
cyber-systemics, systemic governance and disruption - The ...
Upload
independentCategory
view
0download
0
Mouse model of N-acetylgalactosamine-6-sulfatesulfatase deficiency (Galns�/�) produced bytargeted disruption of the gene defective inMorquio A disease
Shunji Tomatsu1,3,*, Koji O. Orii1,4, Carole Vogler2, Jun Nakayama5, Beth Levy2, Jeffrey
H. Grubb1, Monica A. Gutierrez1, Soomin Shim1, Seiji Yamaguchi3, Tatsuo Nishioka1,
Adriana Maria Montano6, Akihiko Noguchi7, Tadao Orii4, Naomi Kondo4 and William S. Sly1
1Edward A. Doisy Department of Biochemistry and Molecular Biology and 2Department of Pathology, Saint Louis
University School of Medicine, St Louis, MO, USA, 3Shimane Medical University, Shimane, Japan, 4Department
of Pediatrics, Gifu University School of Medicine, Gifu, Japan, 5Department of Pathology, Shinshu University School
of Medicine, Matsumoto, Japan, 6Department of Pediatrics, Cardinal Glennon Children’s Hospital, Saint Louis
University, St Louis, MO, USA and 7Department of Biosystems Science, Graduate University for Advanced Studies,
Kanagawa, Japan
Received August 13, 2003; Revised October 10, 2003; Accepted October 21, 2003
Mucopolysaccharidosis IVA is an autosomal recessive disorder caused by a deficiency of N-acetylgalacto-samine-6-sulfate sulfatase (GALNS), a lysosomal enzyme required for the stepwise degradation of keratansulfate (KS) and chondroitin-6-sulfate (C6S). To generate a model for studies of the pathophysiology and ofpotential therapies, we disrupted exon 2 of Galns, the homologous murine gene. Homozygous Galns�/�micehave no detectable GALNS enzyme activity and show increased urinary glycosaminoglycan (GAGs) levels.These mice accumulate GAGs in multiple tissues including liver, kidney, spleen, heart, brain and bonemarrow. At 2 months old, lysosomal storage is present primarily within reticuloendothelial cells such asKupffer cells and cells of the sinusoidal lining of the spleen. Additionally, by 12 months old, vacuolar changeis observed in the visceral epithelial cells of glomeruli and cells at the base of heart valves but it is notpresent in parenchymal cells such as hepatocytes and renal tubular epithelial cells. In the brain, hippocampaland neocortical neurons and meningeal cells had lysosomal storage. KS and C6S were more abundant in thecytoplasm of corneal epithelial cells of Galns�/� mice compared with wild-type mice by immunohistochemi-stry. Radiographs revealed no change in the skeletal bones of mice up to 12 months old. Thus, targeteddisruption of the murine Galns gene has produced a murine model, which shows visceral storage of GAGsbut lacks the skeletal features. The complete absence of GALNS in mutant mice makes them useful forstudies of pharmacokinetics and tissue targeting of recombinant GALNS designed for enzyme replacement.
INTRODUCTION
Mucopolysaccharidosis type IVA (MPS IVA: Morquio type A)is an autosomal recessive disorder caused by the deficiency oflysosomal N-acetylgalactosamine-6-sulfate sulfatase (GALNS:E.C.3.1.6.4). GALNS is one of several sulfatases required todegrade glycosaminoglycans (GAGs), keratan sulfate (KS) and
chondroitin-6-sulfate (C6S). As in other mucopolysacchari-doses, MPS IVA patients show a broad spectrum of clinicalseverity. Phenotypes vary from the classical form with severebone dysplasia (spondyloepiphyseal dysplasia), short trunkdwarfism, hearing loss, heart valve involvement, corneal opacity,and a life span of 20–30 years, to milder forms with fewermanifestations. Patients with mild forms can have a close to
*To whom correspondence should be addressed at: E.A. Doisy Department of Biochemistry and Molecular Biology, Saint Louis University School ofMedicine, 1402 S. Grand Blvd, St Louis, MO 63104, USA. Tel: þ1 3145778131, Fax: þ1 3147761183; Email: [email protected]
Human Molecular Genetics, 2003, Vol. 12, No. 24 3349–3358DOI: 10.1093/hmg/ddg366
by guest on Decem
ber 10, 2013http://hm
g.oxfordjournals.org/D
ownloaded from

normal quality of life with little bone and visceral organinvolvement. Even in severe forms of the disease, there hasbeen no evidence of mental retardation or description oflysosomal storage in brain.
The broad range of clinical phenotypes seen in MPS IVA ispresumed to be the result of many different GALNS mutations.Purification of the enzyme, followed by isolation and character-ization of the full-length cDNA encoding the human GALNSprotein and genomic sequence (1–3) has facilitated investiga-tions into the molecular genetic heterogeneity in MPS IVA.
We have carried out molecular analyses of over 200 MPS IVApatients from 25 ethnic or geographic origins (4–17). To date,over 100 different mutations have been identified. Genotype–phenotype correlation exists for many of these mutations. Twolarge deletions lead to the severe form of MPS IVA and manypoint mutations produce a broad range of phenotypes.
GALNS is encoded by a member of the sulfatase gene familyof which 10 different sulfatase human genes have been cloned.All of these sulfatase genes and their gene products are closelyrelated, showing 20–35% similarity at the amino acid level. Theamino acid residue C79 in exon 2 of human GALNS gene wasidentified as a catalytic site conserved among all knownsulfatases (18).
Although deficiency of GALNS has been described inhumans, it has never been reported in other species. The lack ofan animal model has limited the development and testing ofnovel enzyme replacement and/or gene therapy regimes forMPS IVA. The development of a murine model for MPS IVAcould potentially serve as an important experimental system forthe development of both enzyme replacement therapy (ERT)and gene therapy regimens. Information obtained from treatingthe mouse model could be important for developing treatmentsfor the human disease.
This report describes the targeted disruption of murine Galnsgene, characterization of the resulting deficiency of GALNS,and pathological findings. Furthermore, the initial phenotypicobservations of our model are compared with the other murinemodels of MPS disorders.
RESULTS
Targeting of Galns in ES cells and generation ofGalns knock-out mice
GALNS-deficient mice with a deletion of portions of intron 1 andexon 2 were generated by gene targeting in murine embryonicstem cells as described in Methods and shown schematicallyin Figure 1. This strategy generated mice heterozygous for themutation that no longer contained the neor marker, withthe expected Mendelian segregation at birth (19). Combineddata from crosses between these heterozygous Galnsþ/� miceshowed a normal distribution of 25% (þ/þ), 53% (þ/�), and22% (�/�), in over 100 offspring analyzed, suggesting that thesurvival rate of Galns�/� offspring is not reduced. Total cellularRNA was isolated from tissues of homozygous Galns�/�,heterozygotes and wild-type mice using a guanidinium/phenolsolution. No Galns transcript was observed in Galns�/�mice byNorthern blot (Fig. 2) and RT–PCR before or after removal ofthe neor gene by Cre (data not shown).
Phenotype of the GALNS-deficient mice
Galns�/� mice were normal in appearance and were generallyhealthy. Both males and females were fertile at least up to 12months of age, as indicated by the normal litter sizes producedfrom mating Galns�/� males to Galnsþ/� females or vice versa.No obvious difference in weight or mortality rate was seen inthe first 12 months of life. At 12 months of age, radiographicanalysis of the axial and appendicular skeleton of Galns�/�
mice did not reveal any obvious abnormalities of long bones,thorax, or calvaria. Thus, homozygous mice carrying Galns�/�
have no obvious phenotype and lack the skeletal features ofhuman MPS IVA patients.
