
Mutational analysis and NMR spectroscopy of quail cysteine and glycine-rich protein CRP2 reveal an...
16
Mutational Analysis and NMR Spectroscopy of Quail Cysteine and Glycine-rich Protein CRP2 reveal an Intrinsic Segmental Flexibility of LIM Domains Karin Kloiber 1 , Ralf Weiskirchen 2 , Bernhard Kra ¨ utler 1 , Klaus Bister 2 and Robert Konrat 1 * 1 Institute of Organic Chemistry 2 Institute of Biochemistry University of Innsbruck A-6020, Innsbruck, Austria The LIM domain is a conserved cysteine and histidine-containing structural module of two tandemly arranged zinc fingers. It has been identified in single or multiple copies in a variety of regulatory proteins, either in combi- nation with defined functional domains, like homeodomains, or alone, like in the CRP family of LIM proteins. Structural studies of CRP proteins have allowed a detailed evaluation of interactions in LIM-domains at the mol- ecular level. The packing interactions in the hydrophobic core have been identified as a significant contribution to the LIM domain fold, whereas hydrogen bonding within each single zinc binding site stabilizes zinc finger geometry in a so-called ‘‘outer’’ or ‘‘indirect’’ coordination sphere. Here we report the solution structure of a point-mutant of the carboxyl-terminal LIM domain of quail cysteine and glycine-rich protein CRP2, CRP2(LIM2)R122A, and discuss the structural consequences of the disrup- tion of the hydrogen bond formed between the guanidinium side-chain of Arg122 and the zinc-coordinating cysteine thiolate group in the CCHC rubredoxin-knuckle. The structural analysis revealed that the three-dimen- sional structure of the CCHC zinc binding site in CRP2(LIM2)R122A is adapted as a consequence of the modified hydrogen bonding pattern. Additionally, as a result of the conformational rearrangement of the zinc binding site, the packing interactions in the hydrophobic core region are altered, leading to a change in the relative orientation of the two zinc fin- gers with a concomitant change in the solvent accessibilities of hydro- phobic residues located at the interface of the two modules. The backbone dynamics of residues located in the folded part of CRP2(LIM2)R122A have been characterized by proton-detected 15 N NMR spectroscopy. Analysis of the R 2 /R 1 ratios revealed a rotational correlation time of 6.2 ns and tum- bling with an axially symmetric diffusion tensor (D k /D ? 1.43). The relax- ation data were also analyzed using a reduced spectral density mapping approach. As in wild-type CRP2(LIM2), significant mobility on a pico- second/nanosecond time-scale was detected, and conformational exchange on a microsecond time-scale was identified for residues located in loop regions between secondary structure elements. In summary, the relative orientation of the two zinc binding sites and the accessibility of hydro- phobic residues is not only determined by hydrophobic interactions, but can also be modified by the formation and/or breakage of hydrogen bonds. This may be important for the molecular interactions of an adaptor- type LIM domain protein in macromolecular complexes, particularly for the modulation of protein-protein interactions. # 1999 Academic Press E-mail address of the corresponding author: [email protected] Abbreviations used: CRP, cysteine and glycine-rich protein; LIM, specific double zinc finger domain; LIM1, amino- terminal LIM domain of CRP protein; LIM2, carboxyl-terminal LIM domain of CRP protein; CSRP, gene encoding CRP protein; CRIP, cysteine-rich intestinal protein; PCR, polymerase chain reaction; NOE, nuclear Overhauser effect; HSQC, heteronuclear single-quantum correlation spectroscopy; T 1 , longitudinal relaxation time; T 2 , transverse relaxation time. Article No. jmbi.1999.3118 available online at http://www.idealibrary.com on J. Mol. Biol. (1999) 292, 893–908 0022-2836/99/390893–16 $30.00/0 # 1999 Academic Press
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Mutational analysis and NMR spectroscopy of quail cysteine and glycine-rich protein CRP2 reveal an...

Article No. jmbi.1999.3118 available online at http://www.idealibrary.com on J. Mol. Biol. (1999) 292, 893±908
Mutational Analysis and NMR Spectroscopy of QuailCysteine and Glycine-rich Protein CRP2 reveal anIntrinsic Segmental Flexibility of LIM Domains
Karin Kloiber1, Ralf Weiskirchen2, Bernhard KraÈutler1, Klaus Bister2
and Robert Konrat1*
1Institute of Organic Chemistry2Institute of BiochemistryUniversity of InnsbruckA-6020, Innsbruck, Austria
E-mail address of the correspond
Abbreviations used: CRP, cysteinterminal LIM domain of CRP proteCRP protein; CRIP, cysteine-rich intHSQC, heteronuclear single-quanturelaxation time.
0022-2836/99/390893±16 $30.00/0
The LIM domain is a conserved cysteine and histidine-containing structuralmodule of two tandemly arranged zinc ®ngers. It has been identi®ed insingle or multiple copies in a variety of regulatory proteins, either in combi-nation with de®ned functional domains, like homeodomains, or alone, likein the CRP family of LIM proteins. Structural studies of CRP proteins haveallowed a detailed evaluation of interactions in LIM-domains at the mol-ecular level. The packing interactions in the hydrophobic core have beenidenti®ed as a signi®cant contribution to the LIM domain fold, whereashydrogen bonding within each single zinc binding site stabilizes zinc ®ngergeometry in a so-called ``outer'' or ``indirect'' coordination sphere. Here wereport the solution structure of a point-mutant of the carboxyl-terminalLIM domain of quail cysteine and glycine-rich protein CRP2,CRP2(LIM2)R122A, and discuss the structural consequences of the disrup-tion of the hydrogen bond formed between the guanidinium side-chain ofArg122 and the zinc-coordinating cysteine thiolate group in the CCHCrubredoxin-knuckle. The structural analysis revealed that the three-dimen-sional structure of the CCHC zinc binding site in CRP2(LIM2)R122A isadapted as a consequence of the modi®ed hydrogen bonding pattern.Additionally, as a result of the conformational rearrangement of the zincbinding site, the packing interactions in the hydrophobic core region arealtered, leading to a change in the relative orientation of the two zinc ®n-gers with a concomitant change in the solvent accessibilities of hydro-phobic residues located at the interface of the two modules. The backbonedynamics of residues located in the folded part of CRP2(LIM2)R122A havebeen characterized by proton-detected 15N NMR spectroscopy. Analysis ofthe R2/R1 ratios revealed a rotational correlation time of �6.2 ns and tum-bling with an axially symmetric diffusion tensor (Dk/D? � 1.43). The relax-ation data were also analyzed using a reduced spectral density mappingapproach. As in wild-type CRP2(LIM2), signi®cant mobility on a pico-second/nanosecond time-scale was detected, and conformational exchangeon a microsecond time-scale was identi®ed for residues located in loopregions between secondary structure elements. In summary, the relativeorientation of the two zinc binding sites and the accessibility of hydro-phobic residues is not only determined by hydrophobic interactions, butcan also be modi®ed by the formation and/or breakage of hydrogenbonds. This may be important for the molecular interactions of an adaptor-type LIM domain protein in macromolecular complexes, particularly forthe modulation of protein-protein interactions.
# 1999 Academic Press
ing author: [email protected]
e and glycine-rich protein; LIM, speci®c double zinc ®nger domain; LIM1, amino-in; LIM2, carboxyl-terminal LIM domain of CRP protein; CSRP, gene encodingestinal protein; PCR, polymerase chain reaction; NOE, nuclear Overhauser effect;m correlation spectroscopy; T1, longitudinal relaxation time; T2, transverse
# 1999 Academic Press

894 Conformational Flexibility of LIM Domains
Keywords: zinc ®nger; hydrophobic interaction; hydrogen bonding;protein dynamics; protein-protein interaction
*Corresponding authorIntroduction
The LIM domain contains a cysteine-rich motifthat was ®rst identi®ed in, and named after, theprotein products of three regulatory genes, lin-11,isl1, and mec-3 (Freyd et al., 1990; Karlsson et al.,1990). The LIM motif is basically composed of twozinc ®nger structures separated by a two-aminoacid spacer and conforms to the consensus seq-uence CX2CX16-23HX2CX2CX2CX16-21CX2(C/H/D)(Sanchez-Garcia & Rabbits, 1994; Dawid et al.,1995, 1998; Taira et al., 1995). Spectroscopic studiesof LIM domains derived from different LIM pro-teins revealed that each double ®nger LIM domainspeci®cally binds two zinc ions, containing two tet-rahedral Zn(II)-coordinating sites of the CCHC andCCCC type, respectively (Michelsen et al., 1993,1994; Kosa et al., 1994). Although originally recog-nized as a distinct protein motif within LIM home-odomain (LIM-HD) proteins, LIM domains havenow been identi®ed in a variety of proteins thatlack homeodomains, either being composed almostentirely of LIM domains (LIM-only proteins) orcontaining additional functional domains of var-ious types (e.g. kinase domains) (Sanchez-Garcia &Rabbits, 1994; Dawid et al., 1995, 1998; Taira et al.,1995). Thus, the LIM domain proteins form asuperfamily of proteins sharing a commonsequence motif but displaying diverse biochemicalfunctions.
The biological importance of LIM-HD and LIM-only proteins in fundamental developmental pro-cesses has spurred research into the molecularmechanisms through which they function (Dawidet al., 1998). The common theme of all the familymembers seems to be the mediation of protein-pro-tein interactions. Speci®c interactions betweenLIM-HD as well as LIM-only proteins and otherDNA-binding proteins have been demonstrated. Itwas shown that in erythroid cells Lmo2 forms anovel DNA-binding complex with GATA-1, TAL1,E2A and Ldb1/NLI (Wadman et al., 1997). TheLIM-HD protein Lmx1 cooperates with the basichelix-loop-helix (bHLH) protein E47/Pan-1 to acti-vate the insulin promoter in transfected ®broblasts(Johnson et al., 1997). Further evidence for theassembly of LIM domain proteins into nuclearcomplexes that regulate gene expression is theassociation of the LIM-HD protein Lim-3 with thePOU-domain protein Pit-1 (Bach et al., 1995).Recently, a nuclear LIM interactor (Ldb1/NLI) wasidenti®ed in the nuclei of developing embryonicneuronal cells, which is coexpressed with Isl-1early in motor neuron differentiation (Jurata et al.,1996) and binds to the LIM homeodomain andrhombotin-type LIM-only proteins, includingLmo2 and the related protein Lmo1. Cooperativity
and synergistic activation of transcription betweenthe POU-type homeodomain protein UNC-86 andthe LIM-HD protein Mec-3 have been demon-strated in the nematode Caenorhabditis elegans (Xueet al., 1993; Lichtsteiner & Tijan, 1995). Interest-ingly, the LIM domains display a dual functional-ity. They positively regulate LIM-HD activity bypromoting protein-protein interactions that allowcooperative binding to regulatory regions of tissue-speci®c promoters. They also negatively regulateLIM-HD activity, presumably by preventing home-odomain association with DNA. Interaction of LIMdomains with other proteins relieves this interfer-ence, permitting DNA binding and providing amechanism for re®ning LIM-HD activity. Speci®-cally, there is evidence that in LIM-HD proteinsthe LIM domains interact intramolecularly with thehomeodomain to impair its DNA binding, therebyproviding a means to regulate the transcriptionalactivity of the LIM-HD protein. For example, themouse Isl-1 HD binds to the TAAT consensusbinding sequence. In the full-length Isl-1 proteinthis DNA binding capability is lost but can berestored by removal of the Isl-1 LIM domains orby disruption of the global fold stabilizing zinc®nger structure (SaÂnchez-Garcia et al., 1993).Similarly, LIM domains inhibit in vitro binding ofLim-3 to the aGSU and bTSH promoters (Bachet al., 1995), and of Mec-3 to the mec-3 promoter(Xue et al., 1993). Again, removal or disruption ofthe LIM domains restores promoter binding. Thenature of LIM inhibition of DNA binding by HDproteins is still unknown and could result eitherfrom direct intramolecular interaction between theLIM domain and the homeodomain or, indirectly,from association with a repressor protein. In bothcases interference might be relieved by binding toanother protein, such as Ldb-1, a LIM-bindingprotein which has been demonstrated to increaseLim-1 homeodomain association with speci®c pro-moters (Agulnick et al., 1996). Other examples ofspeci®c LIM homeodomain binding partners arePan-1 (German et al., 1992), CLIM-1 (Bach et al.,1997) and Chip (Morcillo et al., 1997). The partici-pation of LIM homeodomain proteins in functionaltranscription complexes, however, is more com-plex. For example, the transcriptional activity ofLim-3 is enhanced by the HD protein P-Otx butonly in the presence of CLIM-1 (Bach et al., 1997)and it has been shown that the structural integrityof the ternary complex is LIM-dependent.
A distinct family of genes, the CSRP genes,encodes a speci®c class of LIM proteins, termedcysteine-rich proteins (CRP) that contain two LIMdomains but no other apparent protein sequencemotif (Weiskirchen et al., 1995). The expression pat-tern of CSRP genes and the structural properties of

