
Mucin 1 (MUC1) signalling contributes to increase the resistance to cell death in human bronchial...
10
Mucin 1 (MUC1) signalling contributes to increase the resistance to cell death in human bronchial epithelial cells exposed to nickel acetate Alessandro Castorina • Salvatore Giunta Received: 8 April 2014 / Accepted: 9 July 2014 Ó Springer Science+Business Media New York 2014 Abstract We have previously reported that nickel acetate (Ni 2? ), a well-known human carcinogenic agents, differentially affected apoptosis in two differ- ent airway epithelial cell lines derived from the human respiratory tract (A549 and Beas-2B, respectively), suggesting a potential involvement of epidermal growth factor receptor (EGFR)/Neu receptors in mediating this effect. Since ErbBs are closely associ- ated to Mucin 1 (MUC1), a glycoprotein component of airway mucus that is overexpressed in lung tumors, we have investigated the role of this signaling system in the survival response of airway epithelial cells against Ni 2? -induced cell death. We found that A549 cells exposed to Ni 2? do not show any significant increase of MUC1 levels. Conversely, Beas-2B cells exposed to equivalent concentrations of Ni 2? showed increased expression of MUC1 levels and this correlated with increased phosphorylation of both EGFR and of the extracellular-regulated kinase 1/2 (ERK1/2) and increase resistance to apoptosis, as indicated by cell viability assessments and DNA damage. Interestingly, suppression of MUC1 by small interfering RNA inhibited the EGFR activation in Beas-2B cells, leading to a significant decrease of survival and enhancement of DNA fragmentation and cleaved Caspase-3 expression. These results strongly suggest a role for MUC1 in Ni 2? -induced neoplastic transfor- mation, which likely involves the activation of the EGFR-mediated cell survival pathway, highlighting new avenues in the molecular approach to lung cancer prevention. Keywords Nickel Lung cancer Apoptosis Mucin-1 EGFR Introduction Professional exposure to nickel compounds is associ- ated with increased incidence of certain human cancer, including lung and nasal cancers (Costa 1991). Several mechanisms have been proposed for nickel-induced carcinogenesis. These include production of reactive oxygen species (ROS) (Capasso et al. 2014), induction of DNA damage (Costa 1991; Lu et al. 2005), alteration of epigenetic changes such as histone modifications (Salnikow and Zhitkovich 2008; Salni- kow and Costa 2000), disruption of cellular iron homeostasis (Davidson et al. 2005), and activation of oncogenic pathways (Pan et al. 2011). It has been reported that nickel compounds can regulate the expression of specific genes related to tumor devel- opment (Cangul et al. 2002; Salnikow and Costa 2000), however, the molecular mechanisms involved have not been clearly identified. A. Castorina S. Giunta (&) Department of Bio-Medical Sciences, Section of Anatomy and Histology, University of Catania, Via S.Sofia, 87, 95123 Catania, Italy e-mail: [email protected] 123 Biometals DOI 10.1007/s10534-014-9776-x
Transcript of Mucin 1 (MUC1) signalling contributes to increase the resistance to cell death in human bronchial...

Mucin 1 (MUC1) signalling contributes to increasethe resistance to cell death in human bronchial epithelialcells exposed to nickel acetate
Alessandro Castorina • Salvatore Giunta
Received: 8 April 2014 / Accepted: 9 July 2014
� Springer Science+Business Media New York 2014
Abstract We have previously reported that nickel
acetate (Ni2?), a well-known human carcinogenic
agents, differentially affected apoptosis in two differ-
ent airway epithelial cell lines derived from the human
respiratory tract (A549 and Beas-2B, respectively),
suggesting a potential involvement of epidermal
growth factor receptor (EGFR)/Neu receptors in
mediating this effect. Since ErbBs are closely associ-
ated to Mucin 1 (MUC1), a glycoprotein component of
airway mucus that is overexpressed in lung tumors, we
have investigated the role of this signaling system in
the survival response of airway epithelial cells against
Ni2?-induced cell death. We found that A549 cells
exposed to Ni2? do not show any significant increase
of MUC1 levels. Conversely, Beas-2B cells exposed
to equivalent concentrations of Ni2? showed increased
expression of MUC1 levels and this correlated with
increased phosphorylation of both EGFR and of the
extracellular-regulated kinase 1/2 (ERK1/2) and
increase resistance to apoptosis, as indicated by cell
viability assessments and DNA damage. Interestingly,
suppression of MUC1 by small interfering RNA
inhibited the EGFR activation in Beas-2B cells,
leading to a significant decrease of survival and
enhancement of DNA fragmentation and cleaved
Caspase-3 expression. These results strongly suggest
a role for MUC1 in Ni2?-induced neoplastic transfor-
mation, which likely involves the activation of the
EGFR-mediated cell survival pathway, highlighting
new avenues in the molecular approach to lung cancer
prevention.
Keywords Nickel � Lung cancer � Apoptosis �Mucin-1 � EGFR
Introduction
Professional exposure to nickel compounds is associ-
ated with increased incidence of certain human cancer,
including lung and nasal cancers (Costa 1991). Several
mechanisms have been proposed for nickel-induced
carcinogenesis. These include production of reactive
oxygen species (ROS) (Capasso et al. 2014), induction
of DNA damage (Costa 1991; Lu et al. 2005),
alteration of epigenetic changes such as histone
modifications (Salnikow and Zhitkovich 2008; Salni-
kow and Costa 2000), disruption of cellular iron
homeostasis (Davidson et al. 2005), and activation of
oncogenic pathways (Pan et al. 2011). It has been
reported that nickel compounds can regulate the
expression of specific genes related to tumor devel-
opment (Cangul et al. 2002; Salnikow and Costa
2000), however, the molecular mechanisms involved
have not been clearly identified.