Biochemical findings
Enzyme activity. GALNS enzyme activity was determined inhomogenates of liver, kidney, brain, spleen, lung, heart, muscle,bone, bone marrow cells from femur, and serum. In wild-typemice, the activity was highest in the kidney, bone marrow,spleen and liver and lowest in brain, skeletal muscle and heart.Homozygous mutant mice had no activity, confirming that thetargeted allele was null. Heterozygotes had half the normalactivity (Table 1). The gene dosage response was seen in alltissues tested.
Secondary elevation. Several other lysosomal enzymesincluding a-galactosidase and b-galactosidase exhibited slightbut not statistically significant secondary elevations in severaltissues including liver, spleen, brain and serum (Fig. 3).Elevations of b-glucuronidase and b-hexosaminidase activities(data not shown) were even less pronounced, but the trend wasthe same. In contrast, a-mannosidase activity was not elevatedin the GALNS-deficient mice.
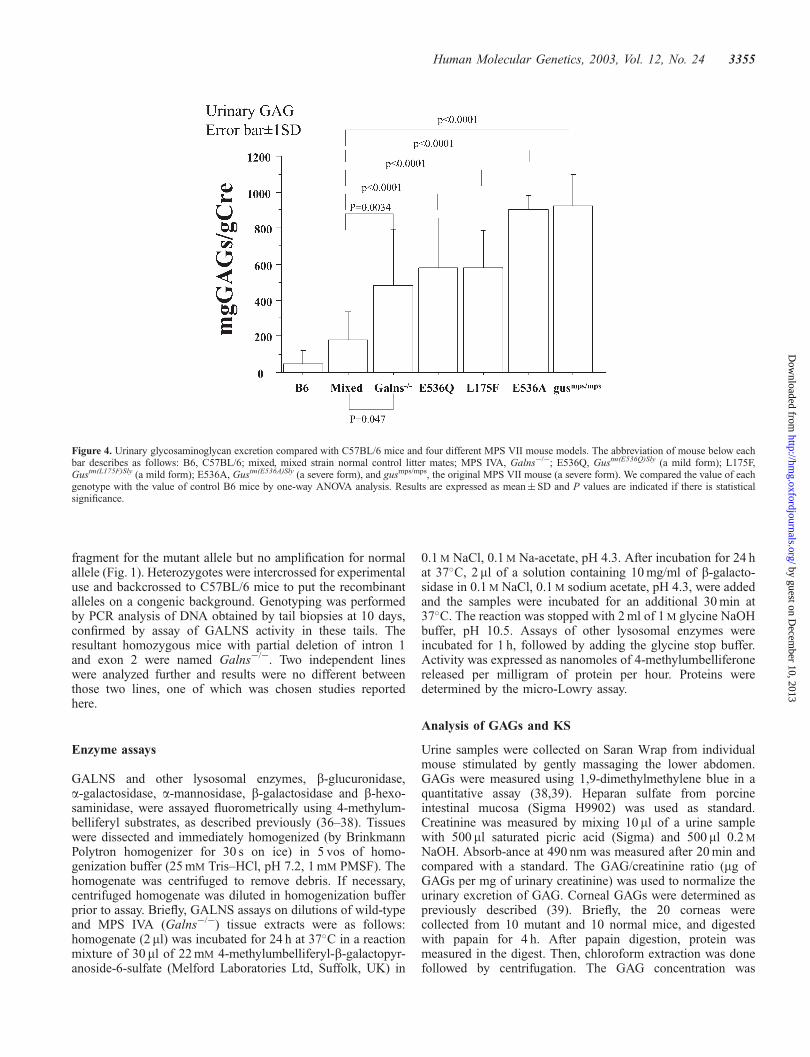
Urinary GAG excretion (Fig. 4). Urine was collected fromGalns�/� (n¼ 10), þ/þ and þ/� littermates (mixed back-ground; n¼ 12), and C57BL/6 mice (n¼ 16). Additionally,urine was collected from four different MPS VII mouse strains;gusmps/mps (n¼ 7), Gustm(E536A)Sly (n¼ 8), Gustm(L175F)Sly (n¼ 7),and Gustm(E536Q)Sly (n¼ 5) (20). Total urinary GAG excretionexpressed as a ratio of milligrams GAGs to grams creatinineexcretion was elevated in Galns�/� mice when compared withB6 mice (P¼ 0.034) and their littermates (P¼ 0.063). Galns�/�
mice at 2–6 months of age excreted 387� 275 mg GAG/g Cre,which was on average 2- and 6-fold greater than their littermates(142� 142 mg GAG/g Cre) and normal control B6 mice(60� 55 mg GAG/g Cre), respectively. Severe murine MPSVII models (gusmps/mps and Gustm(E536A)Sly) excreted 5-foldgreater levels of GAGs than control littermates. The level ofincrease in Galns�/� mice was more comparable to that ofmilder murine MPS VII models (Gustm(L175F)Sly andGustm(E536Q)Sly) (20). Urinary KS was not detectable inGalns�/� mice or normal control mice.
Corneal GAGs and KS. The level of total GAGs in mutantcornea (extracted from 10 mice and pooled) was 2.3-fold higherthan that in littermate corneas (2.94 versus 1.23 mg GAG/g wet
3350 Human Molecular Genetics, 2003, Vol. 12, No. 24
by guest on Decem
ber 10, 2013http://hm
g.oxfordjournals.org/D
ownloaded from

tissue weight) while the corneal KS level per mg protein inpooled extracts of corneal tissue from 10 mutant mice was 1.7fold of littermates (176 versus 109 ng KS/mg protein).