Conformational Flexibility of LIM Domains 895
their protein products suggest that these genesmay play important roles in the regulation of celldifferentiation and proliferation. The CSRP1 genewas shown to have properties typical for a primaryresponse gene (Liebhaber et al., 1990). The CSRP2gene encoding the CRP2 protein was discoveredon the basis of its strong suppression in avian®broblasts transformed by retroviral oncogenes orchemical carcinogens (Weiskirchen & Bister, 1993).The CSRP3 gene encoding the CRP3 protein (ormuscle Lim protein, MLP) was shown to beinvolved in muscle differentiation (Arber et al.,1994, 1997). CRP3 was shown to be associatedwith the cytoskeletal components (Arber & Caroni,1996) but was also reported to enhance the myo-genic program by physical interaction with themuscle basic helix-loop-helix (bHLH) transcriptionfactors MyoD, MRF4 and myogenin (Kong et al.,1997). A consistent picture of the biochemical func-tion of CRP proteins is still lacking, but there isexperimental evidence that CRP LIM domains playa role in protein-protein interactions. Beckerle andco-workers have shown that CRP1 co-localizeswith zyxin in vivo (Sadler et al., 1992), and in vitrostudies indicate that zyxin-CRP1 interactions aremediated by the N-terminal LIM domain(Schmeichel & Beckerle, 1994), demonstrating thata solitary LIM domain in a CRP protein de®nes adiscrete functional unit that mediates protein-pro-tein interactions. The amino-terminal LIM1 domainof CRP1 localizes along the actin stress ®bers andassociates with the cytoskeleton by direct inter-actions with the actin-binding protein a-actinin(PomieÁs et al., 1997). Based on these ®ndings that asingle LIM domain acts as a speci®c and distinctprotein-binding interface, it was suggested thatproteins with multiple LIM domains may functionas adaptor molecules arranging two or more pro-tein constituents into a macromolecular complex,either as part of a nuclear transcription complex orof a cytoskeletal complex involving zyxin, actin,a-actinin and other yet unidenti®ed protein bind-ing partners.
Three-dimensional structures of LIM domainshave been determined for the carboxyl-terminalLIM2 domains of chicken CRP1 (PeÂrez-Alvaradoet al., 1994) and of quail CRP2 (Konrat et al., 1997),for the single LIM domain of the cysteine-richintestinal protein (CRIP) (PeÂrez-Alvarado et al.,1996), for the CCHC subdomain of the single LIMdomain from Lasp-1 (HammarstroÈm et al., 1996),and for the amino-terminal LIM1 domain of quailCRP2 (Kontaxis et al., 1998). Recently, the solutionstructures of uniformly 13C,15N-enriched full-lengthquail CRP2 (Konrat et al., 1998a) and 15N-enrichedfull-length chicken CRP1 (Yao et al., 1999) weredetermined. In both studies, it was demonstrated,that the global folds of the amino and carboxyl-terminal LIM domains in the context of the full-length proteins are identical to the previouslydetermined solution structures of the isolated indi-vidual LIM domains of CRP2 (Konrat et al., 1997;Kontaxis et al., 1998) and CRP1 (PeÂrez-Alvarado
et al., 1994). The NMR dynamic data unambigu-ously showed that the tandemly arrayed LIMdomains in full-length CRP2 do not show tightdirect interactions, but instead exhibit independentdomain motion. The two autonomously foldedstructural domains are connected via a ¯exiblelinker region. Further insight into the structuralcharacteristics of a LIM domain was obtained froma recent NMR structural study of a point-mutantof CRP2(LIM2), CRP2(LIM2)E155G, where wehave delineated an extended H-bonding networkin which all the directly ligating residues of thetwo Zn-®nger structures of LIM2 are engaged asH-bonding partners in interactions with other resi-dues in the same Zn-®nger unit, extending thestructuring role of the Zn ion via the directlybound ligands to a second, outer coordinationsphere (Konrat et al., 1998b). The observed H-bond-ing pattern de®ned the protonation states of themetal-coordinating ligands and characterized all ofthe coordinating cysteine residues as thiolates. Atthe CCCC site, four thiolates, and at the CCHCsite, three thiolates are direct ligands of the Znions. Electroneutrality is closely preserved in thesezinc binding centers by H-bonding interactions ofsome metal coordinating thiolates with positivelycharged basic amino acid residues. Based on theseand other structural studies of LIM domains in sol-ution, it was argued that H-bonding does play apivotal role in de®ning the conformation of theCCHC and CCCC Zn2�-binding sites (``rubredoxin(Rd) knuckles'') (PeÂrez-Alvarado et al., 1994, 1996;Konrat et al., 1997, 1998b). On the other hand,based on structural comparisons of the carboxyl-terminal LIM domain of CRP1 and the single LIMdomain protein CRIP, it was concluded that therelative orientation of the CCHC and CCCC mod-ules is stabilized only by hydrophobic interactions,but not by speci®c hydrogen bonding, and thatmutations in the hydrophobic core region couldlead to both a structural and functional switch ofthe LIM domain (PeÂrez-Alvarado et al., 1996).
Here we report a three-dimensional structuraland dynamical study of the point mutantCRP2(LIM2)R122A, in which the hydrogen bonddonor Arg122 of wild-type CRP2 is substituted byalanine. The structural and possibly functional rel-evance of this residue is indicated by the fact thatArg122 is conserved in all family members of theCRP protein family (Weiskirchen et al., 1995, 1997).In contrast, in the amino-terminal LIM domains ofthe CRP protein family, the analogous position isinvariantly occupied by a hydrophobic residue(e.g. alanine or valine) (Weiskirchen et al., 1995,1997). The primary goal of the investigationdescribed below was to elucidate the structuralconsequences of the disruption of the hydrogenbond formed between the guanidinium side-chainof the conserved Arg122 and the zinc-coordinatedcysteine thiolate group in the CCHC Rd-knuckle.Additionally, the backbone dynamics ofCRP2(LIM2)R122A were investigated using two-dimensional 15N-1H NMR methods to determine

