
Mitochondrial energy metabolism is markedly impaired by d-2-hydroxyglutaric acid in rat tissues
12
Molecular Genetics and Metabolism 86 (2005) 188–199 www.elsevier.com/locate/ymgme 1096-7192/$ - see front matter 2005 Elsevier Inc. All rights reserved. doi:10.1016/j.ymgme.2005.05.002 Mitochondrial energy metabolism is markedly impaired by D-2-hydroxyglutaric acid in rat tissues Alexandra Latini a,b , Cleide Gonçalves da Silva a , Gustavo C. Ferreira a , Patrícia F. Schuck a , Karina Scussiato a , João J. Sarkis a , Carlos S. Dutra Filho a , Angela T.S. Wyse a , Clóvis M.D. Wannmacher a , Moacir Wajner a,b,c,¤ a Departamento de Bioquímica, Instituto de Ciências Básicas da Saúde, Universidade Federal do Rio Grande do Sul, Porto Alegre—RS, Brazil b Serviço de Genética Médica, Hospital de Clínicas de Porto Alegre, Porto Alegre—RS, Brazil c Universidade Luterana do Brasil, Canoas—RS, Brazil Received 1 April 2005; accepted 7 May 2005 Available online 15 June 2005 Abstract Tissue accumulation of high amounts of D-2-hydroxyglutaric acid (DGA) and L-2-hydroxyglutaric acid (LGA) is the biochemical hallmark of the inherited neurometabolic disorders D-2-hydroxyglutaric aciduria (DHGA) and L-2-hydroxyglutaric aciduria (LHGA), respectively. Patients aVected by DHGA predominantly present neurological and cardiomuscular symptoms, while those with LHGA have mainly severe neurological symptoms. Lactic aciduria and/or lactic acidemia may also occur in both disorders, suggesting mitochondrial dysfunction. We have previously reported that cytochrome c oxidase (COX) activity is severely inhibited by DGA in rat cerebral cortex and human skeletal muscle. In the present study, we initially evaluated the role of DGA and LGA on the mitochondrial respiratory chain complex activities, as well as CO 2 on production in cardiac and skeletal muscle from 30-day-old Wistar rats. DGA sig- niWcantly inhibited COX and ATP synthase (F 0 F 1 -ATP synthase) activities, in contrast to the other activities of the respiratory chain enzymes which were not aVected by DGA in both muscular tissues. In addition, CO 2 production was also markedly reduced by DGA in rat skeletal and cardiac muscles. On the other hand, LGA did not interfere with any of the respiratory chain complex activities studied, neither with CO 2 generation. We also measured mitochondrial respiratory parameters in rat brain mitochondrial preparations in the presence of DGA and LGA. Both metabolites signiWcantly lowered the respiratory control ratio in the presence of glutamate/malate and succinate. Since the metabolites stimulated oxygen consumption in state IV and compromised ATP formation, it can be presumed that these organic acids might act as endogenous uncouplers of mitochondria respiration. Moreover, COX activity linked to TMPD-ascor- bate was signiWcantly reduced by DGA in the brain mitochondrial enriched fractions. Finally, DGA and LGA reduced cell viability of rat cerebral cortex slices, as determined by the MTT assay. In case our in vitro data also occur in vivo, it may be presumed that impair- ment of energy metabolism may contribute to the understanding of the clinical features mainly of patients aVected by DHGA. 2005 Elsevier Inc. All rights reserved. Keywords: D-2-Hydroxyglutaric acid; D-2-Hydroxyglutaric aciduria; Respiratory chain; Cytochrome c oxidase; F 1 F 0 -ATP synthase; Skeletal muscle; Cardiac muscle Introduction The D and L enantiomers of 2-hydroxyglutaric acid are excreted in small amounts in urine of normal individuals. However, excessive excretion of D-2-hydroxyglutaric acid (DGA) and L-2-hydroxyglutaric acid (LGA) is character- istically found in the metabolic disorders D-2-hydroxyglu- taric aciduria (DHGA) and L-2-hydroxyglutaric aciduria (LHGA), respectively [1–4]. Increased amounts of DGA have also been demonstrated in a murine model of succi- nic semialdehyde dehydrogenase deWciency [5]. * Corresponding author. Fax: +55 51 3316 5540. E-mail address: [email protected] (M. Wajner).
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Mitochondrial energy metabolism is markedly impaired by d-2-hydroxyglutaric acid in rat tissues

Molecular Genetics and Metabolism 86 (2005) 188–199
www.elsevier.com/locate/ymgme
Mitochondrial energy metabolism is markedly impaired by D-2-hydroxyglutaric acid in rat tissues
Alexandra Latini a,b, Cleide Gonçalves da Silva a, Gustavo C. Ferreira a,Patrícia F. Schuck a, Karina Scussiato a, João J. Sarkis a, Carlos S. Dutra Filho a,
Angela T.S. Wyse a, Clóvis M.D. Wannmacher a, Moacir Wajner a,b,c,¤
a Departamento de Bioquímica, Instituto de Ciências Básicas da Saúde, Universidade Federal do Rio Grande do Sul, Porto Alegre—RS, Brazilb Serviço de Genética Médica, Hospital de Clínicas de Porto Alegre, Porto Alegre—RS, Brazil
c Universidade Luterana do Brasil, Canoas—RS, Brazil
Received 1 April 2005; accepted 7 May 2005Available online 15 June 2005
Abstract
Tissue accumulation of high amounts of D-2-hydroxyglutaric acid (DGA) and L-2-hydroxyglutaric acid (LGA) is the biochemicalhallmark of the inherited neurometabolic disorders D-2-hydroxyglutaric aciduria (DHGA) and L-2-hydroxyglutaric aciduria (LHGA),respectively. Patients aVected by DHGA predominantly present neurological and cardiomuscular symptoms, while those with LHGAhave mainly severe neurological symptoms. Lactic aciduria and/or lactic acidemia may also occur in both disorders, suggestingmitochondrial dysfunction. We have previously reported that cytochrome c oxidase (COX) activity is severely inhibited by DGA in ratcerebral cortex and human skeletal muscle. In the present study, we initially evaluated the role of DGA and LGA on the mitochondrialrespiratory chain complex activities, as well as CO2 on production in cardiac and skeletal muscle from 30-day-old Wistar rats. DGA sig-niWcantly inhibited COX and ATP synthase (F0F1-ATP synthase) activities, in contrast to the other activities of the respiratory chainenzymes which were not aVected by DGA in both muscular tissues. In addition, CO2 production was also markedly reduced by DGA inrat skeletal and cardiac muscles. On the other hand, LGA did not interfere with any of the respiratory chain complex activities studied,neither with CO2 generation. We also measured mitochondrial respiratory parameters in rat brain mitochondrial preparations in thepresence of DGA and LGA. Both metabolites signiWcantly lowered the respiratory control ratio in the presence of glutamate/malate andsuccinate. Since the metabolites stimulated oxygen consumption in state IV and compromised ATP formation, it can be presumed thatthese organic acids might act as endogenous uncouplers of mitochondria respiration. Moreover, COX activity linked to TMPD-ascor-bate was signiWcantly reduced by DGA in the brain mitochondrial enriched fractions. Finally, DGA and LGA reduced cell viability ofrat cerebral cortex slices, as determined by the MTT assay. In case our in vitro data also occur in vivo, it may be presumed that impair-ment of energy metabolism may contribute to the understanding of the clinical features mainly of patients aVected by DHGA. 2005 Elsevier Inc. All rights reserved.