Pathological features
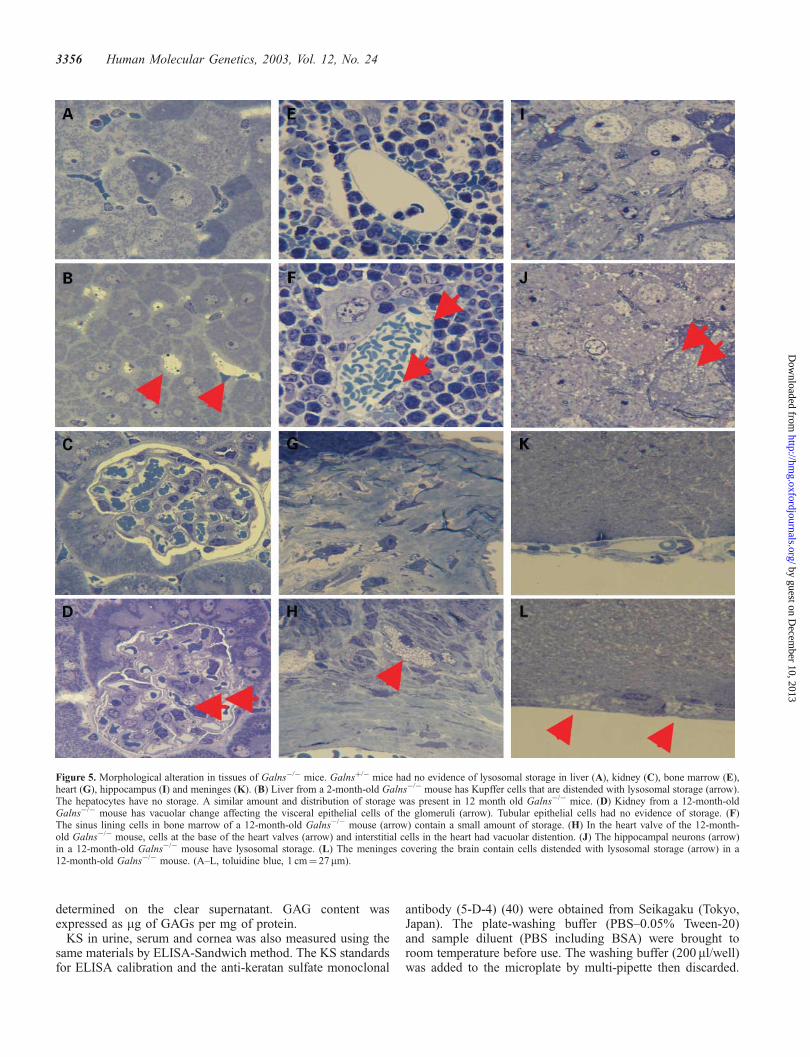
The Galns�/� mice evaluated were 2 months (n¼ 3), 7 months(n¼ 1) and 12 months (n¼ 3) of age. Histological evidence ofstorage was apparent in most of the affected mice (Fig. 5)although there was variability from mouse to mouse that didnot always correlate with the animal’s age. In the liver (Fig. 5B)and the spleen (not shown), storage was restricted to the sinuslining cells. There was no storage in the hepatic parenchymalcells. In the kidney (Fig. 5D), particularly in the 12-month-oldmice, there was a vacuolar change affecting the visceralepithelial cells of the glomeruli. Tubular epithelial cells had noevidence of storage. Cells at the base of the heart valves(Fig. 5H) and interstitial cells in the heart had vacuolardistention. Three of seven mice had slight distention of thesinus lining cells in the bone marrow (Fig. 5F). There was noapparent storage in the chondrocytes or in the osteoblasts. Inthe brain, hippocampal neurons (Fig. 5J) and neocortical glialcells from five Galns�/� mice had storage. In four, neocortical
neurons also had minimal evidence of lysosomal storage. Therewas a small amount of storage in meningeal cells (Fig. 5L)in six Galns�/� mice. The cornea from three mice had veryrare stromal fibroblasts containing cytoplasmic vacuoles (datanot shown). The long bones from the Galns�/� showed nomorphological alteration when compared as a group with thebones from control mice.
In order to examine the distribution patterns of KS and C6Sin the corneas of wild-type and Galns�/� mice, immunohisto-chemistry was performed using specific antibodies against KSand C6S (Fig. 6). In the wild-type mice, KS was expressed inthe corneal epithelium, stroma and endothelium as shownpreviously (21) (Fig. 6A). In Galns�/� mice, by contrast, KSwas more abundant in the perinuclear cytoplasm of cornealepithelial cells (Fig. 6D). Although the amount of KS stainingappeared slightly increased in the stroma of Galns�/� mice, noincrease in KS was apparent in corneal endothelial cells(Fig. 6D). C6S was less abundant than KS in the cornea ofthe wild-type mice (Fig. 6B). However, in the Galns�/� mice,C6S was also increased in the cytoplasm of corneal epithelialcells (Fig. 6E). These results suggest that both KS and C6Sare increased in the cytoplasm, possibly in lysosomes in thecorneal epithelium of Galns�/� mice. Immunofluorescencestudies were also carried out on sections of costal cartilage andthe growth plate in the femur. KS and C6S were detected insome chondrocytes of costal cartilage and growth plate in thefemur, but no significant differences were seen between thewild-type mice and Galns�/� mice (data not shown).
DISCUSSION
We describe here the generation of a GALNS-deficient mouseproduced by targeted disruption of murine Galns gene usinghomologous recombination in embryonic stem cells. Disruptionof Galns gene was complete, as shown by the lack ofexpression of Galns mRNA and the absence of detectableGALNS enzyme activity in Galns�/� mice. Clinical and patho-logical characterization of Galns�/� mice revealed evidenceof lysosomal storage in visceral organs, brain and the corneaand increased excretion of urinary GAGs as is found inMPS IVA in humans. It will ultimately be important for theGalns�/� mutation to be bred onto a uniform genetic back-ground and possibly even evaluate clinical phenotypes ondifferent mouse strain backgrounds since strain specificmodifier genes could influence the phenotype and contributeto mouse-to-mouse variability in mice on a mixed strainbackground.
Between 2 and 12 months of age in MPS IVA mice, weobserved storage with involvement of reticuloendothelial cellsin spleen and liver, epithelial cells in kidney, cells at the base ofthe heart valves, neurons and meningeal cells. Despite thehistological and histochemical evidence of storage, the micehad a normal phenotype, appeared healthy and were fertile upto an age of 12 months. They had neither clinical nor radiologicevidence of the skeletal abnormalities characteristic of severehuman MPS IVA patients.