896 Conformational Flexibility of LIM Domains
steady-state 1H-15H NOE values, and the T1 and T2
relaxation times of 15N magnetization. The resultsprovide valuable information on the anisotropy ofthe rotational diffusion of CRP2(LIM2) and on thelocal motion dynamics of the LIM2 domain.
Results and Discussion
Preparation of mutant recombinantprotein CRP2(LIM2)R122A
Using a site-directed mutagenesis procedurebased on the method developed by Deng &Nickoloff (1992), a derivative of the pET3dexpression plasmid (Studier et al., 1990) was con-structed that directs synthesis of a 113 amino acidpeptide encompassing amino acid residues 82-194of the quail LIM domain protein CRP2 includingthe carboxyl-terminal LIM2 domain in which thearginine at position 122 within the CCHC zinc-binding site of the wild-type protein is substitutedby alanine (Figure 1(a)). The mutation was veri®edby comparison of the nucleotide sequences of theexpression plasmids encoding the wild-typeCRP2(LIM2) protein (Konrat et al., 1997) and themutant CRP2(LIM2)R122A protein, respectively(Figure 1(b)). The mutant recombinant protein hasa calculated Mr value of 12,020 and an estimatedisoelectric point of 9.10, in comparison to a calcu-lated Mr value of 12,105 and an isoelectric point of9.35 for the wild-type protein (Konrat et al., 1997).The molecular masses determined by mass spec-troscopy of the puri®ed recombinant proteins were12,106.1(�0.5) for the wild-type protein and12,020.9(�0.5) for the mutant protein. Usingstandard SDS-polyacrylamide gel electrophoresis(Bister et al., 1977; Jansen et al., 1983; Hartl &Bister, 1998), both the wild-type and the mutantrecombinant protein migrated as a single bandwith an apparent Mr value of 14,700, as deter-mined by co-electrophoresis of chicken lysozyme(Mr 14,300) and other molecular mass markers(Figure 1(c)).
NMR signal assignment and identification ofsecondary structure
It could be inferred from the chemical shift dis-persion in the sensitivity enhanced PFG two-dimensional 15N-HSQC spectrum shown inFigure 2 that CRP2(LIM2)R122A adopts a wellde®ned structure in aqueous solution. As in wild-type CRP2(LIM2), only a fraction of all NH signalsare visible due to fast exchange with bulk waterand conformational exchange. A total of 52 second-ary backbone amides could be assigned, all ofthem within the structured region which comprisesresidues Lys119-Gly177. Speci®cally, residuesLys119, Asn143 and residues Asn175-Gly177 wereassigned based on their NOESY patterns and bychemical shift comparison with 13C,15N-labeledfull-length CRP2. As was the case for wild-typeCRP2(LIM2), residues which could not be ident-
i®ed by sequential assignment encompass the ¯ex-ible parts of the peptide (residues 82-118 and 178-194) as well as turn and loop regions (Ala129-Ala130, Ala136, Glu155-Thr158, Glu163). Sinceexamination of the spectrum yielded only onecross-peak for the amide of non-proline residues, itcould be concluded that only one structural formof the protein was present in solution. Sequentialassignment was carried out using the establishedstrategy (WuÈ thrich, 1986) to identify spin systemsby the means of the three-dimensional TOCSY-HSQC and combining this information with dis-tance information provided by three-dimensionalNOESY-HSQC and two-dimensional NOESY spec-tra. Side-chain protons could not always beassigned unambiguously due to spin system over-lap and signal degeneracy of TOCSY peaks in theup®eld region.
Secondary structure elements were identi®edprimarily on the basis of characteristic cross-peakpatterns in three-dimensional NOESY-HSQC andtwo-dimensional NOESY spectra. Strong sequentialHa(i ÿ 1)-HN(i)-NOE connectivities indicatingextended antiparallel b-sheet conformation werefound for residues (Lys119-Ala122), (Asp125-Tyr128), (Val133-Gly135), (Lys138-W140), (Phe145-Ala148), (Thr160-Lys162), and (Ile166-Lys169),comprising b-strands bI-bV and bVII-bVIII whichhave also been found in wild-type CRP2(LIM2).Residues Lys152-Leu154, which make up b-strandVI in wild-type CRP2(LIM2), showed almostdegenerate Ha chemical shifts in the mutantCRP2(LIM2)R122A, thus leading to an overlap ofthe strong sequential inter-residue with the intra-residue NOE. Interstrand NOEs have been foundbetween bI and bII (HN(Asp125)-Hb1(Cys120) andHb2(Cys120), HN(Val127)-Ha(Lys119), HN(Val127)-HN(Cys120), and HN(Cys120)-Ha(Ser126)), betweenbIII and bIV (HN(Val133)-HN(Trp140), HN(Gly135)-HN(Lys138), and HN(Gly135)-Ha(Lys138)), andbetween bVII and bVIII (Ha(Glu161)-Ha(Ile166),Ha(Glu161)-HN(Tyr167), Hg(Lys162)-HN(Tyr167),HN(Lys162)-HN(Glu165), HN(Lys162)-Ha(Ile166),Ha(Glu163)-HN(Glu165)). Turn regions were identi-®ed by strong sequential HN(i)-HN(i � 1) andHN(i)-HN(i � 2)-NOEs as found for Ser121-Gly124,Lys149-Gly151. A carboxyl-terminal a-helix (aI)was identi®ed by HN(i)-HN(i � 1) NOEs (Cys171-Lys174). Flexible loop and turn regions compriseresidues Ala129-Ala130, Glu155-Thr158, Ala136,Asn143, and Glu163 which could not be assignedby the means of sequential NOEs.
Hydrogen bonds between b-strands and to themetal coordinating thiolates could be identi®ed onthe basis of amide proton attenuation factors andcharacteristic NOE patterns (Konrat et al., 1997).Additional information on secondary structure wasprovided by the means of proton chemical shiftanalysis (Wishart et al., 1991, 1992) and 3J(HNHa)coupling constants. Chemical shift analysis of Ha
protons reveals the existence of secondary struc-ture elements. In a-helices they are generallyshifted up®eld, whereas in b-sheets they exhibit a

Figure 2. 15N-1H HSQC of CRP2(LIM2)R122A. Assigned cross-peaks for backbone amide proton and nitrogenatoms are labeled by amino acid type and sequence number. Cross-peaks marked with ? correspond to unassignedresidues which did not show any inter-residue NOE connectivities and displayed smaller than average 15N T2 valuesand/or negative heteronuclear 1H-15N NOEs and are thus located in predominantly unstructured regions of theprotein.
Figure 1. Generation and charac-terization of mutant recombi-nant protein CRP2(LIM2)R122A.(a) Amino acid sequence of the 113amino acid peptide encoded byexpression plasmid pET3d-qCRP2(LIM2)R122A. Numbering inthe margin corresponds to aminoacid positions in the full-length 194amino acid quail CRP2 protein(Weiskirchen & Bister, 1993;Weiskirchen et al., 1995). The resi-dues coordinating the two zincions of the CCHC and CCCC zinc-binding sites of the LIM2 domainare shown in reversed type, andthe alanine for arginine substitutionis indicated by a box. (b) Veri®ca-tion of site-directed mutagenesisby nucleotide sequence analysisof expression plasmids pET3d-qCRP2(LIM2)R122A (left) andpET3d-qCRP2(LIM2) (right) encod-ing mutant or wild-type (Konratet al., 1997) CRP2(LIM2) recombi-
nant protein, respectively. (c) SDS-polyacrylamide (13 %, w/v) gel electrophoresis of wild-type CRP2(LIM2) (1),mutant CRP2(LIM2)R122A (2), and chicken egg white lysozyme (Mr 14,300) (3). Molecular mass markers (M) were(in the order of decreasing mass): albumin (bovine serum), albumin (chicken egg white), glyceraldehyde-3-phosphate-dehydrogenase (rabbit muscle), carbonic anhydrase (bovine erythrocytes), trypsinogen (bovine pancreas), trypsininhibitor (soy bean), and a-lactalbumin (bovine milk). Proteins were visualized by staining with Coomassie brilliantblue.
Conformational Flexibility of LIM Domains 897
898 Conformational Flexibility of LIM Domains
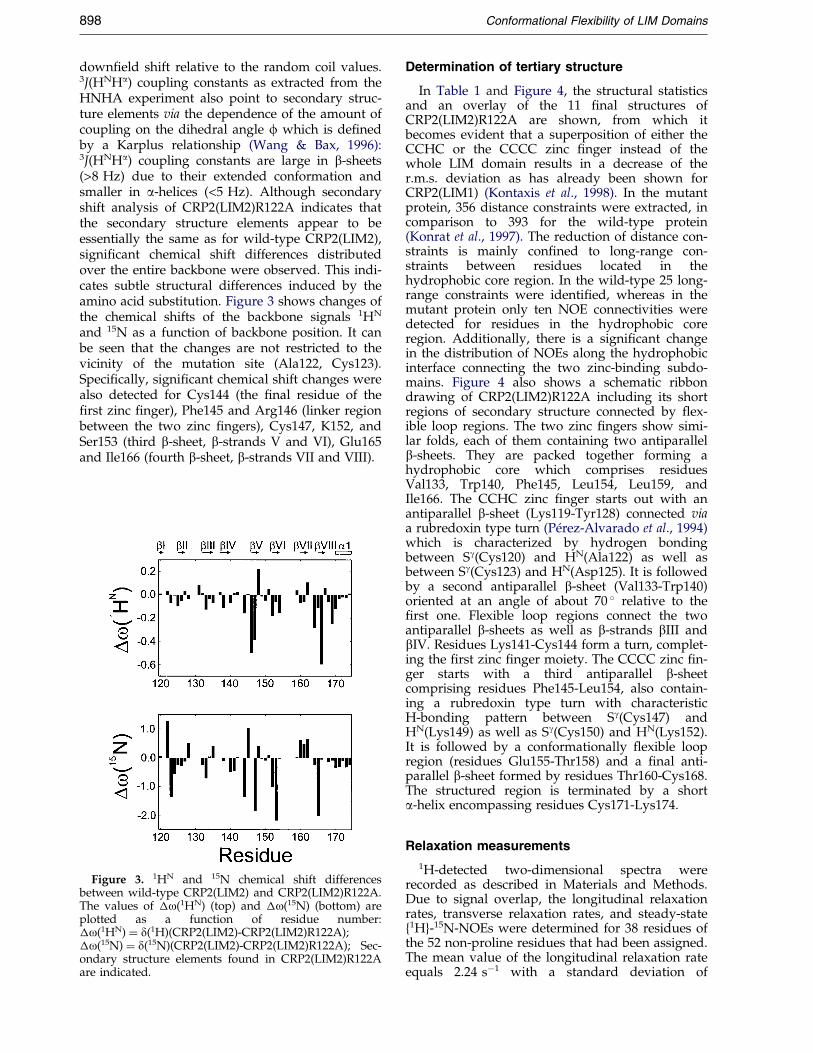
down®eld shift relative to the random coil values.3J(HNHa) coupling constants as extracted from theHNHA experiment also point to secondary struc-ture elements via the dependence of the amount ofcoupling on the dihedral angle f which is de®nedby a Karplus relationship (Wang & Bax, 1996):3J(HNHa) coupling constants are large in b-sheets(>8 Hz) due to their extended conformation andsmaller in a-helices (<5 Hz). Although secondaryshift analysis of CRP2(LIM2)R122A indicates thatthe secondary structure elements appear to beessentially the same as for wild-type CRP2(LIM2),signi®cant chemical shift differences distributedover the entire backbone were observed. This indi-cates subtle structural differences induced by theamino acid substitution. Figure 3 shows changes ofthe chemical shifts of the backbone signals 1HN
and 15N as a function of backbone position. It canbe seen that the changes are not restricted to thevicinity of the mutation site (Ala122, Cys123).Speci®cally, signi®cant chemical shift changes werealso detected for Cys144 (the ®nal residue of the®rst zinc ®nger), Phe145 and Arg146 (linker regionbetween the two zinc ®ngers), Cys147, K152, andSer153 (third b-sheet, b-strands V and VI), Glu165and Ile166 (fourth b-sheet, b-strands VII and VIII).
Figure 3. 1HN and 15N chemical shift differencesbetween wild-type CRP2(LIM2) and CRP2(LIM2)R122A.The values of �o(1HN) (top) and �o(15N) (bottom) areplotted as a function of residue number:�o(1HN) � d(1H)(CRP2(LIM2)-CRP2(LIM2)R122A);�o(15N) � d(15N)(CRP2(LIM2)-CRP2(LIM2)R122A); Sec-ondary structure elements found in CRP2(LIM2)R122Aare indicated.
Determination of tertiary structure
In Table 1 and Figure 4, the structural statisticsand an overlay of the 11 ®nal structures ofCRP2(LIM2)R122A are shown, from which itbecomes evident that a superposition of either theCCHC or the CCCC zinc ®nger instead of thewhole LIM domain results in a decrease of ther.m.s. deviation as has already been shown forCRP2(LIM1) (Kontaxis et al., 1998). In the mutantprotein, 356 distance constraints were extracted, incomparison to 393 for the wild-type protein(Konrat et al., 1997). The reduction of distance con-straints is mainly con®ned to long-range con-straints between residues located in thehydrophobic core region. In the wild-type 25 long-range constraints were identi®ed, whereas in themutant protein only ten NOE connectivities weredetected for residues in the hydrophobic coreregion. Additionally, there is a signi®cant changein the distribution of NOEs along the hydrophobicinterface connecting the two zinc-binding subdo-mains. Figure 4 also shows a schematic ribbondrawing of CRP2(LIM2)R122A including its shortregions of secondary structure connected by ¯ex-ible loop regions. The two zinc ®ngers show simi-lar folds, each of them containing two antiparallelb-sheets. They are packed together forming ahydrophobic core which comprises residuesVal133, Trp140, Phe145, Leu154, Leu159, andIle166. The CCHC zinc ®nger starts out with anantiparallel b-sheet (Lys119-Tyr128) connected viaa rubredoxin type turn (PeÂrez-Alvarado et al., 1994)which is characterized by hydrogen bondingbetween Sg(Cys120) and HN(Ala122) as well asbetween Sg(Cys123) and HN(Asp125). It is followedby a second antiparallel b-sheet (Val133-Trp140)oriented at an angle of about 70 � relative to the®rst one. Flexible loop regions connect the twoantiparallel b-sheets as well as b-strands bIII andbIV. Residues Lys141-Cys144 form a turn, complet-ing the ®rst zinc ®nger moiety. The CCCC zinc ®n-ger starts with a third antiparallel b-sheetcomprising residues Phe145-Leu154, also contain-ing a rubredoxin type turn with characteristicH-bonding pattern between Sg(Cys147) andHN(Lys149) as well as Sg(Cys150) and HN(Lys152).It is followed by a conformationally ¯exible loopregion (residues Glu155-Thr158) and a ®nal anti-parallel b-sheet formed by residues Thr160-Cys168.The structured region is terminated by a shorta-helix encompassing residues Cys171-Lys174.
Relaxation measurements
1H-detected two-dimensional spectra wererecorded as described in Materials and Methods.Due to signal overlap, the longitudinal relaxationrates, transverse relaxation rates, and steady-state{1H}-15N-NOEs were determined for 38 residues ofthe 52 non-proline residues that had been assigned.The mean value of the longitudinal relaxation rateequals 2.24 sÿ1 with a standard deviation of