A. Castorina � S. Giunta (&)
Department of Bio-Medical Sciences, Section of Anatomy
and Histology, University of Catania, Via S.Sofia, 87,
95123 Catania, Italy
e-mail: [email protected]
123
Biometals
DOI 10.1007/s10534-014-9776-x

Several studies have reported that nickel com-
pounds may either promote apoptosis or increase cell
resistance in different cell types (Ahamed et al. 2011;
Ding et al. 2006; Pan et al. 2011). These opposite
effects might depend on the different activation of
specific target membrane receptors and/or molecular
pathways. In a previous work (Giunta et al. 2012) we
have shown that nickel acetate (Ni2?) exposure
differentially affected apoptosis in two distinct airway
epithelial cell lines derived from the human respira-
tory tract. In particular, we observed a decreased
susceptibility of bronchial (Beas-2B) compared to
alveolar (A549) epithelial cells to Ni2?-induced
apoptosis which was correlated to an increased
activation of both epidermal growth factor receptor
(EGFR) and neuregulin (Neu) receptor. Such recep-
tors have been shown to be differently regulated after
exposure to different metals both in A549 and Beas-
2B cells, thus activating molecular pathways that
either promoted proliferation or induced cell death
(Wu et al. 1999; Castorina et al. 2008; Kundu et al.
2011, Mosesson and Yarden 2004). Both EGFR and
Neu receptor are situated at the cell membrane and
have an extracellular ligand-binding region, a trans-
membrane region and a cytoplasmic tyrosine-kinase
domain. Ligand binding to the receptors lead to the
activation of a variety of intracellular signaling
pathways that promote cell growth, proliferation,
differentiation, and migration (Zandi et al. 2007;
Baselga and Arteaga 2005). The EGFR downstream
signaling pathways include components of the Ras/
Raf/MAPK (ERK, JNK and p38) and PI3 K/Akt, of
which ERK represents the main kinase responsible for
EGFR-mediated cell survival (Chen et al. 1998 Li
et al. 1998). Although the role of EGFR in lung cancer
progression is well-known, how EGFR is activated by
this specific carcinogen in lung epithelial cell is not
well understood.
Mucin 1 (MUC1) is present on the surface of
various mucosal epithelial cells and is overexpressed
in various adenocarcinomas (Baldus et al. 2004).
MUC1 is synthesized as a single peptide and then
undergoes autocleavage into two subunits, subse-
quently forming a stable non-covalent heterodimer
consisting of an extracellular domain and a cytoplas-
mic tail (Macao et al. 2006). The extracellular domain
of MUC1 is composed of variable number tandem
repeats (VNTR) that consist of 20-amino acids
enriched in serine, threonine, and proline residues
modified by extensive O-glycans, which is thought of
as a physical barrier against the extracellular milieu.
Enhanced expression of MUC1 by tumor microenvi-
ronment has been associated with tumor progression
(Schroeder et al. 2004). In addition, in vitro studies
have demonstrated that the expression of MUC1 in
cancer cells is involved in the invasion (Kohlgraf et al.
2003; Tsutsumida et al. 2006), potentiation of cellular
signaling (Wei et al. 2006) and resistance to genotoxic
anticancer reagents (Ren et al. 2004; Wei et al. 2005),
suggesting that the expression of MUC1 is closely
associated with malignancy of tumor, ultimately
leading to poor prognosis. Recently, It has been shown
that MUC1 interacts with EGF receptors on the cell
surface (Schroeder et al. 2001; Pochampalli et al.
2007a), leading to the sustenance of cell signaling by
ligand binding.
In the present study we have hypothesized that the
higher resistance of Beas-2B to Ni2?-induced apop-
tosis could be due to an enhancement of MUC1
expression, which in turn stimulates the transactiva-
tion of EGF receptors. Furthemore, since MAPKK/
ERK signaling is the major pathway involved in
EGFR-mediated cell survival, we also investigated
total and phosphorylated protein levels of ERK1/2.
The results show that MUC1 confers resistance to
Ni2?-induced apoptosis through EGFR/ERK1/2 sig-
naling in human bronchial epithelial cells (Beas-2B),
highlighting the role of MUC1 and EGFR as potential
molecular targets in lung cancer prevention.
Materials and methods
Cell cultures
Human alveolar type 2-like (A549) and bronchial
epithelial cells (Beas-2B) were purchased from ATCC
(cat no. CCL-185 and CRL-9609, respectively). Both
cell lines were maintained in epidermal growth factor
(EGF)-free Dulbecco modified Eagle medium (DMEM)
containing 500 mg/L glucose, L-glutamine, 100 mg/L
sodium pyruvate supplemented with 10 % FBS,
0.02 M/L HEPES, 100 U/mL penicillin, and 50 ng/mL
streptomycin. Cells were grown in an incubator at 37 �C
in a humidified atmosphere with 5 % CO2 and passaged
at 80 % confluence.