Keywords: D-2-Hydroxyglutaric acid; D-2-Hydroxyglutaric aciduria; Respiratory chain; Cytochrome c oxidase; F1F0-ATP synthase; Skeletal muscle;Cardiac muscle
Introduction
The D and L enantiomers of 2-hydroxyglutaric acid areexcreted in small amounts in urine of normal individuals.
* Corresponding author. Fax: +55 51 3316 5540.E-mail address: [email protected] (M. Wajner).
1096-7192/$ - see front matter 2005 Elsevier Inc. All rights reserved. doi:10.1016/j.ymgme.2005.05.002
However, excessive excretion of D-2-hydroxyglutaric acid(DGA) and L-2-hydroxyglutaric acid (LGA) is character-istically found in the metabolic disorders D-2-hydroxyglu-taric aciduria (DHGA) and L-2-hydroxyglutaric aciduria(LHGA), respectively [1–4]. Increased amounts of DGAhave also been demonstrated in a murine model of succi-nic semialdehyde dehydrogenase deWciency [5].

A. Latini et al. / Molecular Genetics and Metabolism 86 (2005) 188–199 189
DHGA was Wrst recognized by Chalmers and co-workers in 1980, and has now been reported in over 30patients [1,2,6–9]. The clinical condition in DHGA isheterogeneous and varies from neonatal-onset intracta-ble epilepsy to mild developmental delay [1,2]. The mostseverely aVected patients suVer from a rather uniformencephalopathy with developmental delay, epilepsy, andhypotonia. Enlarged frontal subarachnoid spaces andsubdural eVusions and signs of cerebral delayed matura-tion with subependymal cysts in the caudate nucleousare generally seen in the magnetic resonance imaging(MRI). Dystonia and choreoathetotic movement disor-ders and cardiomyopathy with cardiomegaly, hypertro-phy and reduced contractility are also observed in manypatients aVected by this disorder [10,11]. The otherDHGA variant is characterized by a mild phenotypewhich essentially shares the characteristics of the severephenotype [1]. Biochemically, aVected patients withDHGA present large urinary excretion and accumula-tion of DGA in plasma and CSF [1,2].
The underlying DHGA molecular defect was recentlyreported to be caused by missense mutations in the geneof D-2-hydroxyglutarate dehydrogenase, as detected intwo unrelated patients aVected by DGHA, being this dis-ease catalogued as an autosomal recessive inheritancedisorder [12].
LHGA was Wrst described in 1980 by Duran and co-workers (1980) and has now been described in about 75patients reported from diVerent countries [3,4,13–21].Clinical symptoms and disease course are homogeneousamong the patients aVected by LHGA, usually presentingin early to mild childhood psychomotor retardation,mental deterioration, seizures, pyramidal, and extrapyra-midal symptoms. Ataxia, tremor, choreiform movements,and hypotonia are also observed [3,4]. Neuroimagingreveals subcortical leukoencephalopathy, progressive lossof myelinated arcuate Wbres, alteration in basal ganglia,and cerebellar atrophy [18,19,21]. The diagnosis dependsupon increased levels of LGA in urine, plasma andCSF [19].
Mutations in LHGA patients were recently identiWedin a gene called duranin, localized on chromosome14q22.1 and encoding a mitochondrial L-2-hydroxyglut-arate dehydrogenase linked to FAD [22,23]. The exactrole of this enzyme in the intermediary metabolism inhumans remains to be established. However, subcellularfractioning indicated that the liver enzyme is present inmitochondria, where it is bound to membranes suggest-ing that it transfers its reducing equivalents to therespiratory chain, most probably at the level of ubiqui-none [23].
Besides the high urinary excretion of the D and L
enantiomers of 2-hydroglutaric acid, elevated amountsof lactate, 2-ketoglutarate and others citric acid cycleintermediates in urine are also found in some DHGA [2]and LHGA [17,24] patients, indicating that mitochon-
drial dysfunction may be implicated in the pathophysiol-ogy of this disease. In this context, recent studiesdemonstrated that DGA markedly inhibited the activi-ties of complex IV in rat cerebral cortex and human skel-etal muscle homogenates [25], as well as of complex V insubmitochondrial particles from bovine heart [26] andcreatine kinase in rat cerebellum, heart, and skeletalmuscle [27,28]. Interestingly, cardiomyopathy is alsocommonly found in mitochondrial diseases, either inassociation with neuromuscular symptoms or as themain clinical feature [29–33]. This is the case for DHGA,in which, besides the predominant central nervous sys-tem dysfunction, myopathy with hypotonia, muscleweakness and atrophy, and cardiomyopathy are fre-quent features [1,2,34]. However, to date the exact under-lying mechanisms contributing to the muscle pathologyin this disorder are still poorly established. Therefore, weextended our previous studies [25,27,28] by investigatingthe in vitro eVects of DGA, and also its enantiomerLGA, on the respiratory chain enzyme complex I–Vactivities and on CO2 production in rat heart and skele-tal muscle. We also tested whether DGA and LGA couldalter oxygen consumption by puriWed mitochondrialfractions from rat brain, as well as cell viability of ratcerebral cortex slices, determined by the reduction ofMTT, in the hope to determine at what level theseorganic acids could compromise energy metabolism inthese high energy demanding tissues.
