
Γομάτος Λ., Αρμακόλας Σ., Μπακόπουλος Ν., «Παιδαγωγική-Διδακτική προετοιμασία των Εκπαιδευτικών της

Molecular adsorption of small alkanes on a PdO„101… thin film:Evidence of �-complex formation
Jason F. Weaver,a� Can Hakanoglu, Jeffery M. Hawkins, and Aravind AsthagiriDepartment of Chemical Engineering, University of Florida, Gainesville, Florida 32611, USA
�Received 27 October 2009; accepted 4 December 2009; published online 12 January 2010�
We investigated the molecular adsorption of methane, ethane, and propane on a PdO�101� thin filmusing temperature programmed desorption �TPD� and density functional theory �DFT� calculations.The TPD data reveal that each of the alkanes adsorbs into a low-coverage molecular state onPdO�101� in which the binding is stronger than that for alkanes physically adsorbed on Pd�111�.Analysis of the TPD data using limiting values of the desorption prefactors predicts that the alkanebinding energies on PdO�101� increase linearly with increasing chain length, but that the resultingline extrapolates to a nonzero value between about 22 and 26 kJ/mol at zero chain length. Thisconstant offset implies that a roughly molecule-independent interaction contributes to the alkanebinding energies for the molecules studied. DFT calculations predict that the small alkanes bind onPdO�101� by forming dative bonds with coordinatively unsaturated Pd atoms. The resultingadsorbed species are analogous to alkane �-complexes in that the bonding involves electrondonation from C–H � bonds to the Pd center as well as backdonation from the metal, whichweakens the C–H bonds. The binding energies predicted by DFT lie in a range from 16 to 24 kJ/mol,in good agreement with the constant offsets estimated from the TPD data. We conclude that both thedispersion interaction and the formation of �-complexes contribute to the binding of small alkaneson PdO�101�, and estimate that �-complex formation accounts for between 30% and 50% of thetotal binding energy for the molecules studied. The predicted weakening of C–H bonds resultingfrom �-complex formation may help to explain the high activity of PdO surfaces toward alkaneactivation. © 2010 American Institute of Physics. �doi:10.1063/1.3277672�
I. INTRODUCTION
The initial C–H bond activation of alkanes on metal-based catalysts has attracted widespread interest due to thedesire to more effectively utilize saturated hydrocarbons asenergy sources as well as feedstocks for chemical produc-tion. Because initial C–H bond cleavage is often a rate-determining step in the catalytic processing of alkanes, ex-tensive efforts have been devoted toward understandingalkane adsorption in detail, with most studies focusing onclean transition metal surfaces.1 In contrast, the interactionsof alkanes with metal oxide surfaces have not been widelyexplored, largely because alkanes interact weakly with manyoxide surfaces and are hence difficult to activate under ultra-high vacuum �UHV� conditions. Palladium oxide �PdO� is animportant exception as this material is highly active towardthe complete oxidation of alkanes. In fact, prior studies con-ducted at commercially relevant pressures demonstrate thatthe formation of PdO is responsible for the exceptional ac-tivity of supported Pd catalysts in the catalytic combustion ofnatural gas in excess oxygen.2–13 This finding provides sub-stantial motivation for studying the surface chemistry of PdOin detail. In particular, investigations with well-defined PdOsurfaces provide opportunities for gaining insights into the
mechanisms for alkane activation on transition metal oxidesas well as clarifying the oxide surface properties that en-hance C–H bond activation.
In recent work, we observed that the initial C–H bondcleavage of propane is highly facile on a PdO�101� thin filmgrown on Pd�111� in UHV.14 Our results demonstrate that theinitial dissociation of propane on the PdO�101� surface oc-curs by a precursor-mediated mechanism in which a molecu-larly adsorbed state of propane acts as the precursor to initialC–H bond cleavage. Temperature programmed desorption�TPD� measurements reveal that the molecular precursor isstrongly bound relative to physisorbed propane on Pd�111�,leading us to suggest that dative bonding interactions con-tribute to the binding of the alkane precursor on PdO�101�.In the present study, we examined the adsorption of methane,ethane, and propane on PdO�101� both experimentally andcomputationally, and present further evidence that dativebonding strengthens the binding of small alkanes onPdO�101�.
Dative bonding between alkane molecules and transitionmetal complexes is well known.15–17 These bonding interac-tions produce compounds known as alkane �-complexes thatcan serve as key intermediates in the initial C–H bond cleav-age of alkanes by mononuclear transition metal compounds.Bond strengths for alkane �-complexes are typically between20 and 60 kJ/mol.15 The dative bonding interaction that pro-duces an alkane �-complex generally involves electron do-nation from C–H � bonds of the alkane molecule into empty
a�Author to whom correspondence should be addressed. Electronic mail:[email protected]. Tel.: 352-392-0869. FAX: 352-392-9513.
THE JOURNAL OF CHEMICAL PHYSICS 132, 024709 �2010�
0021-9606/2010/132�2�/024709/10/$30.00 © 2010 American Institute of Physics132, 024709-1

d-orbitals of the metal center, and, in some cases, backdona-tion of electrons from filled d-orbitals of the metal into C–H�� bonds. Among the few examples that have been reported,the adsorption of H2 on RuO2�110� provides the most com-prehensive example of �-complex formation on a solidsurface.18,19 Recent work by Blanco-Rey et al.20 as well asour group21 also demonstrates that dihydrogen experiencesrelatively strong dative bonding on PdO�101�, which sug-gests the possibility that the surfaces of late transition metaloxides have properties that generally favor coordinate bondformation with saturated molecules.
While it is reasonable to expect that dative interactionswould also influence the bonding of alkanes on transitionmetal or transition metal compound surfaces, clear evidencefor such bonding is rather limited. For example, several stud-ies have reported softening of C–H vibrational modes of al-kanes upon adsorption on transition metal surfaces.22–31
However, work by Fosser et al.30 provides evidence thatC–H mode softening can occur even when the attractive dis-persion interaction dominates the alkane-surface binding. Inthis case, the dispersion interaction can bring the moleculeclose enough to the surface to cause charge flow from metalstates into Rydberg molecular orbitals, resulting in C–Hmode softening. Thus, the C–H mode softening seen onmetal surfaces is not necessarily a consequence of the type ofdative bonding that characterizes alkane �-complexes.
Recent studies have established systematic trends in thedesorption kinetic parameters for alkane molecules that arephysically adsorbed on solid surfaces.32,33 As demonstratedin the present study, these trends provide a benchmark forcomparison with systems in which dative bonding may affectthe alkane-surface binding. At low coverage, straight chainalkane molecules adopt a flat-lying geometry when phys-isorbed on a close-packed surface.1 This configuration maxi-mizes the attractive interaction between the molecule andsurface by allowing each CHn group to reside within theattractive region of the molecule-surface potential. In thissimplified model, each CHn group contributes equally to themolecule-surface interaction, resulting in alkane binding en-ergies E that increase linearly with the chain length N. Fur-ther, a plot of E versus N should have a zero intercept if eachCHn group makes an additive contribution to the total bind-ing energy.
Recently, Tait et al.32,33 reported extensive investigationsof the desorption of physisorbed alkanes from MgO�100�,Pt�111�, and C�0001�, and confirmed that the E versus Nrelationships are linear with zero intercepts. These workersdeveloped a so-called inversion-optimization method to de-termine both the prefactor and �coverage-dependent� activa-tion energy for desorption from TPD spectra obtained at dif-ferent initial coverages. A key finding from their analysis isthat the prefactors for desorption increase significantly withincreasing chain length, which is consistent with earlier mo-lecular dynamics simulations of alkane desorption.34,35 Com-pared with an analysis that assumes a molecule-independentprefactor, the effect of the increasing prefactors is to increasethe slope of the E versus N curve, causing this curve toexhibit the expected zero intercept. In the context of transi-tion state theory, the prefactors for desorption increase with
chain length because the densities of translational and rota-tional states of gaseous alkane molecules increase substan-tially with increasing molecular size, resulting in larger en-tropy changes upon desorption. Tait et al.33 also reportformulas, derived from transition state theory, that give lim-iting values of the desorption prefactors for several alkanesas a function of the surface temperature. The minimum andmaximum prefactors correspond to adsorbed molecules thatare fully mobile versus completely immobile, respectively.Overall, the work of Tait et al.32,33 provides important guid-ance in interpreting alkane TPD spectra, and identifying sys-tems that deviate from the systematic trends established forlinear alkanes physisorbed on close-packed surfaces.