Biometals
123

siRNA mediated knock down of MUC1
MUC1-specific siRNA was generated to the following
target sequence of the MUC1 extracellular domain:
(MUC1: 50-AAGACTGATGCCAGTAGCACT-30)(Lan et al. 1990), and a non-silencing siRNA was
designed to the following target sequence, which lacks
homology to any known mammalian gene:
AATTCTCCGAACGTGTCACGT (Qiagen). Trans-
fections were performed with Lipofectamine 2000
(Invitrogen) following the manufacturer’s suggestions,
4–6 h post-transfection the medium was changed. The
effective gene silencing was confirmed by Western blot
using the appropriate antibody.
Cell viability (MTT assay)
To assess cell viability, we used the cell proliferation
kit I (MTT) following manufacturer’s instructions
(Roche). Cells were seeded into 96-well plates at a
concentration of 1 9 104 cells/well and allowed to
adhere for 24 h. Cells were then treated with 0.1 mM
of nickel acetate (Ni2?) (Sigma Aldrich) for 24 and
48 h. EGF-free DMEM containing 0.5 mg/mL 3-[4,5-
dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bro-
mide (MTT) (Sigma–Aldrich) was added in each
well. Following incubation for 4 h at 37 �C, medium
was removed, and 100 lL of DMSO was added.
Formazan formed by the cleavage of the yellow
tetrazolium salt MTT was measured spectrophotomet-
rically by absorbance change at 550–600 nm using a
microplate reader.
Apoptotic assay by immunodetection
of oligonucleosomes
Mononucleosomes and oligonucleosomes released
from the nucleus into the cytoplasm of apoptotic cells
were detected with the use of a sandwich enzyme-
linked immunosorbent assay (The Cell Death Detec-
tion) ELISAPLUS 10 9 (Roche Applied Sciences). For
sample preparation, after the treatments, Beas-2B cells
were harvested by trypsinization. Cells were lysed in
incubation buffer for 30 min. The lysate was then
centrifuged at 20,000g9 10 min. Supernatant was
diluted to yield 1 9 104 cell equivalents/mL and used
for immunodetection. The assay was performed as
follows: (1) an antibody that react with the histone H1,
H2A, H2B, H3 and H4 was fixed on the wall of the
microplate module provided with the kit; (2) samples
prepared as described above were added to the plate
containing the immobilized anti-histone antibody; (3)
anti-DNA monoclonal antibodies conjugated to per-
oxidase were added, to allow their binding to the DNA
part of nucleosomes; and (4) after removal of unbound
peroxidase conjugate, the amount of peroxidase
retained in the immunocomplex was determined
photometrically with 2,20-azino-di(3-ethylbenzthiaz-
oline sulfonate) as a substrate.
Hoechst 33258 nuclear staining
The typical morphological features of apoptotic
degeneration were analyzed by the use of fluorescence
microscopy with the nuclear dye Hoechst 33258
(Forloni et al. 1993). Cells were fixed with a solution
of methanol/acetic acid (3:1 v/v) for 30 min, washed
three times in PBS and incubated for 15 min at 37 �C
with 0.4 lg/mL Hoechst 33258 dye. After being
rinsed in water, cells were visualized for determination
of nuclear chromatin morphology with the use of an
Axiovert 40 fluorescence microscope (Carl Zeiss Inc.).
Apoptotic cells were recognized on the basis of
nuclear condensation and/or fragmented chromatin.
Each condition was reproduced in three dishes per
experiment. Both apoptotic and normal cells were
determined by analyzing at least three different fields
per dish in a fixed pattern as previously described
(Castorina et al. 2012).
Western blot analysis
Lysates were prepared from subconfluent cells as
previously described (Giunta et al. 2012). Immunoblot
analysis was performed by using antibodies listed
below: MUC1 mouse monoclonal antibody (cell
signaling, #4538), p-EGF Receptor (Tyr 1173) rabbit
polyclonal antibody (sc-03, Santa Cruz Biotechnology,
Inc.), p-Neu Receptor (Tyr 1248) rabbit polyclonal
antibody (sc-12352, Santa Cruz Biotechnology, Inc.),
cleaved Caspase-3 rabbit polyclonal antibody (sc-
22171-R, Santa Cruz Biotechnology, Inc), ERK1/2
Antibody (MK1) mouse monoclonal antibody (sc-
135900, Santa Cruz Biotechnology, Inc.), p-ERK1/2
(Thr 202/Tyr 204) rabbit polyclonal antibody (sc-
16982, Santa Cruz Biotechnology, Inc) and b-tubulin
rabbit polyclonal antibody (sc-9104, Santa Cruz Bio-
technology, Inc.) which was used as loading control.
Biometals
123

All primary antibodies were diluted 1:200, while
secondary antibodies (HRP-conjugated goat anti-
mouse and anti-rabbit antibodies, Amersham Biosci-
ences) were used at 1:10,000. Blots were developed
using enhanced chemiluminescence technique (Amer-
sham Biosciences). No signal was detected when the
primary antibody was omitted (data not shown).
Statistical analysis
All statistical analyses were performed using Graph-
Pad InStat version 3.00, GraphPad Software Inc., San
Diego CA, USA). Data are reported as mean ± SEM.