Materials and methods
Subjects and reagents
Male Wistar rats of 30 days of life obtained from theCentral Animal House of the Department of Biochemis-try, ICBS, Universidade Federal do Rio Grande do Sul,Porto Alegre, RS—Brazil, were used. Rats had freeaccess to water and a 20% (w/w) protein commercialchow and were kept in a room with a 12:12 h light/darkcycle and temperature of 24 § 1 °C. The experimentalprotocol was approved by the Ethics Committee for ani-mal research of the Federal University of Rio Grande doSul, Porto Alegre, Brazil and followed the “Principles ofLaboratory Animal Care (NIH Publication No. 85–23,revised 1985). All chemicals were purchased from SigmaChemical, St. Louis, MO, USA. DGA and LGA weredissolved in the speciWc buVer used for each techniqueand the pH of these solutions was adjusted to 7.4.
Tissue preparation for measurement of respiratory chain complex activities
Animals were killed by decapitation without anesthe-sia; skeletal muscle (soleus) and heart were dissectedonto an ice-cold glass plate and homogenized using an

190 A. Latini et al. / Molecular Genetics and Metabolism 86 (2005) 188–199
ice-chilled glass homogenizing vessel at 1300 rpm. Ratskeletal and cardiac muscles were homogenized in 20volumes of SETH buVer, pH 7.4 (250 mM sucrose, 2 mMEDTA, 10 mM Tris, and 50 UI/mL heparin). Thehomogenates were centrifuged at 800g for 10 min at 4 °C,the pellet was discarded and the supernatants were keptat ¡70 °C until enzyme activity determination.
Mitochondria from skeletal and cardiac muscles werealso puriWed to measure the activities of complexes I andV. BrieXy, the tissues were homogenized in 10 volumes ofphosphate buVer pH 7.4 containing 0.3 M sucrose, 5 mMMops, 1mM EGTA, and 0.1% bovine serum albumin.The homogenates were centrifuged at 1500g for 10 min at4 °C and the pellet was discarded. The supernatant wascentrifuged at 15,000g to isolate the mitochondrial frac-tion present in the pellet, which was Wnally dissolved in thesame buVer. These enriched mitochondrial fractions werealso used for the oxygen consumption experiments [35].
Tissue slices preparation
Skeletal (soleus) and cardiac muscles of rats were cutto produce slices of 100 and 50 mg, respectively, and usedfor CO2 production assays. Four hundred micrometerswide rat cerebral cortex slices obtained by using a McIl-wain chopper were used for the viability assays, assessedby the MTT assay.
Respiratory chain enzyme activities
The activities of complexes II, II–III, and succinatedehydrogenase (SDH) were determined in musclehomogenates according to the method of Fischer et al.[36]. The activities of complexes IV and V were measuredaccording to Rustin et al. [37], whereas that of complexI–III was assayed according to the method described bySchapira et al. [38]. Finally, complex I activity was per-formed according to Cassina and Radi [35].
Respiratory chain enzyme activities were deter-mined in the presence of various concentrations ofDGA (0.05–7.5 mM) or LGA (5 mM), whereas controlgroup did not contain these organic acids in themedium. Some of the methods above described to mea-sure the various activities were slightly modiWed, asdescribed in details in a previous report [25].
Target molecule reduction and oxidation assays
DGA (1.0 mM) was Wrst incubated for 5 min in amedium devoid of homogenates in the presence of theelectron acceptors cytochrome, phenazine methasulfate(PMS) and 2,6-dichlorophenolindophenol (DCPIP) atthe same concentrations used for the determination ofthe activities of the various complexes of the respiratorychain. Reduction of these acceptors was measured byabsorbance changes at 550 and 600 nm.
GSH (0.4 mM) and protein-bound sulfhydryl groups(PBSG) from mitochondrial membrane preparations(50 �g protein) were also exposed to 1.0 mM DGA for 0–30 and 60 min, respectively. The sulfhydryl groupoxidants N-ethylmaleimide (NEM) and p-hydroxy-mer-curybenzoic acid (PHMB) were also used in the assaysas positive controls. Oxidation of the reduced sub-strates was determined by Xuorescence measurementusing o-phthaldialdehyde (OPA) and by the 5,5�-dithiobis-(2-nitrobenzoic acid) DTNB assay, respectively [39,40].
14CO2 production assays in slices from rat muscle
CO2 production was determined in the presence5.0 mM DGA or LGA in rat muscles. Skeletal andcardiac muscle slices (100 and 50 mg, respectively) wereadded to small Xasks (11 cm3) containing 0.45 mlKrebs–Ringer bicarbonate buVer, pH 7.4. Flasks werepre-incubated in a metabolic shaker at 37 °C for 15 min(90 oscillations min¡1). After pre-incubation, 0.2 �Ci[U-14C]glucose and 5.0 mM of the unlabeled glucosewere added to the Xasks containing the skeletal muscleslices whereas, 0.2 �Ci [U-14C]lactate and 1.0 mM of theunlabeled lactate to the Xasks containing the cardiacslices. The Xasks were gassed with an O2/CO2 (95:5) mix-ture and sealed with rubber stoppers and ParaWlm M.Glass center wells containing a folded 65/5 mm piece ofWhatman 3 Wlter paper were hung from the stoppers.After 60 min of incubation at 37 °C, 0.1 ml of 50% tri-chloroacetic acid was added to the medium and 0.1 mlbenzethonium hydroxide was added to the center wellswith needles introduced through the rubber stopper. TheXasks were left to stand for 30 min to complete CO2 trap-ping and then opened. The Wlter papers were removedand added to vials containing Opti Phase “Hi-safe” 3scintillation Xuid, and radioactivity was measured [41].Results are expressed as nanomole CO2/h/g tissue.
Mitochondrial respiratory parameters
The rate of oxygen consumption was measuredpolarographically using a Clark-type electrode in a ther-mostatically controlled (37 °C) and magnetically stirredincubation chamber of 1.6 mL capacity [35]. The assay wasperformed with 1.0 mg/mL of mitochondrial protein incu-bated in the same buVer used for mitochondrial isolationin the presence of NAD-linked (glutamate/malate, 2.5 mMof each) and FAD-linked (5 mM succinate) substrates.The rate of oxygen consumption in these conditions corre-sponded to state IV mitochondrial respiration. State IIIwas initiated by adding 250 nmol ADP. The respiratorycontrol ratio (RCR; state III/state IV) was then calcu-lated. State IV was also measure by using N,N,N�,N�-tetra-methyl-p-phenylenediamine (TMPD; 250�M) andascorbate (500�M) that donate electrons directly tocytochrome c bypassing the bc1 complex [35]. These

A. Latini et al. / Molecular Genetics and Metabolism 86 (2005) 188–199 191
experiments were performed using digitonine at a Wnalconcentration of 1.8/10 mg mitochondrial protein.
