
MODELISATION DE L'EXTRACTION DU NITRATE DE ...
244
THESE de DOCTORAT de l'UNIVERSITE PARIS Spécialité: CHIMIE ANALYTIQUE présentée par Christian SOREL Pour obtenir le grade de DOCTEUR de l'UNIVERSITE PARIS VI MODELISATION DE L'EXTRACTION DU NITRATE DE CESIUM PAR UN CALIXARENE Application à la modélisation du transport à travers des membranes liquides supportées Soutenue le 12 décembre 1996 devant la commission: M. B. TREMILLON M. M.J.F. LEROY M. C. MADIC M. P. DANNUS M. G. DURAND M. C. POITRENAUD M. J. SARRAZIN Mme. N. SIMON Président Rapporteurs Examinateurs
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of MODELISATION DE L'EXTRACTION DU NITRATE DE ...

THESE de DOCTORAT de l'UNIVERSITE PARIS
Spécialité:CHIMIE ANALYTIQUE
présentée parChristian SOREL
Pour obtenir le grade de DOCTEUR de l'UNIVERSITE PARIS VI
MODELISATION DE L'EXTRACTION DU NITRATE DE CESIUMPAR UN CALIXARENE
Application à la modélisation du transport à traversdes membranes liquides supportées
Soutenue le 12 décembre 1996 devant la commission:
M. B. TREMILLON
M. M.J.F. LEROYM. C. MADIC
M. P. DANNUSM. G. DURANDM. C. POITRENAUDM. J. SARRAZINMme. N. SIMON
Président
Rapporteurs
Examinateurs

RESUME
Ce travail s'inscrit dans le cadre des études menées au CEA sur la décontaminationd'effluents liquides contenant des radioisotopes à vie longue. Une classe de moléculesorganiques appartenant à la famille des calixarènes s'est révélée très efficace pour extrairesélectivement le césium (dont l'isotope !35Cs possède une période de 2 300 000 années) desolutions aqueuses dont la composition simule celle des effluents à traiter. Devant lacomplexité des mécanismes d'extraction étudiés, une modélisation physico-chimique s'estavérée nécessaire. Le système pris en compte lors de notre étude comprend:
- une phase aqueuse concentrée en acide nitrique et/ou en nitrate de sodium,
- une phase organique composée du diluant 1,2-nitrophényloctyléther et du 1,3-diisopropoxy-calix[4]arène-couronne-6 à 10"2 mol.L'1.
La mise en jeu de solutions aqueuses concentrées nécessite la prise en compte desécarts à l'idéalité par le calcul des coefficients d'activité. Les différentes théories permettant lecalcul des écarts à l'idéalité dans les phases aqueuses et organiques sont présentées dans lapremière partie.
Le calcul du coefficient d'activité des nitrates de césium et de sodium dans desmatrices très concentrées a nécessité un travail théorique et expérimental important détaillédans la seconde partie. Le but de cette étude était de compléter les données thermodynamiquesde solutions aqueuses de CsNO3 et de NaNO3. Nous avons également présenté les outilsinformatiques élaborés pour atteindre notre objectif de modélisation.
Dans un troisième temps, la mise en œuvre de ces outils théoriques et informatiquesnous a permis de déterminer la stœchiométrie des espèces extraites en phase organique ainsique les constantes associées. La modélisation des isothermes d'extraction de l'eau, de l'acide etdes différents nitrates alcalins par une solution organique du diluant seul puis du calixarène anécessité la prise en compte d'un grand nombre de complexes. La validité d'une tellereprésentation a été ensuite éprouvée dans des conditions très variées de salinité et d'acidité.
La technique des membranes liquides supportées (MLS) est une méthode originale deséparation par extraction liquide-liquide. Dans la dernière partie, nous avons développé unmodèle de transport adapté aux caractéristiques propres à notre système. La mise en équations'appuie sur les connaissances acquises lors de la modélisation des systèmes d'extraction. Lemodèle proposé permet de décrire les phénomènes de transport à travers les MLS dès lors quela représentation proposée de la phase organique est satisfaisante.

THESE de DOCTORAT de 1TJNIVERSITE PARIS VI
Spécialité:CHIMIE ANALYTIQUE
présentée parChristian SOREL
Pour obtenir le grade de DOCTEUR de l'UNIVERSITE PARIS VI
MODELISATION DE L'EXTRACTION DU NITRATE DE CESIUMPAR UN CALIXARENE
Application à la modélisation du transport à traversdes membranes liquides supportées
Soutenue le 12 décembre 1996 devant la commission:
M. B. TREMILLON Président
M. M.J.F. LEROY RapporteursM. C. MADIC
M. P. DANNUSM. G. DURANDM. C. POITRENAUD ExaminateursM. J. SARRAZINMme. N. SIMON

AVANT-PROPOS
Ces travaux ont été réalisés au Commissariat à l'Energie Atomique, dans leslaboratoires de l'Unité d'Enseignement Chimie et Cycle du Combustible de l'INSTN-SACLAY,et ceux du Laboratoire des Procédés de Traitement des Effluents du CEA-CADARACHE.
Monsieur le Professeur Claude POITRENAUD a dirigé mes travaux de recherchependant ces trois années. La qualité de son encadrement a contribué pour une large part à laréalisation de cette thèse. Je tiens à lui exprimer toute ma gratitude pour la confiance dont ila bien voulu faire preuve à mon égard.
Nicole SIMON et Pascal DANNUS ont assuré le suivi quotidien tout au long dudéroulement de ce travail. J'ai pu apprécier leurs compétences, leurs disponibilités et leursqualités humaines durant ces trois années. Je leur en suis profondément reconnaissant.
Monsieur le Professeur B. TREMILLON a accepté de faire partie de ce jury, c'est pourmoi un grand honneur. Qu'il me soit également permis de lui exprimer toute mon admiration.
Je suis très sensible à l'honneur que me font Messieurs les Professeurs M. LEROY etC. MADIC d'avoir bien voulu juger ce travail.
Messieurs les Professeurs G. DURAND et J. SARRAZIN se sont intéressés à mestravaux, je les remercie de cet intérêt.
Je garderai un très bon souvenir des personnes qui m'ont accueilli au sein de l'INSTNet, pour n'en citer qu'une, Nadia NOWACKIpour sa gentillesse et son efficacité.
Monsieur J.F. DOZOL m'a accueilli au sein de son équipe, je le remercie ainsi queMademoiselle V. LAMARE qui a participé au suivi de cette thèse.
Durant ma présence au sein du Service d'Etude des Procédés, j'ai apprécié l'ambiancede travail avec l'ensemble des doctor ants. La gentillesse de Nathalie REYNIER et de LaetitiaDELMAU, ainsi que l'amitié de Christophe BRESSOT et de Philippe BEUDAERT resterontpour moi un agréable souvenir. J'associe bien-sûr à ces remerciements l'équipe de techniciensqui m'ont été d'une aide précieuse. Je retiendrai, en particulier, les conseils prodigués parSerge EYMARD et nos discussions enrichissantes.

Sommaire
INTRODUCTION GENERALE.
1. La gestion des déchets radioactifs. 11
2. Le développement d'extractants spécifiques du césium. 14
3. La mise en œuvre des membranes liquides supportées. 19
CHAPITRE 1TRAITEMENT DES EFFETS DE MILIEU DANS LES SYSTEMES
D'EXTRACTION LIQUIDE-LIQUIDE.
Introduction. 27
1. Rappels thermodynamiques. 28
1.1. Potentiel chimique. 28
1.2. Activité et état standard. 29
1.3. Coefficient d'activité et comportement de référence. 30
1.4. Coefficient d'activité des electrolytes. 33
2. Les deux approches possibles de calcul des coefficients d'activité. 36
2.1. L'approche analytique. 36
2.2. L'approche thermodynamique. 39
3. Calcul des coefficients d'activité et de l'activité d'eau dans les solutions aqueuses. 40
3.1. Solutions binaires (électrolyte/eau). 40
3.2. Mélanges d'electrolytes. 423.2.1. L'approche thermodynamique de McKAY-PERRING. 42
3.2.2. L'apport du comportement dit "simple" au calcul du coefficient d'activité. 44
4. Calcul des coefficients d'activité dans les solutions organiques. 46
4.1. La présence d'eau en phase organique. 46
4.2. La relation de SERGIEVSKII-DANNUS. 46
Conclusion. 49
CHAPITRE 2ACQUISITION DES OUTILS DE BASE NECESSAIRES A LA MODELISATION DU
SYSTEME D'EXTRACTION HNO3/NaNO3/CsNO3/H2O | Calixarène/NPOE.Introduction. 57
1. Etude critique et comparative de différents modèles d'écart à l'idéalité dans le butd'acquérir les données binaires de solutions aqueuses sursaturées fictives. 58
1.1. Présentation de la problématique. 59

Sommaire
1.2. L'approche mathématique. 601.2.1. Ajustement polynomial. 601.2.2. Méthode "curvilinéaire". 611.2.3 Méthode de HIDALGO et ORR. 62
13. L'approche physico-chimique théorique. 631.3.1. Les modèles de BROMLEY et de PITZER. 65
1.3.1.1. Le modèle de BROMLEY. 671.3.1.2. Le modèle de PITZER. 69
1.3.2. Le modèle de CHEN. 741.3.3. Etude du comportement "en extrapolation" des modèles de PITZER et
de CHEN. 791.3.3.1. Comportement "en extrapolation" de la relation de PITZER. 811.3.3.2. Comportement "en extrapolation" de la relation de CHEN. 83
1.4. L'approche physico-chimique expérimentale. 841.4.1. La méthode de KUSSIK-MEISSNER-TESTER. 841.4.2. La méthode de ZDANOVSKII-STOCKES-ROBINSON. 90
1.5. Comparaison de la méthode "ZSR inverse" et de l'extrapolation de la relationde CHEN. 93
1.6. Acquisition des données binaires sursaturées fictives de NaNO3 et de CsNO3. 941.6.1. Mise en œuvre expérimentale de la relation "ZSR inverse". 951.6.2. Extrapolation de la relation de CHEN. 97
2. Quantification des systèmes d'extraction de type solvatant. 100
2.1. Mise en équation des équilibres d'extraction. 100
2.2. Le logiciel "EXTRACTION". 1032.2.1 Saisie des données nécessaires à l'optimisation. 1032.2.2. Optimisation des paramètres du système. 104
2.2.3. Présentation des résultats. 104
2.3. Le logiciel "PREVISION". 106
Conclusion. 109
CHAPITRE 3MODELISATION PHYSICO-CHIMIQUE DU SYSTEME D'EXTRACTION
HNO3/NaNO3/CsNO3/H2O | Calixarène/NPOE
Introduction. 117
1. Conditions expérimentales. 118
1.1. Préparation des solutions aqueuses. 118
1.2. Préparation des solutions organiques. 118
1.3. Techniques analytiques. 1191.3.1. Mesure de la masse volumique. 119

Sommaire
13.2. Dosages d'eau par Karl FISCHER. 1191.3.3. Dosages acide-base. 1191.3.4. Chromatographie ionique. 1191.3.5. Spectrométrie gamma. 120
1.4. Manipulations d'extraction liquide-liquide. 120
2. Résultats 121
2.1. Extraction de l'acide nitrique et de l'eau 1212.1.1. Extraction de l'eau par le NPOE. 1212.1.2. Extraction de l'eau par le calixarène dilué dans le NPOE. 1232.1.3. Extraction de l'acide nitrique par le NPOE. 1252.1.4. Extraction de l'acide nitrique par le calixarène dilué dans le NPOE. 129
2.2. Extraction des nitrate alcalins 1342.2.1. Extraction du nitrate de sodium par le calixarène dilué dans le NPOE. 1342.2.2. Extraction du nitrate de césium par le calixarène dilué dans le NPOE. 1392.2.3. Extraction du nitrate de potassium et de rubidium par le calixarène dilué
dans le NPOE. 1432.2.4. Système NaNOJCsNO^/H.O 1 Calixarène/NPOE. 145
2.3. Système HNCyCsNOj/HzO | Calixarène/NPOE. 146
3. Prévision de l'extraction du nitrate de césium à partir de matrices concentrées. 150
3.1. Prévision de l'extraction du nitrate de césium à partir de solutions fortementsalines. 152
3.2. Prévision de l'extraction du nitrate de césium à partir de solutions fortementacides. 156
3.3. Prévision de l'extraction du nitrate de césium à partir de solutions
concentrées en acide et en sels. 160
Conclusion. 163
CHAPITRE 4MODELISATION DU TRANSPORT A TRAVERS DES MEMBRANES LIQUIDES
SUPPORTEES
Introduction. 169
1. Techniques et mécanismes de séparation membranaire. 170
1.1. Membranes liquides supportées. 1711.1.1. Caractéristiques du support. 1711.1.2. Caractéristiques du diluant. 172
1.2. Mécanismes du transport à travers les membranes liquides. 1721.2.1. Transports passifs. 1721.2.2. Transports actifs. 173

Sommaire
2. Les principaux modèles de transport facilité. 175
2.1. Caractéristiques du système à modéliser. 176
2.2. Présentation des principaux modèles de transport facilité. 1762.2.1. Le modèle de WARP et ses développements. 1762.2.2. Le modèle de DANESI. 1792.2.3. Le modèle de REINHOUDT. 183
3. Proposition d'un modèle de transport facilité. 187
4. Conditions expérimentales. 191
4.1. Préparation des solutions et techniques analytiques. 191
4.2. Manipulations de transport. 1914.2.1. Dispositif expérimental. 191
4.2.2. Protocole expérimental. 192
5. Résultats. 194
5.1. Transport de l'acide nitrique par le NPOE. 194
5.2. Transport de l'acide nitrique par le calixarène dilué dans le NPOE. 196
5.3. Transport du nitrate de sodium par le calixarène dilué dans le NPOE. 198
5.4. Transport du nitrate de césium par le calixarène dilué dans le NPOE. 200
6. Prévision du transport du nitrate de césium à partir de matrices concentrées. 2036.1. Prévision du transport du nitrate de césium à partir de solutions fortement
salines. 203
6.2. Prévision du transport du nitrate de césium à partir de solutions concentréesen nitrate de sodium et en acide nitrique. 204
6.3. Prévision du transport du nitrate de césium à partir de solutions concentrées
en acide nitrique. 207
Conclusion. 209
CONCLUSION GENERALE. 215
ANNEXES 217
Annexe 1. 219
Annexe 2. 221
Annexe 3. 233
Annexe 4. 239

Liste des figures
LISTE DES FIGURES.
INTRODUCTION GENERALE.
Figure 1: Cycle simplifié du combustible nucléaire.Figure 2a: Tert-butyl-benzo-21-couronne-7.Figure 2b: Décyl-benzo-21-couronne-?.Figure 3a: Bis(hydroxyheptyl-benzo)-18-couronne-6.Figure 3b: Bis(tert-butyl-benzo)-24-couronne-8.Figure 4: Formule générale d'un calixarène.Figure S: Les différentes conformations des calix[4]arènes parents.Figure 6: l,3-calix[4]arène-couronne-6.Figure 7: l,3-calix[4]arène-bis-couronne-6.Figure 8: 1,3-diisopropoxy calix[4]arène-couronne-6.Figure 9: 1,3-calix[4]arène-bis-couronne-6.Figure 10 1,2-nitrophényloctyléther.
121515151516171818181819
CHAPITRE 1TRAITEMENT DES EFFETS DE MILIEU DANS LES SYSTEMES
D'EXTRACTION LIQUIDE-LQUIDE.
Figure 1-la: Ecart à l'idéalité positif.Figure 1-lb: Ecart à l'idéalité négatif.Figure l-2a: Activité de i (écart à l'idéalité positif).Figure l-2b: Activité de i (écart à l'idéalité négatif).Figure l-3a: Coefficient d'activité de i (écart à l'idéalité positif).Figure l-3b: Coefficient d'activité de i (écart à l'idéalité négatif).Figure 1-4: Représentation graphique de l'intégration de McKAY-PERRING.
31313232323243
CHAPITRE 2ACQUISITION DES OUTILS DE BASE NECESSAIRES A LA MODELISATION DU
SYSTEME D'EXTRACTION HNO3/NaNO3/CsNO3/H2O | Calixarène/NPOE.
Figure 2-1:Figure 2-2:Figure 2-3:Figure 2-4:Figure 2-5:Figure 2-6:Figure 2-7:Figure 2-8:Figure 2-9:Figure 2-10:Figure 2-11:Figure 2-12:
Extrapolation polynomiale de la variation de l'activité d'eau de NaNO3.Extrapolation curvilinéaire de la variation de l'activité d'eau de CsNO3.Méthode de HIDALGO-ORR appliquée au cas des nitrates alcalins.Variation du paramètre AB en fonction de la molalité pour RbNO3.Coefficient d'activité de NaNO3, NH4NO3, AgNO3, CsNO3.Variation du coefficient d'activité de 21 electrolytes.Abaques F ^ ^ I ) pour le domaine de force ionique 0-2 moLkg"1.Abaques F ^ ^ I ) pour le domaine de force ionique 0-20 moLkg"1.Abaques F^H^I) .Comparaison des relations de CHEN et "ZSR inverse".Activité d'eau de solutions binaires sursaturées fictives de NaNO3.Coefficient d'activité de solutions binaires sursaturées fictives de CsNO3.
606263678084868688949999

Liste des figures
CHAPITRE 3MODELISATION PHYSICO-CHIMIQUE DU SYSTEME D'EXTRACTION
HNO3/NaNO3/CsNO3/H2O | Calixarène/NPOE.
Figure 3-1: Isotherme d'extraction de l'eau par le NPOE à 25°C. 121Figure 3-2: Isotherme d'extraction de l'eau par le calixarène à trois concentrations différentes
dansleNPOEà25°C. 123
Figure 3-3: Variation des quantités d'eau extraites par le calixarène (à aH 0 = 1 ) en fonction
de sa concentration. 124Figure 3-4: Isothermes d'extraction de l'acide nitrique et de l'eau par le NPOE à 25°C 126Figure 3-5a: Isothermes expérimentales et calculées de l'acide nitrique et de l'eau (espèce
(HNO3)(NPOE)). 127Figure 3-5b: Isothermes expérimentales et calculées de l'acide nitrique et de l'eau (espèce
(HNO3)(H2O)(NPOE)). 127Figure 3-6: Isothermes expérimentales et calculées de l'acide nitrique et de l'eau (espèces
(HNO3)(NPOE) et (HNO3)(H2O)(NPOE)). 128Figure 3-7: Diagramme de répartition de l'acide entre les deux complexes retenus lorsde la
modélisation du système d'extraction HNO3/H2O | NPOE à 25°C 129Figure 3-8: Isothermes d'extraction de l'acide nitrique et de l'eau par le calixarène à 0,00769
moLkg-1 dilué dans le NPOE à 25°C. 130Figure 3-9: Isothermes d'extraction de l'acide nitrique et de l'eau par le calixarène seul à 25°C. 131Figure 3-10: Isothermes d'extraction expérimentales et calculées de l'acide nitrique et de l'eau par le
calixarène à 0,00769 mol.kg"1 à 25°C. 132Figure 3-11: Diagramme de répartition du calixarène en phase organique. 133Figure 3-12: Isothermes d'extraction du nitrate de sodium et de l'eau par le calixarène à 0,00769
mol.kg1 dilué dans le NPOE à 25°C. 134Figure 3-13: Double calix^Jarène-double-couronne-S. 136Figure 3-14: Isothermes d'extraction expérimentales et calculées du nitrate de sodium et de l'eau
par le calixarène à 0,00769 mol.kg' à 25°C (complexe (Calix)(NaNO3) ). 137Figure 3-15: Isothermes d'extraction expérimentales et calculées du nitrate de sodium et de l'eau
par le calixarène à 25°C (complexes (Calix)2 (NaNO3 ) (Calix)(NaNO3 ) ). 138Figure 3-16: Diagramme de répartition du calixarène en phase organique. 138Figure 3-17: Isothermes d'extraction du nitrate de césium et de l'eau par le calixarène à 0,00769
moLkg-1 dilué dans le NPOE à 25°C.Figure 3-18a: Isothermes d'extraction du nitrate de césium par le calixarène à 0,00769 moLkg"1 dilué
dans le NPOE.Figure 3-18b: Coefficient de distribution du nitrate de césium.Figure 3-19: Isothermes d'extraction expérimentales et calculées du nitrate de césium et de l'eau par
le calixarène à 0,00769 mol.kg1 dans le NPOE à 25°C.Figure 3-20: Diagramme de répartition du calixarène en phase organique.Figure 3-21: Isothermes d'extraction expérimentales et calculées du nitrate de potassium et de l'eau
par le calixarène à 0,00769 moLkg"1 dans le NPOE à 25°C.Figure 3-22: Isothermes d'extraction expérimentales et calculées du nitrate derubidium et de l'eau
par le calixarène à 0,00769 mol.kg"1 dans le NPOE à 25°C.Figure 3-23: Isothermes d'extraction expérimentales et calculées des nitrates de sodium et de césium
par le calixarène à 0,00769 mol.kg1 dans le NPOE à 25°C. 146Figure 3-24: Isothermes d'extraction de l'acide nitrique et de l'eau par le calixarène à 25°C (avec ou
sans césium saturant) par le calixarène à 0,00769 mol.kg1 dans le NPOE. 147Figure 3-25: Isothermes d'extraction expérimentales et calculées de l'acide nitrique et de l'eau à 25°C
(avec césium saturant) par le calixarène à 0,00769 mol.kg1 dans le NPOE. 148Figure 3-26: Prévision de l'extraction du nitrate de césium (10'5 mol.kg1) par le calixarène (0,00769
mol.kg'1) à partir de solutions concentrées en nitrate de sodium à 25°C. 152Figure 3-27: Calcul du coefficient d'activité et de l'activité à l'équilibre du nitrate de césium
(initialement à 10'5 moLkg"1) dans des solutions concentrées en nitrate de sodium. 153
139
141141
142143
144
144

Liste des figures
Figure 3-28: Diagramme de répartition du calixarène entre les différentes espèces présentes àl'équilibre en phase organique.
Figure 3-29: Prévision de l'extraction du nitrate de césium (10"5 mol.kg"') par le calixarène (0,00769moLkg"1) à partir de solutions concentrées en acide nitrique à 25°C.
Figure 3-30: Calcul du coefficient d'activité et de l'activité à l'équilibre du nitrate de césium(initialement à 10'5 mol.kg"') dans des solutions concentrées en acide nitrique.
Figure 3-31 : Diagramme de répartition du calixarène entre les différentes espèces présentes àl'équilibre en phase organique.
154
156
157
158
CHAPITRE 4MODELISATION DU TRANSPORT A TRAVERS DES MEMBRANES LIQUIDES
SUPPORTEES.
Figure 4-1:Figure 4-2:Figure 4-3a:Figure 4-3b:Figure 4-4a:Figure 4-4b:Figure 4-5:Figure 4-6:Figure 4-7:Figure 4-8:Figure 4-9:Figure 4-10:Figure 4-11:Figure 4-12:Figure 4-13:
Figure 4-14:
Figure 4-15:
Figure 4-16:
Figure 4-17:
Figure 4-18:
Figure 4-19:
Figure 4-20:
Figure 4-21:
Figure 4-22:
Figure 4-23:
Figure 4-24:
de
Membranes liquides épaisses et supportées.Schéma d'une membrane liquide émulsionnée.Mécanisme de transport simple.Mécanisme de transport facilité.Mécanisme de contre-transport.Mécanisme de co-transport.Mécanisme de transport couplé avec une transformation de l'extractant.Etapes successives du transport membranaire.Etapes considérées dans le modèle de WARD.Etapes considérées dans le modèle de DANES1.Mécanisme de transport proposé par STOLWIJK.Dispositif expérimental de transport par MLS.Transport de l'acide nitrique par le NPOE vers la solution de réception (t=25°C).Modélisation du transport de l'acide nitrique par le NPOE (t=25°C).Transport de l'acide nitrique par une solution organique contenant 0,00769 moLkg"1
calixarène dilué dans le NPOE (t=25°C).Modélisation du transport de l'acide nitrique par une solution organique contenant0,00769 mol.kg"1 de calixarène dilué dans le NPOE (t=25°C).Répartition de l'acide nitrique dans les solutions organiques membranaires à l'équilibred'extraction (t=25°C).Transport du nitrate de sodium par une solution organique contenant 0,00769 rn.ol.kg'1
de calixarène dilué dans le NPOE (t=25°C).Modélisation du transport du nitrate de sodium par une solution organique contenant0,00769 mol.kg"1 de calixarène dilué dans le NPOE (t=25°C).Transport du nitrate de césium par une solution organique contenant 0,00769 moLkg"1
de calixarène dilué dans le NPOE (t=25°C).Modélisation du transport du nitrate de césium par une solution organique contenant0,00769 mol.kg"1 de calixarène dilué dans le NPOE (t=25°C).Prévision du transport des nitrates de césium et de sodium à partir d'une solutioncontenant 3,5 moLL"1 deNàNO3 et 1,1.10"4 mol.L"1 de CsNO3.Prévision du transport de l'acide nitrique à partir d'une solution contenant 1 moLL"1
de HNO3, 4 mol.L"1 de NaNO3 et 10^ mol.L' de CsNO3.Prévision du transport des nitrates de césium et de sodium à partir d'une solutioncontenant 1 mol.L"1 de HNO3, 4 mol.L"1 de NaNO3 et 10"4 mol.L"' de CsNO3.Prévision du transport du nitrate de césium à partir d'une solution contenant 3 moLL"1
de HNO3 et 7,52.10"" mol.L"1 de CsNO3.Variation de la concentration de CsNO3 en phase organique membranaire sur les facesd'alimentation et de réception.
170171173173173173174175177180183192194195
196
197
198
199
200
201
202
204
205
206
207
208

Liste des tableaux
LISTE DES TABLEAUX.
CHAPITRE 1TRAITEMENT DES EFFETS DE MILIEU DANS LES SYSTEMES D'EXTRACTION
LIQUIDE-LIQUIDE.
Tableau 1-1: Principales théories des solutions aqueuses.Tableau 1-2: Principales théories des solutions organiques.
3738
CHAPITRE 2ACQUISITION DES OUTILS DE BASE NECESSAIRES A LA MODELISATION DU
SYSTEME D'EXTRACTION HNO^aNO^ CsNO3/H2O | Calixarène/NPOE.
Tableau 2-1:Tableau 2-2:Tableau 2-3:Tableau 2-4:Tableau 2-5:Tableau 2-6:
Traitement des interactions dans les deux cellules.Mélanges NaNO3/LiNO3/H2O (1èr6 série).Mélanges NaNO3/LiNO3/H2O (2*"" série).Mélanges NaNO3/NH4NO3/H2O (3èm* série).Paramètres de la relation de CHEN pour NaNO3 et CsNO3.Les différentes combinaisons des coefficients stœchiométriques.
97
779696
98101
CHAPITRE 3MODELISATION PHYSICO-CHIMIQUE DU SYSTEME D'EXTRACTION
HNO3/NaNO3/ CsNO3/H2O | Calixarène/NPOE.
Tableau 3-1: Paramètres optimisés pour les complexes acides. 132Tableau 3-2: Paramètres optimisés pour les complexes du potassium et du rubidium. 144Tableau 3-3: Paramètres optimisés pour les deux complexes mixtes. 148Tableau 3-4: Caractéristiques des effluents à traiter. 150Tableau 3-5: Simulation de la décontamination d'un effluent contenant initialement 5.10"6 mol.kg"' de
césium et de 1 à 5 mol.L'1 de nitrate de sodium. 155Tableau 3-6: Simulation de la décontamination d'un effluent contenant initialement 5.10"3 mol.kg"1 de
césium et 3 mol.L1 d'acide nitrique par une solution de calixarène à 0,00769 mol.kg"1. 159Tableau 3-7: Prévision de l'extraction du nitrate de césium (10"5 mol.kg"1) par le calixarène (0,00769
moLkg"1) à partir de matrices concentrées en acide nitrique et en nitrate de sodium.Tableau 3-8: Répartition du calixarène (en %) entre les espèces présentes en phase organique lors de
l'extraction du nitrate de césium (10'5 mol.kg"') à partir de matrices concentrées enacide nitrique et en nitrate de sodium.
Tableau 3-9: Simulation de la décontamination par le calixarène à 0,00769 moLkg' d'un effluentcontenant initialement 5.10"* mol.kg'1 de césium, 1 mol.L' d'acide nitrique et 2,5 à 4mol.L"1 de nitrate de sodium. 162
160
161

INTRODUCTION GENERALE

Introduction générale
1. La gestion des déchets radioactifs.
L'industrie nucléaire française assure pour plus de 75% la production d'énergieélectrique, ce qui constitue la plus forte proportion parmi les grands pays nucléarisés. Al'heure actuelle, le combustible nucléaire est constitué d'oxyde d'uranium enrichi en moyenneà 3,5% (combustible UOX) et peut également contenir une faible proportion d'oxyde deplutonium (combustible MOX: Mixed OXydes). Après trois années d'irradiation dans unréacteur élecîronuciéaire, ce combustible s'est transformé et contient [1]:
- 95,5% d'uranium,
- 1 % de plutonium,
- 3,4% de produits de fission,
- 0,1% d'actinides mineurs.
Le combustible usé est ainsi constitué de deux classes d'éléments: les actinides et les produitsde fission. Les actinides sont des radioéléments, principalement émetteurs alpha, caractériséspar leur longue durée de vie. Les plus abondants sont l'uranium et le plutonium. D'autres sonten plus faible quantité, d'où leur dénomination d'actinides mineurs: il s'agit du neptunium, del'américium et du curium. Les produits de fission (PF) contiennent des radioélémentsémetteurs bêta et gamma dont la majorité possèdent une durée de vie courte. Ils représentent99% de la radioactivité du combustible.
L'uranium et le plutonium sont potentiellement recyclables en vue de la fabrication denouveaux assemblages combustibles. C'est une des raisons pour lesquelles la France a optépour une politique de retraitement, plutôt que pour le stockage direct du combustible aprèsdéchargement du cœur du réacteur.
Pour le moment, aucune valorisation des produits de fission et des actinides mineurs n'estenvisagée, ils constituent donc un déchet nucléaire.
Les opérations de retraitement du combustible irradié mettent en œuvre des procédéshydrométallurgiques fondés sur l'extraction liquide-liquide. Après extraction de l'uranium etdu plutonium selon le procédé PUREX (Plutonium Uranium Refining by Extraction),l'effluent aqueux contient l'essentiel des PF et des actinides mineurs. Cette solution est ensuitevitrifiée.
Le cycle du combustible nucléaire que nous avons décrit jusqu'à présent peut être résumé parla figure 1 :
11

Introduction générale
Unnium aoumvri k.
IXh **. Oiom/e d'uranium A
duranyie
Figure 1: Cycle sii du combustible nucléaire [2].
Les verres issus du retraitement du combustible nucléaire ne sont pas les seuls déchetsradioactifs produits. D'autres résidus non utilisables sont générés par l'industrie nucléaire lorsde l'exploitation des centrales et des usines de retraitement ou, plus généralement, lors del'utilisation de radioéléments dans l'industrie, la recherche et la santé. Une classification trèsprécise de l'ensemble de ces déchets a été établie par l'Agence Nationale pour la gestion desDéchets Radioactifs (ANDRA) [3], Trois catégories différentes ont ainsi été définies:
Catégorie A: Déchets contenant des éléments à vie courte (émetteurs bêta et gamma) dont lapériode n'excède pas trente ans. L'activité massique ne doit pas dépasser 3,7GBq.f ' en émetteurs alpha au bout de 300 ans et 4,8 GBq.t*1 en émetteurs bêta-gamma à l'arrivée des colis. Ces déchets proviennent surtout de l'exploitationdes centrales nucléaires. Ils sont conditionnés sous la forme de conteneursbétons et entreposés en surface dans les centres de stockage de la Manche et del'Aube. Leur production annuelle est de 30 000 m3 par an.
Catégorie B: Déchets de moyenne activité contenant peu d'éléments à vie longue. Leurstockage est inacceptable en surface du fait de la présence d'actinides et de PF àvie longue (**Tc et iï5Cs), ou bien de PF irradiants comme le ^Sr et le 137Cs.Leur production, provient essentiellement des usines de retraitement et defabrication de combustibles, elle correspond à un volume annuel de 3000 m3
soit un volume cumulé de 88 000 m3 en l'an 2000.
Catégorie C: Déchets de haute activité chargés en éléments à vie longue (émetteurs alpha) etpossédant un haut pouvoir calorifique. Ces déchets correspondent aux verresissus du retraitement du combustible irradié. Ils représentent un volume annuelde 200 m3 soit un volume cumulé de 2000 m3 en l'an 2000.
12

Introduction générale
Etant donnée la très longue durée de vie des radioéléments contenus dans les déchetsde moyenne et haute activité, leur stockage pose un véritable problème de société. C'estpourquoi, le gouvernement a légiféré le 30 décembre 1991 en prévoyant d'orienter lesrecherches selon trois axes [4]:
© Recherche de solutions permettant la séparation et la transmutation (réduction de lapériode radioactive) des éléments radioactifs à vie longue présents dans les déchets.
© Etude des possibilités de stockage réversible ou irréversible dans les formationsgéologiques profondes, notamment grâce à la réalisation de deux laboratoiressouterrains.
(D Etude de procédés de conditionnement et d'entreposage de longue durée en surface.
Pour répondre au premier axe de la loi, le CEA a lancé un programme de recherche désignésous l'acronyme SPIN (SéParation et INcinération). Les éléments à vie longue à séparer desdéchets sont les actinides et certains PF. Nous rappelons les périodes (en années) desprincipaux radioéléments à considérer:
Actinides:
Produits de fission:
Tu2 3 7Np2 4 1 Am2 4 4Cm
"TeI 3 5CsI29T
24 000,2 140 000,432,18,
213 000,2 300 000,15 700 000
C'est dans le cadre de la séparation du césium à partir d'effluents liquides de catégorieB et C que se situe notre travail de thèse.
Les déchets de catégorie B à décontaminer contiennent de faibles quantités d'éléments à vielongue (135Cs et actinides) et de PF (137Cs, 90Sr). La présence de ces radioéléments interdit toutstockage en surface et nécessite un entreposage en profondeur beaucoup plus onéreux. Lesétudes menées au laboratoire portent sur deux types d'effluents:
- Les concentrats d'évaporateurs de la Station de Traitement des Effluents Liquides deMARCOULE (STEL). Ils proviennent du retraitement du combustible usé de lafilière "Uranium Naturel Graphite Gaz". Ces effluents sont caractérisés par laprésence de fortes concentrations de nitrate de sodium (> 200 g.L"1 ou 2,5 M) etd'acide nitrique (> 1 M).
- Les effluents salins issus de la décontamination d'installations nucléaires. Cessolutions contiennent entre 100 et 400 g.L'1 de nitrate de sodium, leur pH est comprisentre 7 et 12.
Comme nous l'avons déjà vu, les effluents C issus directement du retraitement du combustibleirradié sont des déchets de haute activité chargés en éléments à vie longue et possédant un
13

Introduction générale
haut pouvoir calorifique. Après trente années de stockage, le césium 137 et le strontium 90contribuent pour 98% à l'énergie thermique dégagée et pour 97% au flux des radiationsémises. Dans une optique de stockage géologique à long terme, la présence de fortes quantitésde césium 135 est très pénalisante du fait de sa très grande période et de sa mobilitéimportante. L'extraction du césium permettrait donc de diminuer l'activité (contribution du137Cs) puis, à plus long terme, l'impact à l'exutoire des verres (contribution du 135Cs).
2. Le développement d'extractants spécifiques du césium.
Les caractéristiques propres à chacun des effluents que nous venons de présenterbrièvement impliquent des contraintes très strictes quant au choix et au développementd'extractants du césium.
En effet, la présence de fortes quantités de sodium et d'acide dans les effluents de catégorie Bnécessite une très grande sélectivité vis-à-vis du césium. La décontamination en césium dessolutions de produits de fission se heurte aux problèmes de résistance à la chaleur et à laradiolyse. Ces effluents de catégorie C sont également caractérisés par une acidité compriseentre 3 et 4 mol.L"1 en acide nitrique, ce qui implique un bon comportement de l'extractant visà vis de l'hydrolyse.
De telles contraintes ont nécessité le développement de molécules extradantes trèssophistiquées. Jusqu'en 1991, trois familles d'extractants avaient été testées: les éthers-couronne, les dicarbollides et les échangeurs minéraux, la classe des éthers-couronne étant deloin la plus étudiée.
© De bons résultats ont été obtenus par BLASIUS [5] lors de la décontaminationd'effluents acides légèrement salins par un mélange synergique contenant un dérivé dubisbenzo-21-couronne-7 associé à un hétéropolyanion (acide d'hexachlorure d'antimoine V).Des molécules voisines ont été étudiées au Service d'Etudes Procédés (CEA-CADARACHE)pour permettre la décontamination en césium et en strontium de concentrats d'évaporateurs[6]. Toutefois, les extractants les plus performants: tert-butyl-benzo-21-couronne-7 et décyl-benzo-21-couronne-7 (Figures 2a et 2b), ne possédaient pas une sélectivité Cs/Na suffisantepour permettre le traitement de solutions fortement salines.
14

Introduction générale
Figure 2a: Tert-butyl-benzo-21-couronne-7.
C10H21
Figure 2b: Décyl-benzo^l-couronne-T.
Des molécules extractantes de la classe des éthers-couronne ont été également mises en œuvreafin d'extraire le césium à partir de solutions de produits de fission fortement acides. Lesmeilleurs résultats ont été obtenus avec le bis(hydroxyheptyl-benzo)-18-couronne-6 [7] et lebis(tert-butyl-benzo)-24-couronne-8 [8] (Figures 3a et 3b). Les performances de l'extractantsont améliorées par la présence d'un acide organique (acide didodécylnaphtalènesulfonique:HDDNS) jouant le rôle d'un échangeur cationique et induisant une synergie.Toutefois, les performances de tels mélanges diminuent fortement avec l'acidité de la phaseaqueuse, ce qui limite leur application pratique.
C7H15OH
C7H15OH
Figure 3a: bis(hydroxyheptyl-benzo)-18-couronne-6.
Figure 3b: bis(tert-butyI-benzo)-24-couronne-8.
© Les bisdicarbollylcobaltates (dicarbollides) ont été développés afin d'extraire lecésium et le strontium de solutions acides de produits de fission [9]. Il s'agit de complexesanioniques du cobalt (par exemple le (7i-(3)-l,2-C2B9Hn)2Co") pouvant former des pairesd'ions dans des diluants organiques polaires comme le nitrobenzene. Comme dans le cas deséthers-couronne, les performances du système d'extraction diminuent lorsque l'acidité del'effluent aqueux dépasse 0,5 mol.L'1.
15

Introduction générale
(D La mise en œuvre d'échangeurs d'ions minéraux a été également envisagée afin deséparer le césium de solutions de produits de fission. L'élution sur une colonne contenant lephosphate de zirconium d'ammonium phosphotungstate (Zr-P-APW) a permis d'extraire entotalité le césium d'efïluents de haute activité d'acidité maximale de 2 mol.L'1 [10]. Unecolonne garnie de micro-sphères d'hexacyanoferrate de potassium-titane (KTiFC) a été mise àprofit pour décontaminer des solutions de haute activité contenant 58.g.L"' de sodium et 1mol.L"1 d'acide nitrique [11]. Un tel procédé de traitement génère un volume de déchetsimportant. Ces déchets correspondent au garnissage de la colonne après sorption ou bien à lasolution aqueuse de lavage après désorption.
Les recherches menées au sein du SEP sur la décontamination des concentratsd'évaporateurs en strontium et en césium par des éthers-couronne n'avaient pas atteint en 1992la totalité des objectifs fixés [12]. En effet, le facteur de décontamination obtenu pour lecésium restait faible du fait de l'extraction compétitive du sodium. C'est pourquoi, il a étédécidé de poursuivre la recherche d'extractants du césium plus spécifiques par rapport ausodium. Ce travail a fait l'objet d'une collaboration étroite entre le SEP et quatre laboratoiresuniversitaires européens (BELFAST, PARME, STRASBOURG, TWENTE) spécialisés dansla synthèse de macrocycles de la classe des calixarènes. Plusieurs dizaines de produitsdifférents ont ainsi été testées dans le cadre de la thèse de C. HILL [13].
Les calixarènes sont des macrocycles formés de 4 à 8 unités phénoliques reliées entre elles pardes ponts méthyléniques au niveau des positions ortho de la fonction hydroxyle (Figure 4).
Figure 4: Formule générale d'un calixarène.
La mobilité des unités phénoliques autour des groupes méthyléniques ponteurs permet à lamolécule d'adopter différentes conformations en solution. Pour les composés tétramères,quatre configurations différentes ont ainsi été mises en évidence [14], leur dénominationportée sur la figure 5 est attribuée à GUTSCHE [15] auquel on doit également le termegénérique de "calixarène".
16

Introduction générale
RO
cône partiel
RO
1,2- alte rné e 1,3- alte niée
Figure 5: Les différentes conformations des calix[4] arènes parents.
Les calixarènes les plus simples (calixarènes "parents") ne possèdent aucune propriétéextractante vis à vis du césium excepté à des pH élevés (> 11-12). Afin de permettrel'extraction du nitrate de césium de solutions simulant les concentrats d'évaporateurs (HNO3
1M et NaNO3 4M), une étude poussée a été menée sur la fonctionnalisation de la molécule,sur sa conformation ainsi que sur le nombre optimal d'unités phénoliques. Les calixarènesprésentant une bonne affinité et une sélectivité élevée pour le césium sont:
- composés de quatre unités phénoliques en conformation 1,3-alternée,
- fonctionnalisés par une ou deux couronnes étheroxyde comportant six atomesd'oxygène.
Les figures 6 et 7 réunissent les deux séries de calixarènes couronne retenues par C. HILL envue de la décontamination en césium des concentrats d'évaporateurs [16, 17].
17

Introduction générale
Figure 6: l,3-calix[4]arène-couronne-6. Figure 7: l,3-calix[4]arène-bis-couronne-6.
La faisabilité de l'extraction du césium par des calixarènes couronne à partir de solutions deproduits de fission a été démontrée plus récemment [18]. Les tests ont porté sur des raffînatsréels issus du retraitement d'un combustible MOX. Les molécules extradantes retenues lorsde ces manipulations étaient le 1,3-diisopropoxy calix[4]arène-couronne-6 et le calix[4]arène-bis-couronne-6 dont les formules sont présentées figures 8 et 9.
Figure 8: 1,3-diisopropoxycalix[4]arène-couronne-6.
Figure 9: l,3-calix[4]arène-bis-couronne-6.
Dans le cadre de notre travail de thèse, l'extractant retenu est le 1,3-diisopropoxycalix[4]arène-couronne-6. Ce choix a été guidé par trois critères principaux:
18

Introduction générale
- une solubilité relativement élevée de la molécule,
- un rendement d'extraction du césium de 95% à partir des concentrats d'évaporateurset de 90% pour les solutions de produits de fission,
- une sélectivité Cs/Na parmi les plus élevées mesurées jusqu'à présent (>28 500).
De plus, la disponibilité d'une assez grande quantité d'extractant permettait de mener à bien unnombre important d'expériences d'extraction liquide-liquide.
3. La mise en œuvre des membranes liquides supportées.
La technique des membranes liquides a été utilisée au SEP dans le cadre desrecherches sur la décontamination en césium des concentrats d'évaporateurs et des solutionsde produits de fission. Ce choix s'explique par les avantages que présentent ce dispositif parrapport à des techniques d'extraction liquide-liquide classiques, à savoir:
- la mise en œuvre de très faibles quantités de solutions organiques extractantes,
- la combinaison des étapes d'extraction et de désextraction.
Parmi les différents types de procédés membranaires, les membranes liquides supportées(MLS) ont été retenues. A l'échelle du laboratoire, ces membranes se présentent sous la formede disques plans de faible épaisseur préparés à partir de films polymériques utilisés enultrafiltration. La membrane est placée entre deux solutions aqueuses: une phased'alimentation contenant l'élément à extraire dans une matrice plus ou moins complexe et unephase de réception constituée d'eau déminéralisée.
La solution extradante est incorporée à l'intérieur des pores du support par simple trempage.Les différents paramètres influençant la stabilité de la membrane et assurant ainsil'immobilisation de la phase organique ont été longuement étudiés au sein de notre équipe[6,12]. Ces études ont débouché sur le choix d'un support et d'un diluant. Les filmspolymériques CELGARD 2500 ont été retenus comme support des MLS. Le diluantpermettant le meilleur compromis entre la solubilisation des calixarènes et la stabilité desmembranes appartient à la famille des 1,2-nitrophénylalkyléthers. Dans le cadre de notreétude, nous avons utilisé le 1,2-nitrophényloctyléther (NPOE) dont la formule est présentéesur la figure 10.
OC 8H 1 7
Figure 10: 1,2-nitrophényloctyléther.
19

Introduction générale
A l'échelle industrielle, les volumes de solutions à traiter étant plus importants, il estnécessaire d'augmenter la surface d'échange entre les phases d'alimentation et de réception.Pour ce faire, on fait appel à des assemblages compacts contenant des fibres creuses. La phaseorganique imprègne le support polyménque, une des phases aqueuses circule à l'intérieur de lafibre, alors que l'autre circule à l'extérieur. Un tel dispositif est intéressant économiquementdu fait de l'utilisation de faibles quantités d'extradants et d'une consommation d'énergieréduite.
Les études menées jusqu'à présent ont permis de démontrer l'efficacité élevée descalixarènes lors de l'extraction et du transport du césium à partir de solutions fortement salineset acides. Toutefois, la complexité du système a rendu impossible une compréhensioncomplète des différents phénomènes mis en jeu. En effet, il est nécessaire de prendre encompte un grand nombre d'équilibres correspondant à l'extraction simultanée de deux sels(NaNO3 et CsNO3) et de l'acide nitrique par le diluant et par le calixarène. De plus, les phasesaqueuses mises enjeu sont des solutions concentrées nécessitant la prise en compte des effetsde milieu par le calcul des coefficients d'activité.
C'est pourquoi, une modélisation physico-chimique du système complet s'avèrenécessaire. Il s'agit de déterminer le nombre et la stoechiométrie des différentes espècesformées en phase organique ainsi que leur constante d'extraction associée.
Pour assurer la validité d'une telle approche, il est nécessaire de prendre en compte les effetsde milieu dans les deux phases. L'objectif final est de fournir une représentation précise desdifférents phénomènes gouvernant l'extraction de faibles quantités de césium à partir dematrices concentrées en acide et/ou en sels.
Les renseignements qualitatifs et quantitatifs ainsi obtenus permettront de prévoir lesconditions optimales de traitement d'effluents issus du cycle du combustible nucléaire. Notredémarche de modélisation permettra alors d'apporter les premiers éléments de réponse auxbesoins pratiques de l'industriel.
20

Introduction générale
BIBLIOGRAPHIE.
[ 1 ] Commissariat à l'Energie Atomique,Informations utiles,Direction de la Communication, (1995).
[2] J. ROBERT,Gestionnaire des matières nucléaires: un métier du groupe COGEMA,COGEMA Magazine, 58, (février, mars, avril 1996), 40-42.
[3] Y. MARQUE,La gestion à long terme des déchets radioactifs du cycle du combustible,Communication personnelle, (1995).
[4] Loi n° 91-1381 du 30 décembre 1991 relative aux recherches sur la gestion des déchetsradioactifs.
[5] (a) E. BLASIUS, K.H. MLLES,The removal of Cesium from Medium-Active Waste Solutions, part I,Radiochimica Acta, 35, (1984), 173-182.
(b) E. BLASIUS, K.H. NILLES,The removal of Cesium from Medium-Active Waste Solutions, part II,Radiochimica Acta, 36, (1984), 207-214.
[6] J. CASAS i GARCIA,Décontamination sélective du césium et du strontium des concentrats d'évaporationprovenant des usines de retraitement de combustibles nucléaires avec des éthers-couronnes par transport à travers des membranes liquides supportées,Thèse de Doctorat de l'Université de BARCELONE, (1991).
[7] I.H. GEROW, M.W. DAVIS Jr,The Use of 24-Crown-8's in the Solvent Extraction of CsNO3 and Sr(NO3)2,Sep. Sci. Technol., 14, (1979), 395-414.
[8] W.J. Me DOWELL, G.N. CASE, J.A. DONOUGH, R.A. BARTSCH,Selective Extraction of Cesium from Acidic Nitrate Solutions with Didodecylnaphta-lenesulfonic Acid Synergized with Bis(tert-ButylBenzo)-21-crown-7,Anal. Chem., 64, (1992), 3013-3017.
[9] (a) J. RAIS, J. PLESEK, P. SELUCKY, M. KYRS, L. KADLECOVA,Extraction of cesium with Derivatives of Carborane into Nitrobenzene,J. of Radioanalytical andNucl. Chem., articles, 148, (1991), 349-357.
21

Introduction générale
(b) T.H. HA, J. RAIS, J. PLESEK, P. SELUCKY, M. KYRS,Extraction of Cs, Sr and Ba into Nitrobenzene in the Presence of Cobalt Dicarbollideand Mono-n-Dodecylethers of Polyethylene Glycol,Nucléon, 2, (1993), 6-9.
[10] V.N. REDDY, J. SATYANARAYANA, G.S. MURTY, A. DASH,Studies on the Separation of Cesium-137 from the Acidic Fission Product WasteSolutions on a new Complex Inorganic Exchanger (Zr-P-APW),J. Radioanal. Nucl. Chem., 183, (1994), 371-377.
[11] S. CHONGLI, X. SHIPING, S. YONGXIA, J. RONGZHOU,Separation of Cesium from Highly Saline HLLW with Potassium TitaniumHexacyanoferrate,Extented Abstracts, Vol. II, G-O7, 4th International Conference on Nuclear andRadiochemistry, S'Malo, FRANCE, (8-13 Septembre 1996).
[12] J.F. DOZOL, S. EYMARD, R. GAMBADE, G. La ROSA, J. CASA i GARCIA,Décontamination des concentrats d'évaporateurs en Cs, en Sr et transuraniens,Rapport EUR 13887 FR, (1992).
[13] CHILL,Application des calixarènes fonctionnalisés au traitement des effluents radioactifs parmembranes liquides supportées,Thèse de Doctorat de l'Université Louis PASTEUR de STRASBOURG, (1994).
[14] J.W. CORNFORTH, P. D'ARC Y HART, G. A. NICHOLLS, R.J.W. REES, J.A.STOCK,Antituberculous Effects of Certain Surface-Active polyoxyethylene Ethers,Br. J. Pharmacol., 10, (1955), 73-86.
[15] CD. GUTSCHE,Calixarènes,Royal Society of Chemistry, Thomas Graham House, Science park, CAMBRIDGE CB44WF, (1989).
[16] J.F. DOZOL, H. ROUQUETTE, R. UNGARO, A. CASNATI,Calix[4]arènes-couronnes, leur procédé de préparation et leur utilisation pourl'extraction sélective du césium et des actinides,Brevet français 93 04566, (1993).
[17] J.F. DOZOL, Z. ASFARI, C. HILL, J. VICENS,Calix [4] arènes-bis-couronnes, leur procédé de préparation et leur utilisation pourl'extraction sélective du césium et des actinides,Brevet français 92 14245, (1992).

Introduction générale
[18] J.F. DOZOL, N. SIMON, H. ROUQUETTE, S. EYMARD, B. TOURNOIS, V.LAMARE, M. LECOMTE, M. MASSON, C. VIALLESOUBRANNE,Extraction and Transport of Long Lived Radionuclides with Functionalized Calixarenes,GLOBAL, Vol. l,pp. 1024-1031, International Conference on Evaluation of EmergingNuclear Fuel Cycle Systems, Versailles, FRANCE, 11-14 septembre 1995.
23

CHAPITRE 1
TRAITEMENT DES EFFETS DE MILIEU DANS LESSYSTEMES D'EXTRACTION LIQUIDE-LIQUIDE.