Table 1. Structural statistics of the ®nal 11 structures of CRP2(LIM2)R122A
A. Experimental restraints 356Intra-residue 124Inter-residue sequential (jiÿjj�1) 81Inter-residue sequential (jiÿjj�2) 13Inter-residue sequential (jiÿjj�3) 13Inter-residue sequential (jiÿjj�4) 4Inter-residue long range (jiÿjj>4) 105Hydrogen bonds 16B. Average r.m.s. deviations from experimental restraintsDistance restraints (AÊ ) 0.037 � 0.001C. Average r.m.s. deviations from idealized covalent geometryBonds (AÊ ) 0.011 � 1.2e-04Angles (degrees) 2.55 � 0.050D. X-PLOR energiesAverage EL(J (kcal molÿ1)a ÿ574.7 � 15.4E. PROCHECK% Residues in allowed regions of Ramachandran plot 98Atomic r.m.s. deviation (AÊ ) Backboneb Heavy atomsc
Residues 120-145, 146-174d 1.58 � 0.63 2.10 � 0.59Residues 120-145 0.78 � 0.16 1.59 � 0.32Residues 146-174 0.59 � 0.21 1.32 � 0.28
a EL-J is the Lennard Jones van der Waals enenrgy calculated with X-PLOR using the CHARMMforce ®eld parameters.
b Pairwise r.m.s. deviations calculated from the superposition of backbone heavy atoms (C0, Ca
and N) of the 11 X-PLOR structures.c Pairwise r.m.s. deviations calculated from all non-hydrogen atoms of the 11 X-PLOR struc-
tures.d Numbering of residues is as for Figure 1. Residues 120-145 and 146-174 encompass the CCHC
and CCCC zinc ®nger subdomains, respectively, and residues 120-174 represent the entire LIM2domain of CRP2(LIM2)R122A. The conformationally ¯exible loop region between residues 145 and146 was excluded from the structural analysis.
Conformational Flexibility of LIM Domains 899
0.24 sÿ1. Residues signi®cantly deviating from themean value are Cys120, with a lower than averageR1 value, and Ser121, Ala148, Lys162, and Lys174
Figure 4. Solution structure of CRP2(LIM2)R122A. Ribbonthe calculated set for CRP2(LIM2)R122A (residues 118 to 1sites encompassing residues 120 to 145 (middle) or residuesbetween the two zinc ®nger subdomains. The diagrams wer1996).
with a higher than average R1 value. The meantransverse relaxation rate is 8.31 sÿ1 with a stan-dard deviation of 1.82 sÿ1. Residue Cys123 exhibits
diagram (left) of a selected representative structure from74). Separate matches for each of the two zinc binding146 to 173 (right), respectively, indicate residual mobilitye produced using the program MOLMOL (Koradi et al.,

Figure 5. Reduced spectral densities as a function ofresidue number. The values of J(0), J(oN) and J(0.87oH)calculated from the relaxation data acquired at 11.74 Tare plotted as a function of residue number. Secondarystructure elements found in CRP2(LIM2)R122A areindicated.
900 Conformational Flexibility of LIM Domains
a lower than average R2 value. In general, residueslocated in turn or loop regions connecting second-ary structure elements show faster than averagetransverse relaxation. Interestingly, Leu154 whichis at the beginning of the ¯exible loop region com-prising residues up to the position Leu159 showsclose to average R1 and R2 relaxation rates,whereas Ser153, which is part of b-strand VI,shows a much higher transverse relaxation rateand R2/R1 ratio. This ®nding, however, is inaccordance with evidence from analysis of relax-ation data of the wild-type protein, pointing toconformational exchange on a microsecond tomillisecond time scale at this molecular site(Ser153). In addition to the larger deviations, R2
values are quite low for residues Phe145 toCys150, a region comprising the short linkerbetween the two zinc ®nger moieties and the ®rstrubredoxin knuckle of the second zinc ®nger. Thetendency in R2/R1 ratios follows the R2 rates quiteclosely, being low in the vicinity of the mutationand as well as in the linker region and high in theabove-mentioned more disordered regions.
Reduced spectral density function mapping
Because anisotropic tumbling of the peptideaffects substantially the usual model-free analysisof 15N-spin relaxation data, the reduced spectraldensity mapping formalism (Farrow et al., 1995a,b)has been applied instead. An estimate of the over-all correlation time was provided by the gridsearch procedure supplied by Farrow et al. (1994)in the course of which an error function of calcu-lated and experimental relaxation data is mini-mized. The values of the spectral density functionswere determined for the frequencies 0, (oN), and(0.87oH) from R1, R2, and steady-state NOE valuesat 11.7 T (Farrow et al., 1995b). This approachis based on the assumption that the dynamics ofan amide bond vector can be expressed as a sumof exponentially decaying functions and thatoti41 for overall tumbling and oti51 for internalmotion; there is, however, no need for the moleculeto tumble isotropically. The results forCRP2(LIM2)R122A are shown in Figure 5. The dis-tribution of J(0) along the protein backbone followsclosely the trend observed in wild-typeCRP2(LIM2) (Konrat et al., 1997). There is a goodcorrelation between J(0) and secondary structure.Residues within loop regions exhibit signi®cantlyincreased J(0) values. As was found for wild-typeCRP2(LIM2), there is also a systematic increase inJ(0) towards the end of the C-terminal helix, pre-sumably as a result of conformational isomeriza-tion processes due to reversible hydrogen bonding(e.g. helical fraying). The signi®cantly reducedvalue of J(0) for residue Cys123 was also detectedin wild-type CRP2(LIM2) and is thus an inherentdynamic feature of the LIM2 domain and not aconsequence of the amino acid substitution. Inter-estingly, the mutant does not show an increasedJ(0) value of Cys144 which had been found for
CRP2(LIM2) (Konrat et al., 1997). The high-frequency components J(oN) and J(0.87oH),however, show trends similar to those ofCRP2(LIM1). It was observed experimentally(Kontaxis et al., 1998) that CRP2(LIM1) exhibits sig-ni®cant intramodular dynamics (Gly11, Val17,Glu22-Cys25, Ser29, Phe30, Leu36, Val39, Arg41,Lys42, Ala50, His52, Val56, Cys58, Ser60-Gly63).Speci®cally, residues involved in the formation ofthe hydrophobic core show internal mobility in thenanosecond time range, superimposed on smallangle ¯uctuations in the picosecond time scale.Given the sparsity of NOE connectivities withinthe hydrophobic core in CRP2(LIM1), this intra-

Conformational Flexibility of LIM Domains 901
modular mobility was attributed to the time-dependent orientation of the two zinc-bindingsites. Residues in CRP2(LIM2)R122A located atsites corresponding to those of CRP2(LIM1) show-ing intramodular dynamics are: Ser121, Val127,Lys132-Gly135, Pro139, Trp140, Arg146, Lys149,Gly151, Lys152, Thr160, Lys162, Ile166, Cys168,Gly170-A173. The data shown in Figure 5 alsoreveal that signi®cant high-frequency motions aredetected for residues in the mutant proteinCRP2(LIM2)R122A located at identical or adjacentbackbone sites, respectively. Together with thedecrease of aliphatic NOE connectivities in thehydrophobic core and the above-mentioned struc-tural statistics, this intramodular dynamic isadditional evidence for a less dense interface pack-ing in CRP2(LIM2)R122A compared to wild-typeCRP2(LIM2).
Rotational diffusion tensor
As relaxation parameters are sensitive to aniso-tropic overall tumbling, it was of interest to deter-mine the rotational diffusion tensor. This wasaccomplished by the local diffusion approximation(BruÈ schweiler et al., 1995) using an initial estimatefor Diso corresponding to the global correlationtime as analyzed by the means of the model-freeapproach. The values of Dk/D? for the grid searchprocedure ranged from 0.4 to 2. For the determi-nation of the inertia tensor and the rotational diffu-sion tensor, an atom type representing the zincions had to be added prior to analysis. The methodanalyses local diffusion coef®cients derived fromrelaxation rate constants to determine the diffusiontensor assuming isotropic, axially symmetric, andasymmetric diffusion. The inertia tensor was calcu-lated for the 11 ®nal structures from their respect-ive coordinate ®les. The average ratio of theirprincipal values was 0.41:0.97:1. For the analysis ofthe relaxation data, residues signi®cantly deviatingfrom the mean values of J(0) (Cys123, Gly135,Gly137, Ser153, Leu159, Lys162, Gly170) and/orJ(oN) (Ser121, Ala148, Lys162, Lys174) (indicatingeither fast motion on a picosecond to nanosecondtime-scale and/or conformational exchange on amillisecond to microsecond time-scale) wereexcluded, as well as residues with high uncertain-ties in relaxation parameters. The analysis wastherefore performed with 15N-relaxation data of 22
Table 2. Diffusion parameters for CRP2(LIM2)R122A
Tensor Diso(10ÿ7 sÿ1) 2Dzz/(Dxx � Dyy)
Isotropic 2.67 -Axialc 2.69 (0.013) 1.43 (0.04)
Values of Di for 22 residues were ®t using the local diffusion appra The angles y and f de®ne the orientation of the symmetry axis ob The F-statistic compares the quality of the ®t for the axially symmc The weighted mean values and uncertainties from nine individ
deviations calculated over the ensemble of NMR structures.
residues. The quality of the ®t was characterizedby a statistical F-test. There were two structures inthe ®nal bundle which showed a positive corre-lation between Di and the projection onto theprincipal axis of the diffusion tensor due to a ratioDk/D? < 1. These structures were omitted in calcu-lating the mean values given below. For theremaining structures, the procedure yields F-valuesranging from 5.8 to 9.0 and from 0.42 to 3.85 forthe axial and the anisotropic models, respectively,indicating signi®cant improvement in the ®t of thedata going from an isotropic model to axial modelbut not to the totally anisotropic model (Table 2).Thus, the results of the axial model were taken foranalysis of diffusion. The average calculated isotro-pic correlation time is 6.2(�0.06) ns. Dk/D? valuesrange from 1.38 to 1.50 with an average value of1.43(�0.037), the average value of the angle ybetween the principal axis of the inertia tensor andthe axial diffusion tensor is y � 51.4(�7.7)�. Theorientational dependence of the local diffusion con-stants is shown in Figure 6. For example, the N-Hbond vectors for residues Cys120 and Ala173 areoriented preferentially along the symmetry axis ofthe diffusion tensor (Cys120: Y20 � 0.88; Ala173:Y20 � 0.74). These residues are thus relaxed mainlyby relatively slow motions about the perpendicularaxes.
Structural comparison of CRP2(LIM2)R122Awith CRP2(LIM2) and CRP2(LIM1)
A comparison of the local backbone geometryand of the tertiary structure of wild-typeCRP2(LIM2) and mutant CRP2(LIM2)R122Ashows signi®cant differences for some regions ofthe peptide which largely correspond to regionswith the chemical shift alterations mentionedabove. The differences in the backbone dihedralangles between wild-type CRP2(LIM2) and mutantCRP2(LIM2)R122A as a function of residue pos-ition are given in Figure 7. The point mutation ispositioned in the turn region (Rd-knuckle) betweenthe strands bI and bII. Noticeable backbone geome-try differences occur within the ®rst CCHC zincbinding site. Dihedral angles (f and c) associatedwith residues Cys123 and Gly124 in the Rd-knuckle show signi®cant deviations compared tothe wild-type CRP2(LIM2). Additionally, structuralanalysis of the obtained ensemble of NMR struc-
y(degree)b f(degree)a Fb
- - -51.4 (7.7) 48.6 (11.2) 7.58 (0.98)
oach.f the diffusion tensor.
etric tensor with that of the isotropic tensor.ual ®ts are shown. The values in parentheses are the sample