One-way analysis of variance (ANOVA) was used to
compare differences among groups and statistical
significance was assessed by Tukey–Kramer post hoc
test, unless otherwise indicated. The level of signif-
icance accepted for all statistical tests was p B 0.05.
Results
Ni2? treatment increases MUC1 protein expression in
Beas-2B but not in A549 cells
In our previous report we have demonstrated that
Beas-2B are more resistant to Ni2?–induced cell death
than A549 cells. To investigate whether such resistance
was correlated to increased MUC1 expression, both
A549 and Beas-2B were treated with 0.1 mM Ni2? for
24 and 48 h (these experimental conditions were
chosen according to our previous work, Giunta et al.
2012) and subsequently MUC1 protein levels were
measured by Western blot analyses. Our findings
indicate that MUC1 protein expression was differen-
tially affected by Ni2? exposure in either cell line after
24 and 48 h (Fig. 1). Data obtained from these analyses
showed that, as compared to untreated controls, MUC1
protein levels were significantly increased with treat-
ment in Beas-2B cells both after 24 and 48 h
(**p \ 0.01 vs Control) (Fig. 1). Conversely, Ni2?-
treated A549 cells did not show any significant increase
in MUC1 protein expression at both times tested. These
results demonstrated that resistance to Ni2?-induced
apoptosis is correlated with increased MUC1 levels.
Therefore, subsequent experiments were performed
only on Beas-2B cells.
MUC1 suppression inhibits Ni2?-induced EGFR
activation
As previously shown the resistance of Beas-2B cells to
Ni2? is associated to increased phosphorylation levels
of both EGFR and Neu. Therefore, we sought to
determine whether MUC1 plays a role in the activation
of both EGFR and Neu receptors in this cell line. To
this end, before Ni2? treatment, Beas-2B were trans-
fected with either MUC1-specific siRNA to knock-
down MUC1 protein expression (MUC1-siRNA) or
with a non-targeted sequence, which was used as
control siRNA (Control-siRNA), and changes in
Fig. 1 Ni2? treatment
increased MUC1 protein
expression in Beas-2B but
not in A549 cells. MUC1
expression was measured by
Western blot analyses in
A549 and Beas-2B treated
with 0.1 mM Ni2? for 24
and 48 h. Bands were
quantified using
ImageQuantTL software
and normalized values were
plotted in the histogram
shown below; (**p \ 0.01
vs Ctrl). Each condition was
reproduced at least three
times with similar results
using three different batches
of cells (n = 3)
Biometals
123

expression of phosphorylated proteins (p-EGFR and
p-Neu, respectively) were evaluated by Western Blot
analyses. Our findings demonstrated that the basal
activation levels of EGFR were significantly lower in
MUC1-siRNA than in control-siRNA cells (*p \ 0.05
vs Control-siRNA). Moreover, loss of MUC1 inhibits
Ni2?-induced EGFR activation at both time consid-
ered (**p \ 0.01 vs Control-siRNA), (Fig. 2a). Oppo-
sitely, the rate of Neu activation was not affected by
MUC1 silencing neither after 24 and 48 h (Fig. 2a).
The association of MUC1 expression and EGFR
activation suggests that MUC1 plays a role in Ni2?-
induced EGFR activation.
MUC1 attenuation partially blocks EGF-induced
EGFR transactivation in Beas-2B cells
To confirm the mechanism of MUC1-mediated EGFR
activation, we examined the effects of MUC1 silencing
on EGFR activation in response to exogenous EGF
treatment. Both MUC1-siRNA and Control-siRNA
cells were maintained either in EGF-free medium and
stimulated with 50 or 100 ng/mL EGF (Sigma) for 24 h
and subsequently p-EGFR protein levels were mea-
sured by Western Blot analyses. Data obtained from
these analyses showed that EGFR phosphorylation
induced by EGF treatment after 24 h was significantly
lower in MUC1-siRNA as compared to control-siRNA
cells at both doses tested (#p \ 0.05, ##p \ 0.01,
###p \ 0.001 vs EGF-unstimulated cells) (Fig. 2b).
These findings corroborate the idea that EGFR is, at
least in part, activated through a MUC1-dependent
mechanism.
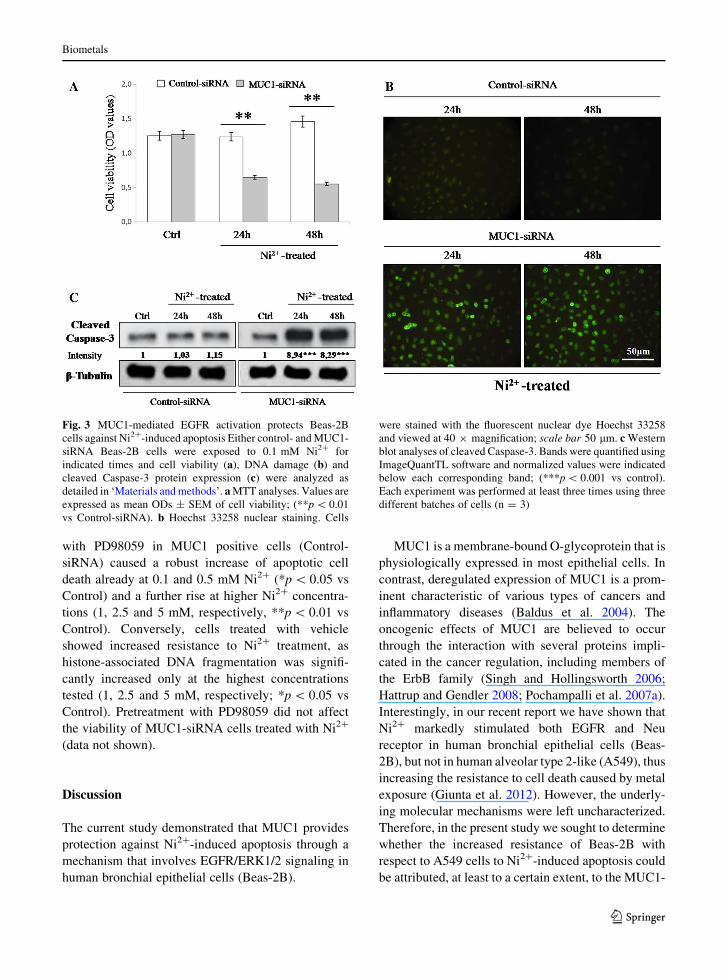
MUC1-driven EGFR activation protects Beas-2B
cells against Ni2?-induced apoptosis
To investigate whether MUC1-mediated EGFR acti-
vation prevents the induction of apoptosis by Ni2?