In the present study, we investigated the molecular ad-sorption of methane, ethane, and propane on PdO�101� usingTPD measurements and density functional theory �DFT� cal-culations. The TPD results show that each of these smallalkanes adsorbs in a molecular state that is more stronglybound than alkanes physically adsorbed on Pd�111�, and thatthe E versus N relationship has a nonzero intercept between22 and 26 kJ/mol, which is consistent with dative bondingcontributing to the alkane binding energy on PdO�101�. DFTcalculations predict that these small alkanes bind to coordi-natively unsaturated �cus� Pd atoms through a donor-acceptor interaction, resulting in an adsorbed state that isanalogous to an alkane �-complex.
II. EXPERIMENTAL DETAILS
Previous studies36,37 provide details of the three-levelUHV chamber utilized for the present experiments. ThePd�111� crystal employed in this study is a circular disk �8� �1 mm� spot-welded to W wires and attached to a cop-per sample holder in thermal contact with a liquid nitrogencooled reservoir. A type K thermocouple spot-welded to thebackside of the crystal allows sample temperature measure-ments. Resistive heating, controlled using a proportional-integral-derivative controller that varies the output of a pro-grammable dc power supply, supports maintaining or linearlyramping the sample temperature from 81 to 1250 K. Initially,sample cleaning consisted of sputtering with 600 eV Ar+ ionsat a surface temperature of 900 K, followed by annealing at1100 K for several minutes. Subsequent cleaning involvedroutinely exposing the sample held at 856 K to an atomicoxygen beam for several minutes, followed by flashing thesample to 923 K to desorb oxygen and carbon oxides. Asdiscussed previously,38 we limited the sample temperature to923 K to maintain oxygen saturation in the subsurface reser-voir, and thereby ensure reproducibility in preparing thePdO�101� thin films used in this study. We considered thePd�111� sample to be clean when we could no longer detectcontaminants with x-ray photoelectron spectroscopy, ob-tained sharp low energy electron diffraction patterns consis-tent with the Pd�111� surface, and did not detect CO produc-tion during flash desorption after oxygen adsorption.
A two-stage differentially pumped chamber attached tothe UHV chamber houses the inductively coupled rf plasmasource �Oxford Scientific Instruments� utilized to generatebeams containing oxygen atoms for this study. We refer the
024709-2 Weaver et al. J. Chem. Phys. 132, 024709 �2010�

reader to prior work for details of the beam system.36,37 Toproduce a PdO�101� thin film, we expose a Pd�111� sampleheld at 500 K to an �12 ML dose of gaseous oxygen atomssupplied in a beam, where we define 1 ML as equal to thePd�111� surface atom density of 1.53�1015 cm−2. This pro-cedure generates a high-quality PdO�101� film that has astoichiometric surface termination, contains �3.0 ML ofoxygen atoms and is about 12 Å thick.39,40 The structure ofthe PdO�101� surface is discussed in detail below.
Alkanes were delivered to the sample from a calibratedbeam doser at incident fluxes between 5�10−4 and 5�10−3 ML s−1. We set the sample-to-doser distance to about50 mm to ensure uniform impingement of the alkanes acrossthe sample surface. After the alkane exposures, we conductedTPD experiments by positioning the sample in front of themass spectrometer and then heating at a constant rate of1 K s−1 until the sample temperature reached 923 K. ThePdO thin films completely decompose when heated to 923 Kso it was necessary to prepare fresh PdO films for each al-kane adsorption experiment. In a few experiments withmethane and ethane, we terminated the TPD experiments at asample temperature of 550 K to avoid decomposition of thePdO film. The resulting TPD spectra differ negligibly fromthose obtained from fresh PdO films, most likely becausemethane and ethane dissociate to an immeasurable extent onPdO�101� �vide infra�.
We estimated ethane and propane coverages by scalingintegrated desorption spectra obtained from PdO�101� withintegrated TPD spectra collected from monolayers of ethaneand propane adsorbed on Pd�111� at 95 and 110 K, respec-tively, and assuming that these monolayers saturate at0.227 ML versus 0.205 ML on Pd�111�. These values corre-spond to the saturation coverages of ethane and propanemonolayers on Pt�111� as determined from calibrated mo-lecular beam experiments by Carlsson and Madix.41 Kao andMadix42 showed that the binding energies of ethane and pro-pane are only slightly higher on Pd�111� compared withPt�111� so it seems reasonable to assume that the monolayerssaturate at the same coverages on each metal surface.
Since only low coverages of methane can be generatedon Pd�111� at 85 K, we estimated methane coverages byscaling the mass spectrometer signals obtained from knowngaseous flow rates generated with a calibrated leak. Our ap-proach involves separately admitting methane and ethaneinto the UHV chamber using a calibrated leak and measuringmass spectrometer signals of these molecules. This proce-dure provides a relation between the ratio of CH4 and C2H6
leak rates into the chamber and the corresponding ratio ofmass spectrometer signals. We then collect a few CH4 andC2H6 TPD spectra by facing the sample away from the massspectrometer so that the resulting signals represent back-ground partial pressures. This approach simulates the condi-tions employed during the calibrated leak measurements. Us-ing the absolute coverage of a saturated monolayer of C2H6
on Pd�111�, we express the background TPD spectra ofethane in terms of absolute ethane coverage. Finally, with theethane background TPD spectra given in units of ML s−1, we
use the relation between CH4 and C2H6 flow rates and massspectrometer signals to estimate the absolute methane cover-ages.
III. COMPUTATIONAL DETAILS
All of the DFT calculations in this paper are performedusing Vienna ab initio simulation package �VASP�.43–46 Weuse the projector augmented wave pseudopotentials47,48 pro-vided in the VASP database. Calculations have been done us-ing the generalized gradient approximation Perdew–Burke–Ernzerhof �PBE� exchange-correlation functional.49 We havetested select configurations with the Perdew–Wang func-tional, which yields adsorption energies that are 2–4 kJ/molhigher than those obtained with PBE, but the differences areinsufficient to affect the conclusions discussed in this paper.Additionally, we are mindful that these exchange-correlationfunctionals fail to describe long-range van der Waals ener-gies, which are important for organic molecules such asalkanes.50 As discussed in further detail in the presentation ofthe DFT results, we use DFT to probe the dative bondingbetween alkane molecules and the PdO�101� surface. Whilethe dispersive interactions provide a substantial portion ofthe adsorption energy, we would expect these interactions tobe less sensitive to the molecule-surface geometry comparedwith the dative bonding interaction. Therefore, the adsorp-tion geometry is expected to be relatively accurate, while theDFT adsorption energy can be approximately assigned to theentire dative bonding contribution and will be a lower bound.A plane-wave expansion with a cutoff of 400 eV is used, andthe total energy calculations are done using a residual mini-mization method with direct inversion in the iterative sub-space for electronic relaxations, accelerated usingMethfessel–Paxton Fermi-level smearing with a Gaussianwidth of 0.1 eV.51 The positions of the atoms are relaxedusing a limited memory Broyden–Fletcher–Goldfarb–Shanno optimization method52 until the forces on all uncon-strained atoms are less than 0.03 eV/Å. While experimentallythe PdO�101� film is grown on the Pd�111� surface, the oxidefilm is sufficiently thick �13 Å� that we assume the Pd�111�substrate may be ignored in our DFT calculations. Inclusionof the Pd�111� substrate would require large supercells due tothe lattice mismatch between the oxide film and Pd�111� sur-face. Instead, we represent the PdO�101� film with four lay-ers, which corresponds to a thickness of approximately 9 Å.The bottom layer is fixed, but all other atoms in the oxidefilm and the alkane adsorbate are allowed to relax. We use avacuum spacing of 20 Å, which is sufficient to minimize anyspurious periodic interactions in the surface normal direc-tion. The PdO�101� film is obtained from the relaxed PdObulk structure, but the film is strained �a=3.057 Å, b=6.352 Å� to match the reported experimental structure.39 A4�2�1 �2�4�1� Monkhorst–Pack k-point mesh53 wasused for the 1�4 �2�2� surface unit cell. We have evalu-ated the zero point corrections for methane in its favoredconfiguration on PdO�101� and find the contribution is lessthan 2 kJ/mol. The zero point correction is sufficiently smallthat it does not affect the conclusions drawn in this paper andis excluded in our DFT values for adsorption energy.