Cell viability
Cell viability was determined in cerebral cortex slicesby the tetrazolium salt 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazoliumbromide (MTT) method. After 3 hDGA and LGA incubation at 35 °C, The cerebral cortexslices were pre-incubated for 3 h in the presence of5.0 mM DGA or LGA, after which incubation for45 min at 35 °C occurred in the presence 45 �g/ml MTT.Active mitochondrial dehydrogenases of living cellscause cleavage and reduction of the soluble yellow MTTdye to the insoluble purple formazan [42] in dimethylsulfoxide. Extinction was measured at 570 and 630 nmand the net �A(570–630) was taken as an index of cell via-bility. Results were compared to control samples towhich 100% viability was attributed.
Protein determination
Protein was measured by the method of Lowry et al.[43] using serum albumin bovine as standard.
Statistical analysis
Results are presented as means § standard deviation.Assays were performed in duplicate or triplicate and themean was used for statistical analysis. Data were ana-lyzed using one-way analysis of variance (ANOVA) fol-lowed by the post hoc Duncan multiple range test whenF was signiWcant. Only signiWcant F values are given inthe text. For analysis of dose-dependent eVect, linearregression was used. DiVerences between the groupswere rated signiWcant at P < 0.05. All analyses were car-ried out in an IBM-compatible PC computer using theStatistical Package for the Social Sciences (SPSS) soft-ware.
Results
Inhibition of the respiratory chain by DGA in rat muscles
We Wrst investigated the eVect of DGA and LGA onthe activities of the respiratory chain enzyme com-plexes in homogenates and in mitochondrial enrichedfractions from rat skeletal and cardiac muscles. Fig. 1shows that DGA strongly reduced the activity ofcomplex IV (up to 86%) [F(7, 32) D 17.80, P < 0.001] andcomplex V (up to 40%) [F(3, 12) D 4.30, P < 0.05] in adose-dependent manner [complex IV: � D ¡0.75,P < 0.001; complex V: � D ¡0.71, P < 0.01] in rat skele-tal muscle. Furthermore, DGA also inhibited complexIV (up to 82%) [F(7, 31) D 54.74, P < 0.001] and complex
V (up to 43%) [F(4, 15) D 16.32, P < 0.001] activities in ratcardiac muscle in a dose-dependent manner [complexIV: � D ¡0.89, P < 0.001; complex V: � D ¡0.87,P < 0.001] (Fig. 2). In contrast, LGA did not alter anyof the other activities of the respiratory chain in bothmuscular tissues.
Searching for DGA reducing and oxidant activities
Then, it was tested whether 1.0 mM DGA could perse aVect COX reaction by incubating the metabolitewith reduced cytochrome in identical conditions asthose of the assay, but without adding homogenates inthe medium. We observed that the measured absor-bance (550 nm), which reXects the amount of reducedcytochrome, was not altered by DGA per se, suggest-ing that this organic acid does not interfere with theassay and does not exchange electrons with cyto-chrome. Therefore, it is unlikely that the inhibitoryeVect of DGA on COX activity was an in vitro artifact.We further investigated whether DGA could behave asa reducing or oxidant agent, by incubating this organicacid at 1.0 mM concentration with various substrates,such as the common electron acceptors PMS andDCPIP, as well as GSH and protein-bound sulfhydrylgroups (PBSG) from mitochondrial membranepreparations. The presence of 1.0 mM DGA in amedium devoid of homogenates did not modify theparameters analyzed, strongly indicating that DGAdid not behave as a reducing or oxidant agent. How-ever, NEM and PHMB (positive controls) were able tosigniWcantly oxidize GSH and PBSG, respectively(data not shown).
IC50 and kinetics of the DGA interaction with complexes IV and V in rat muscles
The IC50 (inhibitor concentration necessary to reduce50% of the enzyme activity) [44] was determined forcomplexes IV and V in both rat muscular tissues (Figs. 1and 2). The IC50 values obtained for the complex IVinhibition by DGA were 0.30 § 0.04 and 0.79 § 0.02 mM(means § SD; n D 3) in skeletal muscle and heart, respec-tively, suggesting that complex IV activity in skeletalmuscle was more susceptible to DGA action. In addi-tion, the IC50 values for complex V were 9.85 § 2.70 and9.45 § 1.11 mM (means § SD; n D 3) in skeletal muscleand heart, respectively, reXecting the lower inhibitionprovoked by DGA on this complex.
The kinetics of the interaction of DGA with COX insupernatant from skeletal muscle and heart was thendetermined. The Lineweaver–Burk double reciprocal plotwas analyzed over a range of cytochrome c concentrations(6–25nM) in the absence or presence of DGA (0.5–3mM)[45]. The data indicate that the inhibition of complex IVactivity by DGA is uncompetitive in both tissues (Fig. 3).

192 A. Latini et al. / Molecular Genetics and Metabolism 86 (2005) 188–199
The Km calculated was 0.06§0.02 and 0.05§0.01 mM(means§SD; nD3) for skeletal muscle and cardiac mus-cle, respectively. The Ki value (the dissociation constant ofthe enzyme–substrate–inhibitor complex) was calculatedby the method of Dixon [44], which provides a simple wayof determining the inhibitor constant (Ki) for uncompeti-tive inhibitors. The Ki calculated was 0.56§0.02 mM forskeletal muscle and 0.47§0.18 mM for cardiac muscle(means§SD; nD3) (Fig. 3). Km values for F1F0-ATP syn-thase activity were not determined because this enzymepresents a complex kinetics with multiple interactions,presenting three diVerent Km [46,47].
EVect of DGA on CO2 production in rat muscles
CO2 production from glucose and lactate assubstrates was further studied in rat skeletal and cardiacmuscle slices, respectively, in the presence of DGA andLGA. Fig. 4 shows that 5.0 mM DGA signiWcantlyreduced CO2 production (up to 42%) from [U-14C]glucose [F(2,12) D 4.66; P < 0.05] in skeletal muscle (A)and from [U-14C]lactate [F(2,12) D 5.76; P < 0.05] (up to
38%) in rat cardiac muscle (B). In contrast, 5.0 mM LGAdid not cause any eVect on this measurement.