It is not uncommon for mouse models of human lysosomalstorage diseases to have a milder phenotype than their humancounterpart. Examples include murine GM1 gangliosidosis
Table 1. Tissue levels of GALNS activity (units/mg)
Tissue Animal (n¼ 8) GALNS (� 1SD)(units/mg)
Kidney Control 22.4� 3.61Galnsþ/� 13.8� 9.46Galns�/� 0.011� 0.013
Spleen Control 11.6� 5.07Galnsþ/� 5.42� 1.87Galns�/� 0.17� 0.23
Liver Control 7.86� 2.39Galnsþ/� 3.73� 0.91Galns�/� 0.21� 0.49
Lung Control 2.28� 0.93Galnsþ/� 1.24� 0.27Galns�/� 0.037� 0.052
Brain Control 1.73� 0.37Galnsþ/� 0.96� 0.29Galns�/� 0.089� 0.069
Heart Control 0.51� 0.22Galnsþ/� 0.33� 0.19Galns�/� 0.009� 0.015
Muscle Control 0.64� 0.27Galnsþ/� 0.36� 0.32Galns�/� 0.024� 0.027
Tail Control 19.4� 4.7Galnsþ/� 7.89� 3.42Galns�/� 0.01� 0.019
Leg Control 0.64� 0.27Galnsþ/� 0.36� 0.32Galns�/� 0.025� 0.029
Rib Control 1.73� 0.37Galnsþ/� 0.96� 0.29Galns�/� 0.025� 0.025
Bone marrow Control 0.51� 0.22Galnsþ/� 0.33� 0.19Galns�/� 0.021� 0.031
Serum (units/ml) Control 11.7� 1.31Galnsþ/� 7.03� 2.25Galns�/� 0� 0
Human Molecular Genetics, 2003, Vol. 12, No. 24 3351
by guest on Decem
ber 10, 2013http://hm
g.oxfordjournals.org/D
ownloaded from

(22), a-mannosidosis (23), Tay–Sachs disease (24), Sanfilipposyndrome type B (25). In murine Tay–Sachs disease a seconddegradative pathway has been found to be the basis of the verymild phenotype (24). Although some ancillary pathway ofdegradation may exist in the Galns�/� mouse, a more likelyexplanation for the milder phenotype than that found inMorquio A patients lies in differences in the amount ofsynthesis and distribution of KS between murine and humantissue. Specifically, two different types of KS are found in thecornea (KS I) and skeleton (KS II) and the synthesis of KS II inthe mouse and rat is much reduced compared with other animal
species in most tissues except the cornea (26). The cartilagecells in rodents do not have KS chains in their aggrecan, whichis a KS proteoglycan in other mammals (26). We report herethat KS in urine and serum was actually undetectable innormal and MPS IVA mice, while KS in other species likehuman, dog and cat was easily detectable (data not shown).Histopathological examinations in MPS IVA mice also failedto show progressive GAG storage in cartilage cells and thebones. Taken together, these findings suggest that KS II and KSturnover is less important in skeletal tissues of mouse possiblybecause KS can be replaced by other GAGs such as chondroitin
Figure 1. Targeted disruption of the Galns gene. (A) The structure of the endogenous wild-type gene, the targeting construct, the disrupted allele, and the neor-excised allele are presented schematically on successive lines. The shadow bars above lines represent the 50 probe used for Southern blots. The large arrows showthe loxP sequences. Abbreviations for restriction enzymes are: RI, EcoRI; S, SacI; X, XbaI; B, BamHI; H, HindIII; K, KpnI. (B) Southern blot identification ofthe disrupted gene is shown (bottom): the wild-type and targeted mutant alleles were identified by hybridization of the 50 probe to KpnI restriction fragments.The hybridized fragment is smaller in the disrupted allele than in the normal allele (7.1 versus 12.0 kb) because of the presence of the additional KpnI site inthe multicloning site of targeting vector. Lanes 1 and 6, wild-type mouse (normal alleles); lane 2, targeted ES cell line (one wild-type, one mutant allele); lanes3–5, Galnsþ/� mice produced from initial chimeras (both bands present). (C) Detection of the knockout allele at the murine Galns gene by genomic PCR ampli-fication. Two sets of primers produce a single 1.2 kb fragment for homozygotes, two fragments of 1.2 and 1.8 kb for heterozygotes, and a single 1.8 kb fragment forwild-type mice. Lane 1, Galns�/� (1.2 kb); lane 2, Galnsþ/� (1.2 and 1.8 kb); lane 3, a normal control, Galnsþ/þ (1.8 kb).
3352 Human Molecular Genetics, 2003, Vol. 12, No. 24
by guest on Decem
ber 10, 2013http://hm
g.oxfordjournals.org/D
ownloaded from

sulfate. In this view, the absence of KS II in murine aggrecanexplains why MPS IVA mice have no skeletal phenotype.On the other hand, the expression of KS I in the cornea mayaccount for the modest amount of storage detected in thistissue.
Storage material was found mainly in cells of the macrophagelineage in liver, heart, bone marrow and spleen, suggesting thatthese cells may act as a clearing house for circulating KS and/orC6S. These KS and/or C6S fragments would be taken upthrough the macrophage mannose receptor, which is known toalso recognize terminal fucose and N-acetylglucosamine (27).Specifically, vacuoles were seen in sinusoidal cells but few ifany were seen in hepatocytes in MPS IVA patients. Thispredominant deposition of GAG storage in reticuloendothelialcells has also been noted in human MPS IVA patients (personalcommunication, Dr T. Orii, Gifu University, Japan).
In humans, KS and C6S are abundant in bones and cornea butless prominent in visceral organs, brain, vessels and muscles.The dysfunction in the heart valves in MPS IVA patients canlead to death. The deposition of storage materials at the baseof heart valves of MPS IVA mice is thus of interest. MPSIVA patients retain normal intelligence throughout their lives.There has been no description of storage material in brain.In fact, to our knowledge, no studies of brain in MPS IVApatients have been reported. The novel finding that MPS IVAmice have vacuoles in parts of the brain needs further investi-gation regarding its relevance to human MPS IVA disease.
The deficiency of one enzyme involved in degradation ofGAG is often accompanied by increases in other lysosomalenzymes. This had been reported in the MPS I (28), II, IIIA(29), IIIB (25), VI (30) and VII murine models (20,31) butthe elevations are very modest in most tissues in the MPSVIA mouse. Such increases may reflect an increased numberof lysosomes, increased biosynthesis, and/or stabilization ofthe enzymes by storage material. The reason the secondaryelevation of other lysosomal enzymes is less prominent in MPSIVA mice compared with other MPS murine models could bedue to the lower amount of GAG storage in this disorder orthe milder secondary effect of the KS/C6S stored. Declinesin the level of secondary elevations of lysosomal enzyme
activities have been proposed as indicators of effectivenessof treatments such as ERT and gene therapy (20). The verymodest secondary elevations of other lysosomal enzymes inthis model will make this a less useful marker of therapeuticresponse.
In other murine models, the level of urinary GAG excretioncorrelates with severity of visceral and bone involvement. Forexample, the level of GAG was in proportion with the clinicalseverity in the murine MPS VII model [this study, Fig. 6, and(20,31)]. MPS I mice have a mild phenotype and have aboutthe same level of GAG excretion in urine (3-fold that of controlmice) as mild MPS VII mice (28). MPS IVA mice showed onlya 2-fold increase in total GAG excretion compared with normalcontrol mice, which is consistent with their mild phenotype andapparent absence of bone involvement.
Potential therapies for lysosomal storage disorders includebone marrow transplantation, somatic gene therapy and ERT.These therapeutic approaches depend in part at least on theexpectation that lysosomal enzymes can be secreted and takenup through a mannose-6-phosphate receptor mediated mecha-nism (32,33). The reversibility of some of the features ofhuman MPS IVA was suggested by the results of bonemarrow transplantation (unpublished data, Dr T. Orii, GifuUniversity, Japan). Factors that influence the responsiveness tothese forms of therapy relate to the age of the patient and thedegree of progression prior to the commencement of therapyor to efficiency of enzyme delivery to the skeleton. Until now,there has been no animal model for MPS IVA reported whichwould permit evaluation of experimental therapies for thisdisease. The GALNS deficient mouse described here, eventhough it lacks the skeletal phenotype of the human disease,should prove useful for evaluation of pharmacokinetics andtargeting of various forms of GALNS in experimental ERTprotocols.
MATERIALS AND METHODS
Construction of the Galns gene targeting vector
The Galns gene was cloned from a 129/Sv mouse genomiclibrary (Stratagene), as previously described (34). The cysteineresidue found in the catalytic domain of all sulfatases is criticalfor the enzyme activity (18), and thus exon 2 encoding theCys79 residue of human GALNS was selected for replacementin the gene disruption construct. After digestion with therestriction endonuclease HindIII, the 1.8 kb fragment contain-ing part of intron 1 and exon 2 was replaced by a cassettecontaining the neor gene under control of the mouse phos-phoglycerate kinase promoter (20). A cassette containing theHerpes simplex virus thymidine kinase gene under control ofthe mouse phosphoglycerate kinase promoter was cloned intothe vector upstream of the Galns gene (Fig. 1). The loxPsequences are positioned at both ends of the neor gene. Thecontinued presence of neomycin-resistant gene (neor) in theconstruct may affect the clinical phenotype in the mouse model(35). This lox–neo–lox cassette can be eliminated by matingthe heterozygotes with transgenic mice expressing the Crerecombinase enzyme. After neor is removed, this deletion
Figure 2. Expression of murine Galns mRNA. (A) Northern blot analysis ofmurine Galns mRNA from the livers (A), spleens (B), kidney (C), and lungs(D) of wild type (þ/þ) and homozygote (�/�) mice. Twenty micrograms oftotal RNA from each tissue were analyzed by northern blotting with murineGalns cDNA 32P-labeled probe. The 2.3 kb murine Galns mRNA transcriptwas expressed in wild-type tissues (lanes 1 and 2) and was not detected inGalnsþ/þ tissues (lane 3).
