
Model study of the impact of orbital choice on the accuracy of coupled-cluster energies. I....
15
— — < < Model Study of the Impact of Orbital Choice on the Accuracy of Coupled-Cluster Energies. I. Single-Reference-State Formulation K. JANKOWSKI, 1 K. KOWALSKI, 1 K. RUBINIEC, 1 J. WASILEWSKI 2 1 Institute of Physics, Nicholas Copernicus University, 87-100 Torun, Poland ´ 2 Department of Computer Methods, Nicholas Copernicus University, 87-100 Torun, Poland ´ Received 13 January 1997; revised 19 September 1997; accepted 15 October 1997 ABSTRACT: The impact of the choice of molecular orbital sets on the results of Ž . single-reference-state coupled-cluster CC methods was studied for the H4 model. This model offers a straightforward way of taking into account all possible symmetry-adapted orbitals. Moreover, the degree of quasi-degeneracy of its ground state can be varied over a wide range by changing its geometry. The CCD, CCSD, and CCSDT approaches are considered. Surfaces representing the dependence of the energy on the parameters defining the orbitals are obtained. It is documented that for every method there exist alternative orbital sets which allow one to obtain more accurate energies than the Ž . standard HF, BO, and NO ones. However, for many of the former orbital sets, one obtains relatively large one-body amplitudes or one may encounter problems with solving the CC equations by conventional methods. An interesting variety of orbitals which might be useful for studies of quasi-degenerate states by the CCD method was found. Q 1998 John Wiley & Sons, Inc. Int J Quant Chem 67: 205]219, 1998 Key words: coupled-cluster theory; electron correlation effects; molecular orbital choice; reference determinant choice; quasi-degenerate states Correspondence to:K. Jankowski. Contract grant sponsor: Polish Committee for Scientific Re- Ž . search KBN . Contract grant number: 2P30301407. Contract grant sponsor: Rector of the Nicholas Copernicus University. ( ) International Journal of Quantum Chemistry, Vol. 67, 205 ]219 1998 Q 1998 John Wiley & Sons, Inc. CCC 0020-7608 / 98 / 040205-15
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Model study of the impact of orbital choice on the accuracy of coupled-cluster energies. I....

— —< <
Model Study of the Impact of OrbitalChoice on the Accuracy ofCoupled-Cluster Energies. I.Single-Reference-State Formulation
K. JANKOWSKI,1 K. KOWALSKI,1 K. RUBINIEC,1 J. WASILEWSKI2
1Institute of Physics, Nicholas Copernicus University, 87-100 Torun, Poland´2Department of Computer Methods, Nicholas Copernicus University, 87-100 Torun, Poland´
Received 13 January 1997; revised 19 September 1997; accepted 15 October 1997
ABSTRACT: The impact of the choice of molecular orbital sets on the results ofŽ .single-reference-state coupled-cluster CC methods was studied for the H4 model. This
model offers a straightforward way of taking into account all possible symmetry-adaptedorbitals. Moreover, the degree of quasi-degeneracy of its ground state can be varied overa wide range by changing its geometry. The CCD, CCSD, and CCSDT approaches areconsidered. Surfaces representing the dependence of the energy on the parametersdefining the orbitals are obtained. It is documented that for every method there existalternative orbital sets which allow one to obtain more accurate energies than the
Ž .standard HF, BO, and NO ones. However, for many of the former orbital sets, oneobtains relatively large one-body amplitudes or one may encounter problems withsolving the CC equations by conventional methods. An interesting variety of orbitalswhich might be useful for studies of quasi-degenerate states by the CCD method wasfound. Q 1998 John Wiley & Sons, Inc. Int J Quant Chem 67: 205]219, 1998
Key words: coupled-cluster theory; electron correlation effects; molecular orbitalchoice; reference determinant choice; quasi-degenerate states
Correspondence to: K. Jankowski.Contract grant sponsor: Polish Committee for Scientific Re-
Ž .search KBN .Contract grant number: 2P30301407.Contract grant sponsor: Rector of the Nicholas Copernicus
University.
( )International Journal of Quantum Chemistry, Vol. 67, 205]219 1998Q 1998 John Wiley & Sons, Inc. CCC 0020-7608 / 98 / 040205-15

JANKOWSKI ET AL.
Introduction
he formulations of the vast majority of meth-T ods of the many-electron theory of atoms andmolecules are based on the use of orbitals, definedin terms of finite sets of primitive basis functions.The latter determine certain standards of accuracyfor the energy and wave function called ‘‘basis setlimits,’’ which can be obtained by performing full
Ž .configuration interaction FCI calculations. Prop-erties calculated at this level of accuracy are inde-pendent of the choice of orbitals. Unfortunately,FCI results can only be obtained for small systemswhen using very limited basis sets. For the de-scription of realistic systems, approximate meth-ods, far less demanding than the FCI one, have tobe employed.
In implementations of individual approximatemethods of the many-electron theory, the choice oforbitals may be of crucial importance, which iscaused mainly by the following factors: First—themethodological factor: By a proper choice of theorbitals, the formal structure of the equations ofthe theory as well as the final results may besimplified, e.g., the use of HF orbitals in many-
Ž . w xbody perturbation theory MBPT 1 eliminatesmany of the possible diagrams, whereas the useof single-excitation-adapted molecular orbitalsŽ . w xSEAMO 2 causes a reduction of the number ofsignificant configurations in the description of ex-cited states dominated by singly excited configura-tions. Second—the practical factor: The accuracy ofthe desired result may strongly depend on theorbitals employed, e.g., by a proper choice of or-bitals, one may minimize the energy obtainedwithin the CI method restricted to all singly and
Ž .doubly excited configurations CISD for a givenw xprimitive basis set 3 or obtain reliable values ofw xenergy differences 4 . Third—the numerical fac-
tor: The choice of the orbital sets may influence theconvergence characteristics of the numerical proce-dures employed for solving the working equationsof individual methods.
The factors just mentioned have resulted in thatthe difficult problem of generating orbitals that areoptimum from certain points of view has attractedconsiderable interest also among researchers de-
Ž . w xveloping coupled-cluster CC -type approaches 5Ž .both within the single-reference SR and multiref-
Ž . Žerence MR formulations extensive reviews of thepresent situation and references can be found inw x.6 . At early stages of the development of the CCtheory, its computational implementations werealmost exclusively based on reference states de-
Ž .fined in terms of the restricted Hartree]Fock RHForbitals. This situation is caused mainly by the factthat attention was focused on closed-shell groundstates where the RHF approach provides a reason-able starting point for more accurate methods.However, even for these states, there has beenattempts to make alternative choices of the refer-
Ž w x.ence-state orbitals see, e.g., 7 . So far, most atten-tion has been attracted to the Brueckner-type
Ž . w xorbitals BO 8 , which yield the reference deter-minant of maximum overlap with the FCI wavefunction and allow one to suppress the role ofone-electron cluster amplitudes. The latter fact isemployed to eliminate these amplitudes from theworking equations which simplifies their formalstructure. This simplification is to a large degreepaid off by the additional numerical effort re-quired for the generation of BOs. Computationalstudies along this line were pioneered by Chiles
w x w xand Dykstra 9 as well as Purvis and Bartlett 10and have been continued by the Cambridge groupŽ w x.for details and references, see 11 .
Another interesting class of reference-state or-bitals that have been employed in CC studies ofclosed-shell ground states are localized orbitalsŽ .LO not derivable by localization of canonical HF
w xorbitals. It was demonstrated by Laidig et al. 12that when taking LOs that are simple linear combi-nations of hybrid atomic orbitals, which yieldrather poor single-determinant energies, one canobtain CCSD energies very close to those obtainedfor RHF orbitals. LOs later were used by several
Ž w x.groups in CC-type studies see, e.g., 13, 14 . Veryrecently, the problem of generating LOs to be usedin CC approaches was undertaken again by the
w xGainesville group 15 who focused their attentionon orthogonalized Hartree product orbitals.
The significance of the problem of choosingorbitals best suited for effective CC calculations
Žseems to increase when non-closed-shell i.e.,.quasi-degenerate and open-shell states are consid-
ered. This situation is caused mainly by the factthat for these states HF-type procedures may nolonger provide orbitals disclosing the desirableproperties. The list of orbitals currently employedis rather long. It includes, first of all, the RHF and
Ž .unrestricted HF UHF orbitals. The latter ones
VOL. 67, NO. 4206

ACCURACY OF COUPLED-CLUSTER ENERGIES. I
have been used in SR-CC calculations for open-w xshell systems for almost 20 years 16 . The for-
malisms based on UHF reference functions haveŽtheir advantages and shortcomings for a relevant
w x.discussion, see 17 . The most serious limitation ofthe UHF-based CC methods seems to be theirinability to handle other than high-spin states. Theproblems encountered when using UHF orbitalshave resulted in that they are often replaced by
Ž . w xrestricted-open-shell HF ROHF 17 or quasi-re-Ž . w xstricted HF QRHF 17 orbitals. The later are
generated by an RHF method for a near-lyingclosed-shell state. As recently shown by Li and
w xPaldus 18 , the ROHF reference functions maybecome doublet instable in certain regions of thepotential energy surface, which may cause theneed of changing the orbitals when proceeding toanother region.