DGA and LGA eVects on respiratory parameters in brain mitochondrial enriched fractions
The respiratory state IV (NAD and FAD-linkedsubstrates), state III (ADP stimulated respiration) andrespiratory control ratio (RCR) parameters werethen measured in rat cerebrum enriched mitochondriafractions. Fig. 5 shows that the RCR values calculatedfrom glutamate/malate- and succinate-respiring mito-chondria were signiWcantly reduced by 1.0 mM DGA(up to 38 and 31 %, respectively) and by 1.0 mM LGA(up to 43 and 50 %, respectively) as compared tocontrols [RCR(glutamate/malate): F(2,9) D 29.33; P < 0.001;RCR(succinate): F(2,9) D 14.03; P < 0.01]. Fig. 5 also showsthat state IV was signiWcantly increased by DGA withthe NAD-linked substrates (up to 68%) and moderatelyincreased by LGA (up to 34%) [F(2,9) D 5.82; P < 0.05],whereas state III respiration was slightly reduced byLGA (up to 8%). State IV linked to succinate was also
Fig. 1. EVect of D-2-hydroxyglutaric acid (DGA) and L-2-hydroxyglutaric acid (LGA) on the activities of complexes IV (A) and V (B) of the respira-tory chain in rat skeletal muscle. Values are means § SD for four to Wve (animals) independent experiments per group measured in the presence orabsence of the metabolites. *P < 0.05, ***P < 0.001, compared to controls (Duncan multiple range test). The inset shows the Dixon plot for IC50determination (n D 3) and the data presented are representative of one experiment.
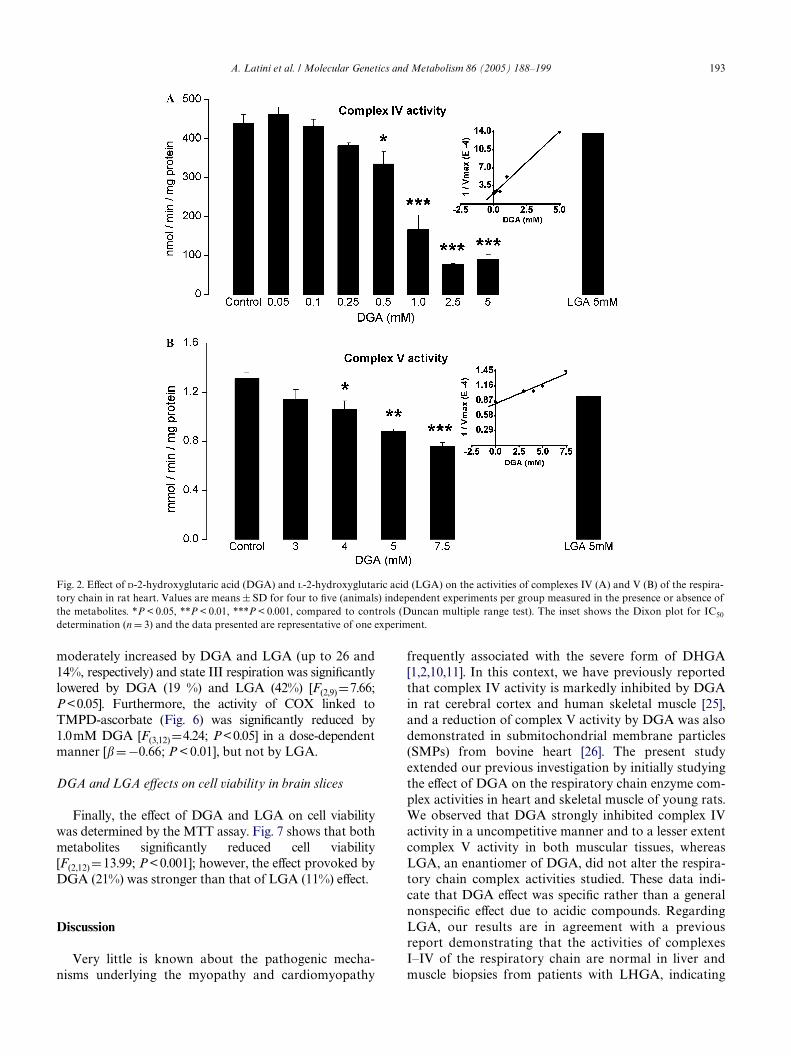
A. Latini et al. / Molecular Genetics and Metabolism 86 (2005) 188–199 193
moderately increased by DGA and LGA (up to 26 and14%, respectively) and state III respiration was signiWcantlylowered by DGA (19 %) and LGA (42%) [F(2,9)D7.66;P<0.05]. Furthermore, the activity of COX linked toTMPD-ascorbate (Fig. 6) was signiWcantly reduced by1.0mM DGA [F(3,12)D4.24; P<0.05] in a dose-dependentmanner [�D ¡0.66; P < 0.01], but not by LGA.
DGA and LGA eVects on cell viability in brain slices
Finally, the eVect of DGA and LGA on cell viabilitywas determined by the MTT assay. Fig. 7 shows that bothmetabolites signiWcantly reduced cell viability[F(2,12) D13.99; P< 0.001]; however, the eVect provoked byDGA (21%) was stronger than that of LGA (11%) eVect.
Discussion
Very little is known about the pathogenic mecha-nisms underlying the myopathy and cardiomyopathy
frequently associated with the severe form of DHGA[1,2,10,11]. In this context, we have previously reportedthat complex IV activity is markedly inhibited by DGAin rat cerebral cortex and human skeletal muscle [25],and a reduction of complex V activity by DGA was alsodemonstrated in submitochondrial membrane particles(SMPs) from bovine heart [26]. The present studyextended our previous investigation by initially studyingthe eVect of DGA on the respiratory chain enzyme com-plex activities in heart and skeletal muscle of young rats.We observed that DGA strongly inhibited complex IVactivity in a uncompetitive manner and to a lesser extentcomplex V activity in both muscular tissues, whereasLGA, an enantiomer of DGA, did not alter the respira-tory chain complex activities studied. These data indi-cate that DGA eVect was speciWc rather than a generalnonspeciWc eVect due to acidic compounds. RegardingLGA, our results are in agreement with a previousreport demonstrating that the activities of complexesI–IV of the respiratory chain are normal in liver andmuscle biopsies from patients with LHGA, indicating
Fig. 2. EVect of D-2-hydroxyglutaric acid (DGA) and L-2-hydroxyglutaric acid (LGA) on the activities of complexes IV (A) and V (B) of the respira-tory chain in rat heart. Values are means § SD for four to Wve (animals) independent experiments per group measured in the presence or absence ofthe metabolites. *P < 0.05, **P < 0.01, ***P < 0.001, compared to controls (Duncan multiple range test). The inset shows the Dixon plot for IC50
determination (n D 3) and the data presented are representative of one experiment.