treatment in Beas-2B cells, we studied the effects of
MUC1-siRNA on Ni2?–induced cell death. As previ-
ously indicated, both MUC1-siRNA and Control-
siRNA cells were exposed to 0.1 mM Ni2? for 24 and
48 h and assayed for cell viability and DNA fragmen-
tation. In agreement with our previous work (Giunta
et al. 2012) the survival rate in control-siRNA cells
was not significantly affected by Ni2? treatment at
both times tested (p [ 0.05 vs untreated controls).
Interestingly, the same Ni2? exposure caused a
significant decline in cell viability in MUC1-siRNA
cells both after 24 and 48 h when compared to control-
siRNA (**p \ 0.01 vs Control-siRNA) (Fig. 3a).
Representative images displayed in Fig. 3b show
the morphological signs of nuclear damage (as deter-
mined by Hoechst 33258 staining) in response to Ni2?
treatment both in MUC1-siRNA and Control-siRNA
cells. As shown, After 24 and 48 h treatment, MUC1-
siRNA cells showed the typical morphological fea-
tures of apoptotic degeneration, whereas apparent
normal morphology persisted in Ni2?-treated Control-
siRNA cells (Fig. 3b).
In the light of these results, to further confirm the
activation of an apoptotic process in MUC1-siRNA
cells, the expression levels of the apoptotic cleaved
Caspase-3 protein was also measured after metal
treatment by Western blot analysis. As demonstrated
in Fig. 3c, Ni2?-treated MUC1-siRNA cells showed a
dramatic increase of cleaved Caspase-3 expression
(***p \ 0.001 vs control), inferring on the existence
of a potential correlation between MUC1 levels and
the activation of apoptotic pathway. Conversely, any
significant changes in the expression of cleaved
Caspase-3 protein were observed in Ni2?-treated
Control-siRNA cells.
MUC1 induces EGFR-mediated ERK1/2
activation
The common cell signalling pathways for EGFR-
mediated cell survival is the MAPKK/ERK1/2 signaling
cascade. To determine whether these downstream
effectors are involved to Ni2?-induced resistance to
apoptosis by EGFR/MUC1 signaling, we used Western
Blot analyses to detect changes in ERK1/2 protein levels
as well as in their phosphorylated forms both in control-
and MUC1-siRNA cells treated with Ni2?. Our findings
indicate that, ERK1/2 phosphorylation was triggered by
Ni2? treatment in MUC1-expressing bronchial epithe-
lial cells (Control-siRNA) (***p \ 0.001 vs control),
but not in MUC1 knockdown cultures (MUC1-siRNA)
(Fig. 4a), suggesting that the activation of ERK1/2
signalling by EGFR may be MUC1-dependent.
To confirm whether resistance to Ni2?-induced
apoptosis of Beas-2B cells is attributable to MUC1/
EGFR-mediated activation of ERK1/2 signalling,
Beas-2B cells were pretreated with the specific
MEK1 inhibitor (PD98059, 25 lM) for 30 min prior
to exposure to Ni2?. As shown in Fig. 4b, pretreatment
Biometals
123

Fig. 2 MUC1 suppression inhibits EGFR activation induced by
Ni2? in Beas-2B cells. Phosphorylated EGFR and Neu receptor
protein was measured by Western blot analyses both in control
and MUC1-siRNA Beas-2B cells exposed to 0.1 mM Ni2? at
the indicated times (a) or EGF (b). Bands were quantified using
ImageQuantTL software and normalized values were plotted in
the histogram shown on the right; (*p \ 0.05, **p \ 0.01,
***p \ 0.001 vs control-siRNA), (#p \ 0.05, ##p \ 0.01,
###p \ 0.001 vs EGF-unstimulated cells). Each condition was
reproduced at least three times with similar results using three
different batches of cells (n = 3)
Biometals
123
with PD98059 in MUC1 positive cells (Control-
siRNA) caused a robust increase of apoptotic cell
death already at 0.1 and 0.5 mM Ni2? (*p \ 0.05 vs
Control) and a further rise at higher Ni2? concentra-
tions (1, 2.5 and 5 mM, respectively, **p \ 0.01 vs
Control). Conversely, cells treated with vehicle
showed increased resistance to Ni2? treatment, as
histone-associated DNA fragmentation was signifi-
cantly increased only at the highest concentrations
tested (1, 2.5 and 5 mM, respectively; *p \ 0.05 vs
Control). Pretreatment with PD98059 did not affect
the viability of MUC1-siRNA cells treated with Ni2?