Figure 6. Rotational diffusion anisotropy ofCRP2(LIM2)R122A. The local diffusion anisotropyconstants, Di, are plotted versus the Legendre poly-nomial Y20 � 1/2(3cos2yi ÿ 1), in which yi is the polarangle between the ith N-H bond vector and the sym-metry axis of the diffusion tensor. The error bars rep-resent sample deviations calculated over the ensembleof nine NMR solution structures. The linear least-squares ®t to the data is shown for illustration.
Figure 7. Backbone geometry differences betweenwild-type and mutant CRP2(LIM2) as a function of resi-due number. The values of �(fc) � j(fwt
ÿ fR122A)j � j(cwt ÿ cR122A)j calculated from the ensem-ble of NMR structures are plotted as a function of resi-due number.
902 Conformational Flexibility of LIM Domains
tures (e.g. His141-Cys144), together with thechemical shift changes observed for the ®nal zinccoordinating residue Cys144, indicate that themutation causes conformational changes in theentire CCHC zinc binding site. In an NMR studyof the point-mutant CRP2(LIM2)E155G and fromstructural comparison with wild-type CRP2(LIM2)we have characterized the H-bonding network inthe coordination polyhedra around the Zn ion inthe CCHC zinc ®nger and could experimentallyidentify the zinc-coordinating cysteines as thiolates(Konrat et al., 1998b). It was shown that in thewild-type structure, the guanidinium group ofArg122 shows hydrogen bonding to Sg(Cys144)and is thus part of an integral proteinic outer-coordination sphere stabilizing the zinc ®ngergeometry. The observed conformational rearrange-ment of the CCHC zinc binding site due to the lossof a hydrogen bond donor can therefore beregarded as a consequence of the preservation ofthis extended hydrogen bonding network. Theturn region between Lys142 and Cys144 completesthe amino-terminal CCHC zinc ®nger and showsconformational ¯exibility. A similar observationwas made for wild-type CRP2(LIM2), and thisintramolecular dynamic could be a necessary pre-requisite for the conformational adjustement of theturn in order to select a different hydrogen bond-ing donor for the thiolate group of Cys144. How-ever, as was the case for CRP2(LIM1), thehydrogen bonding partner of Sg(Cys144) could notunambiguously be identi®ed. Most likely thehydration site is occupied by a coordinating watermolecule which could be bound by additionalhydrogen bonds to side-chain functional groups ofadjacent residues.
The structural consequences of the mutation,however, are not con®ned to the CCHC zinc bind-ing site. It has been veri®ed by NOEs betweenHb(Ala122) and aromatic signals of Trp140 that theside-chain of Ala122 comes closer to the aromaticring of Trp140, due to both its reduced stericdemand and its hydrophobic nature. As a result,the ®rst b-sheet seems to adopt a more compactconformation than in the wild-type structure, com-ing closer to the hydrophobic core and also alteringits position relative to the second b-sheet fromalmost 90 � to about 70 �. The perturbation intro-duced by the mutation also extends to the hydro-phobic core which shows alterations in thearrangement of some side-chains (Figure 8). Thestacking of the two aromatic side-chains of Trp140and Phe145 has become more pronounced, as indi-cated by NOEs between them which have nocounterpart in wild-type CRP2(LIM2). The proxi-mity of these two side-chains does not allow theHd1 methyl group of Ile166 to be stacked betweenthem as was the case for CRP2(LIM2) (Figure 8).This is additionally corroborated by its relativedown®eld shift (from ÿ0.8 ppm to ÿ0.2 ppm)when compared to the wild-type protein. Thechemical shift changes of the backbone signals (15Nand 1HN) of Phe145 and Arg146 (Figure 3) alsore¯ects, at least in part, this modi®ed arrangementof the two aromatic side-chains. In the C-terminalCCCC zinc binding site, residues Cys150-Lys152(Figure 7) display altered backbone dihedralangles, and residues Cys147 and Ser153 exhibitsigni®cant chemical shift changes, presumably dueto different hydrogen bonding. In wild-typeCRP2(LIM2), there was evidence for a hydrogenbond between Cys147 and Ser153. A consistentup®eld shift for both Cys147 and Ser153 would bein agreement with a breakage (or loosening) of thishydrogen bonding interaction. Interestingly, Ser153also shows an increased R2 relaxation rate due to

Figure 8. Structural comparison of hydrophobic corepacking between wild-type and mutant CRP2(LIM2).Orthogonal views (top and bottom) of differential steri-cal interactions in the hydrophobic core of wild-type(left, blue) and mutant (right, yellow) CRP2(LIM2). Theside-chains of the hydrophobic core residues Trp140,Phe145 and Ile166 are shown. The diagram of thesolution structure was produced with the programMOLMOL (Koradi et al., 1996).
Figure 9. Schematic representation showing thechange in the relative orientation of the two CCHC andCCCC zinc-binding sites in CRP2(LIM2)R122Aintroduced by the point mutation at residue position122. (a) Ribbon drawing showing the superposition of arepresentative structure taken from the ensembleof 11 energy-minimized NMR structures ofCRP2(LIM2)R122A (yellow) and the wild-typeCRP2(LIM2) structure with the lowest residual restraintviolations (Konrat et al., 1997) (blue). Backbone atoms ofthe N-terminal CCHC zinc ®nger encompassing residuesCys120-Cys144 were matched. (b) Orthogonal view dis-playing only the C-terminal CCCC zinc binding site ofmutant CRP2(LIM2)R122A (yellow) and wild-type pro-tein (blue). The pictures were produced with theprogram MOLMOL (Koradi et al., 1996).
Conformational Flexibility of LIM Domains 903
conformational exchange processes. This indicatesthat loosening of the hydrogen bond could lead tochanges in the populations of the two (or more)states interconverting on a milli- to microsecondtime scale (e.g. hydrogen bond isomerization pro-cesses). Hence, the slight modi®cation of the back-bone geometry of the peptide linker necessitates acorresponding minor conformational rearrange-ment of the adjacent CCCC zinc binding site.
Another region which shows alterations in back-bone chemical shifts (Figure 3) compared to thewild-type peptide ranges from residues 164 to 166.These changes whithin the fourth b-sheet(b-strands VII to VIII) are partly due to the reorien-tation of the hydrophobic core side-chain of Ile166,which is accompanied by a local adjustement ofthe secondary structure (Figures 7 and 9). Inaddition, the aromatic side-chain of Y167 exhibits adifferent position in the mutant protein where itlies almost parallel to the sheet, whereas in wild-type CRP2(LIM2) it is oriented almost perpendicu-lar to the sheet such that the anisotropy of the aro-matic ring current causes an up®eld shift of amideprotons of residues 164 and 166 relative to theirwild-type counterparts. In fact, not only local struc-tural changes can be observed. A comparison ofthe wild-type and the mutant peptide structuresreveals a signi®cant difference in the orientation ofthe zinc ®ngers with respect to each other: whereasthe second (b2) and the fourth b-sheet (b4) arestacked in CRP2(LIM2), they are rotated againsteach other about an average angle of about 30 � inCRP2(LIM2)R122A (Figure 9), which is very simi-
lar to CRP2(LIM1), which also shows a rotatedrelative orientation of the two zinc ®ngers whencompared to CRP2(LIM2). This is even moresigni®cant, as CRP2(LIM1) also contains an alanineresidue at the position corresponding to position122 of the LIM2 domain. Interestingly, thissimilarity between CRP2(LIM2)R122A andCRP2(LIM1) is also re¯ected in an analogousincrease in intramodular mobility as compared to