194 A. Latini et al. / Molecular Genetics and Metabolism 86 (2005) 188–199
that the sustained tissue elevation of LGA does not pro-voke inhibition of these activities [24].
We also demonstrated that DGA does not act as areducing or oxidant agent when incubated with variouselectron acceptors, including reduced cytochrome c,which is the substrate for COX reaction, or with GSHand protein-bound sulfhydryl groups (PBSG). In addi-tion, the inhibitory property of DGA on COX activitywas further conWrmed in rat brain mitochondrialenriched preparations by measuring the rate of oxygenconsumption linked to TMDP-ascorbate, which wasfound to be also inhibited by 1.0 mM DGA. Therefore,the reduction of complex IV activity induced by DGAwas not an artiWcial in vitro result, but rather a true inhi-bition of this activity.
On the other hand, the inhibitory eVect of DGA onCOX activity was not observed by Kölker et al. [26], whoonly found that complex V (ATP synthase) activity was
reduced by this organic acid in SMPs from bovine heart.This discrepancy may be due in part to the experimentalmethodological procedures utilized since we used in ourassays whole homogenates instead of SMPs.
Regarding to the mechanisms underlying the inhibi-tion of COX activity, it is well established that this respi-ratory complex is reversibly regulated by nitric oxide[48,49] and that 4-hydroxynonenal and malondialdehyde(MDA), products of lipid peroxidation, inhibit the activ-ity of puriWed COX by forming adducts with the enzyme[50,51]. In addition, nitration of cytochrome c modiWes itsredox properties, such as resistance to reduction byascorbate [52]. In this scenario, it has been reported thatDGA induces reactive species formation and lipid perox-idation [26,53]. These observations may explain whyDGA inhibits complex IV in brain [25] and also in musclehomogenates, as demonstrated here, which contain thewhole cell machinery necessary to induce free radical
Fig. 3. Kinetic analyses of the inhibition of cytochrome c oxidase (COX) from skeletal muscle (A) and cardiac muscle (B) of rats by D-2-hydroxyglu-taric acid (DGA). The graphs show a double plot of COX activity for cytochorme c concentrations (8–25 �M) [S] in the absence (�, controls) and inthe presence of 1.0 mM (�), 2 mM (�), 2.5 mM (�), and 3 mM (�) DGA [I]. The insert shows the Dixon plot for Ki determination. All experimentswere repeated at least three times and similar results were obtained. Data presented are representative of one experiment.

A. Latini et al. / Molecular Genetics and Metabolism 86 (2005) 188–199 195
Fig. 4. EVect of D-2-hydroxyglutaric acid (DGA) and L-2-hydroxyglu-taric acid (LGA) on CO2 production from [U-14C]glucose and [U-14C]lactate in rat skeletal muscle and heart, respectively. Values aremeans § SD for Wve (animals) independent experiments per groupmeasured in the presence or absence of the metabolites. *P < 0.05,**P < 0.01, compared to controls (Duncan multiple range test).
generation and lipid peroxidation, but not in puriWedSMPs [26]. It may be also presumed that DGA does nothave a direct action on COX, since no eVect could bedetected when SMPs were used to measure COX activity.
On the other hand, considering that a respiratorychain blockage could lead to other mitochondrial func-tion alterations, such as inhibition of the citric acid cycle,we also assessed CO2 production from glucose and lac-tate in skeletal and cardiac muscles, respectively, in thepresence of 5.0 mM DGA and LGA. The observed lowerrate of CO2 production from glucose in skeletal muscle
Fig. 6. EVect of D-2-hydroxyglutaric acid (DGA) and L-2-hydroxyglu-taric acid (LGA) on cytochrome c oxidase (COX) activity linked toTMPD-ascorbate. See Material and methods for details. Values aremeans § SD for four (animals) independent experiments per groupmeasured in the presence or absence of the metabolites.*P < 0.05 com-pared to controls (Duncan multiple range test).
Fig. 5. EVect of D-2-hydroxyglutaric acid (DGA) and L-2-hydroxyglutaric acid (LGA) on respiratory parameters in rat brain mitochondrialpreparations. Values are means § SD for four (animals) independent experiments per group measured in the presence or absence of the metabolites.Glutamate plus malate (2.5 mM) and succinate (5 mM) were used as substrates for measuring state IV respiration. State III was measured after addi-
tion of 250 nmol ADP to the mitochondrial preparations. Respiratory control ratio (RCR) was determined dividing state III by state IV. **P < 0.01, ***P < 0.001, compared to controls (Duncan multiple range test).
196 A. Latini et al. / Molecular Genetics and Metabolism 86 (2005) 188–199
indicate a deWcient aerobic glycolysis, consistent with thehypotonia and increased levels of lactate found in serumand urine of some patients aVected by DHGA [1,2,24]. Inaddition, the cardiomyopathy with cardiomegaly, hyper-trophy and reduced heart contractility presented inmany of these patients [10,11] might possibly be due to alower aerobic respiration.
Furthermore, DGA and also LGA decreased cell via-bility in cerebral cortex slices as assessed by the MTTmethod, indicating that both metabolites aVect mito-chondrial function, leading to increased nonviable cells.The lower cell viability detected in our study in brainslices was similar to that found by other investigators incultured neural cells [26]. Although we cannot establishat the present the underlying toxic mechanisms relatedto the eVect of LGA reducing neural cell viability, thismay have occurred because this organic acid is able toinduce free radicals generation in the brain [54]. Regard-ing DGA, its severe eVect on energy metabolism, asobserved here and in a previous paper [25], allied to itsability to elicit free radicals and excitotoxicity [26,53]may possibly explain the reduced cell viability found incerebral cortex slices.