Relatively little is known about the impact ofthe orbital choice on the performance of MR-CCmethods. For the valence-universal CC approachesw x19 , some attempts to explore this problem were
w xundertaken by Haque and Kaldor 20 andw xJankowski et al. 21 . Recently Balkova and Bartlett
w x22 addressed the same problem for the statew xuniversal CC theory 23 .
As one can see from this short review, a consid-erable experience on choosing orbitals for CC cal-culations has been already collected as a result ofthe effort of several groups when studying theeffectiveness of various orbitals for a variety ofmany-electron systems. It seems to us that thisexperience would be further enriched if all theorbitals discussed so far were employed for thesame system. Since the realization of such a projectfor systems of practical interest may encountervarious barriers, including the financial one, itmight be worthwhile to perform comparative stud-ies at least for model systems.
The object of this article was to present resultsof comprehensive model studies of the impact ofthe orbital choice on the results of several SR-CCapproaches formulated in terms of these orbitals.One of the problems addressed is whether thereexist orbital sets that provide interesting alterna-tives to the traditional canonical HF ones. We
w xemployed the H4 model 24 which offers theunique possibility of simply representing all possi-ble orbital sets. The CCD, CCSD, and CCSDT ap-proaches are considered. Special attention was paidto the dependence of the accuracy attainable bythe individual approaches on the orbital set em-
ployed. Some considerations are also given to thedependence of the structure of the relevant sets ofcluster amplitudes on the approximation imposedon the cluster operator.
Model and Orbital Description
There exists a small model system especiallysuitable for studies of the impact of the choice oforbital sets on the performance of various methodsbecause for it one can define any orbital set by anappropriate choice of a small number of parame-ters. This model, referred to as the H4 model, wasintroduced by Paldus and one of the present au-
w xthors 24 and has since been used by severalŽ w x.authors see, e.g., 25 in reliability studies of
various CC and many-body perturbation theoryapproaches. The H4 model consists of two stretchedhydrogen molecules in an isosceles trapezoidal ar-
Ž .rangement see Fig. 1 . All nearest-neighbor inter-nuclear separations are fixed at 2 au, while the
1Ž .angle f varies in the interval f g 0, p . The2
model is characterized by the parameter a defin-ing the angle f s ap . By varying this parameter,it is possible to continuously change the degree ofquasi-degeneracy of the ground state from a fully
Ž .degenerate case a s 0.0 to a nondegenerate caseŽ .a s 0.5 . Although the model system consideredis relatively small, it is known to epitomize manyof the essential difficulties encountered in quan-tum chemical computations.
Ž .We employ the same minimum basis set MBSw xas in 24 . The orbital space may be spanned by
four real MOs whose spatial symmetry group is
FIGURE 1. Nuclear configurations and definition of themodel parameter a for the H4 model.
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 207

JANKOWSKI ET AL.
C . We shall assume that the reference wave2 v< :function F for the ground state is defined in
terms of doubly occupied symmetry-adapted MOs.Let us notice, however, that instead of these MOsone may employ basis sets consisting of orbitalswithout definite symmetry characteristics, as, e.g.,
w xlocalized orbitals 26 . The latter can be obtainedby appropriate unitary transformations of the for-mer ones.
For H4, one has two orbitals of the irreducibleŽ .representation irrep a , which can be written in1
terms of Gaussian functions x , centered at atom kkŽ .see Fig. 1 , as
a aŽ . aŽ .w s c x q x q d x q xi i 1 4 i 2 3
Ž . Ž .i s 1, 2 1
and two orbitals of the irrep b ,2
b b Ž . b Ž .w s c x y x q d x y xi i 1 4 i 2 3
Ž . Ž .i s 1, 2 . 2
We assume that i s 1 for the orbital correspondingto the lower expectation value of the one-electronHamiltonian. Hence, the reference wave functionof definite symmetry for the ground state is de-fined as
a a b b< : < < Ž .F s w w w w . 31 1 1 1
The normalization and orthogonality conditionsresult in that for each irrep all four coefficients in
Ž . Ž .Eqs. 1 and 2 can be expressed in terms of aŽ .single parameter. For the reference function 3 , it
is convenient to use the parameters
a a b b Ž .x s d rc and x s d rc 4a 1 1 b 1 1
for the a and b irreps, respectively. Varying1 2Ž .these parameters in the range 0, ` allows one to
define a vast variety of orbital set for the H4model. Hence, for this model, every reference
Ž .function 3 as well as the relevant set of MOs areŽ .defined by a point of the x , x plain.a b
Ž .In Table I, the x , x points are given fora bseveral orbitals employed so far in quantum chem-ical calculations. These orbitals include the stan-dard HF orbitals both for the 1a2 2b2 configuration,1 2which has been used in most studies using the H4model, and for the 1a2 2 a2 configuration employed1 1
w xin our calculations for excited states 27 . Bothconfigurations just mentioned define the MCSCForbitals shown in the table. The table also includes
TABLE IValues of the x and x parameters for orbitals,a bused in previous quantum chemical studies.
ax and x parametersa b
Orbitals a = 0.005 a = 0.1 a = 0.5
2 2 b( )HF 1a 1b 1.038 1.511 1.6261 20.983 0.823 0.823
Brueckner 1.053 1.506 1.6830.973 0.835 0.794
Natural 1.051 1.506 1.6820.974 0.823 0.793
MCSCF 1.088 1.693 1.8970.948 0.754 0.727
cKohn]Sham 1.070 1.691 2.0060.977 0.766 0.699
dg-Hartree 1.038 1.608 2.0040.980 0.747 0.653
2 2( )HF 1a 2a 1.040 1.757 3.7371 10.968 0.599 0.437
aFor each orbital, the x parameters are listed above the xa bparameters.bOrbital configuration given in parentheses.cOrbitals for the BLYP potential.d [ ]See 27 .
w xthe ‘‘exact’’ BOs 8]10 obtained for the FCI func-Ž . w xtion, the natural orbitals NO 28 , and the
w xKohn]Sham orbitals 29 . The latter orbitals wererecently used in the MR-CI calculations by Warkenw x30 . The BO, NO, and Kohn]Sham orbitals weregenerated for the ground state by means of the
w xGAUSSIAN-92 system of programs 31 . Finally,we also include in Table I the g-Hartree orbitals
w xdefined by Dietz et al. 32 which were later rec-w xommended by Dietz and Hess 33 to be used in
fast-converging configuration interaction calcula-tions. The g-Hartree orbitals were obtained whenusing the program developed by those authors.
We note from the results of Table I that theform of the individual orbitals depends on thegeometry of the system. For a s 0.005, all orbitalsconsidered are very similar. They are determinedby points belonging to the small rectangle definedby 1.038 F x F 1.088 and 0.948 F x F 0.983. Thisa b
similarity seems to be a consequence of the factthat the geometry considered is close to that for
Ž .a s 0 the square arrangement when the orbitalsare determined by symmetry and x s x s 1. Fora ba s 0.1 and 0.5, the differences between the indi-vidual orbitals are more pronounced, i.e., the dis-
VOL. 67, NO. 4208

ACCURACY OF COUPLED-CLUSTER ENERGIES. I
Ž .persion of the x , x points defining the individ-a bual orbitals is larger, e.g., for a s 0.5, one finds1.625 F x F 3.737. As may be seen in the table,a
Ž 2 2 .the points corresponding to the HF 1a 1b ,1 2Brueckner, and natural orbitals are very close toeach other. Let us remind the reader that theseorbitals provide single-configuration descriptionsof the ground state that are optimum with respectto various criteria. We shall refer to these orbitalsas standard orbitals. Another group of close pointsdefines the MCSCF, Kohn]Sham, and g-Hartreeorbitals, which we shall term second-group orbitals.Let us make the observation that in the weakquasi-degeneracy region the Kohn]Sham and g-Hartree orbitals are fairly well alike. Finally, the
Ž 2 2 .points representing the HF 1a 2 a orbitals are1 1rather far away from the points belonging to thetwo groups just mentioned, which is caused by thefact that the 1a2 2 a2 configuration dominates in1 1excited states and is of little importance in theground-state wave function.