904 Conformational Flexibility of LIM Domains
wild-type CRP2(LIM2) (Table 1 and Figure 5), dueto less sterical strain in the hydrophobic core.
It had been deduced from the structures andamino acid sequences of chicken CRP1(LIM2) andCRIP that the nature of the amino acid residuescorresponding to the positions Ala136, Phe145,Leu159, and Ile166 in CRP2(LIM2) mainly deter-mines the packing interactions within the hydro-phobic core and the relative orientation of the twozinc binding sites (PeÂrez-Alvarado et al., 1996).CRP1(LIM2) has exactly the same hydrophobicresidues as CRP2(LIM2) and also the same orien-tation of zinc ®ngers. In contrast, in the CRIPprotein these residues are replaced by Leu19,Leu28, His42, and Pro49 which results in a relativezinc ®nger orientation similar to CRP2(LIM1).However, investigation of the mutant CRP2(LIM2)-R122A protein provided evidence that hydro-phobic core packing is also affected by hydrogenbonding interactions within a particular zinc bind-ing site, as the hydrogen bond donor Arg122 isabsolutely conserved in the LIM2 domains andinvariantly replaced by a hydrophobic residue (Valor Ala) in the LIM1 domains of the CRP family(Weiskirchen et al., 1995, 1997). It can thus be con-cluded that a subtle sequence of delicate inter-actions (hydrophobic as well as hydrogen bondinginteractions) de®ne a complex energy landscapeand allow for multiple alternative structural realiz-ations of the LIM domain fold with only marginaldifferences in energy.
Functional implications of LIMdomain structure
NMR structure analyses of three isolated LIMdomains from avian CRP family members CRP1(LIM2, PeÂrez-Alvarado et al., 1994) and CRP2(LIM2, Konrat et al., 1997; LIM1, Kontaxis et al.,1998) revealed the structural conservation andautonomous folding of the LIM protein motif. Thiswas corroborated by NMR studies on full-lengthCRP2 (Konrat et al., 1998a) and chicken CRP1 (Yaoet al., 1999) demonstrating that in both CRP pro-teins the two LIM domains are independent ofeach other and do not exhibit a preferred relativeorientation in solution. The CCHC and CCCCzinc-binding subdomains of the LIM motif containtwo orthogonally arranged antiparallel b-sheets.The carboxyl-terminal end of the CCHC site con-tains a tight turn, and the carboxyl terminus of theCCCC site forms an a-helix. The two zinc-bindingsites pack together via hydrophobic interactionsbetween highly conserved residues, forming ahighly compact structure. Although the variousLIM domains within the CRP protein familydisplay a nearly identical global fold, they differ interms of the relative orientations of the CCHC andCCCC subdomains. In the LIM2 domains, thesecond (b2, b-strands III and IV) and the fourthb-sheet (b4, b-strands VII and VIII) are aligned in aparallel fashion (PeÂrez-Alvarado et al., 1994; Konratet al., 1997). In LIM1, the fourth b-sheet is rotated
clockwise relative to the second (Kontaxis et al.,1998). However, the relative orientation of the twozinc-binding sites in LIM1 is less static but rathertime-dependent, apparently as a consequence ofweaker packing interactions in the hydrophobiccore. This difference in orientation leads to awidening of the hydrophobic core region with anextended hydrophobic groove, and hence to adifferent exposure and accessibility of residueslocated at the interface of the CCHC and CCCCsubdomains. In view of the pronounced bindingspeci®cities of LIM domains, it is thus conceivablethat this structural feature may provide a mecha-nism to modulate the protein binding capacity ofthe LIM domain. Additionally, the solution struc-ture of the point-mutant CRP2(LIM2)R122Ademonstrates that packing interactions in thehydrophobic core and the relative orientation ofthe two CCHC and CCCC subdomains in an intactLIM domain is not an intrinsic rigid feature of thedomain but can be modi®ed by, for example, theformation or breakage of hydrogen bonds and/orsalt bridges.
LIM domains interact speci®cally with otherLIM domains and with many different proteindomains and are thus thought to function as pro-tein interaction modules, mediating speci®c con-tacts between members of functional complexesand modulating the activity of the macromolecularprotein constituents. The participation of LIMdomains in physiologically important macromol-ecular (oligomeric) complexes appears to require atleast two coordinated activities: in the case of LIM-HD proteins, the modulation of LIM domain-homeodomain association and the assembly of atranscription complex or, in the case of LIM-onlyproteins, the reversible association with diverseprotein binding partners. In order to ful®ll thesediverse regulatory functions, it seems plausiblethat the interacting LIM domain makes use of aninherent conformational domain ¯exibility (e.g.switch of subdomain orientation) by which homeo-domain association and binding of other authenticprotein binding partners can be modulated, eitherpermitting and facilitating, or impairing the rel-evant interaction. The present study demonstratesthat packing in the hydrophobic core and the rela-tive orientation of the two zinc-binding CCHC andCCCC sites is not an invariant feature of the LIMdomain but might be modi®ed with only smallexpenditures of energy in the course of speci®cinteractions with authentic binding partners. Sucha conformational switch could be used synergisti-cally to modify binding af®nities of LIM domainsin the context of a macromolecular complex.
Materials and Methods
Synthesis, isotopic labeling, and purification ofmutant recombinant protein CRP2(LIM2)R122A
The expression plasmid pET3d-qCRP2(LIM2) encodesa 113 amino acid CRP2(LIM2) peptide encompassing

Conformational Flexibility of LIM Domains 905
amino acid residues 82-194 of quail CRP2 including thecarboxyl-terminal LIM domain (LIM2) (Konrat et al.,1997). In order to substitute alanine for the arginine resi-due in CRP2(LIM2) at position 122 (numbering refers tothe position in the full-sized CRP2 protein), oligonucleo-tide-directed in vitro mutagenesis of the expression plas-mid DNA was performed using the Transformer site-directed mutagenesis kit (Clontech) based on the methodof Deng & Nickoloff (1992). This procedure of intro-ducing mutations in double-stranded plasmid DNAemploys a mutagenic oligonucleotide for the introduc-tion of the desired mutation in the cloned genesequences and an additional selection oligonucleotidethat destroys a unique restriction site in the plasmid vec-tor sequences. The two oligonucleotides are simul-taneously annealed to one strand of the denaturedplasmid DNA, followed by DNA elongation and lig-ation. After transformation into Escherichia coli strainBMH 71-18 mutS defective in mismatch repair, themutated plasmid is selected from isolated plasmid DNAby its resistance to digestion by the selection restrictionenzyme. Here, the mutagenic oligonucleotide50-CAGAATCCCCACACGCAGAACATTTCTCCGCACC-30 (the base substitutions are underlined) complementaryto the CRP2(LIM2) coding strand was used to introducethe desired R122A substitution. The selection oligo-nucleotide 50-CAGGAAAGAAGATCTGAGCAAAAG-30(the base substitutions are underlined) was used todestroy the unique A¯III site (A'CATGT) in the pET3dvector sequences and transform it into a BgIII site(A'GATCT). For veri®cation of the mutagenesis pro-cedure, the nucleotide sequence of the entire insert DNAof the pET3d-qCRP2(LIM2)R122A mutant plasmid wasdetermined by the dideoxynucleotide chain terminationmethod (Sanger et al., 1977) using a T7 sequencing kit(Pharmacia) and pET vector-speci®c primers. Indepen-dent veri®cation of the mutagenesis procedure wasobtained by mass spectroscopy (LC-MS) of the recombi-nant protein product (see below) using a MarinerESI-TOF mass spectrometer (PE Biosystems) (kindlyperformed by F. Lottspeich, Max-Planck-Institute ofBiochemistry, Martinsried).
Synthesis of the mutant recombinant proteinCRP2(LIM2)R122A in the T7 RNA polymerase directedbacterial expression system (Studier et al., 1990) and itspuri®cation by cation exchange chromatography wereperformed essentially as described for the wild-typeCRP2(LIM2) protein (Konrat et al., 1997). The homogen-eity of the ®nal product was veri®ed by SDS-polyacryl-amide gel electrophoresis (Bister et al., 1977; Jansen et al.,1983; Hartl & Bister, 1998) and by amino-terminalsequencing using a Procise 492 sequencer (PE Biosys-tems) (kindly performed by F. Lottspeich). Wild-typeCRP2(LIM2) protein used for comparison was preparedas described (Konrat et al., 1997). 15N-labeling of themutant recombinant protein by growing bacteria inminimal medium supplemented with 15NH4Cl was car-ried out as described for the wild-type protein (Konratet al., 1997). The ®nal yield of 15N-labeled CRP2(LIM2)-R122A protein was approximately 4 mg per liter of bac-terial culture.
Nuclear magnetic resonance spectroscopy
All NMR experiments were recorded at 26 �C on anVarian Unity Plus 500 MHz spectrometer equipped witha PFG unit using a triple resonance probe with activelyshielded z-gradients. The sample contained 1.1 mM15N-labeled CRP2(LIM2)R122A, 20 mM potassium phos-
phate, 50 mM potassium chloride and 0.5 mM dithio-threitol in 90 % H2O/10 % 2H2O at pH 7.0. Spectrarecorded for spin system identi®cation and sequentialassignment include two-dimensional SS-NOESY (100 ms,64 scans per increment) (Smallcombe, 1993), watergateNOESY (64 scans per increment) (Piotto et al., 1992),TOCSY (75 ms), sensitivity enhanced 15N-HSQC (64scans per increment), three-dimensional 15N-TOCSY-HSQC (70 ms), and 15N-NOESY-HSQC (150 ms) (Marionet al., 1989). The SS-NOESY used a 330 ms readout pulsewith an excitation null at the water frequency. Sensitivityenhancement and coherence selection of the 15N-HSQC,15N-NOESY-HSQC and the 15N-TOCSY-HSQC wereachieved by the means of pulsed ®eld gradients (Kayet al., 1992). Both three-dimensional spectra were per-formed with a water ¯ip back pulse to minimize theeffects of radiation damping and solvent exchange(Grzesiek & Bax, 1993; Stonehouse et al., 1994). Decou-pling during acquisition of heteronuclear spectra wasachieved by a 15N GARP sequence (Shaka et al., 1985)using a 1.5 kHz radio frequency ®eld. Isotropic mixingwas performed using a DIPSI-2 sequence (Rucker &Shaka, 1989). The FIDs were processed using shifted sinebell functions. The sizes of the data matrices of the two-dimensional spectra after zero®lling in both dimensionswere 900 � 1984 for the SS-NOESY, 512 � 1984 forthe watergate NOESY, 512 � 1984 for the TOCSYand 2 � 64 � 128 for the 15N-HSQC spectrum. Thethree-dimensional spectra resulted from data matricesof 256 � 64 � 2048 (15N-NOESY-HSQC) and256 � 64 � 2048 data points (15N-TOCSY-HSQC),respectively, after linear prediction and zero-®lling inboth indirect dimensions and zero-®lling in the directdimension. FID processing and Fourier transformationwas performed using NMRPipe software (Delaglio et al.,1993), analysis of all spectra was carried out usingANSIG 3.3 software (Kraulis, 1989) and followed theestablished strategy of combining spin system infor-mation provided by the 15N-TOCSY-HSQC sequenceswith sequential information extracted from the three-dimensional 15N-NOESY-HSQC spectrum. 3J(HNHa)coupling constants which served as a means to deter-mine secondary structure elements were determined bythe three-dimensional HNHA-experiment (Kubinowaet al., 1994) with an evolution delay of 13.4 ms. Inte-gration of HNHA peaks was performed with NMRpipesoftware. Backbone dynamic information was obtainedby measuring 15N longitudinal and transverse relaxationas well as heteronuclear {1H}-15N-NOE spectra (Farrowet al., 1994). T1 spectra were recorded with delays of0, 111, 222, 444 and 666 ms; T2 values were derived fromspectra recorded with delays of 0, 31.4, 62.8, 109.9 and157 ms. Heteronuclear NOE values were determinedfrom spectra recorded with and without proton satur-ation of three seconds. For equilibrium nitrogen magneti-zation spectra (spectra without NOE), a relaxation delayof eight seconds was employed, whereas in spectra witha three second proton saturation a relaxation delay of®ve seconds was used prior to the saturation. The size ofthe data matrices for all spectra was 2 � 64 � 128, with64 scans per increment. Integration of peaks (T1 and T2)was performed with either FELIX software (Biosym/MSI, 1995) or (NOE) NMRpipe software. The relaxationrates were determined by employing a non-linear least-squares ®t of experimental data to an exponentiallydecaying function (Farrow et al., 1994). Steady-stateNOEs were calculated as the ratios of peak volumes inthe spectra with and without proton presaturation. ForT1, T2, and NOE measurements, the uncertainties in the