Human Molecular Genetics, 2003, Vol. 12, No. 24 3353
by guest on Decem
ber 10, 2013http://hm
g.oxfordjournals.org/D
ownloaded from

eliminates exon 2, causing a frame shift. The final constructcontains 4.2 and 4.5 kb of 50 and 30 homology, respectively.
Homologous recombination in ES cells andgeneration of germline chimeras
The vector (25 mg) was linearized with NotI restriction endo-nuclease before electroporation into embryonic stem (ES) cells(Incyte Genome Systems, St Louis, MO, USA), which arederived from mouse strain 129SvJ. The ES cells, 107, wereelectroporated in a 1 ml cuvette at 240 V and 500 mF by aBio-Rad gene pulser. After 24 h, the cells were placed underselection with 200 mg/ml G418 (GIBCO/BRL, Gaithersburgh,MD, USA) and 2 mM gangciclovir (Syntex, Palo Alto, CA,USA) for 6 days. ES cell colonies resistant to double selectionwere isolated and subjected to PCR, followed by Southern blotanalysis. Surviving colonies were subjected to PCR analyses toidentify clones that had undergone homologous recombination.The PCR method utilizes a forward primer in the loxP–neor–loxP cassette sequence [LoxL(2)R: 50-CTTCGTATAGCATACATTACGAAGTTATGCGA-30]. and a reverse primer in intron4 outside the targeting sequences [MOex5-1R: 50-ACAGGGATGTTGGGCTTGGCCTTGTTGTCATACGG-30]. PCR amplifi-cation from the disrupted allele produces a 4.5 kb fragment.Positive clones identified by PCR were then confirmed bySouthern analysis. Genomic DNAs of the positive clones byPCR screening were digested with KpnI, transferred to nylonmembranes and hybridized with the 50 probe (a 1.3 kb SacI–BamHI genomic fragment) generating 7.1 kb fragment in the
recombinants instead of 12.0 kb fragment in the normal alleleshown in Figure 1. About 10% of the survivors of the drugselection were found to have a disrupted Galns gene.
Two independent targeted ES clones were injected intoC57BL/6J blastocysts and three chimeric mice were generated.Chimeric males were back-crossed for germline transmission toC57BL/6J females. Germline transmission was achieved fromall three chimeric mice. The resultant F1 mice transmitted tothe germline from all three chimeric mice were crossed withmice expressing Cre enzyme to remove the neor gene, and theresultant neo-excised heterozygous mice were mated to producehomozygous mutant mice. This excision leaves only 34 bp ofone loxP site in intron 1. The removal of neor was diagnosed byPCR of tail DNA using a forward primer in the loxP–neor–loxPcassette sequence (VDE: 50-GGATCCTCTAGAAATGCCATTTCATTACCTCTTT-30) and a reverse primer in intron 3(mGMO4R: 50-GGCTTAGGAAAGCATCTTTCCTAGGCTAGA-30), which amplified a 1.3 kb fragment while non neor-excised allele did not reveal a detectable fragment under theseconditions. After excision of neor, we performed multiple PCRusing two sets of primers to distinguish Galns�/�, Galnsþ/�
and wild-type (Galnsþ/þ) mice. A combination of a forwardprimer in the deleted portion of intron 1 (mGMO1’: 50-GCTCTTCCTTGTCACAGAGGAACC-30) and a reverse pri-mer in exon 4 (MOex4-1R: 50-CTTGTTGGTATAGCCTTCTTCAGGAGCTC-30) shows 1.8 kb fragment for normal allelebut no amplification for mutant allele. PCR using another set ofprimers, mGALNS2 (50-CCCCTGGAGTGTAGTCACAGACAGTTGCAAGC-30) and VDE 1 vector sequence (50-GGAGAAAGAGGTAATGAAATGGCATTTCTAGA-30), reveals a 1.2 kb
Figure 3. Secondary elevation of a-galactosidase and b-galactosidase activities in mutant mice compared with heterozygotes and control littermates. Enzyme activ-ities were assayed in tissues of homozygous mutant, heterozygous and wild-type mice. Enzyme activities were expressed as nmol/h/mg (units/mg) protein fromtissue homogenates prepared from Galnsþ/þ, Galnsþ/� and Galns�/� control mice. Six mice were analyzed for each group. Activities are expressed as the meanwith observed range. The average b-galactosidase levels of normal control in serum, kidney, spleen, liver, lung and brain are 94 units/ml, 339, 614, 117, 72 and 86units/mg protein, respectively. The average a-galactosidase levels of normal control in serum, kidney, spleen, liver, lung and brain are 13.8 units/ml, 24, 71, 37, 14and 20 units/mg protein, respectively.
3354 Human Molecular Genetics, 2003, Vol. 12, No. 24
by guest on Decem
ber 10, 2013http://hm
g.oxfordjournals.org/D
ownloaded from
fragment for the mutant allele but no amplification for normalallele (Fig. 1). Heterozygotes were intercrossed for experimentaluse and backcrossed to C57BL/6 mice to put the recombinantalleles on a congenic background. Genotyping was performedby PCR analysis of DNA obtained by tail biopsies at 10 days,confirmed by assay of GALNS activity in these tails. Theresultant homozygous mice with partial deletion of intron 1and exon 2 were named Galns�/�. Two independent lineswere analyzed further and results were no different betweenthose two lines, one of which was chosen studies reportedhere.
Enzyme assays
GALNS and other lysosomal enzymes, b-glucuronidase,a-galactosidase, a-mannosidase, b-galactosidase and b-hexo-saminidase, were assayed fluorometrically using 4-methylum-belliferyl substrates, as described previously (36–38). Tissueswere dissected and immediately homogenized (by BrinkmannPolytron homogenizer for 30 s on ice) in 5 vos of homo-genization buffer (25 mM Tris–HCl, pH 7.2, 1 mM PMSF). Thehomogenate was centrifuged to remove debris. If necessary,centrifuged homogenate was diluted in homogenization bufferprior to assay. Briefly, GALNS assays on dilutions of wild-typeand MPS IVA (Galns�/�) tissue extracts were as follows:homogenate (2 ml) was incubated for 24 h at 37�C in a reactionmixture of 30 ml of 22 mM 4-methylumbelliferyl-b-galactopyr-anoside-6-sulfate (Melford Laboratories Ltd, Suffolk, UK) in
0.1 M NaCl, 0.1 M Na-acetate, pH 4.3. After incubation for 24 hat 37�C, 2 ml of a solution containing 10 mg/ml of b-galacto-sidase in 0.1 M NaCl, 0.1 M sodium acetate, pH 4.3, were addedand the samples were incubated for an additional 30 min at37�C. The reaction was stopped with 2 ml of 1 M glycine NaOHbuffer, pH 10.5. Assays of other lysosomal enzymes wereincubated for 1 h, followed by adding the glycine stop buffer.Activity was expressed as nanomoles of 4-methylumbelliferonereleased per milligram of protein per hour. Proteins weredetermined by the micro-Lowry assay.