Outline of SR-CC Theory andComputational Aspects
The SR-CC theory has been presented in detailŽ w x.in the literature see, e.g., 5, 6, 16 . For the sake of
completeness, let us recall the essential ideas andnotation relevant to the subject of this note. In the
< :SR-CC method, the exact wave function C isobtained from the independent particle reference
< :wave function F by means of the exponentialwave operator:
< : T < : Ž .C s e F , 5
with the cluster operator T defined in terms ofk-electron components T ask
N
Ž .T s T , 6Ý kks1
where N designates the number of electrons in thesystem.
In the second quantization formalism, T iskgiven in the form
y2 a ??? a r ??? r1 k 1 kŽ . Ž .T s k ! T e , 7k r ??? r a ? ? ? a1 k 1 k
with
r1 ??? rk q q Ž .e s X ??? X X ??? X , 8a ??? a r r a a1 k 1 k k 1
where antisymmetric cluster amplitudes are em-ployed and the Einstein summation convention isimplied. The indices a and r denote the hole andi i
< :particle spin]orbital states defined by F , respec-Ž .tively. Substituting Eq. 5 into the Schrodinger¨
Žequation, and eliminating disconnected terms cf.w x.e.g., 5, 6 , we find that
Ž T . < : < : Ž .H e F s De F , 9N C
² < < :where De s E y E , E s F H F , H s H y E0 0 N 0is the Hamiltonian in the normal product formw x5, 6 , and C indicates the connected part of theoperator product. The energy De is obtained by
Ž .the projection of Eq. 9 on the reference wavefunction,
² <Ž T . < :De s F H e FN C
12² < < : Ž .s F H T q T q T F , 10N 1 2 1ž /2
whereas the cluster amplitudes satisfy the systemof the equation
²r1 ??? rk <Ž T . < :H e F s 0a ??? a N C1 k
Ž . Ž .k s 1, 2, . . . , N , 11
where all possible excited configurations
<r1 ??? rk : r1 ??? rk < : Ž .s e F , 12a ??? a a ??? a1 k 1 k
for k s 1, . . . , N, are accounted for. Arranging thecluster amplitudes T a1, T a1 a2 , . . . T a1 ??? aN in some ar-r r r r ??? r1 1 2 1 N
bitrary but fixed order and designating them sim-ply as the unknowns by t and taking into accountjthat for up to two-particle interactions in H oneN
Ž w x.can write see, e.g., 6
4y1T mŽ . Ž . Ž . Ž .H e s m! H T , 13ÝN NC C
ms0
Ž .the system 11 takes the form
a q b t q c t t q d t t tÝ Ý Ýi i j j i jk j k i jk l j k lj jFk jFkFl
Ž .q e t t t t s 0. 14Ý i jk lm j k l mjFkFlFm
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 209

JANKOWSKI ET AL.
In practical applications, restrictions have to beimposed on the form of the cluster operator which
Ž .consist in retaining in Eq. 6 only few-electronterms. Here, we shall be concerned with the fol-lowing approximate forms:
Ž . Ž .T f T CCD 152
Ž . Ž .T f T q T CCSD 161 2
and
Ž . Ž .T f T q T q T CCSDT . 171 2 3
The set of equations routinely used for definingthe cluster amplitudes for a given approximate
Ž .form can be obtained from the set 11 by takinginto account the equations corresponding to pro-jections only on such excited configuration whichare used in the given approximation of T and byomitting terms including amplitudes not consid-ered in this approximation. The most drastic ap-proximation is implemented at the CCD level
Ž .where the set 14 contains only up to quadraticterms.
The present numerical studies are based on ourcomputer code for performing up to CCSDTQ cal-
w xculations for small systems 27 . The set of nonlin-ear equations for the cluster amplitudes is solvedby means of a Newton]Raphson-type algorithmw x34 . Considerable attention is paid to the choice ofappropriate starting approximations, which mini-mizes convergence problems and makes it possibleto obtain both standard and nonstandard solutionsw x35 . To examine the accuracy of the latter, they are
Ž .used to calculate the left-hand sides of the set 14 .In all cases presented below, numerical zeros wereobtained for all equations of the set.
Results and Discussion
Taking advantage of the possibility of definingarbitrary symmetry-adapted orbital sets for the H4model by means of two parameters x and x , wea bapplied the CCD, CCSD, and CCSDT methodsdefined by a large variety of orbitals to the groundstate for several geometries of the H4 model. Theseorbitals are defined by points of a two-dimensionalgrid with 15 points ranging from 0.001 to 100 ineach of the x and x directions. In the ensuinga btext, this grid is referred to as an orbital grid. It
seems to us that the area covered by this grid issufficient for defining reference states suitable fora reliable description of the ground state. An indi-cation of this fact may be the expectation value ofthe Hamiltonian calculated for reference functionsdefined by points from the outer regions of thearea. These values are close to the energies of thesecond and third excited states, e.g., for a s 0.005
Ž .and the point 0.1, 0.1 , one obtains 0.561H,whereas E s 0.447H and E s 0.644H. Hence, for3 4the reference functions represented by points fromthe outer regions, the results for the ground statedo not correspond to the standard solutions of the
w xCC equations 35 .We present in the tables only a small subset of
the results that has been chosen to represent theessential features of the whole set. However, theenergy surfaces shown in Figures 2]5 are obtainedwhen using the complete set of energies. We shallrefer to the neighborhood of the points on the x xa b
plane corresponding to the standard orbitals as the‘‘standard region.’’ Our attention is concentratedon two geometries of the H4 model for which the
Ž .ground state is strongly a s 0.005 and weaklyŽ .a s 0.5 quasi-degenerate. The standard regionfor a s 0.005 and a s 0.5 are close to the grid
Ž . Ž .points 1.0, 1.0 and 1.5, 1.0 , respectively.This section is devoted mainly to the presenta-
tion of the ground-state energies. Limitations ofspace preclude a satisfactory discussion of thestructure of the cluster operator. We shall only beconcerned with the maximum magnitudes of theamplitudes of individual T operators. We use thei
following notation to denote these amplitudes:
< a1 ??? ai < Ž .t s max T . 18� 4i r ??? r1 i
Ž .All energies are given relative the full CI FCIvalues, which represent the exact results for themodel.
To provide some reference points for the analy-sis of the results obtained for the orbitals definedby the orbital grid, we display in Table II the CCD,CCSD, and CCSDT energies and the correspondingt values obtained for the orbitals presented ini
second section. For the strong quasi-degeneracyregion, the calculated energies corresponding tovarious orbitals and all versions of the CC methoddiffer very little and the t and t values remain2 3almost constant. Only the very small t ampli-1tudes vary within the range 0.0]0.0066. Notice that
VOL. 67, NO. 4210

ACCURACY OF COUPLED-CLUSTER ENERGIES. I
TABLE II( )Differences of the CC and FCI energies in mH and maximum cluster amplitudes for several orbital sets
obtained for the ground state of the H4 model.
a = 0.005 a = 0.5a 4 a 4( ) ( )Orbitals DE t / t =10 t DE t / t =10 t1 3 2 1 3 2
2 2( )HF 1a 1b CCD y2.159 .856 0.118 .1961 2CCSD y2.167 29. .856 0.001 71. .200CCSDT y2.169 27./ 11. .856 y0.056 69./ 35. .200
BO CCD y2.165 .856 y0.006 .200CCSD y2.165 0.0 .856 y0.006 0.2 .200CCSDT y2.169 1.1 / 11. .856 y0.056 3.9 / 36. .200
NO CCD y2.165 .856 y0.006 .200CCSD y2.165 3.3 .856 y0.006 2.6 .200CCSDT y2.169 2.1 / 11. .856 y0.056 2.3 / 36. .200
MCSCF CCD y2.115 .858 0.800 .212CCSD y2.161 65. .856 y0.028 244. .201CCSDT y2.171 66./ 10. .856 y0.057 248./ 39. .200
Kohn-Sham CCD y2.146 .857 1.689 .217CCSD y2.163 44. .856 y0.040 353. .201CCSDT y2.169 45./ 10. .856 y0.058 358./ 41. .200
g-Hartree CCD y2.159 .856 2.601 .221CCSD y2.167 30. .856 y0.056 360. .201CCSDT y2.169 29./ 11. .856 y0.059 361./ 42. .200
2 2( )HF 1a 2a CCD y2.165 .856 26.0 .2751 2CCSD y2.165 34. .856 y0.293 1355. .208CCSDT y2.169 33./ 11. .856 y0.070 1363./ 57. .206
aThe FCI energies amount to y1.980593 and y2.168926 H for a = 0.005 and 0.5, respectively.