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
Introduction.
La mise en équation des équilibres d'extraction liquide-liquide et, en particulier,récriture de la constante d'équilibre nécessitent en toute rigueur de raisonner dans les deuxphases en terme d'activité thermodynamique. Dans notre domaine qui est celui del'hydrométallurgie, la mise en œuvre de solutions complexes ne permet pas de fairel'approximation selon laquelle les activités sont prises égales aux concentrations.
Il est toujours possible de s'affranchir du calcul explicite des activitésthermodynamiques si l'on procède à un développement purement mathématique de laconstante d'extraction. Cette démarche totalement empirique aboutit à des relations souventtrès complexes où les coefficients ne possèdent aucun sens physico-chimique. De plus, lesrésultats obtenus ne renseignent absolument pas le chimiste sur la nature des espèces extraitesen phase organique.
En revanche, une approche physico-chimique des écarts à l'idéalité doit permettred'expliciter le calcul des activités thermodynamiques. Ainsi, ce chapitre consiste en unesynthèse bibliographique qui nous conduit des bases de thermodynamique aux modèlesphysico-chimiques de calcul des coefficients d'activité retenus pour aborder notre étude.
Le premier paragraphe rappelle les bases thermodynamiques nécessaires à la compréhensionde notre étude. Le second paragraphe apporte une réflexion sur les différentes écoles dontémanent les modèles physico-chimiques. Les deux dernières parties présentent de façondétaillée les modèles mis en œuvre dans notre étude pour calculer les coefficients d'activité enphase aqueuse et en phase organique.
27

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
1. Rappels thermodynamiques.
Avant de détailler les méthodes de calcul des coefficients d'activité, il est utile derappeler les relations thermodynamiques qui s'appliquent à un équilibre chimique. Ainsi, nousprésentons dans la suite de ce texte, les notions fondamentales de potentiel chimique, d'étatstandard, d'activité, d'état de référence et de coefficient d'activité. Nous précisons égalementles différentes échelles de concentration à la disposition du physico-chimiste et les relations deconversion entre ces différentes bases de concentration.
1.1. Potentiel chimique.
L'état macroscopique d'un système chimique à l'équilibre est défini par la connaissanced'un certain nombre de paramètres indépendants que l'on désigne sous le terme de "variablesd'état" [1]. On appelle "fonction d'état", toute grandeur qui dépend exclusivement des valeursdes variables d'état. Ce sont par exemple, l'énergie interne U, l'entropie S, l'enthalpie H etl'enthalpie libre G.
L'état macroscopique d'un système chimique est souvent décrit grâce à la fonction enthalpielibre G définie par:
G = H - T . S (1).
En 1885, VAN'T HOFF [2] montre que le signe de la variation d'enthalpie libre AG permet dedéterminer le sens d'évolution d'une réaction chimique.
AG = AH-T.AS (2)
Cette relation montre que la variation d'enthalpie libre d'un système est la combinaison dedeux contributions: une d'origine enthalpique et l'autre de nature entropique.
Pour un système homogène, l'enthalpie libre est fonction uniquement de la température T, dela pression P et du nombre de moles nj de chacun des constituants (autrement dit sacomposition). G étant une fonction d'état, on démontre que sa dérivée peut s'exprimer sous laforme:
Gj est l'enthalpie libre molaire partielle du constituant i, on emploie plus souvent ladénomination de potentiel chimique noté \i\.
On démontre que:
28

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
(4)
Le potentiel chimique se décompose donc, comme nous l'avons déjà noté pour l'enthalpie
libre, en une contribution enthalpique Hj et une contribution entropique T. S; .
Un système ne peut être à l'équilibre que s'il est isotherme et isobare [1], ceci signifieque les deux premiers termes de l'expression (3) de l'enthalpie libre sont nuls. Lorsquel'équilibre est atteint, G est minimale, c'est à dire que son gradient est nul, soit finalement:
i .dn i=0 (5)
Lorsque le système est composé de plusieurs phases (cp) en équilibre, la relation précédente(5) s'exprime sous la forme suivante:
On démontre alors que, dans les conditions d'équilibre, le potentiel chimique de chacun desconstituants est le même dans toutes les phases. En particulier, dans le cas d'un équilibreliquide-gaz, le potentiel chimique d'un constituant a la même valeur dans la phase liquide et laphase gazeuse. Tant que la pression totale reste faible, (inférieure à 10 ou 20 atmosphères), onpeut admettre que les gaz ont un comportement de gaz parfaits. Le potentiel chimique estalors de la forme:
(7)
\x\ est le potentiel chimique du composé i gazeux à la pression de 1 bar et à la température T,Pi est la pression partielle de i au-dessus de la solution.
1.2. Activité et état standard.
A l'équilibre entre une solution liquide et un gaz, nous avons vu que le potentielchimique de chacun des constituants est le même dans les deux phases, soit:
L'étude de cette relation montre que le potentiel chimique est maximal lorsque la pression
partielle de i est la plus élevée, c'est à dire lorsque i est pur ( p, = p° ).
Dans une solution différente du composé pur, la connaissance de la pression partielle de ipermet de calculer une valeur absolue du potentiel chimique. Lorsque l'on désire connaître laréactivité de i dans cette solution, la valeur de Uj n'apporte en elle même, aucune information.
29

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
Par contre, si l'on dispose également de la valeur de p°, la connaissance de ps permet parcomparaison de situer la réactivité de i dans le système.
Il est donc nécessaire de définir une "réactivité relative" ou "activité" qui compare la réactivitédu constituant dans la solution à la réactivité du composé dans un état particulier dénommé"état standard".L'activité de i dans la solution est définie comme le rapport de la pression partielle de i au-dessus de la solution à la pression partielle de i au-dessus d'une solution de i dans l'étatstandard, soit:
En pratique, l'état standard est choisi parmi les deux états suivants:
© le composé pur, soit: pf = p°
© le composé pur ayant le comportement d'une solution infiniment diluée, c'est à dire
qui suit la loi de HENRY (pf = kj.Xj, Xj étant la fraction molaire), soit: pf = k;.
1.3. Coefficient d'activité et comportement de référence.
L'activité du composé i est reliée à sa concentration dans la solution par l'intermédiaired'un coefficient de proportionnalité, désigné sous le terme de "coefficient d'activité".
Suivant l'échelle de concentration retenue, les notations suivantes sont adoptées pour lecoefficient d'activité.
Echelle des fractions molaires X; (moles par mole de solution): f< = —'- (10)
Echelle des molalités nij (moles par kilogramme de solvant): y i = —*- (11)
Echelle des molarités Cj (moles par litre de solution): y; = — (12)Ci
Les formules de conversion entre les différentes échelles de concentration ainsi qu'entre lescoefficients d'activité exprimés dans ces trois bases sont regroupées dans l'Annexe 1.
Suivant l'état standard retenu pour définir l'activité (cf. 1.2.), on peut écrire lesrelations suivantes (présentées ici dans l'échelle des fractions molaires):
© fj = ' „ = i? , p^oult est la pression partielle de i, si i se comporte suivant laXj-Pi Pi °u
loi de RAOULT (comportement idéal parfait).
30

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
® fj = = H y , p"enry est la pression partielle de i, si i se comporte suivant la loi
de HENRY (comportement infiniment dilué).
Le coefficient d'activité est donc défini comme le rapport de la pression partielle de i au-dessus de la solution, à la pression partielle de i au-dessus d'une solution possédant uncomportement limite (ou comportement de référence).
f; = Pi
Piréf.
(13)
Nous avons réuni sur les Figures 1-1 a et 1-lb, les définitions de l'état standard et ducomportement de référence, dans le cas d'un écart à l'idéalité (écart à la loi de RAOULT)positif et négatif.
Il est important de noter que, l'état standard correspond à une seule solution (représentée parun point), tandis que le comportement de référence se rapporte à un ensemble de solutions(représenté par une droite).
•ë«
.1s
I
Etat standard CDSolution de référence ©Etat standard ©Solution de référence ©Solution réelle
0.4 0.6
Fraction molaire de i
•8
"S
0.4 0.6
Fraction molaire de i
1.0
Figure 1-la; Ecart à l'idéalité positif. Figure 1-lb: Ecart à l'idéalité négatif.
31

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
La compréhension des définitions de l'activité et du coefficient d'activité que nousavons présentées, est également facilitée grâce aux quatre représentations graphiquessuivantes (figures 1-2 et 1-3).
i -
1 1
Etat standard ®Etat standard @
yy
yy
/ . - • • •
/ . • • • • •
i i
i i
yy
y
y
y
y ..-••''
0.0 0.2 0.4 0.6 0.8 1.0
1 -
0.0 0.2
1 1
Etat standard ©Etat standard ®
y•y
_.-y
//
//
/• /
//
//
Fraction molaire de i
0.4 0.6
Fraction molaire de i
0.8 1.0
Figurel-2a: Activité de i(écart à l'idéalité positif).
Figure l-2b: Activité de i(écart à l'idéalité négatif).
tivité
de
i
i
I,
1 1 1
Comportement de référence ©Comportement de référence ©
\ \ \ \ \ \
1 ! 1 1
0.0
• 1
0.2 0.4 0.6
Fraction molaire de i
0.8 1.0
•u
h
0.0 0.2
1
s*
l 1 [.
Comportement de référence ©Comportement de référence ©
y
—
0.4 0.6
Fraction molaire de i
0.8 1.0
Figure l-3a: Coefficient d'activité de i(écart à l'idéalité positif).
Figure l-3b: Coefficient d'activité de i(écart à l'idéalité négatif).
32

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
On retiendra que:
- l'activité du composé i est maximale lorsque celui-ci est pur,
- le coefficient d'activité tend vers 1 lorsque le comportement réel de la solution serapproche du comportement de référence,
- dans le cas d'un écart à l'idéalité négatif, le coefficient d'activité est une fonctionstrictement croissante,
- dans le cas d'un écart à l'idéalité positif, le coefficient d'activité est une fonctionstrictement décroissante.
1.4. Coefficient d'activité des electrolytes.
Jusqu'à présent, nous avons présenté les raisonnements thermodynamiques permettantde définir le coefficient d'activité d'un composé i dans une solution. Les phases aqueuses quenous devons mettre en œuvre sont des solutions d'électrolytes (acide ou sels). Si l'on considèreun electrolyte MpXq en phase aqueuse, on peut écrire l'équilibre suivant:
MpXq ^ p . M m + + q . X x - (14)
L'activité globale de l'électrolyte s'écrit par définition de la manière suivante dansl'échelle des molalités:
Y «m* et yvX. : coefficients d'activité des ions libres,
m. _„„. et mv!1. : molalités des ions libres.
Pour simplifier récriture de la relation (15), le coefficient d'activité ionique moyen del'électrolyte est défini tel que:
V(P+<0 _ V P .v<! H'où a - v ( p + q ) -m p -mq CI6Ïy±,Mpxq - T M « 'x*- a o u aMpx, -Y±,Mpxq m
M-"+ " V - v l o J
Le coefficient d'activité ionique moyen y± M x englobe l'ensemble des écarts à l'idéalité qui
résultent des interactions dans la solution entre les ions ou bien, entre les ions et les moléculesneutres.
Le calcul de l'activité globale de l'électrolyte nécessite de connaître la valeur de y+ et lesconcentrations des ions libres en solution. Dans le cas d'une dissociation totale, cesconcentrations sont égales à celle de l'électrolyte MpXq (affectée des coefficientsstœchiométriques respectifs p et q). Lorsque l'électrolyte n'est pas totalement dissocié, il est
33

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
nécessaire de prendre en compte d'autres espèces que les ions libres Mm+ et Xx~, ces espècesprovenant de:
- la dissociation incomplète de l'électrolyte,
- la formation d'espèces chargées ou neutres,
- la formation de paires d'ions.
Dans le domaine particulier des solutions d'électrolytes le phénomène d'association doit êtregénéralement pris en compte [3]. Par exemple, les données expérimentales disponibles pourl'acide nitrique [4], montre que l'association ne peut plus être négligée (association > 5%) àpartir d'une concentration de 1,5 mol.L'1. Pourtant, les phénomènes d'association dans lessolutions de sels restent quant à eux peu étudiés.
Pour contourner ce manque de données, il est intéressant de substituer lesconcentrations des ions constitutifs aux concentrations des ions libres. Cette démarcheconsiste à regrouper en deux termes, l'ensemble des espèces contenant les ions constitutifs del'électrolyte, soit:
mM = 2_, Molalités des espèces contenant MM
mx = ^ Molalités des espèces contenant Xx
L'activité globale de l'électrolyte s'exprime sous la forme:
(17)
yM, YX coefficients d'activité des ions constitutifs M et X,mM, mx: molalités des ions constitutifs M et X.
Le coefficient d'activité stœchiométrique moyen est défini de la manière suivante:
V ( P + I ) _ V P . v q n o \Yst,Mpxq ~ Y M Y X v 1 0 ;
Lorsque l'on introduit la notation de coefficient d'activité stœchiométrique moyen, la relation(17) s'écrit:
- n p - a q
PI
A la différence de la relation (16), le calcul de l'activité globale ne nécessite pas de connaîtrela répartition des différentes espèces en solution. Il suffit d'avoir accès à la concentrationtotale de l'électrolyte par une méthode analytique globale. L'éventuelle formation d'espèces estdonc implicitement prise en compte par le coefficient d'activité stœchiométrique moyen.Lorsque de telles réactions ont lieu, il est clair que les deux coefficients d'activité y± et Yst
34

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
prennent des valeurs différentes. Il n'y a égalité entre y± et de yst que dans le cas où ladissociation de l'électrolyte est totale. La majorité des données tabulées dans la littératurecorrespond à yst, même si cela n'est pas clairement explicité. Dans la suite de ce mémoire,c'est la définition du coefficient d'activité stœchiométrique moyen qui est retenu et, afind'alléger les écritures, le suffixe "st" est omis.
35

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
2. Les deux approches possibles de calcul des coefficients d'activité.
Après avoir présenté les relations thermodynamiques nous permettant d'appréhender lanotion d'écart à l'idéalité et, par là même, la grandeur "coefficient d'activité", nous présentonsdans ce qui suit les deux démarches permettant de quantifier cette variable.
2.1. L'approche analytique.
Dès le début du siècle, les physico-chimistes ont tenté d'analyser les interactionsresponsables des écarts à l'idéalité. La démarche suivie lors de cette analyse peut êtredécomposée en trois étapes successives:
© Un inventaire des espèces présentes dans la solution est effectué. Les auteursassimilent les différentes espèces chimiques à des "analogues géométriques" (point, sphère...).Il s'agit donc de proposer une représentation schématique du système.
© Les interactions qui peuvent exister entre les espèces sont recensées. A ce niveauseules les forces jugées responsables des écarts à l'idéalité sont retenues.
<D Enfin, l'énergie mise en jeu dans ces interactions est quantifiée. On distingue deuxtypes de démarche:
- une première basée sur un traitement purement mathématique des différents typesd'interactions,
- une seconde s'appuyant sur des équations issues de l'électrostatique ou de lamécanique statistique.
La progression chronologique non exhaustive de ces théories des solutions peut être présentéesous la forme de deux tableaux. Le premier (tableau 1-1) regroupe les théories successivesdéveloppées pour aboutir à une expression théorique du coefficient d'activité en phaseaqueuse.
36

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
Tableau 1-1: Principales théories des solutions aqueuses.
Auteurs
DEBYE-HÛCKEL(1923) [5]
GUGGENHEIM(1935) [6]
BROMLEY (1973)[7]
PITZER(1973)[8]
CHEN (1982) [9]
© Représentationschématique retenue
Ions = chargesponctuelles
Ions = sphèresincompressibles
Cellules constituéesd'ions et de molécules
d'eau
® Influencesconsidérées
Attraction entre ions
de charges opposées
Répulsion et attraction
entre ions
Répulsion et attractionentre les ions et les
molécules d'eau
® Traitement desinteractions
Electrostatique
Electrostatique
+ mathématique
Mécanique statistique+ mathématique
Mécanique statistique+ composition locale
Le second tableau (tableau 1-2) concerne les différentes théories des solutions appliquées autraitement des écarts à l'idéalité dans les solutions de non-électrolytes. Pour tous ces modèles,la quantification des différentes interactions existant entre les molécules est basée sur deséquations issues de la mécanique statistique.
L'aptitude de tels modèles à rendre compte de la réalité dépend de la qualité de ladescription et du traitement des différents types d'interactions existant dans les solutions.Cette démarche qui s'applique donc à rendre compte des phénomènes physiques, peut êtredésignée sous le terme de démarche "phénoménologique" [10].
37

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
Tableau 1-2: Principales théories des solutions organiques.
Auteurs
HILDEBRAND (1929) [11]Théorie des solutions régulières.
FLORY-HUGGINS (1941) [12]Théorie des solutions
athermales.
GUGGENHEIM (1952) [13]Théorie quasi-chimique.
WILSON (1964) [14].
RENON-PRAUSNITZ (1968)[15] Théorie NRTL.
ABRAMS-PRAUSNITZ (1975)[16]UNIQUAC.
FREDENSLUND-PRAUSNITZ(1975) [17] UNIFAC.
© Représentation schématique retenue
o Molécules sphériques de même taille,
o Mélange au hasard.
o Molécules de formes et tailles variées => mélangenon au hasard.
o Molécules sphériques de même taille,
o Mélange non au hasard.
o Molécules de formes et tailles variées => mélangenon au hasard,
o Solution décomposée en "cellules".
o Molécules de formes et tailles variées => mélangenon au hasard,
o Solution décomposée en deux types de "cellules"organisées autour d'une molécule centrale (A ou B).
o Molécules de formes et tailles variées => mélangenon au hasard,
o Solution décomposée en "cellules".
o Molécules de formes et tailles variées => mélangenon au hasard,
o Solution décomposée en "cellules" et moléculesdécomposées en groupes fonctionnels.
© Influences considérées
o Interactions mutuelles A-A et B-B différentes =>interaction A-B => ÀtteO.
o AS=0.
o Interactions mutuelles A-A et B-B identiques =>AH=0. AS*0.
o Interactions mutuelles A-A et B-B différentes =>interaction A-B => AH^O.
o AS*0 (terme de non hasard*).
o Interactions mutuelles A-A et B-B différentes =>interaction A-B => AH*0.
o AS*0 (terme de non hasard + paramètresmorphologiques).
•Terme de non hasard: facteur entropique qui n'est pas lié à des différences morphologiques mais induit par les interactions de type A-B.
38

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
2.2. L'approche thermodynamique.
Comme nous l'avons vu précédemment (§1.1.)> l'état d'un système chimique àl'équilibre (T et P constants) est totalement décrit par sa composition (nombre de moles nj dechacun des éléments). Ainsi, il existe des relations thermodynamiques reliant les expressionsde l'activité ou du coefficient d'activité aux concentrations. La relation la plus utilisée est celledémontrée par GIBBS-DUHEM [18]:
_ tj.dlnfa^O (20)i
a;: activité du constituant i.
En 1949, GUGGENHEIM [19] démontre un autre type de relation que l'on désigne sous leterme de "relation des dérivées partielles croisées":
En 1952, McKAY [20] présente une série de développements mathématiques, basés surl'intégration de la relation des dérivés partielles croisées pour un système ternaire. La relationde base s'écrit sous la forme:
(22)
Les numéros 1 et 2 représentent deux solutés, le solvant étant désigné par le numéro 3. Lepassage du nombre de moles ni de chacun des trois constituants à leur molalité mj (expriméepar rapport au solvant), permet de simplifier la relation (22). En effet, lorsque l'on exprime lesconcentrations de 1 et de 2 par rapport au solvant 3, l'intégration à (ni, n3) ou (n2, n3) constantsest ramenée à une intégration à m! ou m2 constant:
(23)
Cette relation différentielle constitue alors un moyen simple de relier entre elles, les activitésdes constituants du mélange.
Il existe donc des relations thermodynamiques permettant d'expliciter le calcul desactivités dans les solutions sans avoir recours à un inventaire laborieux et non exhaustif desdifférentes causes des écarts à l'idéalité. Cette démarche thermodynamique peut égalementêtre désignée sous le terme d'"approche globale" [10]. La mise en œuvre de ces relations, pourle calcul des coefficients d'activité dans les solutions aqueuses et organiques, est présentéedans les deux paragraphes suivants. Lors de cette présentation, une attention particulière estportée à la signification physico-chimique des calculs d'intégration.
39

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
3. Calcul du coefficient d'activité et de l'activité d'eau dans les solutionsaqueuses.
3.1. Solutions binaires (électrolyte/eau).
Le terme de solution binaire désigne une solution à deux constituants. Les deuxconstituants d'une solution aqueuse binaire d'électrolyte sont l'eau et le constituant"electrolyte" MpXq.
Par définition, l'activité d'un constituant dans une solution est égale au rapport de lapression partielle au-dessus de la solution à la pression partielle au-dessus d'une solution dansl'état standard. L'activité d'eau d'une solution est définie par rapport à l'état standard "solutionpure":
a H o = PM. ( 2 4 )PH2O
La relation de GIBBS-DUHEM (16) s'écrit dans le cas d'une solution binaire selon:
nH,o-dln(aH,o) + nMnxn -d l n (Vx n ) = 0 (25)
L'intégration de cette relation est le plus souvent réalisée en introduisant la variable"coefficient osmotique" O définie dans l'échelle des molalités selon:
, _ 1°2£_ . to^) (26)v -mM p V
M H 2 o
v: nombre d'ions libérés par la dissociation supposée totale d'une mole de MpXq,MH2O : masse molaire de l'eau,
mM x : molalité de l'électrolyte MpXq.
Le résultat final de l'intégration de la relation de GIBBS-DUHEM est:
(27)
Nous rappelons que y M x représente le coefficient d'activité stcechiométrique moyen de
l'électrolyte MpXq.
Pratiquement, le calcul du terme intégral ne peut pas être effectué, comme il est decoutume, par une méthode graphique. En effet, lorsque la molalité tend vers zéro, le quotient
O - ltend vers l'infini. Pour contourner cette difficulté de calcul, on a recours à une
expression analytique de O obtenue par ajustement des valeurs expérimentales de
40

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
0 = f( mM x ). La fonction le plus souvent proposée, dans le cas des electrolytes, est de la
forme:
> = l - 7 - V ^ V ^ + EAi-mMPx, (28)• i
avec rj=l+0,5.i
1 est le degré du polynôme retenu, A; désignent les coefficients du polynôme.
A correspond au paramètre de l'équation de DEBYE-HÛCKEL-ONSAGER, à 25 °C pour unelectrolyte 1-1 [21]:
A = 1,1778 mof2.L2
Finalement, le coefficient d'activité de l'électrolyte s'exprime selon:
(29)
La connaissance de l'activité d'eau d'une solution binaire d'électrolyte et par voie deconséquence de son coefficient osmotique, permet donc de calculer l'activité de l'électrolyteMpXq.
Il existe plusieurs techniques expérimentales permettant la détermination de l'activitéd'eau de solutions aqueuses. Parmi les dispositifs expérimentaux les plus connus et les plusadaptés à l'étude de solutions concentrées, nous pouvons retenir:
- la méthode isopiestique,
- la méthode par "evaporation",
- la méthode électrochimique.
Une description détaillée des deux premières techniques a été présentée par MAJIMA et Coll.[22]. Ces deux méthodes sont caractérisées par une grande précision (de l'ordre de 0,01%),mais nécessitent des conditions opératoires fastidieuses et très rigoureuses. Nous retiendronsque la méthode isopiestique est réservée à la mesure de la pression partielle au-dessus desolutions d'électrolytes non volatils [22, 23]. Au contraire, la méthode par evaporation estdestinée à l'étude d'électrolytes volatils comme les solutions d'acide nitrique ou chlorhydrique[4, 22].
La dernière technique est retenue dans le cadre de cette étude. Le dispositif expérimental meten œuvre un capteur électrolytique dont l'impédance varie avec la tension de la vapeur d'eau àson contact. Ce montage expérimental permet la détermination de l'activité d'eau de solutionsaqueuses avec une précision maximale de 0,1%.
41

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
Un grand nombre de déterminations expérimentales de l'activité d'eau (et par suite, desvaleurs du coefficient d'activité) de solutions binaires d'électrolytes est compilé dans lalittérature. Les valeurs nécessaires à notre étude sont publiées par HAMER et WU [24, 25]dans l'échelle des molalités et pour une température de 25°C. Le calcul de l'activité d'eaud'une solution d'électrolyte de molalité connue se fait par interpolation de ces donnéestabulées. La détermination du coefficient d'activité suit naturellement la même démarche.Cette interpolation des données binaires est informatisée dans le logiciel AQUACALC,développé initialement par LY [26].
3.2. Mélanges d'électrolytes.
3.2.1. L'approche thermodynamique de McKAY-PERRING.
Lors de la présentation de l'approche thermodynamique de calcul des coefficientsd'activité, il est apparu que la mise en œuvre de la relation des dérivées partielles croiséesconstitue une alternative élégante à la relation de GIBBS-DUHEM. Ainsi, les développementseffectués par McKAY [20] montrent que la mesure de l'activité d'eau d'une solution deplusieurs electrolytes permet de calculer les activités de l'ensemble des constituants de lasolution.
Malheureusement, à la différence des solutions binaires, très peu de mesuresexpérimentales sont publiées sur les mélanges d'électrolytes. En particulier, les mélangesacide nitrique/sels de nitrate restent peu étudiés.Pour pallier ce manque d'informations, il est bien sûr envisageable d'acquérir les donnéesnécessaires à l'étude de notre système. Il s'agirait de mesurer l'activité d'eau de mélangesH2O/HNO3/NaNO3/CsNO3 dont les compositions doivent couvrir le domaine de concentrationde nos expériences d'extraction liquide-liquide. Le nitrate de césium étant en faibleconcentration dans des matrices concentrées en acide et/ou en sels, l'activité d'eau du mélangeest donc voisine de celle obtenue pour la solution ternaire H2O/HNO3/NaNO3. L'étudeexpérimentale se limiterait alors aux mesures d'activité d'eau de ces mélanges ternaires.
Le résultat de l'intégration de la relation des dérivées partielles croisées réalisée parMcKAY [27] pour un mélange ternaire (MpXq)i/(MpXq)2/H2O est présenté, sous une formelégèrement modifiée, par MAJIMA [28]:
0,018 -vMX,i v lMX,
V M Y V M Y
avec: m* = mMX + - ^ - mMX et x, = ^ ^MX, ._ . . ,
v M X v M X m
VMX, e t VMX2 : nombres d'ions libérés par la dissociation supposée complète des electrolytes
•(MpXq)i et (MpXq)2 du mélange ternaire,mMX| et mMXz : molalités des electrolytes (MpXq)i et (MpXq)2 dans le mélange,
YMX[ : coefficient d'activité de l'électrolyte (MpXq)i dans le mélange,
42
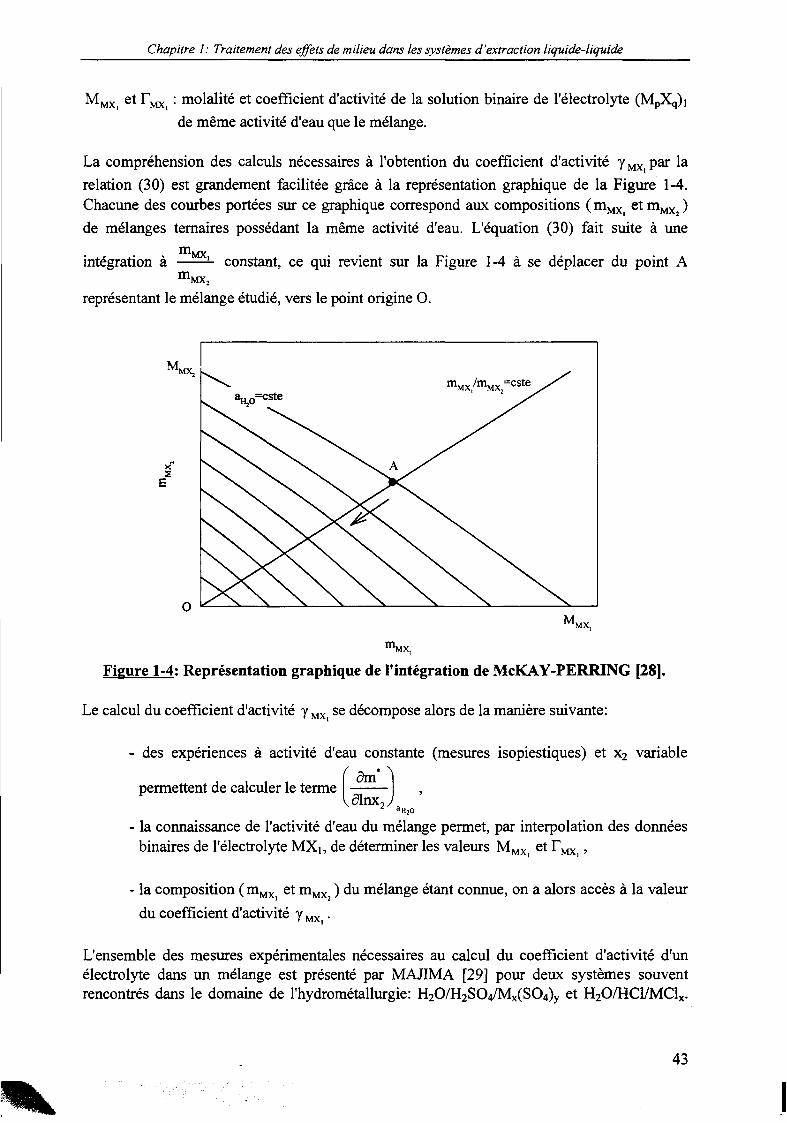
Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
MMXi et FMX : molalité et coefficient d'activité de la solution binaire de l'électrolyte (MpXq)i
de même activité d'eau que le mélange.
La compréhension des calculs nécessaires à l'obtention du coefficient d'activité yMX par larelation (30) est grandement facilitée grâce à la représentation graphique de la Figure 1-4.Chacune des courbes portées sur ce graphique correspond aux compositions ( mMX et m,^ )de mélanges ternaires possédant la même activité d'eau. L'équation (30) fait suite à une
intégration à m.MX,
mconstant, ce qui revient sur la Figure 1-4 à se déplacer du point A
MX,
représentant le mélange étudié, vers le point origine 0.
mMX,
Figure 1-4: Représentation graphique de l'intégration de McKAY-PERRING [28].
Le calcul du coefficient d'activité y MX| se décompose alors de la manière suivante:
- des expériences à activité d'eau constante (mesures isopiestiques) et x2 variable
permettent de calculer le terme.dlnx,
"H2O
- la connaissance de l'activité d'eau du mélange permet, par interpolation des donnéesbinaires de l'électrolyte MX1; de déterminer les valeurs MMXi et FMX| ,
- la composition ( mMX et mMX ) du mélange étant connue, on a alors accès à la valeur
du coefficient d'activité y MX|.
L'ensemble des mesures expérimentales nécessaires au calcul du coefficient d'activité d'unelectrolyte dans un mélange est présenté par MAJIMA [29] pour deux systèmes souventrencontrés dans le domaine de l'hydrométallurgie: HbO/F^SCVM^SO^y et H2O/HC1/MC1X.
43

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
Cette étude nous indique que la mise en œuvre expérimentale de la relation de McKAY-PERRING nécessite un nombre élevé de mesures.
Le calcul du coefficient d'activité d'un electrolyte dans un mélange selon la relation deMcKAY-PERRING fait suite à des développements thermodynamiques rigoureux. Nousavons pu souligner que cette relation nécessite l'acquisition d'un nombre très élevé de donnéesexpérimentales. Une telle charge expérimentale n'est pas envisageable dans le temps imparti àun travail de thèse.
La relation (30) sous sa forme complète ne peut donc pas être mise en œuvre pour notre étude.C'est pourquoi, nous avons cherché à simplifier cette relation grâce à l'introduction d'unehypothèse extra-thermodynamique. L'hypothèse retenue est désignée sous le terme de "règlede ZDANOVSKII".
3.2.2. L'apport du comportement dit "simple" au calcul du coefficient d'activité.
En 1936, ZDANOVSKII [30], à la suite d'observations expérimentales, publie la règleempirique suivante:
Lorsque l'on mélange plusieurs solutions aqueuses binaires d'électrolyte de même activitéd'eau (solutions isopiestiques), le mélange obtenu a une activité d'eau identique à celle dessolutions initiales s'il n'existe pas d'interaction chimique entre les electrolytes en son sein.
La règle de ZDANOVSKII se traduit mathématiquement sous la forme suivante:
Y - % = 1 (31)
C; : concentration de l'électrolyte i dans le mélange,Cf1 : concentration de l'électrolyte i dans la solution binaire de même activité d'eau que celledu mélange.
En 1962, TIMOFEEV [31] propose de désigner les systèmes qui obéissent à la règle deZDANOVSKII sous le nom de "systèmes simples".
En 1966, la relation (31) est obtenue par STOCKES-ROBINSON [32]. Ces auteurs ont réalisédes mélanges de solutions binaires (H2O-sucre) en équilibre isopiestique. C'est pourquoi, larelation (31) est souvent désignée sous le terme de relation de ZDANOVSKII-STOCKES-ROBINSON (relation ZSR).
Nous nous limiterons à l'étude de mélanges ternaires d'électrolytes. La relation ZSRs'écrit alors, dans l'échelle des molalités, de la manière suivante:
(32)m MX, m M X 2
- mMX et mMX : concentrations des electrolytes dans le mélange,
44

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
- m^X[, m ^ : concentrations des electrolytes dans leur solution binaire respective de même
activité d'eau que celle du mélange (notées MMXi et MMXî par MAJIMA),
Si les données binaires des deux electrolytes sont connues, ainsi que la composition dumélange, l'activité d'eau du mélange peut être calculée selon une méthode itérative. A ladifférence de la relation de McKAY-PERRING, il n'est donc pas nécessaire de mesurer lavaleur de l'activité d'eau.
L'introduction de la relation (32) dans l'équation de McKAY-PERRING (30) annule le termeintégral. L'expression du coefficient d'activité est alors fortement simplifiée et se met sous laforme suivante:
_y MX, -
: coefficient d'activité de i dans le mélange,: coefficient d'activité de i dans la solution binaire de même activité d'eau que le mélange
(noté FMX| par MAJIMA),
' et de " : nombre d'ions libérés lors de la dissociation supposée totale de i et de k,mMK, m MX k
et : molalité de i et de k dans le mélange,m^x : molalité de i dans la solution binaire de même activité d'eau que le mélange.
Cette expression a été obtenue, pour la première fois, en 1968 par MIKULIN [33]. Elle esthabituellement désignée sous le nom de "relation de MIKULIN". Dans la suite de notre étude,les coefficients d'activité des electrolytes dans les mélanges seront calculés à l'aide de cetterelation.
45

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
4. Calcul des coefficients d'activité dans les solutions organiques.
Nous avons pu voir dans le paragraphe précédent que la quantification des écarts àl'idéalité dans les solutions aqueuses peut être menée selon une approche thermodynamique.En effet, l'intégration de la relation des dérivés partielles croisées permet, moyennant unehypothèse extra-thermodynamique, d'avoir accès à la valeur du coefficient d'activité dans lesmélanges d'électrolytes.
Une démarche similaire doit permettre de quantifier l'écart à l'idéalité dans les solutionsorganiques.
Après avoir présenté l'origine privilégiée des écarts à l'idéalité en phase organique,nous détaillons l'approche thermodynamique retenue pour développer le coefficient d'activitédes constituants puis des espèces. Les hypothèses ainsi que la signification des différentstermes introduits lors des développements thermodynamiques sont précisées.
4.1. La présence d'eau en phase organique.
La plupart des auteurs soulignent que la prise en compte de l'eau extraite en phaseorganique est nécessaire lors de la description des systèmes d'extraction.
Le plus souvent la présence d'eau en phase organique est attribuée à la formation d'espèceshydratées. Les écarts à l'idéalité provenant de la formation de tels complexes sont alors dus àl'existence d'interactions fortes entre les constituants extraits et l'eau, le terme "d'interactionsordonnées" est parfois employé [10].
Cependant, la mise en œuvre de méthodes spectrales (RMN, IR) permet de mettre en évidencela présence d'eau "non-stœchiométrique" en interaction faible avec les constituantsorganiques. Par conséquent, les quantités d'eau dosées en phase organique ne peuvent pas êtreattribuées à la seule formation de ces espèces hydratées. Le terme "d'interactions non-ordonnées" permet également de désigner ces forces [10]. La prédominance de ce typed'interactions a pu être confirmée par ROZEN [34, 35] lors de la modélisation de l'extractionde l'acide nitrique et du nitrate d'uranyle par le TBP par la théorie des solutions régulières
Afin de s'affranchir de tous les inconvénients inhérents à une approche analytique,nous avons adopté un traitement thermodynamique des écarts à l'idéalité. Nous présentonsdans ce qui suit la relation de SERGIEVSKII, modifiée par DANNUS.
4.2. La relation de SERGIEVSKII-DANNUS [10].
Lors de la présentation générale de l'approche thermodynamique (cf §2.2.), la relationdes dérivées partielles croisées est apparue comme une alternative élégante à l'intégration deGIBBS-DUHEM.
L'intégration de cette relation est effectuée par KLOFUTAR [36] puis par SERGIEVSKII [37]dans le cas d'un système ternaire Diluant/Extractant/Eau. Lors de ce calcul intégral, les auteurs
46

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
cherchent à simplifier les expressions mathématiques par l'introduction d'hypothèses extra-thermodynamiques. Ainsi, deux hypothèses simplificatrices sont introduites, à savoir:
© Hypothèse de linéarité: la concentration de l'eau est une fonction linéaire de laconcentration des constituants de la phase organique.
© Hypothèse d'idéalité: l'hydratation suit une loi de HENRY (cf §1.3.)-
Le résultat final permet de relier les écarts à l'idéalité de l'extractant à l'hydratation de la phaseorganique:
(34)
a2: activité du constituant extractant dans le mélange,
a® : activité du constituant extractant dans une solution saturée d'eau d'activité d'eau égale à 1(état hypothétique),
h® : degré d'hydratation du constituant extractant pour une activité d'eau égale à 1.
L'intégration étant réalisée à concentration d'extractant (1112) constante, l'introduction descoefficients d'activité dans la relation (34) permet d'écrire:
d'où finalement:
y2=y®-exp[h®-(l-aH2O)] (36)
y2: coefficient d'activité du constituant extractant dans le mélange,
y® : coefficient d'activité du constituant extractant dans la solution d'activité d'eau égale à 1,c'est à dire en équilibre avec une solution d'eau pure.
Cette relation est désignée sous le terme de "relation de SERGIEVSKII" et son applicationpeut être généralisée à l'ensemble des constituants d'un mélange.
Nous disposons donc d'une relation permettant de développer le coefficient d'activitéd'un constituant organique dans un mélange. Seulement, à la différence du raisonnementsuivi pour la phase aqueuse, l'un des buts de la modélisation des systèmes d'extraction est dedéterminer le nombre et la stœchiométrie des espèces extraites. Ainsi, il est nécessaire deraisonner en termes d'espèces chimiques et non plus de constituants. Cette transposition estprésentée d'une manière très détaillée par DANNUS [10]. Nous exposons ici les principalesmodifications apportées.
47

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
L'intégration de la relation des dérivés partielles croisées pour un système composéd'espèces est en tout point semblable à celle développée pour un mélange de constituants. Laseule précaution à prendre se situe au niveau des hypothèses simplificatrices. Les conditionsde linéarité et d'idéalité sont énoncées en termes d'espèces et peuvent être formuléesmathématiquement de la manière suivante:
hx = h®.aH20 Vmx (X étant l'espèce considérée) (37)
Les écarts à l'idéalité correspondant à la formation des espèces sont quantifiés parl'intermédiaire de la loi d'action de masse. Le calcul du coefficient d'activité de ces espècespeut être dégagé de l'ensemble des interactions fortes existant entre les différentes espèces etl'eau.
Le nombre d'hydratation hx correspond au nombre de molécules d'eau en interaction non-ordonnée avec chacune des espèces. Il s'agit d'eau solubilisée par les espèces que nousnoterons par la suite sx. Quant à l'eau entrant dans la composition des complexes hydratés,elle est naturellement désignée sous le terme d'eau d'hydratation.
La relation de SERGIEVSKII modifiée que nous utiliserons dans la suite de noscalculs est exprimée de la manière suivante:
Yx=Yx-exp[s®-(l-aH20)] ( 3 8 )
Yx: coefficient d'activité de l'espèce X dans le mélange,
y® : coefficient d'activité de l'espèce X dans la solution d'activité d'eau égale à 1,
s® : nombre de molécules d'eau solubilisée par l'espèce X à activité d'eau égale à 1.
La quantification de l'influence de l'eau d'hydratation étant prise en compte via la loi d'actionde masse, le coefficient yx englobe les écarts à l'idéalité liés:
- aux interactions entre les différentes espèces et les molécules d'eau solubilisée =>
terme expjs® -(l-aH i 0)],
- aux interactions faibles entre les différentes espèces et le diluant => terme y®.
Nous retiendrons que l'approche de SERGIEVSKII-DANNUS fait suite à undéveloppement thermodynamique rigoureux dans lequel sont introduites deux hypothèsessimplificatrices (conditions de linéarité et d'idéalité). Ces hypothèses ne sont vérifiablesqu'avec les solutions organiques les plus simples, à savoir le diluant seul ou bien le mélange[extradant (à concentration fixée) / diluant]. Pour ces solutions organiques simples, leséventuels écarts à la relation (37) peuvent facilement être mis en évidence si l'on effectue desdosages d'eau à différentes valeurs d'activités d'eau. Pratiquement, les expériences à activitéd'eau variable sont menées en utilisant des solutions aqueuses contenant un sel non-extractiblepar le diluant et par l'extractant. Dès l'instant où l'on étudie l'extraction d'acide et de sels, il estde plus en plus difficile de discerner la part de l'eau apportée par chacune des espècessupplémentaires (complexes acides ou salins) présentes en phase organique. La vérification de
48

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
la relation (37) est alors impossible, les hypothèses de linéarité et d'idéalité doivent êtreadmises a priori.
Conclusion.
Au cours de ce premier chapitre, nous avons pu montrer qu'une approche basée sur desrelations thermodynamiques permet un traitement rigoureux des effets de milieu dans lessolutions. A la différence d'une démarche analytique, les interactions responsables des écartsà l'idéalité sont traitées d'une manière globale. Une identification laborieuse et souventincomplète de chaque type de force n'est donc pas nécessaire.
Toutefois, la quantification des écarts à l'idéalité par ces expressions thermodynamiquesrigoureuses nécessite l'acquisition d'un nombre important de données expérimentales. Cettecharge de travail n'est pas envisageable dans le cadre d'un travail de thèse. C'est pourquoi,nous avons cherché à simplifier ces relations grâce à l'introduction d'un minimumd'hypothèses extra-thermodynamiques.
Nous avons retenu les relations de MIKULIN et de SERGIEVSKII-DANNUS. Ces relationsnous permettront, dans la suite de notre étude, de développer les coefficients d'activité desconstituants et des espèces respectivement dans les mélanges aqueux et organiques.
49

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
BIBLIOGRAPHIE.
[I] R. GABORIAUD,Thermodynamique appliquée à la chimie des solutions,Ed. MARKETING (1988).
[2] M. KARAPETIANZ, N. BASILINE,Thermodynamique Chimique,Editions MIR, MOSCOU, (1978).
[3] G.W. DAVIS,Ion Association,BUTTERWORTH., (1962)
[4] W. DAVIS, H.J. De BRUIN,New Activity Coefficients of 0-100 per cent Aqueous nitric Acid,J. Inorg. Nucl. Chem., 26, (1964), 1069-1083.
[5] P. DEB YE, E. HUCKEL,Physik. Z., 24, (1923), 185-206.
[6] E.A. GUGGENHEIM,The Specific Thermodynamic Properties of Aqueous Solutions of Strong Electrolytes,Phil. Mag., 19, (1935), 588-643.
[7] L. A. BROMLEY,Thermodynamic Properties of Strong Electrolytes in Aqueous Solutions,AIChEJ., 19, (1973), 313-320.
[8] K.S. PITZER,Thermodynamics of Electrolytes. I. Theorical Basis and General Equations,J. Phys. Chem., 77, (1973), 268-277.
[9] C.C. CHEN, H.I. BRITT, J.F. BOSTON, L.B. EVANS,Local Composition Model for Excess Gibbs Energy of Electrolyte Systems,AIChE J., 28, (1982), 588-596.
[10] P.DANNUS,Modélisation physico-chimique de l'extraction de constituants inorganiques par desréactifs solvatants,Thèse de Doctorat de l'Université PARIS VI (1991).
[II] J.H. HILDEBRAND,Solubility. XII. Regular Solutions,J. Am. Chem. Soc, 51, (1929), 66-80.
51

[12] (a) M.L. HUGGINS,Solutions of Long Chain Compounds,J.Chem.Phys., 9, (1941), 440.
(b) P.J. FLORY,Thermodynamics of High Polymer Solutions,J.Chem.Phys., 9, (1941), 660-661.
(c) P.J. FLORY,Thermodynamics of High Polymer Solutions,J.Chem.Phys., 10, (1942), 51-61.
[13] E.A. GUGGENHEIM,MIXTURES. The Theory of the Equilibrium Properties of some Simple Classes ofMixtures Solutions and Alloys,OXFORD, CLARENDON PRESS, (1952).
[14] G.M. WILSON,Vapor-Liquid Equilibrium. XI. A New Expression for the Excess Free Energy ofMixing,J. Am. Chem. Soc. 86, (1964), 127-130.
[15] H. RENON, J.M. PRAUSNITZ,Local Compositions in Thermodynamic Excess Functions for Liquid Mixtures,AIChE J., 14, (1968), 135-144.
[16] D.S. ABRAMS, J.M. PRAUSNITZ,Statistical Thermodynamics of Liquid Mixtures: A New Expression for the ExcessGibbs Energy of Partly or Completely Miscible Systems,AIChE J., 21, (1975), 116-128.
[17] A. FREDENSLUND, R.L. JONES, J.M. PRAUSNITZ,Group-Contribution Estimation of Activity Coefficients in Nonideal Liquid Mixtures,AIChE J., 21, (1975), 1086-1099.
[18] P.DUHEM,Le potentiel thermodynamique et ses applications à la mécanique chimique et à l'étudedes phénomènes électriques,A. HERMANN, PARIS, (1886).
[19] E.A.GUGGENHEIM,Thermodynamics and advanced treatment for Chemists and Physicists,North-Holland Publishing Compagny, (1949).
[20] H.A.C.McKAY,Thermodynamics of Three-Component Systems,Nature, 169, (1952), 464-465.
52-

Chapitre 1: Traitement des effets de milieu dans les systèmes d'extraction liquide-liquide
[21] R.A. ROBINSON, R.H. STOCKES,Electrolyte Solutions,BUTTERWORTH Ed., p.468, revised (1970).
[22] H. MAJIMA, Y. AWAKURA,Water and Solute Activities of H2SO4-Fe2(SO4)2-H2O and HCl-FeCl3-H2O SolutionSystems: Part I. Activities of Water,Metal. Trans. B, 16B, (1985), 433-439.
[23] A.N. KIRGINTSEV, LUK'YANOV,Non-vacuum apparatus for determining Vapour Pressure by the Isopiestic Method,Russ. J. of Phys. Chem., 37, (1963), 121-122.
[24] W.J. HAMER, Y.C. WU,Osmotic Coefficients and Mean Activity Coefficients of Uni-univalent Electrolytes inWater at 25°C,J. Phys. Chem. Ref. Data, 1, (1972), 1047-1099.
[25] W.J. HAMER, Y.C. WU,Revised Values of the Osmotic Coefficients and Mean Activity Coefficients of SodiumNitrate in Water at 25 °C,J. Phys. Chem. Ref. Data, 9, (1980), 513-515.
[26] J. LY,Le logiciel AQUACALC,Communication personnelle, (janvier 1989).
[27] H.A.C. Me KAY, J.K. PERRING,Calculation of the Activity Coefficients of Mixed Aqueous Electrolytes from VapourPressures,Trans. Faraday Soc, 49, (1953), 163-165.
[28] H. MAJIMA, Y. AWAKURA,Water and Solute Activities of H2SO4-Fe2(SO4)2-H2O and HCl-FeCl3-H2O SolutionSystems: Part II. Activities of Solutes,Metal. Trans. B, 17B, (1986), 621-627.
[29] H. MAJIMA, Y. AWAKURA, Y. KAWASAKI,Activities of Water and Solutes in the Aqueous Systems H2SO4-Mx(SO4)y-H2O andHCl-MCly,AGNE SHOFU Publishing Inc., Tokyo (1988).
[30] A.B. ZDANOVSKII,Trudy Solyanoï Lab. Akad. Nauk. SSSR, 6, (1936).
[31] V.I. TIMOFEEV,Vest. Univ. Leningrad, 4, (1960), 15.
53

[32] R.H. STOCKES, R.A. ROBINSON,Interactions in Aqueous Nonelectrolyte Solutions. I. Solute-Solvent Equilibria,J. Phys. Chem., 70, (1966), 2126-2130.
[33] G.I.MIKULIN,Voprosy Fizicheskoî Khimii Rastvorov Electrolitov (Les questions de chimie physiquedes solutions d'électrolytes),Izdatelstvo "Khimiya" Lenigrad Otdelenie, (1968).
[34] A.M. ROZEN, L.P. KHORKHORINA, G.D. AGASKINA, E.G. TETERIN, A.N.MAL'CEVA, E.V. VORONECKASA,Extraction de l'eau par le TBP pur et dilué,Radiochimie, 12, (1970), 324-333.
[35] A.M. ROZEN, L.G. ANDRUTSKII,A Mathematical Model of the Extraction of Water by Tributyl Phosphate and theMonosolvate of Nitric Acid in Diluents,Russ. J. Inorg. Chem., 26, (1981), 1502-1506.
[36] C. KLOFUTAR, S. PALJK, M. OSTANEK,Hydration of Tri-n-Hexyl Amine Hydrochloride, Tri-n-Octyl Amine Hydrochloride, Tri-n-Decyl Amine Hydrochloride and Tri-n-Dodecyl Amine Hydrochloride BenzeneSolutions,J. Inorg. Nucl. Chem., 38, (1976), 1045-1048.
[37] A.M. CHEKMAREV, A.V. OCHKIN, V.V. SERGIEVSKII, V.V. TARASOV, G.A.YAGODIN,Acid Extraction by Tertiary Amines,Int. Solv. Extract. Conf., (1980), 80-158.
54

CHAPITRE 2
ACQUISITION DES OUTILS DE BASE NECESSAIRES A LAMODELISATION DU SYSTEME D'EXTRACTION
HNO3/NaNO3/CsNO3/H2O | Calixarène/NPOE.

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
Introduction.
Ce chapitre présente les "outils" nécessaires pour atteindre notre objectif demodélisation physico-chimique. Ces outils, spécialement mis au point au cours de ce travail,sont totalement nouveaux par rapport à la situation antérieure.
Le premier paragraphe présente l'étude originale effectuée dans le but d'acquérir les donnéesthermodynamiques de solutions binaires "sursaturées" de NaNO3 et de CsNO3. Ces donnéessont nécessaires pour la modélisation physico-chimique du système d'extractionHNO3/NaNO3/CsNO3/H2O | Calixarène/NPOE. Cette étude a été menée à la fois sur un planthéorique et expérimental.
Le second paragraphe détaille le travail informatique. En particulier, on insiste sur lesmodifications profondes apportées au logiciel "EXTRACTION" et sur les atouts du nouveaucode de calcul "PREVISION" pour une application à visée industrielle.
57

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
1. Etude critique et comparative de différents modèles d'écarts à l'idéalitédans le but d'acquérir les données binaires de solutions aqueusessursaturées fictives.
Lorsque l'on désire calculer le coefficient d'activité d'un constituant dans un mélanged'électrolytes, il est nécessaire de disposer des données thermodynamiques des solutionsaqueuses binaires de chacun des constituants du mélange et ce, quelle que soit la relationutilisée. Les données binaires nécessaires dépendent du modèle adopté lors du calcul descoefficients d'activité.
© Les théories de BROMLEY, PITZER et CHEN nécessitent la connaissance ducoefficient d'activité de chacun des constituants dans leur solution binaire respective de mêmeforce ionique que le mélange.
© Les équations de McKAY-PERRING et de MIKULIN mettent en œuvre lecoefficient d'activité et la concentration des constituants dans leur solution binaire respectivede même activité d'eau que le mélange.
Les systèmes d'extraction que nous devons modéliser mettent en jeu des solutionsquaternaires H2O/HNO3/NaNO3/CsNO3 dans lesquelles le nitrate de césium est en faibleconcentration. Le mélange type possède une composition voisine de celle des concentratsd'évaporateurs de la Station de Traitement des Effluents Liquides de MARCOULE (STEL-MARCOULE) soit: HNO3 1 mol.L"1, NaNO3 4mol.L"1. Le calcul des coefficients d'activitédes constituants nécessite alors de connaître les données binaires thermodynamiques dechacun des electrolytes, sur une gamme de force ionique de 0 à 5 mol.L"1, soit sur une gammed'activité d'eau de 1 à 0,829. Ceci est réalisé dans le cas de l'acide nitrique et du nitrate desodium, mais pas dans celui du nitrate de césium. Le fichier de données binaires de CSNO3,publié par HAMER et WU [1], couvre un domaine de concentration peu étendu du fait de lafaible solubilité de CsNO3: la saturation est atteinte pour une molalité de 1,4 mol.kg"1,l'activité d'eau est alors égale à 0,965. Le calcul du coefficient d'activité du nitrate de césiumest donc, pour le moment, impossible dans des solutions aqueuses de force ionique supérieureà 1,4 mol.kg"1, ou d'activité d'eau inférieure à 0,965.
La détermination de l'effet de milieu sur l'électrolyte CsNO3 au moyen d'une équationfournie par un modèle, passe donc par l'acquisition de données binaires dans une zone deconcentration qui se situe au-delà de la saturation de ce sel et dénommée zone fictive.
Afin de déterminer le plus grand nombre d'espèces susceptibles de se former en phaseorganique dans un système (H2O/HNO3/NaNO3/CsNO3 | Calix/NPOE), nous balayerons unelarge gamme de concentration en acide nitrique et/ou en sel NaNO3. Les solutions aqueusesalors mises en jeu pourront présenter des valeurs d'activité d'eau inférieures à 0,829(concentrats d'évaporateurs), voire même inférieures à 0,700 (solutions salines > 4 mol.L" etfortement acides > 2 mol.L"1).Dans ce cas, le fichier des données binaires du nitrate de sodium devient à son tour limitantpuisque la valeur d'activité d'eau obtenue à saturation de ce sel dans l'eau est de 0,741 [2].
58

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
Nous allons donc chercher à mettre au point une méthode capable de nous fournir des donnéesthermodynamiques (mi5 yi5 aH 0 ) de solutions binaires de NaNO3 et de CsNO3 au-delà de lasaturation.
1.1.Présentation de la problématique.
Le problème de limitation par la saturation d'un sel dans l'eau pour le calcul deparamètres physico-chimiques a déjà été soulevé dans la littérature. L'idée d'acquérir desdonnées binaires dans une zone "fictive" de concentration a motivé plusieurs études [3, 4, 5].
Nous distinguons trois approches:
© l'approche mathématique,
© l'approche physico-chimique théorique,
® l'approche physico-chimique expérimentale.
© La première approche est fondée sur un traitement purement mathématique desvariations du coefficient d'activité ou de l'activité d'eau dans la zone accessible àl'expérience. Les données binaires dans la zone de "sursaturation" sont obtenues parextrapolation de la fonction mathématique ajustée.
© II est possible de s'appuyer sur des théories des solutions qui mettent en jeu desparamètres d'interactions entre ions ou entre ions et molécules. Les valeurs des paramètressont déterminées par ajustement des données expérimentales. Comme précédemment, laméthode garde un aspect mathématique (l'étape d'ajustement) qui est toutefois nuancé parl'apport d'une théorie physico-chimique.Les théories de DEBYE-HÛCKEL, BR0NSTED-GUGGENHEIM, BROMLEY, PITZER etCHEN sont les plus connues.
<3> Les approches de ZDANOVSKII-STOCKES-ROBINSON (ZSR) et de KUSSIK-MEISSNER-TESTER (KMT) s'appuient sur l'analyse du comportement physico-chimiqueexpérimental des solutions d'électrolytes.
ZDANOVSKII observe que lorsque l'on mélange plusieurs solutions aqueuses binairesd'électrolytes de même activité d'eau (solutions isopiestiques), le mélange obtenu a uneactivité d'eau identique à celle des solutions initiales. STOCKES et ROBINSON ont fait lesmêmes observations pour des mélanges de solutions de non-électrolytes.
KUSSIK-MEISSNER-TESTER remarquent que lorsque l'on trace les variationsexpérimentales du coefficient d'activité d'un grand nombre d'électrolytes en fonction de laconcentration, on aboutit à une famille de courbes possédant des caractéristiques communes.
59

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
1.2.1. Ajustement polynomial.
Dans le domaine de solubilité d'un electrolyte, il est toujours possible d'ajuster lesparamètres b; des polynômes à partir des valeurs expérimentales de l'activité d'eau:
aH2o = m: molalité (39)i=0
Pour aboutir aux valeurs de l'activité d'eau de solutions sursaturées fictives, on pourraitenvisager l'extrapolation de ces polynômes au delà de la saturation.Cette méthode a été appliquée à l'extrapolation des fichiers de données binaires de CsNO3 et deNaNO3, l'ajustement des paramètres des polynômes étant réalisé par la méthode des moindrescarrés. Il apparaît clairement sur la Figure 2-1 que l'utilisation de ces polynômes au-delà dudomaine de solubilité aboutit à des valeurs totalement arbitraires puisqu'elles varient selon ledegré du polynôme envisagé.
1,00
0,95
0,90
0,85 -
2 0»80 "S
X)
S 0^5
^ 0,70 -
0,65 -
0,60 -
0,550,50
0
1 1
1 1
1 1
1
° ExpérienceaHO=a+b.m+c.m2+d.m3+e.m4+f.m5+g.m6
a« O=a+b.m+c.m2+d.m3+e.m4+f.m5
aj, O=a+b.m+c.m2+d.m3+e.m4
— a» O=a+b.m+c.m2+d.m3
aj, o=a+b.m+c.m2
aj,0=a+b.m
V\
\i i i i i i
/
-^
\
\
6 8 10 12 14
Molalité (mol.kg"1)
16 18 20
Figure 2-1: Extrapolation polynomiale de la variation de l'activité d'eaude solutions aqueuses de NaNO3.
60

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
1.2.2. Méthode "curvilinéaire".
Cette méthode a été développée par TRAN et Coll. [5] en 1974. Elle peut êtredécomposée en six étapes successives:
© On recherche le polynôme qui permet d'ajuster le mieux possible la variationexpérimentale de l'activité d'eau.
© Ce polynôme est utilisé pour calculer les valeurs de l'activité d'eau à différentesconcentrations: ms, 0,9.ms, 0,8.ms, et 0,7.ms, (ms: molalité à saturation).
® L'activité d'eau à 1,1.ms est calculée par régression linéaire sur les quatre pointsobtenus précédemment.
© La valeur ainsi calculée est reportée dans le fichier de données binaires utilisé lorsde la première étape.
© La première étape est répétée en intégrant cette dernière donnée.
© Les séquences 2 à 4 sont alors reprises.
Les auteurs ont utilisé cette méthode de calcul pour accéder aux valeurs d'activité d'eaude solutions sursaturées d'une dizaine d'électrolytes. Les valeurs ainsi calculées sontcomparées à celles obtenues à l'aide d'autres méthodes (en particulier la méthode ZSR).L'étude conclut que la méthode "curvilinéaire" ne peut être étendue à des concentrationssupérieures à 1,5 fois la molalité de saturation.
Dans le but d'étendre le fichier de données binaires de CsNO3 jusqu'à des activitésd'eau de 0,800, douze itérations seraient nécessaires. La concentration de la "solutionsursaturée fictive" correspondante serait alors de 5 mole.kg-' soit, plus de 3 fois la molalité desaturation.
Cette méthode "curvilinéaire" tend rapidement vers une variation linéaire de l'activitéd'eau, comme le montre la Figure 2-2. Le processus de calcul imaginé par TRAN et Coll.converge donc vers l'ajustement polynomial précédemment présenté pour un degré dupolynôme pris égal à 1. Le choix de cette variation linéaire ne s'appuie sur aucune théoriephysico-chimique, elle reste donc totalement arbitraire.
61
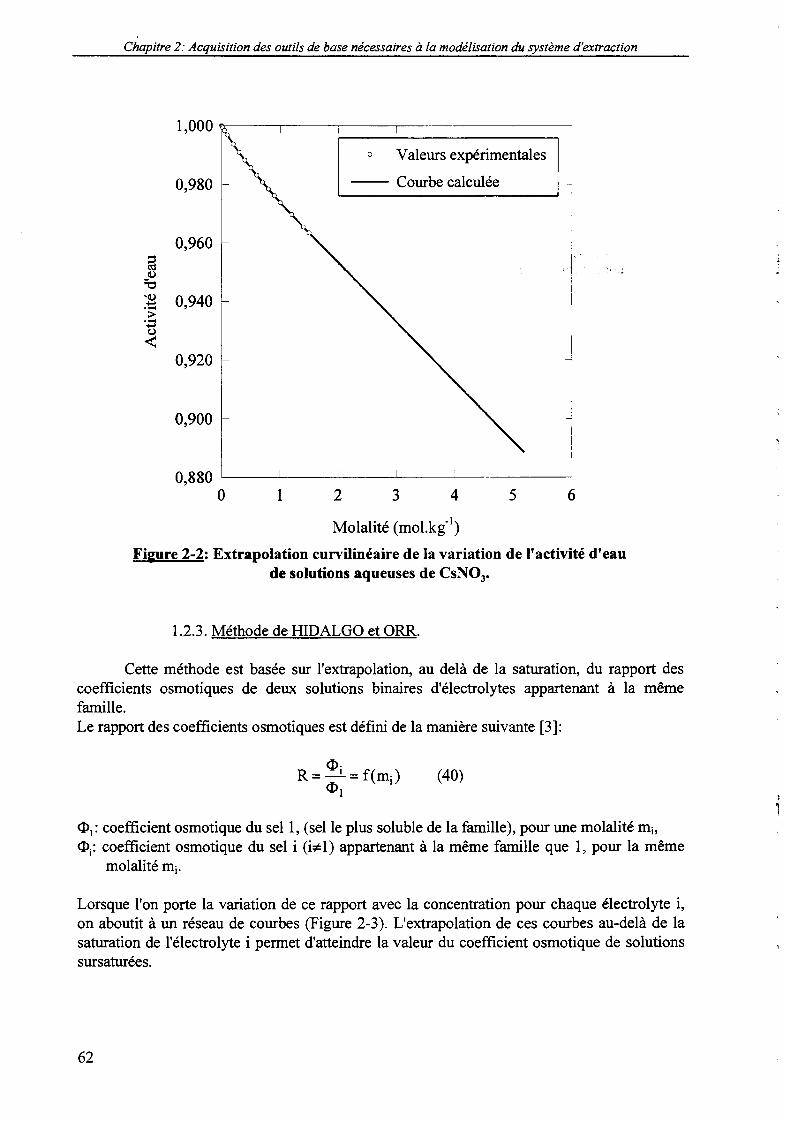
Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
1,UUU "
0,980
0,960
| 0,940'•5
0,920
0,900
n «en
\
1
/
-
-
:° Valeurs expérimentales
Courbe calculée
\
1 :
0 1 4
Molalité (mol.kg"1)
Figure 2-2: Extrapolation curvilinéaire de la variation de l'activité d'eaude solutions aqueuses de CsNO3.
1.2.3. Méthode de HIDALGO et ORR.
Cette méthode est basée sur l'extrapolation, au delà de la saturation, du rapport descoefficients osmotiques de deux solutions binaires d'électrolytes appartenant à la mêmefamille.Le rapport des coefficients osmotiques est défini de la manière suivante [3]:
(40)
O,: coefficient osmotique du sel 1, (sel le plus soluble de la famille), pour une molalité m;,OJ: coefficient osmotique du sel i (i*l) appartenant à la même famille que 1, pour la même
molalité m\.
Lorsque l'on porte la variation de ce rapport avec la concentration pour chaque electrolyte i,on aboutit à un réseau de courbes (Figure 2-3). L'extrapolation de ces courbes au-delà de lasaturation de l'électrolyte i permet d'atteindre la valeur du coefficient osmotique de solutionssursaturées.
62
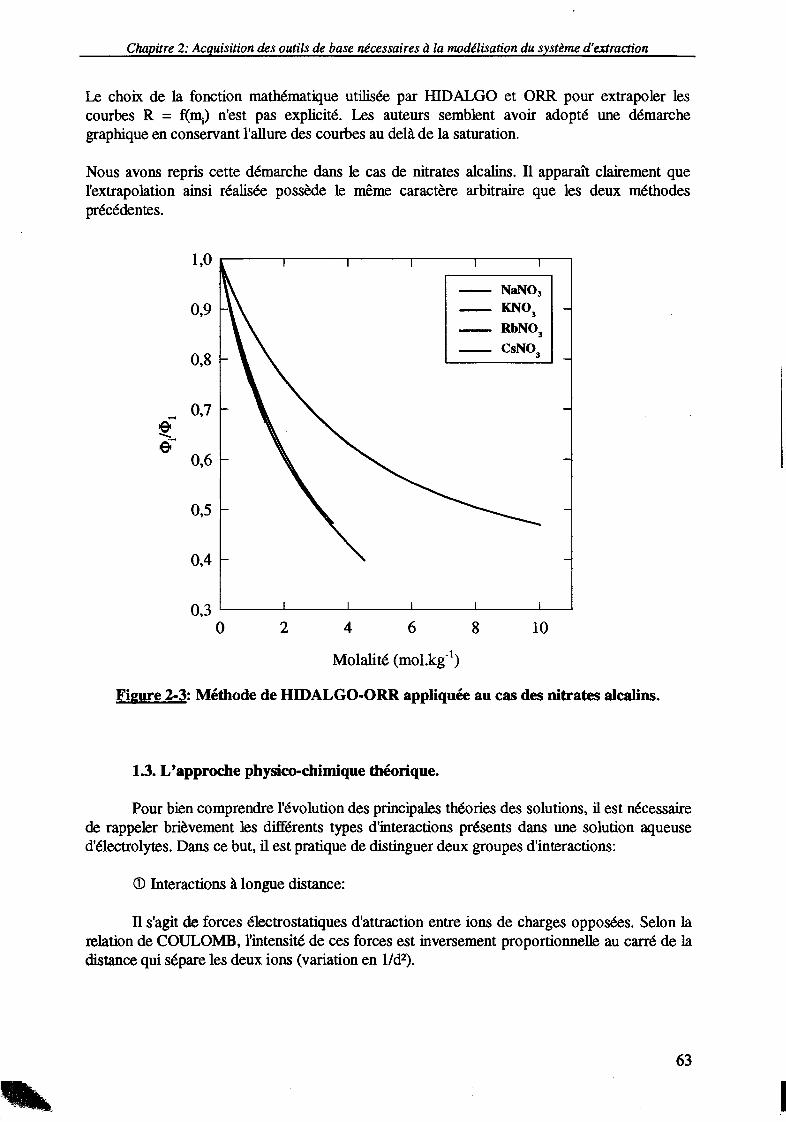
Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
Le choix de la fonction mathématique utilisée par HIDALGO et ORR pour extrapoler lescourbes R = f(m;) n'est pas explicité. Les auteurs semblent avoir adopté une démarchegraphique en conservant l'allure des courbes au delà de la saturation.
Nous avons repris cette démarche dans le cas de nitrates alcalins. Il apparaît clairement quel'extrapolation ainsi réalisée possède le même caractère arbitraire que les deux méthodesprécédentes.
0 2 4 6 8 10
Molalité (molJcgf1)
Figure 2-3: Méthode de HIDALGO-ORR appliquée au cas des nitrates alcalins.
13. L'approche physico-chimique théorique.
Pour bien comprendre l'évolution des principales théories des solutions, il est nécessairede rappeler brièvement les différents types d'interactions présents dans une solution aqueused'électrolytes. Dans ce but, il est pratique de distinguer deux groupes d'interactions:
© Interactions à longue distance:
H s'agit de forces électrostatiques d'attraction entre ions de charges opposées. Selon larelation de COULOMB, l'intensité de ces forces est inversement proportionnelle au carré de ladistance qui sépare les deux ions (variation en 1/d2).
63