Analysis of GAGs and KS
Urine samples were collected on Saran Wrap from individualmouse stimulated by gently massaging the lower abdomen.GAGs were measured using 1,9-dimethylmethylene blue in aquantitative assay (38,39). Heparan sulfate from porcineintestinal mucosa (Sigma H9902) was used as standard.Creatinine was measured by mixing 10 ml of a urine samplewith 500 ml saturated picric acid (Sigma) and 500 ml 0.2 M
NaOH. Absorb-ance at 490 nm was measured after 20 min andcompared with a standard. The GAG/creatinine ratio (mg ofGAGs per mg of urinary creatinine) was used to normalize theurinary excretion of GAG. Corneal GAGs were determined aspreviously described (39). Briefly, the 20 corneas werecollected from 10 mutant and 10 normal mice, and digestedwith papain for 4 h. After papain digestion, protein wasmeasured in the digest. Then, chloroform extraction was donefollowed by centrifugation. The GAG concentration was
Figure 4. Urinary glycosaminoglycan excretion compared with C57BL/6 mice and four different MPS VII mouse models. The abbreviation of mouse below eachbar describes as follows: B6, C57BL/6; mixed, mixed strain normal control litter mates; MPS IVA, Galns�/�; E536Q, Gustm(E536Q)Sly (a mild form); L175F,Gustm(L175F)Sly (a mild form); E536A, Gustm(E536A)Sly (a severe form), and gusmps/mps, the original MPS VII mouse (a severe form). We compared the value of eachgenotype with the value of control B6 mice by one-way ANOVA analysis. Results are expressed as mean� SD and P values are indicated if there is statisticalsignificance.
Human Molecular Genetics, 2003, Vol. 12, No. 24 3355
by guest on Decem
ber 10, 2013http://hm
g.oxfordjournals.org/D
ownloaded from
determined on the clear supernatant. GAG content wasexpressed as mg of GAGs per mg of protein.
KS in urine, serum and cornea was also measured using thesame materials by ELISA-Sandwich method. The KS standardsfor ELISA calibration and the anti-keratan sulfate monoclonal
antibody (5-D-4) (40) were obtained from Seikagaku (Tokyo,Japan). The plate-washing buffer (PBS–0.05% Tween-20)and sample diluent (PBS including BSA) were brought toroom temperature before use. The washing buffer (200 ml/well)was added to the microplate by multi-pipette then discarded.
Figure 5. Morphological alteration in tissues of Galns�/� mice. Galnsþ/� mice had no evidence of lysosomal storage in liver (A), kidney (C), bone marrow (E),heart (G), hippocampus (I) and meninges (K). (B) Liver from a 2-month-old Galns�/� mouse has Kupffer cells that are distended with lysosomal storage (arrow).The hepatocytes have no storage. A similar amount and distribution of storage was present in 12 month old Galns�/� mice. (D) Kidney from a 12-month-oldGalns�/� mouse has vacuolar change affecting the visceral epithelial cells of the glomeruli (arrow). Tubular epithelial cells had no evidence of storage. (F)The sinus lining cells in bone marrow of a 12-month-old Galns�/� mouse (arrow) contain a small amount of storage. (H) In the heart valve of the 12-month-old Galns�/� mouse, cells at the base of the heart valves (arrow) and interstitial cells in the heart had vacuolar distention. (J) The hippocampal neurons (arrow)in a 12-month-old Galns�/� mouse have lysosomal storage. (L) The meninges covering the brain contain cells distended with lysosomal storage (arrow) in a12-month-old Galns�/� mouse. (A–L, toluidine blue, 1 cm¼ 27mm).
3356 Human Molecular Genetics, 2003, Vol. 12, No. 24
by guest on Decem
ber 10, 2013http://hm
g.oxfordjournals.org/D
ownloaded from

This washing procedure was repeated three times. Samples(50 ml/well) of KS standards and diluted unknown sampleswere added to the well and incubated at 37�C for 60 min. Theplate wells were then washed four times as described above.Next, 25 ml/well of horseradish peroxidase conjugated strep-tavidin and 25 ml/well of biotinylated antibody were added tothe plate and the plate again incubated for 60 min at 37�C.After washing the plate four times, 50 ml/well of substratesolution (tetramethylbenzidine) was is added to each well andthe plate incubated for 10 min at room temperature. Finally, thereaction is stopped with 50 ml of stop solution (1 N HCl). Theabsorbance was measured at 450 nm with microplate spectro-photometer reference to 630–650 nm. The KS concentrationwas read by applying the absorbances of each sample to thecalibration curve.
Pathological examinations
Liver, spleen, kidney, heart, rib, eye and brain were examinedfrom seven Galns�/� mice from 2 to 12 months of age.Tissues fixed in 4% paraformaldehyde/2% glutaraldehyde,and embedded in Spurr’s resin were sectioned, stained withToluidine blue, evaluated by light microscopy, and comparedwith age matched control mice. The long bones were fixedin formalin, embedded in paraffin, sectioned, stained withhematoxylin and eosin and examined by light microscopy.Skeletons from 7- and 12-month-old Galns�/� mice wereradiographed and compared to those of normal mice, aspreviously described (41).
Immunohistochemical detection of KS and C6S in the corneaof 2-month-old Galns�/� mice as well as the age-matched wild-type mice was carried out using monoclonal antibodies, 5D4and MC21C specific for KS and C6S, respectively, as describedpreviously (21). These primary antibodies were purchased fromSeikagaku (Tokyo, Japan), and horseradish peroxidase-labeledanti-mouse IgG was used for the secondary antibody. Theperoxidase reaction was developed with a diaminobenzidine/H2O2 solution, and counter-staining was performed with
hematoxylin. A control experiment was done by omitting theprimary antibody from the staining procedure and no specificstaining was found.
ACKNOWLEDGEMENTS
This work was supported by grants from the Austrian MPS, theGerman MPS, the International Morquio Organization and theGenzyme Corporation.
REFERENCES
1. Masue, M., Sukegawa, K., Orii, T. and Hashimoto, T. (1991)N-acetylgalactosamine-6-sulfate sulfatase in human placenta: purificationand characteristics. J. Biochem. (Tokyo), 110, 965–970.
2. Tomatsu, S., Fukuda, S., Masue, M., Sukegawa, K., Fukao, T.,Yamagishi, A., Hori, T., Iwata, H., Ogawa, T., Nakashima, Y. et al. (1991)Morquio disease: isolation, characterization and expression of full-lengthcDNA for human N-acetylgalactosamine-6-sulfate sulfatase. Biochem.Biophys. Res. Commun., 181, 677–683.
3. Nakashima, Y., Tomatsu, S., Hori, T., Fukuda, S., Sukegawa, K., Kondo, N.,Suzuki, Y., Shimozawa, N. and Orii, T. (1994) Mucopolysaccharidosis IVA: molecular cloning of the human N-acetylgalactosamine-6-sulfatasegene (GALNS) and analysis of the 50-flanking region. Genomics, 20,99–104.