024709-3 Alkane �-complexes on PdO�101� J. Chem. Phys. 132, 024709 �2010�
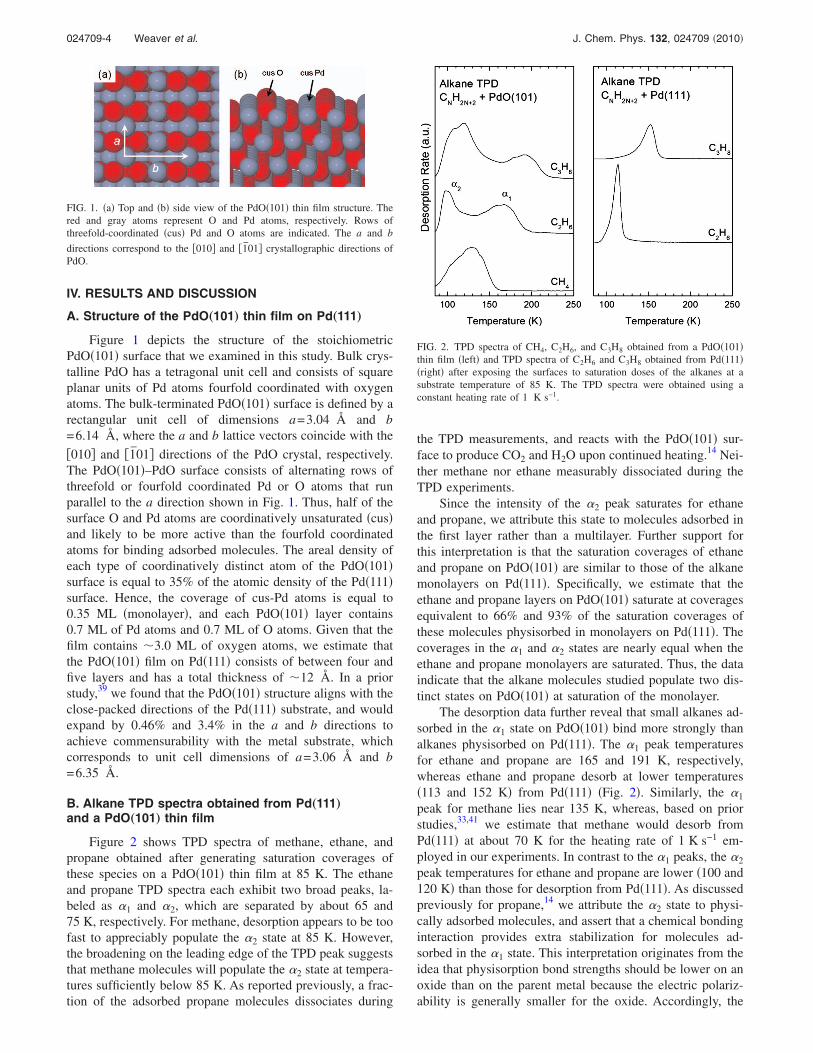
IV. RESULTS AND DISCUSSION
A. Structure of the PdO„101… thin film on Pd„111…
Figure 1 depicts the structure of the stoichiometricPdO�101� surface that we examined in this study. Bulk crys-talline PdO has a tetragonal unit cell and consists of squareplanar units of Pd atoms fourfold coordinated with oxygenatoms. The bulk-terminated PdO�101� surface is defined by arectangular unit cell of dimensions a=3.04 Å and b=6.14 Å, where the a and b lattice vectors coincide with the
�010� and �1̄01� directions of the PdO crystal, respectively.The PdO�101�–PdO surface consists of alternating rows ofthreefold or fourfold coordinated Pd or O atoms that runparallel to the a direction shown in Fig. 1. Thus, half of thesurface O and Pd atoms are coordinatively unsaturated �cus�and likely to be more active than the fourfold coordinatedatoms for binding adsorbed molecules. The areal density ofeach type of coordinatively distinct atom of the PdO�101�surface is equal to 35% of the atomic density of the Pd�111�surface. Hence, the coverage of cus-Pd atoms is equal to0.35 ML �monolayer�, and each PdO�101� layer contains0.7 ML of Pd atoms and 0.7 ML of O atoms. Given that thefilm contains �3.0 ML of oxygen atoms, we estimate thatthe PdO�101� film on Pd�111� consists of between four andfive layers and has a total thickness of �12 Å. In a priorstudy,39 we found that the PdO�101� structure aligns with theclose-packed directions of the Pd�111� substrate, and wouldexpand by 0.46% and 3.4% in the a and b directions toachieve commensurability with the metal substrate, whichcorresponds to unit cell dimensions of a=3.06 Å and b=6.35 Å.
B. Alkane TPD spectra obtained from Pd„111…and a PdO„101… thin film
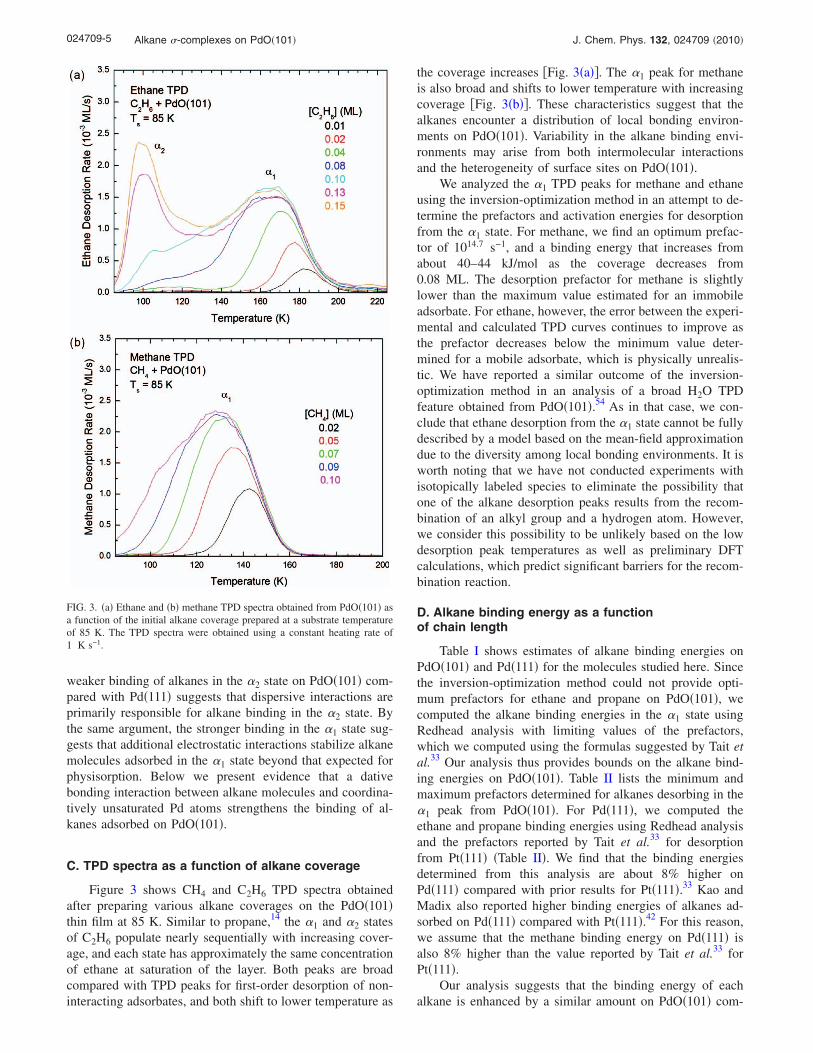
Figure 2 shows TPD spectra of methane, ethane, andpropane obtained after generating saturation coverages ofthese species on a PdO�101� thin film at 85 K. The ethaneand propane TPD spectra each exhibit two broad peaks, la-beled as �1 and �2, which are separated by about 65 and75 K, respectively. For methane, desorption appears to be toofast to appreciably populate the �2 state at 85 K. However,the broadening on the leading edge of the TPD peak suggeststhat methane molecules will populate the �2 state at tempera-tures sufficiently below 85 K. As reported previously, a frac-tion of the adsorbed propane molecules dissociates during
the TPD measurements, and reacts with the PdO�101� sur-face to produce CO2 and H2O upon continued heating.14 Nei-ther methane nor ethane measurably dissociated during theTPD experiments.