in this region the BOs and NOs yield at the CCDŽ 2 2 .level less accurate results than for the HF 1a 1b ,1 1
MCSCF, Kohn]Sham, and g-Hartree orbitals.For the weakly quasi-degenerate ground state
Ž .a s 0.5 , the energies depend both on the CCmethod and the orbitals considered. The CCD en-ergies are very accurate for the standard orbitals.Unlike the situation found in the strong quasi-de-generacy region, the accuracy is especially high forthe BO and NO orbitals. Due to the smallness ofthe t amplitudes, the CCD and CCSD results are1practically equal. Notice that for these orbitalsproceeding to the CCSDT method causes a littleloss of accuracy. For the second-group orbitalsdefined in the second section, the CCD energiesdisclose an error of the order of 1 milliHartreeŽ .mH . The inclusion of the T operator results in a1reduction of the energy deviation by a factor of 40.This reduction is accompanied by a change of t2amounting up to 10%. The resulting t values are2very close to their equivalents obtained for the firstgroup of orbitals. One can also see that the t1
values for the second-group orbitals are muchlarger than for the standard one. These results canbe rationalized by means of the Thouless theoremw x36 , which states that any two nonorthogonal de-
< : < :terminantal wave functions F and F9 are mu-tually related in the following way:
< : T1 < : Ž .F9 s e F . 19
Now, the CCSD wave function defined for the< :reference state F can be represented as a CCD
< :wave function for a modified reference state F9 :
T1qT 2 < : T2 T1 < : T2 < :C s e F s e e F s e F9 « C .CCSD CCD
Ž .20
Hence, the main effect of T is to replace the1original reference state by a better suited one. As aresult, we find that for the second-group orbitalsthe significant improvement of the CCSD energytakes place for relatively large t amplitudes. The1
Ž 2 2 .results for the HF 1a 2 a orbitals shown in Table1 1II disclose a behavior similar to their second-group
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 211
JANKOWSKI ET AL.
TABLE IIIDifferences of the CC energies and FCI energya obtained for various orbital sets for the ground state of the
( )H4 model for a = 0.005 in mH .
xb
x Method 0.001 0.1 0.5 1.0 1.5 2.0 5.0 100.0a
0.001 CCD 33.4 36.5 51.3 58.7 55.4 52.6 52.6 58.5CCSD 0.81 y0.10 y1.98 y3.26 y4.08 y4.66 y6.09 y7.25CCSDT 0.35 0.33 y0.81 y2.39 y3.66 y4.66 y7.73 y11.5
0.1 CCD 37.6 39.3 49.0 50.7 45.2 42.9 48.0 58.3CCSD y0.09 y0.69 y1.98 y2.97 y3.67 y4.17 y5.49 y6.65CCSDT 0.35 0.12 y1.04 y2.32 y3.31 y4.08 y6.47 y9.46
0.5 CCD 56.5 53.1 40.3 16.6 12.7 18.8 44.2 63.2CCSD y1.75 y1.81 y2.01 y2.37 y2.71 y2.99 y3.84 y4.74CCSDT y0.76 y1.01 y1.68 y2.20 y2.58 y2.86 y3.74 y4.89
1.0 CCD 66.0 58.4 22.2 y2.08 8.88 23.0 50.2 64.6CCSD y2.74 y2.56 y2.19 y2.17 y2.28 y2.40 y2.85 y3.39CCSDT y2.28 y2.23 y2.16 y2.17 y2.19 y2.22 y2.34 y2.53
1.5 CCD 65.2 55.5 14.9 3.79 20.9 33.8 50.9 59.8CCSD y3.35 y3.07 y2.39 y2.15 y2.12 y2.15 y2.34 y2.60CCSDT y3.44 y3.12 y2.48 y2.16 y2.00 y1.90 y1.66 y1.42
2.0 CCD 62.8 52.7 15.4 13.7 30.7 39.8 49.3 54.8CCSD y3.77 y3.43 y2.56 y2.18 y2.06 y2.02 y2.04 y2.09CCSDT y4.31 y3.79 y2.71 y2.16 y1.87 y1.70 y1.26 y0.82
5.0 CCD 57.3 49.2 30.3 40.4 47.3 47.6 43.6 41.8CCSD y4.84 y4.38 y3.09 y2.36 y2.01 y1.82 y1.37 y0.80CCSDT y6.81 y5.71 y3.37 y2.19 y1.59 y1.23 y0.44 0.20
100.0 CCD 56.3 52.1 48.6 57.6 56.7 52.5 40.8 34.2CCSD y5.80 y5.28 y3.68 y2.63 y2.06 y1.70 y0.73 0.63CCSDT y9.57 y7.85 y4.12 y2.23 y1.33 y0.83 0.09 0.43
aThe FCI energy amounts to y1.980593 H.
equivalents. Here, in turn, the inclusion of T im-3
proves the agreement of the energy with the FCIvalues. Notice that for all orbitals displayed inTable II the t amplitudes take close values.3
The subset of results obtained for selected pointsof the orbital grid are presented in Tables III]V.The energies obtained for the CCD, CCSD, andCCSDT methods for the strongly quasi-degenerate
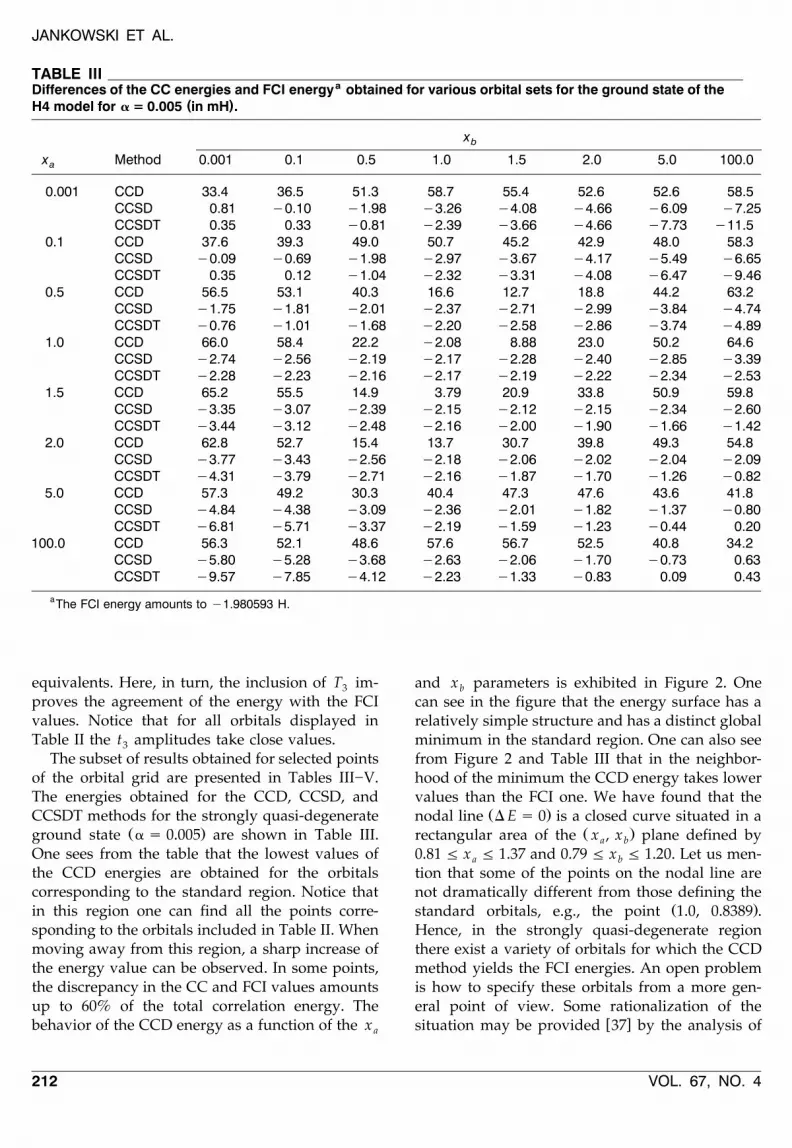
Ž .ground state a s 0.005 are shown in Table III.One sees from the table that the lowest values ofthe CCD energies are obtained for the orbitalscorresponding to the standard region. Notice thatin this region one can find all the points corre-sponding to the orbitals included in Table II. Whenmoving away from this region, a sharp increase ofthe energy value can be observed. In some points,the discrepancy in the CC and FCI values amountsup to 60% of the total correlation energy. Thebehavior of the CCD energy as a function of the xa
and x parameters is exhibited in Figure 2. Oneb
can see in the figure that the energy surface has arelatively simple structure and has a distinct globalminimum in the standard region. One can also seefrom Figure 2 and Table III that in the neighbor-hood of the minimum the CCD energy takes lowervalues than the FCI one. We have found that the
Ž .nodal line D E s 0 is a closed curve situated in aŽ .rectangular area of the x , x plane defined bya b
0.81 F x F 1.37 and 0.79 F x F 1.20. Let us men-a b
tion that some of the points on the nodal line arenot dramatically different from those defining the
Ž .standard orbitals, e.g., the point 1.0, 0.8389 .Hence, in the strongly quasi-degenerate regionthere exist a variety of orbitals for which the CCDmethod yields the FCI energies. An open problemis how to specify these orbitals from a more gen-eral point of view. Some rationalization of the
w xsituation may be provided 37 by the analysis of
VOL. 67, NO. 4212
ACCURACY OF COUPLED-CLUSTER ENERGIES. I
TABLE IVDifferences of the CC energies and FCI energya obtained for various orbital sets for the ground state of the
( )H4 model for a = 0.500 in mH .