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
L'influence de ces interactions électrostatiques sur l'écart à l'idéalité des solutions aqueusesd'électrolytes a été quantifiée pour la première fois par DEBYE et HÛCKEL [6] en 1923, enassimilant les ions à des charges ponctuelles.
© Interactions à courte distance:
II s'agit de forces attractives ou répulsives, de nature électrostatique ou non-électrostatique, à faible rayon d'action (par exemple, certaines de ces forces ont une intensitéqui varie en 1/d6). Par conséquent, on comprend que si dans les solutions diluées lesinteractions au sein de la solution sont imposées par les seules forces coulombiennes, lesinteractions à courte distance ne peuvent plus être négligées dès que la concentration enelectrolyte augmente.Elles interviennent entre les ions (aussi bien entre ions de charges opposées qu'entre ions demême charge), entre ions et molécules et entre molécules elles-mêmes.
C'est BR0NSTED [7] en 1922 qui a cherché le premier à quantifier ces interactions. Sa"théorie des interactions spécifiques" ne prend en compte que les interactions à courtedistance entre les ions de charges opposées.
En 1935, GUGGENHEIM [8] propose une équation du coefficient d'activité d'un electrolyteMX dans un mélange où apparaissent:
- le terme de DEBYE-HÙCKEL qui quantifie les interactions électrostatiques à longuedistance,
- un terme qui quantifie les interactions à courte distance selon le principed"interaction spécifique" défini par BR0NSTED.
L'équation est obtenue en introduisant certaines équations de mécanique statistique dans desrelations thermodynamiques.
L'évolution des théories des solutions est caractérisée par la prise en compte d'unnombre croissant d'interactions. Au terme initial de DEBYE-HUCKEL sont venues s'ajouterdes fonctions caractérisant les interactions à courte distance. L'élaboration de ces nouveauxmodèles est toujours motivée par la volonté d'atteindre un système d'équations utilisable sur leplus large domaine possible de concentration. Dans ce qui suit, nous présentons succinctementles principaux modèles rencontrés dans la littérature:
- modèle de BROMLEY (1973),
- modèle de PITZER (1973 à 1980),
- modèle de CHEN (1982).
Les deux premiers sont apparentés au modèle de GUGGENHEIM. Dans ses travaux,GUGGENHEIM développe les interactions à courte distance à l'aide d'un paramètre constant,indépendant de la concentration.
64

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
PITZER et BROMLEY, quant à eux, développent ce terme d'interactions à courte distancepour prendre en compte sa variation avec la concentration en electrolyte.
Le modèle de CHEN prend en compte les interactions à courte distance en faisant appel auconcept de "composition locale". Ce concept a déjà montré son intérêt dans le cadre de lamodélisation des écarts à l'idéalité dans les mélanges de non-électrolytes depuis 1964(modèles de WILSON, NRTL, UNIQUAC, UNIFAC).
Note:
A l'origine, tous ces modèles ont été élaborés pour représenter la variation desdonnées binaires (m^ yp aH 0) d'electrolytes. Plus tard, ils ont été proposés et utilisés pour
calculer les coefficients d'activité des constituants dans les mélanges d'électrolytes.Bien-sûr, les principes et les hypothèses de base sont inchangés lorsque l'on passe du systèmebinaire au mélange. Néanmoins, la complexité des équations et la multiplicité des paramètresd'interaction les rendent d'usage peu pratique.Par conséquent, la présentation suivante se limite au cas des solutions binaires.
1.3.1. Les modèles de BROMLEY et de PITZER.
Les travaux de BROMLEY et de PITZER datent du début des années 1970 et ont étémenés au sein d'une même équipe.
Ces travaux s'inspirent de la démarche de GUGGENHEIM [8], selon laquelle l'équation ducoefficient d'activité s'écrit comme la somme de deux contributions:
- la contribution des interactions à longue distance,
- la contribution des interactions à courte distance.
Cette équation, proposée en 1935 par GUGGENHEIM, s'écrit:
l n Y M X =
] , e t l1000 ; D.k.T
yMX: coefficient d'activité de l'électrolyte MpXq,Ay : coefficient de DEBYE-HÛCKEL pour le coefficient d'activité, dépend de la nature dusolvant et de la température,ZM, Zx: charge du cation, de Fanion,I: force ionique molale,PMX: paramètre d'interactions à courte distance indépendant de la molalité,mMX: molalité de l'électrolyte MpXq,D: constante diélectrique de la solution,No: nombre d'AVOGADRO,
65

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
dw: masse volumique du solvant,1: distance minimale d'approche entre M et X,e: charge de l'électron,k: constante de BOLTZMANN,T: température en Kelvin.
Le premier terme de cette équation correspond à la contribution des interactions à longuedistance au coefficient d'activité. Il s'agit de la relation de DEBYE-HUCKEL.Le second terme correspond à la contribution des interactions à courte distance.L'ajout du paramètre (PMX) d'interactions à courte distance permet d'ajuster les valeursexpérimentales du coefficient d'activité jusqu'à 0,1 mol.L"1, contre 1O3 mol.L-' pour l'équationde DEBYE-HUCKEL.
Dans le but d'élargir le domaine d'application de la relation de GUGGENHEIM,PITZER et BREWER [9] s'intéressent aux variations du terme d'interactions à courte distanceavec la concentration. Pour appréhender cette variation, les auteurs calculent la différence descoefficients d'activité entre deux electrolytes 1-1, MX et M'X' à la même concentration m.Dans ce cas, le premier terme de l'équation (41) s'annule:
lnYMX-lnyM.x. =2.m.(pMX-PM.x.) (42)
Le sel M'X' constitue le sel de référence; les auteurs choisissent le chlorure de potassium.
Le coefficient d'activité d'un electrolyte MX quelconque s'écrit alors selon:
(43)
Les valeurs de AB sont ainsi tabulées en fonction de la concentration pour quelqueselectrolytes. L'influence de la molalité sur la valeur de pMX est alors évidente.Dans le domaine des faibles concentrations, on observe une forte décroissance de la valeurabsolue de AB avec la concentration, tandis que pour des concentrations plus élevées, on tendvers une valeur constante (voir Figure 2-4 avec l'exemple du nitrate de rubidium).
66

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
-0,036
-0,038 -
-0,040 -
PQ<
-0,042 -
-0,044 -
-0,046 -
-0,048 -
-0,0500
Molalité (moLkg'1)
Figure 2-4: Variation du paramètre AB en fonction de la molalité pour RbNO3.
La variation du paramètre d'interactions PMX avec la concentration d'électrolyte ayantété mise en évidence, BROMLEY [10] d'une part, et PITZER [11] d'autre part, vont chercherà la mettre en équation. C'est à ce niveau que leurs approches divergent:
- BROMLEY se contente d'une approche purement empirique.
- PITZER utilise une théorie de mécanique statistique [12, 13], la théorie des "sphèresdures", pour suggérer plusieurs formes possibles de la fonction (3MX(m).
1.3.1.1. Le modèle de BROMLEY.
Pour ajuster au mieux les données expérimentales logyMX = f(I) de tout sel MpXq
totalement dissocié, BROMLEY propose l'équation:
(44)(1 + a.I)2
yMX: coefficient d'activité de l'électrolyte MpXq,A : coefficient de DEBYE-HUCKEL pour le coefficient d'activité,
67

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
ZM, Zx: charge du cation, de l'anion,I: force ionique molale,b: paramètre ajustable, dépendant de la distance minimale d'approche,a, Bo et B: paramètres ajustables.
La forme de cette fonction permet un bon ajustement des données jusqu'à une force ionique de6 mole.kg-1.
Les paramètres a, Bo et B n'ont aucune signification physico-chimique. Le paramètre b estintroduit pour prendre en compte la distance de moindre approche.
A partir d'un grand nombre d'essais d'ajustement BROMLEY conclut que:
•=> La valeur de b dépend de la valence du sel,
- b=l pour les sels 1-1,1-2,2-1,2-2, 3-2 et 1-4,
- b=l,4 pour les sels 3-1 et 1-3,
- b=l,6 pour les sels 4-1.
<=> Le paramètre a est inversement proportionnel au produit des charges ZM et Zx. Leproduit a.|ZM.Zx | peut varier entre 0,8 et 3, la valeur de 1,5 étant le plus souvent relevée.
^ Les valeurs de Bo et de B sont reliées par la relation suivante:
R ~ R =0,06 + 0,6.B (45)
L'équation empirique finale (pour b=l et a.|ZM.Zx| = l) ne comporte plus qu'un seulparamètre ajustable (B) par electrolyte:
-Ay.\ZM.Zx\.ï'2 (0,06 + 0,6.B).|ZM.Zxl.I
—I :— r a. 11+1» „ . W ,
7 7
Les valeurs de B ont été obtenues par ajustement de logyMX jusqu'à une force ioniquede 6 mole.kg1, (ou moins si la saturation est atteinte avant), selon la méthode des moindrescarrés.Les valeurs du coefficient d'activité calculées à l'aide de cette équation s'écartent au maximumde 5,1% des valeurs expérimentales dans cette zone de force ionique.L'extrapolation au-delà de 6 mole.kg-1 conduit à de forts écarts entre les valeurs calculées etexpérimentales en raison de la nature exponentielle de l'équation utilisée.Pour réduire cette erreur, BROMLEY propose de réaliser un ajustement sur tout le domainede solubilité en introduisant deux termes supplémentaires à l'équation précédente:
C.I3/2+D.I2 (47)
68

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
L'introduction de ces deux paramètres ajustables supplémentaires, C et D, est de nouveautotalement empirique.
Commentaires:
Le calcul du coefficient d'activité de solutions binaires sursaturées fictives de NaNO3
et de CsNO3 par l'équation de BROMLEY, reviendrait à extrapoler cette relation au-delà dudomaine d'ajustement des paramètres. Une telle extrapolation a été réalisée par TRAN et Coll.[5], dans le cas de l'acquisition de données sursaturées de KNO3. L'activité d'eau calculée àpartir de cette extrapolation est totalement erronée. En effet, la valeur de l'activité d'eauobtenue pour une solution sursaturée fictive est supérieure à la valeur expérimentale àsaturation, or l'activité d'eau est une fonction strictement décroissante de la concentration desel.
Cette limitation s'explique par le caractère purement "mathématique" de la fonction proposéepour quantifier les interactions à courte distance.
1.3.1.2. Le modèle de PITZER.
La démarche suivie par PITZER est plus rigoureuse que celle adoptée par BROMLEY.En effet, si le traitement des interactions à courte distance réalisé par BROMLEY s'est limité àune mise en équation mathématique, les investigations menées par PITZER possèdent unfondement physico-chimique. La mise en équation s'appuie sur un développement demécanique statistique qui bénéficie de l'apport du concept de "sphères dures".
Le concept de "sphères dures" [12,13] consiste à assimiler les particules à des sphèresrigides entre lesquelles n'existe aucune interaction. Seuls des chocs élastiques peuvent seproduire entre les sphères, ce qui constitue une des contributions à l'énergie du système (cettecontribution n'est pas prise en compte dans le modèle de DEBYE-HÙCKEL).En d'autres termes, ce concept permet de considérer le phénomène d'ordre et de désordre ausein d'un système, c'est à dire de quantifier, approximativement, la contribution entropique àl'enthalpie libre du système.
L'utilisation d'équations de la mécanique statistique et du concept de "sphères dures" conduitPITZER à établir une expression de l'enthalpie libre d'excès de la forme:
— =nHO.fex(I) + -.VX..(I).n..n.+-4-.yKi.k.n..n..nk (48)R-T 2 nH2O y nH20 ijk
Gex : enthalpie libre d'excès,R: constante des gaz parfaits,T: température,nH20, ns, n_j, nk : nombre de molécules d'eau, d'ions i, j ,k,
où: => £"(1) est une fonction de la force ionique qui quantifie l'influence des interactionsélectrostatiques à longue distance,
69

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
est une fonction de la force ionique qui quantifie les interactions à courtedistance entre deux ions de charges opposées ou identiques,
•=> nijk est un terme indépendant de la force ionique qui quantifie les éventuellesinteractions à courte distance entre trois ions.
PITZER est donc le premier à valider la démarche jusqu'alors arbitraire (et réalisée pour lapremière fois par GUGGENHEIM en 1935) qui consiste à représenter l'enthalpie libre d'excèscomme la somme de deux contributions (longue distance, courte distance).
L'expression du coefficient d'activité est obtenue par dérivation de la relation (48), soit,pour une solution binaire d'un electrolyte quelconque MpXq:
R.T dn(49)
9.vM.vx.m2
f-^- etddl * dl
lnyMX m: coefficient d'activité dans l'échelle des molalités de l'électrolyte MpXq,m: molalité de l'électrolyte MpXq,ZM, Zx: charge du cation, de l'anion,vM, vx: nombre d'ions M, X libérés lors de la dissociation totale d'une molécule de MpXq,V = V M+ V X-
Pour alléger l'écriture de la relation (48), PITZER opère le changement de variables suivant:
Yf = -.fex' (50a)
"MX
9( V M- U MMX + V X 4*MXX) ( 5 O C )
La relation (48) se réduit alors à l'expression:
x,m=Tf + m-TBMX(I) + m2:CMX (51)
70

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
Une expression en tout point semblable (dérivée de l'équation (48)) a été développéepour le coefficient osmotique. Cette relation fait intervenir les fonctions <l>f(I)et <PBMX(I),ainsi que le terme ""C,^. Ces fonctions sont reliées aux expressions (50a), (50b) et (50c) de lamanière suivante:
(52a)
(52b)
-2. , (52c)
La relation (48) et, par suite, les relations (49) et (51), correspondent à une expressionsynthétique de la variable "coefficient d'activité".En fait, au cours de son développement théorique, PITZER a obtenu une expressionanalytique des fonctions pf(I) et (pBMx(I) dans lesquelles apparaissent plusieurs paramètresd'interaction.
> Conceptuellement, la fonction %ITZER se distingue de la fonction de DEBYE-HUCKELfo.H par la prise en compte de la contribution entropique. Il semblait donc intéressant de
comparer la qualité des ajustements (sur le coefficient d'activité ou le coefficient osmotique)obtenus avec ces deux expressions. PITZER a ainsi testé l'ajustement des valeursexpérimentales du coefficient osmotique à l'aide des fonctions:
<Pf _ _ AAPITZER ~ ^ <
V'2(53a)
avec: a(b.II/2) =
1 fonction proposée par PITZER pour quantifier l'influence des interactions à longuedistance sur le coefficient osmotique,vfD.H'- fonction proposée par DEBYE-HUCKEL pour quantifier l'influence des interactions àlongue distance sur le coefficient osmotique,A^: coefficient de DEBYE-HUCKEL pour le coefficient osmotique,I: force ionique molale,b: paramètre dépendant de la distance minimale d'approche,CT(X): fonction empirique définie par DEBYE et HUCKEL.
> Quant aux fonctions ^BMX^) et YBMx(I)> c'est la première fois qu'une expression théoriquequantifiant les interactions électrostatiques à courte distance est proposée.
71

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
On ne peut donc la comparer qu'à une représentation purement "mathématique" desinteractions à courte distance (type démarche de BROMLEY).Il est important de remarquer que les représentations "mathématiques" englobent l'ensembledes interactions à courte distance, c'est-à-dire de nature électrostatique et non-électrostatique.Les fonctions comparées par PITZER sont:
PITZER (54a)
(54b)
p(0), P(1) et a : paramètres ajustables.
La fonction ^ B ^ ^ proposée par PITZER pour quantifier les interactions à courte distanceutilise trois paramètres, tandis que la fonction proposée par BROMLEY ne mettait en jeuqu'un seul paramètre.
PITZER a ainsi réalisé une série de tests portant sur l'ajustement des données expérimentalesdu coefficient osmotique relatives à 13 electrolytes dans une gamme de concentrationssupérieures à 2 molckg-1. Les quatre combinaisons suivantes ont été comparées:
4- "PRPITZER DPITZER
XPITZER °Math.
4 RD-H ^ DPITZER
La combinaison © a permis le meilleur ajustement. Les termes yf(I) et TBMX(I) calculés àpartir des fonctions Tpn-zgR et ^ B ^ ^ (utilisation des relations (52a) et (52b)) retenuesprécédemment sont de la forme:
1 2_
b(55a)
+ oc.I1/2 - -
Ap : coefficient de DEBYE-HÛCKEL pour le coefficient osmotique,I: force ionique molale,b, p(0), P(1), a : paramètres ajustables.
<=> A^ est égal à 0,392 à 25 °C pour l'eau.
(55b)
72

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
"=> Le paramètre b dépend de la distance de moindre approche entre les ions de mêmecharge. Pour réduire le nombre de paramètres ajustables, PITZER a cherché une valeurmoyenne de b conduisant à un ajustement acceptable pour un grand nombre d'électrolytes. Lavaleur b=l ,2 (pour T=25°C) a été retenue.Dès l'instant où ce paramètre est maintenu constant, le terme d'interactions à longue distanceYf(I) ou ^(1) est le même pour tous les electrolytes (à même force ionique).
•=> Les paramètres P(0), P(I), a et VCMX permettent de décrire les interactions à courtedistance. Ils doivent normalement être ajustés pour chaque electrolyte. Toujours dans le but deréduire le nombre de paramètres ajustables, une valeur de a égale à 2,0 (pour T=25°C) a étéretenue pour l'ensemble des electrolytes.
Les paramètres P(0), P(1) et <I>CMX ont été tabulés pour un grand nombre d'électrolytes parajustement des valeurs expérimentales du coefficient osmotique molal à 25°C, dans la gammede concentrations 0-6 mole.kg-1 [14]. L'utilisation de ces valeurs permet de calculer lecoefficient d'activité et le coefficient osmotique de solutions binaires d'électrolytes pour desconcentrations inférieures à 6 mole.kg-1.
Commentaires:
O Nous avons remarqué précédemment que la fonction f(I) (Yf ou vf) reste inchangéed'un composé à l'autre, lorsque le paramètre b est maintenu constant. Cela signifie donc queles éventuelles variations de la distance de moindre approche d'un electrolyte à l'autre sontenglobées dans la fonction B(I). Le choix d'une valeur unique du paramètre b conduit donc àfaire une confusion entre les interactions à longue et courte distance. Ce traitement n'est pasacceptable, d'autant que les deux types d'interactions possèdent des lois de variationdifférentes. Il apparaît donc nécessaire que b soit un paramètre à ajuster pour chaqueelectrolyte.
O La fonction retenue pour décrire les interactions à courte distance provient d'untraitement purement mathématique réalisé sur des données expérimentales. Cette démarchesouffre de toutes les limitations rencontrées lors d'un traitement mathématique des propriétésdes solutions d'électrolytes.
O L'équation développée par PITZER pour le coefficient osmotique a été utilisée parTRAN et Coll. [5] pour accéder aux valeurs de l'activité d'eau de solutions sursaturées. Lecomportement "en extrapolation" de la relation de PITZER a été jugé, selon les auteurs, bienmeilleur que celui de l'équation de BROMLEY. En effet, les valeurs d'activité d'eau calculéespar la relation de PITZER pour des solutions sursaturées sont, le plus souvent, en accord aveccelles obtenues par d'autres méthodes.
Malgré tout, il est légitime de mettre en doute l'extrapolation d'une telle équation, procédantpar ajustement mathématique des interactions à courte distance, dans un domaine deconcentrations où ce type d'interactions est majoritaire.
73

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
1.3.2. Le modèle de CHEN.
Le concept de composition locale pour modéliser les propriétés physico-chimiques dessolutions d'électrolytes a été mis en œuvre pour la première fois par CRUZ et RENON en1978 [15].
En 1982, CHEN [16] propose à son tour un jeu d'équations obtenu en utilisant ce mêmeconcept. Toutefois, des hypothèses de départ différentes permettent à CHEN de réduire lenombre de paramètres ajustables et d'élargir le domaine de concentrations accessibles.Dans ce modèle, les propriétés thermodynamiques des solutions d'électrolytes sont denouveau étudiées en distinguant les contributions des interactions à longue et courte distance.Le traitement des interactions à longue distance effectué par PITZER [11] est reprisintégralement.Les interactions électrostatiques et non-électrostatiques à courte distance sont abordées par lebiais du concept de composition locale.Les travaux de CHEN permettent d'aboutir à une relation de l'enthalpie libre d'excès et à uneexpression du coefficient d'activité d'un electrolyte fort en solution.Ces deux expressions sont exprimées dans l'échelle des fractions molaires. Cette échelle deconcentration est inhérente au modèle de composition locale utilisé pour développer lesinteractions à courte distance.
Le traitement des interactions à longue distance reprend les résultats du calcul demécanique statistique effectué par PITZER.La relation originale proposée en 1973 [11] dans l'échelle des molalités a ensuite ététransposée par PITZER [17] dans l'échelle des fractions molaires. La relation reliant lesexpressions des coefficients d'activité dans les deux échelles de concentration est:
. . . .(56)V 1000
y MX x : coefficient d'activité dans l'échelle des fractions molaires,YMx,m: coefficient d'activité dans l'échelle des molalités,Ms: masse molaire du solvant,m: molalité de l'électrolyte MX.
Dans l'échelle des fractions molaires, la contribution des interactions à longue distance(suffixe "ld") au coefficient d'activité d'une solution d'un electrolyte uni-univalent s'écrit:
1/2 r2 / inv ( i f - 2 i f y-•lnl + p.lf A .,*/ (57)P
A^: coefficient de DEBYE-HUCKEL pour le coefficient osmotique,p: paramètre dépendant de la distance de moindre approche,
Ix = — .]jr Zf .xs : force ionique dans l'échelle des fractions molaires,^ i
x{: fraction molaire de l'anion, du cation,
74

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
Z{. charge de l'anion, du cation.
Le paramètre de moindre approche, (noté p dans l'échelle des fractions molaires et b dansl'échelle des molalités), varie d'un electrolyte à l'autre. Comme nous l'avons déjà dit, et dans lebut de conserver une relation simple, les auteurs ont l'habitude de maintenir ce paramètreconstant.PITZER [11] retient une valeur de b égale à 1,2, ce qui correspond dans l'échelle des fractionsmolaires à une valeur de 8,94.Lors de l'étude des propriétés thermodynamiques de solutions de sels fondus, PITZER [17] arevu l'ajustement de ce paramètre. L'étude portait alors sur deux solutions de nitrate à unetempérature de 100°C. La valeur de p alors ajustée était égale à 14,9 (soit b=2 dans l'échelledes molalités), indiquant que p dépend de la température (voir relation (41)).Curieusement, c'est cette dernière valeur qui est retenue par CHEN bien que l'ensemble destravaux de CHEN soit effectué à une température de 25°C.
Une valeur de A^ égale à 0,391 (pour l'eau et à une température de 25°C) est retenue parCHEN.
Les interactions à courte distance sont considérées en mettant en œuvre le concept decomposition locale.A l'origine, plusieurs modèles de composition locale ont été développés pour étudier lespropriétés thermodynamiques des solutions de non-électrolytes (cf Chap.l §2.1.). Citons:
- le modèle de WILSON,
- le modèle NRTL de RENON-PRAUSNITZ,
- le modèle UNIQUAC d'ABRAMS-PRAUSNITZ,
- le modèle UNIFAC de FREDENSLUD-PRAUSNITZ.
Selon ce concept, il est possible de distinguer au sein d'un mélange de non-électrolytes,plusieurs types de "cellules" différant par leur composition.
Dans le but d'étendre ce concept aux solutions d'électrolytes, CHEN a édicté deux règles:
© La composition locale des cations (des anions) autour des cations (respectivementdes anions) est nulle. Cette hypothèse signifie que les forces de répulsion entre les ions demême charge sont très importantes.
© La distribution des anions et des cations autour d'une molécule neutre de solvant esttelle que la charge locale est nulle. C'est le principe d'électroneutralité locale.
Afin de satisfaire à ces deux règles, il est possible de distinguer trois types de "cellules" tellesque:
75

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
Les cellules © et © répondent à la règle de répulsion entre deux ions de même charge.Les cellules ©+© et la cellule (D répondent au principe d'électroneutralité.
Parmi les différents modèles de composition locale existants, CHEN adopte la démarcheretenue par RENON et PRAUSNITZ [18] lors de l'élaboration de leur modèle NRTL ("NonRandom Two-Liquids").
Selon le type de cellules étudié, CHEN introduit différents paramètres d'interactions àcourte distance.
> Dans le cas de la cellule (D, CHEN définit les paramètres d'interactions à courte distance dela manière suivante:
= (gji —So)
R.T(58)
i correspond à la molécule d'eau centrale (indice m),j correspond à l'espèce périphérique, c'est à dire l'un des deux ions (indice a ou c) ou unemolécule d'eau.Les termes gjj et g représentent les énergies d'interactions à courte distance entre lesdifférentes espèces présentes dans la "cellule". Ces énergies sont bien-sûr symétriques, onpeut donc écrire l'égalité suivante: gj; = g~.
Au sein de cette cellule, le traitement des interactions à courte distance fait intervenir les deuxparamètres xam et xcm (Tmm = 0).
76

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
> Dans le cas des cellules © et ©, CHEN définit les paramètres d'interactions à courtedistance en employant une notation similaire:
T =1Ji.ki R.T
(59)
i correspond à l'ion central, (le cation dans la cellule ©, l'anion dans la cellule ©).j correspond à une des deux espèces périphériques (l'anion ou la molécule d'eau dans la cellule©, le cation ou la molécule d'eau dans la cellule ©).k correspond à la seconde espèce périphérique.
Note:
La notation employée pour définir les paramètres d'interactions à courte distance dans la"cellule" Œ> ne peut être étendue aux "cellules" (Det ®. En effet, afin de satisfaire la règle derépulsion entre ions de même charge, nous avons vu précédemment qu'au sein des cellules (Det ©, la charge de l'ion périphérique est forcément différente de celle de l'ion central. Dèsl'instant où deux ions de même charge ne peuvent être présents dans la même "cellule",l'énergie d'interaction gu ne peut intervenir dans l'expression du paramètre d'interactions àcourte distance r^. Les auteurs choisissent une nouvelle notation, et introduisent l'indice k.
Le traitement des interactions à courte distance effectué dans les cellulesrésumé sous la forme du tableau suivant:
et © peut être
Tableau 2-1: Traitement des interactions dans les deux cellules.
Type de "cellule"
i
j
k
gk.
Tji,ki
©
cation (c)
anion (a) ou H2O (m)
H2O (m) ou anion (a)
g» ou gmc
gmc on gac
ac,mc mc,ac
©
anion (a)
cation (c) ou H2O (m)
H2O (m) ou cation (c)
gca OU g™
gma O U gca
ca,ma ma,ca
Le traitement des interactions à courte distance dans ces deux cellules fait intervenir les deuxparamètres: TaC)mc (= Tmc ac) et xa>n> (= xma,ca).
La règle d'électroneutralité locale édictée par CHEN se traduit par une égalité entre lesénergies d'interactions g m et gcm. Cette égalité permet de réduire le nombre de paramètresd'interactions de quatre à deux:
mc.ac l ma.ca m.ca
77

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
am cm ca,m
Les paramètres d'interactions Tm ca et x^m sont introduits par CHEN dans le but d'allégerl'écriture.
Ces paramètres d'interactions sont introduits dans les équations thermodynamiquesdéveloppées par CHEN grâce au modèle de composition locale. La contribution desinteractions à courte distance (suffixe "cd") au coefficient d'activité d'un electrolyte uni-univalent s'exprime alors de la manière suivante:
j X .T . G X.T .X . ( Ji cd m ca,m ca,m m,ca m m,cam JMX,x ~ / \2 ~i vT
(2.x.Gcam+xm) (x + xm.Gmca)
/ r^. \ m,ca ca.m" ca,m
Gca,m=exp(-a.Tcam)
Gm,ca=exp(-a.Tmca)
x, x,,,: fraction molaire d'un ion, de l'eau,xcam, Tmca: paramètres d'interactions à courte distance,a: terme de "non hasard".
Le terme de "non hasard" trouve son origine dans le modèle NRTL développé par RENON etPRAUZNITZ en 1968 pour des solutions de non-électrolytes. L'arrangement des molécules(eau et soluté) dans de telles solutions ne se fait pas au hasard, et satisfait à un certain nombrede règles.De même, c'est la distribution non aléatoire des solutés qui prévaut dans les solutionsd'électrolytes, et les principes qui conditionnent l'arrangement des ions et molécules d'eaudans le modèle de CHEN sont les deux règles édictées par l'auteur (répulsion entre les ions demême charge et électroneutralité locale). Le terme de "non hasard" prend donc en compte ceslois de distribution dans l'expression de la contribution des interactions à courte distance auxpropriétés thermodynamiques des solutions d'électrolytes.
A la suite d'un ajustement, CHEN retient une valeur de a égale à 0,2 pour l'ensemble deselectrolytes, à une température de 25°C.
L'expression finale du coefficient d'activité d'un electrolyte en solution est obtenue enadditionnant les deux contributions ; ln yld et ln fà.
l n Y MX,x = l n Y MX.x + l n Y MX.x ( 6 1 )
L'utilisation de cette expression passe par l'ajustement des deux paramètres d'interactions àcourte distance; xcam et tmca. D'autre part, et comme nous l'avons déjà souligné, il estnécessaire d'ajuster le paramètre de moindre approche p.
78

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
Commentaires:
O Une des principales qualités du modèle de CHEN par rapport aux autres travauxdéjà présentés, réside dans le traitement des interactions à courte distance. En effet,l'utilisation du concept de composition locale permet de prendre en compte à la fois:
- des interactions entre les ions,
- des interactions entre un ion et une molécule d'eau,
- des interactions entre les molécules d'eau.
Les interactions à courte distance sont majoritaires dans le domaine des fortes concentrations,aussi un meilleur traitement de ce type d'interactions permet d'envisager l'étude de solutionsfortement concentrées. CHEN a ainsi appliqué avec succès son modèle aux solutions de selsfondus.
O Le modèle de composition locale de CRUZ et RENON [15] n'est pas applicable surun domaine de concentrations aussi étendu. En effet, lors des développements du modèleNRTL, les auteurs font l'hypothèse que les ions sont entourés d'une coquille de solvatation.Cette hypothèse semble inaproppriée dans le domaine des fortes concentrations. En effet, lenombre de molécules d'eau n'est plus suffisant pour assurer la solvatation complète des ions.D'autre part, ce modèle fait intervenir quatre paramètres ajustables par electrolyte (contre troispour le modèle de CHEN).; deux pour les interactions à longue distance et deux pour lesinteractions à courte distance.
O Nous avons vu que le concept de composition locale employé par CHEN permet unmeilleur traitement des interactions à courte distance. Ce type d'interactions étantprépondérant au sein des solutions fortement concentrées (et, qui plus est, "sursaturées")l'utilisation de la relation de CHEN en mode "extrapolation" semble être a priori tout à faitindiquée pour répondre à notre problème d'obtention de données binaires de solutions binairessursaturées.Aussi, parmi l'ensemble des méthodes semi-empiriques présentées, la relation de CHENconstitue le moyen le plus prometteur d'acquisition des données binaires de solutionssursaturées de NaNO3 et de CsNO3.
1.3.3. Etude du comportement "en extrapolation" des modèles de PITZER et deCHEN
Afin de déterminer la ou les méthodes de calcul les plus appropriées pour acquérir desdonnées binaires, nous avons effectué une série de tests consistant à comparer les valeurscalculées aux valeurs expérimentales publiées dans la littérature.
Ces tests portent sur des electrolytes uni-univalents dont les données binaires couvrent unlarge domaine d'activité d'eau. Il s'agit des nitrates d'ammonium (NH4NO3), d'argent (AgNO3)et de sodium (NaNO3).La similitude des courbes yMX = f(mMX) que présentent ces sels par rapport à CsNO3 (voirFigure 2-5) en fait également des electrolytes de choix dans le cadre de cette étude.
79

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
8 10 12 14 16
Molalité (mol.kg1)
18 20 22 24
Figure 2-5: Coefficient d'activité de NaNO3, NH4NO3, AgNO3, CsNO3 [1,2].
La démarche suivie lors des tests peut être décomposée de la manière suivante :
© Les différentes méthodes envisagées sont mises en œuvre (étape d'ajustement desparamètres) à partir de données expérimentales tronquées, c'est-à-dire ne couvrant pas latotalité du domaine de concentration publié.Pour éprouver la méthode et déceler ses limites, plusieurs domaines d'ajustement serontétudiés. En effet, une théorie des solutions fiable doit permettre d'atteindre des valeursconstantes des paramètres d'interactions et ce, quel que soit le domaine de concentrationretenu lors de l'ajustement.
® Les données binaires correspondant au domaine précédemment omis sont calculées àl'aide des différentes méthodes et de leurs paramètres associés ajustés au cours de l'étape 0 ; ils'agit donc bien d'une extrapolation.Cette étape simule l'acquisition de données binaires de solutions sursaturées.
G) Les valeurs ainsi calculées sont comparées aux valeurs expérimentales effectivementdisponibles.
Le comportement "en extrapolation" des relations de PUZER et de CHEN dépend :
0 de la qualité du traitement théorique des interactions. Ce point a déjà été commentélors de la présentation de chacune des méthodes. On sait que les deux modèles ne se
80

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
distinguent que par la relation mise en jeu pour quantifier les interactions à courte distance.Différentes remarques ont permis de conclure, qu'a priori, le modèle de CHEN repose sur desfondements plus solides que ceux adoptés par PITZER.
© de la qualité et de la répartition des interactions effectivement présentes dans lessolutions MNO3 / H2O considérées lors de l'étape d'ajustement des paramètres. Dans lessolutions infiniment diluées, seules les interactions à longue distance entre les ions existent.Lorsque la concentration augmente, la contribution de ce type d'interactions aux propriétésphysico-chimiques de la solution augmente jusqu'à atteindre une valeur constante. En mêmetemps, une contribution supplémentaire apparaît ; les interactions à courte distance.Au sein des solutions concentrées (et à plus forte raison sursaturées), ces interactions à courtedistance deviennent majoritaires. A domaine de concentration identique, l'ajustement desparamètres conduit à des valeurs d'autant plus "justes" que la proportion de ce typed'interactions est plus élevée. En effet, il n'est pas raisonnable de penser atteindre une valeur"juste" des paramètres représentatifs des interactions à courte distance si ce type d'interactionsest tout-à-fait mineur dans les solutions MNO3/H2O considérées. La valeur obtenue aprèsajustement n'aurait alors plus qu'un sens mathématique.En d'autres termes, on peut s'attendre à ce que les écarts entre les valeurs calculées etexpérimentales soient d'autant plus faibles que la nature des interactions présentes dans ledomaine d'ajustement des paramètres est proche de celles rencontrées dans le domained'extrapolation (fortes concentrations).
Pour les trois electrolytes choisis pour effectuer les tests, la part des interactions à courtedistance croît dans l'ordre suivant [19] :
N a N O 3 , NH4NO3, AgNO 3
On s'attend donc à un meilleur comportement en extrapolation dans le cas des solutions denitrate d'argent. Les figures sont reportées dans l'annexe 2 (figures A2-1 à A2-12).
1.3.3.1. Comportement "en extrapolation" de la relation de PITZER.
On a comparé deux situations :
© Ajustement des paramètres d'interactions à courte distance (5^, P ^ e t ^ C ^ , leparamètre d'interactions à longue distance, b, étant fixé à 1,2 (valeur préconisée par PITZER).
® Ajustement des paramètres d'interactions à courte distance $°\ $^' et ""C^ et duparamètre d'interactions à longue distance b.
© L'analyse des figures A2-1 à A2-3 met en évidence le mauvais comportement enextrapolation de la relation de PITZER et ce, quel que soit le domaine d'ajustement desparamètres.Le mauvais comportement en extrapolation de la relation de PITZER peut provenir de lafaiblesse théorique du traitement des interactions à courte distance fait par l'auteur.
81

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
D'autre part, contrairement au comportement attendu, l'amplitude des écarts entre les valeursexpérimentales et calculées varie dans le sens :
NaNO3 < NH4NO3 < AgNO3
L'évolution des écarts peut s'expliquer par la confusion faite entre les interactions à longue etcourte distance lorsque le paramètre b reste fixé (cf § 1.3.1.2.).
© L'ajustement du paramètre de moindre approche devrait permettre d'éviter cettecause éventuelle d'erreur.
Dans le cas de NaNO3 et de NH4NO3, on constate que le comportement en extrapolation n'estpas amélioré (Figures A2-4 et A2-5). Les valeurs de b obtenues par ajustement des donnéesexpérimentales de NaNO3 et de NH4NO3 restent proches de 1,2 quel que soit le domaine
d'ajustement. Quant aux paramètres P ^ , P^ et PCMX, ils conservent eux aussi des valeursproches de celles ajustées précédemment.Le mauvais comportement en extrapolation ne peut donc s'expliquer que par l'approchemédiocre utilisée par PITZER pour quantifier les interactions à courte distance (approchepurement mathématique).
Dans le cas de AgNO3, l'ajustement du paramètre de moindre approche permet de diminuerconsidérablement les écarts entre valeurs calculées et expérimentales du coefficient d'activité(Figure A2-6). Les valeurs de b ajustées sont comprises entre 0,35 et 0,50 lorsque le domained'ajustement est réduit jusqu'à 3 mol.kg-1. Cependant, lorsque l'ajustement des paramètres esteffectué dans le domaine de concentration 0-1,4 mol.kg1, la valeur de b est multipliée par unfacteur 4 (b=l,2).Les écarts relevés entre les valeurs du paramètre b mettent ainsi en évidence l'erreurimportante que l'on peut commettre lors du traitement des différents types d'interactions aumoyen de la relation de PITZER.
Toutefois, on remarque que pour l'ensemble des electrolytes testés, les écarts relevés restentfaibles tant que le domaine d'ajustement des paramètres correspond à un domaine deconcentration supérieur ou égal à la moitié de la molalité à saturation. L'ensemble desparamètres ajustés conservent alors des valeurs proches de celles déterminées par ajustementsur tout le domaine de concentration disponible.
Le comportement en extrapolation de la relation de PITZER semble donc autoriserl'acquisition de données binaires sursaturées jusqu'à des concentrations égales à deuxfois la molalité à saturation.
82

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
1.3.3.2. Comportement "en extrapolation" de la relation de CHEN.
Les paramètres ajustés lors des tests sont :
© Les deux paramètres d'interactions à courte distance TcametTmca, le paramètred'interactions à longue distance, p, étant fixé par CHEN à 14,9.
© Les deux paramètres d'interactions à courte distance Tcam etxmca , et le paramètred'interactions à longue distance p.
© Les figures A2-7 à A2-9 présentent un comportement en extrapolation de la relationde CHEN décevant. Les écarts entre les valeurs calculées et expérimentales du coefficientd'activité des trois electrolytes sont, dans tous les cas, supérieurs aux valeurs mesurées lors del'extrapolation de la relation de PITZER.
© Comme nous l'avons vu précédemment, le comportement en extrapolation doit êtreamélioré lorsque le traitement des différents types d'interactions inclut l'ajustement duparamètre de moindre approche.
L'examen des Figures A2-10 à A2-12 indiquent que l'ajustement du paramètre p permet dediminuer fortement les écarts entre les valeurs calculées et expérimentales.Comme attendu, les écarts sont d'autant plus faibles que la part des interactions à courtedistance est importante. Dans tous les cas, le comportement en extrapolation de la relation deCHEN est plus satisfaisant que celui obtenu avec la relation de PITZER.
L'acquisition des données binaires de solutions sursaturées de NaNO3 et de CsNO3 aumoyen de la relation de CHEN semble donc être la méthode de choix.
Malgré tout, lorsque le domaine d'ajustement des paramètres est réduit à 0-1,4 mol.kg-1,l'incertitude calculée sur les valeurs des différents paramètres d'interactions est élevée. Dans lebut de limiter l'erreur commise lors de l'extrapolation de la relation de CHEN, il apparaît doncplus raisonnable de réaliser l'ajustement des paramètres du modèle sur des domaines deconcentration au moins égaux à 0-3 mol.kg-'.
83

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
1.4. L'approche physico-chimique expérimentale.
1.4.1. La méthode de KUSSIK-MEISSNER-TESTER.
Les travaux de KUSSIK-MEISSNER-TESTER (KMT) débutent en 1972 [20], c'est-à-dire en même temps que les études de BROMLEY et de PITZER.
Le but initial de l'étude menée par KMT est de mettre au point une méthode graphique simple,permettant de représenter les variations du coefficient d'activité d'électrolytes forts surl'ensemble de leur domaine de solubilité.
L'élaboration de cette méthode graphique s'appuie, à l'origine, sur les valeursexpérimentales du coefficient d'activité d'électrolytes en solution. Dans un premier temps, lesauteurs étudient les variations expérimentales du coefficient d'activité de 21 electrolytes enfonction de la force ionique. Ces electrolytes sont de type 1-1, 2-1, 1-2, 3-1 et 3-2, la forceionique variant de 0 à 2 mol.kg-1.
La figure obtenue est assez confuse, les courbes y MX = f(i) présentant de nombreusesintersections (voir Figure 2-6). Cependant, les auteurs remarquent qu'à l'intérieur d'une mêmeclasse d'électrolytes (même stœcbiométrie), les différentes courbes ne se croisent pas etpossèdent des variations voisines.
2,0
"3
oU
1- 2
9- 4
5- 6
7- 8
9
II
-
21-.
"l9>s
i i
HCIHCI04
LiBr
KOHNH4N0,AflNO3
LiCI04
Co(CIO4L
Sr(CI04)e
CeBre
i i | i i
I 2 C O I £
13 Mld2
MCuCle
15 C«(NO^2KCflfNO,^«7LI2SO4
W (NH4)8S04 /19 C«S04 /
2?ÎICIS°4)* / ^ * 4
—^ ^l^Jy
_———^^^^ ————M
^ ~ 16 •
"
1 1
* /
/
/ y/ 7JL
f / y f%%1 s /
^ ^
14
17——^———
1,0Force ionique (mol/kg)
2,0
Figure 2-6: Variation du coefficient d'activité de 21 electrolytes [20].
Afin de s'affranchir de la différence de stoechiométrie entre les différents electrolytesprésents en solution, KUSSIK introduit une nouvelle variable nommée "coefficient d'activitéréduit". Pour un electrolyte MpXq, le coefficient d'activité réduit FMX est défini comme:
rMx=(ï MX (62)
84

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
: coefficient d'activité de l'électrolyte MV MXV ,ZM, Zx: charge du cation, de l'anion.
Le tracé des courbes FMX = f(l) permet d'aboutir à une seule famille de courbes: quel que soit
le type de l'électrolyte, les courbes FMX = f(l) ne se croisent plus.D'autre part, pour des forces ioniques inférieures à 0,01 mol.kg-1, l'ensemble des courbesconvergent vers une même fonction. Cette fonction correspond à la loi limite de DEBYE-HÛCKEL qui, changement de variable opéré, s'exprime de la manière suivante:
logrMX = -0,5107.I1/2 (63)
Selon KMT, la mise en évidence d'une telle similitude entre les variations expérimentales ducoefficient d'activité réduit permet d'envisager une généralisation à l'ensemble deselectrolytes. Les auteurs généralisent avec succès leur étude graphique à plus de 120electrolytes. Seules les courbes relatives à une dizaine d'électrolytes présentent desintersections avec les courbes voisines.
La connaissance d'une seule valeur expérimentale du coefficient d'activité réduit d'unelectrolyte en solution doit permettre de localiser la courbe FMX = f (i) de cet electrolyte.
Il est alors possible:
© d'accéder à la valeur du coefficient d'activité pour des concentrations allant de 0 à lamolalité de saturation,
© d'envisager d'accéder à la valeur du coefficient d'activité d'un electrolyte dans unesolution binaire sursaturée.
Dans le but de faciliter l'exploitation graphique des fonctions F ,^ = f(i), les auteurschoisissent de présenter un nombre limité de courbes régulièrement espacées. Un réseaud'abaques est ainsi élaboré à partir des courbes expérimentales des 110 electrolytesprécédemment retenus. Les tracés obtenus ne correspondent donc plus nécessairement à lavariation expérimentale du coefficient d'activité réduit d'un electrolyte précis.
KMT proposent deux abaques. L'un permettant de couvrir le domaine de force ionique 0 à 2mol.kg-1, l'autre le domaine 0 à 20 mol.kg-1 (Figures 2-7 et 2-8).
85

00
n
crM
ffi«a
i- 6
o»t
ao3
na
©•
HS
3e
I
ooCD
gc
e
00O
K)O
o
iogr
to o\
,i#
yf
/
1t
/i
j/lt
1 V-TTXUn yï/ïm itv/y/y/nJWSJw
N\luAL InlSflc;
1 /fl/lJUJL
Tn7 / nni/NI /f\l\ I / 1/ fl I» / " "F ;i /n u/ "L
/I 1// r/ fr i/ fl7 /n \f\\ \
u " iuf/1fl///
tiii
/1/iu1I1111
T1 I1 I1 In 1n 1! 111 '
TIfL
i
vI1
liraPwu\l|V\II\\\\\\
\\W\YA\\\
"s\w
\
j \K\\A
1 |\ l X \n \\\\n i \ i\ i\
I
rttTT
1 \ \ \
JLV
>^
\
v\
y\\I u\ \ \1M i \ \
111 R n \[
V
,\\\ ii\
i
SS
y.sv'V
\
\\\
\i 1 \ i \ \ \ l \U 1 \ \ u i1 l \ ï \1 i l X 1 X11KlII1 11
U 11 1
U 1 1 |\
\SssA\
.
\i\
\\\
\
\\
1 l B \ \i R \ \ M1 i \ iI \ u IA l l Rn ni i il
\
V
s
L
\
\
\
y\\\i\\\
\\.
\
c*\
\
rT
\\
\
vs\\\\\\} i
1\
\S\
ssv
y\
\l
\
\<
s
\>
v
V
V
y
V\
V
i t
*
.
\
\
\
v
\y
\
\
i\
L
\
\
i
\
*
s
\>
i
\
I
II
o"J
m"
ao3»anan0aSn
3o
I
îogr
o
nS's
• S ' • -B on O
?o
oK)O
f
A
À
i
J
JA
à
i
1
'Ai
nm\ïhim \Nm\\\fli\\\\\WiW////1/lllllllM/y/l/l/IIIIIIIUM Md//rl / l l l l l l lM\IN
i /I l/l 1 1/1 n 1' 1 i 1 fl • 1 il I1 / M ri 11/È f Lr F NI II
/ / / M/ / n\\\i h Mu1 \ F
\I I
nuI1HK\ U >
\
l i i KM NH\\H\ \\n\\N\
\
\\
\\>

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
L'utilisation de ces abaques se décompose de la manière suivante:
© Une valeur expérimentale du coefficient d'activité est utilisée pour calculer la valeurdu coefficient d'activité réduit de l'électrolyte en question.
© Cette valeur "expérimentale" est reportée sur l'un des abaques.
® La courbe log FMX = f (i) se situant le plus près de ce point expérimental est retenuepour décrire la variation du coefficient d'activité réduit de l'électrolyte sur le domaine de forceionique couvert par le graphique.
Ainsi mise en œuvre, la méthode graphique de KMT reste d'un usage peu pratique. Lorsquel'on désire accéder à la valeur du coefficient d'activité réduit à différentes forces ioniques, ilest nécessaire de relever graphiquement chacune des valeurs de logFMX. Cette démarche serévèle rapidement fastidieuse et reste peu précise.
Afin de permettre une utilisation plus facile de sa méthode, KUSSIK [21] propose uneéquation assurant un bon ajustement des courbes logFMX = f(l) précédemment retenues.
L'équation proposée ne comporte qu'un seul paramètre ajustable (q) par electrolyte:
logF^ =log[l + B.(l + 0,l.l)q -BJ + logr* (64)
B = 0,75-0,065.q
. _ . -O,51O7.I'/2
avec log F = ^—* 1 + C.I1/2
C = l + 0,055.q.exp(-0,023.I3)
F: coefficient d'activité réduit,I: force ionique molale,q: paramètre ajustable qui dépend de l'électrolyte et de la température,log F est égal à log F* dans le domaine des solutions infiniment diluées soit:
lim(log F) = limflog F * ) = -0,5107.11/2.
Les courbes F ,^ = f(l) obtenues à partir de la relation (64) ont été regroupées par KUSSIKsur une seule figure (Figure 2-9).
87

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
es
•**
'3
oU
H // M////
0,1
Force ionique (mol.kg" )
Figure 2-9; Abaques TMX = f (i) [21].
88

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
KMT [21] ont également tabulé les valeurs du paramètre q à 25°C pour 120 electrolytes. Cesont les valeurs expérimentales du coefficient d'activité de ces electrolytes qui ont étéprécédemment utilisées pour tracer les abaques (2-7) et (2-8). Les auteurs n'indiquent pas laméthode de calcul de ces paramètres. Vraisemblablement, les valeurs de q ont été obtenuespar ajustement des valeurs expérimentales disponibles du coefficient d'activité et selon laméthode des moindres carrés.Signalons qu'aujourd'hui de nouvelles valeurs expérimentales des coefficients d'activitéd'électrolytes peuvent être disponibles (avec, en principe, des précisions améliorées parrapport aux valeurs anciennes). D'autre part, des outils informatiques plus performantsdevraient permettre d'améliorer l'ajustement du paramètre q. Par conséquent, nousrecommandons de reprendre la démarche proposée par les auteurs en 1978:
© Une valeur expérimentale du coefficient d'activité est utilisée pour calculer la valeurdu coefficient d'activité réduit de l'électrolyte en question.
© La résolution de l'équation (64) à l'aide de cette valeur "expérimentale" permetd'accéder à une valeur de q.
(D Cette valeur du paramètre q permet de calculer (équation (64)) le coefficientd'activité réduit quelle que soit la force ionique.
La méthode ainsi définie par KMT ne suit donc plus une démarche graphique. La relation (64)permet de s'affranchir de tous les problèmes rencontrés lors de la détermination graphique, àsavoir ; choix de la courbe logFMX = f(l) et lecture des valeurs de logFMX.
Commentaires:
O En 1974, la méthode graphique de KMT a été mise en œuvre par TRAN [5] pourcalculer les données binaires de solutions sursaturées. Les valeurs d'aH2Û ainsi calculées ontété comparées à celles obtenues par d'autres méthodes. Pour les electrolytes les moinssolubles, l'utilisation de la méthode graphique de KMT se caractérise par une incertitudeélevée due à la lecture graphique.
O KMT, tout comme BROMLEY, proposent une équation du coefficient d'activité nefaisant intervenir qu'un seul paramètre ajustable par electrolyte.Cependant, la démarche suivie par KMT fait suite à une étude générale sur le comportementthermodynamique d'un très grand nombre de solutions binaires d'électrolytes. Tous leselectrolytes étudiés ont présenté une caractéristique commune lorsque les études decomportement portaient sur la variable "coefficient d'activité réduit". De plus, la fonctionretenue par KMT permet de modéliser la variation du coefficient d'activité réduit sur undomaine beaucoup plus étendu de force ionique, que celui pris en compte par BROMLEY.
O L'utilisation de la méthode KMT "révisée" de 1978 devrait limiter l'incertitude surles valeurs des données binaires sursaturées calculées par cette méthode.Les courbes FMX = f(l) présentées sur la Figure 2-9 divergent lorsque la force ioniqueaugmente. L'utilisation d'une donnée expérimentale du coefficient d'activité à une faible
89

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
valeur de la force ionique risque donc d'entraîner une erreur importante lorsque l'on sedéplace vers des solutions concentrées.
L'acquisition de données binaires sursaturées par la méthode KMT semble donc plusprometteuse dans le cas des solutions de NaNO3 que de CsNO3, les données expérimentalesdu coefficient d'activité de CsNO3 n'étant disponibles que sur un domaine étroit de forceionique.
1.4.2. La méthode de ZDANOVSKII-STOCKES-ROBINSON.
Dans le cas de mélanges ternaires d'électrolytes M pX q ( l ) /M pX q (2) /H 2O. La
relation ZSR s'écrit, dans l'échelle des molalités, de la manière suivante (cf chapl. §3.2.2):
2±L + .mwv
MXi
On dénombre cinq variables expérimentales ;
- mMX), mMXj : les concentrations des electrolytes dans le mélange,
- m^x , m^x : les concentrations des electrolytes dans leur solution binaire respective
de même activité d'eau que celle du mélange,
- aH 0 : l'activité d'eau du mélange.
Si la composition du mélange ( mMX|, mMXj ) est connue (de par le mode de préparation du
mélange ou à la suite d'un dosage analytique) le nombre d'inconnues est ramené à trois. Onpeut alors distinguer deux situations qui correspondent aux deux modes possibles d'utilisationde la relation ZSR ;
© Lorsque l'on dispose des données binaires des deux electrolytes, un processus decalcul itératif permet de calculer l'activité d'eau du mélange à partir de la relation (32).
© Lorsque l'on dispose des données binaires d'un seul electrolyte (par exemple cellesde l'électrolyte 1), la connaissance de la valeur de l'activité d'eau du mélange permet de
calculer la valeur de m ^ . En effet, dans ce cas, la relation (32) est une équation à une
inconnue telle que:
Dl I MA) (ft^}
Cette utilisation de la relation ZSR est souvent désignée sous le terme de "ZSR inverse".
90

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
On notera que si l'activité d'eau du mélange est inférieure à l'activité d'eau à saturationde la solution binaire de l'électrolyte 2, le couple de valeurs (m^Xj, aHO) correspond àdes données binaires de solutions sursaturées.
Dans le but d'acquérir les données binaires de solutions "sursaturées" de CsNO3
(electrolyte 2), on doit par conséquent:
- choisir l'électrolyte associé (electrolyte 1) de telle sorte que ses données binairessoient disponibles sur un domaine d'activité d'eau étendu,
- mesurer l'activité d'eau de mélanges ternaires contenant le CsNO3 (electrolyte 2) etl'électrolyte 1.
La procédure est identique dans le cas de l'acquisition des données binaires "sursaturées" deNaNO3.
Une revue bibliographique des données expérimentales disponibles pour de nombreuxsystèmes ternaires indique que peu de systèmes vérifient exactement la règle ZSR. C'estpourquoi, en 1981, FROLOV [22] présente la relation ZSR comme une loi limite que lessystèmes réels approchent plus ou moins bien.
Toutefois, une prise en compte des écarts à la simplicité des mélanges ternaires d'électrolytesest toujours possible.
Ainsi, en 1963, KIRGINTSEV-LUK'YANOV [23] proposent de quantifier les écarts à lasimplicité au moyen d'un paramètre b défini selon:
1 1 1 , „ , ,—=—s—-yi -1—w— •y2+t>-yi-y2 (66)m mMX, mMX2
m: molalité totale du mélange = mMX| + mMX2,
y,: fraction molaire de l'électrolyte 1 dans le mélange =m MX,
mm...,
y2: fraction molaire de l'électrolyte 2 dans le mélange =m
Lorsque b est nul, le mélange possède un comportement simple. Dans le cas contraire, lesystème n'est pas simple, la valeur absolue de b mesure l'amplitude de l'écart à la simplicité.
En 1966, les valeurs du paramètre b de 34 systèmes ternaires d'électrolytes ont étéregroupées par KIRGINTSEV-LUK'YANOV [24]. En 1973, cette étude a été étendue à 117mélanges d'électrolytes et de non-électrolytes [25]. Des études plus récentes ont été menées en1980 par l'équipe de MAJIMA [26] sur des mélanges H2SO4/MX(SO4) /H2O et
HC1/MC1X/H2O.
91

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
Les auteurs calculent les valeurs du paramètre b à l'aide de la relation (32) dans laquelle lesdonnées expérimentales du mélange (mMX|, mMX2 et aH 0 ) , ainsi que les données binaires
( m^X|, m^x2 ) des deux electrolytes, sont connues.
L'analyse des valeurs ainsi calculées permet de distinguer trois cas de figure:
© b = 0 (67a),
H20 (67b),
H2o + C.y1 (67c).
La relation (67c) est la plus indiquée lorsque l'on veut modéliser le plus précisément possibleles écarts à la simplicité des mélanges.
On remarquera que la valeur de b est caractéristique d'un couple d'électrolytes.D'autre part, la valeur absolue de b est d'autant plus faible que les electrolytes constituant lemélange possèdent des propriétés physico-chimiques voisines (même famille chimique, mêmedensité de charge...).
L'acquisition des données binaires sursaturées au moyen de la relation ZSR doitprendre en compte ces éventuels écarts à la simplicité.
Nous avons pu remarquer précédemment que le calcul du paramètre b n'est possible que sil'on dispose des données binaires des deux electrolytes. Or, dans le cas de l'acquisition dedonnées binaires sursaturées, m\' étant une inconnue, le calcul de b ne peut être mené à bien.
LENZI [25] a effectué ce calcul en extrapolant les lois de variation de b publiées dans lalittérature. Les éventuelles erreurs commises lors de cette extrapolation mathématique peuventêtre appréhendées si l'on compare les données binaires sursaturées obtenues en choisissantdifférents electrolytes associés à l'électrolyte 2.
Commentaires:
O La méthode "ZSR inverse" constitue la seule méthode permettant d'acquérir lesdonnées binaires de solutions sursaturées à partir de mesures expérimentales.
O Nous avons vu précédemment que l'acquisition des "données binaires sursaturées"avec la plus grande justesse possible, doit prendre en compte les écarts à la règle ZSR.Dans le but d'acquérir les "données binaires sursaturées" de NaNO3 et de CsNO3, il est doncnécessaire de connaître les lois de variation du paramètre b pour les mélanges ternairesMNO3 / NaNO3 / H2O et MNO3 / CsNO3 / H2O.
> Dans le cas des systèmes MNO3 / NaNO3 / H2O, b est calculé au moyen des donnéesexpérimentales publiées par KIRGINTSEV-LUK'YANOV. Ces calculs peuvent être menéssur un large domaine d'activité d'eau (1-0,760) et de composition. Le domaine d'activité d'eau
92

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
couvert autorise une extrapolation des lois de variation de b jusqu'à des activités d'eauinférieures à 0,700, ce qui constitue notre objectif.
> Dans le cas des mélanges MX(NO3) /CsNO3/H2O (M=Na ou UO^), les données
publiées par KIRGINTSEV-LUK'YANOV se limitent à des activités d'eau supérieures à0,960. L'extrapolation des lois de variation de b jusqu'à des activités d'eau de 0,700 n'est doncpas raisonnable.L'acquisition des données binaires sursaturées de CsNO3 ne pourrait donc être envisagée quesi b était supposé nul. En choisissant un electrolyte associé à CsNO3 possédant des propriétésphysico-chimiques voisines (RbNO3 ou KNO3), on peut limiter au maximum l'écart à la règleZSR (b ->• 0). Toutefois, le domaine d'activité d'eau couvert par de tels mélanges se limite à0,920 et ne permet toujours pas d'atteindre les valeurs d'activité d'eau voulues (< 0,700).
1.5. Comparaison de la méthode "ZSR inverse" et de l'extrapolation de larelation de CHEN.
Il ressort de l'ensemble de cette étude critique que l'acquisition des "données binairessursaturées" fictives de NaNC>3 et de CSNO3 peut être envisagée par l'extrapolation de larelation de CHEN pour les deux sels, ou bien par la méthode expérimentale "ZSR inverse"pour NaNO3 seulement.
Il est intéressant à ce niveau de comparer les résultats obtenus par ces deux méthodes.Nous avons choisi comme electrolyte test: KNO3 dont le comportement physico-chimique estvoisin de celui de NaNO3 et de CSNO3.
Les paramètres de la relation de CHEN sont ajustés en utilisant les données binaires (m et y)disponibles sur le domaine de solubilité de KNO3 (0-3,5 mol.kg"1) [1].Nous disposons de mesures isopiestiques très précises de l'activité d'eau de mélangesH2O/NaNO3/KNO3 [27]. De tels mélanges présentent de très faibles écarts à la règle deZDANOVSKII, les valeurs de b calculées pour ce système n'excèdent pas 0,007 [24].L'utilisation de la relation "ZSR inverse" ne nécessite donc pas la prise en compte d'éventuelsécarts à la simplicité.
Les valeurs du coefficient d'activité de solutions sursaturées fictives de KNO3, obtenues par lamise en œuvre des deux méthodes sont regroupées sur la figure 2-10. Il est à noter que lesrésultats sont tout à fait comparables sur un domaine de concentration très étendu.
93

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
1,0
0,9
0,1
0,0
Données expérimentalesZSR inverseCHEN
0 10 15 20 25 30
Molalité (mol.kg )
Figure 2-10; Comparaison des relations de CHEN et "ZSR inverse".
1.6. Acquisition des "données binaires sursaturées" fictives de NaNO3 et deCsNO3.
L'ensemble des expériences ou des calculs nécessaires à l'acquisition des valeurs del'activité d'eau et du coefficient d'activité de solutions sursaturées fictives de NaNC>3 et deCSNO3 est présenté dans ce chapitre.
La méthode expérimentale "ZSR inverse" n'a été mise en œuvre que dans le cas deNaNC>3. Selon cette procédure, l'acquisition des "données binaires sursaturées" se décomposeen trois étapes:
© Acquisition de valeurs expérimentales d'activité d'eau de mélanges ternairescontenant NaNCV
© Calcul de la molalité de la solution binaire sursaturée de NaNO3 par la relation"ZSR inverse".
(D Calcul du coefficient d'activité de la solution binaire sursaturée de NaNC>3 parl'intégration de la relation de GIBBS-DUHEM (Chap. 1 §3.1).
94