Since the intensity of the �2 peak saturates for ethaneand propane, we attribute this state to molecules adsorbed inthe first layer rather than a multilayer. Further support forthis interpretation is that the saturation coverages of ethaneand propane on PdO�101� are similar to those of the alkanemonolayers on Pd�111�. Specifically, we estimate that theethane and propane layers on PdO�101� saturate at coveragesequivalent to 66% and 93% of the saturation coverages ofthese molecules physisorbed in monolayers on Pd�111�. Thecoverages in the �1 and �2 states are nearly equal when theethane and propane monolayers are saturated. Thus, the dataindicate that the alkane molecules studied populate two dis-tinct states on PdO�101� at saturation of the monolayer.
The desorption data further reveal that small alkanes ad-sorbed in the �1 state on PdO�101� bind more strongly thanalkanes physisorbed on Pd�111�. The �1 peak temperaturesfor ethane and propane are 165 and 191 K, respectively,whereas ethane and propane desorb at lower temperatures�113 and 152 K� from Pd�111� �Fig. 2�. Similarly, the �1
peak for methane lies near 135 K, whereas, based on priorstudies,33,41 we estimate that methane would desorb fromPd�111� at about 70 K for the heating rate of 1 K s−1 em-ployed in our experiments. In contrast to the �1 peaks, the �2
peak temperatures for ethane and propane are lower �100 and120 K� than those for desorption from Pd�111�. As discussedpreviously for propane,14 we attribute the �2 state to physi-cally adsorbed molecules, and assert that a chemical bondinginteraction provides extra stabilization for molecules ad-sorbed in the �1 state. This interpretation originates from theidea that physisorption bond strengths should be lower on anoxide than on the parent metal because the electric polariz-ability is generally smaller for the oxide. Accordingly, the
FIG. 1. �a� Top and �b� side view of the PdO�101� thin film structure. Thered and gray atoms represent O and Pd atoms, respectively. Rows ofthreefold-coordinated �cus� Pd and O atoms are indicated. The a and b
directions correspond to the �010� and �1̄01� crystallographic directions ofPdO.
FIG. 2. TPD spectra of CH4, C2H6, and C3H8 obtained from a PdO�101�thin film �left� and TPD spectra of C2H6 and C3H8 obtained from Pd�111��right� after exposing the surfaces to saturation doses of the alkanes at asubstrate temperature of 85 K. The TPD spectra were obtained using aconstant heating rate of 1 K s−1.
024709-4 Weaver et al. J. Chem. Phys. 132, 024709 �2010�
weaker binding of alkanes in the �2 state on PdO�101� com-pared with Pd�111� suggests that dispersive interactions areprimarily responsible for alkane binding in the �2 state. Bythe same argument, the stronger binding in the �1 state sug-gests that additional electrostatic interactions stabilize alkanemolecules adsorbed in the �1 state beyond that expected forphysisorption. Below we present evidence that a dativebonding interaction between alkane molecules and coordina-tively unsaturated Pd atoms strengthens the binding of al-kanes adsorbed on PdO�101�.
C. TPD spectra as a function of alkane coverage
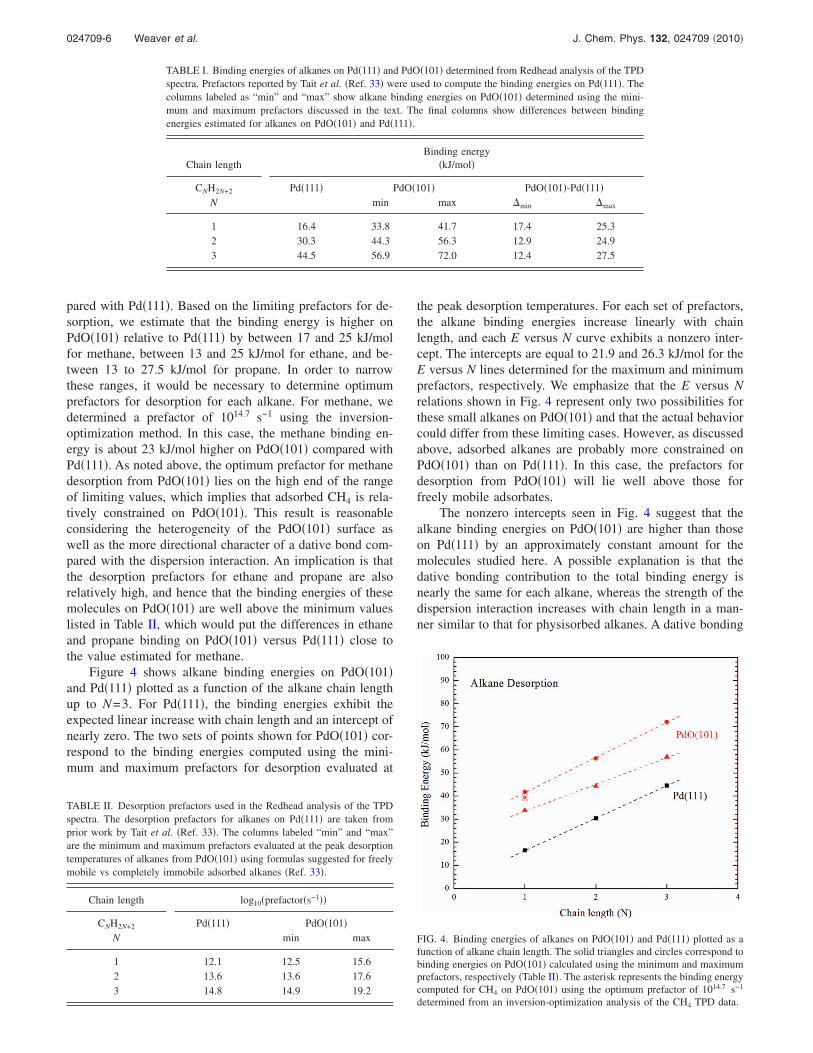
Figure 3 shows CH4 and C2H6 TPD spectra obtainedafter preparing various alkane coverages on the PdO�101�thin film at 85 K. Similar to propane,14 the �1 and �2 statesof C2H6 populate nearly sequentially with increasing cover-age, and each state has approximately the same concentrationof ethane at saturation of the layer. Both peaks are broadcompared with TPD peaks for first-order desorption of non-interacting adsorbates, and both shift to lower temperature as
the coverage increases �Fig. 3�a��. The �1 peak for methaneis also broad and shifts to lower temperature with increasingcoverage �Fig. 3�b��. These characteristics suggest that thealkanes encounter a distribution of local bonding environ-ments on PdO�101�. Variability in the alkane binding envi-ronments may arise from both intermolecular interactionsand the heterogeneity of surface sites on PdO�101�.