xb
x Method 0.001 0.1 0.5 1.0 1.5 2.0 5.0 100.0a
0.001 CCD 403.5 401.8 401.4 417.7 418.2 422.6 460.3 476.34CCSD 1.68 0.80 y1.51 y3.56 y5.35 y6.96 y13.4 y25.2CCSDT 0.05 0.08 0.05 y0.08 y0.22 y0.34 y0.82 y1.75
0.1 CCD 433.2 428.5 393.6 337.2 330.8 342.2 419.0 472.7CCSD 0.33 y0.11 y1.18 y2.15 y3.10 y4.02 y8.17 y16.8CCSDT 0.04 0.04 0.00 y0.09 y0.16 y0.23 y0.49 y1.01
0.5 CCD 289.2 246.4 137.4 98.5 107.7 133.1 274.6 456.1CCSD y0.43 y0.46 y0.42 y0.38 y0.45 y0.58 y1.59 y4.60CCSDT 0.00 y0.02 y0.05 y0.07 y0.08 y0.10 y0.14 y0.22
1.0 CCD 167.5 129.3 38.6 17.0 38.2 72.1 238.5 460.4CCSD y0.43 y0.36 y0.15 y0.02 0.02 0.00 y0.32 y1.53CCSDT y0.03 y0.04 y0.06 y0.06 y0.06 y0.06 y0.06 y0.05
1.5 CCD 127.8 92.8 14.3 3.85 32.4 71.5 250.4 469.3CCSD y0.52 y0.40 y0.11 0.03 0.07 0.07 y0.11 y0.74CCSDT y0.05 y0.06 y0.06 y0.05 y0.05 y0.05 y0.03 y0.01
2.0 CCD 113.5 80.9 10.4 7.16 40.5 82.8 269.3 470.4CCSD y0.65 y0.48 y0.12 0.02 0.06 0.05 y0.09 y0.46CCSDT y0.07 y0.07 y0.06 y0.05 y0.04 y0.04 y0.02 0.02
5.0 CCD 110.1 82.8 31.0 44.0 88.0 137.4 339.0 469.7CCSD y1.25 y0.94 y0.32 y0.10 y0.06 y0.08 y0.20 y0.21CCSDT y0.11 y0.10 y0.07 y0.05 y0.03 y0.02 0.00 0.05
100.0 CCD 132.5 109.6 72.3 97.4 149.0 203.2 411.0 473.0CCSD y2.22 y1.73 y0.70 y0.31 y0.20 y0.19 y0.26 y0.06CCSDT y0.17 y0.14 y0.08 y0.04 y0.02 y0.01 0.02 0.05
aThe FCI correlation energy amounts to y2.168926 H.
the breakdown of the one-electron picture in thestrong-degeneracy region, which is manifested by
w xinstabilities of the HF solutions 38 . It should benoticed that due to the nonvariational character ofthe CCD method the orbitals corresponding tovery good energies do not have to be optimal forother quantities.
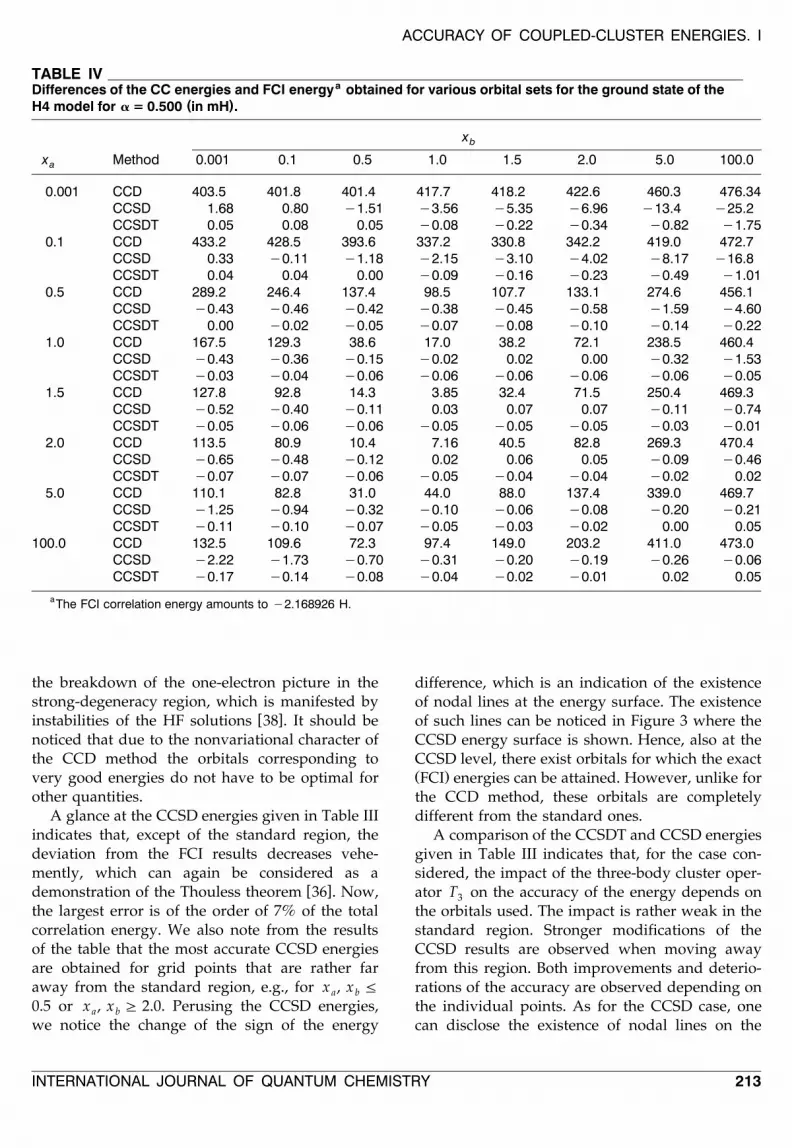
A glance at the CCSD energies given in Table IIIindicates that, except of the standard region, thedeviation from the FCI results decreases vehe-mently, which can again be considered as a
w xdemonstration of the Thouless theorem 36 . Now,the largest error is of the order of 7% of the totalcorrelation energy. We also note from the resultsof the table that the most accurate CCSD energiesare obtained for grid points that are rather faraway from the standard region, e.g., for x , x Fa b
0.5 or x , x G 2.0. Perusing the CCSD energies,a b
we notice the change of the sign of the energy
difference, which is an indication of the existenceof nodal lines at the energy surface. The existenceof such lines can be noticed in Figure 3 where theCCSD energy surface is shown. Hence, also at theCCSD level, there exist orbitals for which the exactŽ .FCI energies can be attained. However, unlike forthe CCD method, these orbitals are completelydifferent from the standard ones.
A comparison of the CCSDT and CCSD energiesgiven in Table III indicates that, for the case con-sidered, the impact of the three-body cluster oper-ator T on the accuracy of the energy depends on3
the orbitals used. The impact is rather weak in thestandard region. Stronger modifications of theCCSD results are observed when moving awayfrom this region. Both improvements and deterio-rations of the accuracy are observed depending onthe individual points. As for the CCSD case, onecan disclose the existence of nodal lines on the
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 213

JANKOWSKI ET AL.