4. Fukuda, S., Tomatsu, S., Masue, M., Sukegawa, K., Iwata, H., Ogawa, T.,Nakashima, Y., Hori, T., Yamagishi, A., Hanyu, Y. et al. (1992)Mucopolysaccharidosis type IVA. N-acetylgalactosamine-6-sulfate sulfa-tase exonic point mutations in classical Morquio and mild cases. J. Clin.Invest., 90, 1049–1053.
5. Ogawa, T., Tomatsu, S., Fukuda, S., Yamagishi, A., Rezvi, G.M.,Sukegawa, K., Kondo, N., Suzuki, Y., Shimozawa, N. and Ori, T. (1995)Mucopolysaccharidosis IVA: screening and identification of mutationsof the N-acetylgalactosamine-6-sulfate sulfatase gene. Hum. Mol. Genet., 4,341–349.
6. Hori, T., Tomatsu, S., Nakashima, Y., Uchiyama, A., Fukuda, S.,Sukegawa, K., Shimozawa, N., Suzuki, Y., Kondo, N., Horiuchi, T. et al.(1995) Mucopolysaccharidosis type IVA: common double deletion in theN-acetylgalactosamine-6-sulfatase gene (GALNS). Genomics, 26,535–542.
Figure 6. Detection of KS and C6S in the cornea of 2-month-old wild-type and age-matched Galns�/� mice. Distribution patterns of KS and C6S in the cornea ofwild-type and Galns�/� mice were analyzed by immunohistochemistry. Immunostaining for both KS and C6S is more prominent in the cytoplasm of cornealepithelial cells of Galns�/� mice (D and E) than those of wild-type mice (A and B). C and F indicate control experiments using second antibody alone forwild-type and Galns�/� mice, respectively. The clefts in the stroma are artifacts from tissue processing. Immunostaining was performed using 5D4 antibody(A and D) and MC21C antibody (B and E). The corneal epithelium, stroma, and endothelium are indicated as (Ep), (St) and (En) in the figure. Bar¼ 100mm.The corneal stroma fibroblasts in the mutant mice contained a small amount of vacuolar storage. Neither corneal epithelial nor endothelial cells had any lysosomaldistention apparent by light microscopy.
Human Molecular Genetics, 2003, Vol. 12, No. 24 3357
by guest on Decem
ber 10, 2013http://hm
g.oxfordjournals.org/D
ownloaded from

7. Tomatsu, S., Fukuda, S., Cooper, A., Wraith, J.E., Rezvi, G.M.,Yamagishi, A., Yamada, N., Kato, Z., Isogai, K., Sukegawa, K. et al. (1995)Mucopolysaccharidosis IVA: identification of a common missense mutationI113F in the N-acetylgalactosamine-6-sulfate sulfatase gene. Am. J. Hum.Genet., 57, 556–563.
8. Tomatsu, S., Fukuda, S., Cooper, A., Wraith, J.E., Rezvi, G.M.,Yamagishi, A., Yamada, N., Kato, Z., Isogai, K., Sukegawa, K. et al. (1995)Mucopolysaccharidosis type IVA: identification of six novel mutationsamong non-Japanese patients. Hum. Mol. Genet., 4, 741–743.
9. Tomatsu, S., Fukuda, S., Cooper, A., Wraith, J.E., Yamada, N.,Isogai, K., Kato, Z., Sukegawa, K., Kondo, N., Suzuki, Y. et al. (1995)Two new mutations, Q473X and N487S, in a Caucasian patient withmucopolysaccharidosis IVA (Morquio disease). Hum. Mutat., 6,195–196.
10. Fukuda, S., Tomatsu, S., Masuno, M., Ogawa, T., Yamagishi, A.,Rezvi, G.M., Sukegawa, K., Shimozawa, N., Suzuki, Y., Kondo, N. et al.(1996) Mucopolysaccharidosis IVA: submicroscopic deletion of 16q24.3and a novel R386C mutation of N-acetylgalactosamine-6-sulfate sulfatasegene in a classical Morquio disease. Hum. Mutat., 7, 123–134.
11. Fukuda, S., Tomatsu, S., Cooper, A., Wraith, J.E., Kato, Z., Yamada, N.,Isogai, K., Sukegawa, K., Kondo, N. and Orii, T. (1996)Mucopolysaccharidosis IVA (Morquio A): three novel small deletions inthe N-acetylgalactosamine-6-sulfate sulfatase gene. Hum. Mutat., 8,187–190.
12. Tomatsu, S., Fukuda, S., Yamagishi, A., Cooper, A., Wraith, J.F., Hori, T.,Kato, Z., Yamada, N., Isogai, K., Sukegawa, K. et al. (1996)Mucopolysaccharidosis IVA: four new exonic mutations in patients withN-acetylgalactosamine-6-sulfate sulfatase deficiency. Am. J. Hum. Genet.,58, 950–962.
13. Tomatsu, S., Fukuda, S., Cooper, A., Wraith, J.E., Ferreira, P., Di Natale, P.,Tortora, P., Fujimoto, A., Kato, Z., Yamada, N. et al. (1997) Fourteen novelmucopolysaccharidosis IVA producing mutations in GALNS gene. Hum.Mutat., 10, 368–375.
14. Tomatsu, S., Fukuda, S., Cooper, A., Wraith, J.E., Yamagishi, A., Kato, Z.,Yamada, N., Isogai, K., Sukegawa, K., Suzuki, Y. et al. (1998) Fifteenpolymorphisms in the N-acetylgalactosamine-6-sulfate sulfatase (GALNS)gene: diagnostic implications in Morquio disease. Hum. Mutat., Suppl. 1,S42–S46.
15. Kato, Z., Fukuda, S., Tomatsu, S., Vega, H., Yasunaga, T., Yamagishi, A.,Yamada, N., Valencia, A., Barrera, L.A., Sukegawa, K. et al. (1997)A novel common missense mutation G301C in the N-acetylgalactosamine-6-sulfate sulfatase gene in mucopolysaccharidosis IVA. Hum. Genet., 101,97–101.
16. Bunge, S., Kleijer, W.J., Tylki-Szymanska, A., Steglich, C., Beck, M.,Tomatsu, S., Fukuda, S., Poorthuis, B.J., Czartoryska, B., Orii, T. et al.(1997) Identification of 31 novel mutations in the N-acetylgalactosamine-6-sulfatase gene reveals excessive allelic heterogeneity among patients withMorquio A syndrome. Hum. Mutat., 10, 223–232.
17. Yamada, N., Fukuda, S., Tomatsu, S., Muller, V., Hopwood, J.J., Nelson, J.,Kato, Z., Yamagishi, A., Sukegawa, K., Kondo, N. et al. (1998) Molecularheterogeneity in mucopolysaccharidosis IVA in Australia and NorthernIreland: nine novel mutations including T312S, a common allele thatconfers a mild phenotype. Hum. Mutat., 11, 202–208.
18. Schmidt, B., Selmer, T., Ingendoh, A. and von Figura, K. (1995) A novelamino acid modification in sulfatases that is defective in multiplesulfatase deficiency. Cell, 82, 271–278.
19. Schwenk, F., Baron, U. and Rajewsky, K. (1995) A cre-transgenic mousestrain for the ubiquitous deletion of loxP-flanked gene segmentsincluding deletion in germ cells. Nucl. Acids Res., 23, 5080–5081.