We analyzed the �1 TPD peaks for methane and ethaneusing the inversion-optimization method in an attempt to de-termine the prefactors and activation energies for desorptionfrom the �1 state. For methane, we find an optimum prefac-tor of 1014.7 s−1, and a binding energy that increases fromabout 40–44 kJ/mol as the coverage decreases from0.08 ML. The desorption prefactor for methane is slightlylower than the maximum value estimated for an immobileadsorbate. For ethane, however, the error between the experi-mental and calculated TPD curves continues to improve asthe prefactor decreases below the minimum value deter-mined for a mobile adsorbate, which is physically unrealis-tic. We have reported a similar outcome of the inversion-optimization method in an analysis of a broad H2O TPDfeature obtained from PdO�101�.54 As in that case, we con-clude that ethane desorption from the �1 state cannot be fullydescribed by a model based on the mean-field approximationdue to the diversity among local bonding environments. It isworth noting that we have not conducted experiments withisotopically labeled species to eliminate the possibility thatone of the alkane desorption peaks results from the recom-bination of an alkyl group and a hydrogen atom. However,we consider this possibility to be unlikely based on the lowdesorption peak temperatures as well as preliminary DFTcalculations, which predict significant barriers for the recom-bination reaction.
D. Alkane binding energy as a functionof chain length
Table I shows estimates of alkane binding energies onPdO�101� and Pd�111� for the molecules studied here. Sincethe inversion-optimization method could not provide opti-mum prefactors for ethane and propane on PdO�101�, wecomputed the alkane binding energies in the �1 state usingRedhead analysis with limiting values of the prefactors,which we computed using the formulas suggested by Tait etal.33 Our analysis thus provides bounds on the alkane bind-ing energies on PdO�101�. Table II lists the minimum andmaximum prefactors determined for alkanes desorbing in the�1 peak from PdO�101�. For Pd�111�, we computed theethane and propane binding energies using Redhead analysisand the prefactors reported by Tait et al.33 for desorptionfrom Pt�111� �Table II�. We find that the binding energiesdetermined from this analysis are about 8% higher onPd�111� compared with prior results for Pt�111�.33 Kao andMadix also reported higher binding energies of alkanes ad-sorbed on Pd�111� compared with Pt�111�.42 For this reason,we assume that the methane binding energy on Pd�111� isalso 8% higher than the value reported by Tait et al.33 forPt�111�.
Our analysis suggests that the binding energy of eachalkane is enhanced by a similar amount on PdO�101� com-
FIG. 3. �a� Ethane and �b� methane TPD spectra obtained from PdO�101� asa function of the initial alkane coverage prepared at a substrate temperatureof 85 K. The TPD spectra were obtained using a constant heating rate of1 K s−1.
024709-5 Alkane �-complexes on PdO�101� J. Chem. Phys. 132, 024709 �2010�
pared with Pd�111�. Based on the limiting prefactors for de-sorption, we estimate that the binding energy is higher onPdO�101� relative to Pd�111� by between 17 and 25 kJ/molfor methane, between 13 and 25 kJ/mol for ethane, and be-tween 13 to 27.5 kJ/mol for propane. In order to narrowthese ranges, it would be necessary to determine optimumprefactors for desorption for each alkane. For methane, wedetermined a prefactor of 1014.7 s−1 using the inversion-optimization method. In this case, the methane binding en-ergy is about 23 kJ/mol higher on PdO�101� compared withPd�111�. As noted above, the optimum prefactor for methanedesorption from PdO�101� lies on the high end of the rangeof limiting values, which implies that adsorbed CH4 is rela-tively constrained on PdO�101�. This result is reasonableconsidering the heterogeneity of the PdO�101� surface aswell as the more directional character of a dative bond com-pared with the dispersion interaction. An implication is thatthe desorption prefactors for ethane and propane are alsorelatively high, and hence that the binding energies of thesemolecules on PdO�101� are well above the minimum valueslisted in Table II, which would put the differences in ethaneand propane binding on PdO�101� versus Pd�111� close tothe value estimated for methane.
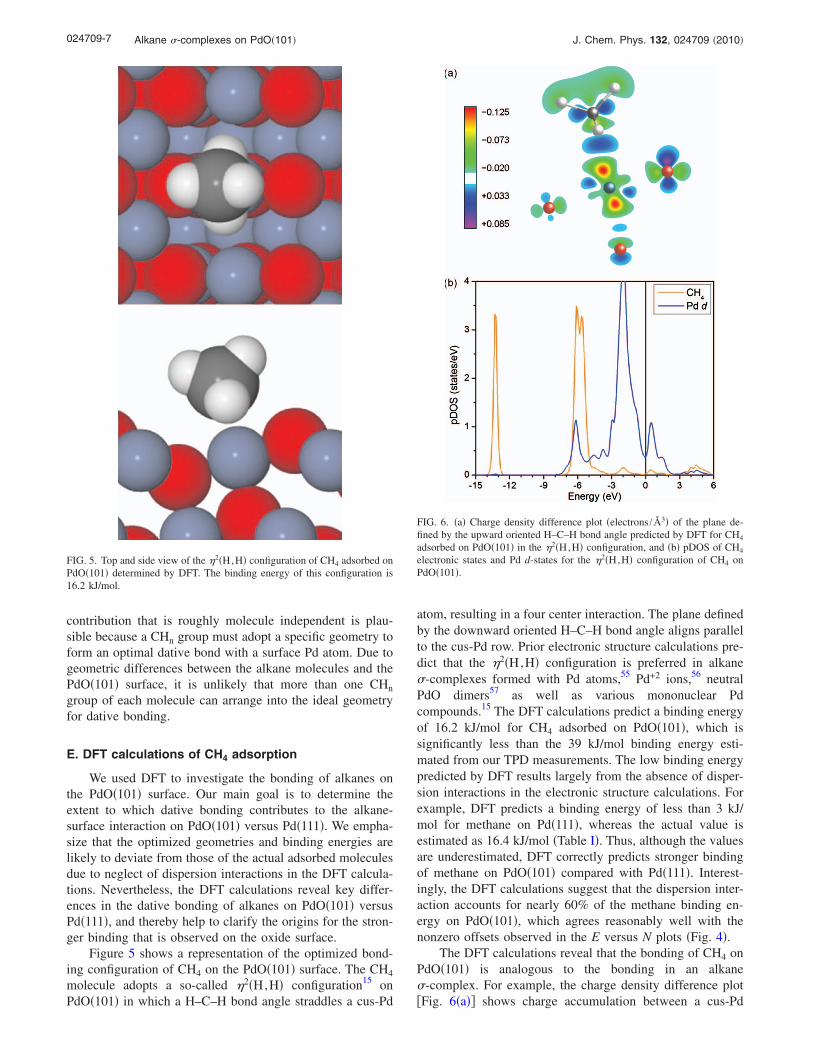
Figure 4 shows alkane binding energies on PdO�101�and Pd�111� plotted as a function of the alkane chain lengthup to N=3. For Pd�111�, the binding energies exhibit theexpected linear increase with chain length and an intercept ofnearly zero. The two sets of points shown for PdO�101� cor-respond to the binding energies computed using the mini-mum and maximum prefactors for desorption evaluated at
the peak desorption temperatures. For each set of prefactors,the alkane binding energies increase linearly with chainlength, and each E versus N curve exhibits a nonzero inter-cept. The intercepts are equal to 21.9 and 26.3 kJ/mol for theE versus N lines determined for the maximum and minimumprefactors, respectively. We emphasize that the E versus Nrelations shown in Fig. 4 represent only two possibilities forthese small alkanes on PdO�101� and that the actual behaviorcould differ from these limiting cases. However, as discussedabove, adsorbed alkanes are probably more constrained onPdO�101� than on Pd�111�. In this case, the prefactors fordesorption from PdO�101� will lie well above those forfreely mobile adsorbates.