To further assess the overall consequences of DGAinhibitory eVect on the electron transfer chain on mito-chondrial respiration we tested the eVect of 1.0 mM DGAon the rate of mitochondrial oxygen consumption in thepresence of glutamate/malate or succinate as substratesin rat cerebrum mitochondrial preparations. LGA wasalso used in these experiments. To assure the entrance ofthese organic acids into the mitochondria, preparationswere treated with digitonine, which makes the outermitochondrial membrane permeable but preserves theinner membrane and mitochondria function [55]. DGA
Fig. 7. EVect of D-2-hydroxyglutaric acid (DGA) and L-2-hydroxyglu-taric acid (LGA) on MTT reduction in rat cerebral cortex slices. Val-ues are means § SD for Wve (animals) independent experiments pergroup measured in the presence or absence of the metabolites.*P < 0.05; ***P < 0.001 compared to controls (Duncan multiple rangetest).
and LGA signiWcantly lowered the RCR values by sig-niWcantly reducing state III respiration in the presence ofsuccinate and signiWcantly increasing state IV respirationin the presence of NAD+-linked substrates, indicatingthat ATP synthesis (reduction of state III respiration) iscompromised in mitochondria exposed to DGA andLGA and that these metabolites are able to uncouple oxi-dative phosphorylation (reduction of RCR values). Thus,we might conclude that DGA and LGA might act asendogenous uncouplers of mitochondrial respiration.
The defective response to ADP in activating state IIIrespiration may be attributed either to a less active ATPsynthase, as shown for DGA here and in a previousreport [26], or alternatively to alterations of the ADP/ATP translocator, which were not investigated in thepresent study. In addition, the increased state IV respira-tion caused by DGA and LGA may have occurredbecause these compounds are weak organic acids(pKa V 5), which may potentially dissipate the protongradient across the mitochondrial inner membrane bybinding and transferring protons to the mitochondrialmatrix and decreasing the electrochemical gradient.
DeWciency of human COX activity is frequently asso-ciated with lactic acidemia, being clinically manifestedby hypotonia, lethargy, feeding diYculties, failure tothrive, psychomotor delay, seizures, and progressivedeterioration of basal ganglia and brain stem, as well asby myopathy and cardiomyopathy [56–60]. In addition,Wbroblasts from aVected patients present raised lactateto pyruvate ratio and reduced COX activity of 35–45%of normal [61–63]. These laboratorial and clinical signsare similar to those found in DHGA, a fact that possiblyindicates that the marked inhibition (86%) of COXactivity observed in the present study may potentially beimplicated in the pathophysiology of the neuromuscularWndings presented by patients aVected by DHGA. This isfurther reinforced by the concentration of DGA neces-sary to decrease 50% (IC50) of COX activity in skeletal(0.3 mM) and cardiac (0.7 mM) muscles, which werelower or similar than the concentrations usually found inplasma and CSF of DHGA patients (up to 700 �M) [1,2].Therefore, considering that the rate of the respiratorychain and ATP synthesis can be eVectively regulated bya mechanism based on the allosteric inhibition of COXat a high intramitochondrial ATP/ADP ratio [64–66] itis expected that the degree of COX inhibition found inthe present study would probably disrupt energy metab-olism in muscular tissues of DGHA aVected patients.Similarly, because myocardium depends heavily on oxi-dative metabolism, it is feasible that the COX inhibitionby DGA demonstrated in this work might also be associ-ated with the cardiomyopathy characteristic of thesevere form of DHGA. Furthermore, the tight control ofrespiration by COX probably explains the phenotype ofmild COX deWciency in muscle observed in several mito-chondriopathies [67] or in aging [68].

A. Latini et al. / Molecular Genetics and Metabolism 86 (2005) 188–199 197
On the other hand, genetic defects in complex V havebeen related with the pathogenesisis of several condi-tions such us maternally inherited Leigh syndrome,MELAS syndrome (mitochondrial encephalomyopathy,lactic acidosis, and stroke-like symptoms), NARP syn-drome (neurogenic muscle weakness, ataxia, and retinitispigmentosa) and in a fatal case with neonatal lacticacidaemia, cardiomegaly and hepatomegaly [69–75].These disorders associated with reduced complex Vactivity are clinically characterized by lactic acidemia,hypotonia, lethargy, ataxia, seizures, dilated and hyper-trophic cardiomyopathies, and degeneration of speciWcbrain regions. Therefore, it may be presumed that ourresults of reduced complex V activity caused by DGA incardiac and skeletal muscles may also, at least in part,contribute to the pathogenesis of the cardiomyopathyencountered in DHGA patients. In this scenario, itshould be stressed that muscular tissues require a highsupply of ATP, which is mainly obtained in the mito-chondrial OXPHOS [76].
A recent report from our laboratory has shown thatDGA signiWcantly inhibits creatine kinase (CK) activity,predominantly the mitochondrial isoform (Mi–CK), inrat cardiac, and skeletal muscle [28]. This reinforces theassumption that DGA strongly disturbs energy metabo-lism since CK, particularly Mi–CK activity, is function-ally coupled to the mitochondrial adenine nucleotidetranslocator and oxidative phosphorylation [77].
In conclusion, although it is diYcult to extrapolateour Wndings to the human condition, if our results areconWrmed in vivo by measuring complexes IV and Vactivities in human tissues from DHGA patients, it isfeasible that a signiWcant inhibition of the respiratorychain at two critical steps as found here, may result inreduced ATP generation, leading to muscular dysfunc-tion in these patients. Our present Wndings may alsoexplain the elevated levels of lactate and citric acid inter-mediates in the body Xuid of some of these patients,which may occur secondarily to the inhibition of therespiratory chain electron Xux leading to an increase ofNADH and FADH2 concentrations. Elevation of thesereduced nucleotides could result in increased lactate dueto the high NADH/NAD+ ratio.
Acknowledgment
This work was partly supported by Grants fromCNPq, PRONEX II, and FAPERGS.
References
[1] M.S. van der Knaap, C. Jakobs, G.F. HoVman, M. Duran, A.C.Muntau, S. Schweitzer, R.I. Kelley, F. Parrot-Roulaud, J. Amiel, P.De Lonlay, D. Rabier, O. Eeg-Olofsson, D-2-Hydroxyglutaric
aciduria: further clinical delineation, J. Inherit. Metab. Dis. 22(1999) 404–413.
[2] M.S. van der Knaap, C. Jakobs, G.F. HoVmann, W.L. Nyhan,W.O. Renier, J.A. Smeitink, C.E. Catsman-Berrevoets, O. Hjalm-arson, H. Vallance, K. Sugita, C.M. Bowe, J.T. Herrin, W.J.Craigen, N.R. Buist, D.S. BrookWeld, R.A. Chalmers, D-2-Hydrox-yglutaric aciduria: biochemical marker or clinical disease entity?Ann. Neurol. 45 (1999) 111–119.
[3] P.G. Barth, G.F. HoVman, J. Jaeken, W. Lehnert, F. Hanefeld,A.H. van Gennip, M. Duran, J. Valk, R.B. Schutgens, F.K. Trefz,et al., L-2-Hydroxyglutaric acidaemia: a novel inherited neuromet-abolic disorder, Ann. Neurol. 32 (1992) 66–71.