(data not shown).
Discussion
The current study demonstrated that MUC1 provides
protection against Ni2?-induced apoptosis through a
mechanism that involves EGFR/ERK1/2 signaling in
human bronchial epithelial cells (Beas-2B).
MUC1 is a membrane-bound O-glycoprotein that is
physiologically expressed in most epithelial cells. In
contrast, deregulated expression of MUC1 is a prom-
inent characteristic of various types of cancers and
inflammatory diseases (Baldus et al. 2004). The
oncogenic effects of MUC1 are believed to occur
through the interaction with several proteins impli-
cated in the cancer regulation, including members of
the ErbB family (Singh and Hollingsworth 2006;
Hattrup and Gendler 2008; Pochampalli et al. 2007a).
Interestingly, in our recent report we have shown that
Ni2? markedly stimulated both EGFR and Neu
receptor in human bronchial epithelial cells (Beas-
2B), but not in human alveolar type 2-like (A549), thus
increasing the resistance to cell death caused by metal
exposure (Giunta et al. 2012). However, the underly-
ing molecular mechanisms were left uncharacterized.
Therefore, in the present study we sought to determine
whether the increased resistance of Beas-2B with
respect to A549 cells to Ni2?-induced apoptosis could
be attributed, at least to a certain extent, to the MUC1-
Fig. 3 MUC1-mediated EGFR activation protects Beas-2B
cells against Ni2?-induced apoptosis Either control- and MUC1-
siRNA Beas-2B cells were exposed to 0.1 mM Ni2? for
indicated times and cell viability (a), DNA damage (b) and
cleaved Caspase-3 protein expression (c) were analyzed as
detailed in ‘Materials and methods’. a MTT analyses. Values are
expressed as mean ODs ± SEM of cell viability; (**p \ 0.01
vs Control-siRNA). b Hoechst 33258 nuclear staining. Cells
were stained with the fluorescent nuclear dye Hoechst 33258
and viewed at 40 9 magnification; scale bar 50 lm. c Western
blot analyses of cleaved Caspase-3. Bands were quantified using
ImageQuantTL software and normalized values were indicated
below each corresponding band; (***p \ 0.001 vs control).
Each experiment was performed at least three times using three
different batches of cells (n = 3)
Biometals
123

mediated activation of the EGFR/ERK1/2 signaling
cascade.
In the first part of this paper, we revealed that Ni2?
exposure significantly increased MUC1 expression in
Beas-2B but not in A549 cells (Fig. 1). The following
inhibition of MUC1 expression by siRNA approach in
Beas-2B suppressed the previously observed activa-
tion of EGFR caused by Ni2?, whereas any effect was
observed in the activation state of Neu (Fig. 2a).
Finally, MUC1 silencing also provoked a marked
overall decrease in EGFR activation after exogenous
stimulation with EGF (Fig. 2b). These data are
consistent with other studies demonstrating an onco-
genic role of MUC1 through its ability to activate
EGFR signalling. In particular, using in vivo models,
several research groups reported that overexpression
of MUC1 is associated with increased binding of EGF
to its high affinity EGFR in mammary glands and in
the malignant progression of breast tumors (Schroeder
et al. 2004). In contrast, MUC1 depletion has been
shown to decrease cyclin D1 expression and delayed
the onset of breast tumors (Pochampalli et al. 2007b).
Moreover, in vitro studies have demonstrated that
MUC1 promotes the EGFR-dependent activation of
Fig. 4 MUC1 induces EGFR-mediated ERK1/2 activation in
Beas-2B cells. a Either control- and MUC1-siRNA Beas-2B
cells were exposed to 0.1 mM Ni2? for indicated times and
changes in ERK1/2 protein levels as well as in their
phosphorylated forms was measured by Western blot analyses;
Bands were quantified using ImageQuantTL software and
normalized values were plotted in the histogram shown on the
right; (***p \ 0.001 vs control). b Effect of Ni2? exposure on
apoptosis in Beas-2B cells as determined by oligonucleosomes
detection. After pretreatment with vehicle or 25 lm PD98059
for 30 min, Beas-2B cells were exposed to increasing concen-
trations of Ni2? (0.1, 0.5, 1, 2.5 and 5 mM, respectively) for
24 h and processed for oligonucleosomes detection as described
in ‘Materials and methods’; Values are expressed as mean
ODs ± SEM; (*p \ 0.05 or **p \ 0.01 vs untreated cells).
Each experiment was performed at least three times using three
different batches of cells (n = 3)
Biometals
123

the PI3 K/AKT pathway in non-small cell lung cancer
cells (Raina et al. 2011), further supporting the
involvement of this molecule in cancer development.
In the second part of this study we discovered that
attenuation of MUC1 expression through siRNA
delivery, and hence the reduced the interaction with
EGFR, resulted in a significant decrease of ERK1/2
transactivation (Fig. 4a) when cells were exposed to
Ni2?, and this correlated with enhancement of cell
death, cleaved Caspase-3 expression and DNA dam-
age (Fig. 3a–c). We also found that the formerly
observed effects were partially reversed by treatment
with the MEK1 inhibitor (Fig. 5b).