The nonzero intercepts seen in Fig. 4 suggest that thealkane binding energies on PdO�101� are higher than thoseon Pd�111� by an approximately constant amount for themolecules studied here. A possible explanation is that thedative bonding contribution to the total binding energy isnearly the same for each alkane, whereas the strength of thedispersion interaction increases with chain length in a man-ner similar to that for physisorbed alkanes. A dative bonding
TABLE I. Binding energies of alkanes on Pd�111� and PdO�101� determined from Redhead analysis of the TPDspectra. Prefactors reported by Tait et al. �Ref. 33� were used to compute the binding energies on Pd�111�. Thecolumns labeled as “min” and “max” show alkane binding energies on PdO�101� determined using the mini-mum and maximum prefactors discussed in the text. The final columns show differences between bindingenergies estimated for alkanes on PdO�101� and Pd�111�.
Chain lengthBinding energy
�kJ/mol�
CNH2N+2 Pd�111� PdO�101� PdO�101�-Pd�111�N min max �min �max
1 16.4 33.8 41.7 17.4 25.32 30.3 44.3 56.3 12.9 24.93 44.5 56.9 72.0 12.4 27.5
TABLE II. Desorption prefactors used in the Redhead analysis of the TPDspectra. The desorption prefactors for alkanes on Pd�111� are taken fromprior work by Tait et al. �Ref. 33�. The columns labeled “min” and “max”are the minimum and maximum prefactors evaluated at the peak desorptiontemperatures of alkanes from PdO�101� using formulas suggested for freelymobile vs completely immobile adsorbed alkanes �Ref. 33�.
Chain length log10�prefactor�s−1��
CNH2N+2 Pd�111� PdO�101�N min max
1 12.1 12.5 15.62 13.6 13.6 17.63 14.8 14.9 19.2
FIG. 4. Binding energies of alkanes on PdO�101� and Pd�111� plotted as afunction of alkane chain length. The solid triangles and circles correspond tobinding energies on PdO�101� calculated using the minimum and maximumprefactors, respectively �Table II�. The asterisk represents the binding energycomputed for CH4 on PdO�101� using the optimum prefactor of 1014.7 s−1
determined from an inversion-optimization analysis of the CH4 TPD data.
024709-6 Weaver et al. J. Chem. Phys. 132, 024709 �2010�
contribution that is roughly molecule independent is plau-sible because a CHn group must adopt a specific geometry toform an optimal dative bond with a surface Pd atom. Due togeometric differences between the alkane molecules and thePdO�101� surface, it is unlikely that more than one CHn
group of each molecule can arrange into the ideal geometryfor dative bonding.
E. DFT calculations of CH4 adsorption
We used DFT to investigate the bonding of alkanes onthe PdO�101� surface. Our main goal is to determine theextent to which dative bonding contributes to the alkane-surface interaction on PdO�101� versus Pd�111�. We empha-size that the optimized geometries and binding energies arelikely to deviate from those of the actual adsorbed moleculesdue to neglect of dispersion interactions in the DFT calcula-tions. Nevertheless, the DFT calculations reveal key differ-ences in the dative bonding of alkanes on PdO�101� versusPd�111�, and thereby help to clarify the origins for the stron-ger binding that is observed on the oxide surface.
Figure 5 shows a representation of the optimized bond-ing configuration of CH4 on the PdO�101� surface. The CH4
molecule adopts a so-called �2�H,H� configuration15 onPdO�101� in which a H–C–H bond angle straddles a cus-Pd
atom, resulting in a four center interaction. The plane definedby the downward oriented H–C–H bond angle aligns parallelto the cus-Pd row. Prior electronic structure calculations pre-dict that the �2�H,H� configuration is preferred in alkane�-complexes formed with Pd atoms,55 Pd+2 ions,56 neutralPdO dimers57 as well as various mononuclear Pdcompounds.15 The DFT calculations predict a binding energyof 16.2 kJ/mol for CH4 adsorbed on PdO�101�, which issignificantly less than the 39 kJ/mol binding energy esti-mated from our TPD measurements. The low binding energypredicted by DFT results largely from the absence of disper-sion interactions in the electronic structure calculations. Forexample, DFT predicts a binding energy of less than 3 kJ/mol for methane on Pd�111�, whereas the actual value isestimated as 16.4 kJ/mol �Table I�. Thus, although the valuesare underestimated, DFT correctly predicts stronger bindingof methane on PdO�101� compared with Pd�111�. Interest-ingly, the DFT calculations suggest that the dispersion inter-action accounts for nearly 60% of the methane binding en-ergy on PdO�101�, which agrees reasonably well with thenonzero offsets observed in the E versus N plots �Fig. 4�.
The DFT calculations reveal that the bonding of CH4 onPdO�101� is analogous to the bonding in an alkane�-complex. For example, the charge density difference plot�Fig. 6�a�� shows charge accumulation between a cus-Pd
FIG. 5. Top and side view of the �2�H,H� configuration of CH4 adsorbed onPdO�101� determined by DFT. The binding energy of this configuration is16.2 kJ/mol.
FIG. 6. �a� Charge density difference plot �electrons /Å3� of the plane de-fined by the upward oriented H–C–H bond angle predicted by DFT for CH4
adsorbed on PdO�101� in the �2�H,H� configuration, and �b� pDOS of CH4
electronic states and Pd d-states for the �2�H,H� configuration of CH4 onPdO�101�.
024709-7 Alkane �-complexes on PdO�101� J. Chem. Phys. 132, 024709 �2010�

atom and the CH4 molecule as well as depletion of chargenear the Pd atom and the H atoms of the methane molecule.The decrease in electron density near the H-atoms suggeststhat electron donation occurs from bonding orbitals of theCH4 molecule toward the cus-Pd atom, with charge accumu-lating between the molecule and the cus-Pd atom. In addi-tion, the pronounced charge depletion around the Pd atomimplies that the bonding also involves backdonation from thePd atom to antibonding orbitals of the CH4 molecule.
The partial density of states �pDOS� plot confirms thatthe CH4–PdO�101� bonding interaction involves both dona-tion and backdonation �Fig. 6�b��. A relatively strong inter-action between the highest occupied molecular orbitals �2t1�and the d-states is evident from the overlap of peaks at �6.1eV. Inspection of the orbital-decomposed DOS reveals that apreferred interaction between Pd d-states and the C 2py and2pz orbitals breaks the degeneracy of the three 2t1 hybridmolecular orbitals and causes the associated peak in thepDOS to split into two components centered at �6.1 and�5.6 eV. The C and H s states and the C 2pz state contributeto both peaks, whereas the C 2px state contributes mainly tothe peak at �5.6 eV and the C 2py state contributes mainlyto the peak at �6.1 eV. The latter peak overlaps a sharp peakin the d-band, indicating an interaction between these states.The pDOS also shows that the d-states of the surface interactonly weakly with the 2a1 orbital of methane located at�13.1 eV.
Several small peaks between �3 and +6 eV are alsoevident in the pDOS of CH4 and overlap with Pd d-states.The CH4 states between +3 and +6 eV appear to arise pri-marily from the original antibonding molecular orbitals.However, the peaks at about �2 and +0.5 eV likely repre-sent new antibonding states that result from the relativelystrong interactions between filled d-states and the occupied2t1 orbitals of the molecule. In this case, the pDOS calcula-tion indicates that one of antibonding states, centered at�2 eV, is occupied due to backdonation from the surface.
