. Nouailles, H. J. Mollenkopf, D. Oberbeck-M€uller, S. Jorg, et al. 2013.MicroRNA-223 controls susceptibility to tuberculosis by regulating lung neu-trophil recruitment. J. Clin. Invest. 123: 4836–4848.
31. Ma, F., S. Xu, X. Liu, Q. Zhang, X. Xu, M. Liu, M. Hua, N. Li, H. Yao, andX. Cao. 2011. The microRNA miR-29 controls innate and adaptive immuneresponses to intracellular bacterial infection by targeting interferon-g. Nat.Immunol. 12: 861–869.
32. Liu, Y., X. Wang, J. Jiang, Z. Cao, B. Yang, and X. Cheng. 2011. Modulation ofT cell cytokine production by miR-144* with elevated expression in patientswith pulmonary tuberculosis. Mol. Immunol. 48: 1084–1090.
33. Liu, Y., J. Jiang, X. Wang, F. Zhai, and X. Cheng. 2013. miR-582-5p is up-regulated in patients with active tuberculosis and inhibits apoptosis of monocytesby targeting FOXO1. PLoS ONE 8: e78381.
34. Kumar, R., P. Halder, S. K. Sahu, M. Kumar, M. Kumari, K. Jana, Z. Ghosh,P. Sharma, M. Kundu, and J. Basu. 2012. Identification of a novel role of ESAT-
4546 miR-132 AND miR-26a ALTER HUMAN MACROPHAGE IFN-g RESPONSES
by guest on April 15, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

6-dependent miR-155 induction during infection of macrophages with Myco-bacterium tuberculosis. Cell. Microbiol. 14: 1620–1631.
35. Wang, J., K. Yang, L. Zhou, Y. Minhaowu, Y. Wu, M. Zhu, X. Lai, T. Chen,L. Feng, M. Li, et al. 2013. MicroRNA-155 promotes autophagy toeliminate intracellular mycobacteria by targeting Rheb. PLoS Pathog. 9:e1003697.
36. Gagneux, S. 2012. Host-pathogen coevolution in human tuberculosis. Philos.Trans. R. Soc. Lond. B Biol. Sci. 367: 850–859.
37. Das, K., S. Saikolappan, and S. Dhandayuthapani. 2013. Differentialexpression of miRNAs by macrophages infected with virulent and avir-ulent Mycobacterium tuberculosis. Tuberculosis (Edinb.) 93(Suppl.):S47–S50.
The Journal of Immunology 4547
by guest on April 15, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from




