Previous findings by Schroeder et al. (2001) have
demonstrated the role of MUC1 in mediating ERK1/2
activation using MUC1 transgenic mice. Interestingly,
other studies have shown that overexpression of
MUC1 in mouse breast cancer cells (Horn et al.
2009) and in human lung cancer cells (Yao et al. 2011)
results in ERK activation, although whether MUC1
directly contributes to the activation of ERK1/2
signalling remains to be investigated.
In conclusion, we have demonstrated that Ni2?
exposure induced MUC1 expression to confer resis-
tance to apoptotic cell death through the activation of
the EGFR/ERK1/2 signaling pathway in human
bronchial epithelial cells (Beas-2B). In the light of
these data, we hypothesize that persistent MUC1
expression during chronic exposure to nickel com-
pounds might contribute to increase epithelial cell
resistance, thereby promoting carcinoma development
through inhibition of the physiological apoptotic
machinery.
Acknowledgments The present research was partially
supported by grants from the Department of Bio-Medical
Sciences, Section of Anatomy and Histology, University of
Catania. We would like to thank Mr. P. Asero for his technical
support.
References
Ahamed M, Akhtar MJ, Siddiqui MA, Ahmad J, Musarrat J, Al-
Khedhairy AA, AlSalhi MS, Alrokayan SA (2011) Oxi-
dative stress mediated apoptosis induced by nickel ferrite
nanoparticles in cultured A549 cells. Toxicology 283:
101–108
Baldus SE, Engelmann K, Hanisch FG (2004) MUC1 and the
MUCs: a family of human mucins with impact in cancer
biology. Crit Rev Clin Lab Sci 41:189–231
Baselga J, Arteaga CL (2005) Critical update and emerging
trends in epidermal growth factor receptor targeting in
cancer. J Clin Oncol 23:2445–2459
Cangul H, Broday L, Salnikow K, Sutherland J, Peng W, Zhang
Q, Poltaratsky V, Yee H, Zoroddu MA, Costa M (2002)
Molecular mechanisms of nickel carcinogenesis. Toxicol
Lett 127:69–75
Capasso L, Camatini M, Gualtieri M (2014) Nickel oxide
nanoparticles induce inflammation and genotoxic effect in
lung epithelial cells. Toxicol Lett 226:28–34
Castorina A, Tiralongo A, Cavallo D, Loreto C, Carnazza ML,
Iavicoli S, D’Agata V (2008) Expression profile of ErbB
receptor’s family in human alveolar type 2-like cell line
A549 exposed to hexavalent chromium. Toxicol In Vitro
22:541–547
Castorina A, Giunta S, Scuderi S, D’Agata V (2012) Involve-
ment of PACAP/ADNP signaling in the resistance to cell
death in malignant peripheral nerve sheath tumor
(MPNST) cells. J Mol Neurosci 48:674–683
Chen W, Martindale JL, Holbrook NJ, Liu Y (1998) Tumor
promoter arsenite activates extracellular signal-regulated
kinase through a signaling pathway mediated by epidermal
growth factor receptor and Shc. Mol Cell Biol 18:
5178–5188
Costa M (1991) Molecular mechanisms of nickel carcinogene-
sis. Annu Rev Pharmacol Toxicol 31:321–337
Davidson T, Chen H, Garrick MD, D’Angelo G, Costa M (2005)
Soluble nickel interferes with cellular iron homeostasis.
Mol Cell Biochem 279:157–162
Ding J, Zhang X, Li J, Song L, Ouyang W, Zhang D, Xue C,
Costa M, Melendez JA, Huang C (2006) Nickel com-
pounds render anti-apoptotic effect to human bronchial
epithelial Beas-2B cells by induction of cyclooxygenase-2
through an IKKbeta/p65-dependent and IKKalpha- and
p50-independent pathway. J Biol Chem 281:39022–39032
Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M,
Bugiani O, Tagliavini F (1993) Neurotoxicity of a prion
protein fragment. Nature 362:543–546
Giunta S, Castorina A, Scuderi S, Patti C, D’Agata V (2012)
Epidermal growth factor receptor (EGFR) and neuregulin
(Neu) activation in human airway epithelial cells exposed
to nickel acetate. Toxicol In Vitro 26:280–287
Hattrup CL, Gendler SJ (2008) Structure and function of the
cellsurface (tethered) mucins. Annu Rev Physiol 70:431–457
Horn G, Gaziel A, Wreschner DH, Smorodinsky NI, Ehrlich M
(2009) ERK and PI3 K regulate different aspects of the epi-
thelial to mesenchymal transition of mammary tumor cells
induced by truncated MUC1. Exp Cell Res 315:1490–1504
Kohlgraf KG, Gawron AJ, Higashi M, Meza JL, Burdick MD,
Kitajima S, Kelly DL, Caffrey TC, Hollingsworth MA
(2003) Contribution of the MUC1 tandem repeat and
cytoplasmic tail to invasive and metastatic properties of a
pancreatic cancer cell line. Cancer Res 63:5011–5020
Kundu S, Sengupta S, Bhattacharyya A (2011) EGFR upregu-
lates inflammatory and proliferative responses in human
lung adenocarcinoma cell line (A549), induced by lower
dose of cadmium chloride. Inhal Toxicol 23:339–348
Lan MS, Batra SK, Qi WN, Metzgar RS, Hollingsworth MA
(1990) Cloning and sequencing of a human pancreatic
tumor mucin cDNA. J Biol Chem 265:15294–15299
Biometals
123

Li X, Lee JW, Graves LM, Earp HS (1998) Angiotensin II
stimulates ERK via two pathways in epithelial cells: pro-
tein kinase C suppresses a G protein-coupled receptor-EGF
receptor transactivation pathway. EMBO J 17:2574–2583
Lu H, Shi X, Costa M, Huang C (2005) Carcinogenic effect of
nickel compounds. Mol Cell Biochem 279:45–67
Macao B, Johansson DG, Hansson GC, Hard T (2006) Auto-
proteolysis coupled to protein folding in the SEA domain
of the membrane-bound MUC1 mucin. Nat Struct Mol Biol
13:71–76
Mosesson Y, Yarden Y (2004) Oncogenic growth factor
receptors: implications for signal transduction therapy.