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
Les données binaires de solutions sursaturées fictives de NaNC>3 et de CSNO3 sontégalement calculées par l'intermédiaire de la relation de CHEN. La mise en œuvre de cetteméthode comporte deux étapes successives:
® L'utilisation "en extrapolation" des paramètres d'interactions ajustés sur le domainedisponible à l'expérience permet d'avoir accès aux couples de données binairessursaturées ( mbl, ybl ).
© L'activité d'eau des solutions binaires sursaturées est ensuite calculée parl'intégration de la relation de GIBBS-DUHEM.
1.6.1. Mise en œuvre expérimentale de la relation "ZSR inverse".
Les mélanges retenus pour acquérir l'activité d'eau de solutions sursaturées fictives deNaNO3 sont: NaNO3/LiNO3/H2O et NaNO3/NH4NO3/H2O.Ces solutions sont obtenues par mélange des solutions binaires d'électrolytes. Lesconcentrations des constituants du mélange sont contrôlées, dans l'échelle des molalités, par lamesure de la masse de chacune des solutions mères binaires versées. Nous présentons dansl'annexe 3, les relations mathématiques permettant de calculer la molalité d'un constituantdans un mélange, à partir de la masse de chacune des solutions servant à constituer lemélange.
L'activité d'eau de ces mélanges est mesurée avec l'Aw CENTER à une température de25°C. Le principe de fonctionnement détaillé de cet appareil est présenté dans l'annexeregroupant l'ensemble des techniques analytiques (Annexe 4). Nous retiendrons quel'incertitude sur le résultat des mesures d'activité d'eau a pu être minimisée grâce à l'attentionparticulière portée aux phases de linéarisation et d'étalonnage. Par rapport à des manipulationsantérieures [28], l'incertitude a ainsi été ramenée de 0,005 à 0,002 unité d'activité d'eau.
Nous présentons les résultats expérimentaux dans les trois tableaux suivants. La connaissancede l'activité d'eau du mélange permet d'aboutir par interpolation des données expérimentalesdisponibles (cf Chap 1. §3.1.) à la valeur de la molalité de la solution binaire (de LiNO3 ou deNH4NO3) de même activité d'eau que le mélange. L'utilisation de la relation "ZSR inverse"permet ensuite de calculer la valeur de la molalité de la solution binaire de NaNO3 de mêmeactivité d'eau que le mélange. Ce calcul est effectué dans les deux cas suivants:
- les écarts à la simplicité sont supposés nuls (b=0),
- les lois de variation de b calculées par KIRGINTSEV-LUK'YANOV sont prises encompte et extrapolées en dehors de leur domaine d'ajustement.
95

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
Tableau 2-2: Mélanses NaNO^LiNO^H,O flère série).
mNaNO,
3,34
3,49
3,46
3,62
3,79
3,93
3,92
4,11
4,31
4,51
4,75
mLiNO,
3,28
3,43
3,39
3,55
3,72
3,86
3,85
4,04
4,23
4,43
4,66
(aH2oL,
0,771
0,759
0,763
0,753
0,742
0,732
0,733
0,719
0,705
0,692
0,676
5,29
5,53
5,45
5,66
5,88
6,08
6,06
6,35
6,34
6,90
7,24
/ b. \b=0
8,81
9,18
9,17
9,75
10,30
10,76
10,72
11,29
11,88
12,57
13,34
/ bi \ b *°lmN>NoJ c a l c
9,00
9,33
9,34
9,89
10,39
10,79
10,76
11,23
11,71
12,28
12,87
Tableau 2-3: Mélanges NaNO,/LiNOVH,O r2ème série).
mNaNO,
2,21
2,23
2,37
2,29
2,33
2,37
2,43
2,52
2,54
2,59
2,64
2,67
2,75
2,83
2,91
2,99
mLiNO,
6,04
6,12
6,13
6,25
6,36
6,47
6,65
6,73
6,87
7,09
7,23
7,30
7,52
7,74
7,96
8,19
( a H 2 o) e x ,
0,677
0,676
0,672
0,668
0,662
0,658
0,649
0,644
0,639
0,628
0,619
0,618
0,606
0,596
0,587
0,577
K^Lp.7,22
7,24
7,32
7,41
7,53
7,61
7,80
7,91
8,02
8,25
8,45
8,47
8,74
8,96
9,16
9,39
lmNaNO,Lc.
13,59
14,34
14,58
14,65
15,00
15,77
16,39
16,84
17,78
18,42
18,20
19,26
19,82
20,86
22,18
23,41
/ y \b»0lmN3NO3Lo.
13,10
13,77
13,95
13,95
14,18
14,81
15,22
15,52
16,23
16,55
16,20
17,02
17,21
17,77
18,50
19,10
96

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
Tableau 2-4: Mélanges NaNO*/NIi,NO,/H,O r3ème sériel
m NaNO 3
3,13
3,38
3,43
3,50
3,60
3,63
3,61
3,74
3,93
4,10
4,33
4,56
m NH 4 NO 3
13,44
14,55
14,79
15,04
15,45
15,57
15,62
16,15
16,92
17,71
18,68
19,70
0,698
0,683
0,680
0,677
0,673
0,669
0,669
0,662
0,652
0,642
0,628
0,617
(mNH4NO,) e x p
17,90
19,20
19,46
19,72
20,07
20,44
20,44
21,11
22,14
23,23
25,05
26,83
/ bi \ b = 0
12,55
13,94
14,31
14,75
15,62
15,25
15,31
15,93
16,67
17,28
17,02
17,15
/™bi \b*°
12,30
13,63
13,99
14,40
15,23
14,88
14,94
15,52
16,23
16,81
16,56
16,69
Les écarts à la simplicité peuvent dans la plupart des cas être négligés. Ils ne doivent être prisen compte que dans les mélanges NaNO3/LiNO3/H2O les plus concentrés. Les mélangesNaNOs/NRiNOs/HaO présentent de très faibles écarts à la règle de ZDANOVSKII même auxfaibles valeurs d'activité d'eau. Cependant, nous avons choisi de retenir dans tous les cas lesdonnées binaires sursaturées obtenues en prenant en compte le terme b d'écart à la simplicité.On aboutit alors à un couple de valeurs (mJ,'aN0), aH2Û) correspondant à des données binaires
sursaturées fictives. Le coefficient d'activité correspondant est ensuite obtenu par intégrationde la relation de GIBBS-DUHEM.
1.6.2. Extrapolation de la relation de CHEN.
Les trois paramètres de la relation de CHEN sont ajustés sur le domaine de solubilitédes electrolytes NaNO3 et CSNO3. L'extrapolation de ces paramètres au delà du domained'ajustement permet d'aboutir à des couples de données (mbl, ybl), le calcul de l'activité d'eauest ensuite effectué par intégration de la relation de GIBBS-DUHEM. Cette relation s'écrit,dans le cas d'une solution binaire d'un electrolyte MpXq, sous la forme suivante (cf Chap 1.§3.1.):
nMX.dln(aMX) + nH2O.dln(aH2O) = 0 (25)
Le résultat de l'intégration s'écrit:
Y
QMX -0 .0360. JmMX.dlog(yMX) (68)
97

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
A la différence de l'intégration "classique" permettant de calculer le coefficient d'activité àpartir des variations de l'activité d'eau, aucun problème lié aux bornes d'intégration ne se poseici, il est donc possible de calculer le terme intégral par une méthode graphique.
Les valeurs des trois paramètres ajustés sont données dans le tableau 2-5.
Tableau 2-5: Paramètres de la relation de CHEN pour NaNO3 et CsNO3.
Electrolyte
considéré
NaNO3
CSNOB
Paramètres ajustés
ca,m
-3,66 ± 0,01
-2,97 ± 0,08
m,ca
7,39 ± 0,03
6,62 ±0,17
b
15,34 ±0,30
9,78 ± 0,28
La figure 2-11 regroupe les valeurs d'activité d'eau de solutions binaires de NaNO3, surl'ensemble du domaine de solubilité et dans une zone sursaturée. Les valeurs obtenues par laméthode "ZSR inverse" se placent de part et d'autre de la courbe calculée par extrapolation dela relation de CHEN. La dispersion des mesures est liée au cumul des erreurs expérimentaleset de l'erreur commise en extrapolant les lois de variation du paramètre d'écart à la simplicitéb. Les valeurs du coefficient d'activité de CsNO3 dans des solutions binaires sont regroupéessur la figure 2-12. On retiendra que l'extrapolation de la relation de CHEN a dû être effectuéesur un domaine de concentration très étendu, afin d'atteindre des valeurs d'activité d'eau de0,720.
98
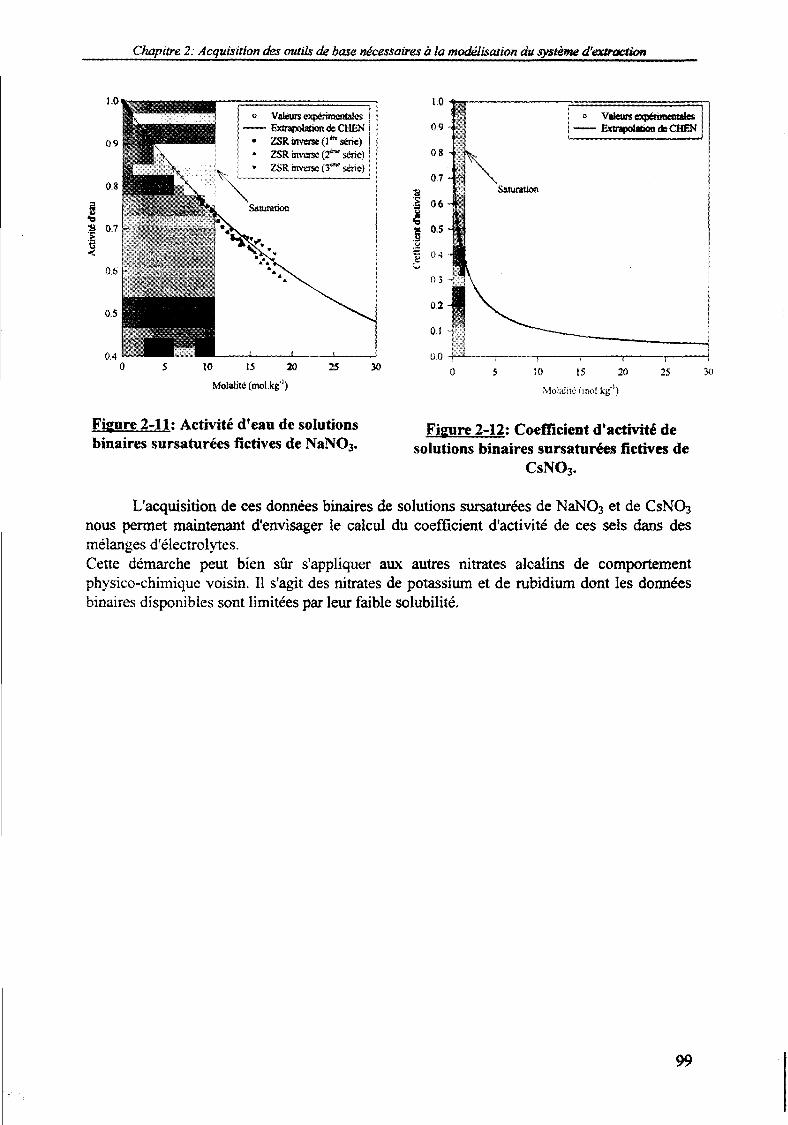
Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
mm \
o Valeurs expérimentales— Exttapolatioij<feCHBN• ZSR inverse (1** série)• 2SR inverse (2*** série)• ZSR inverse (3*"* série) |
Valeurs expérimentalesExtrapolation de CHEN
10 15 20
MoîalitsKmoUg4)
Figure 2-11: Activité d'eau de solutionsbinaires sursaturées fictives de
S Î0 15 20
Molalitcfmol.kg" )
25
Figure 2-12: Coefficient d'activité desolutions binaires sursaturées fictives de
CsNO3.
L'acquisition de ces données binaires de solutions sursaturées de NaNO3 et de CSNO3nous permet maintenant d'envisager le calcul du coefficient d'activité de ces sels dans desmélanges d'électrolytes.Cette démarche peut bien sûr s'appliquer aux autres nitrates alcalins de comportementphysico-chimique voisin. Il s'agit des nitrates de potassium et de rubidium dont les donnéesbinaires disponibles sont limitées par leur faible solubilité.
99

Chapitre 2; Acquisition des outils de base nécessaires à la modélisation du système d'extraction
2. Quantification des systèmes d'extraction de type solvatant.
Jusqu'à présent, nous nous sommes attachés à présenter les théories de calcul descoefficients d'activité dans les solutions aqueuses et organiques. La mise en œuvre de cesoutils théoriques, dans le but de modéîiser notre système d'extraction, est détaillée dans lasuite de ce paragraphe. Nous insisterons plus particulièrement sur l'aspect informatique, lesmodifications apportées au logiciel existant "EXTRACTION" puis le développement dunouveau code de calcul "PREVISION" sont successivement présentés.
2.1. Mise en équation des équilibres d'extraction.
Le système d'extraction complet que nous avons à étudier est composé:
- d'une matrice aqueuse concentrée en acide nitrique et en nitrate de sodium contenantde faibles quantités de nitrate de césium,
- d'un calixarène dilué dans le 1,2-mtrophényIoctyîéther.
L'équilibre d'extraction peut s'écrire d'une manière générale selon (les barres au dessus desespèces signalent leur présence en phase organique):
O + dDil«*(E)i(HNO3)j(XNO,)k(HîO)h(Dil)(l (69)
E, Dil : molécule d'extractant et de diluant,X+: cation alcalin.
Suivant la valeur des coefficients stœchiométriques (i, j , k, h et d), il est possible de définirdifférentes familles d'équilibres d'extraction:
CD Polymérisation de la molécule d'extractant.
© Hydratation de la molécule d'extractant ou de diluant,
® Partage de l'acide ou du sel entre la phase aqueuse et le diluant,
® Extraction de l'acide ou du sel, avec ou sans hydratation, par l'extractant ou lediluant,
® Extraction mixte (acide+sel), avec ou sans hydratation, par l'extractant ou le diluant.
Les différentes combinaisons de coefficients stecMométriques correspondant à ces cinqfamilles peuvent être résumées sous la forme du tableau suivant:
100
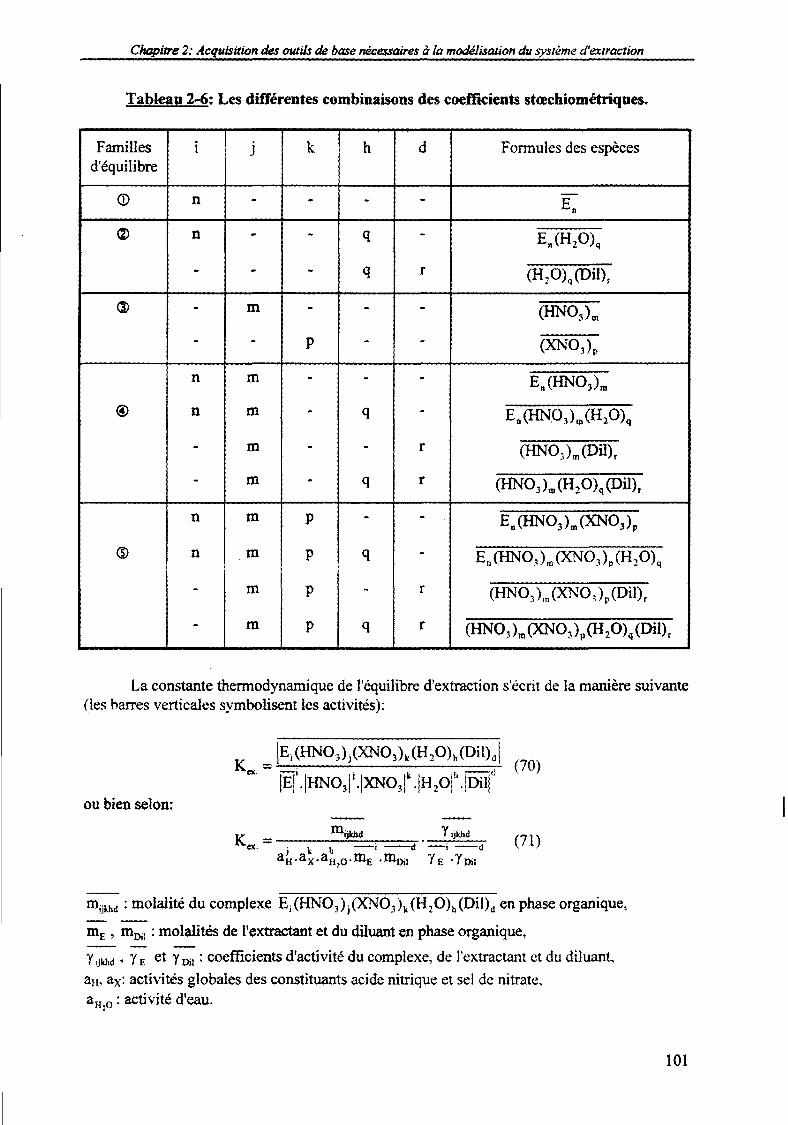
Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
Tableau 2-6: Les différentes combinaisons des coefficients stœchiométriques.
Famillesd'équilibre
G>
©
•
CD
i
n
n
-
n
n
n
n
j
-
-
m
m
m
m
m
m
m
m
m
k
-
-
-
P
-
•B
P
P
P
P
h
-
q
q
-
-
q
q
-
q
q
d
-
-
r
-
-
r
r
-
r
r
Formules des espèces
i;Eft(H2O)q
(H2O)q(Dil)f
(HNO3)m
(XNO3)p
En(HNO3)m
E^HNCg^O),
(HNO3)ro(Diï)r
(HNO3)ffl(H2O)<1(DU)r
Ett(HNO3)œ(XNO3)p
E,(HNO,)i,(XNO,) | l(H2O),
(HNO3)ra(XNOî)p(Dil)f
(HNOO.CXNO^^^CDil),
La constante thermodynamique de l'équilibre d'extraction s'écrit de la manière suivante(les barres verticales symbolisent les activités):
ou bien selon:
Ej(HNO3)j(XNO3)k(H2O)h(Dil)
E|l.|HNOJ|j.|XNO3f.|H2O|h.|Dil
(70)
a a
_ _ ; d-_j , (71)
mijkhd : molalité du complexe Ei(HNO3)i(XNO3)k(H2O)h(Dil)li en phase organique,
mE , 11%, : molalités de l'extractant et du diluant en phase organique,
Tijkhd ' T E e t YD»Ï : coefficients d'activité du complexe, de l'extractant et du diluant,aH. ax: activités globales des constituants acide nitrique et sel de nitrate,aH 0 : activité d'eau.
101

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
L'écriture de cette constante d'extraction permet à nouveau d'insister sur le fait que nousraisonnons en termes de constituants en phase aqueuse, et en termes d'espèces en phaseorganique. Le calcul des activités des constituants de la phase aqueuse a été détaillé dans lechapitre 1. Le développement des coefficients d'activité des espèces en phase organique par larelation de SERGIEVSKII (cf §4.2.) conduit à l'expression:
XV ""~ .... .1 . .. • .4 VAL/1 Mk>:xLj «I A * • « fï I I V * « Z
avec ASJM = SJM ~lst ~â-
y ma " efficient d'activité du complexe dans la solution d'activité d'eau égale à 1,
yf, y£j, : coefficient d'activité de l'extractant et du diluant dans la solution d'activité d'eauégale à 1,
sjkhd, sf et SQJJ : nombre de molécules d'eau solubilisées par le complexe, l'extractant et lediluant à activité d'eau égale à 1.
Comme il a déjà été souligné, les coefficients y j ^ , yf et de y^, regroupent l'ensemble desécarts à l'idéalité exceptés ceux dus à la présence d'eau et à la formation d'espèces en phaseorganique. Les valeurs de ces termes vont certainement varier le long de l'isothermed'extraction. Cependant, on peut supposer que leurs variations restent voisines dès lors que leseffets de milieu subis par les différentes espèces sont voisins.
Y-Le rapport ;.:;;:.:::::•;"• 4 peut donc être considéré constant le long de l'isotherme. CetteYE -Y«s
hypothèse permet d'écrire une constante effective d'extraction selon la terminologie introduitepar SERGIEVSKII.
Kf- _ K YE -YDJI . mijM>4 expfAs® ( î - a )1 (73)YijldKi aH-ax-aHî0-mE -mDil
L'écriture de la constante effective d'extraction fait intervenir cinq grandeurs expérimentales:
- mB et mm : obtenus directement par des dosages.
- an, ax et aHi0 : calculés à partir des résultats de dosages (mu, mx et mH o ),
Apparaissent également une variable m ^ calculée à partir de la loi d'action de masse et
deux paramètres K^,a et A s ^ à déterminer.
102

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
Les deux paramètres K ^ et As®^ sont calculés à partir de l'ajustement des isothermes
d'extraction. L'ensemble des calculs est informatisé dans ïe logiciel "EXTRACTION".
22. Le logiciel "EXTRACTION11,
Le code de calcul "EXTRACTION" a été développé initialement par R. ROZOT [29] àl'INSTN à partir d'un cahier des charges établi par P. DANNUS [28]. Il permettait à l'originede modéliser l'extraction d'acide, d'un sel et de l'eau par un réactif de type solvatant dilué dansun solvant inerte. Le langage de programmation choisi est le TURBO PASCAL Version 7pour compatible PC.
Nous avons apporté des modifications importantes à ce logiciel, en particulier afin d'enétendre les possibilités de traitement à notre système d'extraction.Ainsi» les propriétés extractantes du diluant vis-à-vis de l'acide et de l'eau sont maintenantintégrées dans le code de calcul. La prise en compte de l'extraction simultanée de l'acide, dedeux sels (dans notre cas NaN03 et CSNO3) et de l'eau par l'extradant et par le diluant estégalement nécessaire.En même temps, nous avons amélioré le calcul de la répartition des différents constituantsentre chacune des espèces présentes en phase organique à l'équilibre. 0 est maintenantpossible de générer un fichier regroupant la répartition de l'acide, des sels, de l'eau, del'extractant et du diluant entre les différents complexes en phase organique. Le format de cefichier est choisi de manière à permettre une exportation vers des logiciels graphiques ducommerce (EXCEL de Microsoft, SIGMAPLOT de Jandel Scientific...).
Le code peut être décomposé selon trois phases successives où sont regroupées:
CD la saisie des caractéristiques du système d'extraction modélisé,
® l'optimisation des paramètres Kjjjj , et s ^ ,
CD l'ensemble des résultats des calculs.
2.2.1. Saisie des données nécessaires à l'optimisation.
L'utilisateur doit introduire l'ensemble des données nécessaires aux calculs, il faut pourcela saisir:
- le nom des constituants en phase aqueuse (acide et sels), de l'extractant et du diluant,
- le nombre de molécules d'eau solubilisée par molécule d'extractant ( sf ) et de diluant ( s^, ),- l'échelle de concentration, molalité ou molarité,
- la concentration de l'extractant et la masse molaire du diluant,- le fichier de points expérimentaux qui regroupe les activités des constituants en phaseaqueuse et les concentrations de l'acide, des sels et de l'eau en phase organique à l'équilibre,l'acquisition de ces données expérimentales nécessitant un grand nombre de manipulations,- un fichier regroupant les coefficients stœchiométriques des différents complexes testés en
phase organique, ainsi que les valeurs initiales de K^k|iM_ et de s®.kh(L associées.
103

Chapitre 2: Acquisition des outils de hase nécessaires à la modélisation du système d'extraction
2.2.2. Optimisation des paramètres du système.
La deuxième phase regroupe l'ensemble des calculs permettant d'aboutir aux valeursoptimisées de la constante effective d'extraction et du nombre de molécules d'eau solubiliséepar chaque complexe. Dans le but d'alléger les expressions mathématiques, nous limitons laprésentation des calculs à un système d'extraction dont la phase aqueuse contient un acide etun seul sel, l'échelle de concentration présentée étant l'échelle des molalités.
La mise en équation est réalisée à partir de l'écriture de la loi d'action de masse et du bilanmatière sur chaque constituant:
Loi d'action de masse:
K fif?" ! k îî f T**°î * ï*T*v* tH * i ® / 1 1 i / •? il \
m *ï »8 *S| R ** ft I i*( I i l \ i I I £*W\I \ O 1 *2t ,w,i, f i l i / Z t ï
•.j.*.».». i.J,k.M,'aH-aX •Zn,o\i^\ -L"11] •eXP[ASi.jok>h.d.-\aH3O lJJ V/4->
Bilans matières relatifs à chacun des constituants:
(75)
(76)
l'acide:
le sel:
l'eau:N
l'extractant:
le diluant:
m H —
mx =
N
mE =
mDil. =
N
H
1000 ^
A
«An+
(77)
(78)
[DÛ] (79)
L'optimisation des paramètres est basée sur la minimisation de l'écart entre l'isothermecalculée et l'isotherme expérimentale. La méthode des moindres carrés en mode itératif(NEWTON, RAPHSON et MARQUARD [30]) est retenue pour effectuer ce calcul. Le testd'arrêt est celui de HOOKE et JEEVES [31]. Le détail de ces calculs mathématiques estprésenté en Annexe 3.
2.2.3. Présentation des résultats.
Les résultats des calculs d'optimisation peuvent être consultés selon deux formesdifférentes. La première présenMion consiste en une série de tableaux de résultats, la secondeforme reprend les résultats des calculs sous la forme de représentations graphiques.
Dans le premier cas, l'édition des résultats est présentée à l'utilisateur sous la forme desept écrans successifs qui regroupent:
104

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
© les données initiales, à savoir, le nom des constituants du système d'extraction, lenombre de molécules d'eau solubilisée par l'extractant et le diluant, les concentrations del'extractant et du diluant,
<D la liste des complexes et de leurs paramètres initiaux et optimisés,
<D les concentrations expérimentales et calculées de l'extractant, de l'acide et des selsextraits en phase organique,
® ies concentrations expérimentales et calculées de l'eau et du diluant en phaseorganique,
<S> les concentrations calculées de chacun des complexes en phase organique,
© les concentrations calculées de l'extractant et du diluant libres en phase organique,
® les activités expérimentales en phase aqueuse de l'acide, des sels extraits et de l'eau.
Ces sept écrans peuvent également être sauvegardés dans un fichier.
Pour une meilleure visualisation, l'ensemble des résultais des calculs de modélisationpeuvent être présentés sous la forme de courbes à l'écran ou sur un traceur. Il est possibled'accéder à des représentations graphiques très complètes.
Les représentations graphiques auxquelles l'utilisateur peut accéder permettent de comparerles isothermes expérimentales et calculées afin de juger de la bonne adéquation avec le jeud'espèces proposées. Il est ainsi possible de visualiser les variations des concentrations enphase organique de l'acide, des deux sels et de l'eau en fonction de:
- l'activité de l'acide, des sels ou de l'eau en phase aqueuse,
- les concentrations de l'acide ou des sels en phase aqueuse après les avoir saisies(jusqu'à présent, seules ies activités en phase aqueuse sont connues).
Il est également possible de tracer la répartition de l'ensemble des constituants entre iesdifférents complexes en phase organique.
105

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
23 . Le logiciel "PREVISION".
Nous avons vu que le logiciel de modélisation "EXTRACTION" répond à desquestions fondamentales sur la chimie de l'extraction d'acide et de sels par un extractant detype solvatant
A partir des connaissances acquises grâce à cette modélisation, le logiciel "PREVISION"entièrement développé durant cette étude, permet de nous placer dans une optique de mise aupoint de procédés. Il permet donc d'atteindre des objectifs beaucoup plus pratiques pourl'industriel. Le but des calculs est de déterminer les concentrations à l'équilibre desconstituants extractibles dans les deux phases à partir de leurs concentrations initiales enphase aqueuse. Par conséquent, cet outil prédictif facilite grandement la recherche desconditions optimales d'extraction du constituant d'intérêt (le nitrate de césium dans notreétude).
Comme dans le logiciel "EXTRACTION", l'extraction simultanée d'acide, de deux sels (Xi etX2) et de l'eau par l'extractant et le diluant peut être étudiée.
Ces calculs sont menés en prenant en compte les écarts à l'idéalité:
- en phase aqueuse, par le calcul des activités grâce à la relation de MIKULIN,
- en phase organique, par la loi d'action de masse et le développement des coefficientsd'activité par la relation de SERGIEVSKII.
Le déroulement de "PREVISION" se décompose en deux phases.
Une première partie regroupe la saisie des caractéristiques du système d'extraction. Il s'agit:
- des constantes effectives d'extraction K^M et du nombre de molécules d'eau
solubilisée s®khd pour chacun des complexes, paramètres optimisés par le logiciel
"EXTRACTION",
- de la composition des phases aqueuse et organique initiales, à savoir:
- le nombre et la nature des constituants dans les deux phases,- les molalités des constituants en phase aqueuse, mH jm, mX) mj et mXj >mt,- la moïalité de l'extractant en phase organique, rmym ,
- les masses molaires de l'ensemble des constituants du système, MH, MXj,
MX i , ME et Mou.,
- les masses (m*,., m ^ ) ou les volumes (V^., V^g) initiaux des phasesaqueuses et organiques.
Une seconde phase regroupe l'ensemble des calculs permettant d'accéder aux concentrations àl'équilibre.
Î06

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
L'activité d'eau est calculée par une procédure itérative basée sur la relation deZDAN0VSKI1.Les activités des constituants (cas de stœchiométries 1-1) en phase aqueuse s'expriment selon:
ax, = Y x , - m v ( m H + m x , + m x i )
axs = Y x, mx2 .(mH + mx
Les coefficients d'activité des constituants en phase aqueuse sont déterminés grâce à larelation de MIKULIN et nécessitent donc de faire appel aux fichiers de données binaires.
Les moiaiités des différents soivates sont calculées par la loi d'action de masse en prenant encompte les écarts à l'idéalité dans les deux phases:
(80)
Les concentrations de chaque constituant en phase organique sont obtenues en additionnanties concentrations des solvates affectées de leur coefficient stœchiométrique respectif.Ce processus de calcul suit un mode itératif. Les concentrations des constituants dans la phaseaqueuse, de I'extractant libre et du diluant libre en phase organique sont, pour la première,itération prises égales aux concentrations initiales. Un bilan matière sur chacun desconstituants en phase aqueuse et organique constitue le critère d'arrêt. Si l'écart entre lenombre de moles initial et le nombre de moles total à l'équilibre pour chaque constituant estsupérieur à une précision donnée, les concentrations à l'équilibre sont incrémentées et leprocessus de calcul répété.
Le détail des calculs est présenté ci-après:
Calcul des masses d'eau et de diluant présentes dans le système d'extraction:
2 > U l J j . M t ™m 1000+mE,i»..Mï
ou bien
Ecriture de la conservation de masse:
Pour les constituants présents initialement en phase aqueuse (C=acide ou sels):
nC.ini = mC,im - m H , 0 = *«<:*,.• mH,O
Pour I'extractant:
107

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
Pour le diluant:
Le résultat final regroupe sous la forme d'un tableau, les concentrations initiales et àl'équilibre des constituants dans les deux phases ainsi que l'activité d'eau. A partir de cesvaleurs, il est alors possible de calculer le coefficient de distribution, le rendement d'extractionou bien le facteur de decontamination.
108

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
Conclusion.
A l'issu de ce chapitre, nous disposons de l'ensemble des outils de base nécessaires à lamodélisation physico-chimique de notre système d'extraction.
L'étude critique et comparative de différentes théories des solutions a permis d'acquérirles données binaires de solutions sursaturées fictives des nitrates de césium et de sodium. Laconnaissance de ces données thennodynamiques permet maintenant de calculer le coefficientd'activité de CSNO3 et de NaN03 dans des matrices concentrées.
La recherche du nombre et de la stœchiométrie des espèces extraites en phaseorganique sera menée grâce à un outil informatique élaboré. La mise en œuvre du logiciel"EXTRACTION" va nous permettre de répondre aux questions fondamentales sur la chimiede l'extraction de l'acide et des sels par un calixarène. Afin de nous placer dans une optique demise au point de procédé, nous avons développé le code de calcul "PREVISION". Cet outilprédictif permettra, à partir des connaissances acquises lors de la mise en œuvre du logiciel"EXTRACTION" de prévoir les conditions optimales d'extraction du césium d'efïluentsliquides issus du cycle du combustible nucléaire.
109

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
BIBLIOGRAPHIE.
[I] WJ. HAMER, Y.C. WU,Osmotic Coefficients and Mean Activity Coefficients of Uni-univalent Electrolytes inWater at 25°C,J. Phys. Chem. Ref. Data, 1, (1972), 1047-1099.
[2] W J . HAMER, Y.C. WU,Revised Values of the Osmotic Coefficients and Mean Activity Coefficients of SodiumNitrate in Water at 25°C,J. Phys. Chem. Ref. Data, % ( 1980), 513-515.
[3] A.F. HIDALGO, Cl. ORR Jr,Method for Predicting the Properties of Supersaturated Solutions of the Alkali ChloridesJ. of Chem. and Eng. Data, 13, (1968), 49-53
[4] J. SANGSTER, T.T. TENG, F. LENZI,A General Method of Calculating the Water Activity of Supersaturated AqueousSolutions from Ternary Data,Can. J. Chem., 51, (1973), 2626-2631.
[5] T.T. TRAN, F. LENZI,Methods of Estimating the Water Activity of Supersaturated Aqueous Solutions,Can. J. Chem. Eng., 52, (1974), 798-802.
[6] P. DEBYE, E. HÛCKEL,Z. Physik., 25, (1923), 185-206.
[7] J.N. BR0NSTED,Studies of Solubility. IV. The Principle of the Specific Interaction of Ions,J. Amer. Chem. Soc, 44, (1922), 877-898.
[8] E.A. GUGGENHEIM,The Specific Thermodynamic Properties of Aqueous Solutions of Strong Electrolytes,Phil. Mag., 19, (1935), 588-643.
[9] K.S. PITZER, L. BREWER,Thermodynamics,McGraw-Hill, New York, (1961).
[10] L.A. BROMLEY,Thermodynamic Properties of Strong Electrolytes in Aqueous Solutions,AJChE J., 19, (1973), 313-320.
[II] K.S.PITZER.Thermodynamics of Electrolytes. 1. Theorical Basis and General Equations,J. Phys. Chem., 77, (1973), 268-277.
I l l

Chapitre 2: Acquisition des outils de hase nécessaires à la modélisation du système d'extraction
[12] J.C. RASAIAH, H.L. FRIEDMAN,Integral Equation Methods in the Computation of Equilibrium Properties of IonicSolutions,J. Chem. Phys., 48, (1968^2742-2752
[13] J.C. RASAIAH, ILL. FRILDMAN,Integral Equation Computation for Aqueous 1-1 Electrolytes. Accuracy of the Method,J. Chem. Phys., 50, (1969), 3965-3976.
[14] K.S.PITZER,Thermodynamics of Electrolytes. II. Activity and Osmotic Coefficients for StrongElectrolytes with One or Both Ions Univalent,J. Phys. Chem., 77, (1973), 2300-2308
[15] J.L. CRUZ, H. RENON,A New Thermodynamic Representation of Binary Electrolyte Solutions Nonideality inthe Whole Range of Concentrations.AlChE J.» 24, (1978), 817-830.
[16] C.C. CHEN, H.I. BRITT, J.F. BOSTON, L.B. EVANS.Local Composition Model for Excess Gibbs Energy of Electrolyte Systems,AIChE J., 28, ( 1982), 588-596.
[17] K.S.PITZER,Electrolytes. From Dilute Solutions to Fused Salts.J. of Am. Chem. Soc., 102, (1980), 2902-2906.
[18] H. RENON, J.M. PRAUSNITZ,Local Compositions in Thermodynamic Excess Functions for Liquid Mixtures,AIChE J.. 14, (1968), 135-144.
[19] Y. MARCUS, A.S. KERTES,Ion Exchange and Solvent Extraction of Metal Complexes,WILEY-INTERSCIENCE, New York-London, (1969), 233-234.
[20] H.P. MEISSNER, J.W. TESTER,Activity Coefficients of Strong Electrolytes in Aqueous Solutions,Ind. Eng. Process. Develop., 11, (1972). 128-133.
[21] H.P. MEISSNER,Thermodynamics of Aqueous Systems with Industrial Applications.ACS Symp. Ser. 133. (1980), 495-511.
[22] Yu.G. FROLOV,Theory of Mixed Isoactive Electrolyte Solutions,Russ. Chem. Rev. 50, (1981), 232-247.
112

Chapitre 2: Acquisition des outils de base nécessaires à la modélisation du système d'extraction
[23] A.N. KIRGINTSEV, A.V. LUK'YANOV,Isopiestic Investigation of Ternary Solutions. I,Russ. J. Phys. Chem., 37, (1963), 1501-1502.
[24] A.N. KIRGINTSEV, A.V. LUK'YANOV,Isopiestic Investigation of Ternary Solutions. VIII. General Treatment of the Results for34 Ternary Aqueous Salt Solutions,Russ. J. Phys. Chem., 40, (1966), 953-956.
[25] H. CHEN, J. SANGSTER, T.T. TENG. F. LENZI.A General Method of Predicting the Water Activity of Ternary Aqueous Solutions fromBinary Data,Can. J. Chem. Eng., 51, (1973), 234-241.
[26] H. MAJIMA, Y. AWAKURA, Y. KAWASAKI,Activities of Water and Solutes in the Aqueous Systems H2SO4-Mx(SO4)y-H2O andHCl-MCly,AGNE SHOFU Publishing Inc., Tokyo (1988),
[27] G.I.MIKULIN,Voprosy Fiacheskoï KMmii Rastvorov Elektrolitov (Les questions de chimie physiquedes solutions d'électrolytes),Izdatelstvo "Khimiya" Leningrad Otdelenie, (1968), 279-319.
[28] P.DANNUS,Modélisation physico-chimique de l'extraction de constituants inorganiques par desréactifs solvatants,Thèse de Doctorat de l'Université PARIS VI (1991 ).
[29] R.ROZOT,Des logiciels en chimie analytique.Communication personnelle, INSTN/UECCC, (1989).
[30] D.W. MARQUARDT,An Algorithm for Least-squares Estimation of Nonlinear Parameters.J. Soc. Indust. Appl. Math., 11, (1963), 431-441.
[31] R. HOOKE, T.A. JEEVES,"Direct Search" Solution of Numerical and Statistical Problems,J. Ass. Comput. Mach., 8, (1961), 212-229.
113

CHAPITRE 3
MODELISATION PHYSICO-CHIMÏQUE DU SYSTEMED'EXTRACTION HNOa/NaNOyCsNO^O | Calixarène/NPOE.

Chapitre 3: Modélisation physico-chimique du système d'extraction
Introduction.
Le processus de calcul adopté dans le logiciel "EXTRACTION" est basé sur laminimisation de l'écart entre les isothermes d'extraction expérimentale et calculée. Lamodélisation de notre système d'extraction nécessite donc l'acquisition de donnéesexpérimentales d'extraction.
Nous cherchons à modéliser l'extraction du nitrate de césium à partir de matricesconcentrées en acide nitrique et en nitrate de sodium. La mise en œuvre du logiciel"EXTRACTION" sur les données expérimentales du système complet est délicate. En effet, lamodélisation correcte des isothermes d'extraction peut nécessiter la prise en compte d'unnombre important d'équilibres. L'optimisation des deux paramètres ( K ^ d , As®^ ) associés à
chacune des espèces envisagées risque alors de se heurter à des problèmes de convergence descalculs (le terme de "dégénérescence mathématique" est parfois employé pour décrire ceproblème).
Afin de préserver la qualité des calculs, nous préconisons d'adopter une démarche analytiqueen divisant le système d'extraction en sous-systèmes simples. Les équilibres permettantd'obtenir le meilleur ajustement entre les isothermes expérimentales et calculées sont retenus.Il peut arriver que des configurations différentes aboutissent à une bonne description dusystème d'extraction, dans ce cas, la représentation la plus simple est retenue.
La description des systèmes plus complexes est effectuée à partir des résultats obtenus sur lessous-systèmes d'extraction. Lorsque la description finale n'est pas satisfaisante, les écarts sontinterprétés par la formation de nouvelles espèces.
Ainsi, nous présentons successivement l'extraction de l'eau, de l'acide et des nitratesalcalins par le diluant NPOE puis par le calixarène. Ces études nous permettent de quantifierl'hydratation du diluant et du calixarène, ainsi que leurs propriétés extradantes vis-à-vis del'acide et des sels. L'extraction simultanée de deux sels ou de l'acide et d'un sel est ensuiteenvisagée.
Enfin, la mise en œuvre de l'ensemble des résultats de modélisation par l'intermédiaire dulogiciel "PREVISION" permet de prévoir le comportement du calixarène vis-à-vis del'extraction de faibles quantités de CsNO3 à partir de matrices concentrées en acide et en sel.Ces calculs de prévision ont pour but de préciser les conditions optimales de décontaminationd'effluents liquides radioactifs.
117

Chapitre 3: Modélisation physico-chimique du système d'extraction
1. Conditions expérimentales.
1.1. Préparation des solutions aqueuses.
La préparation des solutions binaires se fait par dilution de solutions mèresconcentrées. Le contrôle de la concentration de ces solutions est effectué par mesure de lamasse volumique à 25°C. Cette mesure permet d'accéder, par interpolation des donnéesbinaires disponibles dans la littérature [1], à la concentration de l'électrolyte dans lesdifférentes échelles de concentration à 25 °C. La procédure d'interpolation est informatiséedans le logiciel AQUACALC [2].
Les solutions ternaires et quaternaires sont obtenues par mélange des solutions binairesde chacun des electrolytes. Les concentrations des constituants minéraux dans le mélange sontcalculées à partir des masses et des molalités de chacune des solutions binaires utilisées (voirannexe 4-1).
1.2. Préparation des solutions organiques.
Le diluant 1,2-Nitrophényloctyléther (NPOE) est synthétisé par la société ERAS Labo.Deux lavages successifs sont effectués avec de l'eau déminéralisée afin d'extraire leséventuelles impuretés solubles en phase aqueuse. La solution organique est ensuite filtrée surfiltre MILLIPORE (FH 0,5 um).
Le calixarène utilisé est le 1,3-diisopropoxy calix[4]arène-couronne-6, synthétisé parl'équipe de R. UNGARO à l'Université de Parme [3]. L'extractant se présente sous la formed'une fine poudre grisâtre. La solubilité limite de ce calixarène dans le NPOE est de 0,1mol.L"1. La concentration retenue pour nos solutions extractantes est de 0,01 mol.L"1, ceci afinde limiter la quantité d'extractant utilisée lors de nos manipulations. La mise en solution ducalixarène s'accompagne dans tous les cas de la présence d'insolubles en assez grandesquantités. Deux lavages successifs volume à volume avec de l'eau déminéralisée permettent derassembler ces impuretés à l'interface phase aqueuse/phase organique puis de les éliminer.
La concentration réelle de l'extractant en phase organique n'est donc plus rigoureusementaccessible par la seule pesée du calixarène. Il est alors nécessaire de recourir à une méthode dedosage du calixarène en solution organique.Les méthodes spectrophotométriques nécessitant un étalonnage sont inadaptées car nous nedisposons d'aucun étalon.La concentration du calixarène est déterminée indirectement après une extraction avec unesolution concentrée de nitrate de césium. Des études préalables [3] montrent que pour lesconcentrations les plus importantes de CsNO3 en phase aqueuse, la saturation du calixarèneest atteinte, le complexe formé contenant un atome de césium par molécule d'extractant. Laconnaissance de la concentration du césium en phase organique nous permet alors deconnaître la concentration du 1,3-diisopropoxy calix[4]arène-couronne-6 dans le NPOE.La détermination de cette concentration est réalisée par spectrométrie y ainsi que par
chromatographie ionique. La molalité ainsi déterminée est de 0,00769 mol. kg"1.
118

Chapitre 3: Modélisation physico-chimique du système d'extraction
1.3. Techniques analytiques.
Ce paragraphe présente succinctement l'ensemble des techniques analytiques mises enœuvre pour déterminer la composition des phases aqueuses et organiques. La descriptiondétaillée de chacune des techniques est regroupée dans l'annexe 4.
1.3.1. Mesure de la masse volumique.
La connaissance de la masse volumique des solutions permet la conversion entre leséchelles de concentration (Annexe 1). La mesure de la masse volumique est également utiliséecomme méthode analytique pour déterminer la concentration des solutions binairesd'électrolytes. Cette démarche originale permet de déterminer directement et précisément desmacro-concentrations de sels ou d'acides par interpolation des données binaires disponibles[1]. A la différence des autres techniques disponibles, il est possible de doser sans dilutionpréalable des solutions d'électrolytes très concentrées. Nous avons pu vérifier que cetteméthode analytique aboutit aux mêmes résultats que d'autres techniques (potentiométrie pourl'acide nitrique ou bien chromatographie ionique pour les nitrates alcalins).
1.3.2. Dosages d'eau par Karl FISCHER.
Les concentrations d'eau présente en phase organique à l'équilibre sont mesuréesdirectement selon la méthode de Karl FISCHER. La mise en œuvre de cette méthode dedosage par voie coulométrique permet de s'affranchir de tous les étalonnages de la méthodevolumétrique classique.
1.3.3. Dosages acide-base.
Les concentrations d'acide présent à l'équilibre dans les deux phases sont déterminéespar dosage potentiométrique. Les quantités d'acide en phase organique restant faibles, nousavons porté un soin particulier à la réalisation des dosages en utilisant des burettes de titragede petit volume et des solutions de soude peu concentrées.
1.3.4. Chromatographie ionique.
Le dosage par chromatographie ionique permet de déterminer la concentration descations alcalins:
- dans les phases aqueuses les plus diluées,
- dans les phases aqueuses de désextraction, ce qui permet d'accéder à la concentrationen phase organique à l'équilibre.
Cette méthode est préférée à la spectrométrie gamma dans la mesure où l'absence deradioélément évite la nuclearisation des autres techniques analytiques (densimétrie, KarlFISHER et potentiométrie). De plus, la chromatographie ionique permet d'améliorer enprécision le dosage du nitrate de sodium par rapport aux résultats obtenus précédemment parspectrométrie y [6].
119

Chapitre 3: Modélisation physico-chimique du système d'extraction
1.3.5. Spectrométrie gamma.
Cette méthode est mise en œuvre uniquement lors du dosage de traces de césium enphase organique et en phase aqueuse. Le césium 137 est utilisé dans nos manipulationscomme traceur, ce radioisotope est un émetteur (V dont le fils radioactif est le barium 137métastable, émetteur y.
1.4. Manipulations d'extraction liquide-liquide.
Les expériences sont menées dans tous les cas dans une enceinte thermostatée à 25°Cet elles se décomposent en 5 étapes successives:
© Mise en contact de mex ("ex" symbolise l'étape d'extraction) grammes de phase
aqueuse et de mex grammes de phase organique dans un tube à essai de 15 mL.
© Agitation pendant 15 minutes.
(D Centrifugation à 4000 tr.min'1 pendant 15 minutes.
© Mesure des masses volumiques des deux phases.
© Dosages d'eau en phase organique par coulométrie selon la méthode Karl FISHER.
Après ces cinq étapes, les concentrations des electrolytes dans les phases aqueuses binaires etde l'eau en phase organique sont connues dans les deux échelles de concentration. Lesformules de calcul sont regroupées dans l'annexe 4.
La détermination des concentrations de l'acide nitrique et des nitrates alcalins dans laphase organique à l'équilibre est réalisée en phase aqueuse après désextraction (formules decalcul en annexe 4). Une seule étape suffit pour désextraire quantitativement l'acide nitrique etle nitrate de sodium. Deux étapes sont nécessaires pour obtenir la désextraction totale desautres nitrates alcalins (potassium, rubidium et césium). Dans tous les cas le protocoleexpérimental est le suivant:
© Mise en contact de mdsex ("dsex" symbolise l'étape de désextraction) grammes d'eau
pure et de mdsex grammes de phase organique dans un tube à essai de 15 mL.
© Agitation pendant 15 minutes.
<D Centrifugation à 4000 tr.min"1 pendant 15 minutes.
© Mesure des masses volumiques des deux phases en équilibre.
© Dosage d'acide dans la phase aqueuse de désextraction.
© Dosages des alcalins dans les phases aqueuses de désextraction.
120

Chapitre 3: Modélisation physico-chimique du système d'extraction
2. Résultats.
2.1. Extraction de l'acide et de l'eau.
2.1.1. Extraction de l'eau par le NPOE.
L'utilisation de solutions aqueuses de sels non extractibles à différentes concentrationspermet de faire varier l'activité d'eau du système. Le choix de cet electrolyte est bien sûr guidépar son caractère non extractible, mais également par le domaine d'activité d'eau couvert parses données binaires [4].
Les sels de lithium, LiCl et LiNC>3, répondent favorablement à ces deux critères. En effet,nous avons pu vérifier que ces deux electrolytes ne sont extraits ni par le diluant ni par lecalixarène. Les valeurs d'activité d'eau obtenues pour des solutions saturées sont de 0,4621pour LiNO3 et de 0,1186 pour LiCl (cf. Annexe 4).
Dû
"o
SPoO
0,060
0,050
0,040
0,030
0,020
0,010
0,000
H2O/LiCl | NPOE
HjO/LiNC^ | NPOE
Droite de régression
0,0 0,2 0,8 1,00,4 0,6
Figure 3-1: Isotherme d'extraction de l'eau par le NPOE à 25°C.
L'ensemble des résultats expérimentaux est regroupé sur la Figure 3-1. Chaque point del'isotherme correspond au minimum à dix mesures, le nombre élevé de déterminationsexpérimentales nous permet de procéder à un traitement statistique.
121

Chapitre 3: Modélisation physico-chimique du système d'extraction
II apparaît clairement que les quantités d'eau dosées en phase organique varient linéairementavec l'activité d'eau du système, et ce quel que soit le sel non extractible présent en phaseaqueuse.
Lors de la présentation de la relation de SERGIEVSKII-DANNUS (Chap.l §4.2.), nous avonsdistingué l'eau provenant de l'hydratation des espèces en phase organique de l'eau solubiliséepar ces mêmes espèces. La molalité d'eau totale présente en phase organique est alors donnéepar la relation suivante:
mH n = Vhn.m,- j t h , +Vs®, v h A .aHn .m; , t h H + Sn,,.aH n.[Dill (81)H 2 ° £-i n ^nJnknhndn i—i 'nJnknhiidii « 2 ° 1nJnkn1>,,dn D l 1 H 2 ° L J V '
n n
hn: coefficient stœchiométrique d'hydratation du complexe " injnknhndn ",
««ij.k.n.d. : molalité du complexe " injnknhndn ",sy.knhndn
: nombre de molécules d'eau solubilisée par le complexe " ijnknhndn " à aH2O =1,
s®n : nombre de molécules d'eau solubilisée par le diluant à aHj0 =1,
aH20 : activité d'eau du système,
[Dil] : concentration du diluant libre.
Etant données les faibles concentrations d'eau extraite comparées à la molalité du diluantlibre, nous n'avons pas envisagé la formation d'espèces hydratées avec le NPOE. Les quantitésd'eau présentes en phase organique sont alors attribuées à la solubilisation d'eau par le diluant,d'où:
(82)
Si l'on n'envisage pas la formation d'espèces hydratées, la concentration du diluant libre est
égale à sa molalité totale. La pente de la droite de régression mH2O = f(aH20J portée sur la
Figure 3-1 permet d'accéder au nombre de molécule d'eau solubilisée par molécule de diluantà activité d'eau égale à 1 :
1000'1s®, .[NPOE] = (0,054±0,001) mol.kg'1 et [NPOE] = - ^ ^ - = 3,9789 mol.kg
=> s®, =(0,0136±0,0003)
Cette valeur peut être comparée à celle que nous avons pu calculer dans d'autres solvants àpartir de mesures expérimentales à 25°C [5]. Par exemple, le nombre de molécules d'eausolubilisées par le n-dodécane (diluant couramment utilisé lors des opérations d'extractionliquide-liquide) n'est que de 6,2.10"6. En revanche, dans le cas du nitrobenzene, molécule
voisine du NPOE, la valeur calculée est proche de celle du NPOE avec: s®itr0ben2ène = 0,0164.
122

Chapitre 3: Modélisation physico-chimique du système d'extraction
2.1.2. Extraction de l'eau par le calixarène dilué dans le NPOE.
La démarche suivie lors de cette étude est en tout point identique à celle détailléeprécédemment pour le diluant seul. Le tracé de l'isotherme d'extraction de l'eau par lecalixarène en solution dans le NPOE permet d'étudier l'hydratation de la moléculed'extractant. Ainsi, nous cherchons à mettre en évidence l'éventuelle formation d'hydrates enphase organique.
Les manipulations d'extraction mettent en contact des solutions concentrées de chlorure delithium et une solution organique contenant le calixarène à 0,00769 mol.kg"1. Nous avonschoisi de répéter les manipulations pour deux concentrations supérieures en extractant. Cesexpériences supplémentaires doivent permettre de distinguer plus aisément l'eau extraite par lecalixarène de celle solubilisée par le NPOE (Figure 3-2).
Pour chaque point de l'isotherme nous avons réalisé cinq dosages d'eau. Le nombre demesures est réduit afin de limiter la quantité d'extractant utilisée lors des extractions. Onremarque que la dispersion des résultats est plus importante pour les valeurs d'activité d'eaules plus élevées. Ce problème est lié aux difficultés rencontrées lors de la séparation desphases aqueuse et organique pour des masses volumiques voisines.
Calixarène 0,00769 mol.kgCalixarène 0,0191 mol.kg"1
Calixarène 0,0392 mol.kg"1
NPOE seul
aH2O
Figure 3-2: Isothermes d'extraction de l'eau par le calixarène à trois concentrationsdifférentes dans le NPOE à 25°C.
123

Chapitre 3: Modélisation physico-chimique du système d'extraction
Nous retrouvons sur la Figure 3-2, et ce quelle que soit la concentration de l'extractant, unevariation linéaire de la molalité de l'eau en phase organique avec l'activité d'eau du système.Les quantités d'eau extraite restent très voisines de celles obtenues avec le diluant seul.Lorsque l'on calcule la concentration d'eau apportée par l'extractant seul à activité d'eau égale
à 1 (m®20Calix), il apparaît que l'on a environ une molécule d'eau pour trois molécules de
calixarène (voir Figure 3-3).
0,014
0,012
0,010 -
3
S 0,008
§ 0,0060,004
0,002
0,000
Droite de régression
0,00 0,01 0,02 0,03 0,04
Figure 3-3: Variation des quantités d'eau extraite par le calixarène (à aHO = 1) enfonction de sa concentration.
L'hypothèse la plus simple, c'est-à-dire la solubilisation d'eau par le calixarène ne met en jeuqu'une seule espèce en phase organique. Cette seule contribution permet à nouveau demodéliser correctement l'isotherme d'extraction sur l'ensemble du domaine d'activité d'eau. Lamolalité de l'eau dosée en phase organique s'exprime alors selon:
(83)
ou bien:
mH2o,caiix (84)
124

Chapitre 3: Modélisation physico-chimique du système d'extraction
La pente de la droite de régression portée sur la Figure 3-3 permet d'accéder directement au
nombre de molécules d'eau solubilisée par l'extractant, soit: s®^ = 0,314.
Cette étude expérimentale permet également de valider les conditions de linéarité et d'idéaliténécessaires au développement du coefficient d'activité des espèces organiques selonSERGIEVSKII. Nous avons pu voir que les deux hypothèses se traduisent mathématiquementselon:
hx=hx-aH2o Vmx (37)
hx: degré d'hydratation globale de l'espèce X à activité d'eau quelconque,
h® : degré d'hydratation globale de l'espèce X à activité d'eau égale à 1.
Si l'on admet que le calixarène n'est pas hydraté, la relation (37) s'écrit pour l'espèce"extractant libre" selon:
SCalix=SCalix-aH2O VmCalix (85)
Cette relation linéaire entre l'hydratation du calixarène et l'activité d'eau du système estvérifiée pour trois concentrations mcaiix différentes (voir Figure 3-2). Les conditions delinéarité et d'idéalité sont donc remplies, ce qui permet de valider le développement ducoefficient d'activité de l'extractant selon la relation de SERGIEVSKII.
2.1.3. Extraction de l'acide nitrique par le NPOE.
Des études précédentes [6] ont pu mettre en évidence les propriétés extradantes dudiluant NPOE vis-à-vis de l'acide nitrique. Cependant, aucune conclusion définitive n'a étéjusqu'à présent retenue quant à la nature exacte des espèces extraites en phase organique. Eneffet, l'absence d'informations sur les écarts à l'idéalité en phase organique, en particulier surson éventuelle hydratation, empêchait une modélisation correcte des équilibres d'extraction.
Nous cherchons donc à décrire plus précisément le système d'extraction de l'acide par lediluant grâce à un traitement rigoureux des écarts à l'idéalité en phase organique (loi d'actionde masse et développement des coefficients d'activité selon la relation de SERGIEVSKII).
Les solutions binaires d'acide nitrique mises en œuvre couvrent un large domaine deconcentration. La solution la plus concentrée est une solution contenant 12 mol.kg" d'acidesoit une molarité de 8,6 mol.L"1. De telles concentrations se situent bien au-delà des domainesrencontrés dans l'industrie du retraitement. Cependant nous avons choisi de couvrir une largegamme de concentrations afin de mettre en évidence le plus grand nombre d'espèces en phaseorganique et d'atteindre des activités d'acide nitrique élevées, voisines de celles calculées dansdes matrices concentrées HNC^/NaNC^/H^O.
Les données expérimentales concernant l'acide nitrique et l'eau extraits en phaseorganique sont regroupées dans les figures 2-4. Nous avons choisi de présenter lesconcentrations en phase organique en fonction de la molalité de l'acide en phase aqueuse oubien de son activité thermodynamique globale.
125

Chapitre 3: Modélisation physico-chimique du système d'extraction
s"5S,""S
d"
mol
0,22
0,20
0,18
0,16
0,14
0,12
0,10
0,08
0,06
0,04
0,02
0,00
- 0,14 r
m HNO3
100 200 300 400
60
o.OX
0,22
0,20
0,18
0,16
0,14
0,12
0,10
0,08
0,06
0,04
0,02
0,00500
Figures 3-4a et 3-4b: Isothermes d'extraction de l'acide nitrique et de l'eau par le NPOEà 25°C.
126
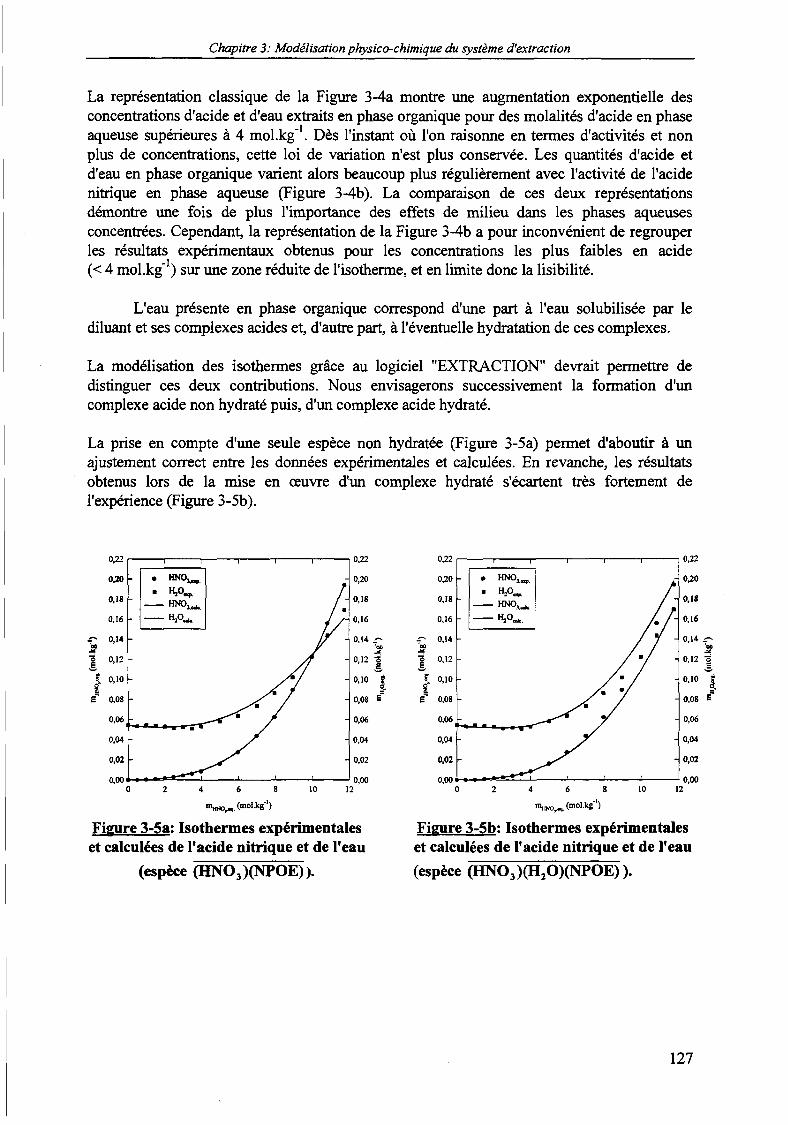
Chapitre 3: Modélisation physico-chimique du système d'extraction
La représentation classique de la Figure 3-4a montre une augmentation exponentielle desconcentrations d'acide et d'eau extraits en phase organique pour des molalités d'acide en phaseaqueuse supérieures à 4 mol.kg"1. Dès l'instant où l'on raisonne en termes d'activités et nonplus de concentrations, cette loi de variation n'est plus conservée. Les quantités d'acide etd'eau en phase organique varient alors beaucoup plus régulièrement avec l'activité de l'acidenitrique en phase aqueuse (Figure 3-4b). La comparaison de ces deux représentationsdémontre une fois de plus l'importance des effets de milieu dans les phases aqueusesconcentrées. Cependant, la représentation de la Figure 3-4b a pour inconvénient de regrouperles résultats expérimentaux obtenus pour les concentrations les plus faibles en acide(< 4 mol.kg' ) sur une zone réduite de l'isotherme, et en limite donc la lisibilité.
L'eau présente en phase organique correspond d'une part à l'eau solubilisée par lediluant et ses complexes acides et, d'autre part, à l'éventuelle hydratation de ces complexes.
La modélisation des isothermes grâce au logiciel "EXTRACTION" devrait permettre dedistinguer ces deux contributions. Nous envisagerons successivement la formation d'uncomplexe acide non hydraté puis, d'un complexe acide hydraté.
La prise en compte d'une seule espèce non hydratée (Figure 3-5a) permet d'aboutir à unajustement correct entre les données expérimentales et calculées. En revanche, les résultatsobtenus lors de la mise en œuvre d'un complexe hydraté s'écartent très fortement del'expérience (Figure 3-5b).
0,22
0,20
0,18
0,16
r* 0,14'S| 0,12
1 0,10
E 0,08
0,06
0,04
0,02
0,00
-
-
• H N O W
• HA^HNOx«k.
HA*.
1 1 l
y.
J.0,22
0,20
0,18
0,16
0,14 s-'oo
0,12 -S
0,10
0,08 S
0,06
0,04
0,02
10 120,00
Figure 3-5a: Isothermes expérimentaleset calculées de l'acide nitrique et de l'eau
(espèce (HNO3)(NPOE)).
0,22
0,20
0,18
0,16
0,14
0,12
0,10
0,08
0,06
0,04
0,02
0,00'
0
_-
••
-
_
« —
1 1
HNO,^
H A ,
•"A*.
y*•
y
/y
i
/
' •
/
V :
-10 12
0,22
0,20
0,1»
0,16
0,14 ~
'S
0,12 "S
0,10 |
0,08 S"
0,06
0,04
0,02
0,00
Figure 3-5b; Isothermes expérimentaleset calculées de l'acide nitrique et de l'eau
(espèce (HNO3)(H2O)(NPOE) ).
127