TABLE VMaximum magnitudes of the one- and two-body cluster amplitudes obtained for the CCD and CCSD methods
( )when using various orbital sets for the ground state of the H4 model a = 0.5 .
xb
ax Amplitude 0.001 0.5 1.0 1.5 5.0 100.0a
0.001 t .88 .84 .82 .80 .76 .721t .37 .34 .34 .34 .41 .522Xt 10. 3.9 1.2 .62 1.2 2.52
0.5 t .36 .33 .32 .31 .39 .631t .25 .22 .22 .22 .26 .322Xt .30 .17 .14 .14 .24 .982
1.0 t .33 .14 .12 .13 .42 .661t .23 .21 .20 .21 .24 .302Xt .30 .20 .16 .15 .18 .922
1.5 t .32 .09 .05 .14 .43 .681t .22 .20 .20 .20 .24 .292Xt .32 .22 .18 .17 .18 1.42
2.0 t .31 .09 .06 .15 .44 .691t .22 .20 .20 .20 .24 .292Xt .34 .24 .20 .18 .19 2.12
5.0 t .29 .18 .19 .20 .46 .711t .23 .21 .21 .21 .25 .312Xt .39 .28 .24 .22 .27 4.22
100.0 t .28 .30 .32 .33 .47 .731t .24 .22 .22 .23 .26 .332Xt .44 .32 .29 .27 .11 6.62
a tX and t denote the amplitudes of maximum magnitudes corresponding to the CCD and CCSD methods, respectively.2 2
CCSDT energy surface. These lines are distinctlydifferent from their CCSD counterparts. As in theCCSD case, the orbitals defined by the nodal linesfor the CCSDT approach differ very much fromthe standard orbitals.
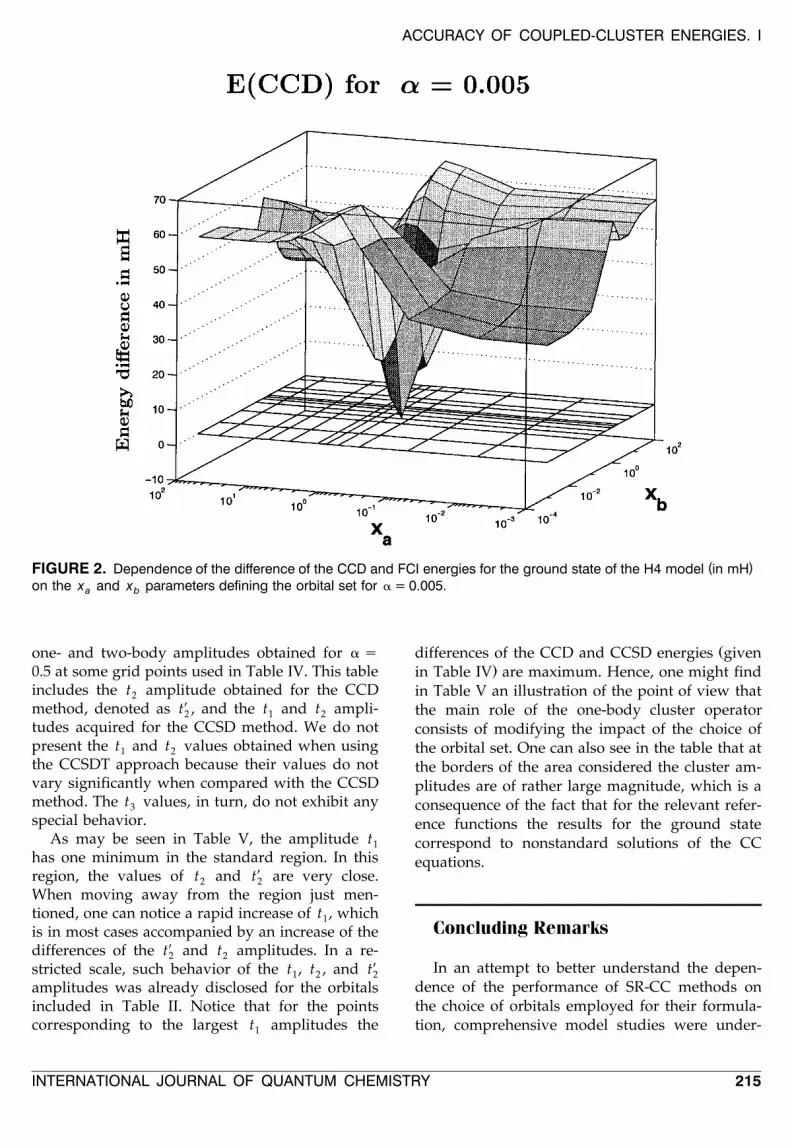
Table IV includes the energies obtained for theorbital subgrid in the case of the weakly quasi-de-
Ž .generate ground state a s 0.5 . As may be seen inthe table and in Figure 4, the error of the energy is
Ž .minimum at the point 1.5, 1.0 close to the stan-dard region. When leaving the neighborhood ofthis point, one can notice a large increase of thedeviation of the energy, which amounts up toabout 900% of the correlation energy. Let us noticethat all CCD energies are larger than the FCI onesand the CCD energy surface has a distinct globalminimum. The accuracy of the energy improvesradically when proceeding to the results of theCCSD method, which again reminds one of thesituation just discussed for the strongly quasi-de-generate case. Comparison of the CCSD energies inTables III and IV indicates that in the weaklyquasi-degenerate region the overall improvement
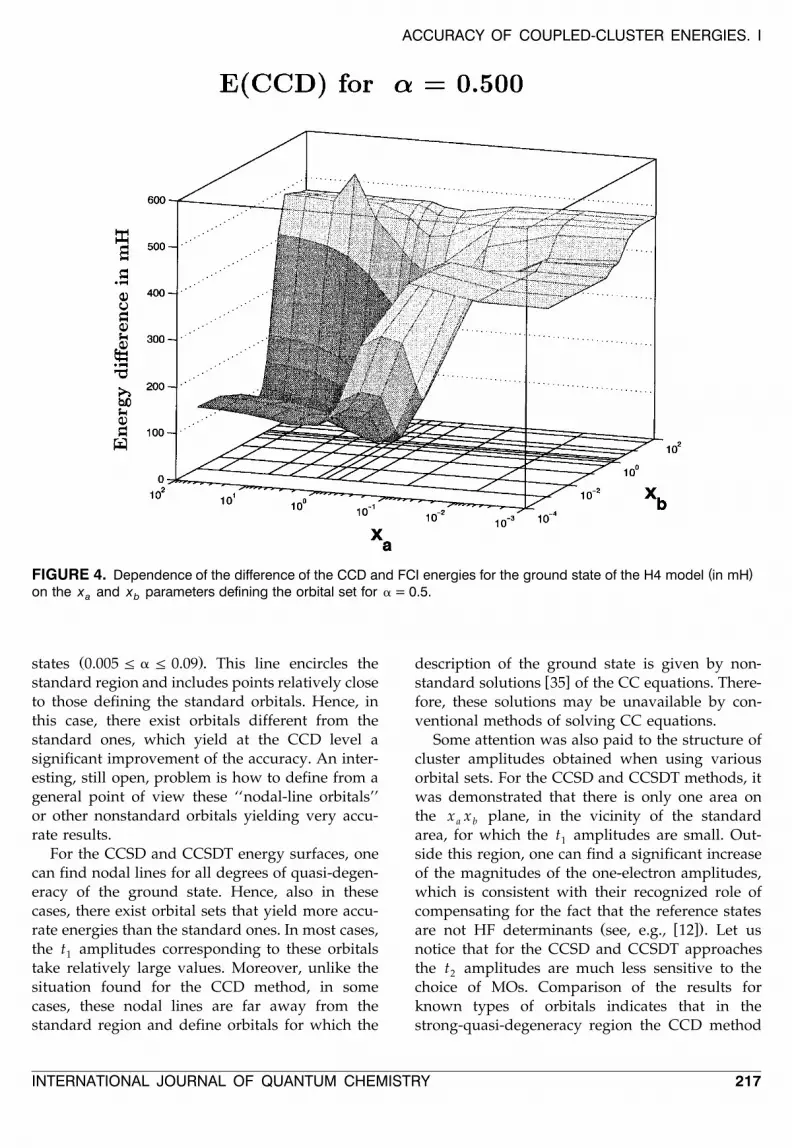
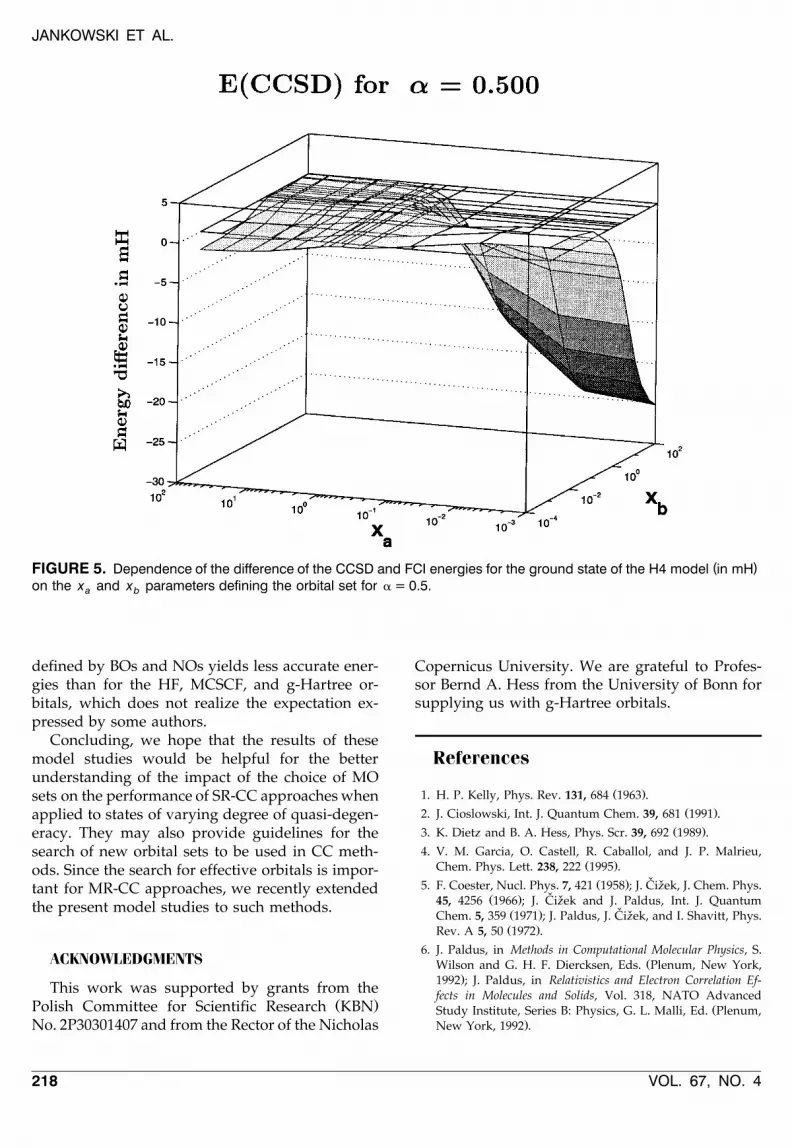
in the present case is more pronounced than in theprevious one. An inspection of the CCSD resultsdiscloses that there are nodal curves at the energysurface. That this is, in fact, the case can also beseen in Figure 5. It is interesting to note that,unlike the situation found for the CCSD method inthe strongly quasi-degenerate region, one of thesecurves passes through the close neighborhood ofthe points defining the standard orbitals.
Comparing the CCSD and CCSDT energiesgiven in Table IV, one can see that the latterdisclose a much better overall agreement with theFCI values than do the former ones. In the domi-
Ž .nating part of the x , x plane, the absolute valuea b
of the discrepancy in the FCI and CCSDT energiesis less than 0.1 mH. The results also indicate thepresence of nodal curves. However, one can seethat for the CCSDT energies they pass throughregions which are rather far away from the stan-dard region.
To partly rationalize the changes of the struc-ture of the energy surfaces obtained for variousCC methods, we present in Table V the maximum
VOL. 67, NO. 4214
ACCURACY OF COUPLED-CLUSTER ENERGIES. I
( )FIGURE 2. Dependence of the difference of the CCD and FCI energies for the ground state of the H4 model in mHon the x and x parameters defining the orbital set for a = 0.005.a b
one- and two-body amplitudes obtained for a s0.5 at some grid points used in Table IV. This tableincludes the t amplitude obtained for the CCD2method, denoted as tX , and the t and t ampli-2 1 2tudes acquired for the CCSD method. We do notpresent the t and t values obtained when using1 2the CCSDT approach because their values do notvary significantly when compared with the CCSDmethod. The t values, in turn, do not exhibit any3special behavior.
As may be seen in Table V, the amplitude t1has one minimum in the standard region. In thisregion, the values of t and tX are very close.2 2When moving away from the region just men-tioned, one can notice a rapid increase of t , which1is in most cases accompanied by an increase of thedifferences of the tX and t amplitudes. In a re-2 2stricted scale, such behavior of the t , t , and tX
1 2 2amplitudes was already disclosed for the orbitalsincluded in Table II. Notice that for the pointscorresponding to the largest t amplitudes the1
Ždifferences of the CCD and CCSD energies given.in Table IV are maximum. Hence, one might find