906 Conformational Flexibility of LIM Domains
measured volumes were estimated from the base-planenoise in the spectra.
Interpretation of 15N relaxation data followed thereduced spectral density mapping approach (Farrowet al., 1995a,b; Ishima & Nagayama, 1995). Standardrelationships between 15N relaxation parameters andspectral density functions for the motion of the NH bondvector have been used (Abragam, 1986). The rotationaldiffusion anisotropy of CRP2(LIM2)R122A was deter-mined using the local diffusion approximation(BruÈ schweiler et al., 1995; Lee et al., 1997). Local diffusionconstants were determined from the R2/R1 ratios for 22well-ordered residues, using an isotropic spectral densityfunction (Fairbrother et al., 1998). The diffusion tensorwas determined by least-squares solution of the equationrelating the local diffusion constant with the diffusiontensor and the transformation matrix relating the mol-ecular reference frame and the principal axes of the dif-fusion tensor. The ensemble of the 11 energy-minimizedNMR solution structures was used and the averagevalues of the diffusion tensor and transformation matrixelements are given. Axially and spherically symmetricdiffusion models were compared using F-statistical test-ing (Lee et al., 1997).
Structure calculations
Structure calculation was performed on SGI work-stations using X-PLOR software (BruÈ nger, 1992) with thestandard simulated annealing/structure re®nement/energy minimization protocols (Nilges et al., 1988).Experimental NOE distance restraints were extractedfrom two and three-dimensional NOESY spectra andclassi®ed as strong (1.8-2.7 AÊ ), medium (1.8-3.3 AÊ ), weak(1.8-5 AÊ ) and very weak (1.8-6 AÊ ). First calculations wereperformed on an covalent linear template structure witheach zinc ion covalently attached to one of its ligands.Distance restraints between the zinc ion and the coordi-nating residues were used to enforce tetrahedral geome-try of residues Cys120, Cys123, His141, and Cys144 andof residues Cys147, Cys150, Cys168 and Cys171, respect-ively. Upper and lower distance limits for the atomsparticipating in coordination were (in AÊ ) 3.30 () Sg-Sg () 3.50 and 3.30 () Sg-Nd1 () 3.50. In thecourse of the calculations, covalent bonds to the zincions were de®ned for all coordinating atoms using stan-dard bond lengths and bond angles, which comprise theequilibrium distances Zn-Sg (2.30 AÊ ), Zn-Nd1 (2.00 AÊ )with force constants of 200 kcal/(mol AÊ 2) as well as equi-librium bond angles centered on the coordinating het-eroatoms and the metal atoms and their correspondingforce constants which were de®ned as follows: Zn-Sg-Cb,107 �, 40 kcal/(mol.rad2), Zn-Nd1-Ce1, 120 �, 40 kcal/(molrad2), Sg-Zn-Sg, 109 �, 40 kcal/(mol.rad2), and Sg-Zn-Nd1,109 �, 40 kcal/(mol.rad2) (Lee et al., 1989). The resultingstructures were ®nally re®ned by energy minimizationusing the CHARMM force ®eld (Brooks et al., 1993).Structure superposition and calculation of r.m.s. devi-ation values was carried out using the software packageMOLMOL (Koradi et al., 1996).
Protein Data Bank Accession Number
The coordinates have been deposited in theBrookhaven Protein Data Bank with accession number1CXX.
Acknowledgments
We thank Friedrich Lottspeich (Max-Planck-Instituteof Biochemistry and TopLab, Martinsried, Germany) forprotein sequencing and mass spectroscopy, N.D.Farrow(DuPont Merck Pharmaceutical Company, Wilmington,DE) for making available numerical procedures toextract dynamical parameters from 15N relaxation data,and Nicole Meier and Sabine Weiskirchen for excellenttechnical assistance. This research was supported bygrants P13486 (to R.K.), P11600 (to B.K.) and SFB-F002/211 (to K.B.) from the Austrian Science Foundation(FWF).
References
Abragam, A. (1986). The Principles of Nuclear Magnetism,Clarendon Press, Oxford, UK.
Agulnick, A. D., Taira, M., Breen, J. J., Tanaka, T.,Dawid, I. B. & Westphal, H. (1996). Interactions ofthe LIM-domain-binding factor Ldb1 with LIMhomeodomain proteins. Nature, 384, 270-272.
Arber, S. & Caroni, P. (1996). Speci®city of single LIMmotifs in targeting and LIM/LIM interactions insitu. Genes Dev. 10, 289-300.
Arber, S., Halder, G. & Caroni, P. (1994). Muscle LIMprotein, a novel essential regulator of myogenesis,promotes myogenic differentiation. Cell, 79, 221-231.
Arber, S., Hunter, J. J., Ross, J., Jr, Hongo, M., Sansig,G., Borg, J., Perriard, J. C., Chien, K. R. & Caroni, P.(1997). MLP-de®cient mice exhibit a disruption ofcardiac cytoarchitectural organization, dilated cardi-omyopathy, and heart failure. Cell, 88, 393-403.
Bach, I., Rhodes, S. J., Pearse, R. V., II, Heinzel, T.,Gloss, B., Scully, K. M., Sawchenko, P. E. &Rosenfeld, M. G. (1995). P-Lim, a LIM homeodo-main factor, is expressed during pituitary organand cell commitment and synergizes with Pit-1.Proc. Natl Acad. Sci. USA, 92, 2720-2724.
Bach, I., CarrieÁre, C., Ostendorff, H. P., Andersen, B. &Rosenfeld, M. G. (1997). A family of LIM domain-associated cofactors confer transcriptional synergismbetween LIM and Otx homeodomain proteins.Genes Dev. 11, 1370-1380.
Bister, K., Hayman, M. J. & Vogt, P. K. (1977). Defective-ness of avian myelocytomatosis virus MC29: iso-lation of long-term nonproducer cultures andanalysis of virus-speci®c polypeptide synthesis. Vir-ology, 82, 431-448.
Brooks, B. R., Bruccoleri, R. E., Olafson, B. D., States,D. J., Swaminathan, S. & Karplus, M. (1983).CHARMM: a program for macromolecular energy,minimization, and dynamics calculations. J. Comput.Chem. 4, 635-637.
BruÈ nger, A. T. (1992). X-PLOR version 3.1: a system forX-ray crystallography and NMR, Yale UniversityPress, New Haven, USA, CT.
BruÈ schweiler, R., Liao, X. & Wright, P. E. (1995). Long-range motional restrictions in a multidomain zinc-®nger protein from anisotropic tumbling. Science,268, 886-889.
Dawid, I. B., Toyama, R. & Taira, M. (1995). LIMdomain proteins. C.R. Acad. Sci. Paris, 318, 295-306.
Dawid, I. B., Breen, J. J. & Toyama, R. (1998). LIMdomains: multiple roles as adapters and functionalmodi®ers in protein interactions. Trends Genet. 14,156-162.