Chapitre 3: Modélisation physico-chimique du système d'extraction
La première configuration permet de modéliser parfaitement l'isotherme d'extraction de l'acidenitrique par le NPOE. Par contre, l'ajustement est moins satisfaisant quant à l'hydratation de laphase organique. Nous avons alors envisagé de modéliser le système en prenant en compte lesdeux espèces simultanément. Les résultats obtenus présentent, sur l'ensemble du domaine deconcentration, une parfaire concordance entre l'expérience et le calcul (Figure 3-6).
(mol
iCl
S
0,22
0,20
0,18
0,16
0,14
0,12
0,10
0,08
0,06
0,04
0,02
0,0010 12
m HNO,,aq.
Figure 3-6: Isothermes expérimentales et calculées de l'acide nitrique et de l'eau
(espèces (HNO3)(NPOE)et (HNO3)(H2O)(NPOE)).
Les concentrations d'acide extrait en phase organique par le NPOE sont faibles par rapport àsa concentration totale, la molécule de NPOE libre reste l'espèce majoritaire en solutionorganique à 95%. Il est alors intéressant de calculer, grâce au logiciel "EXTRACTION", larépartition de l'acide nitrique entre les deux complexes envisagés. Cette représentation estportée figure 3-7 ainsi que les valeurs optimisées des constantes effectives d'extraction et desnombres s®.
L'espèce hydratée est majoritaire, sa constante d'extraction étant 4 fois plus élevée que celledu complexe non hydraté. Lorsque la concentration d'acide en phase aqueuse augmente, la
part de l'espèce (HNO3)(H2O)(NPOE) diminue au profit de l'espèce (HNO3)(NPOE). Laformation du complexe hydraté est en effet défavorisée par la diminution de l'activité d'eau dusystème lorsque l'on se déplace vers les fortes acidités.
128

Chapitre 3: Modélisation physico-chimique du système d'extraction
a»o
O••es
100
90
80
70
60
50
40
30
20
10
0
,-5(NPOE)(HNO3) ; Keff=3,531.10 , s =0,0360
(NPOEXHNOjXHjO) ; K^l.452.10"4, se=0,1146
0 10 12
m HNO3>aq.
Figure 3-7: Diagramme de répartition de l'acide entre les deux complexes retenus lors dela modélisation du système d'extraction HNO3/H2O | NPOE à 25°C.
Comme nous l'avons déjà fait remarquer, dès l'instant où la modélisation de l'extraction del'acide nitrique par le NPOE nécessite la prise en compte de deux espèces, il est impossible devérifier les conditions de linéarité et d'idéalité introduites lors du développement descoefficients d'activité des deux complexes par la relation de SERGIEVSKII. Ces deuxhypothèses extra-thermodynamiques doivent donc être admises.
2.1.4. Extraction de l'acide nitrique par le calixarène dilué dans le NPOE.
Le tracé de l'isotherme d'extraction de l'acide nitrique par le calixarène suit unedémarche identique à celle adoptée précédemment. L'ensemble des résultats expérimentauxobtenus à l'équilibre est présenté sur la Figure 3-8.
129
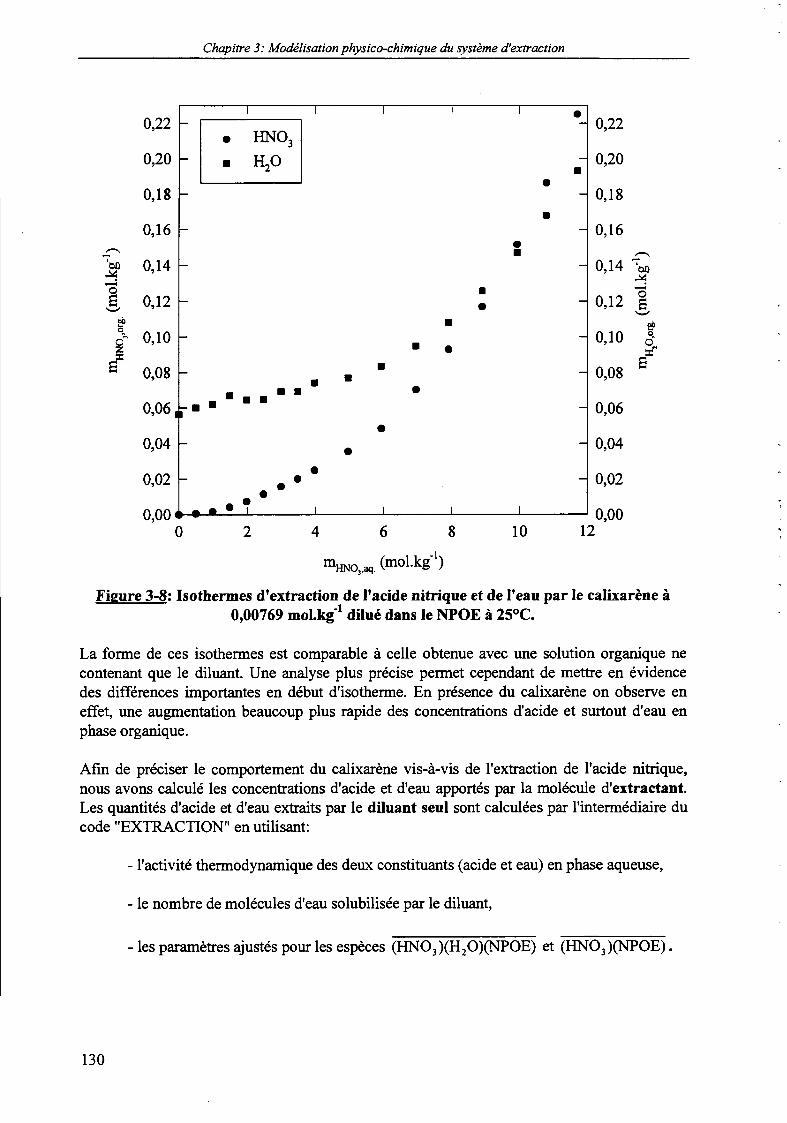
Chapitre 3: Modélisation physico-chimique du système d'extraction
0,22
0,20
0,18
0,16
£ 0,14
I 0,12
£ 0,10
S 0,08
0,06
0,04
0,02
0,00'
1
• HNO3
• HjO
-
—
- • • • • • • •II •
I I ' ' •
•
U
mm
m
• •• _
1 1 1 1
0 10
0,22
0,20
0,18
0,16
- 0,14 \o
0,12 1
0,10 I
0,08 S
0,06
0,04
0,02
0,0012
m HNO3,aq.
Figure 3-8: Isothermes d'extraction de l'acide nitrique et de l'eau par le calixarène à0,00769 moLkg'1 dilué dans le NPOE à 25°C.
La forme de ces isothermes est comparable à celle obtenue avec une solution organique necontenant que le diluant. Une analyse plus précise permet cependant de mettre en évidencedes différences importantes en début d'isotherme. En présence du calixarène on observe eneffet, une augmentation beaucoup plus rapide des concentrations d'acide et surtout d'eau enphase organique.
Afin de préciser le comportement du calixarène vis-à-vis de l'extraction de l'acide nitrique,nous avons calculé les concentrations d'acide et d'eau apportés par la molécule d'extractant.Les quantités d'acide et d'eau extraits par le diluant seul sont calculées par l'intermédiaire ducode "EXTRACTION" en utilisant:
- l'activité thermodynamique des deux constituants (acide et eau) en phase aqueuse,
- le nombre de molécules d'eau solubilisée par le diluant,
- les paramètres ajustés pour les espèces (HNO3)(H2O)(NPOE) et (HNO3)(NPOE).
130

Chapitre 3: Modélisation physico-chimique du système d'extraction
Ces valeurs sont ensuite retranchées des données expérimentales obtenues pour le systèmecomplet HNO3/H2O | Calix/NPOE. Le résultat final correspond donc aux molalités d'acide etd'eau extraites en phase organique par le calixarène seul (voir Figure 3-9).
0,035
0,030 -
0,025 -
1 0,020
t 0,015 -
0,010
0,005
0,000
-
I
II
II •
1
• HNO3
• up
• i
•
••
i•
•
•• i
i i i i ^
• *
•
_ ••
••
••
•. •
• •1
•
/ Calix,totale -
/
1 ! 1 1
0,035
- 0,030
- 0,025
$
0,020 "g
0 10 12
0,015 g;
0,010
0,005
0,000
m HNO3,aq.
Figure 3-9: Isothermes d'extraction de l'acide et de l'eau par le calixarène seul à 25°C.
On observe que la concentration d'acide extrait en phase organique devient supérieure
à celle du calixarène dès mHNC,3 aq > 2,5 mol.kg"1. En fin d'isotherme, on compte jusqu'à cinq
molécules d'acide pour une molécule de calixarène. La modélisation des équilibresd'extraction doit donc mettre en œuvre au minimum cinq complexes successifs destœchiométrie croissante en acide nitrique.
En début d'isotherme, les concentrations d'eau sont supérieures aux molalités d'acide, cecomportement s'inverse pour des concentrations en HNO3 supérieures à 6 moLkg"1 dans laphase aqueuse. Les espèces extraites aux faibles acidités contiennent donc plus d'eau qued'acide. Etant donnée la complexité du système d'extraction, le seul examen des donnéesexpérimentales ne permet pas de distinguer l'eau solubilisée par ces complexes acides, de l'eaudue à leur éventuelle hydratation.
Dans un premier temps, les calculs de modélisation sont effectués en considérant cinqcomplexes non-hydratés. Les concentrations calculées pour l'acide nitrique se rapprochent desdonnées expérimentales. Cependant, la seule prise en compte d'eau solubilisée par ces espècesacides, ne permet pas d'ajuster correctement les quantités d'eau présentes en phase organique.
131
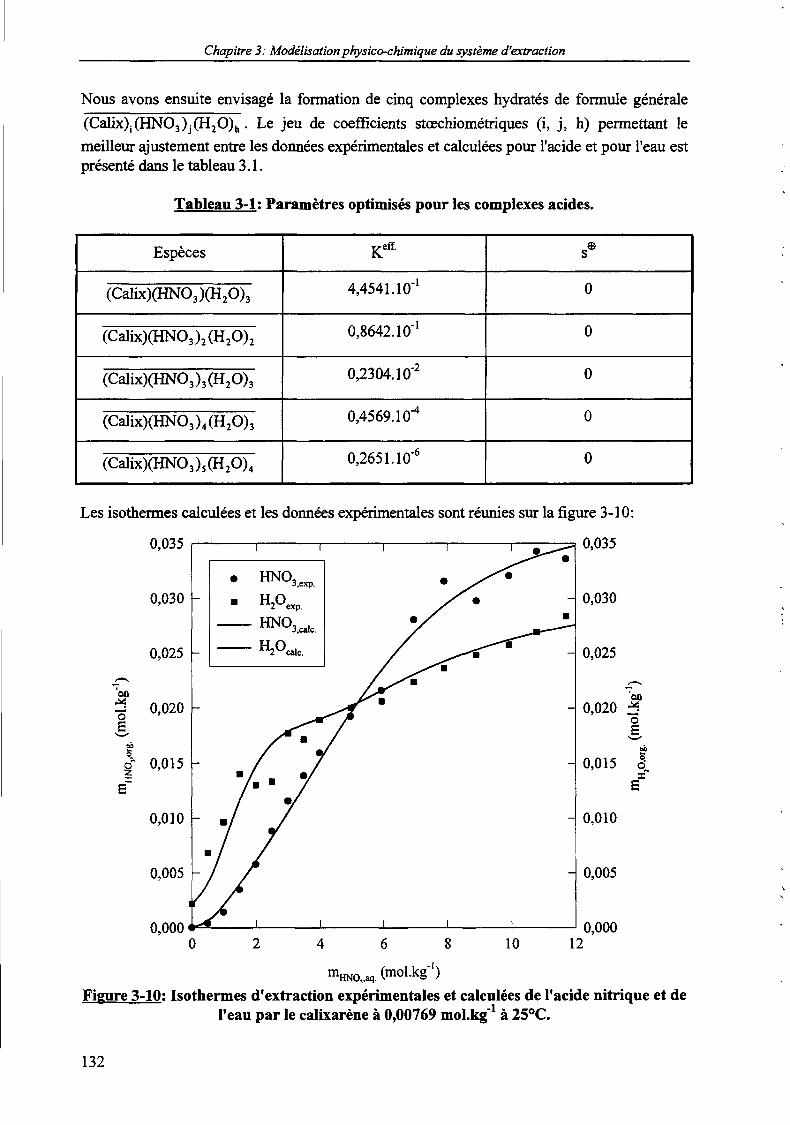
Chapitre 3: Modélisation physico-chimique du système d'extraction
Nous avons ensuite envisagé la formation de cinq complexes hydratés de formule générale
(Calix)i(HNO3)j(H2O)h. Le jeu de coefficients stoechiométriques (i, j , h) permettant le
meilleur ajustement entre les données expérimentales et calculées pour l'acide et pour l'eau estprésenté dans le tableau 3.1.
Tableau 3-1: Paramètres optimisés pour les complexes acides.
Espèces
(Calix)(HNO3)(H2O)3
(Calix)(HNO3)2(H2O)2
(Calix)(HNO3)3(H2O)3
(Calix)(HNO3)4(H2O)3
(Calix)(HNO3)5(H2O)4
Keff.
4,4541.1G"1
0,8642.1G"1
0,2304.10"2
0,4569.1G"4
0,2651.10"6
s®
0
0
0
0
0
Les isothermes calculées et les données expérimentales sont réunies sur la figure 3-10:
0,000 0,00010 12
m HNO,,aq.
Figure 3-10: Isothermes d'extraction expérimentales et calculées de l'acide nitrique et del'eau par le calixarène à 0,00769 mol.kg ' à 25°C.
132

Chapitre 3: Modélisation physico-chimique du système d'extraction
La répartition du calixarène entre chacune des espèces envisagées en phase organique estreportée sur la figure 3-11. Pour des concentrations d'acide nitrique supérieures à 4 moLkg"1 enphase aqueuse à l'équilibre, on note que le calixarène est totalement protoné.
100
80
a»o
60
1
I
40
20
0
Extradant libre
(Calix)(HNO3)2(H2O)2
(Calix)(HNO3)3(H2O)3
(Calix)(HNO3)4(H2O)3
(Calix)(HNO3)5(H2O)4
/.AN /
/
/
o 10 12
Figure 3-11: Diagramme de répartition du calixarène en phase organique.
Une étude similaire sur l'extraction de l'acide nitrique par un calixarène bis-couronne dilué dansle NPOE a été réalisée par HILL [6]. Deux complexes de stœchiométrie 1-1 et 1-2 ont étésuffisants pour modéliser l'isotherme d'extraction jusqu'à des concentrations d'acide de10 moLkg"1 en phase aqueuse. Le calixarène mono-couronne extrait donc des quantités d'acideplus importantes qu'une molécule comportant deux couronnes éther oxyde.
La formation des espèces acides en phase organique peut être interprétée soit par unmécanisme d'inclusion, soit par un mécanisme de protonation de la fonction iso-propoxy oudes chaînes étheroxyde. Cette dernière hypothèse se rapproche du mode de complexationsuggéré lors de l'extraction de l'acide nitrique par des éthers couronne classiques [7]. Dans cecas, l'extraction de l'acide nitrique par le macrocycle ne doit pas défavoriser la complexation decations alcalins par inclusion à l'intérieur de la cavité du calixarène. Nous vérifierons cecomportement dans le paragraphe 3.3. lors de la mise en évidence de la formation de
complexes mixtes du type (Calix).(HNO3).(CsNO3)k.
133

Chapitre 3: Modélisation physico-chimique du système d'extraction
2.2. Extraction des nitrates alcalins.
2.2.1. Extraction du nitrate de sodium par le calixarène dilué dans le NPOE.
Parmi les calixarènes présentant une forte affinité pour le césium, le 1,3-diisopropoxycalix[4]arène-couronne-6 est l'un de ceux qui présentent la plus grande sélectivité Cs/Na [6].Toutefois, il reste que la mise en œuvre de solutions très fortement salines nécessite la priseen compte des propriétés extractantes du calixarène vis-à-vis du sodium.
Les solutions aqueuses utilisées sont des solutions binaires de nitrate de sodium dont lesconcentrations couvrent l'ensemble du domaine de solubilité de 1'electrolyte, soit, à 25°C [8]:[0-10,8 mol.kg"1] ou bien [0-7,8 mol.L"1]. Nous avons pris la précaution d'employer des selsde qualité ultra-pure lors de la préparation des solutions concentrées, ceci dans le but delimiter la présence d'impuretés de césium.
Avant d'acquérir les données expérimentales d'extraction du calixarène dilué dans leNPOE, nous avons vérifié que le diluant n'extrait pas de nitrate de sodium. Les quantités desodium présentes dans le NPOE en équilibre avec une solution aqueuse saturée en mtrate desodium ne dépassent pas 3.10'5 mol.kg"1.
0,0010
0,0008 -
0,0006 -
1j 0,0004a
0,0002 -
0,0000
•II
1 • •• •
• NaNO3
<
•
i
' " " • • .•
••
»
i i i
0 2 4
mNaNO3,aq.
0,06
- 0,05
- 0,04
- 0,03
- 0,02
- 0,01
i
100,00
Figure 3-12: Isothermes d'extraction du nitrate de sodium et de l'eau par le calixarène à1g"1 di0,00769 moLkg"1 dilué dans le NPOE à 25°C.
134

Chapitre 3: Modélisation physico-chimique du système d'extraction
Les données expérimentales d'extraction présentées sur la figure 3-12 possèdent deuxpropriétés remarquables:
- l'isotherme d'extraction du sodium présente un changement de courbure (pourmNaNo3 =1,5 mol. kg-1),
- les quantités d'eau présentes en phase organique diminuent lorsque la concentrationdu nitrate de sodium augmente en phase aqueuse.
La présence d'un changement de courbure semble indiquer que le nitrate de sodium estprésent en phase organique sous la forme de deux complexes différents. Ceux-ci peuventcorrespondre à deux complexes successifs de stcechiométries différentes entre le sodium et le1,3-diisopropoxy calix[4]arène-couronne-6. La formation d'une espèce supplémentaire enphase organique peut également être attribuée à la présence d'une impureté possédant despropriétés extradantes vis-à-vis du sodium.
Nous avons montré que l'extraction du sodium par le diluant reste négligeable devant celle ducalixarène. L'éventuelle présence d'une impureté organique ne peut donc provenir que de lamise en solution de la molécule d'extractant. La solution organique contiendrait alors deuxmolécules extradantes, l'inflexion relevée sur l'isotherme d'extraction du sodium seraitattribuée à la saturation de l'impureté. Si l'on suppose que l'espèce formée possède unestœchiométrie 1-1, l'impureté organique peut être dosée grâce à la position de l'inflexion. Onaboutit alors à une concentration de l'ordre de 2,5.10"4 mol.kg"1 soit 3% de la quantité totaled'extractant. La mise en œuvre de méthodes spectrales comme la RMN ne permet pas demettre en évidence des concentrations d'impuretés inférieures à 5% de la concentration totale.En l'absence de méthode expérimentale directe, nous avons cherché à mettre en évidence laprésence d'une impureté extractante de manière indirecte. Pour cela, nous avons tracél'isotherme d'extraction du sodium par une molécule de calixarène synthétisée selon unschéma différent du 1,3-diisopropoxy calix[4]arène-couronne-6. La molécule choisie est lecalix[4]arène-bis couronne-6 [9] dont la synthèse ne doit logiquement pas aboutir aux mêmesimpuretés. La forme de l'isotherme obtenue est en tout point comparable à celle présentée surla figure 3-12. Il semble donc difficile d'attribuer la formation de deux complexes du sodium àla présence de deux molécules extradantes.
L'autre hypothèse permettant d'expliquer le changement de courbure en début d'isotherme, estla formation de deux complexes successifs entre le calixarène et le sodium. Des étudesprécédentes [3] ont montré que pour les concentrations les plus élevées en nitrate de sodium,le complexe formé comprend un atome de sodium pour une molécule d'extractant. L'espèceprésente en début d'isotherme ne peut donc correspondre qu'à un complexe de stœchiométriesupérieure en extractant, dont la formation est favorisée par la concentration élevée ducalixarène libre en début d'isotherme. Un tel comportement est fréquemment rencontré lors dela mise en œuvre de molécules d'extractant de type solvatant comme le TBP ou les amides.Jusqu'à présent, la formation d'un complexe comportant deux molécules de calixarène n'ajamais été envisagée du fait de l'encombrement stérique important lié à la taille de la moléculed'extractant. Toutefois, la structure d'une telle espèce peut être appréhendée par comparaisonavec les études menées sur la complexation de cations alcalins par des calixarènes couronnedoubles [10] (figure 3-13). La présence du cation alcalin au centre de l'édifice macrocycliquea pu être démontrée grâce à la mise en œuvre de méthodes spectrales.
135

Chapitre 3: Modélisation physico-chimique du système d'extraction
Figure 3-13: Double calix[4]arène-double-couronne-5.
Les quantités d'eau présente en phase organique diminuent lorsque la concentration dusodium augmente. Ce comportement permet d'affirmer que le complexe formé en find'isotherme n'est pas hydraté. La solubilisation d'eau par chacune des espèces présentes enphase organique (extractant, diluant, complexes sodium) permet alors d'expliquer la présenced'eau en phase organique.
Comme on pouvait s'y attendre, la modélisation des isothermes d'extraction à l'aided'un seul complexe de stœchiométrie 1-1 entre le calixarène et le sodium aboutit à un mauvaisajustement des données expérimentales en début et en fin d'isotherme (figure 3-14).
136
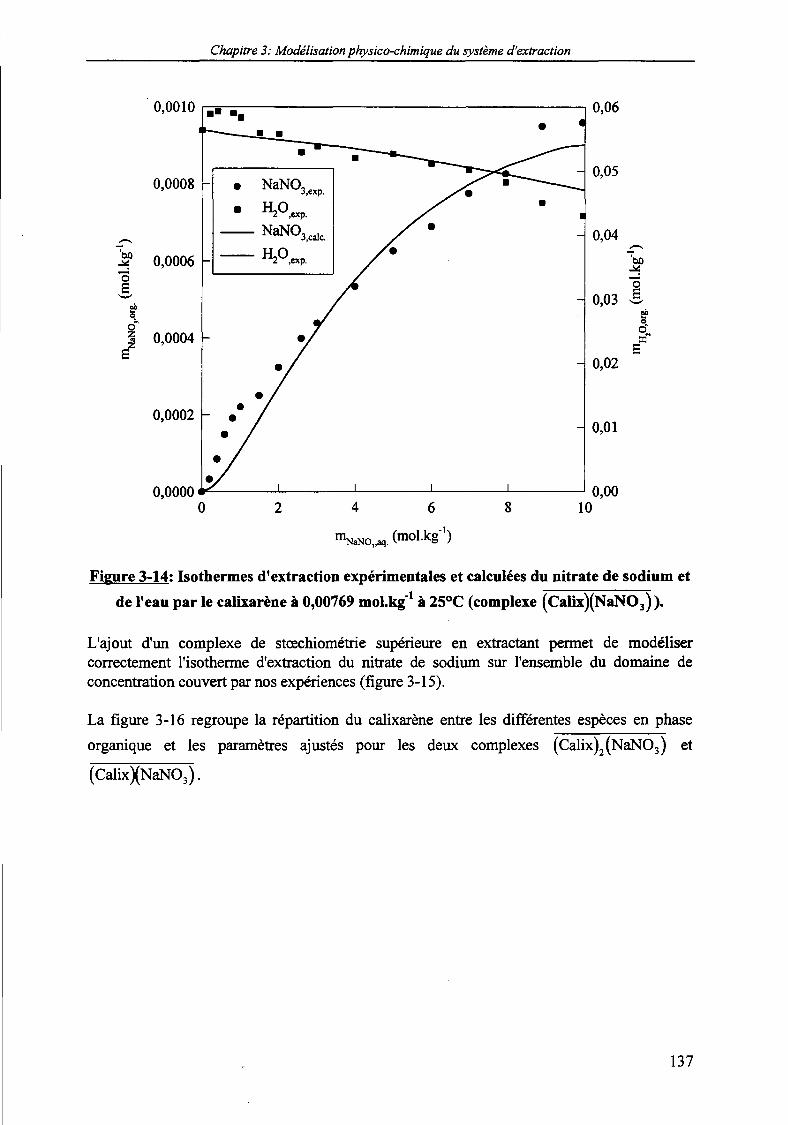
Chapitre 3: Modélisation physico-chimique du système d'extraction
5? 0,0006 -
100
"3
0,0004 -
0,0002 -
0,00000 102 4
mNaNO,,aq.
Figure 3-14: Isothermes d'extraction expérimentales et calculées du nitrate de sodium et
de l'eau par le calixarène à 0,00769 moLkg'1 à 25°C (complexe (Calix)(NaNO3)).
L'ajout d'un complexe de stœchiométrie supérieure en extradant permet de modélisercorrectement l'isotherme d'extraction du nitrate de sodium sur l'ensemble du domaine deconcentration couvert par nos expériences (figure 3-15).
La figure 3-16 regroupe la répartition du calixarène entre les différentes espèces en phase
organique et les paramètres ajustés pour les deux complexes (Calix)2(NaNO3) et
(Calix)(NaNO3).
137

Chapitre 3: Modélisation physico-chimique du système d'extraction
0,06
"oS
I 0,0004 -
0,0002 -
0,0000
"3
s
# •
10
mNaN03,a,.
Figure 3-15: Isothermes d'extraction expérimentales et calculées du nitrate de sodium et
de l'eau par le calixarène à 0,00769 mol.kg ' à 25°C (complexes (CaIix);(NaNO3) et
(Calix)(NaNO3)).
•g.§
I"ao-§§
85
15 -
10
— calixarène libre
- - (Calix)2(NaNO3) ; Krff=l,5124.101 mol"1 .kg, s®=27,1571
• • • (Calix)(NaNO,) ; K -=0,3373.10"', s®=4,0443
10
Figure 3-16: Diagramme de répartition du calixarène en phase organique.
138

Chapitre 3: Modélisation physico-chimique du système d'extraction
2.2.2. Extraction du nitrate de césium par le calixarène dilué dans le NPOE.
Nous avons dans un premier temps pu vérifier que le diluant n'extrait pas de nitrate decésium. Ensuite, l'étude des propriétés extradantes du calixarène vis-à-vis du nitrate decésium a nécessité deux séries de manipulations:
© La mise en œuvre de solutions aqueuses binaires de nitrate de césium dans lagamme [10"3 - 1,2 mol.kg"1] permet de préciser la stœchiométrie et l'hydratation descomplexes formés en phase organique.
© L'utilisation de solutions aqueuses de CsNO3 dans une plus large gamme deconcentration [10~9 - 1 mol.kg"1] permet d'étudier le comportement du calixarène vis-à-vis del'extraction de traces de césium.
© Les isothermes réunies sur la figure 3-17 permettent de mettre en évidence lagrande affinité du calixarène utilisé vis-à-vis du césium. On observe, en pied d'isotherme, unetrès forte augmentation de la concentration du césium en phase organique sans aucunecommune mesure avec le comportement observé lors de l'extraction du nitrate de sodium.
0,070
0,060
- 0,050
1
u,uuo
0,007 '
i
0,006
0,005
0,004
0,003
0,002
0,001
n non (
- •
-
-
• CsNO3
• H2O -
1 !
0,040 ^
g0,030 î
sS
0,020
0,010
0,0 0,2 0,4 0,6 0,8 1,00,000
1,2
m CsNO,,aq.
Figure 3-17: Isothermes d'extraction du nitrate de césium et de l'eau par le calixarène à0,00769 moLkg*1 dilué dans le NPOE à 25°C.
Les quantités d'eau présentes en phase organique sont très proches de celles dosées pour unesolution sans césium. Toutefois, en fin d'isotherme, la quantité d'eau apportée par le calixarène
139

Chapitre 3: Modélisation physico-chimique du système d'extraction
complexé ( mH20jComplexes ) est supérieure à la concentration du césium. Il est alors possible
d'envisager la formation d'une espèce hydratée entre le calixarène et le césium comme unealternative à la solubilisation d'eau par le complexe.
Des études antérieures [3] ont permis de montrer que le complexe formé en fin
d'isotherme est du type (Calix)(CsNO3). Toutefois, aucune information quant à l'hydratationde la phase organique n'était jusqu'à présent disponible. La prise en compte de l'eau présenteen phase organique doit nous permettre de mettre en évidence l'éventuelle formation d'uncomplexe hydraté entre le 1,3-diisopropoxy calix[4]arène-couronne-6 et le césium.
La molalité de l'eau en phase organique est, rappelons-le, exprimée d'une manière généraleselon:
ynknhndnaH2o-miJnknhndn +s* 1 .aH 2 O . [Dil ]+sf .aH 2 O . [E] (86)
n n
Le premier terme représente l'eau provenant de la formation de complexes hydratés. Les troisderniers termes regroupent l'eau solubilisée par chacune des espèces en phase organique, ilsdépendent de l'activité d'eau du système. Il apparaît donc possible de distinguer ces deux
contributions par l'étude des variations mH20 = f(aH2o) •
Le domaine d'activité d'eau couvert lors de nos manipulations est limité à [1-0,97], il est doncimpossible de conclure quant à l'hydratation de l'espèce présente en fin d'isotherme. Afin decouvrir des domaines d'activité d'eau plus étendus, nous avons mis en œuvre des solutionsternaires contenant le nitrate de césium et un sel non extractible. Une concentration en nitratede césium de 1 mol.kg"1 permet d'aboutir à la saturation de l'extractant, et ce quelle que soit laconcentration du sel non-extractible. La molalité du nitrate de lithium varie de 0 à 5 mol.kg"1,ce qui permet d'atteindre des activités d'eau de 0,75.
La molalité de l'eau apportée par la formation de complexes entre le calixarène et le césium(mH2ocomplexes) correspond aux deux premiers termes de la relation (86). Elle est doncdéterminée en déduisant l'eau solubilisée par le NPOE de la concentration totale accessible àl'expérience. Les valeurs calculées à la suite de nos manipulations sont indépendantes del'activité d'eau et l'on a:
= 0,0095 mol.kg'1 pour mCsNO3 = 0,0077 mol.kg-1
On peut donc légitimement conclure à la formation d'un complexe (Calix)(CsNO3)(H2O) enfin d'isotherme.
La mise en œuvre d'un complexe de stœchiométrie 1-1 (hydraté ou non) entre lecalixarène et le nitrate de césium ne permet pas de modéliser correctement les isothermesd'extraction. En début d'isotherme, les concentrations calculées en phase organique pour lecésium sont très largement sous estimées par rapport aux données expérimentales. Comme
140

Chapitre 3: Modélisation physico-chimique du système d'extraction
dans le cas de l'extraction du nitrate de sodium, il semble donc nécessaire d'envisager la
formation d'un deuxième complexe (Calix)2(CsNO3)(H2O)h.
© Le dosage du césium par spectrométrie gamma permet de doser directement enphase organique et en phase aqueuse de faibles concentrations de césium. Le dosage de l'eauselon la méthode Karl FISCHER n'est pas effectué, ceci pour plusieurs raisons:
- l'utilisation d'un traceur radioactif nécessiterait la nuclearisation de la cellulecoulométrique,
- l'activité d'eau de solutions aqueuses diluées (10~9 - 10'3 mol.kg-1) étant très voisinesde 1, les quantités d'eau solubilisée par les espèces en phase organique varient peu,
- les concentrations des complexes formés sont faibles par rapport à la concentrationde l'extractant libre, la molalité de l'eau en phase organique est alors proche de cellede la solution de calixarène initiale.
le-10le-9 le-8 le-7 le-6 le-S le-4 le-3 le-2 le-1 le+O
Figure 3-18a: Isothermes d'extractiondu nitrate de césium par le calixarène à
0,001le-9 le-8 le-7 le-6 le-5 le-4 le-3 le-2 le-1 le+0
mcSNovw,.(m o l kS"')
Figure 3-18b: Coefficient de distributiondu nitrate de césium.
-î0,00769 moLkg"1 dans le NPOE.
Les données expérimentales sont présentées sur les figures 3-18a et 3-18b. Ladécroissance du coefficient de distribution après le premier palier (mCsN03 > 10"5 mol. kg"1 ) nepeut pas être attribuée à la saturation de l'extractant (cf. figure 3-18a). En effet, laconcentration totale du calixarène est de 0,00769 mol.kg"1 et DCsNOj décroît à partir d'unemolalité très inférieure, de 10"6 mol.kg'1 en phase organique. Il est donc nécessaire deconsidérer la formation de deux complexes en phase organique. La présence d'une impuretéen phase organique peut de nouveau être envisagée, mais nous ne retiendrons pas cettehypothèse en accord avec le raisonnement développé au paragraphe précédent.
141
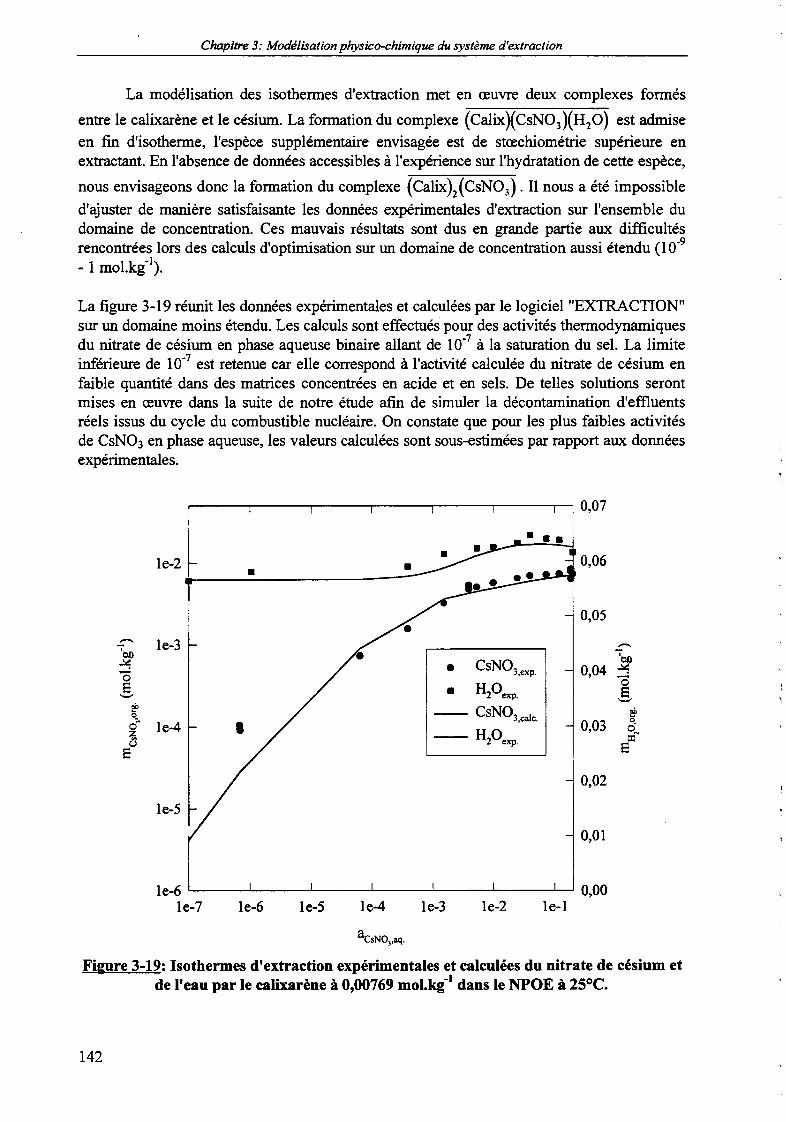
Chapitre 3: Modélisation physico-chimique du système d'extraction
La modélisation des isothermes d'extraction met en œuvre deux complexes formés
entre le calixarène et le césium. La formation du complexe (Calix)(CsNO3)(H2O) est admiseen fin d'isotherme, l'espèce supplémentaire envisagée est de stœchiométrie supérieure enextractant. En l'absence de données accessibles à l'expérience sur l'hydratation de cette espèce,
nous envisageons donc la formation du complexe (Calix)2(CsNO3). Il nous a été impossible
d'ajuster de manière satisfaisante les données expérimentales d'extraction sur l'ensemble dudomaine de concentration. Ces mauvais résultats sont dus en grande partie aux difficultésrencontrées lors des calculs d'optimisation sur un domaine de concentration aussi étendu (10-1 moLkg'1).
-9
La figure 3-19 réunit les données expérimentales et calculées par le logiciel "EXTRACTION"sur un domaine moins étendu. Les calculs sont effectués pour des activités thermodynamiquesdu nitrate de césium en phase aqueuse binaire allant de 10"7 à la saturation du sel. La limiteinférieure de 10~7 est retenue car elle correspond à l'activité calculée du nitrate de césium enfaible quantité dans des matrices concentrées en acide et en sels. De telles solutions serontmises en œuvre dans la suite de notre étude afin de simuler la décontamination d'effluentsréels issus du cycle du combustible nucléaire. On constate que pour les plus faibles activitésde CSNO3 en phase aqueuse, les valeurs calculées sont sous-estimées par rapport aux donnéesexpérimentales.
0,07
le-2 -
"3
§
le-5 -
le-6le-7 le-6 le-5 le-4 le-3 le-2 le-1
aCsNO3,aq.
Figure 3-19: Isothermes d'extraction expérimentales et calculées du nitrate de césium et-1de l'eau par le calixarène à 0,00769 mol.kg ' dans le NPOE à 25°C.
142

Chapitre 3: Modélisation physico-chimique du système d'extraction
La figure 3-20 regroupe la répartition du calixarène entre l'extractant libre et les deux espèces
(Calix)2(CsNO3) et (Calix)(CsNO3)(H2O), ainsi que leurs paramètres optimisés. Les
constantes d'extraction calculées pour les deux complexes du césium sont très largementsupérieures à celles obtenues pour les complexes formés entre le calixarène et le sodium oul'acide nitrique.
cr
12?
8es
•g.
100
80
60 -
£ 40
T3
I 20e2
_LCalixarène libre
(Calix)2(CsNO3) ; Keff =6,4991.105 mol'!.kg, s®=0
(Calix)(CsNO3)(H2O) ; Keff=4,4985.102, s®=0,3812
0le-7
\
le-6 le-5 le-4
aCsNO,,aq.
le-3 le-2 le-1
Figure 3-20: Diagramme de répartition du calixarène en phase organique.
2.2.3. Extraction du nitrate de potassium et de rubidium par le calixarène diluédans le NPOE.
Afin d'étendre notre étude aux autres nitrates alcalins, nous avons étudié l'extraction dunitrate de potassium et de rubidium par le 1,3-diisopropoxy calix^jarène-couronne-ô. Cetravail a été mené par C. BOUZON [11] lors de son stage de DEA Chimie Analytique (juin1996).
Les expériences sont de nouveau réalisées sur l'ensemble du domaine de solubilité des deuxsels. Les résultats expérimentaux présentés sur les figures 3-20 et 3-21 permettent d'établir unlien direct entre la taille du cation alcalin et son affinité pour le calixarène. Le nitrate depotassium est en effet plus extrait que le nitrate de sodium sans toutefois permettre de saturerl'extractant, le comportement du calixarène vis-à-vis de RbNO3 se rapproche quant à lui decelui relevé pour CsNO3.
143

Chapitre 3: Modélisation physico-chimique du système d'extraction
0,007
0,006
Q 0,005
• ^ 0,004
£ 0,003
0,002
0,001
00001
-
_
. — — " • " • — — * "
- /
«A*
/
0,0 0,5 1,0 1,5 2,0 2,5 3,0 3,5
0,07
0,06
0,05
0,04
- 0,03
0,02
0,01
0,00
0,007
«A»RbNO,
HA*
Figure 3-21: Isothermes d'extraction dunitrate de potassium et de l'eau par le
calixarène 0,00769 mol.kg"1 dans leNPOE à 25°C.
0,0 0,5 1,0 1,5 2,0 2,5 3,0 3,5
mRbNO (mol.kg"1)
Figure 3-22: Isothermes d'extraction dunitrate de rubidium et de l'eau par lecalixarène 0,00769 mol.kg"1 dans le
NPOE à 25°C.
0,07
0,06
0,05
0,04 HO
0,03 I
0,02
0,01
0,00
Dans les deux cas, la modélisation des isothermes d'extraction nécessite la prise encompte de deux complexes successifs hydratés entre le calixarène et le nitrate alcalin. Lesparamètres optimisés sont regroupés dans le tableau suivant:
Tableau 3-2: Paramètres optimisés pour les complexes du potassium et du rubidium.
Espèces
(Calix)2(KNO3)(H2O)
(Calix)(KNO3)(H2O)
(Calix)2(RbNO3)(H2O)
(Calix)(RbNO3)(H2O)
Keff.
l,78.102mol-i.kg
2,2
1,33.103 moH .kg
4,2. 101
s®
0
0
0
0
Nous disposons maintenant d'une étude complète sur l'extraction des nitrates alcalinspar le 1,3-diisopropoxy calix[4]arène-couronne-6.
Dans tous les cas, il est nécessaire d'envisager la formation de deux complexes successifsentre la molécule extractante et le sel.
Ces espèces sont toutes hydratées à l'exception des complexes formées avec le nitrate desodium. En l'absence de données sur l'hydratation de la phase organique en début d'isotherme,
nous n'avons pas pu trancher quant à l'éventuelle hydratation de (Calix)2(CsNO3).
144

Chapitre 3: Modélisation physico-chimique du système d'extraction
2.2.4. Système NaNOyCsNCWHoO I Calixarène/NPOE.
Jusqu'à présent, la mise en œuvre de solutions binaires d'acide ou de sel nous a permisde décrire les systèmes d'extraction les plus simples. L'étape suivante de notre démarche demodélisation est la description de systèmes plus complexes mettant en œuvre des solutionsaqueuses ternaires puis quaternaires. La modélisation de ces systèmes peut être effectuée àpartir des résultats obtenus pour les sous-systèmes d'extraction, si aucun nouveau complexe nese forme en phase organique.
Ainsi, nous avons cherché à vérifier si les résultats obtenus précédemment permettent demodéliser le système NaNCVCsNOs/HkO | Calixarène/NPOE. Afin de nous placer dans lesconditions de salinité des effluents de la Station de Traitement des Effluents Liquides (STEL),les phases aqueuses retenues lors de cette étude sont des solutions concentrées en nitrate de
sodium (mNaNOj = 4 mol. kg'1 ).
Les données expérimentales réunies sur la figure 3-23 mettent en évidence la trèsgrande sélectivité Cs/Na du 1,3-diisopropoxy calix[4]arène-couronne-6. L'extraction dunitrate de césium est en effet peu handicapée par la présence de fortes quantités de sodium enphase aqueuse.
Nous avons également porté sur la figure 3-23, les concentrations de sels calculées à partir desparamètres optimisés au cours de la modélisation de l'extraction de NaNC>3 puis de CSNO3 parle calixarène. Les molalités calculées pour le sodium, le césium et l'eau (non représentée) sonttrès proches des données expérimentales. La mise en œuvre des quatre complexes envisagéspour décrire les sous-systèmes simples permet donc une description très satisfaisante dusystème d'extraction complexe.
145

Chapitre 3: Modélisation physico-chimique du système d'extraction
0,008
0,000
0,0006
0,0005
0,0004
0,0003
0,0002
0,0001
0,0 0,1 0,2 0,3
(mol.kg"1
0,40,0000
0,5
Figure 3-23: Isothermes d'extraction expérimentales et calculées des nitrates de sodium-îet de césium par le calixarène à 0,00769 mol.kg dans le NPOE à 25°C.
2.3. Système HNO3/CSNO3/H2O | Calixarène/NPOE.
Comme nous l'avons déjà vu (cf §3.1.4.), l'extraction de l'acide nitrique par lecalixarène peut être interprétée par un mécanisme de protonation de la chaîne étheroxyde. Lacomplexation du césium par inclusion à l'intérieur de la cavité du calixarène ne doit donc pasêtre empêchée par l'extraction de l'acide nitrique. Dans ce cas, la formation de complexes
mixtes du type (Calix)i(HNO3)j(CsNO3)k doit être envisagée. Afin de mettre en évidence laformation de tels complexes, nous avons effectué une série de manipulations mettant enœuvre des solutions aqueuses ternaires contenant:
- du nitrate de césium en concentration élevée (> 0,75 mol.kg"1) permettant d'atteindrela saturation de l'extractant,
- de l'acide nitrique en concentration croissante.
Lorsque la molécule d'extractant est entièrement complexée par le césium, les quantitésd'acide présentes en phase organique ne peuvent être attribuées qu'à l'extraction d'acide par lediluant et à l'éventuelle formation de complexes mixtes.
146

Chapitre 3: Modélisation physico-chimique du système d'extraction
Nous présentons sur la figure 3-24, les quantités d'acide et d'eau apportés par lamolécule d'extractant. Ces concentrations sont obtenues en retranchant les molalités d'acide etd'eau extraits par le diluant aux données expérimentales complètes. Les isothermesd'extraction calculées en l'absence de nitrate de césium sont également réunies sur cette figure.
0,030
0,025 •
0,020 -
3~o
I 0,015
0,010 -
0,005
0,000
•
-
/
- /
HNOs.ex,
HNO3calc (sansCsNOj)n 2 U c a l c l S a n S <~slN<~y
/ / •
Si i i
i
•
•
i
i i
•
•A
1 1
0
0,025
- 0,020
00
0,015 3
- 0,005
0,000
.*
0,010 §sri"
m HNO3,aq.
Figure 3-24: Isothermes d'extraction de l'acide nitrique et de l'eau par le calixarène à0,00769 mol.kg"1 à 25°C (avec et sans césium saturant)
Nous avons pu vérifier au cours des manipulations que la concentration du césium enphase organique est égale à celle de l'extractant. L'extraction de l'acide nitrique peut donc êtreattribuée avec certitude à la formation de complexes mixtes entre l'extractant, le nitrate decésium et l'acide nitrique.Les quantités d'eau extraites par ces espèces ne sont pas suffisantes pour envisager leurhydratation, la présence d'eau solubilisée est donc retenue.
En début d'isotherme, les concentrations d'acide dosées en phase organique sont supérieuresaux molalités calculées en l'absence de nitrate de césium. Dans cette partie de l'isotherme, laprotonation de la molécule d'extraction est donc favorisée par l'inclusion du césium dans lacavité. Ce comportement s'inverse pour les concentrations plus élevées en acide nitrique(mHNo3,aq
>2mol.kg"1), la molalité maximale d'acide est inférieure à 0,01 mol.kg", soitmoins de deux fois la concentration totale d'extractant.
147
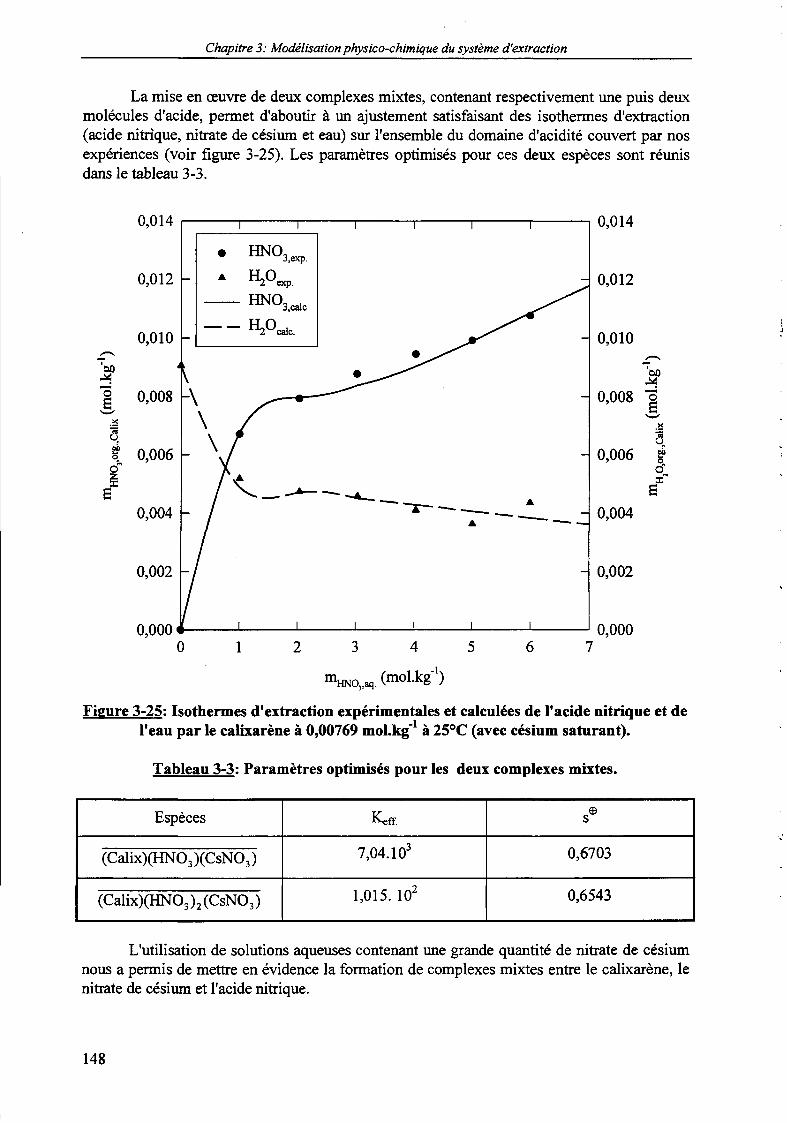
Chapitre 3: Modélisation physico-chimique du système d'extraction
La mise en œuvre de deux complexes mixtes, contenant respectivement une puis deuxmolécules d'acide, permet d'aboutir à un ajustement satisfaisant des isothermes d'extraction(acide nitrique, nitrate de césium et eau) sur l'ensemble du domaine d'acidité couvert par nosexpériences (voir figure 3-25). Les paramètres optimisés pour ces deux espèces sont réunisdans le tableau 3-3.
0,014
0,012
0,010
1 0,008X
3.t 0,006
0,004
0,002 -
0,000 0,000
m HNO3,aq.
Figure 3-25: Isothermes d'extraction expérimentales et calculées de l'acide nitrique et de- î ,l'eau par le calixarène à 0,00769 mol.kg" à 25°C (avec césium saturant).
Tableau 3-3: Paramètres optimisés pour les deux complexes mixtes.
Espèces
(Calix)(HNO3)(CsNO3)
(Calix)(HNO3)2(CsNO3)
Keff.
7,04.103
1,015. 102
s e
0,6703
0,6543
L'utilisation de solutions aqueuses contenant une grande quantité de nitrate de césiumnous a permis de mettre en évidence la formation de complexes mixtes entre le calixarène, lenitrate de césium et l'acide nitrique.
148

Chapitre 3: Modélisation physico-chimique du système d'extraction
La modélisation de l'extraction de faibles quantités de césium à partir de matrices concentréesen acide constitue l'un des objectifs de notre étude. Dans ce cas, la concentration du sel n'estbien sûr pas suffisante pour saturer la molécule d'extractant. La formation des cinq complexes
(Calix)j(HNO3)j(H2O)h doit alors être prise en compte. L'étude physico-chimique complète
du système d'extraction HNO3/CSNO3/H2O | Calixarène/NPOE nécessite la mise en œuvre deonze complexes en phase organique:
- (NPOE)(HNO3), (NPOE)(HNO3)(H2O),
- (Calix)(HNO3)(H2O)3, (Calix)(HNO3)2(H2O)2, (Calix)(HNO3)3(H2O)3
(Calix)(HNO3)4(H2O)3, (Calix)(HNO3)5(H2O)4,
- (Calix)2(CsNO3), (Calix)(CsNO3)(H2O),
- (Calix)(HNO3)(CsNO3), (Calix)(HNO3)2(CsNO3).
Les trois premières séries de complexes ont été envisagées pour modéliser successivementl'extraction de l'acide puis du nitrate de césium à partir de leur solution binaire. Les deuxderniers complexes ont dû être pris en compte lors de l'étude du système ternaire complet.
La formation de complexes mixtes entre le calixarène et les cations alcalins sodium,potassium et rubidium n'a pas été étudiée du fait de la charge de travail expérimental déjàimportante imposée par notre étude. En toute rigueur, une modélisation exhaustive dessystèmes d'extraction mettant en œuvre des solutions aqueuses ternaires HNO3/XNO3/H2O(X=Na, K, Rb) doit intégrer la prise en compte de telles espèces mixtes.
149

Chapitre 3: Modélisation physico-chimique du système d'extraction
3. Prévision de l'extraction du nitrate de césium à partir de matricesconcentrées.
La démarche analytique retenue pour modéliser notre système d'extraction nous apermis de déterminer le nombre, la stœchiométrie ainsi que les constantes d'extraction desdifférentes espèces formées en phase organique. Il est maintenant possible, à partir de laconnaissance de l'activité à l'équilibre de chaque constituant en phase aqueuse de calculer lacomposition de la phase organique.
Afin de s'affranchir de la détermination expérimentale des concentrations en phase aqueuse àl'équilibre, nous avons développé le code de calcul "PREVISION" (cf Chap.2 §2.3). Cenouveau logiciel permet de déterminer les concentrations à l'équilibre des constituantsextractibles dans les deux phases à partir de leur concentration initiale. La détermination desconditions optimales d'extraction du nitrate de césium est ainsi grandement facilitée.
Nous avons choisi de prévoir l'extraction du césium à partir de solutions aqueuses dontla composition correspond à celle d'effluents issus du cycle du combustible nucléaire. Lacomposition de la phase organique est identique à celle mise en œuvre lors de la modélisationdes isothermes. Comme nous l'avons déjà souligné, la recherche d'extractants spécifiques ducésium a pour objectif de développer des procédés de décontamination d'effluents liquidescontenant de faibles quantités de césium radioactif (137Cs, 134Cs et 135Cs). La composition deces solutions aqueuses est caractérisée par une forte salinité et/ou une forte acidité. Le tableau3-4 regroupe les caractéristiques principales de trois catégories d'effluents radioactifs.
Tableau 3-4: Caractéristiques des effluents à traiter.
Type.
Effluents salins.
Raffinats UOX ou MOX.
Concentrats d'évaporateurs.
Composition de la matrice.
NaNO3:100à400g.L'1
(1 à 5 molX"1)
pH de 7 à 12
HNO3: 3 mol.L"1
NaNO3: 200 à 350 g.L"1
(2,5à4mol.L"1)
HNO3:1 mol.L"1
Activité des radioisotopes ducésium et concentration totale.
137Cs: 44 mCi.L'1
Césium total: 5.10"6 mol.L*1
" 'Cs^CLL" 1 ,I34Cs: 3 Ci.L"1
Césium total: 5.10"3 mol.L"1
137Cs: 20 à 90 mCi.L*1,^CsilàSmCiX"1
Césium total: 5.10"6 mol.L"1
Dans un premier temps, nous avons choisi de prévoir l'extraction du nitrate de césiumà une concentration de 10"5 mol.L"1 dans des matrices concentrées en nitrate de sodium et/ouen acide nitrique. Les résultats de ces calculs prédictifs sont comparés aux données
150

Chapitre 3: Modélisation physico-chimique du système d'extraction
expérimentales acquises pour ces systèmes. Cette comparaison doit nous permettre de validerla représentation proposée de la phase organique.
Dans un deuxième temps, nous avons simulé la décontamination d'effluents dont lescompositions reprennent celles du tableau 3-4. L'efficacité de plusieurs étages d'extractionsuccessifs est quantifiée par le logiciel "PREVISION". Pour ce faire, le rendementd'extraction (R) ainsi que le facteur de décontamination (FD) sont calculés à partir desconcentrations du césium initiale et à l'équilibre dans les phases aqueuses et organiques (àvolume égal des deux phases).
R(%) = 100- mCsN0"eq- = 100-( 1 - m C s N ° 3 - e q • ] (87)
mCsNO3,eq.
151

Chapitre 3: Modélisation physico-chimique du système d'extraction
3.1. Prévision de l'extraction du nitrate de césium à partir de solutions fortementsalines.
Le calcul des concentrations de chaque constituant en phase aqueuse et organique est
effectué en considérant la formation des quatre complexes: (Calix)2(CsNO3),
(Calix)(CsNO3)(H2O), (Calix)2(NaNO3), (Calix)(NaNO3). Le coefficient de distributioncalculé pour le césium est comparé à la valeur expérimentale sur la figure 3-26.
8
PrévisionExpérience
0
mNaNO,,ini. (mol .kg ' )
Figure 3-26: Prévision de l'extraction du nitrate de césium (105 mol.kg"1) par lecalixarene (0,00769 mol-kg"1) à partir de solutions concentrées en nitrate de sodium à
25°C.
Le coefficient de distribution du nitrate de césium atteint un maximum pour uneconcentration de NaNO3 de 1 mol.L"1 en phase aqueuse initiale, puis diminue pour dessalinités plus élevées. Ce comportement a déjà été observé par HILL [6] cependant, enl'absence de données précises sur la physico-chimie des solutions, une interprétation définitiven'avait pu être retenue.
L'ensemble des données acquises lors de la modélisation du système d'extraction doitnous permettre d'aboutir à une description précise des différents phénomènes mis enjeu.
152