in Table V an illustration of the point of view thatthe main role of the one-body cluster operatorconsists of modifying the impact of the choice ofthe orbital set. One can also see in the table that atthe borders of the area considered the cluster am-plitudes are of rather large magnitude, which is aconsequence of the fact that for the relevant refer-ence functions the results for the ground statecorrespond to nonstandard solutions of the CCequations.
Concluding Remarks
In an attempt to better understand the depen-dence of the performance of SR-CC methods onthe choice of orbitals employed for their formula-tion, comprehensive model studies were under-
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 215

JANKOWSKI ET AL.
( )FIGURE 3. Dependence of the difference of the CCSD and FCI energies for the ground state of the H4 model in mHon the x and x parameters defining the orbital set for a = 0.005.a b
w xtaken for the MBS H4 system 24 . This modeloffers the unique possibility of defining arbitrarysymmetry-adapted orbitals in terms of two param-eters x and x . The CCD, CCSD, and CCSDTa b
approaches were employed to states disclosingvarious degrees of quasi-degeneracy. Calculationswere performed for a vast variety of MOs definedby nodes of a two-dimensional grid on the x xa b
plane. In addition to these orbitals, we also consid-ered seven MO sets employed so far in quantumchemistry. These sets include the standard RHF,BO, and NO orbitals. The neighborhood of thepoints on the x x plane corresponding to thesea b
orbitals is referred to as the ‘‘standard region.’’Our findings may be conveniently formulated
in terms of the energy surface representing thedependence of the ground-state energy of the H4
Ž .model for a given geometry on the parametersdefining the MOs. It is also interesting to indicatethe positions of nodal lines resulting from the
intersection of the energy surface with the planecorresponding to the FCI energy. For the points ofthe nodal lines, the SR-CC method consideredyields the exact energies.
It was found that the energy surfaces for theCCD method are different from the CCSD sur-faces. The CCSDT surfaces resemble very muchthe latter ones. The CCD surfaces disclose strongvariations for the region of parameters consideredand possess a deep global minimum correspond-ing to a point from the standard region. In turn,the energy surfaces for the CCSD and CCSDTmethods are relatively flat but their structure israther complex. There is no well-pronounced globalminimum.
The structure and position of the nodal linesdepend on the degree of quasi-degeneracy of theground state as well as on the SR-CC methodconsidered. For the CCD approach, a closed nodalline was found for the strongly quasi-degenerate
VOL. 67, NO. 4216
ACCURACY OF COUPLED-CLUSTER ENERGIES. I
( )FIGURE 4. Dependence of the difference of the CCD and FCI energies for the ground state of the H4 model in mHon the x and x parameters defining the orbital set for a = 0.5.a b
Ž .states 0.005 F a F 0.09 . This line encircles thestandard region and includes points relatively closeto those defining the standard orbitals. Hence, inthis case, there exist orbitals different from thestandard ones, which yield at the CCD level asignificant improvement of the accuracy. An inter-esting, still open, problem is how to define from ageneral point of view these ‘‘nodal-line orbitals’’or other nonstandard orbitals yielding very accu-rate results.
For the CCSD and CCSDT energy surfaces, onecan find nodal lines for all degrees of quasi-degen-eracy of the ground state. Hence, also in thesecases, there exist orbital sets that yield more accu-rate energies than the standard ones. In most cases,the t amplitudes corresponding to these orbitals1
take relatively large values. Moreover, unlike thesituation found for the CCD method, in somecases, these nodal lines are far away from thestandard region and define orbitals for which the
description of the ground state is given by non-w xstandard solutions 35 of the CC equations. There-
fore, these solutions may be unavailable by con-ventional methods of solving CC equations.
Some attention was also paid to the structure ofcluster amplitudes obtained when using variousorbital sets. For the CCSD and CCSDT methods, itwas demonstrated that there is only one area onthe x x plane, in the vicinity of the standarda b
area, for which the t amplitudes are small. Out-1
side this region, one can find a significant increaseof the magnitudes of the one-electron amplitudes,which is consistent with their recognized role ofcompensating for the fact that the reference states
Ž w x.are not HF determinants see, e.g., 12 . Let usnotice that for the CCSD and CCSDT approachesthe t amplitudes are much less sensitive to the2
choice of MOs. Comparison of the results forknown types of orbitals indicates that in thestrong-quasi-degeneracy region the CCD method
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 217
JANKOWSKI ET AL.