Changes in the molecular geometry and vibrational fre-quencies demonstrate that the CH4–PdO�101� bonding inter-action weakens the two C–H bonds that coordinate with thecus-Pd atom. For example, the lengths of the downward ori-ented C–H bonds stretch by 1.55% relative to their gas-phasevalue, and the corresponding H–C–H bond angle increasesby 5.6°. The calculations also predict pronounced changes inthe vibrational motions of the downward oriented C–Hbonds. Three modes, an asymmetric stretch, a symmetricstretch, and an asymmetric bend, mainly involve motions ofthe downward oriented C–H bonds and the frequencies ofthese modes are lower by 204, 161, and 107 cm−1, respec-tively, than their gas-phase values. For comparison, the fre-quencies of the remaining vibrational modes change by lessthan 38 cm−1 upon adsorption. Also, the vibrational frequen-cies of CH4 adsorbed on Pd�111� are lower by no more than30 cm−1 from the gas-phase values. The redshifts of thethree strongly perturbed modes of CH4 on PdO�101� are con-siderably larger than those reported for alkanes physisorbedon metal surfaces.1,30
F. DFT calculations of ethane and propaneon PdO„101…
Figure 7 shows the favored �2�H,H� configurations ofethane and propane adsorbed on the PdO�101� surface. Forethane, the �2�H,H� configuration has a binding energy of21.9 kJ/mol and represents the lowest energy configurationthat could be identified with DFT. In this configuration, theupward directed methyl group points toward the neighboringcus-O atom. An �2�H,H� configuration can also be obtainedby rotating the ethane molecule such that the spectator me-thyl group points toward the fourfold O atom. The bindingenergy of this configuration is only 0.4 kJ/mol lower thanthat shown in Fig. 7. According to DFT, the binding energyof C2H6 on PdO�101� is 5.7 kJ/mol higher than that of CH4
adsorbed in the �2�H,H� configuration �Fig. 5�. We attributethis small difference to electrostatic interactions between thespectator methyl group of C2H6 and the neighboring O at-oms.
As seen in Fig. 7, the favored �2�H,H� configuration forpropane involves bonding between the CH2 group and acus-Pd atom, with the molecular plane oriented perpendicu-larly to the cus-Pd row. DFT predicts a binding energy of23.7 kJ/mol for this adsorbed configuration of C3H8, whichis only slightly higher than the binding energy for the fa-vored �2�H,H� configuration of C2H6. For comparison, thebinding energy is about 13.5 kJ/mol when propane binds tothe cus-Pd atom through a CH3 group. A key difference be-tween the CH3 versus CH2 down configurations of propane isthat the latter puts the spectator CH3 groups closer to neigh-boring surface oxygen atoms.
By aligning its molecular plane parallel to the cus-Pdatom row, the propane molecule can bind in a configurationthat is slightly ��3.3 kJ /mol� more favorable than the CH2
�2�H,H� configuration shown in Fig. 7. In the resulting ad-sorbed state �Fig. 8�, one H-atom of each CH3 group coordi-nates with a cus-Pd atom, producing two coordinate bonds
FIG. 7. Top and side view of the favored �2�H,H� configurations of �a�C2H6 and �b� C3H8 adsorbed on PdO�101� determined by DFT. The bindingenergies for ethane and propane are 21.9 and 23.7 kJ/mol, respectively.
024709-8 Weaver et al. J. Chem. Phys. 132, 024709 �2010�

per molecule where each H–Pd bond may be classified as an�1 configuration. The total binding energy for this adsorbedgeometry is 27 kJ/mol. Because their total binding energiesare similar, the DFT results suggest that the CH2 �2�H,H�and �1�2H� configurations will be nearly equally populated.The similar binding energies also indicate that each �1 H–Pdinteraction is weaker than the bonding achieved in the CH2
�2�H,H� configuration. However, the C–H bonds stretch bysimilar amounts �2.2% versus 2.4%� in the CH2 �2�H,H�and �1�2H� configurations, respectively, indicating that thesecondary C–H bonds in the �2�H,H� configuration are per-turbed to a similar extent as the primary C–H bonds involvedin the �1�2H� interaction. An implication is that C–H bondsoftening resulting from the molecule-surface interaction hasa negligible effect on the selectivity of propane C–H bondactivation on PdO�101�.
G. Comparison between experiment and computation
A comparison of the experimentally and computationallydetermined binding energies indicates that the dispersion in-teraction contributes significantly to the alkane-PdO�101� in-teraction, accounting for between 50% and 70% of the total
binding energy for the small alkanes studied here. An impli-cation is that both the dispersion interaction and dative bond-ing are important in determining the optimum binding con-figurations, and hence that the actual configurations maydeviate from those reported here. However, even though thedispersion interaction contributes significantly to the alkanebinding energies, its more diffuse character compared withthe dative interaction may cause only minor perturbationsfrom the optimal configurations determined using DFT. Thegood agreement between the DFT predictions and the experi-mental results generally supports this idea.
The DFT results show that the strength of the coordinatebonding increases only weakly with increasing chain lengthfor the alkanes studied here. In fact, electrostatic interactionsbetween spectator groups and the surface atoms appear to beresponsible for the slight increases that are predicted. Thistrend agrees with the experimental results which suggest anearly constant enhancement in the binding energies ofmethane, ethane, and propane on PdO�101� versus Pd�111�.Furthermore, the DFT-predicted binding energies are similarin magnitude to the differences in alkane binding energies onPdO�101� versus Pd�111� that we estimated from the experi-mental TPD data. Thus, the DFT calculations support theconclusion that the formation of �-complexes is responsiblefor the stronger binding of alkanes on PdO�101� comparedwith Pd�111�, and that the strength of this bonding is similarfor methane, ethane, and propane.
V. SUMMARY
We investigated the molecular adsorption of methane,ethane, and propane on a PdO�101� thin film using TPD andDFT calculations. The TPD data show that alkanes adsorbedin the �1 state on PdO�101� have higher binding energiesthan alkanes physisorbed on Pd�111�. Based on an analysisof the TPD spectra using limiting desorption prefactors, weestimate that the alkane binding energies on PdO�101� in-crease linearly with increasing chain length up to N=3, butthat the E versus N relation has a nonzero intercept betweenabout 22 and 26 kJ/mol. We attribute this constant offset to adative bonding interaction that is similar in magnitude foreach alkane, and suggest that the strength of the dispersioninteraction with PdO�101� increases with alkane chain lengthfor the molecules studied. We estimate that the formation of�-complexes accounts for about 30%–50% of the total bind-ing energy of these small alkanes adsorbed on PdO�101�.
DFT calculations predict that the alkanes bind onPdO�101� by forming coordinate bonds with cus-Pd atoms.The resulting adsorbed species are analogous to alkane�-complexes in that the alkane-Pd coordination involveselectron donation from C–H � bonds to the Pd center as wellas backdonation from Pd d-states into localized antibondingstates. According to DFT, methane and ethane achieve maxi-mum binding energies on PdO�101� by adopting an �2�H,H�configuration on top of a cus-Pd atom. Propane also bindsfavorably in the �2�H,H� configuration through the CH2
group, and can attain a comparable binding energy by ad-sorbing in an �1�2H� configuration along the cus-Pd row.The binding energies for alkanes in the �2 configuration lie
FIG. 8. Top and side view of the �1�2H� configuration of C3H8 adsorbed onPdO�101� determined by DFT. The binding energy of this configuration is27.0 kJ/mol.
024709-9 Alkane �-complexes on PdO�101� J. Chem. Phys. 132, 024709 �2010�

in a range from about 16–24 kJ/mol, which agrees well withthe constant offsets estimated from TPD data. Because�-complex formation weakens alkane C–H bonds, the DFTcalculations suggest that the molecule-surface interactionmay assist in the initial activation of C–H bonds onPdO�101�. This finding is consistent with recent evidencethat the strongly bound molecular state of propane onPdO�101� serves as the precursor to initial bond cleavage.14
Efforts are currently underway to study how surface modifi-cations influence the coordinate bonding of alkanes onPdO�101� and the barriers for C–H bond cleavage.
ACKNOWLEDGMENTS
We gratefully acknowledge financial support for thiswork provided by the Department of Energy, Office of BasicEnergy Sciences, Catalysis Science Division through GrantNo. DE-FG02-03ER15478. We acknowledge the Universityof Florida High-Performance Computing Center �http://hpc.ufl.edu� for providing computational resources for per-forming some of the calculations reported in this paper.