Chapitre 3: Modélisation physico-chimique du système d'extraction
Nous avons reporté sur la figure 3-27 les variations du coefficient d'activité et de l'activitéthermodynamique du nitrate de césium dans les solutions aqueuses à l'équilibre d'extraction.La présence de fortes quantités de nitrate de sodium entraîne une diminution rapide de yCsNO ,
ce qui signifie que les effets de milieu subis par le sel en solution défavorisent sa réactivitéchimique. A l'opposé, l'activité thermodynamique de CsNO3 augmente d'une manièrecontinue avec la salinité de la phase aqueuse. L'effet de milieu négatif est donc minoritaireface à l'augmentation de la concentration des ions nitrate en solution: c'est l'effet de masse quil'emporte. Dans ce cas, l'extraction de CsNO3 doit être favorisée par la mise en œuvre desolutions de salinités croissantes. Cette prévision n'est vérifiée que pour les concentrations deNaNO3 inférieures à 1 mol.kg"1. La diminution du coefficient de distribution observée pourdes molalités supérieures ne peut alors être attribuée qu'à l'extraction compétitive du nitrate desodium.
l,0e-6
0,0e+0
m NaNO3,ini.
Figure 3-27: Calcul du coefficient d'activité et de l'activité à l'équilibre du nitrate decésium (initialement à 10~s mol.kg ') dans des solutions concentrées en nitrate de sodium.
Le calcul de la répartition du calixarène entre les différentes espèces en phase organique(figure 3-28) permet une visualisation de l'extraction compétitive du nitrate de sodium. Laformation des deux complexes entre le calixarène et le sodium est en effet à l'origine d'unediminution de la concentration d'extractant libre. Cette compétition défavorable à l'extractiondu nitrate de césium est le phénomène prépondérant pour des salinités supérieures à 1,5mol.kg'1 en phase aqueuse.
153
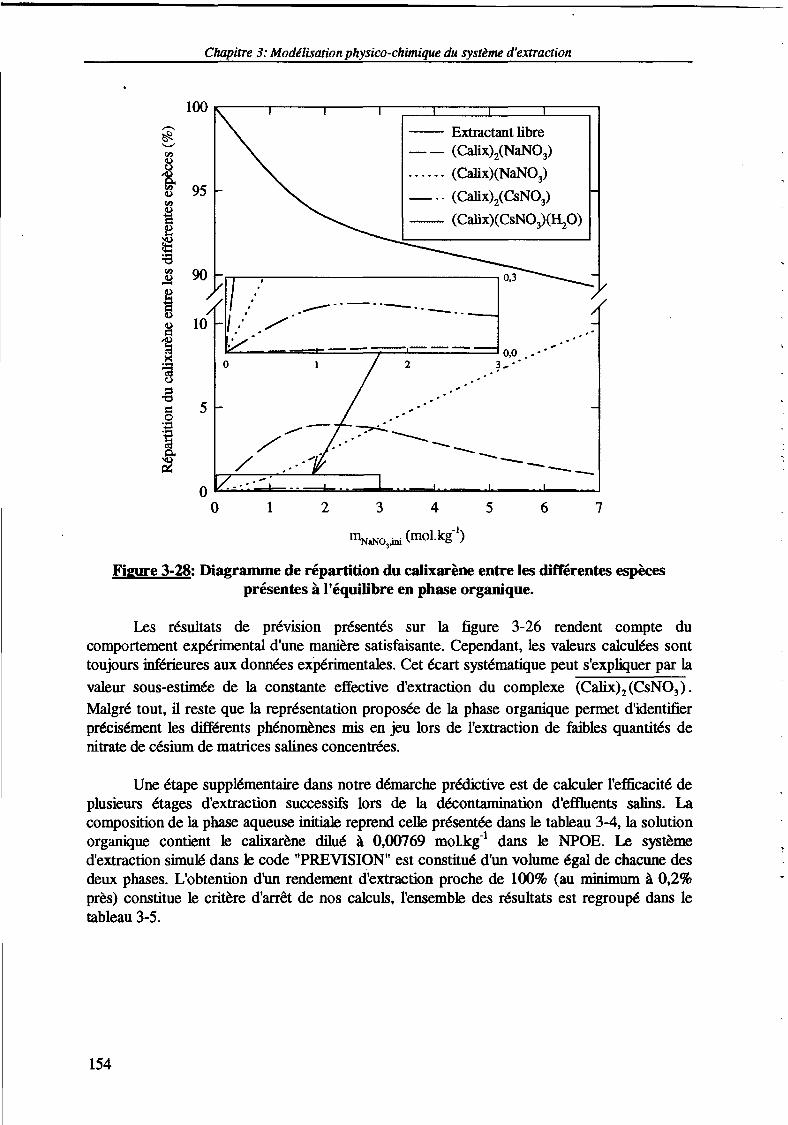
Chapitre 3: Modélisation physico-chimique du système d'extraction
100
S 95CO
II
Extradant libre(Calix)2(NaNO3)
(Calix)(NaNO3)
(Calix)2(CsNO3)
(Calix)(CsNO3)(H2O)
S 90
4>
§••51
Figure 3-28: Diagramme de répartition du calixarène entre les différentes espècesprésentes à l'équilibre en phase organique.
Les résultats de prévision présentés sur la figure 3-26 rendent compte ducomportement expérimental d'une manière satisfaisante. Cependant, les valeurs calculées sonttoujours inférieures aux données expérimentales. Cet écart systématique peut s'expliquer par la
valeur sous-estimée de la constante effective d'extraction du complexe (Calix)2(CsNO3).Malgré tout, il reste que la représentation proposée de la phase organique permet d'identifierprécisément les différents phénomènes mis en jeu lors de l'extraction de faibles quantités denitrate de césium de matrices salines concentrées.
Une étape supplémentaire dans notre démarche prédictive est de calculer l'efficacité deplusieurs étages d'extraction successifs lors de la décontamination d'effluents salins. Lacomposition de la phase aqueuse initiale reprend celle présentée dans le tableau 3-4, la solutionorganique contient le calixarène dilué à 0,00769 moLkg"1 dans le NPOE. Le systèmed'extraction simulé dans le code "PREVISION" est constitué d'un volume égal de chacune desdeux phases. L'obtention d'un rendement d'extraction proche de 100% (au minimum à 0,2%près) constitue le critère d'arrêt de nos calculs, l'ensemble des résultats est regroupé dans letableau 3-5.
154

Chapitre 3: Modélisation physico-chimique du système d'extraction
Tableau 3-5: Simulation de la décontamination d'un effuent contenant initialement5.10 mol.kg" de césium et de 1 à 5 mol.L" de nitrate de sodium.
Nombre d'étagesd'extraction.
1er étage
2ème étage
3ème étage
4èrae étage
Salinité initiale.
1M
5M
1M
5M
1M
5M
1M
5M
mCsNO3,eq.
(mol.kg"1)
6,6.10'7
ÎJ.IO"6
8,8.10"8
5,4.10"7
l,2.10"8
l,8.10"7
l,6.10"9
5,9.10"8
Rendementd'extraction (%)*.
87,5
66,7
98,2
88,9
99,8
96,4
100,0
98,8
Facteur dedécontamination*
8
3
57
9
427
28
3125
85
*: Valeurs cumulées.
Comme nous l'avons déjà vu, la décontamination en césium de solutions fortementsalines est défavorisée par l'extraction compétitive du nitrate de sodium. Ainsi, quatre étagesd'extraction permettent d'extraire quantitativement (à mieux que 0,1% près) le nitrate decésium de solutions molaires en NaNC>3. Le facteur de décontamination est alors supérieur à3000. Par contre, la décontamination de solutions cinq fois plus concentrées en sodium n'estpas aussi performante après quatre opérations d'extraction successives: le FD n'étant que de85.
155

Chapitre 3: Modélisation physico-chimique du système d'extraction
3.2. Prévision de l'extraction du nitrate de césium à partir de solutions fortementacides.
Comme nous l'avons déjà souligné au paragraphe 3.3., l'étude physico-chimique del'extraction du nitrate de césium à partir de solutions acides nécessite la prise en compte deonze complexes. Ce nombre important d'espèces reflète la complexité du système d'extraction.
Les phases aqueuses mises en œuvre lors de cette étude sont des solutions d'acidenitrique à des concentrations comprises dans la gamme [0-4 mol.kg" ] soit [0-3,5 mol.L" ], cequi correspond à l'acidité des raffinats UOX ou MOX. Les résultats expérimentaux ainsi queles valeurs calculées du coefficient de distribution du nitrate de césium sont réunies sur lafigure 3-29.
25
20
15
PrévisionExpérience
mHNO3,ini. (moLkg 1 )
Figure 3-29: Prévision de l'extraction du nitrate de césium (10~5 moLkg"1) par lecalixarène (0,00769 mol.kg"1) à partir de solutions concentrées en acide nitrique à 25°C.
La courbe de distribution de CsNO3 présente un maximum pour une molalité d'acidenitrique légèrement supérieure à 2 mol.kg'1 en phase aqueuse. La position de ce maximumd'extraction est identique à celle relevée lors de l'extraction de traces de césium de solutionsacides par le Calix[4]arène-bis-couronne-6 [6]. Les phénomènes mis en jeu dans ce systèmed'extraction peuvent de nouveau être appréhendés grâce aux connaissances acquises au coursde notre démarche physico-chimique.
156

Chapitre 3: Modélisation physico-chimique du système d'extraction
Dans un premier temps, nous avons quantifié les effets de milieu et de masse encalculant le coefficient d'activité et l'activité du nitrate de césium dans les solutions aqueuses àl'équilibre.
enOu
§
1a
1,0
0,8 -
0,6 -
g 0,4
0,2 -I
0,0
/
/
1
1 1
1
YcsNO,
aHNO,
/
^y
/_
2,5e-7
- 2,0e-7
l,5e-7 g
- l,0e-7
- 5,0e-8
0,0e+0
mHNO,,,ni.
Figure 3-30: Calcul du coefficient d'activité et de l'activité à l'équilibre du nitrate decésium (initialement à 10'5 moLkg"1) pour des solutions concentrées en acide nitrique.
Le coefficient d'activité de CsNO3 dans la phase aqueuse à l'équilibre diminue de manièrecontinue, l'effet de milieu subi par le sel défavorise donc sa réactivité chimique. Toutefois,l'apport d'ions nitrate par l'acide nitrique est à l'origine d'une augmentation de l'activité dunitrate de césium par effet de masse et favorise son extraction. Cependant, à la différence ducomportement observé dans des solutions salines:
© l'activité de CSNO3 atteint un maximum puis décroît pour des acidités supérieures à1 mol.kg"1,
© les valeurs d'activité calculées sont trois à quatre fois inférieures.
On s'attend donc à observer une extraction maximale pour une molalité en acide nitriquevoisine de 1 molal, le coefficient de distribution restant sur tout le domaine d'acidité plusfaible que dans des milieux salins.
157

Chapitre 3: Modélisation physico-chimique du système d'extraction
Comme nous l'avons décrit précédemment (figure 3-29), l'acidité optimale se situe à desconcentrations d'acide nitrique supérieures à 2 mol.kg"1. De plus, le coefficient de distributionalors mesuré est de 22 contre 7 lors de la mise en œuvre de solutions salines. L'environnementthermodynamique du nitrate de césium dans les phases aqueuses acides ne suffit donc pas àexpliquer en totalité les phénomènes mis enjeu lors des équilibres d'extraction.
La formation de complexes acides de stœchiométrie (Calix)(HNO3)j(H2O)h est àl'origine de la diminution de la concentration d'extractant libre (voir figure 3-31), et doit ainsidéfavoriser l'extraction du nitrate de césium. En même temps, l'augmentation de laconcentration d'acide nitrique en solution aqueuse s'accompagne de la formation de
complexes mixtes (Calix)(HNO3)j(CsNO3). Pour des acidités voisines de 2 mol.kg"1, le
nitrate de césium est majoritairement présent en phase organique sous la forme de cescomplexes. La mise en œuvre de solutions plus concentrées en acide nitrique entraîne unediminution de la part de ces espèces au profit des complexes "simples"
(Calix)(HNO3)j(H2O)h. VI
1'•a
Mu
o
.1'•S
I
100
80
60
40
20
0
Extractant libreComplexes acidesComplexes césiumComplexes mixtes
2 3 4
mHNO3,ini.
Figure 3-31: Diagramme de répartition du calixarène entre les différentes espècesprésentes à l'équilibre en phase organique.
La propension du calixarène à former des complexes mixtes permet donc d'expliquer à la foisles valeurs élevées du coefficient de distribution et la présence d'un maximum d'extraction.
158

Chapitre 3: Modélisation physico-chimique du système d'extraction
Les valeurs calculées du coefficient de distribution du nitrate de césium sont trèsvoisines des données expérimentales. Les plus forts écarts, relevés aux faibles acidités, sontattribués une nouvelle fois aux difficultés rencontrées lors de l'optimisation de la constante
effective d'extraction du complexe (Calix)2(CsNO3). La qualité des calculs effectués lors dela description des complexes mixtes permet, quant à elle, de rendre parfaitement compte del'extraction de CSNO3 de solutions acides de 2 à 4 mol.kg"1.
Afin de nous placer dans les conditions réelles de décontamination d'effluents de hauteactivité, nous avons déterminé l'efficacité de plusieurs étages d'extraction. De nouveau,l'utilisation du logiciel "PREVISION" permet de calculer la concentration du césium en phaseaqueuse après chaque opération d'extraction et ainsi d'accéder aux valeurs du rendementd'extraction et du facteur de décontamination.
Tableau 3-6: Simulation de la decontamination d'un effluent contenant 5.10"3 mol.kg"1 decésium et 3 mol.LT1 d'acide nitrique par une solution de calixarène à 0,00769 mol.kg \
Nombre d'étagesd'extraction.
1er étage
2ème étage
3ème étage
mCsNo3>eq. ( m o l . k g ' 1 )
4,6.10"4
2,2.10"5
l,0.10"6
Rendementd'extraction (%)*.
90,9
99,6
100,0
Facteur dedécontamination*.
11
223
4820
*: Valeurs cumulées.
Trois étages d'extraction successifs permettent d'extraire quantitativement (à 0,1% près) lenitrate de césium de solutions très acides, le facteur de décontamination est alors proche de5000. Ce taux de décontamination élevé met une nouvelle fois en évidence l'efficacitéexceptionnelle de la molécule de calixarène vis-à-vis de l'extraction de faibles quantités decésium à partir de matrices concentrées en acide nitrique.
159

Chapitre 3: Modélisation physico-chimique du système d'extraction
3.3. Prévision de l'extraction du nitrate de césium à partir de solutionsconcentrées en acide et en sels.
La validité de nos calculs de prévision appliqués à des phases aqueuses complexes3NaNO3/CsNO3/H2O est confrontée une nouvelle fois à des données expérimentales.
Pour ce faire, nous avons étudié l'extraction du nitrate de césium à 10"5 mol.kg"1 à partir dematrices concentrées en acide nitrique et en nitrate de sodium. Le tableau 3-7 regroupe lesdonnées expérimentales pour trois séries de manipulations:
- une série à concentration de nitrate de sodium constante et à acidité variable (©),
- deux séries à concentration d'acide nitrique constante et à salinité variable (©, (D).
Le calcul du coefficient de distribution du nitrate de césium, par l'intermédiaire du logiciel"PREVISION", nécessite la prise en compte de treize espèces en phase organique. Il s'agit desonze complexes du paragraphe précédent auxquels sont ajoutés les deux complexes:(Calix)2(NaNO3) et (Calix)(NaNO3).
Tableau 3-7: Prévision de l'extraction par le calixarène (0,00769 mol.kg!) du nitrate decésium (10~5 mol.kg"1) à partir de matrices concentrées en acide nitrique et en nitrate de
sodium à 25°C.
Série demanipulation.
©
(D
0,50
1,00
1,40
1,90
0,90
0,90
0,90
1,80
1,80
mNaNO3,rai. (mol .kg ' )
2,90
2,90
2,90
2,90
0,90
1,70
2,50
0,90
:-' " 1,80
^CsNOj.exp.
11,7
13,6
13,8
12,9
15,1
15,0
14,0
18,7
16,2
13,9
D W c .
10,7
14,8
17,7
17,6
12,4
13,6
14,3
- -19'3iv--
18,8
Les variations expérimentales du coefficient de distribution du nitrate de césium sonten accord avec nos études précédentes sur les matrices ternaires NaNC^/CsNC^/H^O etHNO3/CSNO3/H2O. En effet, la mise en œuvre de solutions acides (jusqu'à des molalitésinférieures à 2 mol.kg"1) permet d'augmenter l'extraction du césium. A l'opposé, ladécontamination en césium de solutions de salinité croissante est de plus en plus difficile.
160

Chapitre 3: Modélisation physico-chimique du système d'extraction
La comparaison des valeurs calculées et expérimentales montre que les écarts deviennentimportants dans les solutions les plus concentrées en acide et en sel. Cette erreur de prévisionpeut être attribuée à la non prise en compte des complexes mixtes formés entre le calixarène,l'acide nitrique et le nitrate de sodium. En négligeant la formation de ces espèces, noussurestimons la concentration d'extractant libre et, par là même, la formation des complexes ducésium (voir tableau 3-8).
Toutefois, nos calculs restent proches des données expérimentales lors de la mise en œuvre desolutions peu acides dont la concentration n'excèdent pas 1 mol.kg"1 en HNO3. Dans ce cas,les écarts les plus importants sont relevés aux faibles concentrations de NaNC^. Ils peuventêtre de nouveau attribués aux problèmes rencontrés lors de l'optimisation des paramètres du
complexe (Calix)2(CsNO3). L'augmentation de la salinité de la solution aqueuse favorise la
formation des complexes mixtes (Calix)(HNO3)j(CsNO3), dont les constantes de formation
ont été calculées avec précision. La description faite de la phase organique se rapproche ducomportement expérimental, et ce malgré l'absence de données concernant les espèces mixtes
(Calix)(HNO3)j(NaNO3).
Tableau 3-8: Répartition du calixarène (en %) entre les espèces présentes en phaseorganique lors de l'extraction du nitrate de césium (10~5 mol.kg *) à partir de matrices
concentrées en acide nitrique et en nitrate de sodium.
Série
en
©
®
mHNO3,ini.
(moLkg-1)
0,50
1,00
1,40
1,90
0,90
0,90
0,90
1,80
1,80
1,80
mNaNO3,ini.
(mol.kg'1)
2,90
2,90
2,90
2,90
0.90
1,70
2,50
0,90
1,80
2,60
Extractantlibre
75,33
55,48
36,74
22,90
73,85
65.40
58,57
41,37
32,24
25,30
Complexesacides
19,29
40,99
61,01
75,67
23,82
31,46
37,99
57.60
66,41
73,30
Complexessodium
5,24
3,41
2,13
1,32
2,19
3,02
3,32
0,91
1,22
L29
Complexescésium
0,05
0,01
0,00
0,00
0,07
0,03
0,02
0,01
0,00
0.00
Complexesmixtes
0,08
0,10
0,11
0,12
0,07
0,09
0,10
0,10
0,11
0,11
Etant donnés les domaines d'acidité et de salinité des concentrats d'évaporateurs, ilsemble donc possible de simuler l'efficacité de plusieurs étages d'extraction lors de ladécontamination de tels effluents. Les résultats de nos calculs sont regroupés dans le tableau3-9 ci-dessous:
161
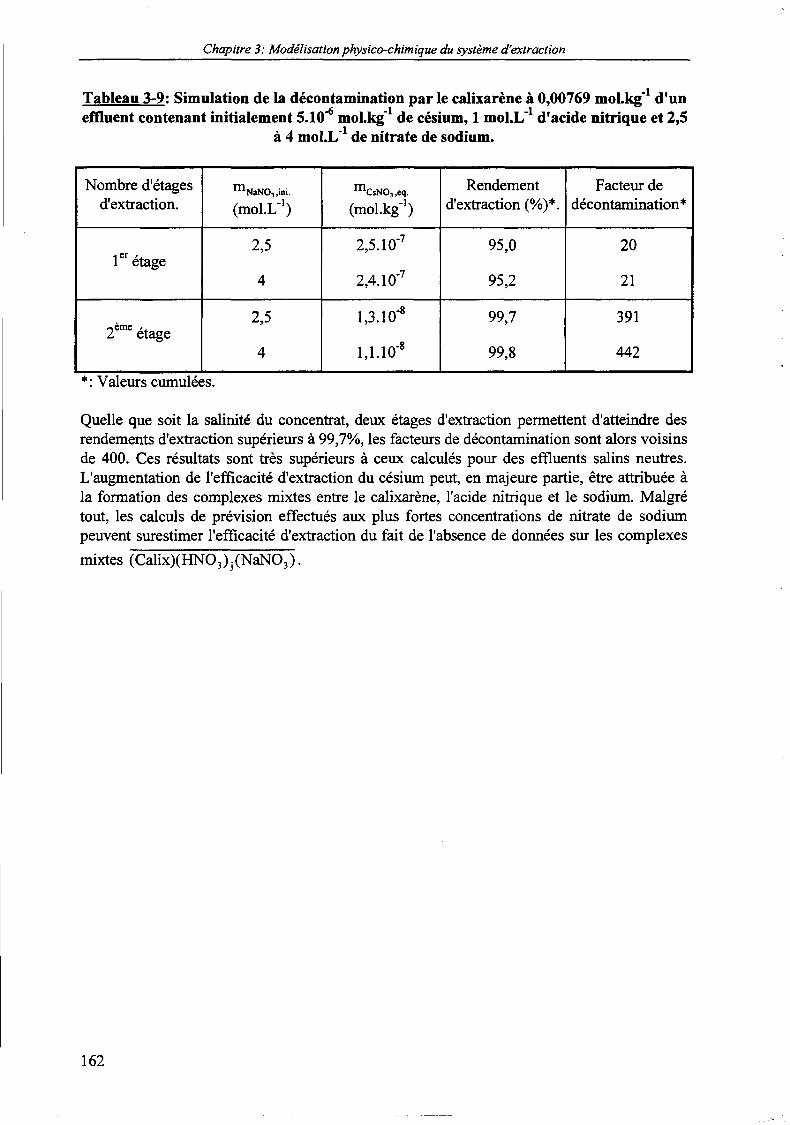
Chapitre 3: Modélisation physico-chimique du système d'extraction
-1Tableau 3-9: Simulation de la decontamination par le calixarène à 0,00769 mol.kg" d'uneffluent contenant initialement 5.10"6 mol.kg"1 de césium, 1 mol.L"1 d'acide nitrique et 2,5
à 4 mol.L"1 de nitrate de sodium.
Nombre d'étagesd'extraction.
1er étage
2ème étage
mNaNO3,ini.
(mol.L"1)
2,5
4
2,5
4
mCsNO3,eq.
(moLkg1)
2,5.10"7
2,4.10"7
l,3.10"8
1,1.10"8
Rendementd'extraction (%)*.
95,0
95,2
99,7
99,8
Facteur dedécontamination*
20
21
391
442
*: Valeurs cumulées.
Quelle que soit la salinité du concentrât, deux étages d'extraction permettent d'atteindre desrendements d'extraction supérieurs à 99,7%, les facteurs de décontamination sont alors voisinsde 400. Ces résultats sont très supérieurs à ceux calculés pour des effluents salins neutres.L'augmentation de l'efficacité d'extraction du césium peut, en majeure partie, être attribuée àla formation des complexes mixtes entre le calixarène, l'acide nitrique et le sodium. Malgrétout, les calculs de prévision effectués aux plus fortes concentrations de nitrate de sodiumpeuvent surestimer l'efficacité d'extraction du fait de l'absence de données sur les complexes
mixtes (Calix)(HNO3)j(NaNO3).
162

Chapitre 3: Modélisation physico-chimique du système d'extraction
Conclusion.
La modélisation physico-chimique des équilibres d'extraction a été l'occasion demettre en œuvre les outils théoriques et informatiques présentés dans les deux premierschapitres. Etant donnée la complexité du système d'extraction à étudier, nous avons choisid'adopter une démarche analytique en divisant le système complet en sous-systèmes plussimples.
L'étude de l'extraction de l'acide nitrique et des nitrates alcalins à partir de leurs solutionsbinaires respectives a permis de préciser la stœchiométrie des espèces extraites en phaseorganique. Ainsi, la modélisation des isothermes d'extraction de l'acide nitrique par unesolution de calixarène dilué dans le NPOE a nécessité la prise en compte de cinq complexeshydratés. La quantification des équilibres d'extraction entre les nitrates alcalins et le calixarènea été menée à bien en envisageant la formation de deux complexes successifs destœchiométrie (Calix)j(XNO3)(H2O)h (i=l ou 2). L'hypothèse de la stœchiométrie supérieureen extradant reste à démontrer par des méthodes spectrales.
La représentation obtenue lors de la modélisation des sous-systèmes binaires NaNO3/H2O etCSNO3/H2O a été mise à profit pour calculer les données relatives à l'extraction simultanée dusodium et du césium. La bonne adéquation entre valeurs calculées et expérimentales permetd'éprouver avec succès la validité des espèces proposées et des paramètres associés (KçX et As
) dans des conditions différentes de celles de l'ajustement.L'étude du système d'extraction HNO3/CSNO3/H2O | Calix/NPOE a permis de mettre enévidence la formation de complexes mixtes entre la molécule d'extractant, l'acide nitrique et lenitrate de césium. Ce comportement se rapproche de celui relevé dans le cas d'autresextradants (amides par exemple). Il permet d'expliquer les performances accrues ducalixarène lors de l'extraction du nitrate de césium de solutions d'acidité voisine de 2 mol.L"1.
La modélisation du système d'extraction complet nécessite de prendre en compte la formationde treize espèces. En dépit d'une telle complexité, nous avons pu valider la représentation faitede la phase organique. C'est ainsi que l'extraction de faibles quantités de nitrate de césium àpartir de matrices concentrées en acide et en sels a pu être prévue. Ces calculs prédictifs ontété menés en prenant soin de décrire les différents phénomènes fondamentaux gouvernantl'extraction du césium, à savoir les effets de milieu et de masse ainsi que les propriétés decomplexation propres à la molécule de calixarène. Une telle description est indispensable afinde permettre une compréhension approfondie du comportement observé expérimentalement.
Le modèle théorique utilisé ainsi que les outils informatiques mis en œuvre ont doncpu être validés sur un système d'extraction comportant le l,3-diisopropoxy-calix[4]arène-couronne-6 dilué dans le NPOE. La modélisation d'autres systèmes engageant une moléculede calixarène diluée dans un autre diluant que le NPOE peut être maintenant envisagéemoyennant l'acquisition des données expérimentales d'extraction.
163

Chapitre 3: Modélisation physico-chimique du système d'extraction
BIBLIOGRAPHIE.
[1] J.A. BEATTIE, B.T. BROOKS, L.J. GILLEPSIE, G. SCATCHARD, W.C. SCHUMB,R.F. TEFFT,Density (Specific Gravity) and Thermal Expansion (under Atmospheric Pressure) ofAqueous Solutions of Inorganic Substances and of Strong Electrolytes,International Critical Tables of Numerical Data, Physics, Chemistry and Technology,Vol. Ill, (1928), 51-111,441-444.
[2] J.LY,Le logiciel AQUACALC,Communication personnelle, (janvier 1989).
[3] A. CASNATI, A. POCHINI, R. UNGARO, F. UGOZZOLI, F. ARNAUD, S. FANNI,M.J. SCHWING, R.J.M. EGBERING, F. DE JONG and D.N. REINHOUDT,Synthesis, Complexation, and Membrane Transport Studies of 1,3-AlternateCalix[4]arene-crown-6 Conformers: A New Class of Cesium Selective Ionophores,J. Am. Chem. Soc, 117, (1995), 2767-2777.
[4] W.J. HAMER, Y.C. WU,Osmotic Coefficients and Mean Activity Cofficients of Uni-univalent Electrolytes inWater at 25°C,J. Phys. Chem. Ref. Data, 1, (1972), 1047-1099.
[5] J. RYDBERG, C. MUSIKAS, G.R. CHOPPPIN,Principles and Practices of Solvent Extraction,Marcel Dekker, Inc., New York-Basel-Hong Kong, (1992), 31-32.
[6] C. HILL,Application des calixarènes fonctionnalisés au traitement des effluents radioactifs parmembranes liquides supportées,Thèse de Doctorat de l'Université Louis PASTEUR de STRASBOURG, (1994).
[7] (a) I.H. GEROW, M.W. DAVIS Jr,The use of 24-Crown-8's in the Solvent Extraction of CsNO3 and Sr(NO3)2,Sep. Sci. Technol., 14, (1979), 395-414.
(b) I.H. GEROW, J.E. SMITH Jr, M.W. DAVIS Jr,Extraction of Cs+ and Sr2+ from HNO3 Solution Using Macrocyclic Polyethers,Sep. Sci. Technol., 16, (1981), 519-548.
[8] W. LINKE, A SEIDELL,Solubilities, Inorganic and Metal-Organic Compounds, K-Z, Vol. I, Fourth Edition,American Chemical Society, Washington D.C., (1965).
165

Chapitre 3: Modélisation physico-chimique du système d'extraction
[9] J.F. DOZOL, Z. ASFARI, C. HILL, J. VICENS,Calix[4]arènes-bis-couronne, leur procédé de préparation et leur utilisation pourl'extraction du césium et des actinides,Brevet français N°9214245, (1992).
[10] Z. ASFARI, R. ABIDI, F. ARNAUD, J. VICENS,Synthesis and Complexing Properties of a Double-Calix[4]arene Crown Ether.J. Incl. Phenom. Mol. Recog. Chem., 13, (1992), 163-169.
[11] C. BOUZON,Modélisation de l'extraction liquide-liquide des nitrates de potassium et de rubidium parun calixarène couronne,Communication interne, DEA de Chimie Analytique, INSTN/UPMC (juin 1996).
166

CHAPITRE 4
MODELISATION DU TRANSPORT A TRAVERS DESMEMBRANES LIQUIDES SUPPORTEES.

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
Introduction.
Les membranes liquides supportées (MLS) constituent un moyen original deséparation par extraction liquide-liquide, Cette technique a été mise en œuvre au sein duService d'Etudes Procédés (SEP) lors des études portant sur la décontamination sélective desconcentrais d'évaporateurs en césium, en strontium et éléments transuraniens [1, 2]. Plusrécemment, le traitement des effluents salins selon un procédé de séparation par MLS a étéenvisagé.
Dans ce dispositif, la phase organique est immobilisée à rintérieur d'un support poreux puismise en contact simultanément avec l'effluent aqueux à traiter et la solution de désextraction.Les mécanismes d'extraction et de désextraction sont ainsi combinés aux phénomènes dediffusion à travers la membrane.
La démarche de modélisation suivie jusqu'à présent nous a permis de fournir une descriptionprécise des mécanismes fondamentaux gouvernant les équilibres d'extraction. Dans la suite dece chapitre, notre objectif est de déterminer puis de quantifier les différents paramètressupplémentaires nécessaires à la description des phénomènes de diffusion.
L'ensemble des résultats ainsi obtenus est ensuite mis à profit pour prévoir le transport defaibles quantités de nitrate de césium à partir de matrices concentrées dont la compositionsimule les effluents à traiter.
169

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
1. Techniques et mécanismes de séparation membranaires.
Les procédés de séparation membranatre peuvent être classés en deux catégories.
© Les membranes solides semi-perméables sont utilisées dans les domaines derultrafiltration, de la dialyse ou de l'osmose inverse. Le mécanisme de séparationest fondé sur l'exclusion par la taille en exploitant les caractéristiques physiques dusupport.
© Les membranes liquides mettent à profit des mécanismes d'extraction par solvant. Ilest ainsi possible, grâce à la mise en œuvre d'extractants très sélectifs, d'aboutir àdes efficacités de séparation élevées. Dans ce cas, ce sont les propriétés de la phaseorganique et non du support qui sont exploitées.
On distingue parmi la classe des membranes liquides plusieurs techniques différentes [3], Lesplus usitées sont les membranes liquides épaisses, supportées et émulsionnées. Le dispositifexpérimental (figure 4-1) correspondant à une membrane liquide épaisse comprend la phaseorganique en contact avec les phases aqueuses d'alimentation et de réception, elles-mêmesséparées par une barrière solide imperméable. L'ensemble des trois solutions est agitémodérément de manière à éviter un éventuel mélange. La technique des membranes liquidessupportées (MLS) met en œuvre une membrane solide polymère dans laquelle est immobiliséela phase organique. Comme le montre la figure 4-1, les phases aqueuses d'alimentation et deréception sont placées de part et d'autre de la membrane. L'intérêt principal d'un tel dispositiftient à l'utilisation de très faibles volumes de solution extractante.
Alimentation*• — Réception
Alimentation
Phase organique mcmbranaire
Réception
Phase organique rnembran aire
Figure 4-1: Membranes liquides épaisses et supportées.
A la différence des deux précédentes techniques, les membranes liquides émulsionnées (MLE)ont recours à une dispersion des trois phases. En effet, la première étape de cette techniqueconsiste à obtenir une emulsion très fine de la solution de réception dans la phase organique(figure 4-2). Cette emulsion est stabilisée par l'emploi d'un tensioactif, puis mise en contactavec la solution à traiter. Une fois les équilibres d'extraction et de désextraction atteints,l'émulsion est séparée puis cassée. On récupère alors la phase de réception chargée, la phaseorganique étant recyclée dans le procédé.
170

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
Réception
Alimentation
Phase organique membranaire Y '• • - r •
Figure 4-2: Schéma d'une membrane liquide émulsionnée.
1.1. Membranes liquides supportées.
Comme nous l'avons déjà dit en introduction, la technique des membranes liquidessupportées a été retenue dans le cadre des études sur la décontamination d'effluents radioactifspar des macrocycles. Ce choix s'explique par la facilité de mise en œuvre ainsi que par lafaible quantité d'extractant nécessaire pour imprégner le support.
1.1.1. Caractéristiques du support.
L'élaboration des MLS met en œuvre des supports polymériques destinés initialementà des techniques d'ultrafiitration. La structure poreuse de ces matériaux est obtenue parlaminage ou bien par attaque chimique d'un film dense.
Chaque support est caractérisé par un ensemble de propriétés structurales, à savoir: lagéométrie, la porosité, la tortuosité, le diamètre des pores et la tension superficielle critique.
La géométrie de la membrane dépend du dispositif expérimental retenu. La configuration laplus simple est constituée par des disques pians de faible épaisseur, découpés dans des filmsdestinés à l'ultrafiltration. Un tel dispositif nécessite de faibles volumes de phase organique etconstitue ainsi une méthode de choix au laboratoire. Les modules de fibres creuses sont desassemblages compacts permettant de multiplier le rapport "surface d'échange/volume à traiter"jusqu'à 1000 m2/m3. Ils sont donc destinés à la réalisation de pilotes préfigurant l'installationindustrielle.
La porosité du support définit le pourcentage d'espace libre dans le réseau par rapport auvolume total de la membrane. Sa connaissance permet donc de déterminer le volume de phaseorganique effectivement admissible dans la membrane.
La tortuosité est la mesure de l'allongement du chemin réel de diffusion par rapport àl'épaisseur de la membrane. Sa valeur dépend de la nature du support et surtout de sa méthodede préparation.
Le diamètre des pores varie de 0,02 à lum pour les supports les plus courants. Sa valeurconditionne la différence de pression admissible de part et d'autre de la membrane.
171

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
La tension superficielle critique (yc) d'un support est définie par analogie avec la tensionsuperficielle des liquides [4], Tout liquide de tension de surface inférieure à yc permet demouiller complètement le support et assure ainsi la stabilité de la membrane.
1-1-2. Caractéristiques du diluant.
Le diluant organique doit» dans un premier temps, assurer la solubilisation de lamolécule d'extractant. Dans un second temps, les caractéristiques du diluant doivent permettrede stabiliser la membrane liquide. L'immobilisation de la solution organique à l'intérieur despores du support nécessite ainsi:
- un diluant inerte, non volatil,
- une tension superficielle du diluant ("fuum) inférieure à la tension superficiellecritique du support pour répondre au critère de mouiîlabilité, mais égalementsupérieure à la tension superficielle des solutions aqueuses en contact pour éviter ledépart de la phase organique membranaire. Pratiquement, on doit avoir:
15 mN.m4 < ymmat < 35 mN.m"1
1.2. Mécanismes du transport à travers les membranes liquides.
Les phénomènes de transport à travers les membranes liquides peuvent être classésselon deux types: les transports passifs et actifs [5].
1.2.1. Transports passifs.
Le transport d'un soluté par un phénomène de transport passif est gouverné par la seuledifférence de son potentiel chimique (ou activité) entre les phases aqueuses d'alimentation etde réception. Le transport d'une espèce peut être assuré par simple partage avec le diluantimmobilisé dans le support suivi d'une étape de diffusion à travers la membrane. On pariealors de transport simple. En l'absence de cette réaction de partage, il est possible d'avoirrecours à un extractant spécifique dans la phase organique membranaire. Le transport est alorsfacilité par l'équilibre d'extraction entre le soluté et la molécule extractante. Les schémassimplifiés correspondant à ces deux mécanismes sont regroupés sur les figures 4-3a et 4-3b.
172

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
M
Alimentation
M
Membrane
M
- • M
Réception Alimentation
ML-
Membrane
M
Réception
Figure 4-3a: Mécanisme de transportsimple.
Figure 4-3b: Mécanisme de transportfacilité.
Le mécanisme de transport facilité a été proposé pour modéliser le transport de moléculesneutres [6] ou de paires d'ions [7] par des macrocycles. Le transport du nitrate de césium pardes calixarènes à travers des membranes liquides supportées appartient ainsi à cette catégoriede transport passif.
1.2.2. Transports actifs.
Dans le cas d'un transport actif, le gradient de potentiel chimique de l'espèce à séparerne constitue plus la seule force motrice. Le flux d'une seconde espèce ou bien une réactionchimique autre que les réactions d'extraction et de désextraction est mis à profit afind'améliorer les performances de la séparation.
Par exemple, le couplage de deux flux membranaires opposés ou concourants intervientrespectivement dans les mécanismes de contre-transport et de co-transport (Figures 4-4a et 4-4b). Le gradient de potentiel chimique choisi pour la seconde espèce (N) est le plus souventsupérieur à celui du premier soluté M à séparer, il constitue donc la force motrice principaledu transport.
M
N -*r ^-fc*
Alimentation Réception Alimentation
Figure 4-4a: Mécanisme de contre-transport.
Figure 4-4b: Mécanisme de co-transport.
Le mécanisme de transport du nitrate de césium par des calixarènes à partir de matricesconcentrées pourrait être apparenté à un phénomène de co-transport. En effet, le transport ducésium est assuré sous la forme de paire d'ions en phase organique. La force motricemajoritaire serait alors la différence de potentiel chimique de l'anion nitrate. Cependant, à la
173

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
différence du mécanisme décrit sur la figure 4-4b, la molécule de calixarène ne peuttransporter individuellement ni le cation ni l'anion, mais seulement la paire d'ions. Il est doncimpossible de discerner le flux du cation de celui de l'anion, la force motrice du transport dunitrate de césium est donc bien assurée par la différence de son potentiel chimique entre lesdeux phases aqueuses.
Il est également possible d'intervenir sur le transporteur plutôt que sur les espècestransportées. On a alors recours à une réaction chimique (parfois même photochimique) del'extractant en solution organique comme le montre la figure 4-5:
Alimentation
r >,L'
L"
iL'
Membrane
M
Réception
Figure 4-5: Mécanisme de transport couplé avec une transformation de l'extractant.
Dans un tel mécanisme, l'extraction du soluté M par le ligand sous la forme L' est suivie de la
diffusion du complexe ML1 à travers la membrane. Au niveau de la face de réception, le
ligand subit une transformation réversible en sa forme L" qui ne possède aucune propriétécomplexante vis-à-vis de M. La réaction de conversion inverse a lieu au niveau de la faced'alimentation. Un tel mécanisme de transport couplé a été mis à profit au laboratoire lors del'étude du transport de cations alcalins par des calixarènes photoisomérisables [8].
174

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
2. Les principaux modèles de transport facilité.
Le transport d'un soluté à travers les membranes liquides peut être décomposé seloncinq étapes successives schématisées sur la figure 4-6 [9]:
© Diffusion à travers la couche limite de diffusion (couche limite de NERNST) auniveau de la phase d'alimentation,
(D Réaction d'extraction,
(D Diffusion à travers la membrane,
© Réaction de désextraction,
© Diffusion vers la solution de réception.
to
1<DO
c
u-aS£
CL,Solution
d'alimentation
- • K
\
Membrane
Couches limites de diffusion
Solutionde réception
Figure 4-6: Etapes successives du transport membranaire.
Selon ce schéma, les profils de concentration dans les couches limites de diffusion et àl'intérieur de la membrane sont linéaires. On suppose donc que le processus de diffusion aatteint un régime stationnaire. Ces deux hypothèses sont admises par l'ensemble des auteurs,nous les admettrons dans la suite de ce texte.
Le mécanisme ainsi détaillé combine les réactions d'extraction et de désextraction (© et ©)avec les phénomènes de diffusion à travers les couches limites de NERNST (© et (D) et lamembrane organique (<D). La mise en équation d'un tel système aboutit à des relationsmathématiques très compliquées dont la mise en œuvre reste délicate. C'est pourquoi, lamajorité des auteurs a cherché à déterminer le phénomène limitant le transport membranaireafin de réduire le nombre d'étapes à prendre en compte. Cette démarche a été à l'origine d'unnombre important de modèles reflétant la très grande diversité des systèmes à étudier. Dans la
175

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
suite de ce paragraphe, nous avons limité notre présentation aux différentes approchesappropriées à l'étude des mécanismes de transport facilité.
2.1. Caractéristiques du système à modéliser.
Les caractéristiques propres au système à modéliser ont été définies antérieurement, demanière précise, par HILL [2].
Comme nous avons pu le voir dans le chapitre 3, les cinétiques d'extraction et de désextractionsont rapides. Ces deux étapes (© et ©) ne limitent donc pas le transport membranaire.
Dans l'ensemble des manipulations, les phases aqueuses d'alimentation et de réception sontuniformément agitées. Cette agitation permet de maintenir l'épaisseur de la couche limite dediffusion à une valeur constante très inférieure aux dimensions de l'installation.
La mise en œuvre de molécules macrocycliques de grande taille, diluées dans un solvant deviscosité élevée (TINPOE = 12,6 cP), sont à l'origine d'une diffusion membranaire très lente(étape (D). Les coefficients de diffusion calculés par la relation de WILKE-CHANG ou deSTOCKES-EINSTEIN, pour des éthers-couronnes de plus faible encombrement que lescalixarènes, sont en effet compris entre 3 et 7.10"7 cm2.s'' [10]. Ces valeurs sont trèsinférieures à celles mesurées dans les solutions aqueuses (> 10"5 cm2.s').
La diffusion à travers la membrane organique apparaît donc, dans notre cas, commel'étape limitante du transport.
2.2. Présentation des principaux modèles de transport facilité.
Nous présentons dans ce qui suit, les principaux modèles développés pour quantifierles mécanismes de transport à diffusion limitante. Malgré la relative simplicité du phénomèneà étudier, la mise en équation peut différer fortement d'un auteur à l'autre.
2.2.1. Le modèle de WARP et ses développements.
En 1970, WARD [6] a développé le premier modèle permettant de quantifier letransport d'une espèce neutre, facilité par la présence d'une molécule extractante dans la phaseorganique membranaire. REUSCH [7] a étendu ces travaux à l'étude du transport de chloruresalcalins à partir de leur solution binaire. La phase organique membranaire est constituée d'unéther-couronne dilué dans le chloroforme. Le mécanisme de transport met en jeu deuxéquilibres:
- Equilibre de partage du sel MCI entre les phases aqueuses et la phase organique.
M+ + Cl" o (M+, Cl" ) avec k = y—Tf—i (89)[M+][X~]
(M+,cr)
- Equilibre de complexation de la paire d'ions par la molécule d'extractant en phaseorganique.
176

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
+ L<->(L)(M\Cr)avec K = â=S—'=ré± (90)[(M*.cr)].[L]
Le transport de MCI est assuré à la fois par la paire d'ions et par le complexe formés en phaseorganique. Le mécanisme de transport de ces deux espèces est décomposé selon:
Solutiond'alimentation
\
Membrane
Couches limites de diffusion
Solutionde réception
Figure 4-7: Etapes considérées dans le modèle de WARD.
Selon ce schéma, les auteurs supposent donc que:
- les gradients de diffusion à travers les couches limites de diffusion sont négligeables,
- les réactions d'extraction et de désextraction (© et ©) sont rapides,
- le transport est limité par la diffusion membranaire (étape (D).
Le flux du sel à travers la membrane correspond à celui de la paire d'ions et du complexe. Lamise en équation fait appel à la première loi de FICK, soit:
ÔCrJ = _ n ^MCl p ^^ComplexeMCI MCI -\ Complexe
JMC1 : flux total de MCI à travers la membrane (mol.cm^.s"1),DMci » complexe: coefficient de diffusion de la paire d'ions et du complexe (cm2.s''),
CMCI ' -complexe: concentration de la paire d'ion et du complexe en phase organique (mol.cm'3),
x: distance par rapport au plan vertical de la membrane (cm).
En admettant que les gradients de concentration sont constants à l'intérieur de la membrane, larelation (91) devient:
177

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
MC1_ DMCL (Q _Q \ . DCompIexe /p _ c \
d: épaisseur de la membrane,
' CMC1 R • concentration de la paire d'ions dans la phase organique en contact avec la
phase aqueuse d'alimentation et de réception,
CcomPiexe,A , CComplexe R : concentration du complexe dans la phase organique en contact avec la
phase aqueuse d'alimentation et de réception.
Les concentrations dans la phase organique membranaire sont inaccessibles à l'expérience.C'est pourquoi, l'équation finale fait intervenir les concentrations de l'électrolyte dans lesphases aqueuses d'alimentation et de réception. Ces concentrations sont calculées à partir deslois d'action de masse associées aux équilibres d'extraction et de désextraction:
(93)
y _JMC1 ~ j
^Complexe •J S-J S- '- 'Ligand
d l + k .K .C 2M C U l + k.K.C2
MC1 ,R/
CMC1A, CMC1R : concentration du sel dans les solutions d'alimentation et de réception,
CLigand : concentration totale du ligand en phase organique.
Le premier terme correspond au flux de l'électrolyte par transport simple, le second représentele flux du sel par transport facilité. La diffusion des electrolytes dans les solutions organiquesrestant très limitée, le premier terme est légitimement négligé par les auteurs. Deux autreshypothèses simplificatrices sont faites en considérant que:
- la concentration du sel dans la phase d'alimentation est très supérieure à celle dans laphase de réception,
- la concentration de l'électrolyte dans la solution d'alimentation reste assez faible pour
vérifier la relation: k.K.C2vlclA]jm« 1.
La relation simplifiée est alors:
T _ ^Complexe• K ' J S "^LigandJMC1 ~ jMC1
Selon cette expression, le flux de l'électrolyte MCI doit être constant au cours du temps et nedoit dépendre que des concentrations du ligand en phase organique et du sel dans la solutiond'alimentation. L'étude expérimentale du transport de KCl par le dibenzo-18-couronne-6 [7] apermis de valider la relation (94). Toutefois, la mise en œuvre de solutions aqueuses contenant
178

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
plus de 1 mol.L"1 de KC1 nécessite de raisonner en terme d'activité thermodynamique et nonplus de concentration.
C'est ainsi que LAMB [11] a développé un modèle analogue à celui de REUSCH en prenantsoin d'expliciter les effets de milieu dans les phases aqueuses d'alimentation. Les hypothèsessimplificatrices retenues par REUSCH sont reprises intégralement, le flux d'un electrolyte 1-1est alors exprimé selon:
T _ ^Complexe• K - J S " V -Li g a ndJ M X - -j
a,,t . , av. . : activité du cation et de l'anion.
En toute rigueur, le produit aM+.ax_ est égal à l'activité thermodynamique de l'électrolyte MX
). Les auteurs utilisent une notation différente: a . . a v . = (a,,+ ) . Cette écriture inexacte/ M X \ M /
provient de la confusion faite entre les définitions du coefficient d'activité ionique et ducoefficient d'activité ionique moyen. Malgré tout, leurs calculs font intervenir les donnéestabulées dans la littérature pour différents electrolytes dans leur solution binaire et sont, parvoie de conséquence, exacts.
La relation (95) a permis l'étude du transport de différents sels de potassium par deux éthers-couronne. Les auteurs ont pu vérifier que le flux du potassium était proportionnel à l'activitédu sel dans la solution aqueuse d'alimentation sur un large domaine d'activité. Cependant, leflux mesuré expérimentalement s'écarte de la droite calculée pour les plus fortes valeursd'activité du sel en phase aqueuse. Cet écart est attribué à l'erreur commise lorsque l'hypothèse
^ « 1 n'est plus vérifiée, c'est-à-dire aux plus fortes valeurs de CMX A .
Commentaires:
O Comme nous l'avons déjà souligné, le modèle de WARD a été développé pourétudier le transport d'un electrolyte à partir d'une solution d'alimentation binaire concentrée(concentration inférieure à 1 mol.L"1). Cependant, la mise en œuvre de phases aqueusesconcentrées est à l'origine d'écarts importants entre les données expérimentales et les valeurscalculées par la relation (95).
O Seul le transport d'un seul electrolyte à partir de sa solution binaire a été modélisé.La mise en jeu de solutions aqueuses plus complexes contenant des sels ou un acidepotentiellement transportables par le macrocycle ou le diluant n'est pas envisagée.
La modélisation du transport du nitrate de césium par un calixarène à partir de matricesconcentrées en acide et/ou en sels n'est donc pas directement envisageable par l'équation (95).
2.2.2 Le modèle de DANESI.
Le modèle de DANESI a été élaboré pour permettre la modélisation du transport dunitrate d'américium par le Carbamoyl Méthylène Phosphine Oxyde (CMPO). Il a été ensuite
179
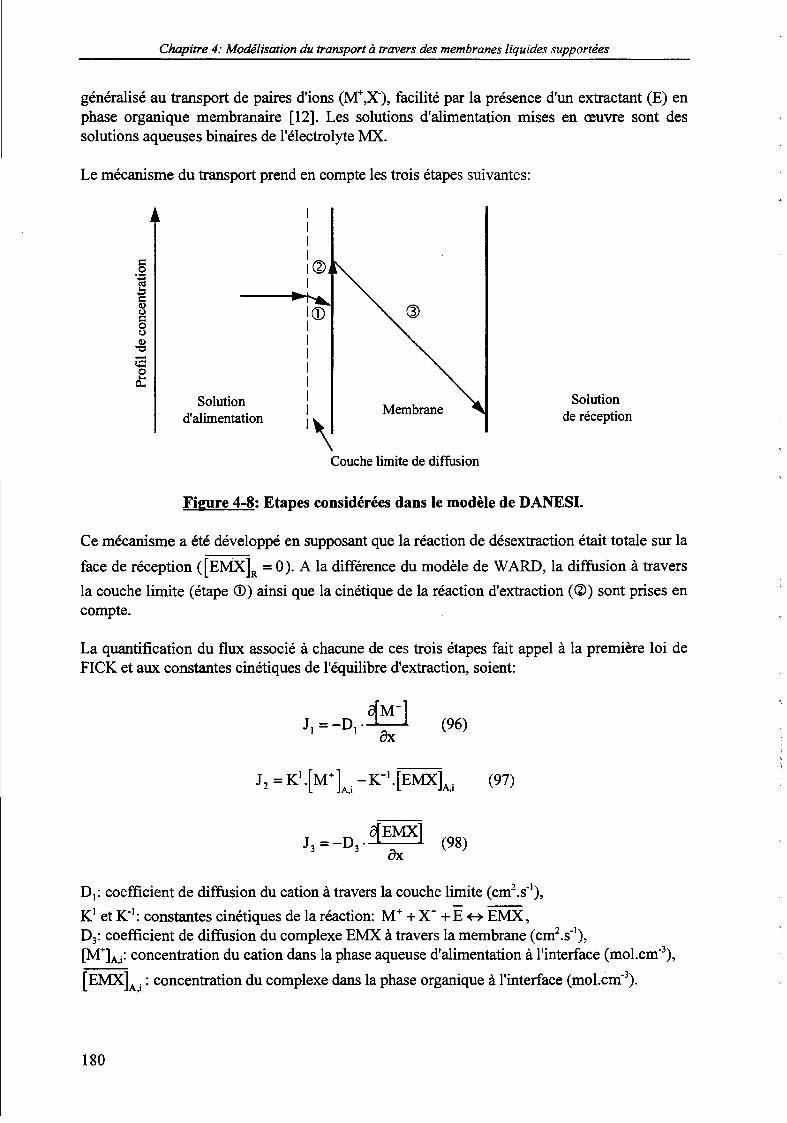
Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
généralisé au transport de paires d'ions (M+,X"), facilité par la présence d'un extractant (E) enphase organique membranaire [12]. Les solutions d'alimentation mises en œuvre sont dessolutions aqueuses binaires de l'électrolyte MX.
Le mécanisme du transport prend en compte les trois étapes suivantes:
ISîo
Solutiond'alimentation
\
MembraneSolution
de réception
Couche limite de diffusion
Figure 4-8: Etapes considérées dans le modèle de DANESI.
Ce mécanisme a été développé en supposant que la réaction de désextraction était totale sur la
face de réception ([EMX]R = 0). A la différence du modèle de WARD, la diffusion à travers
la couche limite (étape ©) ainsi que la cinétique de la réaction d'extraction (©) sont prises en
compte.
La quantification du flux associé à chacune de ces trois étapes fait appel à la première loi deFICK et aux constantes cinétiques de l'équilibre d'extraction, soient:
ôfM+lJ - _ D L JJ l ~ D l dx
(96)
J2 = . - K - \ [ E M X ] A . (97)
dx
D,: coefficient de diffusion du cation à travers la couche limite (cm2.s"'),
K1 et K'1 : constantes cinétiques de la réaction: M+ + X" + E <-» EMX,D3: coefficient de diffusion du complexe EMX à travers la membrane (cmls1),[M+]Ai: concentration du cation dans la phase aqueuse d'alimentation à l'interface (mol.cm"3),
[EMX]A. : concentration du complexe dans la phase organique à l'interface (mol.cm'3).
180

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
L'hypothèse de gradients de concentration linéaires permet de développer les relations (96) et(98):
(98')
d,: épaisseur de la couche limite de diffusion,d3: épaisseur de la membrane,
Lorsque le transport a atteint un régime stationnaire, les trois flux sont égaux. La combinaisondes relations (96'), (97) et (98') et l'introduction du coefficient de perméabilité permettentd'écrire la relation suivante du flux de l'électrolyte MX:
K\C
d, d,avec À, = —L'et À, = —^.1 D, 3 D3
P: coefficient de perméabilité,C: concentration de l'électrolyte dans la phase aqueuse d'alimentation.
Le coefficient de perméabilité est assimilé à la "résistance" au transport de MX à travers lamembrane. Sa valeur est calculée par ajustement des courbes expérimentales JM = f(C). Sil'on introduit le coefficient de distribution de M (DM), et si l'on suppose que K1 » 1,l'expression de P devient:
P =DM.A,+A
3K1 [EMX]avec DM = — r = >** ^
M K"1 [ ]Dans le cas d'un système uniformément agité, l'épaisseur (d,) de la couche limite de diffusionpeut être minimisée. Le coefficient de diffusion (D3) du complexe en phase organique est leplus souvent très inférieur à celui des ions (Dt) dans la solution aqueuse. Il est alors possibled'admettre l'approximation selon laquelle: A, « A3. Cette hypothèse revient à admettre quel'étape limitant le transport est la diffusion membranaire. Le coefficient de perméabilité estalors donné par:
A3
181

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
L'épaisseur de la membrane est une caractéristique géométrique connue et mesurable. Si l'ondispose de la valeur du coefficient de distribution de M, l'ajustement du coefficient deperméabilité à partir des valeurs expérimentales du flux permet d'accéder au coefficient dediffusion du complexe en phase organique membranaire.
Commentaires:
O Nous avons vu que la détermination du coefficient de diffusion des espèces dans laphase organique membranaire nécessite la connaissance du coefficient de distribution de Mdans les conditions précises de la manipulation. L'aspect prédictif du modèle de DANESI estdonc fortement contrarié par la nécessité de procéder à un nombre important de manipulationsd'extraction liquide-liquide.
O Lors de l'établissement de la relation (100), l'auteur fait intervenir le coefficient dedistribution de M tel que:
K1 [EMX= 1AK- 1 " [M+]
Une telle définition est erronée, le rapport des constantes cinétiques de l'équilibre d'extractionétant égal à la constante d'extraction Kex, soit:
K1 „ [EMX]_^ p 1 _ (102)
La relation (101) exprimant le coefficient de perméabilité en fonction des données d'extractionliquide-liquide (Kex ou D ^ devient:
p _ Kex _ DM *^ - dD3 D3
Le coefficient de perméabilité est donc directement proportionnel au coefficient de
distribution lorsque le produit |X~1.[E1 est constant, c'est-à-dire dans le cas du transport de
faibles quantités de sel ne modifiant pas la concentration de l'électrolyte dans la phased'alimentation. HILL [2] a pu mettre en évidence un tel comportement lors de la modélisationdu transport du césium par un calixarène.
O Jusqu'à présent, les effets de milieu dans les solutions d'alimentation concentréesn'ont pas été pris en compte lors de la mise en équation des phénomènes de transport par lemodèle de DANESI.
Le modèle de transport existant n'est donc pas adapté à la description du transport par descalixarènes. Il serait néanmoins possible, à partir de relations modifiées faisant intervenir laconstante d'extraction et les coefficients d'activité, de parvenir à une modélisation correcte.
182

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
2.2.3. Le modèle de REINHOUDT-STOLWICK.
Ce modèle a été développé pour étudier le transport de sels à travers des membranescontenant un éther-couronne dilué dans le nitrophényloctyléther [13]. Par ailleurs, des étudesrécentes ont porté sur la modélisation du transport du nitrate de césium par des calixarènes àpartir de matrices concentrées en acide nitrique et en nitrate de sodium [14]. Ce modèle detransport semble donc, à première vue, parfaitement adapté à notre système complexe.
La nature des espèces transportées est différente de celle proposée dans les modèlesprécédents. L'influence du diluant sur la dissociation du complexe formé en phase organique aété considérée. De plus, l'éventuelle solubilisation du transporteur en phase aqueuse a été priseen compte.
Les auteurs font l'hypothèse que le flux est limité par la diffusion membranaire. Le flux estconsidéré constant pendant l'expérience. La concentration de l'électrolyte dans la phaseaqueuse d'alimentation est donc implicitement très supérieure à la quantité totale transportée.
La caractéristique principale de ce modèle réside dans le mécanisme de transport proposé. Dufait de la constante diélectrique relativement élevée du NPOE (s = 24), les complexes formésen phase organique sont supposés être dissociés en deux ions.
Le mécanisme de transport suppose que la diffusion à travers les couches limites de diffusionest négligeable devant le phénomène de diffusion membranaire. Comme dans le modèle deDANESI, la réaction de désextraction est supposée totale. Les réactions d'extraction proposéespossèdent toutes une cinétique rapide et sont détaillées sur la figure 4-9.
Solutiond'alimentation
Phase organiquemembranaire
Figure 4-9: Mécanisme de transport proposé par STOLWIJK [13].
Afin de conserver une mise en équation homogène, nous avons repris les notations introduitespar REUSCH pour les deux constantes d'équilibre (k et K). La prise en compte de l'éventuelledistribution de l'extradant entre la solution aqueuse et la phase organique nécessitel'introduction de trois équilibres supplémentaires (de constantes kE, kEMX et Kaq ).
183

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
Le flux du soluté est exprimé par l'intermédiaire de la loi de FICK appliquée à l'étape dediffusion membranaire:
— EM" / EM+ EM+ (104)
L'hypothèse d'une réaction de désextraction totale revient à négliger la concentration ducomplexe sur la face de réception.
Le bilan matière de l'extractant s'écrit:
Vm.[E] = Vm.[E] - V B . EM+ -(VA+VR) .[E] (105)
Le bilan matière du cation est largement simplifié grâce à la mise en œuvre de solutionsd'alimentation concentrées dont la composition ne varie pas pendant le processus de transport.
VA.[M+]A=VA.[M+£-Vm.[EM+]-Vm.[M+X-]-VR.[M+]R-(VA+VR).[EM+](106)
Vm, VA, VR: volume de la phase organique membranaire et des phases aqueuses d'alimentationet de réception,
[M+]A, [M+]R: concentration du cation dans la phase aqueuse d'alimentation, de réception.
La combinaison de ces deux bilans avec les lois d'action de masse des équilibres décrits sur lafigure 4-9 permet d'expliciter la concentration du complexe dans la phase organique. De plus,la mise en jeu de solutions aqueuses concentrées nécessite de raisonner en termes d'activitésthermodynamiques et non plus de concentrations. L'expression finale du flux devient alors:
j _ DEM*I J
v_(107)
Une telle expression est très comparable à celle obtenue par REUSCH (relation (95)). Lesdifférences relevées entre ces deux relations tiennent à:
- la nature du complexe en phase organique (associé ou dissocié),
- la prise en compte de la distribution du transporteur entre les deux phases,
Afin de mettre en évidence le caractère dissociant du diluant NPOE, les auteurs remettent enforme la relation (107) selon:
d.J
1 1
EM W K-k V,A(108)
184