Semin Cancer Biol 14:262–270
Pan JJ, Chang QS, Wang X, Son YO, Liu J, Zhang Z, Bi YY, Shi
X (2011) Activation of Akt/GSK3b and Akt/Bcl-2 sig-
naling pathways in nickel-transformed BEAS-2B cells. Int
J Oncol 39:1285–1294
Pochampalli MR, Bitler BG, Schroeder JA (2007a) Trans-
forming growth factor alpha dependent cancer progression
is modulated by Muc1. Cancer Res 67:6591–6598
Pochampalli MR, el Bejjani RM, Schroeder JA (2007b) MUC1
is a novel regulator of ErbB1 receptor trafficking. Onco-
gene 26:1693–1701
Raina D, Kosugi M, Ahmad R, Panchamoorthy G, Rajabi H,
Alam M, Shimamura T, Shapiro GI, Supko J, Kharbanda S,
Kufe D (2011) Dependence on the MUC1-C oncoprotein in
non-small cell lung cancer cells. Mol Cancer Ther
10:806–816
Ren J, Agata N, Chen D, Li Y, Yu WH, Huang L, Raina D, Chen
W, Kharbanda S, Kufe D (2004) Human MUC1 carci-
noma-associated protein confers resistance to genotoxic
anticancer agents. Cancer Cell 5:163–175
Salnikow K, Costa M (2000) Epigenetic mechanisms of nickel
carcinogenesis. J Environ Pathol Toxicol Oncol 19:307–
318
Salnikow K, Zhitkovich A (2008) Genetic and epigenetic
mechanisms in metal carcinogenesis and cocarcinogenesis:
nickel, arsenic, and chromium. Chem Res Toxicol 21:28–
44
Schroeder JA, Thompson MC, Gardner MM, Gendler SJ (2001)
Transgenic MUC1 interacts with epidermal growth factor
receptor and correlates with mitogen-activated protein
kinase activation in the mouse mammary gland. J Biol
Chem 276:13057–13064
Schroeder JA, Masri AA, Adriance MC, Tessier JC, Kotlarczyk
KL, Thompson MC, Gendler SJ (2004) MUC1 overex-
pression results in mammary gland tumorigenesis and pro-
longed alveolar differentiation. Oncogene 23:5739–5747
Singh PK, Hollingsworth MA (2006) Cell surface-associated
mucins in signal transduction. Trends Cell Biol 16:
467–476
Tsutsumida H, Swanson BJ, Singh PK, Caffrey TC, Kitajima S,
Goto M, Yonezawa S, Hollingsworth MA (2006) RNA
interference suppression of MUC1 reduces the growth rate
and metastatic phenotype of human pancreatic cancer cells.
Clin Cancer Res 12:2976–2987
Wei X, Xu H, Kufe D (2005) Human MUC1 oncoprotein reg-
ulates p53-responsive gene transcription in the genotoxic
stress response. Cancer Cell 7:167–178
Wei X, Xu H, Kufe D (2006) MUC1 oncoprotein stabilizes and
activates estrogen receptor alpha. Mol Cell 21:295–305
Wu W, Graves LM, Jaspers I, Devlin RB, Reed W, Samet JM
(1999) Activation of the EGF receptor signaling pathway
in human airway epithelial cells exposed to metals. Am J
Physiol 277:924–931
Yao M, Zhang W, Zhang Q, Xing L, Xu A, Liu Q, Cui B (2011)
Overexpression of MUC1 enhances proangiogenic activity
of non-small-cell lung cancer cells through activation of
Akt and extracellular signal-regulated kinase pathways.
Lung 189:453–460
Zandi R, Larsen AB, Andersen P, Stockhausen MT, Poulsen HS
(2007) Mechanisms for oncogenic activation of the epi-
dermal growth factor receptor. Cell Signal 19:2013–2023
Zhang J, Zhou Y, Wu YJ, Li MJ, Wang RJ, Huang SQ, Gao RR,
Ma L, Shi HJ, Zhang J (2013) Hyper-methylated miR-203
dysregulates ABL1 and contributes to the nickel-induced
tumorigenesis. Toxicol Lett 223:42–51
Biometals
123




