( )FIGURE 5. Dependence of the difference of the CCSD and FCI energies for the ground state of the H4 model in mHon the x and x parameters defining the orbital set for a = 0.5.a b
defined by BOs and NOs yields less accurate ener-gies than for the HF, MCSCF, and g-Hartree or-bitals, which does not realize the expectation ex-pressed by some authors.
Concluding, we hope that the results of thesemodel studies would be helpful for the betterunderstanding of the impact of the choice of MOsets on the performance of SR-CC approaches whenapplied to states of varying degree of quasi-degen-eracy. They may also provide guidelines for thesearch of new orbital sets to be used in CC meth-ods. Since the search for effective orbitals is impor-tant for MR-CC approaches, we recently extendedthe present model studies to such methods.
ACKNOWLEDGMENTS
This work was supported by grants from theŽ .Polish Committee for Scientific Research KBN
No. 2P30301407 and from the Rector of the Nicholas
Copernicus University. We are grateful to Profes-sor Bernd A. Hess from the University of Bonn forsupplying us with g-Hartree orbitals.
References
Ž .1. H. P. Kelly, Phys. Rev. 131, 684 1963 .Ž .2. J. Cioslowski, Int. J. Quantum Chem. 39, 681 1991 .
Ž .3. K. Dietz and B. A. Hess, Phys. Scr. 39, 692 1989 .
4. V. M. Garcia, O. Castell, R. Caballol, and J. P. Malrieu,Ž .Chem. Phys. Lett. 238, 222 1995 .
ˇŽ .5. F. Coester, Nucl. Phys. 7, 421 1958 ; J. Cizek, J. Chem. Phys.ˇˇŽ .45, 4256 1966 ; J. Cizek and J. Paldus, Int. J. Quantumˇ
ˇŽ .Chem. 5, 359 1971 ; J. Paldus, J. Cizek, and I. Shavitt, Phys.ˇŽ .Rev. A 5, 50 1972 .
6. J. Paldus, in Methods in Computational Molecular Physics, S.ŽWilson and G. H. F. Diercksen, Eds. Plenum, New York,
.1992 ; J. Paldus, in Relativistics and Electron Correlation Ef-fects in Molecules and Solids, Vol. 318, NATO Advanced
ŽStudy Institute, Series B: Physics, G. L. Malli, Ed. Plenum,.New York, 1992 .
VOL. 67, NO. 4218

ACCURACY OF COUPLED-CLUSTER ENERGIES. I
7. L. Z. Stolarczyk and H. J. Monkhorst, Int. J. QuantumŽ .Chem. S 18, 267 1984 .
8. K. A. Brueckner, C. A. Levinson, and H. M. Mahmoud,Ž .Phys. Rev. 95, 215 1954 .
9. R. A. Chiles and C. E. Dykstra, J. Chem. Phys. 74, 4544Ž .1981 .
10. G. D. Purvis III and R. J. Bartlett, J. Chem. Phys. 76, 1910Ž .1982 .
11. R. Kobayashi, R. D. Amos, and N. C. Handy, J. Chem. Phys.Ž .100, 1375 1994 .
12. W. D. Laidig, G. D. Purvis, and R. J. Bartlett, Int. J. Quan-Ž .tum Chem. Symp. 16, 561 1982 ; Ibid., Chem. Phys. Lett. 97,
Ž . Ž .209 1983 ; Ibid., J. Phys. Chem. 89, 2161 1985 .Ž .13. S. Saebo and P. Pulay, Chem. Phys. Lett. 131, 384 1984 ; H.
Ž .Sellers, S. Saebo, and P. Pulay, Ibid. 132, 29 1986 .Ž .14. W. Forner, Chem. Phys. 114, 21 1987 .¨
15. S. A. Perera, D. E. Bernholdt, and R. J. Bartlett, Int. J.Ž .Quantum Chem. 49, 559 1994 ; Ibid., J. Chem. Phys. 97,
Ž . Ž .7600 1992 ; Ibid., Int. J. Quantum Chem. 48, 59 1993 .16. R. J. Bartlett and G. D. Purvis, Int. J. Quantum Chem. 14,
Ž .561 1978 ; J. A. Pople, R. Krishnan, H. B. Schlegel, and J. S.Ž .Binkley, Int. J. Quantum Chem. 14, 545 1978 .
Ž .17. M. Rittby and R. J. Bartlett, J. Phys. Chem. 92, 3033 1988 .Ž .18. X. Li and J. Paldus, J. Chem. Phys. 102, 2013 1995 .
Ž .19. B. Jeziorski and J. Paldus, J. Chem. Phys. 90, 2714 1989 .Ž .20. A. Haque and U. Kaldor, Chem. Phys. Lett. 120, 261 1985 .
21. K. Jankowski, J. Paldus, I. Grabowski, and K. Kowalski,Ž . Ž .J. Chem. Phys. 97, 7600 1992 ; Ibid. 101, 3085 1994 .
Ž .22. A. Balkova and J. Bartlett, J. Chem. Phys. 101, 8972 1994 ;Ž .Ibid. 102, 7116 1995 .
23. B. Jeziorski and H. J. Monkhorst, Phys. Rev. A 24, 1668Ž .1981 .
24. K. Jankowski and J. Paldus, Int. J. Quantum Chem. 18, 1243Ž .1980 .
25. S. Wilson, K. Jankowski, and J. Paldus, Int. J. QuantumŽ .Chem. 23, 1781 1983 ; P. Pulay, Int. J. Quantum Chem.
Ž .Symp. 17, 257 1983 ; U. Kaldor, Int. J. Quantum Chem. 28,Ž .103 1985 ; S. Wilson, K. Jankowski, and J. Paldus, Ibid. 28,Ž . Ž .525 1985 ; N. Iijima and A. Saika, Ibid. 27, 481 1985 ; S.
Zarrabian and J. Paldus, Int. J. Quantum Chem. 38, 761Ž .1990 ; A. Balkova, S. A. Kucharski, L. Meissner, and R. J.
Ž .Bartlett, Theor. Chim. Acta 80, 335 1991 ; J. P. Finley, R. K.Ž .Chaudhuri, and K. F. Freed, J. Chem. Phys. 103, 4990 1995 .Ž .26. J. M. Foster and S. F. Boys, Rev. Mod. Phys. 32, 300 1960 ;
Ž .C. Edmiston and K. Ruedenberg, Ibid. 35, 457 1963 .27. K. Jankowski, K. Kowalski, and P. Jankowski, Chem. Phys.
Ž .Lett. 222, 608 1994 .Ž .28. P. O. Lowdin, Phys. Rev. 97, 1474 1955 .¨
Ž .29. W. Kohn and L. J. Sham, Phys. Rev. 140 A 1133 1965 .Ž .30. M. Warken, Chem. Phys. Lett. 237, 256 1995 .
31. M. J. Frisch, G. W. Trucks, M. Head-Gordon, P. M. W. Gill,M. W. Wong, J. B. Foresman, B. G. Johnson, H. B. Schlegel,M. A. Robb, E. S. Replogle, R. Gomperts, J. L. Andres, K.Raghavachari, J. S. Binkley, C. Gonzalez, R. L. Martin, D. J.Fox, D. J. Defrees, J. Baker, J. J. P. Stewart, and J. A. Pople,
ŽGAUSSIAN 92, Revision E.2 Gaussian, Inc., Pittsburgh,.1992 .
32. K. Dietz, O. Lichtenfeld, and G. Weymans, J. Phys. B 15,Ž .4315 1982 .
Ž .33. K. Dietz and B. A. Hess, Phys. Scr. 39, 682 1989 .Ž34. A. Ralston, A First Course in Numerical Analysis McGraw-
.Hill, New York, 1965 .35. K. Jankowski, K. Kowalski, and P. Jankowski, Int. J. Quan-
Ž .tum Chem. 53, 501 1995 .Ž . Ž .36. D. J. Thouless, Nucl. Phys. 21, 225 1960 ; Ibid. 22, 78 1961 .
37. K. Kowalski and K. Jankowski, to be published.38. J. Paldus, in Self-Consistent Field Theory and Applications, R.
ŽCarbo and M. Klobukowski, Eds. Elsevier, Amsterdam,.1990 .
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 219




