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
La fonction y = f(l/ aM+A) doit donc être une droite. Le calcul effectué pour obtenir la valeurde l'activité initiale du cation en phase aqueuse n'est pas clairement explicité. Comme nousl'avons souligné dans le chapitre 1, il est en effet impossible d'avoir accès expérimentalementà l'activité d'un ion individuel. Le calcul de aM+ doit donc faire appel à une théorie dessolutions appropriée. De plus, les notations retenues par les auteurs semblent indiquer uneconfusion entre l'activité du cation M+ (aM+) et l'activité du sel MX (aMX).
La mise en équation a été, par la suite, légèrement modifiée pour permettre lamodélisation complète du transport d'alcalins par des calixarènes dilués dans le NPOE [15].L'expression du flux fait alors intervenir la constante d'extraction Kex, produit des deuxconstantes K et k. En raison de la très grande complexité de cette relation, nous ne ladétaillerons pas dans ce texte. Toutefois, nous retiendrons que la démarche adoptée lors decette modélisation procède par ajustement des valeurs mesurées du flux aux donnéescalculées. Ce calcul est mené par optimisation simultanée du coefficient de diffusion et de laconstante d'extraction.
Plus récemment, une équation simplifiée du flux a permis de quantifier le transport du nitratede césium par différents calixarènes dilués dans le NPOE [14]. La solubilisation dutransporteur dans les solutions aqueuses d'alimentation et de réception n'a pas été prise encompte. L'expression du flux est alors donnée par:
SK-dCsNO, + K e x - a C s N O 3 - J 1 - r ^ ~2^ex-dCsNOj
(109)
De nouveau, on note une confusion entre l'activité du sel et celle du cation. De plus, lescoefficients d'activité sont calculés par l'intermédiaire de la relation de DEBYE-HUCKELbien au-delà du domaine de validité de cette méthode.
Commentaires:
O Comme nous l'avons vu, ce modèle reste limité à la description de phénomènes detransport à flux constant. Dans notre cas, il permettrait donc de modéliser le transport dunitrate de sodium et d'acide nitrique à partir de leur solution binaire concentrée. En aucun cas,son application n'est envisageable pour quantifier le transfert de faibles quantités de césium.
O Le modèle de REINHOUDT permet de décrire le transport d'un electrolyte à partirde sa solution binaire. La mise en œuvre de solutions d'alimentation contenant plusieurssolutés potentiellement extractibles nécessiterait une profonde modification des équations.
O La démarche de calcul adoptée nécessite d'optimiser à la fois le coefficient dediffusion et la constante d'extraction. Cette procédure permet de s'affranchir de l'acquisition dedonnées sur les équilibres d'extraction liquide-liquide. Toutefois, l'ajustement simultané deplusieurs paramètres indépendants peut être à l'origine d'erreurs de calcul importantes. Il seraitpossible de minimiser cette erreur en effectuant un nombre important d'expériences detransport.
185

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
O Enfin, le mécanisme de transport proposé n'a pu être démontré de manièreconvaincante. En effet, la démonstration souffre de la confusion faite entre l'activitéthermodynamique d'un ion et celle d'un electrolyte (cf. Chap. 1 § 1.4.).
L'ensemble de ces limitations s'oppose à la mise en œuvre du modèle développé parREINHOUDT. Il reste que la nature des complexes formés en phase organique doit êtreconfirmée. Cette confirmation nous a été apportée par des mesures de conductivité sur desphases organiques contenant la molécule d'extractant saturée par un electrolyte. L'extractiond'acide et de nitrates alcalins par le calixarène ne s'accompagne pas d'une augmentationsignificative de la conductivité par rapport à la solution organique initiale. Ces donnéesexpérimentales infirment donc la dissociation des différents complexes dans le NPOE.
186

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
3. Proposition d'un modèle de transport facilité.
Aucun des modèles présentés jusqu'à présent ne semble adapté aux caractéristiquespropres à notre système complexe. Les principales difficultés sont liées à la prise en compted'un grand nombre d'équilibres d'extraction et à la présence de fortes concentrations d'acide etde sels. C'est pourquoi, nous avons choisi de développer un modèle répondant aux contraintessuivantes:
- Prise en compte des effets de milieu dans les solutions aqueuses d'alimentation.
- Modélisation du transport simultané de deux sels (NaNO3 et CsNO3) et d'un acide(HNO3) à partir de mélanges d'électrolytes.
- Limitation du transport par l'étape de diffusion membranaire, les gradients deconcentration à travers les couches limites de diffusion sont donc négligés.
Les différents solutés sont transportés à travers la membrane sous la forme decomplexes avec le diluant ou avec la molécule de calixarène. Dans la suite de ce texte, nousdétaillons la mise en équation du transport d'un electrolyte MX par un extractant E présent enphase organique.
L'équilibre d'extraction s'écrit selon:
M++X"+E<-»EMX (110)
Comme nous l'avons vu précédemment (cf Chap.2 §2), la constante d'un tel équilibre s'écrit:
(111)
-. activité de l'électrolyte en phase aqueuse,
H20 : activité d'eau de la solution d'électrolyte,
EMX : nombre de molécules d'eau solubilisées par le complexe EMX à aH 0 = 1.
Le flux du complexe à travers la surface de la membrane peut être exprimé à l'aide de lapremière loi de FICK. Si l'on suppose le gradient de concentration constant, on a:
J = ^ ^ - E M X - E M X l (112)
DEWi: coefficient de diffusion du complexe en phase organique (cnr.s'1),d, s, x: épaisseur (cm), porosité et tortuosité du support,
[EMX]A, [EMX]R : concentration du complexe dans la membrane sur la face d'alimentation,
de réception (mol.cm3).
187

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
Cette expression fait intervenir les caractéristiques géométriques de la membrane et prendainsi en compte le chemin réel de diffusion.
Dans l'hypothèse d'un régime de diffusion stationnaire, la loi d'action de masse permet dedévelopper les concentrations du complexe à l'interface des solutions aqueuses et de la phaseorganique:
[EMX]A = Kex.a^.[¥]A.exp[AsLx-(aH2o,A - l ) ] (113)
[ËMX]R = Kex.aMX,R.[EiR.exp[AsLx.(aH20,R -1)] (114)
En tout point de la membrane, le bilan matière relatif à l'extractant s'écrit:
[E] =[E] + [EMX] (115)
L'expression finale du flux membranaire est obtenue par combinaison linéaire des relations(112) à (115) soit:
d.x
Cette relation complexe peut être décomposée comme le produit de quatre contributionsindépendantes telles que:
~° 017)
© DEMX: contribution diffusionnelle liée à la taille du complexe et à la viscosité du diluant,
® Gviemh = — : contribution géométrique caractéristique du support,mémo. d t
® T E M X M X = r ffi / .\\~~-, 77V1 + ^ex-aMX,A-eXP ZiSEMX-\aH2O,A ~ l)\ l + *S
L v ' J
contribution thermodynamique regroupant les données physico-chimiques du système,o
© [El : concentration initiale du transporteur.
L'écriture du flux fait intervenir cinq grandeurs expérimentales ([E] , aMX^, a^R , aHj0A etaH OR)> tro^s constantes (d, s et t) et trois paramètres (DEMX, Kex et AsfMX). Le nombre de
variables expérimentales peut être réduit à trois ([E] , aMX^ etaH z 0 A), lorsque ladésextraction est totale. L'expression du flux se simplifie selon:
188

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
T __
d . t + K e x .aM X A .exp[AsfM X . (aH 2 0 A -2 /J .[E] (118)
Les valeurs des paramètres Kex et A s ^ ont été ajustées précédemment lors de lamodélisation des équilibres d'extraction. Seul le coefficient de diffusion du complexe reste àdéterminer. Sa valeur peut être optimisée par ajustement des valeurs expérimentales du flux Jaux données calculées par l'intermédiaire des relations (116) ou (118).
Au cours du précédent chapitre, nous avons pu mettre en évidence que chacun deselectrolytes (sels ou acide) est extrait en phase organique sous la forme d'au moins deuxcomplexes. Il est donc nécessaire d'envisager le transport de ces solutés à travers la membranesous la forme de plusieurs espèces. Le flux d'un electrolyte MX est alors donné par:
JMX - ~ J-Ç(DE|(MX)J (\MMX\l -[E^MXjJ J ) (119)
Les complexes Ei(MX)j sont en équilibre dans la phase organique membranaire, leurrépartition évoluant lors du trajet diffusionnel. Les concentrations des différentes espècescontenant MX ne sont donc pas indépendantes. Il est alors impossible de calculer parajustement les coefficients de diffusion individuels. Pour contourner ce problème de calcul,nous avons choisi de définir un coefficient de diffusion global (DMX) tel que:
(120)
La concentration du constituant MX en phase organique est alors calculée par l'intermédiairede la loi d'action de masse associée à l'équilibre d'extraction. Ce calcul est informatisé dans lelogiciel "EXTRACTION".
La diffusion simultanée de deux espèces (A et B) a été mise en équation récemmentpar X. MACHURON-MANDARD [16]. L'auteur a pu montrer que le coefficient de diffusionglobal est relié aux coefficients de diffusion individuels selon:
_ DA.DB.(1 + K)DG>M~ D B + K . D A
avec: A < - ^ B K = j ^ |[A]
Dans le cas particulier d'un équilibre de complexation avec excès de réactif complexant E telque:
189

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
La relation (121) est modifiée de la manière suivante:
DA.DB.(1-, . - ^D Q - DB+K.[E].DA
(122)
Cette équation permettrait d'avoir accès aux coefficients de diffusion des complexes du type(Calix)i(XNO3)(H2O) à partir de la valeur du coefficient de diffusion global. Toutefois, lecalcul nécessiterait l'acquisition de données expérimentales d'extraction (calcul de K) et detransport (détermination de DGloba]) à différentes concentrations d'extractant. Ces manipulationsconstituent une charge de travail supplémentaire que nous n'avons pu mener à bien dans lecadre de cette thèse.
190

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
4. Conditions expérimentales.
4.1. Préparation des solutions et techniques analytiques.
Les solutions aqueuses concentrées d'acide nitrique et de nitrate de sodium sontpréparées par dilution de solutions mères concentrées. Le contrôle de leur concentration esteffectué par mesure de la masse volumique à 25°C. Les concentrations des solutions aqueusesde nitrate de césium mises en œuvre lors de nos expériences de transport sont comprises entre5.10*3 et 5.10*2 mol.kg"'. La densimétrie étant inadaptée à ce domaine de molalité, nous avonschoisi de doser le césium dans les phases aqueuses initiales par chromatographie ionique. Ladescription détaillée de chacune des techniques est regroupée dans l'annexe 4. Le transport del'acide nitrique à travers les MLS est suivi par pH-métrie à l'aide d'une électrode de verre àhaute alcalinité placée directement dans le compartiment de réception.
Les solutions organiques utilisées pour la préparation des membranes sont en toutpoint identiques à celles employées dans les manipulations d(extraction liquide-liquide. Lediluant est le 1,2-nitrophényloctyléther, la concentration du calixarène est de 0,00769 mol.kg"1
4.2. Manipulations de transport.
4.2.1. Dispositif expérimental.
Le dispositif expérimental utilisé lors de nos manipulations de transport reprend leschéma proposé par STOLWIJK [13]. La figure 4-10 présente un module complet de transportà membranes planes. Les phases aqueuses d'alimentation et de réception sont contenues dansdeux demi-cellules en verre. Afin de permettre la thermostatisation du dispositif, ces demi-modules possèdent une double enveloppe. Les solutions aqueuses sont uniformément agitéesgrâce à un aimant interne lui-même fixé sur un arbre d'hélice. L'entraînement de ce dispositifd'agitation est assuré par un aimant externe à champ fort monté sur un moteur électrique dontla vitesse est fixée à 1000 tr.min!. Cette vitesse permet de maintenir l'épaisseur de la couchelimite de diffusion à une valeur constante. La membrane liquide supportée est placée entre lesdeux cellules sans joint intermédiaire» et assure ainsi l'étanchéité du dispositif. Pour éviter unetrop forte evaporation des solutions aqueuses, les orifices de remplissage des modules sontbouchés pendant les manipulations.
Nous disposons de trois modules complets. Le volume des compartimentsd'alimentation et de réception varie de 42 à 45 cm3. La surface d'échange est comprise entre12,6 et 13,9 cm2.
Le support utilisé est un film CELGARD 2500 dont les caractéristiques physiques sontfournies par le fabricant, à savoir: une épaisseur de 25 um, une porosité de 0,45 et unetortuosité estimée de 2.
191

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
Phase aqueused'alimentation
Phase aqueusede réception
Aimant
Membrane imprégnéedu solvant et de fextractant Circuits de thermostatisation Agitation
Figure 4.10; Dispositif expérimental de transport par MLS.
4.2.2. Protocole expérimental.
Dans un premier temps, les membranes sont préparées par trempage du supportpolymérique dans la solution organique. Cette procédure est effectuée sous vide afin depermettre une incorporation rapide et complète dans les pores du polymère. La membraneainsi préparée est placée sur un support absorbant dans le but d'éliminer l'excédent de phaseorganique présent en surface.
Dans un deuxième temps, le module de transport est assemblé. Les compartimentsd'alimentation et de réception peuvent alors être remplis. La phase d'alimentation est unesolution d'électrolyte, la solution de réception est de l'eau déminéralisée. Ce remplissage estcontrôlé par pesée afin de connaître avec précision le volume de solution versé dans chaquedemi-cellule (les masses volumiques étant mesurées par ailleurs). Enfin, l'agitation est mise enroute, la vitesse des moteurs d'entraînement est contrôlée grâce à un tachymètre optique. Ladurée des manipulations de transport varie de 10 à 48 heures. L'ensemble du dispositif estthermostaté à 25°C par l'intermédiaire d'une circulation d'eau.
Comme nous l'avons vu, le suivi du transport de l'acide nitrique est assuré par lamesure du pH dans la solution de réception. Une mesure est effectuée automatiquement toutesles 10 minutes. La valeur du pH est directement liée à l'activité du proton et à sa concentrationpar la relation (123). La loi de DAVIS permet de calculer la valeur du coefficient d'activitéd'un ion. A partir de ces deux valeurs, on a alors accès à la concentration du proton.
(123)
192

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
avec yu. = -0 ,5102~^ 7 *+0,153M (Loi de DAVIS)H l + vl
yH, : coefficient d'activité de l'ion H+,
I: force ionique de la solution.
Le dosage des nitrates alcalins par chromatographie ionique dans la solution deréception permet de suivre leur transfert à travers la membrane. Un prélèvement horaire esteffectué pendant les dix premières heures.
Le calcul du flux est effectué à partir des concentrations des electrolytes dans la phaseaqueuse de réception:
TJ M X W S T îooo.s.t
nMx(t): nombre de moles de l'électrolyte MX dans la solution de réception à l'instant t,QfxCt): concentration de l'électrolyte MX dans la solution de réception à l'instant t (mol.L"1),VR(t): volume de la solution de réception à l'instant t (cm3),S: surface de la membrane (cm2).
193

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
5. Résultats.
La détermination du coefficient de diffusion global de chacun des constituante estréalisée par minimisation des valeurs expérimentales du flux aux données calculées par notremodèle. La modélisation du transport nécessite donc l'acquisition de données expérimentales.Afin de préserver la qualité des calculs, nous adoptons une démarche analytique en étudiantsuccessivement le transfert de l'acide et des sels à partir de leur solution binaire vers unesolution d'eau déminéralisée.
La mise en œuvre de solutions d'alimentations binaires concentrées permet de maintenir unflux constant. Etant donné le rapport élevé (> 2000) entre les volumes de la solution deréception et de la phase organique contenue dans la membrane, on peut admettre que laréaction de désextraction est quantitative. L'expression du flux donnée par la relation (120) sesimplifie alors selon:
(125)
5.1. Transport de l'acide nitrique par le NPOE.
Le transport de l'acide nitrique par le NPOE est étudié à partir de solutions binairesd'alimentation d'acidité comprise entre 1 et 3 mol.kg"'. Les résultats expérimentaux sont réunissur la figure 4-11.
0,06
15
HNO3 l u i
. HNO, 1,5m
: o,o3 -
0,000 1000
Temps (min)Figure 4-11: Transport de l'acide nitrique par le NPOE vers la solution de réception
(t=25°C).
194
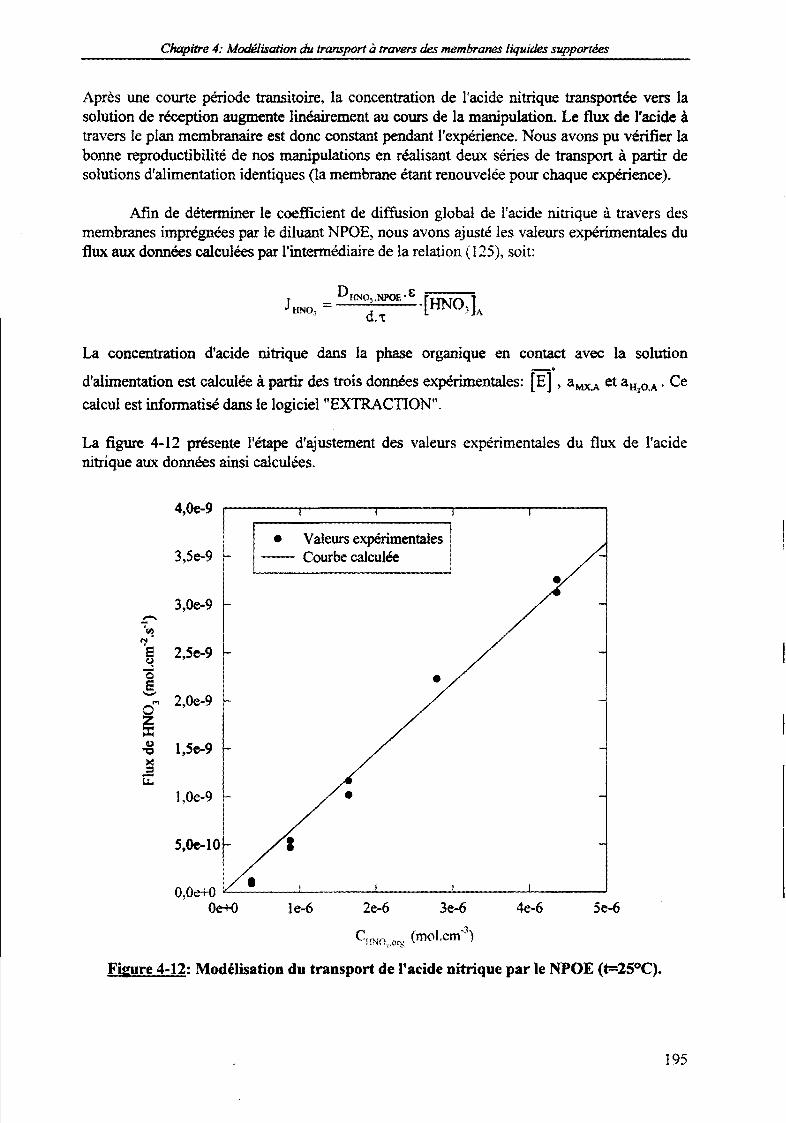
Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
Après une courte période transitoire, la concentration de l'acide nitrique transportée vers lasolution de réception augmente linéairement au cours de la manipulation. Le flux de l'acide àtravers le plan membranaire est donc constant pendant l'expérience. Nous avons pu vérifier labonne reproductibilité de nos manipulations en réalisant deux séries de transport à partir desolutions d'alimentation identiques (la membrane étant renouvelée pour chaque expérience).
Afin de déterminer le coefficient de diffusion global de l'acide nitrique à travers desmembranes imprégnées par le diluant NPOE, nous avons ajusté les valeurs expérimentales duflux aux données calculées par rintermédiaire de la relation (125), soit:
'HNG, aaffi=.[HNo,l,
La concentration d'acide nitrique dans la phase organique en contact avec la solution
d'alimentation est calculée à partir des trois données expérimentales: [E ] , a ^ ^ et aHîO A . Ce
calcul est informatisé dans le logiciel "EXTRACTION".
La figure 4-12 présente l'étape d'ajustement des valeurs expérimentales du flux de l'acidenitrique aux données ainsi calculées.
4,0e-9
3,5e-9
3,0e-9 -»\J
j 2,5e-9 -Î
r 2,0e-9 -
•S l,5e-9 -x
l,0e-9 -
5,0e-10 -
Valeurs expérimentalesCourbe calculée
0,ôe+0 'Oe+0 le-6 2e-6 3e-6 4e-6 5e-6
Figure 4-12: Modélisation du transport de l'acide nitrique par le NPOE (t=25°C).
195

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
Le coefEcient de diffusion global des complexes formés entre l'acide nitrique et le NPOE est
déduit de la valeur ajustée pour la pente de la droite $fm>% = f î jHNO^ I, soit:
Comme on pouvait s'y attendre, étant donnée la viscosité élevée du NPOE, cette valeur estinférieure à celle mesurée pour les solutés dans les phases aqueuses {> 10"5 ernes'1).
5.2. Transport de l'acide nitrique par le calixarène dilué dans le NPOE.
Les expériences de transport mettent en jeu les mêmes solutions binaires d'acidenitrique que précédemment. La membrane liquide supportée est élaborée par trempage dusupport dans une solution organique contenant le calixarène dilué à 0,00769 mol.kg"' dans leNPOE. Les concentrations d'acide nitrique dosé dans la phase aqueuse de réception sontreportées sur la figure 4-13.
0,10
0,08
0,06
1
• HNO3
• HNO3
A HNO3
V HNO3
• HNO3
lm
1,5m
2m2,5m
3m
S 0,04
200 800 1000400 600
Temps (min)
Figure 4-13: Transport de l'acide nitrique par une solution organique contenant0,00769 moLkg1 de calixarène dilné dans le NPOE (t=25°C).
De nouveau, l'augmentation linéaire de la concentration de l'électrolyte dans la solution deréception montre que le flux membranaire est constant. De plus, la reproductibilité de nosmanipulations de transport est une nouvelle fois vérifiée.
196
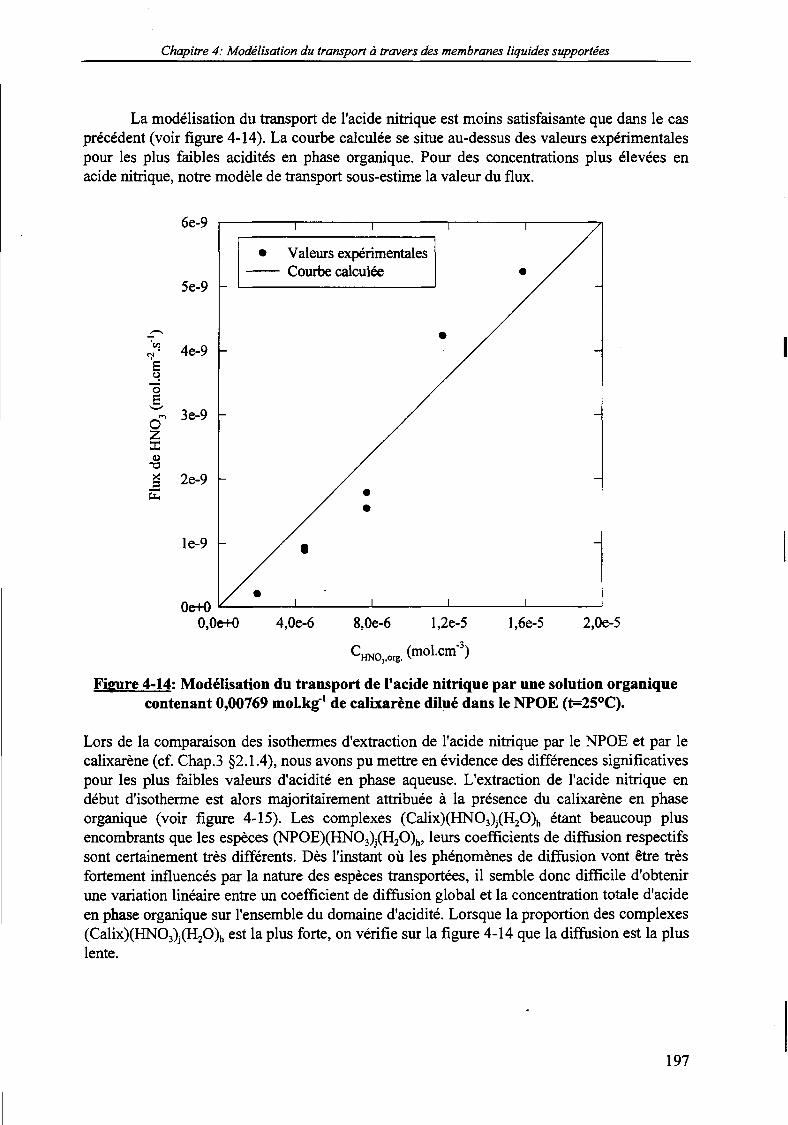
Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
La modélisation du transport de l'acide nitrique est moins satisfaisante que dans le casprécédent (voir figure 4-14). La courbe calculée se situe au-dessus des valeurs expérimentalespour les plus faibles acidités en phase organique. Pour des concentrations plus élevées enacide nitrique, notre modèle de transport sous-estime la valeur du flux.
q~o
6e-9
5e-9
4e-9
3e-9
2e-9
le-9
Oe+0
• Valeurs expérimentalesCourbe calculée
0,0e+0 4,0e-6 8,0e-6
HNo,,org.
l,2e-5 l,6e-5 2,0e-5
Figure 4-14: Modélisation du transport de l'acide nitrique par une solution organiquecontenant 0,00769 raol.kg ' de calixarène dilué dans le NPOE (t=25°C).
Lors de la comparaison des isothermes d'extraction de l'acide nitrique par le NPOE et par lecalixarène (cf. Chap.3 §2.1.4), nous avons pu mettre en évidence des différences significativespour les plus faibles valeurs d'acidité en phase aqueuse. L'extraction de l'acide nitrique endébut d'isotherme est alors majoritairement attribuée à la présence du calixarène en phaseorganique (voir figure 4-15). Les complexes (Calix)(HNO3)j(H2O)h étant beaucoup plusencombrants que les espèces (NPOE)(HNO3)j(H2O)h, leurs coefficients de diffusion respectifssont certainement très différents. Dès l'instant où les phénomènes de diffusion vont être trèsfortement influencés par la nature des espèces transportées, il semble donc difficile d'obtenirune variation linéaire entre un coefficient de diffusion global et la concentration totale d'acideen phase organique sur l'ensemble du domaine d'acidité. Lorsque la proportion des complexes(Calix)(HNO3)j(H2O)h est la plus forte, on vérifie sur la figure 4-14 que la diffusion est la pluslente.
197

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
100
1" 80
03EPouRt
o. 60Complexes (NPOE)(HNO3)j(H2O)h
Complexes (Calix)(HNO3)j(H2O)h
00,0e+0 4,0e-6 8,0e-6 l,2e-5
CHNO3,org. (mol.cm"3)
l,6e-5 2,0e-5
Figure 4-15; Répartition de l'acide nitrique dans les solutions organiques membranairesà l'équilibre d'extraction (t=25°C).
En l'absence de manipulations d'extraction et de transport à concentration d'extractant variableil est pour le moment impossible d'avoir accès au coefficient de diffusion individuel de cesespèces. Aussi, nous retiendrons la valeur globale ajustée à partir des résultats expérimentauxprésentés sur la figure 4-14, soit:
Cette valeur est inférieure à celle ajustée pour les complexes (NPOE)(HNO3)j(H2O)h, ce quicorrespond au ralentissement du processus de transport de l'acide nitrique à travers lamembrane dû à la diffusion lente des espèces (Calix)(HNO3)j(H2O)h.
5.3. Transport du nitrate de sodium par le calixarène dilué dans le NPOE.
Les solutions d'alimentation mises en œuvre sont des solutions binaires de nitrate desodium dont la concentration varie de 2 à 8 mol.kg"1. Deux séries de transport sont effectuéespour chacune de ces solutions. La figure 4-16 réunit l'ensemble des mesures expérimentalessur une durée de 10 heures.
198
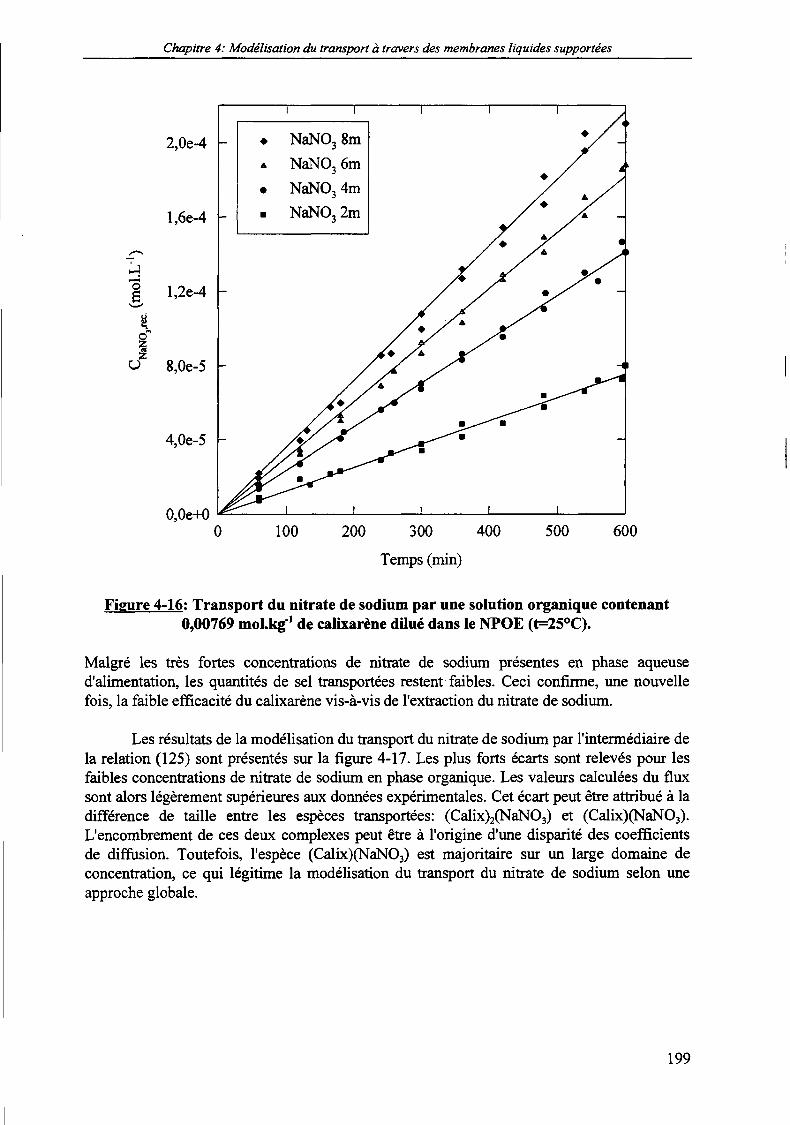
Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
2,0e-4 -
l,6e-4 -
"o| l,2e-4 -
<-> 8,0e-5 -
4,0e-5 -
1 1
• NaNO3 8m
A NaNO3 6m
• NaNO3 4m
• NaNO3 2m
yy-i i i
100 200 300 400
Temps (min)
500 600
Figure 4-16: Transport du nitrate de sodium par une solution organique contenant0,00769 mol.kg ' de calixarène dilué dans le NPOE (t=25°C).
Malgré les très fortes concentrations de nitrate de sodium présentes en phase aqueused'alimentation, les quantités de sel transportées restent faibles. Ceci confirme, une nouvellefois, la faible efficacité du calixarène vis-à-vis de l'extraction du nitrate de sodium.
Les résultats de la modélisation du transport du nitrate de sodium par l'intermédiaire dela relation (125) sont présentés sur la figure 4-17. Les plus forts écarts sont relevés pour lesfaibles concentrations de nitrate de sodium en phase organique. Les valeurs calculées du fluxsont alors légèrement supérieures aux données expérimentales. Cet écart peut être attribué à ladifférence de taille entre les espèces transportées: (Calix)2(NaNO3) et (Calix)(NaNO3).L'encombrement de ces deux complexes peut être à l'origine d'une disparité des coefficientsde diffusion. Toutefois, l'espèce (Calix)(NaNO3) est majoritaire sur un large domaine deconcentration, ce qui légitime la modélisation du transport du nitrate de sodium selon uneapproche globale.
199

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
2,2e-ll
2,0e-11
1,8e-11
0,0e+0
Valeurs expérimentalesCourbe calculée
0e+0 le-7 2e-7 3e-7 4e-7 5e-7 6e-7 7e-7 8e-7 9e-7
NaNO,,org.
Figure 4-17: Modélisation du transport du nitrate de sodium par une solution organiquecontenant 0,00769 mol.kg ' de calixarène dilué dans le NPOE (t=25°C).
Le coefficient de diffusion global calculé pour les complexes (Calix)j(NaNO3) est de:
Cette valeur est tout à fait comparable à celle calculée par STOLWIJK [10] par l'intermédiairedes relations empiriques de WILKE-CHANG ou de STOCKES-EINSTEIN pour des cationsalcalins complexés par des éthers-couronne dans le NPOE (Dcalc = 3 à 7.10"7 cmls"1).
5.4. Transport du nitrate de césium par le calixarène dilué dans le NPOE.
Quatre expériences de transport sont menées à partir de solutions aqueusesd'alimentation contenant 5.10"3 à 5.10'2 mol.kg'1 de nitrate de césium. Les concentrationsmesurées dans la phase de réception sur une durée de 10 heures sont réunies sur la figure 4-18.
La concentration du sel mesurée dans la solution de réception augmente linéairementau cours du transport. Le flux du nitrate de césium à travers la membrane est donc constantsur la durée des manipulations.
200

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
0,0010
0,0008 -
j 0,0006 -o
0,0004 -
0,0002 -
0,0000
• CsNO3 0,05m
. CsNO3 0,025m
CsNO3 0,01m
• CsNO, 0,005m
0 100 200 300 400
Temps (min)
500 600
Figure 4-18; Transport du nitrate de césium par une solution organique contenant0,00769 rnoLkgml de calixarène dilué dans le NPOE (t=25°C).
Les valeurs expérimentales du flux de l'électrolyte à travers la membrane sontreportées sur la figure 4-19 en fonction de la molarité de CsNO3 dans la phase organique enéquilibre avec la solution d'alimentation.
Le flux de CsNO3 est très largement supérieur à celui mesuré lors du transport de NaNO3.Cette augmentation significative des quantités transportées peut être attribuée à l'affinitéélevée du calixarène pour le nitrate de césium.
La modélisation du transport du nitrate de césium selon la relation (125) aboutit à une bonneconcordance entre les données expérimentales et les valeurs calculées. La valeur optimisée ducoefficient de diffusion global des complexes (Calix)i(CsNO3)(H2O)h est voisine de cellecalculée précédemment pour les complexes du sodium, soit:
.2 „-!2,7.1(Tcm'.s
201
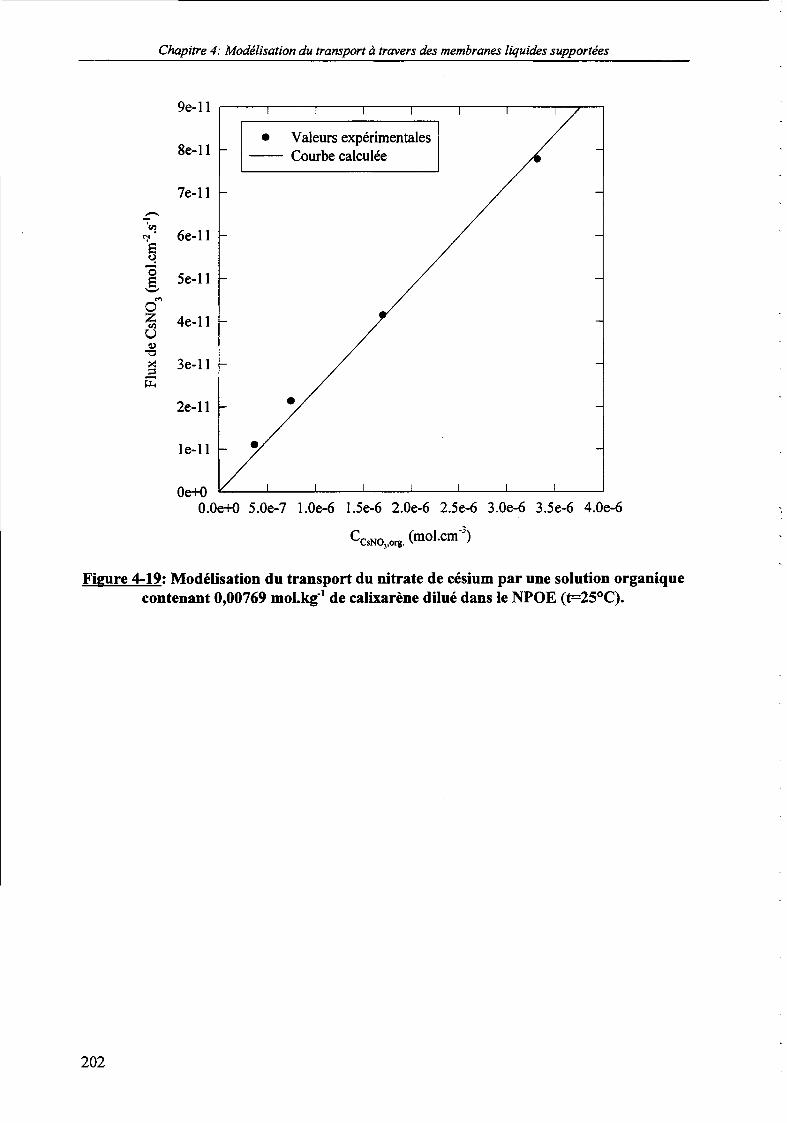
Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
9e-1
8e-ll -
7e-ll -
«?' 6 e - l l
I5e-ll -
O4e-ll -
"* 3e-l 1
2e-ll -
le-11 -
-
1 1
1 1
1 1
1 \
1 1 1 1
• Valeurs expérimentalesCourbe calculée
1 1 \ S
1 1
1 1
1 1
1
0.0e+0 5.0e-7 1.0e-6 1.5e-6 2.0e-6 2.5e-6 3.0e-6 3.5e-6 4.0e-6
ccSNO,,org.
Figure 4-19: Modélisation du transport du nitrate de césium par une solution organiquecontenant 0,00769 moLkg"1 de calixarène dilué dans le NPOE (t=25°C).
202

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
6. Prévision du transport du nitrate de césium à partir de matricesconcentrées.
A partir des connaissances acquises lors de la modélisation des équilibres d'extractionliquide-liquide, nous avons vu qu'il était possible de prévoir la concentration des sels et del'acide extraits par une solution organique constituée du NPOE seul ou bien du calixarènedilué dans le NPOE. A l'issue de la modélisation des mécanismes de transport, nous disposonségalement des valeurs des coefficients de diffusion globaux de chacun des electrolytes àtravers les solutions organiques membranaires.
La relation (120) permet alors de calculer le flux d'un electrolyte à partir de la connaissance:
- des paramètres géométriques de la membrane (d, e, T),
- du coefficient de diffusion global de l'électrolyte (DMX) à travers la solutionorganique membranaire,
- de la concentration du constituant MX dans la phase organique en équilibre avec lessolutions aqueuses d'alimentation et de réception.
II semble donc possible de prévoir le transport de l'acide nitrique et des nitrates alcalins àpartir de solutions d'alimentation de compositions variées.
Afin de valider notre démarche prédictive, nous avons confronté nos calculs deprévision aux données expérimentales acquises lors du transport du nitrate de césium à partirde solutions aqueuses concentrées en nitrate de sodium et/ou en acide nitrique.
6.1. Prévision du transport du nitrate de césium à partir de solutions fortementsalines.
La solution d'alimentation mise en œuvre contient 1,1.10"4 mol.L"1 de CsNO3 dans unematrice concentrée en nitrate de sodium à 3,5 mol.L"1. La figure 4-20 réunit les donnéesexpérimentales et calculées à l'aide de notre modèle. Au bout de 30 heures de manipulation,90% du nitrate de césium sont transportés dans la solution de réception constituée au départd'eau déminéralisée.
203
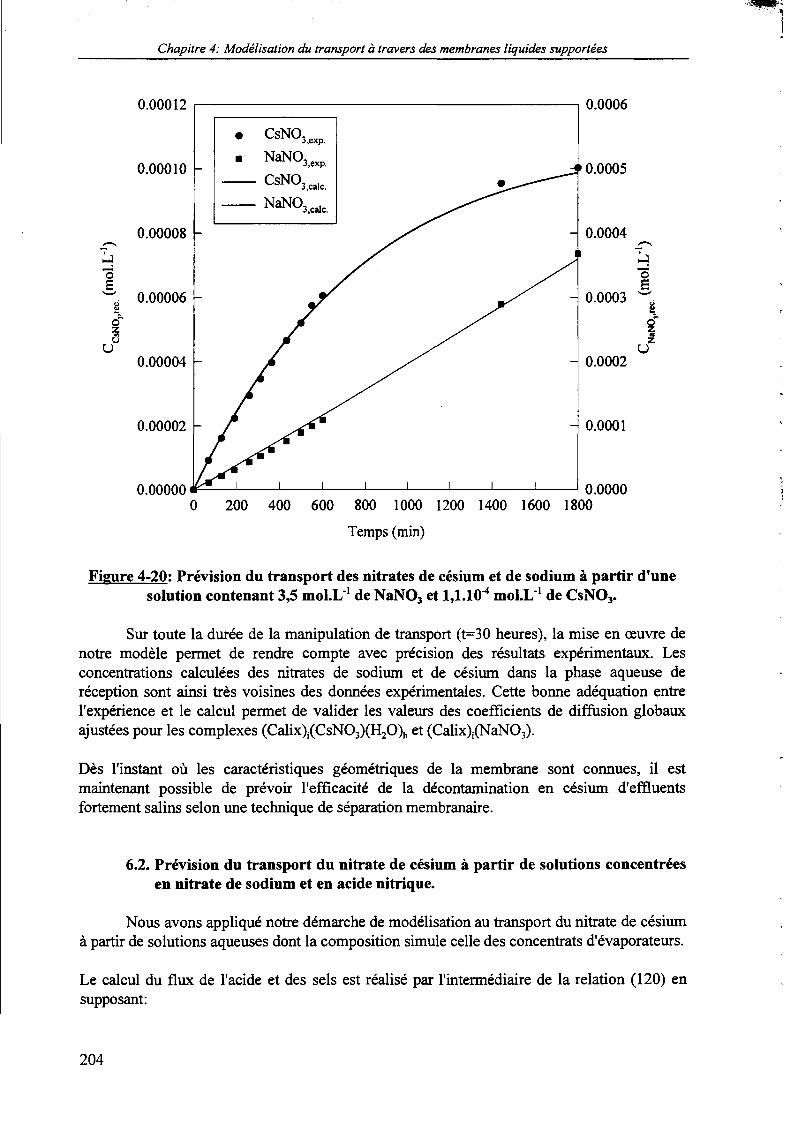
Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
oS
U
0.00012
0.00010 -
0.00008 -
' 0.00006 -
0.00004 -
0.00002 -
0.00000
0.0006
^•0.0005
- 0.0004
- 0.0003
- 0.0002
- 0.0001
§
0.00000 200 400 600 800 1000 1200 1400 1600 1800
Temps (min)
Figure 4-20: Prévision du transport des nitrates de césium et de sodium à partir d'unesolution contenant 3,5 moLL"1 de NaNO3 et 1,1.1c4 moLL"1 de CsNO3.
Sur toute la durée de la manipulation de transport (t=30 heures), la mise en œuvre denotre modèle permet de rendre compte avec précision des résultats expérimentaux. Lesconcentrations calculées des nitrates de sodium et de césium dans la phase aqueuse deréception sont ainsi très voisines des données expérimentales. Cette bonne adéquation entrel'expérience et le calcul permet de valider les valeurs des coefficients de diffusion globauxajustées pour les complexes (Calix)i(CsNO3)(H2O)h et (Calix)j(NaNO3).
Dès l'instant où les caractéristiques géométriques de la membrane sont connues, il estmaintenant possible de prévoir l'efficacité de la décontamination en césium d'effluentsfortement salins selon une technique de séparation membranaire.
6.2. Prévision du transport du nitrate de césium à partir de solutions concentréesen nitrate de sodium et en acide nitrique.
Nous avons appliqué notre démarche de modélisation au transport du nitrate de césiumà partir de solutions aqueuses dont la composition simule celle des concentrats d'évaporateurs.
Le calcul du flux de l'acide et des sels est réalisé par l'intermédiaire de la relation (120) ensupposant:
204

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
- d'une part que la fraction de l'acide transporté par les complexes mixtes(Calix)(HNO3)j(CsNO3) est faible,
- d'autre part que le coefficient de diffusion global du nitrate de césium n'est pasmodifié par la formation des complexes mixtes.
La solution aqueuse d'alimentation contient Î^.IO"4 mol.L"1 de CsNO3, 4 mol.L'1 de NaNO3 et1 mol.L"1 d'acide nitrique. Les résultats expérimentaux sont comparés aux données calculéessur les figures 4-21 et 4-22.
0,040
0,035 -
0,030 -
"o
O
0,025
0,020
0,015
0,010
0,005 -
0,000
1 1 1
-• Valeurs expérimentales
Courbe calculée
-
i i
/ / ;
-
100 200 300 400
Temps (min)
500 600
Figure 4-21: Prévision du transport de l'acide nitrique à partir d'une solution contenant1 moLL'1 de HNO3,4 moLL'1 de NaNO3 et Î .IO"4 moLL'1 de CsNO3.
Les quantités d'acide nitrique transportées vers la solution de réception sont sur-estimées par rapport aux valeurs expérimentales, l'écart restant inférieur à 10%. Cecomportement est comparable à celui relevé lors de l'étape d'ajustement du coefficient dediffusion global de l'acide nitrique à travers une solution organique contenant le calixarène.De plus, nous n'avons jamais tenté de modéliser la période transitoire en début d'expérience.En effet, lors de notre mise en équation, nous nous sommes toujours placés dans lesconditions d'un régime stationnaire.
205

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
0,00012
0,00010 -
0,00008 -
0,00006 -
u 0,00004 -
0,00002 -
0,00000
• / • • •
/ . •
^ •
•
•
•
•t
• CsNO3exp
• NaNO3exp
CsNO3calc
NaNO3calc
— i i
ii
o
0 100 200 300 400
Temps (min)
500
0,0018
0,0016
- 0,0014
- 0,0012
0,0010
0,0008
0,0006
0,0004
0,0002
mol
e.L
id
iu
0,0000600
Figure 4-22; Prévision du transport des nitrates de césium et de sodium à partir d'unesolution contenant 1 moLL"1 de HNO3,4 moLL"1 de NaNO3 et Î^.IO"4 moLL'1 de CsNO3.
Les concentrations calculées des nitrates alcalins dans le compartiment de réceptions'éloignent très fortement des données expérimentales. Ce mauvais résultat peut être attribué àune modélisation incomplète des mécanismes d'extraction.
Nous avons pu voir précédemment (Chap.3 §3.3.) que la représentation faite de la phaseorganique lors de l'extraction du nitrate de césium à partir de solutions fortement salines etacides pouvait s'écarter du comportement expérimental. Cette erreur de prévision pouvait êtreattribuée à la non prise en compte de la formation de complexes mixtes entre le calixarène, lenitrate de sodium et l'acide nitrique. A la vue des résultats présentés sur la figure 4-22, cettehypothèse semble se confirmer. La prise en compte de la formation de complexes du type(Calix)(HNO3)j(NaNO3) permettrait:
- d'une part de rendre compte de l'augmentation de la quantité de sodium transportéepar rapport à une solution neutre (voir figure 4-20),
- et d'autre part de mieux estimer la concentration du césium présent en phaseorganique par un calcul plus juste de la concentration d'extractant libre.
206

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
6.3. Prévision du transport du nitrate de césium à partir de solutions concentréesen acide nitrique.
Nous avons testé la validité de notre modèle de transport en comparant des donnéesexpérimentales acquises au laboratoire aux valeurs calculées. Les manipulations ont étéeffectuées par S. EYMARD et B. TOURNOIS lors d'essais préliminaires sur ladécontamination en césium d'effluents fortement acides par un procédé de séparationmembranaire. Les phases aqueuses d'alimentation contiennent des quantités croissantes denitrate de césium (5.10"4 à 1,5.10"2 mol.L"1) dans une matrice d'acide nitrique à 3,08 mol.L"1.Nous disposons des concentrations du césium dans la phase aqueuse de réception sur les sixpremières heures, puis à 24 heures. La comparaison des résultats expérimentaux et calculéslors du transport du nitrate de césium à partir d'une solution d'alimentation contenantinitialement 7,52.10"4 mol.L"1 est présentée sur la figure 4-23.
0,0008
0,0007
0,0006
0,0005
0,0001
0,0000 4
Valeurs expérimentalesCourbe calculée
0 200 400 600 800 1000
Temps (min)
1200 1400
Figure 4-23: Prévision du transport du nitrate de césium à partir d'une solutioncontenant initialement 3 mol.L ' de HNO3 et 7,52.1G"4 mol.L"1 de CsNO3.
La courbe calculée rend compte d'une manière satisfaisante du comportementexpérimental sur une durée de 24 heures. Cette bonne adéquation permet de valider notreapproche théorique au transport du nitrate de césium à partir de matrices concentrées en acidenitrique. En particulier, nous pouvons vérifier l'hypothèse selon laquelle le coefficient dediffusion global du nitrate de césium n'est pas modifié par la formation des complexes mixtes.
207

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
La mise en œuvre de notre modèle de transport nous a également permis de reproduire unphénomène expérimental parfois relevé, à savoir l'inversion du flux du nitrate de césium. Eneffet, la concentration calculée de CsNO3 dans la phase de réception passe par un maximumpuis diminue au cours du temps. Le flux est directement proportionnel à la différence desconcentrations du sel en phase organique sur les faces d'alimentation et de réception (relation(120)). La diminution puis l'inversion du gradient de concentration de CsNO3 à l'intérieur dela membrane (figure 4-24) est alors à l'origine d'une inversion du flux de ce sel.
o
o
<J
0,005
0,004
0,003
0,002
0,001
0,000
Face d'alimentationFace de réception
200 400 600 800 1000
Temps (min)
1200 1400
Figure 4-24: Variation de la concentration de CsNO3 en phase organique membranairesur les faces d'alimentation et de réception.
L'augmentation de la concentration du sel sur la face de réception indique une réaction dedésextraction incomplète. L'acidité croissante dans la solution aqueuse de réception contribue,en plus du phénomène de transport, à l'augmentation de l'activité du nitrate de césium danscette solution. La réaction de désextraction est alors empêchée par un effet de massedéfavorable.
208

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
Conclusion.
Ce dernier chapitre nous a permis de développer un modèle de transport basé sur notreconnaissance approfondie des équilibres d'extraction. A la différence d'autres modèlesprécédemment mis en œuvre, seuls les coefficients de diffusion des différents constituantstransportés sont déterminés par ajustement des données expérimentales de transport.
Comme dans le précédent chapitre, nous avons de nouveau adopté une démarche analytiqueen étudiant successivement le transport de l'acide nitrique puis des nitrates de sodium et decésium à partir de leurs solutions binaires.
La mise en œuvre conjointe de ces coefficients de diffusion et des paramètres déterminés dansle chapitre 3 nous a permis de décrire d'une manière satisfaisante le transport des nitrates decésium et de sodium à partir d'une solution concentrée en acide ou en sel. La description dutransport à partir de solutions simulant les concentrais d'évaporateurs se heurte, quant à elle,au manque de données concernant la formation de complexes mixtes entre le calixarène,l'acide et le nitrate de sodium.
209

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
BIBLIOGRAPHIE.
[1] J. CASAS i GARCIA,Décontamination sélective du césium et du strontium des concentrats d'évaporationprovenant des usines de retraitement de combustibles nucléaires avec des éthers-couronnes par transport à travers des membranes liquides supportées,Thèse de Doctorat de l'Université de BARCELONE, (1991).
[2] CHILL,Application des calixarènes fonctionnalisés au traitement des effluents radioactifs parmembranes liquides supportées,Thèse de Doctorat de l'Université Louis PASTEUR de STRASBOURG, (1994).
[3] L. BOYADZfflEV, Z. LAZAROVA,Liquid Membranes (Liquid Pertraction),Membrane Separation Technology, Ed. R.D. NOBLE and S.A. STERN, MembraneScience and Technology Series 2, Chap. 7, (1995).
[4] W.A. ZISMAN,Advances in Chemistry, Series N°43,American Chemical Society, Washington D.C., (1964).
[5] Y. INOUE,Mechanistic Principles of Liquid Membrane Transport,Liquid Membranes: Chemical Applications, Ed. T. ARAKI, H. TSUKUBE, CRC Press,Chap. 5, (1990).
[6] W.J. WARD,Analytical and Experimental Studies of Facilitated Transport,AIChE J., 16, (1970), 405-410.
[7] CF. REUSCH, E.L. CUSSLER,Selective Membrane Transport,AIChE J., 19, (1973), 736-741.
[8] N. REYNIER,Etude de la complexation de cations alcalins par des calixarènes photoisomérisables,Thèse de Doctorat de l'Université Louis PASTEUR de STRASBOURG, (1996).
[9] R. MARR, A. KOPP,Liquid Membranes Technology, A Survey of Phenomena, Mechanisms and Models,Intern. Chem. Engin., 22, (1982), 44-60.
211

Chapitre 4: Modélisation du transport à travers des membranes liquides supportées
[10] T.B. STOLWIJK, E.J.R. SUDHÔLTER, D.N. REINHOUDT,Effect of Crown Ether Lipophilicity on the Facilitated Transport of GuadidiniumThiocyanate through an Immobilized Liquid Membrane,J. Am. Chem. Soc, 111, (1989), 6321-6329.
[11] J.D. LAMB, J.J. CHRISTENSEN, S.R. IZATT, K. BEDKE, M.S. ASTIN, R.M.IZATT,Effects of Salt Concentration and Anion on the Rate of Carrier-Facilitated transport ofMetal Cations through Bulk Liquid Membranes Containing Crown Ethers,J. Am. Chem. Soc, 102, (1980), 3399-3403.
[12] P.R.DANESLSeparation of Metal Species by Supported Liquid Membranes,Sep. Sc. Tech., 19, (1984-1985), 857-894.
[13] T.B. STOLWIJK, E.J.R. SUDHÔLTER, D.N. REINHOUDT,Crown Ether Mediated Transport: A Kinetic Study of Potassium Perchlorate Transportthrough a Supported Liquid Membrane Containing Dibenzo-18-crown-6,J. Am. Chem. Soc., 109, (1987), 7042-7047.
[14] A. CASNATI, A. POCHINI, R. UNGARO, F. UGOZZOLI, F. ARNAUD, S. FANNI,M.J. SCHWING, R.J.M. EGBERTNG, F. DE JONG and D.N. REINHOUDT,Synthesis, Complexation, and Membrane Transport Studies of 1,3-AlternateCalix[4]arene-crown-6 Conformers: A New Class of Cesium Selective Ionophores,J. Am. Chem. Soc, 117, (1995), 2767-2777.
[15] W.F. NIJENHUIS, E.G. BUITENHUIS, F. DE JONG, E.J.R. SUDHÔLTER, D.N.REINHOUDT,Calixcrowns as selective Potassium Cation Carriers in Supported Liquid Membranes,J. Am. Chem. Soc., 113, (1991), 7963-7968.
[16] X. MACHURON-MANDARD,Communication personnelle, (juin 1996).
212

CONCLUSION GENERALE

Conclusion générale
L'objectif de ce travail était d'identifier et de comprendre les mécanismesfondamentaux gouvernant l'extraction du nitrate de césium à partir de solutions concentrées enacide et/ou en sels.
La mise en œuvre de solutions aqueuses concentrées a nécessité de raisonner en termed'activité thermodynamique et non plus en terme de concentration. Afin de quantifier lesécarts à l'idéalité dans les phases aqueuses et organiques, nous avons préconisé une approchethermodynamique globale. A la différence d'une démarche analytique procédant par uninventaire laborieux et le plus souvent incomplet des différents types d'interactions existantdans les solutions, les équations de base sont des relations thermodynamiques rigoureuses.Moyennant un nombre réduit d'hypothèses simplificatrices, il est possible de développer desrelations simples permettant le calcul des coefficients d'activité.
La relation de MIKULIN a été retenue pour calculer le coefficient d'activité des constituantsen phase aqueuse. Les coefficients d'activité des espèces en phase organique sont, quant à eux,développés selon la relation de SERGIEVSKII modifiée par DANNUS.
Quelle que soit l'approche retenue pour quantifier les écarts à l'idéalité dans lesmélanges aqueux, il est nécessaire de disposer des données thermodynamiques (concentration,coefficient d'activité et activité d'eau) de chacun des constituants dans leur solution binairerespective. Du fait de la faible solubilité du nitrate de césium, les données relatives à ce sel nesont disponibles que sur un domaine de concentration (d'activité d'eau) limité à 1,2 mol.L"1
(aH2o = 0,965). Le calcul du coefficient d'activité du nitrate de césium dans des mélanges plus
concentrés et de plus faible activité d'eau doit alors faire intervenir des données binaires au-delà de la saturation de CsNO3.
Après une étude critique des différents modèles d'écarts à l'idéalité, les relations de CHEN etde ZDANOVSKII-STOCKES-ROBINSON sont apparues les plus adaptées à l'acquisition dedonnées thermodynamiques de solutions sursaturées fictives de nitrate alcalin. L'extensiond'une telle étude à d'autres familles d'électrolytes est envisageable si l'on prend soin de vérifierles hypothèses inhérentes à chacune des deux relations.
La démarche analytique retenue pour modéliser les isothermes d'extraction de l'eau, del'acide et des nitrates alcalins par le diluant et le calixarène nous a permis de mettre enévidence la complexité du système étudié. Si nous avions tenté de modéliser le systèmecomplet en une seule étape, il est certain que nous n'aurions pu appréhender la totalité deséquilibres d'extraction.
La modélisation de l'extraction des nitrates alcalins a nécessité d'envisager la formation decomplexes de stœchiométrie supérieure en calixarène: (Calix)2(XNO3)(H2O) (XNO3: nitratealcalin). Une telle stœchiométrie reste à confirmer par des méthodes spectrales, mais ellesemble en accord avec des études récentes portant sur d'autres familles de calixarènes. Laformation de complexes mixtes entre le césium, le calixarène et l'acide nitrique a pu être miseen évidence et permet d'expliquer le bon comportement de cet extractant en milieu acide.
Ces résultats nous ont permis, par l'intermédiaire du nouvel outil informatique "PREVISION",de prévoir l'efficacité de l'extraction de faibles quantités de nitrate de césium à partir desolutions concentrées en acide nitrique et/ou en nitrate de sodium. La représentation faite de la
215

Conclusion générale
phase organique est alors validée dans des conditions très éloignées de celles correspondant àl'étape d'ajustement des paramètres (K^ et As®). La compréhension des mécanismesfondamentaux gouvernant l'extraction du césium permet de fournir des indications précisessur les conditions optimales de décontamination d'effluents radioactifs. Ces calculs prédictifsconstituent alors un premier élément de réponse aux besoins pratiques de l'industriel.
La connaissance approfondie des mécanismes d'extraction liquide-liquide a été mise àprofit afin d'étudier les phénomènes de transport à travers des membranes liquides supportées(MLS). Les modèles développés jusqu'à présent étaient handicapés par le manque de donnéesphysico-chimiques concernant à la fois les solutions mises en jeu et les équilibres àconsidérer. Le modèle proposé permet de distinguer les différentes contributionsindépendantes intervenant lors du transport à travers des phases organiques membranaires.L'expression du flux fait ainsi intervenir quatre termes parmi lesquels seul le coefficient dediffusion de l'espèce transportée reste à déterminer. La valeur de ce paramètre est ajustée àpartir des données expérimentales de transport.
La description faite des équilibres d'extraction et des phénomènes de diffusion membranairenous a permis de prévoir d'une manière très satisfaisante le transport du nitrate de césium àpartir de solutions dont la composition simule celle des effluents à traiter. Nous sommes pourle moment en mesure de quantifier la décontamination en césium de solutions concentrées enacide nitrique ou en nitrate de sodium selon un procédé de séparation par MLS. L'étude doitpouvoir être étendue à des mélanges quaternaires HNC^/NaNCyCsNOj/H^O si la formationdes complexes mixtes entre le calixarène, l'acide nitrique et le nitrate de sodium est étudiée.
Les modèles proposés pour décrire les équilibres d'extraction liquide-liquide ainsi queles phénomènes de transport à travers les MLS ont pu être validés dans des conditions trèsvariées. Nous disposons donc de théories ainsi que d'outils informatiques spécifiquementdéveloppés qui doivent nous permettent d'envisager avec confiance la modélisation d'autressystèmes d'extraction de type solvatant.
216