1 J. F. Weaver, A. F. Carlsson, and R. J. Madix, Surf. Sci. Rep. 50, 107�2003�.
2 R. B. Anderson, K. C. Stein, J. J. Feenan, and L. J. E. Hofer, Ind. Eng.Chem. 53, 809 �1961�.
3 R. J. Farrauto, M. C. Hobson, T. Kennelly, and E. M. Waterman, Appl.Catal., A 81, 227 �1992�.
4 R. J. Farrauto, J. K. Lampert, M. C. Hobson, and E. M. Waterman, Appl.Catal. B 6, 263 �1995�.
5 R. Burch and F. J. Urbano, Appl. Catal., A 124, 121 �1995�.6 R. Burch, F. J. Urbano, and P. K. Loader, Appl. Catal., A 123, 173�1995�.
7 J. G. McCarty, Catal. Today 26, 283 �1995�.8 J. N. Carstens, S. C. Su, and A. T. Bell, J. Catal. 176, 136 �1998�.9 A. K. Datye, J. Bravo, T. R. Nelson, P. Atanasova, M. Lyubovsky, and L.Pfefferle, Appl. Catal., A 198, 179 �2000�.
10 P. Salomonsson, S. Johansson, and B. Kasemo, Catal. Lett. 33, 1 �1995�.11 R. S. Monteiro, D. Zemlyanov, J. M. Storey, and F. H. Ribeiro, J. Catal.
199, 291 �2001�.12 H. Gabasch, K. Hayek, B. Kloetzer, W. Unterberger, E. Kleimenov, D.
Teschner, S. Zafeiratos, M. Haevecker, A. Knop-Gericke, R. Schloegl, B.Aszalos-Kiss, and D. Zemlyanov, J. Phys. Chem. C 111, 7957 �2007�.
13 C. F. Cullis and B. M. Willatt, J. Catal. 83, 267 �1983�.14 J. F. Weaver, S. P. Devarajan, and C. Hakanoglu, J. Phys. Chem. C 113,
9773 �2009�.15 C. Hall and R. N. Perutz, Chem. Rev. 96, 3125 �1996�.16 A. E. Shilov and G. B. Shul’pin, Chem. Rev. 97, 2879 �1997�.17 J. A. Labinger and J. E. Bercaw, Nature �London� 417, 507 �2002�.18 J. H. Wang, C. Y. Fan, Q. Sun, K. Reuter, K. Jacobi, M. Scheffler, and G.
Ertl, Angew. Chem., Int. Ed. 42, 2151 �2003�.19 Q. Sun, K. Reuter, and M. Scheffler, Phys. Rev. B 70, 235402 �2004�.
20 M. Blanco-Rey, D. J. Wales, and S. J. Jenkins, J. Phys. Chem. C 113,16757 �2009�.
21 J. M. Hawkins, C. Hakanoglu, J. F. Weaver, and A. Asthagiri, in prepa-ration.
22 J. E. Demuth, H. Ibach, and S. Lehwald, Phys. Rev. Lett. 40, 1044�1978�.
23 F. M. Hoffmann, T. E. Felter, P. A. Thiel, and W. H. Weinberg, Surf. Sci.130, 173 �1983�.
24 F. M. Hoffmann and T. H. Upton, J. Phys. Chem. 88, 6209 �1984�.25 N. R. Avery, Surf. Sci. 163, 357 �1985�.26 R. Raval and M. A. Chesters, Surf. Sci. 219, L505 �1989�.27 S. Lehwald and H. Ibach, Surf. Sci. 89, 425 �1979�.28 M. K. Weldon, P. Uvdal, C. M. Friend, and B. C. Wiegand, Surf. Sci.
355, 71 �1996�.29 K. A. Fosser, R. G. Nuzzo, P. S. Bagus, and C. Woll, Angew. Chem., Int.
Ed. 41, 1735 �2002�.30 K. A. Fosser, R. G. Nuzzo, P. S. Bagus, and C. Woll, J. Chem. Phys. 118,
5115 �2003�.31 M. J. Hostetler, W. L. Manner, R. G. Nuzzo, and G. S. Girolami, J. Phys.
Chem. 99, 15269 �1995�.32 S. L. Tait, Z. Dohnalek, C. T. Campbell, and B. D. Kay, J. Chem. Phys.
122, 164708 �2005�.33 S. L. Tait, Z. Dohnalek, C. T. Campbell, and B. D. Kay, J. Chem. Phys.
125, 234308 �2006�.34 K. A. Fichthorn and R. A. Miron, Phys. Rev. Lett. 89, 196103 �2002�.35 K. E. Becker and K. A. Fichthorn, J. Chem. Phys. 125, 184706 �2006�.36 A. L. Gerrard, J.-J. Chen, and J. F. Weaver, J. Phys. Chem. B 109, 8017
�2005�.37 H. H. Kan, R. B. Shumbera, and J. F. Weaver, J. Chem. Phys. 126,
134704 �2007�.38 H. H. Kan, R. B. Shumbera, and J. F. Weaver, Surf. Sci. 602, 1337
�2008�.39 H. H. Kan and J. F. Weaver, Surf. Sci. 602, L53 �2008�.40 H. H. Kan and J. F. Weaver, Surf. Sci. 603, 2671 �2009�.41 A. F. Carlsson and R. J. Madix, J. Phys. Chem. B 104, 12237 �2000�.42 C. L. Kao and R. J. Madix, J. Phys. Chem. B 106, 8248 �2002�.43 G. Kresse and J. Furthmuller, Comput. Mater. Sci. 6, 15 �1996�.44 G. Kresse and J. Furthmuller, Phys. Rev. B 54, 11169 �1996�.45 G. Kresse and J. Hafner, Phys. Rev. B 47, 558 �1993�.46 G. Kresse and J. Hafner, Phys. Rev. B 49, 14251 �1994�.47 G. Kresse and D. Joubert, Phys. Rev. B 59, 1758 �1999�.48 P. E. Blöchl, Phys. Rev. B 50, 17953 �1994�.49 J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77, 3865
�1996�.50 W. Kohn, Y. Meir, and D. E. Makarov, Phys. Rev. Lett. 80, 4153 �1998�.51 M. Methfessel and A. T. Paxton, Phys. Rev. B 40, 3616 �1989�.52 D. Sheppard, R. Terrell, and G. Henkelman, J. Chem. Phys. 128, 134106
�2008�.53 H. J. Monkhorst and J. D. Pack, Phys. Rev. B 13, 5188 �1976�.54 H. H. Kan, R. C. Colmyer, A. Asthagiri, and J. F. Weaver, J. Phys. Chem.
C 113, 1495 �2009�.55 M. R. A. Blomberg, P. E. M. Siegbahn, and M. Svensson, J. Am. Chem.
Soc. 114, 6095 �1992�.56 M. R. A. Blomberg, P. E. M. Siegbahn, and M. Svensson, J. Phys. Chem.
98, 2062 �1994�.57 D. Y. Hwang and A. M. Mebel, J. Phys. Chem. A 106, 12072 �2002�.
024709-10 Weaver et al. J. Chem. Phys. 132, 024709 �2010�
Copyright © 2022 FDOKUMEN


![Synthesis of Trifluoromethyl-Substituted 3-Azabicyclo[ n .1.0]alkanes: Advanced Building Blocks for Drug Discovery](https://static.fdokumen.com/doc/165x107/63379323d102fae1b6076eda/synthesis-of-trifluoromethyl-substituted-3-azabicyclo-n-10alkanes-advanced.jpg)











![Prime Submodules and Local Gabriel Correspondence in σ[ M ]](https://static.fdokumen.com/doc/165x107/632520837fd2bfd0cb034c8c/prime-submodules-and-local-gabriel-correspondence-in-s-m-.jpg)