Conformational Flexibility of LIM Domains 907
Delaglio, F., Grzesiek, S., Vuister, G., Zhu, G., Pfeifer, J.& Bax, A. (1993). NMRPipe: a multidimensionalspectral processing system based on UNIX pipes.J. Biomol. NMR, 6, 277-293.
Deng, W. P. & Nickoloff, J. A. (1992). Site-directed muta-genesis of virtually any plasmid by eliminating aunique site. Anal. Biochem. 200, 81-88.
Fairbrother, W. J., Liu, J., Pisacane, P. I., Sliwkowski,M. X. & Palmer, A. G., III (1998). Backbonedynamics of the EGF-like domain of heregulin-a.J. Mol. Biol. 279, 1149-1161.
Farrow, N. A., Muhandiram, R., Singer, A. U., Pascal,S. M., Kay, C. M., Gish, G., Shoelson, S. E., Pawson,T., Forman-Kay, J. D. & Kay, L. E. (1994). Backbonedynamics of a free and a phosphopeptide-com-plexed Src homology 2 domain studied by 15NNMR relaxation. Biochemistry, 33, 5984-6003.
Farrow, N. A., Zhang, O., Forman-Kay, J. D. & Kay,L. E. (1995a). Comparison of the backbonedynamics of a folded and an unfolded SH3 domainexisting in equilibrium in aqueous buffer. Biochemis-try, 34, 868-878.
Farrow, N. A., Zhang, O., Szabo, A., Torchia, D. & Kay,L. E. (1995b). Spectral density function mappingusing 15N relaxation data exclusively. J. Biomol.NMR, 6, 153-162.
Freyd, G., Kim, S. K. & Horvitz, H. R. (1990). Novelcysteine-rich motif and homeodomain in the pro-duct of the Caenorhabditis elegans cell lineage genelin-11. Nature, 344, 876-879.
German, M. S., Wang, J., Chadwick, R. B. & Rutter, W. J.(1992). Synergistic activation of the insulin gene bya LIM-homeodomain protein and a basic helix-loop-helix domain protein: building a functional insulinminienhancer complex. Genes Dev. 6, 2165-2176.
Grzesiek, S. & Bax, A. (1993). The importance of notsaturating water in protein NMR. Application tosensitivity enhancement and NOE measurements.J. Am. Chem. Soc. 115, 12593-12594.
HammarstroÈm, A., Berndt, K. D., Sillard, R., Adermann,K. & Otting, G. (1996). Solution structure of a natu-rally-occurring zinc-peptide complex demonstratesthat the N-terminal zinc-binding module of theLasp-1 LIM domain is an independent folding unit.Biochemistry, 35, 12723-12732.
Hartl, M. & Bister, K. (1998). Structure and transcrip-tional regulation of BKJ, a novel AP-1 target geneactivated during jun-or fos-induced ®broblast trans-formation. Oncogene, 17, 2901-2913.
Ishima, R. & Nagayama, K. (1995). Protein backbonedynamics revealed by quasi spectral density func-tion analysis of amide N-15 nuclei. Biochemistry, 34,3162-3171.
Jansen, H. W., Patschinsky, T. & Bister, K. (1983). Avianoncovirus MH2: molecular cloning of proviral DNAand structural analysis of viral RNA and protein.J. Virol. 48, 61-73.
Johnson, J. D., Zhang, W., Rudnick, A., Rutter, W. J. &German, M. S. (1997). Transcriptional synergybetween LIM-homeodomain proteins and basichelix-loop-helix proteins: the LIM2 domain deter-mines speci®city. Mol. Cell. Biol. 17, 3488-3496.
Jurata, L. W., Kenny, D. A. & Gill, G. N. (1996). NuclearLIM interactor, a rhombotin and LIM homeo-domain interacting protein, is expressed early inneuronal development. Proc. Natl Acad. Sci. USA,93, 11693-11698.
Karlsson, O., Thor, S., Norberg, T.Ohlsson, H. &Edlund, T. (1990). Insulin gene enhancer binding
domain Isl-1 is a member of a novel class of pro-teins containing both a homeo- and a Cys-Hisdomain. Nature, 344, 879-882.
Kay, L. E., Keifer, P. & Saarinen, T. (1992). Pure absorp-tion gradient enhanced heteronuclear single quan-tum correlation spectroscopy with improvedsensitivity. J. Am. Chem. Soc. 114, 10663-10665.
Kong, Y., Flick, M. J., Kudla, A. J. & Konieczny, S. F.(1997). Muscle LIM protein promotes myogenesisby enhancing the activity of MyoD. Mol. Cell. Biol.17, 4750-4760.
Konrat, R., Weiskirchen, R., KraÈutler, B. & Bister, K.(1997). Solution structure of the carboxyl-terminalLIM domain from quail cysteine-rich protein CRP2.J. Biol. Chem. 272, 12001-12007.
Konrat, R., KraÈutler, B., Weiskirchen, R. & Bister, K.(1998a). Structure of cysteine and glycine-richprotein CRP2: backbone dynamics reveal motionalfreedom and independent spatial orientation of theLIM domains. J. Biol. Chem. 273, 23233-23240.
Konrat, R., Weiskirchen, R., Bister, K. & KraÈutler, B.(1998b). Bispheric coordinative structuring in a zinc®nger protein: NMR analysis of a point-mutant ofthe carboxy-terminal LIM domain of quail cysteineand glycine-rich protein CRP2. J. Am. Chem. Soc.120, 7127-7129.
Kontaxis, G., Konrat, R., KraÈutler, B., Weiskirchen, R. &Bister, K. (1998). Structure and intramodulardynamics of the amino-terminal LIM domain fromquail cysteine and glycine-rich protein CRP2. Bio-chemistry, 37, 7127-7134.
Koradi, R., Billetter, M. & WuÈ thrich, K. (1996). MOL-MOL: a program for display and analysis of macro-molecular structures. J. Mol. Graph. 14, 51-55.
Kosa, J. L., Michelsen, J. W., Louis, H. A., Olsen, J. I.,Davis, D. R., Beckerle, M. C. & Winge, D. R. (1994).Common metal ion coordination in LIM domainproteins. Biochemistry, 33, 468-477.
Kraulis, P. J. (1989). ANSIG: a program for the assign-ment of protein 1H 2D NMR spectra by interactivegraphics. J. Magn. Reson. 24, 627-633.
Kraulis, P. J. (1991). MOLSCRIPT: a program to produceboth detailed and schematic plot of protein struc-tres. J. Appl. Crystallog. 24, 946-950.
Kuboniwa, H., Grzesiek, S., Delaglio, F. & Bax, A.(1994). Measurement of HNHa J couplings in cal-cium-free calmodulin using new 2D and 3D water-¯ip-back methods. J. Biomol. NMR, 4, 871-878.
Lee, K. L., Rance, M., Chazin, W. & Palmer, A. G., III(1997). Rotational diffusion anisotropy of proteinsfrom simultaneous analysis of 15N and 13Ca nuclearspin relaxation. J. Biomol. NMR, 9, 287-298.
Lee, M. S., Gippert, G. P., Soman, K. V., Case, D. A. &Wright, P. E. (1989). Three-dimensional solutionstructure of a single zinc ®nger DNA-bindingdomain. Science, 245, 635-637.
Lichtsteiner, S. & Tijan, R. (1995). Synergistic activationof transcription by UNC-86 and MEC-3 in Caenor-habditis elegans embryo extracts. EMBO J. 14, 3937-3945.
Liebhaber, S. A., Emery, J. G., Urbanek, M.Wang, X. K.& Cooke, N. E. (1990). Characterization of a humancDNA encoding a widely expressed and highlyconserved cysteine-rich protein with an unusualzinc-®nger motif. Nucl. Acid Res. 18, 3871-3879.
Marion, D., Driscoll, P. C., Kay, L. E., Wing®eld, P. T.,Bax, A., Gronenborn, A. M. & Clore, G. M. (1989).Overcoming the overlap problem in the assignment

908 Conformational Flexibility of LIM Domains
of proton NMR spectra of larger proteins. Biochemis-try, 28, 6150-6156.
Michelsen, J. W., Schmeichel, K. L., Beckerle, M. C. &Winge, D. R. (1993). The LIM motif de®nes aspeci®c zinc-binding protein domain. Proc. NatlAcad. Sci. USA, 90, 4404-4408.
Michelsen, J. W., Sevell, A. K., Louis, H. A., Olsen, J. I.,Davis, D. R., Winge, D. R. & Beckerle, M. C. (1994).The LIM motif de®nes a speci®c zinc-bindingprotein domain. J. Biol. Chem. 269, 11108-11113.
Morcillo, P., Rosen, C., Baylies, M. K. & Dorsett, D.(1997). Chip, a widely expressed chromosomalprotein required for segmentation and activity of aremote wing margin enhancer in Drosophila. GenesDev. 11, 2729-2740.
Nilges, M., Clore, G. M. & Gronenborn, A. M. (1988).Determination of three-dimensional structures ofproteins from interproton distance data by hybriddistance-dynamical simulated annealing calcu-lations. FEBS Letters, 229, 317-324.
PeÂrez-Alvarado, G. C., Miles, C., Michelsen, J. W., Louis,H. A., Winge, D. R., Beckerle, M. C. & Summers,M. F. (1994). Structure of the C-terminal LIMdomain from the cysteine-rich protein, CRP. NatureStruct. Biol. 1, 388-398.
PeÂrez-Alvarado, G. C., Kosa, J. L., Louis, H. A.,Beckerle, M. C., Winge, D. R. & Summers, M. F.(1996). Structure of the cysteine-rich intestinal pro-tein. CRIP. J. Mol. Biol. 257, 153-174.
Piotto, M., Saudek, V. & Sklenar, V. (1992). Gradient-tailored excitation for single-quantum NMR spec-troscopy of aqueous solutions. J. Biomol. NMR, 2,661-665.
PomieÁs, P., Louis, H. A. & Beckerle, M. C. (1997). CRP1,a LIM domain protein implicated in muscle differ-entiation, interacts with a-actinin. J. Cell. Biol. 139,157-168.
Rucker, S. P. & Shaka, A. J. (1989). Broadband homo-nuclear cross polarization in 2D NMR using DIPSI-2. Mol. Phys. 68, 509-517.
Sadler, I., Crawford, A. W., Michelsen, J. W. & Beckerle,M. C. (1992). Zyxin and cCRP: two interactive LIMdomain proteins associated with the cytoskeleton.J. Cell. Biol. 119, 1573-1587.
SaÂnchez-Garcia, I. & Rabbits, T. H. (1994). The LIMdomain: a new structural motif found in zinc-®ngerlike proteins. Trends Genet. 10, 315-320.
SaÂnchez-Garcia, I., Osada, H., Forster, A. & Rabbits,T. H. (1993). The cysteine-rich LIM domains inhibitDNA binding by the associated homeodomain inIsl-1. EMBO J. 12, 4243-4250.
Sanger, F., Nicklen, S. & Coulson, A. R. (1977). DNAsequencing with chain-terminating inhibitors. Proc.Natl Acad. Sci. USA, 74, 5463-5467.
Schmeichel, K. L. & Beckerle, M. C. (1994). The LIMdomain is a modular protein binding interface. Cell,79, 211-219.
Shaka, A. J., Barker, P. B. & Freeman, R. (1985). Com-puter-optimized decoupling scheme for wideband
applications and low-level operation. J. Magn.Reson. 64, 547-552.
Smallcombe, S. (1993). Solvent suppression with symme-trically-shifted pulses. J. Am. Chem. Soc. 115, 4776-4785.
Stonehouse, J., Shaw, G. L., Keeler, J. & Laue, E. D.(1994). Minimizing sensitivity losses in gradient-selected 15N-1H HSQC spectra of proteins. J. Magn.Reson. ser. A, 107, 178-184.
Studier, F. W., Rosenberg, A. H., Dunn, J. J. &Dubendorff, J. W. (1990). Use of T7 RNA polymer-ase to direct expression of cloned genes. MethodsEnzymol. 185, 60-89.
Taira, M., Evrard, J.-J., Steinmetz, A. & Dawid, I. B.(1995). Classi®cation of LIM proteins. Trends Genet.11, 431-432.
Wadman, I. A., Osada, H., GruÈ tz, G. G., Agulnick, A. D.,Westphal, H., Forster, A. & Rabbits, T. H. (1997).The LIM-only protein Lmo2 is a bridging moleculeassembling an erythroid DNA-binding complexwhich includes the TAL1, E47, GATA-1 and Ldb/NLI proteins. EMBO J. 16, 3145-3157.
Wang, A. C. & Bax, A. (1996). Determination of thebackbone dihedral angles f in human ubiquitinfrom reparametrized empirical Karplus equations.J. Am. Chem. Soc. 118, 2483-2494.
Weiskirchen, R. & Bister, K. (1993). Suppression in trans-formed avian ®broblasts of a gene (crp) encoding acysteine-rich protein containing LIM domains.Oncogene, 8, 2317-2324.
Weiskirchen, R., Pino, J. D., Macalma, T., Bister, K. &Beckerle, M. C. (1995). The cysteine-rich proteinfamily of highly related LIM domain proteins.J. Biol. Chem. 270, 28946-28954.
Weiskirchen, R., Erdel, M., Uterman, G. & Bister, K.(1997). Cloning, structural analysis, and chromo-somal localization of the human CSRP2 geneencoding the LIM domain protein CRP2. Genomics,44, 83-93.
Wishart, D. S., Sykes, B. D. & Richards, F. M. (1991).Relationship between nuclear magnetic resonancechemical shift and protein secondary structure.J. Mol. Biol. 222, 311-333.
Wishart, D. S., Sykes, B. D. & Richards, F. M. (1992).The chemical shift index: a fast and simple methodfor the assignment of protein secondary structurethrough NMR spectroscopy. Biochemistry, 31, 1647-1651.
WuÈ thrich, K. (1986). NMR of Proteins and Nucleic Acids,Wiley, New York.
Xue, D., Tu, Y. & Chal®e, M. (1993). Cooperative inter-actions between the Caenorhabditis elegans homeopro-teins UNC-86 and MEC-3. Science, 261, 1324-1328.
Yao, X., PeÂrez-Alvarado, C., Louis, H. A., PomieÁs, P.,Hatt, C., Summers, M. F. & Beckerle, M. C. (1999).Solution structure of the chicken cysteine-rich pro-tein, CRP1, a double-LIM protein implicated inmuscle differentiation. Biochemistry, 38, 5701-5713.
Edited by P. E. Wright
(Received 6 July 1999; received in revised form 11 August 1999; accepted 12 August 1999)




