ANNEXES
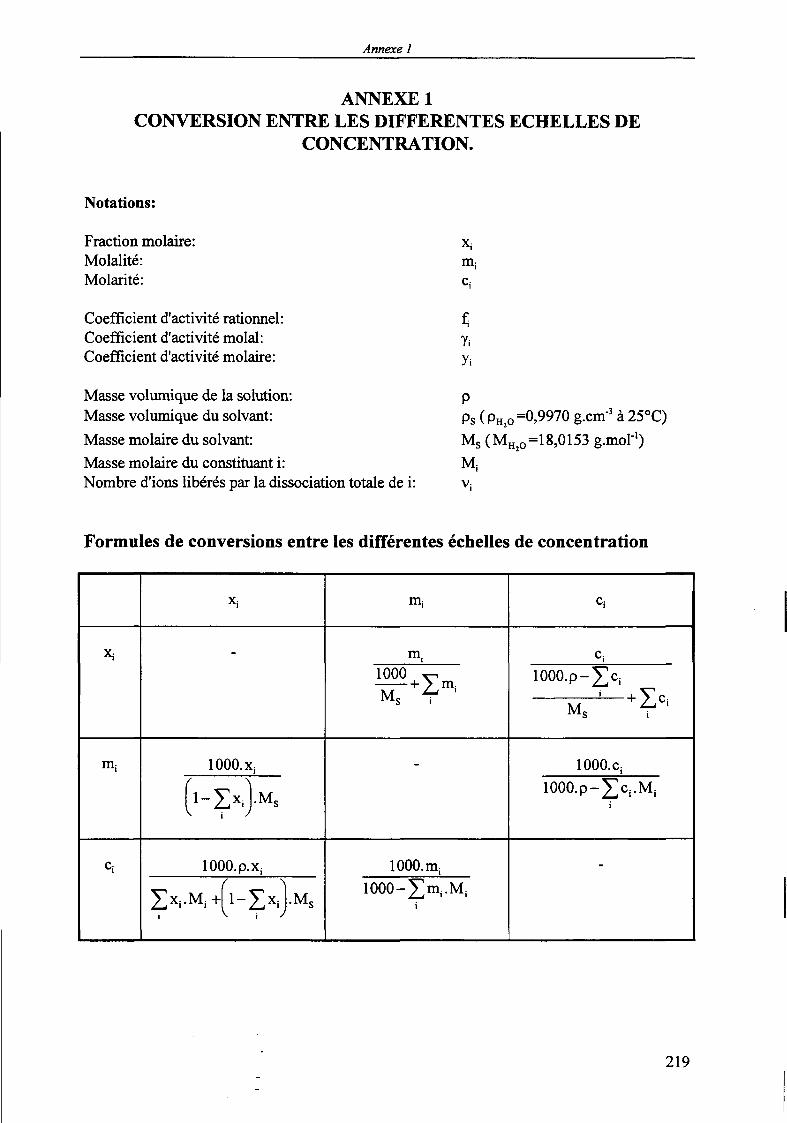
Annexe 1
ANNEXE 1CONVERSION ENTRE LES DIFFERENTES ECHELLES DE
CONCENTRATION.
Notations:
Fraction molaire:Molalité:Molarité:
Coefficient d'activité rationnel:Coefficient d'activité molal:Coefficient d'activité molaire:
Masse volumique de la solution:Masse volumique du solvant:
Masse molaire du solvant:
Masse molaire du constituant i:Nombre d'ions libérés par la dissociation totale de i:
Yi
PS(PH2O =0,9970 g.cm-3 à 25°C)
Ms(MH2O=18,0153g.mor1)
M,V:
Formules de conversions entre les différentes échelles de concentration
Xi
m;
Ci
*
1000. x;
lOOO.p.X;
i ^ i '
1000
Ms
10(i Afvri1 UvU """
mf
m,
i
Ci
Ci
10001000.p-^
-
' +Yc
•C;
i
219

Annexe 1
Formules de conversions entre les expressions des coefficients d'activitédans les trois échelles de concentration
fi
Yi
fi
-
Po f
Ps 'n ^.(v.-Ms-M.)
i ^ i '
1
1 + V i,i
Yi
1+ s ' V i ' m | -YiV 1000 ) '
r i +M s .m iyP sl 1000^ p r '
y.
l 1000.ps ) Ji
l 1000.ps ) "
220

Annexe 2
ANNEXE 2COMPORTEMENT "EN EXTRAPOLATION" DES MODELES DE
PITZER ET DE CHEN
(0-10m)(0-6m)(0-3m)(0-l,4m)
Molalité (mol.kg )
Figure A2-1; Comparaison des valeurs expérimentales et calculées du coefficientd'activité de NaNO3 selon le modèle de PITZER à trois paramètres ajustables.
Domaine
d'ajustement
0-lOmol.kg-1
0-6 moLkg-1
0-3 moLkg-1
Q-l.4mol.kg-1
Paramètres ajustés
p(0)
0,0043 ±0,0001
0,0058 ± 0,0002
0,0086 ±0,0001
0,0089 ± 0,0003
0,1994 ±0,0009
0,1934 ±0,0009
0,1850 ±0,0004
0,1842 ±0,0006
*-MX
-0,00011 ±0,00002
-0,00045 ±0,00004
-0,00140 ±0,00005
-0,0015 ±0,0002
221

Annexe 2
(0-20m)(0-10m)(0-6m)(0-3m)(0-l,4m)Expérience
6 8 10 12 14 16 18 20 22 24
Molalité (mol.kg1)
Figure A2-2: Comparaison des valeurs expérimentales et calculées du coefficientd'activité de NH4NO3 selon le modèle de PITZER à trois paramètres aiustables.
Domaine
d'ajustement
0-20 moLkg-1
0-lOmol.kg-1
0-6 moLkg-1
0-3 moLkg-1
0-l.4mol.kg-1
Paramètres ajustés
p(0)
-0,0163 ±0,0002
-0,0143 ± 0,0002
-0,0114 ±0,0004
-0,0080 ±0,0008
-0,002 + 0,002
pd)
0,085 ± 0,002
0,075 ± 0,002
0,064 ±0,002
0,054 ±0,003
0,040 ±0,005
0,00037 ± 0,00002
0,00005 ±0,00006
-0,0006 ±0,0001
-0,0019 ±0,0003
-0,006 ±0,001
222

Annexe 2
(0-15m)(0-6m)(0-3m)(0-l,4m)Expérience
0 1 5 6 7 8 9 10 11 12 13 14 15
Molalité (mol.kg1)
Figure A2-3: Comparaison des valeurs expérimentales et calculées du coefficientd'activité de AgNO3 selon le modèle de PITZER à trois paramètres ajustables.
Domaine
d'ajustement
0-15 mol-kg-1
0-6 molkg-1
0-3 moLkg-1
0-1,4 molkg-1
p(0)
-0,076 ±0,001
-0,0903 ±0,0008
-0,096 ±0,001
-0,102 ±0,004
Paramètres ajustés
-0,002 ± 0,008
0,051 ±0,003
0,067 ± 0,004
0,080 ±0,009
'"MX
0,0037 ±0,0001
0,0069 ±0,0002
0,0089 ±0,0005
0,013 ±0,003
223

Annexe 2
\ I I I I I T
(0-10m)(0-6m)(0-3m)(0-l,4m)Expérience
4 5 6 7
Molalité (mol.kg1)
10 11
Figure A2-4: Comparaison des valeurs expérimentales et calculées du coefficientd'activité de NaNO3 selon le modèle de PITZER à quatre paramètres ajustables.
Domaine
d'ajustement
0-lOmol.kg-1
0-6 molkg-1
0-3 moLkg-1
0-1,4 moLkg-1
Paramètres ajustés
p(0)
0,004 ±0,002
0,003 ±0,004
0,007 ±0,001
0,0068 ±0,0009
pu
0,20 ±0,01
0,17 ±0,03
0,17 ±0,01
0,16 ±0,01
-0,0001±0,0001
-0,0002±0,0003
-0,0013±0,0001
-0,0016±0,0002
b
1,21 ±0,04
1,28 ±0,09
1,24 ±0,03
1,27 ±0,03
224

Annexe 2
(0-20m)(0-10m)(0-6m)(0-3m)(0-l,4m)Expérience
8 10 12 14 16
Molalité (mol.kg1)
20 22 24
Figure A2-5; Comparaison des valeurs expérimentales et calculées du coefficientd'activité de NH4NO3 selon le modèle de PITZER à quatre paramètres ajustables.
Domaine
d'ajustement
0-20 moLkg-1
0-lOmol.kg-1
0-6 mol-kg-1
0-3 molkg-1
0-1,4 mol.kg-1
Paramètres ajustés
p(0)
-0,023 ±0,001
-0,024 ±0,004
-0,014 ±0,009
-0,001 ±0,037
0,008 ±0,047
pd)
-0,01 ±0,02
-0,03 ±0,05
0,04 ±0,08
0,11 ±0,19
0,10 ±0,24
0,0006±0,0001
0,0006±0,0003
-0,0004±0,0007
-0,003±0,004
-0,006±0,005
b
1,48 ±0,05
1,53 ±0,18
1,27 ±0,25
1,02 ±0,61
1,00 ±0,76
225

Annexe 2
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
Molalité (mol.kg1)
Figure A2-6: Comparaison des valeurs expérimentales et calculées du coefficientd'activité de AgNO3 selon le modèle de PITZER à quatre paramètres ajustables.
Domaine
d'ajustement
0-15 molkg-1
0-6 moLkg-1
0-3 moLkg-1
0-1,4 moLkg-1
Paramètres ajustés
p(0)
-0,013 ±0,002
-0,038 ±0,007
-0,02 ±0,04
-0,106 + 0,004
pd)
0,326 ±0,006
0,28 ±0,02
0,31 ±0,04
-0,20 ±0,05
'-'MX
0,0008±0,0001
0,0026±0,0006
0,0001±0,0032
0,005±0,003
b
0,35 ±0,02
0,51 ±0,05
0,39 ±0,16
2,07 ±0,17
226

Annexe 2
(0-lOm)(0-6m)(0-3m)(0-l,4m)Expérience
0 1 4 5 6 7
Molalité (moLkg1)
10 11
Figure A2-7: Comparaison des valeurs expérimentales et calculées du coefficientd'activité de NaNO3 selon le modèle de CHEN à deux paramètres aiustables.
Domaine
d'ajustement
0-lOmol.kg-1
0-6 moLkgr1
0-3 moLkg-1
0-1,4 moLkg-1
Paramètres ajustés
ca,m
-3,65 ±0,01
-3,60 ±0,02
-3,64 ±0,07
-3,87 ±0,19
m,ca
7,36 ± 0,02
7,26 ±0,04
7,33 ±0,14
7,81 ±0,40
227

Annexe 2
(0.20m)(0-10m)(0-6m)(0-3m)(0-l,4m)Expérience
8 10 12 14 16
Molalité (moLkg1)
18 20 22 24
Figure A2-8: Comparaison des valeurs expérimentales et calculées du coefficientd'activité de NH4NO3 selon le modèle de CHEN à deux paramètres ajustables.
Domaine
d'ajustement
0-20 moLkg-1
0-lOmol.kg-1
0-6 moLkg-1
0-3 moLkg-1
0-1,4 moLkg-1
Paramètres ajustés
"ca,m
-3,34 ±0,01
-3,42 ±0,03
-3,56 + 0,03
-3,87 ±0,05
-4,33 ±0,07
"m,ca
6,97 ±0,03
7,13 ±0,06
7,42 ±0,07
8,06 ±0,10
8,97 ±0,13
228

Annexe 2
(0-15 m)(0-6m)(0-3m)(0-l,4m)Expérience
0 1 5 6 7 8 9 10 11 12 13 14 15
Molalité (moLkg1)
Figure A2-9: Comparaison des valeurs expérimentales et calculées du coefficientd'activité de AgNO3 selon le modèle de CHEN à deux paramètres ajustables.
Domaine
d'ajustement
0-lSmol.kg-1
0-6 moLkg-1
0-3 moLkg-1
0-1,4 mol.kg-1
Paramètres ajustés
"ca,m
-3,08 ±0,02
-3,27 ±0,03
-3,55 ±0,05
-4,03 ±0,06
''m.ca
7,04 ±0,03
7,38 ±0,05
7,87 ±0,09
8,73 ±0,12
229

Annexe 2
(0-10m)(0-6m)(0-3m)(0-l,4m)Expérience
4 5 6 7 8
Molalité (mol.kg1)
10 11
Figure A2-10: Comparaison des valeurs expérimentales et calculées du coefficientd'activité de NaNO3 selon le modèle de CHEN à trois paramètres ajustables.
Domaine
d'ajustement
0-lOmol.kg-1
0-6 molkg-1
0-3 moLkg-1
0-1,4 moLkg-1
Paramètres ajustés
ca,m
-3,66 ±0,01
-3,57 ±0,03
-3,33 + 0,26
-2,62 ±75,13
m,ca
7,39 ±0,03
7,15 ±0,08
6,60 + 0,58
5,11 ±156,40
b
15,2 ±0,3
14,3 ±0,4
13,4 ±0,7
13,1 ±0,7
230
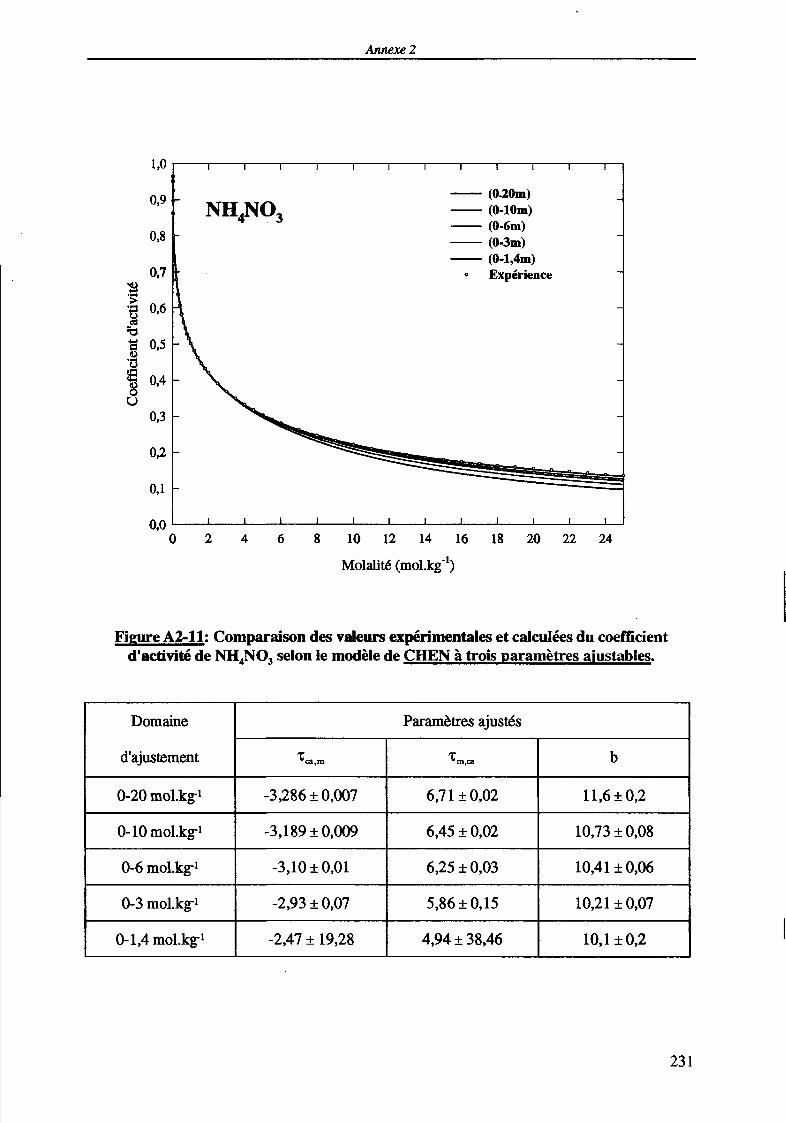
Annexe 2
(0.20m)(0-10m)(0-6m)(0-3m)(0-l,4m)Expérience
8 10 12 14 16 18 20 22 24
Molalité (moLkg1)
Figure A2-11: Comparaison des valeurs expérimentales et calculées du coefficientd'activité de NHLNO, selon le modèle de CHEN à trois paramètres aiustables.
Domaine
d'ajustement
0-20 molkg-1
0-lOmol.kg-1
0-6 moLkg-1
0-3 molkg-1
0-1,4 moLkg-1
Paramètres ajustés
"ca,m
-3,286 ±0,007
-3,189 ±0,009
-3,10 ±0,01
-2,93 ±0,07
-2,47 ± 19,28
m,ca
6,71 ±0,02
6,45 ±0,02
6,25 ±0,03
5,86 ±0,15
4,94 ±38,46
b
11,6 ±0,2
10,73 ±0,08
10,41 ±0,06
10,21 ±0,07
10,1 ±0,2
231

Annexe 2
i i i i i i r i i i
AgNO
0 1
(0-15 m)(0-6m)(0-3m)(0-l,4m)Expérience
6 7 8 9 10 11 12 13 14 15
Molalité (moLkg1)
Figure A2-12: Comparaison des valeurs expérimentales et calculées du coefficientd'activité de AgNO3 selon le modèle de CHEN à trois paramètres ajustables.
Domaine
d'ajustement
0-lSmol.kg-1
0-6 molkg-1
0-3 molkg-1
0-1,4 moLkg-1
Paramètres ajustés
''ca.m
-2,996 ±0,003
-2,96 ±0,01
-2,87 ±0,04
-2,80 ±0,17
"m,ca
6,723 ±0,008
6,65 ±0,03
6,47 ±0,07
6,34 ±0,29
b
11,38 ±0,06
11,16 ±0,08
10,90 ±0,09
10,76 ±0,16
232

Annexe 3
ANNEXE 3METHODE DE NEWTON-RAPHSON-MARQUARDT
TEST D'ARRET DE HOOKE ET JEEVES.
1. Présentation générale.
Dans un premier temps, nous présentons la méthode de calcul appliquée à un système
simple. Nous disposons de R valeurs expérimentales Vjexp, l'expression de y^0' est unefonction f(x,P) de i variables Xj (vecteur colonne x) et de N paramètres Ps (vecteur colonne P).Le problème posé est la détermination de P00 représentant le jeu de paramètres optimumminimisant la somme des moindres carrés g(P):
= (y iexp-f(x i ,P))
Au minimum, le gradient de g doit être nul soit:
OrS
(2)
L'écriture de (2) sous la forme matricielle fait intervenir, H le vecteur colonne des R valeursh(Xj,P) et J le jacobien de la fonction f(Xj,P) sous sa forme transposée lJ:
= -2.'J.H avec J B - ^ S ^ (3)
II est possible de simplifier le système non linéaire de N équations à N inconnues (2) endéveloppant dGs en une série de TAYLOR au premier ordre, ôP étant un vecteur colonne depetite variation de P, on aboutit alors à un système linéaire:
s=i
Si l'on suppose que (P+ôP) est la solution du système, on a dGs(P+ÔP)=0 et l'équation (4)devient, en utilisant la relation (2):
(5)S'=l
ôJiSL'approximation de NEWTON-RAPHSON consiste à négliger les dérivés secondes ——
ôps,lorsque l'on est proche du minimum, d'où:
233

Annexe 3
dC
Soit sous forme matricielle:
L'équation (3) devient alors:
N
S'=l
dG = -2
R
.'J.J.ÔP
* (6)
(7)
= ( tJ.j)"'. tJ.H (8)
Le processus itératif consiste à remplacer P par P+ôP et à répéter l'opération jusqu'à ce que ÔPsoit inférieur à la précision désirée.
En réalité, le terme des dérivés secondes de l'équation (5) n'est pas toujours négligeable etMARQUARD suggère de résoudre le système:
('J.J + WD^ÔP^J.H avec Dss =( ' J . j ) s s +1 (9)
A, est un paramètre généralement initialise à 0,1 au début du processus itératif et qui est, àchaque nouvelle itération:
- soit multiplié par 0,4 si la solution ôP de l'itération précédente vérifie g(P+ôP) < g(P),P est alors remplacé par (P+8P)
- soit multiplié par 10 si g(P+ôP) > g(P) et P n'est pas modifié.
Le test d'arrêt effectivement utilisé ne porte par sur ôP à cause des problèmes d'ordre degrandeur ou de zéro. Il s'agit en fait du test utilisé dans la méthode de HOOKE et JEEVES: si
P est la précision moyenne des R mesures, y?9' (vecteur colonne Yexp ), le processus itératifs'arrêtera lorsque l'on aura obtenu un vecteur P tel que:
m
2. Description des calculs dans EXTRACTION.
Dans ce qui suit la notation suivante sera reprise pour simplifier les écritures mathématiques:
r2n-l J V injnknhndn
P =S®r 2 n S > i n j n k n h n d n
Chaque point 1 de l'isotherme expérimentale qui en compte R est défini par huit grandeurs
accessibles à l'expérience (aHi, aX|, aH 0 | , mH , mx , mE , mH o et mDil ) et deux variables
234

Annexe 3
([E], et [Diljj ). De plus, le système comprend 2N paramètres Ps (deux par solvate au nombre
deN).Les mesures sont effectuées avec une précision moyenne Pc qui dépend du composé C (C=H,X, E, H2O ou Dil.), nous pouvons alors affecter un poids coc à chacune de ces mesures tel que:
1 1coc =
Z_i p 'C=H rC
Une précision moyenne P est définie telle que:
Dil.
(11)
(12)C=H
La fonction g(P) à minimiser correspond à la somme des moindres carrés entre les isothermes
expérimentales et calculées. Cette expression dépend des cinq fonctions mc (aHi, aX|, aH2Û],
[E],, [Dil],, P) dans lesquels P représente un vecteur colonne des deux paramètres P2n et P ^ .
L'expression analytique complète de g(P) est la suivante:
«M-Z1=1
Dil. c, •-mcaH l ,aX i ,aH i O i ,[E]1 ,[DU.] l f
C=E -exp.(13)
avec|2 R—«p|f v/-—«p\
Pour simplifier l'écriture de g(P) nous posons:
mCi = m c ( a H | , aX | , aH2Oi, [E],, [Dil.],,
coc =CO,
-exp.
La composante S du gradient de g(P) s'exprime sous la forme:
R Dil
(14)1=1 C=E
Le système à résoudre est celui proposé par MARQUARD
2N H R Dil.
S'-l L 1=1 C=E
R Dil.
(15)
avec ôss. = 1 si S=S'1=1 C=E
et ôs s. = 0 si S^S'
235

Annexe 3
Dans ce qui suit, nous allons présenter le déroulement des calculs permettant de résoudre cesystème, c'est à dire de déterminer les valeurs de ôPs.. La logique de calcul peut être déclinéeen cinq étapes.
- i. Calcul de la concentration d'extractant et de diluant libre.
La combinaison de la loi d'action de masse et des bilans matière sur l'extractant et le diluantpermet d'écrire deux polynômes dont la résolution permet d'accéder aux concentrationsd'extractant et de diluant libres en chaque point 1 de l'isotherme:
An,.-[Ëf +[Ë] 06)n=l
v l f î ^ ] (17)n=l
avec
An,. = în-
Bn.,=dn.P2n.1.ajî.a^.aî;-iO.exp[p2ll.(aHiO-l)]
Lorsque de degré (i ou d) est inférieur ou égal à 2, le calcul des racines des polynômes estclassique. Pour des degrés supérieurs, les racines sont calculées par la méthode d'interpolationpar fractions continues.
- ii. Calcul de la concentration des complexes en phase organique à l'équilibre.
Pour chaque point de l'isotherme, la concentration de chaque complexe est calculée par la loid'action de masse.
- iii. Calculs des concentrations des constituants en phase organique à l'équilibre.
Les molalités à l'équilibre de chaque constituant en phase organique sont calculées à partir desbilans matières.
- iv. Calculs des jacobiens j£ s .
Pour effectuer le calcul des jacobiens J^s, il est nécessaire de débuter par le calcul de
et de ———. Pour cela, il suffit de dériver les équations (16) et (17) d'où:
n=l
236

Annexe 3
(19)
n=l
Le calcul de Jfs est ensuite effectué par dérivation de la loi d'action de masse:
d?s
- l ) ) (20)
[if (aH20 - l ) )
Le calcul des jacobiens Jj s est enfin réalisé par dérivation des bilans matières.
jH = M ^ H = y . Iw I aps J é - n I
(21)
x _• s " - è n (22)
Jf.= ,,ML44 (23)
( 2 4 )
ap.
n=l
+aH,0n=l '
(25)
+(P 2 n + i n -sf + d n -s®,)-ôm;
- v. Résolution du système (15) suivant la méthode de NEWTON-RAPHSON-MARQUARDT.
ÔPe
237

Annexe 4
ANNEXE EXPERIMENTALE.
1. Préparation de solutions par pesée.
1.1. Préparation d'une solution binaire par dilution d'une solution mère.
p.mxmx = + 0,001.m'x.Mj
mx: molalité du constituant X dans la solution fille (moLkg'1),mx : molalité du constituant X dans la solution mère (moLkg'1),Mx: masse molaire du constituant X (g.mol"1),p: masse versée de la solution mère (g),q: masse d'eau ajoutée (g).
1.2. Préparation d'une solution ternaire par dilution de deux solutions binaires.
p.mXi.(l000+mX2.MX2)mx, = '
X2.MX2)+q.(l000 + m
q.mX2.(l000 + rnXi.MX|)m Y =
mX|, mx : molalités des constituants X,, X2 dans le mélange (mol.kg*1),
mXi, mx : molalités des constituants X,, X2 dans leur solution binaire (mol.kg"1),
MXj , Mx : masses molaires des constituants X,, X2 (g.mol'1),
p: masse versée de la solution binaire de X, (g),q: masse versée de la solution binaire de X2 (g).
1.3. Préparation d'une solution à i constituants par dilution de i solutions binaires.
m x =——;
i ' k*i
mx. : molalité du constituant i dans le mélange (mol.kg1),
mXi : molalité du constituant i dans sa solution binaire (mol.kg"1),
Mx. : masse molaire du constituant i (g.mol"1),
px. : masse versée de la solution binaire de i (g).
239

Annexe 4
2. Mesure de l'activité d'eau avec l'aw-CENTER.
2.1. Principe de la mesure.
Nous avons retenu l'appareil aw-CENTER de la société NOVASINA pour mesurerl'activité d'eau de nos solutions aqueuses. Le signal analogique enregistré correspond àl'impédance d'un capteur électrolytique. Cette grandeur est fonction du taux d'humidité dans lacellule de mesure. C'est la valeur de l'activité d'eau de la solution qui est lue sur un affichagedigital.
L'appareil dont nous disposons est composé de deux parties distinctes: la première regroupel'ensemble de l'électronique de mesure et la seconde est constituée par trois cellules de mesurethermostatées. La thermostatisation des capteurs électrochimiques est assurée par effetPELTIER. Il est théoriquement possible d'effectuer des mesures de 0 à 50°C, toutefois ladifférence entre la température de consigne et la température extérieure ne peut excéder 10°C.
La solution (ou bien le solide à étudier) est placée dans une coupelle PVC puis introduite dansla chambre de mesure constituée d'un réceptacle en acier recouvert par la cellule thermostatée.La mise en équilibre isopiestique (liquide-vapeur ou solide-vapeur) est alors suivie parl'intermédiaire d'un enregistreur.
2.2. Mode opératoire.
La mise en œuvre de l'aw-CENTER se décompose en trois étapes successives:
© La première étape consiste en la linéarisation de la réponse impédanceélectrochimique - activité d'eau. On utilise pour cela deux étalons dont les valeurs d'activitéd'eau encadrent le domaine étudié. La procédure de linéarisation consiste à ajustersuccessivement, à l'aide de deux potentiomètres, les valeurs lues aux deux valeurs étalons.Afin de préserver la qualité de l'ajustement, il est recommandé de réduire le domaine delinéarisation à 0,1 unité d'activité d'eau.
© Dans un deuxième temps, on procède à l'étalonnage de la réponse de l'appareil. Ils'agit de mesurer l'activité d'eau de solutions étalons et de les comparer aux valeurs étalons.
En final, il est possible de tracer une droite d'étalonnage (aHi0] = f(aH20). permettant à
partir d'une valeur mesurée d'accéder à l'activité d'eau de la solution étudiée. La qualité del'étalonnage est bien sûr améliorée lorsqu'un grand nombre de solutions étalons est utilisé.
(D Pour mesurer l'activité d'eau d'une solution, il suffit de remplir la coupelle en PVCavec quelques millilitres de solution et de la placer dans la chambre de mesure. L'équilibre estatteint au bout de trente minutes dans la plupart des cas (sauf pour les solutions saturées).
2.3. Choix des solutions étalons.
Les solutions étalons préconisées et commercialisées par le constructeur sont dessolutions saturées en sels. Ces solutions sont choisies de part leur facilité de préparation etleur conservation prolongée. Les valeurs d'activité d'eau à saturation sont publiées par
240

Annexe 4
GREENSPAN [1] sur une plage étendue de température (0-100°C) pour 28 solutions binaires.Nous avons cherché à recenser les données thermodynamiques à 25 °C pour un nombre plusélevé de solutions saturées d'électrolytes. Cette recherche bibliographique nous a en mêmetemps permis de réviser les valeurs tabulées par GREENSPAN. En effet, les valeurs calculéespar cet auteur s'écartent parfois de celles publiées par le National Bureau of Standards (NBS).Cet écart provient de l'erreur commise lors de l'interpolation des valeurs d'activité d'eau àsaturation à différentes températures. Les données tabulées par le NBS sont obtenues parinterpolation des mesures expérimentales disponibles à 25 °C, elles s'affranchissent donc del'erreur d'ajustement entre différentes températures.
Nous présentons à la fin du texte le résultat final de notre compilation. La molalité àsaturation correspond à la valeur publiée par LINKE [2, 3], retenue par l'ensemble des auteurspubliant pour le NBS.
L'utilisation de solutions saturées lors des étapes de linéarisation et d'étalonnage estfastidieuse, la mise en équilibre isopiestique de ces solutions pouvant atteindre plusieursheures. Le temps d'attente important tient à la présence simultanée de trois phases:
- une phase solide correspondant aux cristaux de sels non dissous,
- une phase liquide constituée de la solution saturée en electrolyte,
- une phase gazeuse composée de la vapeur d'eau et de l'électrolyte lorsque celui-ci estvolatil.
Si l'on veut réduire le temps d'attente, il est possible de conserver les solutions saturées à lamême température que celle de la cellule de mesure. Une autre alternative est d'utiliser dessolutions binaires non-saturées de concentration connue dont les données thermodynamiquesà 25°C sont répertoriées dans la littérature.
Finalement, nous avons réalisé un étalonnage sur un domaine de 0,1 unité d'activité d'eau enutilisant les deux types de solutions (dix en tout). Nous avons atteint une précision maximalede 0,002 unité d'activité d'eau.
3. Mesure de la masse volumique.
L'ensemble des mesures de masse volumique des solutions aqueuses et organiques estréalisé à l'aide d'un densimètre à tube vibrant ANTON-PAAR. Deux appareils sont utilisés, ils'agit du modèle DMA 55 associé au thermostat cryostat JULABO F20 HC et du modèleDMA 48. Le premier modèle possède un affichage à ± 0,00001 g.cm'3 contre 0,0001 g.cm"3
pour le second.
3.1. Principe de la mesure.
La cellule de mesure est composée d'un tube en U (diapason en verre) relié à unsystème oscillant. L'ensemble de ce système est placé dans un cylindre en verre rempli d'ungaz à haute conductivité thermique. Dans le modèle DMA 55, un serpentin placé autour du
241

Annexe 4
diapason permet par circulation d'eau de thermostater la cellule de mesure. Dans le modèleDMA 48, la thermostatisation est assurée par effet PELTIER autour du cylindre en verre etnon plus directement autour du diapason.
La fréquence de résonance du diapason soumis à une vibration entretenue permet d'accéder àla valeur de la masse volumique du liquide ou du gaz contenu dans le tube. En effet, lafréquence F d'oscillations de la cellule de mesure est donnée par:
— avec m = mo+V0.p
c: constante d'élasticité,m: masse totale de la cellule,m,,: masse de la cellule vide,Vo: volume de la cellule,p: masse volumique du liquide ou du gaz contenu dans la cellule.
La période des oscillations T s'exprime selon:
d'où:
La masse volumique de l'échantillon s'exprime alors en fonction des constantes A et B ainsique de la période de vibration, c et Vo dépendent de la température, ce qui signifie que A et Bsont les constantes de l'oscillateur à température donnée. Les valeurs de ces constantes sontdéterminées par la mesure des périodes de vibration obtenues pour des solutions de massevolumique connue. Les masses volumiques de ces étalons doivent bien sûr encadrer la zone demesure parcourue lors de nos expériences. Nous avons choisi de réaliser trois solutionsd'électrolyte dont nous avons fait certifier la masse volumique auprès du Laboratoire Nationald'Essai (LNE) à 25°C:
© p1=(U4795±0,00013) g.cm"3 (electrolyte LiNO3),
© p2=(l,2430±0,0001) g.cm'3 (electrolyte LiNO3),
d> p3=(l,3483±0,0002) g.cm"3 (electrolyte Zn(NO3)2),,
L'eau pure constitue un quatrième étalon: peau=(0,9970429±0,0000001) g.cm3.
Les valeurs des constantes A et B sont calculées directement par l'appareil dans le cas duDMA 48. Lors de l'utilisation du DMA 55, ces valeurs doivent d'abord être calculées parrégression linéaire sur les périodes obtenues pour chacun des étalons, puis consignées sur ledensimètre grâce à deux roues codeuses.
242

Annexe 4
3.2. Mode opératoire.
La solution à analyser est introduite dans la cellule de mesure à l'aide d'une seringue àembout LUER. Le volume minimal nécessaire correspond au volume du diapason c'est à dire0,7 mL. Dans la plupart des cas, un volume de 1 mL est injecté. L'appareil indiquedirectement la valeur de la masse volumique lorsque la température est stabilisée au sein de lacellule. Avec le modèle DMA 55, le contrôle de la température est assuré grâce à unthermomètre digital LERIS associé à une sonde de platine PT100, cette chaine de températureayant été étalonnée par le LNE. Le contrôle de la température est intégré au modèle DMA 48et ne nécessite donc aucun appareil auxiliaire.
4. Titrage coulométrique de l'eau selon la technique Karl FISHER.
La teneur en eau des phases organiques est analysée avec le coulomètre METHROMKF684.
4.1. Principe de la mesure.
Cet appareil est basé sur le dosage de l'eau par l'iode selon la méthode décrite par KarlFISHER [4]. La solution de titrage classiquement utilisée est une solution méthanolique(CHjOH) d'iode I2, de bioxyde de soufre SO2 et d'une base tampon RN. La réaction de titrageglobale est la suivante:
H2O +12 + [RNH]SO3CH3 + 2. RN ->• [RNH]SO4CH3 + 2.[RNH]l
Le point équivalent est détecté grâce à la persistance de la coloration brune de l'iode ensolution. La connaissance du titre en iode et du volume équivalent permet de calculer laconcentration de l'eau présente dans la solution dosée. Cette méthode de titrage volumétriquesouffre de l'instabilité du réactif titrant dont le titre doit être constamment contrôlé à l'aide desolutions étalons.
La mise en œuvre du dosage selon une méthode coulométrique permet de s'affranchirde ce problème. En effet, dans ce cas, la production de l'iode est assurée directement par voieélectrochimique lors du dosage.
L'appareil utilisé est donc constitué d'une cellule d'électrolyse, la production de l'iode a lieu àl'anode lors d'une réaction d'oxydation. Le coulomètre METHROM KF 684 dont nousdisposons est composé de deux compartiments, les deux électrodes génératrices étant séparéespar un fritte. Ce montage expérimental permet d'éviter les éventuelles réactions parasites deréduction à la cathode.
L'utilisation d'un jeu de micro-électrodes indicatrices en platine permet de détecter la fin dutitrage.
Le dispositif est complété par un intégrateur afin de mesurer la charge consommée lors de laproduction d'iode.
243

Annexe 4
II existe une relation strictement quantitative entre la quantité d'iode produite et laquantité d'électricité (2 Faraday par mole d'iode). Il est donc possible de déterminer la quantitéd'eau présente dans un échantillon par l'intermédiaire de la mesure de la charge électriqueconsommée lors du titrage.
Le résultat du titrage est exprimé en ug ou mg d'eau. L'appareil peut détecter des traces d'eaucomprises entre 10 ng et 10 mg.
4.2. Mode opératoire.
L'échantillon est prélevé à l'aide d'une seringue étanche (type chromatographiegazeuse) puis injecté rapidement dans le compartiment anodique à travers un septum. Lavaleur lue après le dosage est exprimée en ug ou mg d'eau. Si l'on connait les masses de laseringue avant et après injection, on a accès à la teneur massique en eau exprimée par unité demasse d'échantillon.
5. Titrages potentiométriques.
Les concentrations d'acide en phase aqueuse et en phase organique à l'équilibre sontobtenues par dosages potentiométriques. Afin de simplifier les protocoles expérimentaux,l'acide nitrique n'est pas dosé directement en phase organique mais dans la phase aqueuse dedésextraction.
5.1. Principe de la mesure.
Le dosage de l'acide est effectué grâce à un dispositif de titrage automatiqueTACUSSEL (TT-Processeur 2). Ce montage expérimental comprend:
- une seringue motorisée contenant le réactif titrant,
- une électrode de verre combinée de haute alcalinité (TACUSSEL XC 250),
- un dispositif électronique relié à la seringue et au capteur permettant de contrôler lesajouts de titrant et les mesures de potentiel.
Le titrant est dans notre cas de la soude, la réaction de dosage correspond donc à untitrage classique acide fort / base forte. Le point équivalent est déterminé automatiquement parle calcul de la dérivée du signal en fonction du volume de réactif ajouté. La connaissance desvolumes d'échantillon dosé et de titrant versé ainsi que le titre de la soude permet de calculerla concentration de l'acide dans la phase aqueuse.
5.2. Mode opératoire.
Le volume de la seringue ainsi que le titre de la soude sont choisis de manière àobtenir des volumes équivalents facilement détectables. Pour le dosage des solutions d'acideles plus concentrées, le volume de la seringue est de 10 mL et le titre de la soude de 1 mol.L"1.Dans le cas du dosage de l'acide dans la phase aqueuse de désextraction, le volume de la
244

Annexe 4
seringue n'est plus que de 2 mL, le titrant choisi étant de la soude 0,01 ou 0,1 moLL"1. Laconcentration de l'ensemble des solutions titrantes utilisées est contrôlée selon la méthodedécrite par VOGEL [5] à l'aide du phtalate acide de potassium.
Le volume de l'échantillon dosé est calculé grâce à la mesure de sa masse et de samasse volumique.
6. Dosages des cations alcalins par chromatographie ionique.
Le dosage des cations alcalins est réalisé par chromatographie ionique dans lessolutions aqueuses de désextraction. Les manipulations sont effectuées sur deux appareilsdifférents: le premier est un appareil WATERS ILC1, le second est de marque DIONEX(DX 100).
6.1. Principe de la mesure.
Pour les deux appareils, le dispositif expérimental est basé sur la séparation des ionssur une colonne échangeuse de cations. Cette colonne est protégée des éventuelles impuretésorganiques par une pré-colonne de même remplissage que la colonne principale.
L'éluant utilisé est une solution acide (HNO3 pour WATERS et acide méthane sulfonique pourDIONEX), le débit retenu étant de 1 mL/min.
La détection est réalisée dans les deux cas par voie conductimétrique, le signal mesuré esttraité par l'intermédiaire d'un intégrateur SPECTRA-PHYSICS.
Cette méthode de dosage nécessite un étalonnage réalisé avec des solutions synthétiques dechacun des cations. La droite d'étalonnage est obtenue en portant l'aire du pic en fonction de laconcentration de la solution injectée.
6.2. Mode opératoire.
Pour chaque cation, cinq solutions étalons différentes sont préparées par dilution d'unesolution mère à 2.10'2 mol.L"1 de sel ultrapur. Les concentrations de ces solutions varient de
Les phases aqueuses à doser sont alors injectées en utilisant une seringue en polypropylene.
245

Annexe 4
7. Dosages du césium par spectrométrie gamma.
Le dosage du césium est également effectué par spectrométrie gamma directement surles phases aqueuses et organiques à l'équilibre. Les manipulations sont réalisées en utilisant leradioisotope 137Cs comme traceur radioactif.
7.1. Principe de la mesure.
La chaîne de mesure est composée d'un analyseur multicanal EGSP-2000-20R de lasociété rNTERTECHNIQUE associé à un détecteur en germanium ultra pur refroidi à l'azoteliquide.
Le traceur radioactif mis en œuvre dans nos manipulations d'extraction liquide-liquide est le137Cs dont le temps de demi-vie est de (30,15 ± 0,06) ans. Ce radioisotope se désintègre parémisssion p' vers le niveau fondamental du 137Ba ainsi que vers le niveau isomère de 661 keV,son schéma de désingration est le suivant:
30,15 ans13755
-2,554 minutes137m-56
537Ba8] stable
C'est la raie du noyau fils 137mBa que l'on utilise pour doser le 137Cs par spectrométrie gamma
L'activité du 137Cs est donnée par:
avec X =In2
M/2A|"csini : a c t ^ t é initiale du 137Cs,
Xm. : constante radioactive du 137Cs,Os
TI/2 : durée de demi-vie.
246
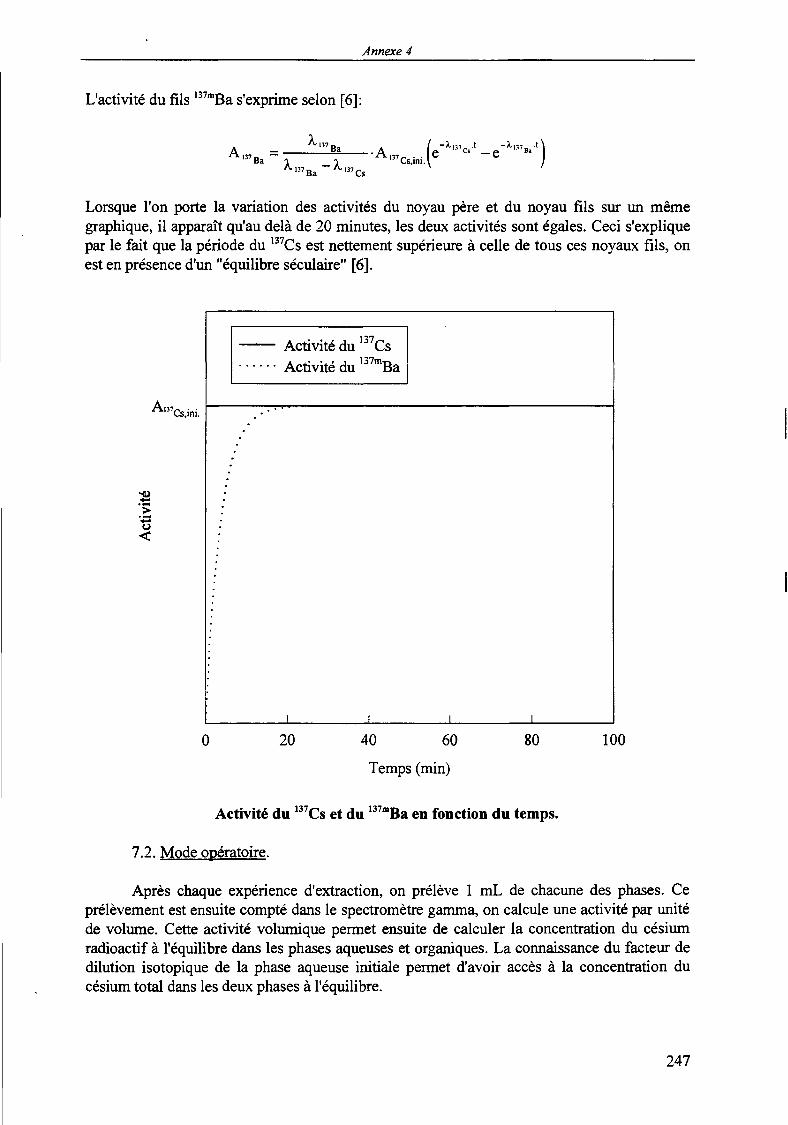
Annexe 4
L'activité du fils I37mBa s'exprime selon [6]:
B*-X,
Lorsque l'on porte la variation des activités du noyau père et du noyau fils sur un mêmegraphique, il apparaît qu'au delà de 20 minutes, les deux activités sont égales. Ceci s'expliquepar le fait que la période du 137Cs est nettement supérieure à celle de tous ces noyaux fils, onest en présence d'un "équilibre séculaire" [6].
A l 3 7 Cs,ini.
1
Activité du 137CsActivité du I37mBa
1 1 1 1
20 40 60
Temps (min)
80 100
Activité du 137Cs et du 137mBa en fonction du temps.
7.2. Mode opératoire.
Après chaque expérience d'extraction, on prélève 1 mL de chacune des phases. Ceprélèvement est ensuite compté dans le spectromètre gamma, on calcule une activité par unitéde volume. Cette activité volumique permet ensuite de calculer la concentration du césiumradioactif à l'équilibre dans les phases aqueuses et organiques. La connaissance du facteur dedilution isotopique de la phase aqueuse initiale permet d'avoir accès à la concentration ducésium total dans les deux phases à l'équilibre.
247

Annexe 4
8. Détail des calculs des concentrations en phase organique.
8.1. Calcul des concentrations d'eau.
La molarité de l'eau présente en phase organique est calculée à partir de la teneur en
eau ( wH 0 ) exprimée en mg d'eau par gramme d'échantillon selon:
w H l o
M H 2 0
cH 0 : molarité de l'eau en phase organique,
wHj0 : teneur en eau de la phase organique,
MH20 : masse molaire de l'eau,
porg : masse volumique de la phase organique.
La molalité de l'eau en phase organique est déduite de cH2O selon les formules de
conversion présentées en annexe 1 soit, dans le cas le plus complexe:
mH,O = ~
1000. Porg"
f1 CCalix *MC a l ix
1000.cH
fcH20.M
2o
H2C.MH4
X•Mx)
H20 : molalité de l'eau en phase organique,ccaiix » CH ' cx : naolarités du calixarène, de l'acide et des nitrates alcalins en phase organique,Mcaiix, MH , M x : masse molaire du calixarène, de l'acide et des nitrates alcalins.
8.2. Calcul de la concentration d'acide nitrique.
La désextraction totale de l'acide nitrique est obtenue après une seule désextraction. Laconcentration de l'acide est déterminée après un dosage acide base par de la soude:
TT 1 Ddes. Porg.-Pdes.
"v«i-~ R A VAq./mdes.,tit. A s PH O
CHNO3 : concentration de l'acide en phase organique à l'équilibre,
cNa0H: titre de ^a soude,Veq : volume équivalent,
mdes_>tit : masse de phase aqueuse de désextraction prélevée pour le titrage,
R eqs / : rapport des masses de phase organique et d'eau utilisées pour la désextraction,
p : masse volumique de la phase organique à l'équilibre,
248

Annexe 4
pdes : masse volumique de la phase aqueuse de désextraction,pH2o : masse volumique de l'eau.
La molalité de l'acide en phase organique se déduit de la valeur de la molarité par laconnaissance des masses volumiques.
m HNO 3
iooo . P o r g -fi CCalix •
I O O O . C H N 0 3
H 2 O - m H 2 O ^ L H N O , M *" / ,CXN(X
V)3-MxNO3J
8.3. Calcul de la concentration des nitrates alcalins.
La désextraction totale du nitrate de sodium est obtenue après une seule désextraction,la désextraction des autres nitrates alcalins nécessite deux désextractions successives. Lesformules de calcul des concentrations en phase organique sont présentées dans le cas le pluscomplexe de deux désextractions successives:
,inj.I -pi p des.l rprg. inj.2 p2 p des.2•"XNO, ' r des / . ' ^-Aq/ ' + CXNO, " r des / . " ^ A q /
/inj. /Org. P J I Q /'nJ- /Org.
CXNO3 : concentration des nitrates alcalins en phase organique à l'équilibre,
Fdés/ : facteur de dilution des phases aqueuses de désextraction,/inj.
V : rapport des masses de phase organique et d'eau utilisées pour les désextractions,
porg : masse volumique de la phase organique à l'équilibre,
pH 0 : masse volumique de l'eau.
La conversion dans l'échelle des molalités est donnée par la relation suivante (cfAnnexe 1):
m X N O ,
iooo . P o r g -f1 CCalix •MCalix+c
1000.
H 2 O - M H 2
CXNO3
0 + CHNO, .MHNOj + cXNO, •MXNO,J
249
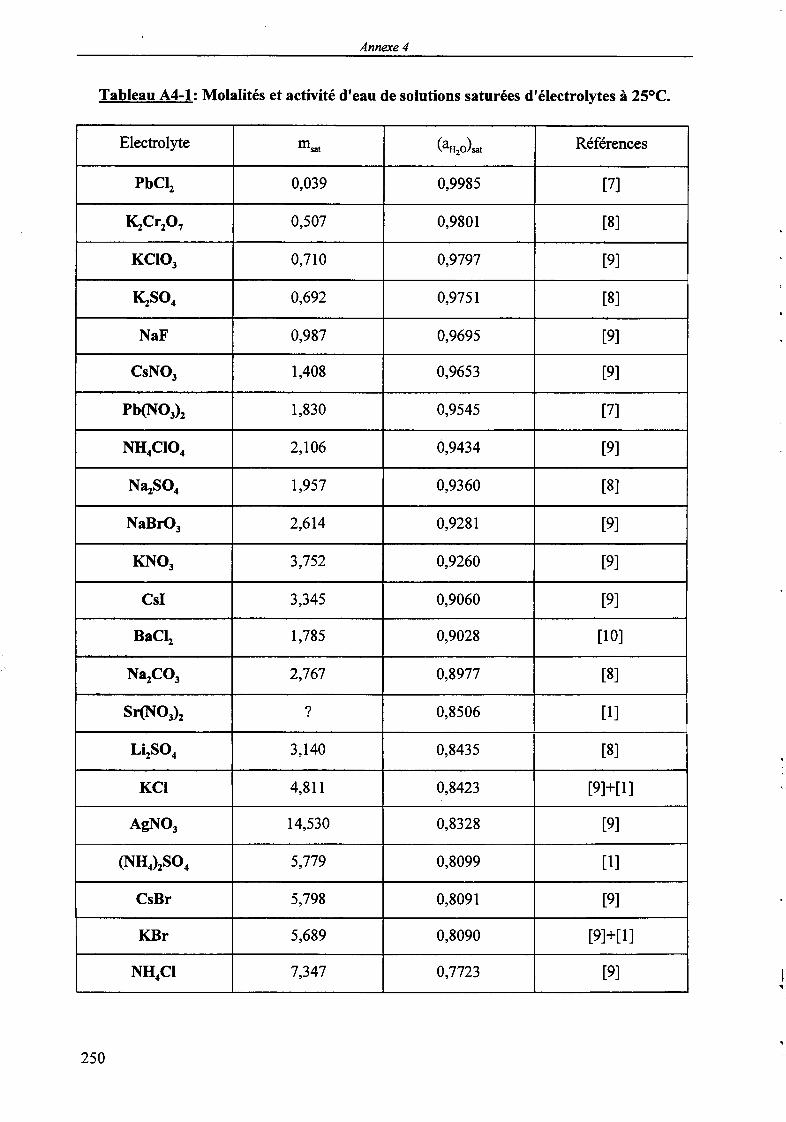
Annexe 4
Tableau A4-1: Molalités et activité d'eau de solutions saturées d'électrolvtes à 25°C.
Electrolyte
PbCl2
KCIO3
KjSO,
NaF
CsNO3
Pb(NO3)2
NH4C1O4
Na2SO4
NaBrO3
KNOj
Csl
BaC^
NajCOj
Sr(NO3)2
LijSO,
KC1
AgNO3
(NH4)2SO4
CsBr
KBr
NH4C1
msat
0,039
0,507
0,710
0,692
0,987
1,408
1,830
2,106
1,957
2,614
3,752
3,345
1,785
2,767
?
3,140
4,811
14,530
5,779
5,798
5,689
7,347
(aH2o)sat
0,9985
0,9801
0,9797
0,9751
0,9695
0,9653
0,9545
0,9434
0,9360
0,9281
0,9260
0,9060
0,9028
0,8977
0,8506
0,8435
0,8423
0,8328
0,8099
0,8091
0,8090
0,7723
Références
[7]
[8]
[9]
[8]
[9]
[9]
[7]
[9]
[8]
[9]
[9]
[9]
[10]
[8]
[1]
[8]
PMI]
[9]
[1]
[9]
[9]+[l]
[9]
250
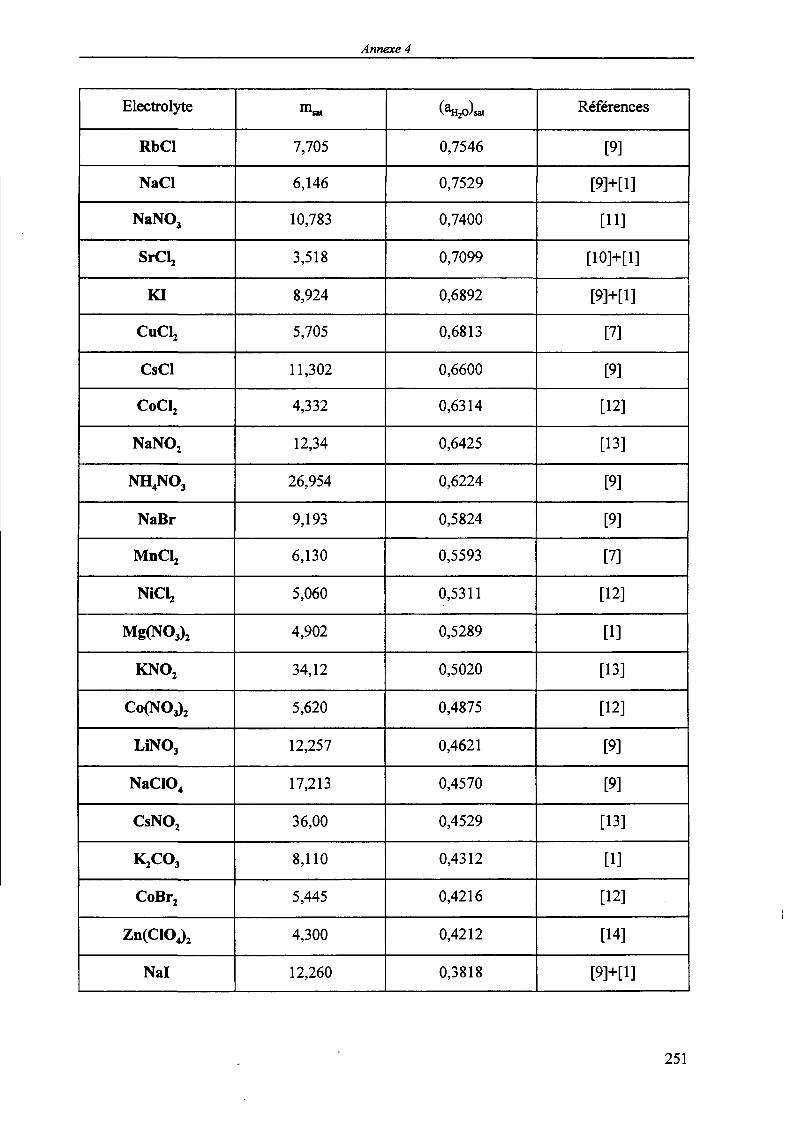
Annexe 4
Electrolyte
RbCl
NaCl
NaNO,
SrCl2
Kl
CuCl2
CsCl
CoCl2
NaNO2
NH4NO3
NaBr
MnCIj
NiClj
Mg(NO3)2
KNO2
Co(N03)2
L1NO3
NaClO4
CsNO2
K2CO3
CoBr2
Zn(ClO4)2
Nal
msat
7,705
6,146
10,783
3,518
8,924
5,705
11,302
4,332
12,34
26,954
9,193
6,130
5,060
4,902
34,12
5,620
12,257
17,213
36,00
8,110
5,445
4,300
12,260
(aH2o)sat
0,7546
0,7529
0,7400
0,7099
0,6892
0,6813
0,6600
0,6314
0,6425
0,6224
0,5824
0,5593
0,5311
0,5289
0,5020
0,4875
0,4621
0,4570
0,4529
0,4312
0,4216
0,4212
0,3818
Références
[9]
[9]+[l]
[11]
[10]+[l]
[9]+[l]
[7]
[9]
[12]
[13]
[9]
[9]
[7]
[12]
[1]
[13]
[12]
[9]
[9]
[13]
[1]
[12]
[14]
[9]+[l]
251

Annexe 4
Electrolyte
Zn(NO3)2
Cu(NO3)2
RbNO2
MgCl2
MgBr2
LiNO2
KF
Col2
CaCl2
KAc
Lil
Pb(ClO4)2
CaBr2
LiCl
KOH
ZnBr2
NaOH
LiBr
CsF
msat
6,750
8,031
62,30
5,780
5,610
19,90
17,496
6,500
7,462
23,601
12,178
10,830
7,660
19,974
21,146
20,877
28,535
19,866
?
(aH2o)sat
0,3780
0,3489
0,3359
0,3357
0,3104
0,3063
0,2920
0,2833
0,2830
0,2251
0,1756
0,1468
0,1433
0,1186
0,0951
0,0749
0,0640
0,0489
0,0339
Références
[14]
[7]
[13]
[10]
[10]
[13]
[9]
[12]
[10]+[15]
[1]
[1]
[7]
[10]
[9]
[9]
[14]
[9]
[9]
[1]
252

Annexe 4
BIBLIOGRAPHIE.
[1] L. GREENSPAN,Humidity Fixed Points of Binary Saturated Aqueous Solutions,J. Res. Nat. Bur. St. A. Phys. Chem., 81 A, (1976), 89.
[2] W. LINKE, A SEIDELL,Solubilities, Inorganic and Metal-Organic Compounds, A-Ir, Vol. I, Fourth Edition,D. Van Nostrand Compagny, Inc. Princeton, New Jersey, (1958).
[3] W. LINKE, A SEIDELL,Solubilities, Inorganic and Metal-Organic Compounds, K-Z, Vol. I, Fourth Edition,American Chemical Society, Washington D.C., (1965).
[4] E. SCHOLZ,KARL FISCHER Titration,Springer Verlag, BERLIN, (1984).
[5] A.I. VOGEL,Quantitative Inorganic Analysis, Chapter III, Volumetric (titrimetric) Analysis,Third Edition Longmans, London, (1962).
[6] J. FOOS,Manuel de radioactivité à l'usage des utilisateurs, Tome 2,FORMASCIENCE, ORSAY, (1994).
[7] R. N. GOLDBERG,Evaluated Activity and Osmotic Coefficients for Aqueous Solutions: Bi-UnivalentCompounds of Lead, Copper, Manganese and Uranium,J. Phys. Chem. Ref. Data, 8, (1979), 1005-1050.
[8] R. N. GOLDBERG,Evaluated Activity and Osmotic Coefficients for Aqueous Solutions : Thirty-six Uni-Bivalent Electrolytes,J. Phys. Chem. Ref. Data, 10, (1981), 671-764.
[9] W. J. HAMER and Y. C. WU,Osmotic Coefficients and Mean Activity Coefficients of Uni-Univalent Electrolytes inWater at 25°C,J. Phys. Chem. Ref. Data, 1, (1972), 1047-1099.
[10] R. N. GOLDBERG and R.L. NUTTALL,Evaluated Activity and Osmotic Coefficients for Aqueous Solutions : The AlkalineEarth Metal Halides,J. Phys. Chem. Ref. Data, 7, (1978), 263-310.
253

Annexe 4
[11] W.J.HAMERandY.C.WU,Revised Values of the Osmotic Coefficients and Mean Activity Coefficients of SodiumNitrate in Water at 25 °C (Comments),J. Phys. Chem. Ref. Data, 9, (1980), 513-515.
[12] R. N. GOLDBERG, R. L. NUTALL and B.R. STAPLES,Evaluated Activity and Osmotic Coefficients for Aqueous Solutions : Iron Chloride andthe Bi-Univalent Compounds of Nickel and Cobalt,J. Phys. Chem. Ref. Data, 8, (1979), 923-1003.
[13] B.R. STAPLES,Activity and Osmotic Coefficients of Aqueous Alkali Metal Nitrites,J. Phys. Chem. Ref. Data, 10, (1981), 765-777.
[14] R. N. GOLDBERG,Evaluated Activity and Osmotic Coefficients for Aqueous Solutions : Iron Chloride andthe Bi-Univalent Compounds of Zinc, Cadnium, and EthyleneBis(Trimethylammonium)Chloride and Iodide,J. Phys. Chem. Ref. Data, 10, (1981), 1-55.
[15] B.R. STAPLES and R.L. NUTALLThe Activity and Osmotic Coefficients of Aqueous Calcium Chloride at 298,15 K,J. Phys. Chem. Ref. Data, 6, (1977), 385.
254**.




