20. Tomatsu, S., Orii, K.O., Vogler, C., Grubb, J.H., Snella, E.M.,Gutierrez, M.A., Dieter, T., Sukegawa, K., Orii, T., Kondo, N. et al. (2002)Missense models [Gustm(E536A)Sly, Gustm(E536Q)Sly, andGustm(L175F)Sly] of murine mucopolysaccharidosis type VII produced bytargeted mutagenesis. Proc. Natl Acad. Sci. USA, 99, 14982–14987.
21. Akama, T.O., Nakayama, J., Nishida, K., Hiraoka, N., Suzuki, M.,McAuliffe, J., Hindsgaul, O., Fukuda, M. and Fukuda, M.N. (2001) Humancorneal GlcNac 6-O-sulfotransferase and mouse intestinal GlcNac6-O-sulfotransferase both produce keratan sulfate. J. Biol. Chem., 276,16271–16278.
22. Hahn, C.N., del Pilar, M.M., Schroder, M., Vanier, M.T., Hara, Y.,Suzuki, K., Suzuki, K. and d’Azzo, A. (1997) Generalized CNS disease andmassive GM1-ganglioside accumulation in mice defective in lysosomalacid beta-galactosidase. Hum. Mol. Genet., 6, 205–211.
23. Stinchi, S., Lullmann-Rauch, R., Hartmann, D., Coenen, R., Beccari, T.,Orlacchio, A., von Figura, K. and Saftig, P. (1999) Targeted disruption ofthe lysosomal alpha-mannosidase gene results in mice resembling a mildform of human alpha-mannosidosis. Hum. Mol. Genet., 8, 1365–1372.
24. Sango, K., Yamanaka, S., Hoffmann, A., Okuda, Y., Grinberg, A.,Westphal, H., McDonald, M.P., Crawley, J.N., Sandhoff, K. and Suzuki, K.(1995) Mouse models of Tay–Sachs and Sandhoff diseases differ inneurologic phenotype and ganglioside metabolism. Nat. Genet., 11,170–176.
25. Li, H.H., Yu, W.H., Rozengurt, N., Zhao, H.Z., Lyons, K.M.,Anagnostaras, S., Fanselow, M.S., Suzuki, K., Vanier, M.T. andNeufeld, E.F. (1999) Mouse model of Sanfilippo syndrome type B producedby targeted disruption of the gene encoding alpha-N-acetylglucosamini-dase. Proc. Natl Acad. Sci. USA, 96, 14505–14510.
26. Venn, G. and Mason, R.M. (1985) Absence of keratan sulphate fromskeletal tissues of mouse and rat. Biochem. J., 228, 443–450.
27. Pontow, S.E., Kery, V. and Stahl, P.D. (1992) Mannose receptor. Int. Rev.Cytol., 137B, 221–244.
28. Clarke, L.A., Russell, C.S., Pownall, S., Warrington, C.L., Borowski, A.,Dimmick, J.E., Toone, J. and Jirik, F.R. (1997) Murine mucopolysacchari-dosis type I: targeted disruption of the murine alpha-L-iduronidase gene.Hum. Mol. Genet., 6, 503–511.
29. Bhaumik, M., Muller, V.J., Rozaklis, T., Johnson, L., Dobrenis, K.,Bhattacharyya, R., Wurzelmann, S., Finamore, P., Hopwood, J.J.,Walkley, S.U. et al. (1999) A mouse model for mucopolysaccharidosistype III A (Sanfilippo syndrome). Glycobiology, 9, 1389–1396.
30. Evers, M., Saftig, P., Schmidt, P., Hafner, A., McLoghlin, D.B.,Schmahl, W., Hess, B., von Figura, K. and Peters, C. (1996) Targeteddisruption of the arylsulfatase B gene results in mice resembling thephenotype of mucopolysaccharidosis VI. Proc. Natl Acad. Sci. USA, 93,8214–8219.
31. Birkenmeier, E.H., Davisson, M.T., Beamer, W.G., Ganschow, R.E.,Vogler, C.A., Gwynn, B., Lyford, K.A., Maltais, L.M. and Wawrzyniak, C.J.(1989) Murine mucopolysaccharidosis type VII. Characterization of amouse with beta-glucuronidase deficiency. J. Clin. Invest., 83, 1258–1256.
32. Sando, G.N. and Neufeld, E.F. (1977) Recognition and receptor-mediateduptake of a lysosomal enzyme, alpha-l-iduronidase, by cultured humanfibroblasts. Cell, 12, 619–627.
33. Montano, A.M., Yamagishi, A., Tomatsu, S., Fukuda, S., Copeland, N.G.,Orii, K.E., Isogai, K., Yamada, N., Kato, Z.I., Jenkins, N.A. et al. (2000)The mouse N-acetylgalactosamine-6-sulfate sulfatase (Galns) gene: cDNAisolation, genomic characterization, chromosomal assignment and analysisof the 50-flanking region. Biochim. Biophys. Acta, 1500, 323–334.
34. Wagner, T.E., Hoppe, P.C., Jollick, J.D., Scholl, D.R., Hodinka, R.L. andGault, J.B. (1981) Microinjection of a rabbit beta-globin gene into zygotesand its subsequent expression in adult mice and their offspring. Proc. NatlAcad. Sci. USA, 78, 6376–6380.
35. Xu, X., Li, C., Garrett-Beal, L., Larson, D., Wynshaw-Boris, A. andDeng, C.X. (2001) Direct removal in the mouse of a floxed neo gene from athree-loxP conditional knockout allele by two novel approaches. Genesis,30, 1–6.
36. Glaser, J.H. and Sly, W.S. (1973) Beta-glucuronidase deficiency muco-polysaccharidosis: methods for enzymatic diagnosis. J. Lab. Clin. Med.,82, 969–977.
37. Peterson, G.L. (1979) Review of the folin phenol protein quantitationmethod of Lowry, Rosebrough, Farr and Randall. Anal. Biochem., 100,201–220.
38. Whitley, C.B., Ridnour, M.D., Draper, K.A., Dutton, C.M. and Neglia, J.P.(1989) Diagnostic test for mucopolysaccharidosis. I. Direct method forquantifying excessive urinary glycosaminoglycan excretion. Clin. Chem.,35, 374–379.
39. Poorthuis, B.J., Romme, A.E., Willemsen, R. and Wagemaker, G. (1994)Bone marrow transplantation has a significant effect on enzyme levels andstorage of glycosaminoglycans in tissues and in isolated hepatocytes ofmucopolysaccharidosis type VII mice. Pediatr. Res., 36, 187–193.
40. Caterson, B., Christner, J.E. and Baker, J.R. (1983) Identification of amonoclonal antibody that specifically recognizes corneal and skeletalkeratan sulfate. Monoclonal antibodies to cartilage proteoglycan. J. Biol.Chem., 258, 8848–8854.
41. Vogler, C., Birkenmeier, E.H., Sly, W.S., Levy, B., Pegors, C., Kyle, J.W.and Beamer, W.G. (1990) A murine model of mucopolysaccharidosis VII.Gross and microscopic findings in beta-glucuronidase-deficient mice. Am.J. Pathol., 136, 207–217.
3358 Human Molecular Genetics, 2003, Vol. 12, No. 24
by guest on Decem
ber 10, 2013http://hm
g.oxfordjournals.org/D
ownloaded from
Copyright © 2022 FDOKUMEN




