[4] P.G. Barth, G.F. HoVmann, J. Jaeken, R.J. Wanders, M. Duran,G.A. Jansen, C. Jakobs, W. Lehnert, F. Hanefeld, J. Valk, et al., L-2-Hydroxyglutaric acidemia: clinical and biochemical Wndings in12 patients and preliminary report on L-2-hydroxyacid dehydroge-nase, J. Inherit. Metab. Dis. 16 (1993) 753–761.
[5] E.A. Struys, N.M. Verhoeven, E.E.W. Jansen, H.J. ten Brink, T.G.Burlingame, M. Grupta, L.S. Quang, T. Maher, A.K. Goodwin,E.M. Weerts, C. Jakobs, K.M. Gibson, Gamma-hydroxybutirate(GHB) metabolism to D-2-hydroxyglutarate (D-2-HG) and 4,5-dihydroxyhexanoate (DHHA): further pathomechanisms in succi-natesemialdehyde dehydrogenase (SSADH) deWciency, J. Inherit.Metab. Dis. 27 (2004) 87–P170.
[6] R.A. Chalmers, A.M. Lawson, R.W.E. Watts, A.S. Tavill, J.P.Kamerling, E. Hey, D. Ogilvie, D-2-Hydroxyglutaric aciduria:case report and biochemical studies, J. Inherit. Metab. Dis. 3(1980) 11–15.
[7] W.L. Nyhan, G.D. Shelton, C. Jacobs, B. Holmes, C. Bowe, C.J.Curry, C. Vance, M. Duran, L. Sweetman, D-2-Hydroxyglutaricaciduria, J. Child Neurol. 10 (1995) 137–142.
[8] M. Wajner, C.R. Vargas, C. Funayama, A. Fernandez, M.L. Elias,S.I. Goodman, C. Jakobs, M.S. van der Knaap, D-2-Hydroxyglu-taric aciduria in a patient with a severe clinical phenotype andunusual MRI Wndings, J. Inherit. Metab. Dis. 25 (2002) 28–34.
[9] S.H. Korman, G.S. Salomons, A. Gutman, R. Brooks, C. Jakobs,D-2-Hydroxyglutaric aciduria and glutaric aciduria type 1 in sib-lings: coincidence or linked disorders?, Neuropediatrics 35 (2004)151–156.
[10] K. Sugita, H. Kakinuma, Y. Okajima, A. Ogawa, H. Watanabe, H.Niimi, Clinical and MRI Wndings in a case report of D-2-hydroxy-glutaric acid uria, Brain Dev. 17 (1995) 139–141.
[11] O. Eeg-Olofsson, D-2-Hydroxyglutaric aciduria with cerebral, vas-cular, and muscular abnormalities in a14-year-old boy, J. ChildNeurol. 15 (2000) 488–492.
[12] E.A. Struys, G.S. Salomons, Y. Achouri, E. Van Schaftingen, S.Grosso, W.J. Craigen, N.M. Verhoeven, C. Jakobs, Mutations inthe D-2-hydroxyglutarate dehydrogenase gene cause D-2-hydroxy-glutaric aciduria, Am. J. Hum. Genet. 76 (2005) 358–360.
[13] M. Duran, J.P. Kamerling, H.D. Barkker, H.A. van Gennip, S.K.Wadman, L-2-Hydroxyglutaric aciduria: an inborn error ofmetabolism, J. Inherit. Metab. Dis. 3 (1980) 11–15.
[14] B. Wilcken, J. Pitt, D. Heath, P. Walsh, G. Wilson, N. Buchanan, L-2-Hydroxyglutaric aciduria: three Australian cases, J. Inherit.Metab. Dis. 16 (1993) 501–504.
[15] P. Divry, C. Jakobs, C. Vianey-Saban, K.M. Gibson, H. Michel-akakis, A. Papadimitriou, R. Divari, B. Chabrol, M.A. Cournelle,M.O. Livet, L-2-hydroxyglutaric aciduria: two further cases, J.Inherit. Metab. Dis. 16 (1993) 505–507.
[16] A. Larnaout, F. Hentati, S. Belal, C. Ben Hamida, N. Kaabachi,M. Ben Hamida, Clinical and pathological study of three Tunisiansiblings with L-2-hydroxyglutaric aciduria, Acta Neuropathol.(Berl) 88 (1994) 367–370.
[17] G.F. HoVmann, C. Jakobs, B. Holmes, L. Mitchell, G. Becker, H.P.Hartung, W.L. Nyhan, Organic acids in cerebrospinal Xuid andplasma of patients with L-2-hydroxyglutaric aciduria, J. Inherit.Metab. Dis. 18 (1995) 189–193.

198 A. Latini et al. / Molecular Genetics and Metabolism 86 (2005) 188–199
[18] E. Chen, W.L. Nyhan, C. Jakobs, C.M. Greco, A.J. Barkovich,V.A. Cox, S. Packman, L-2-Hydroxyglutaric aciduria: neuropatho-logical correlations and Wrst report of severe neurodegenerativedisease and neonatal death, J. Inherit. Metab. Dis. 19 (1996) 335–343.
[19] C. Barbot, I. Fineza, L. Diogo, M. Maia, J. Melo, A. Guimaraes,M.M. Pires, M.L. Cardoso, L. Vilarinho, L-2-Hydroxyglutaric aci-duria: clinical, biochemical and magnetic resonance imaging in sixPortuguese pediatric patients, Brain Dev. 19 (1997) 268–273.
[20] J. Fujitake, Y. Ishikawa, H. Fujii, K. Nishimura, K. Hayakawa, F.Inoue, N. Terada, M. Okochi, Y. Tatsuoka, L-2-Hydroxyglutaricaciduria: two Japanese adult cases in one family, J. Neurol. 246(1999) 378–382.
[21] A. Mahfoud, C.L. Dominguez, A. Perez, T. Rodriguez, E. Caniz-ales, V.H. Jaimes, A. Abadi, E.M. Perez, H.B. Belisario, L-2-Hydroxyglutaric aciduria: clinical, biochemical and neuroradio-logical Wndings in two Venezuelan patients, Rev. Neurol. 39 (2004)343–346.
[22] M. Topçu, F. Jobard, S. Halliez, T. Coskun, C. Yalcinkayal, F.O.Gerceker, R.J. Wanders, J.F. Prud’homme, M. Lathrop, M. Ozguc,J. Fischer, L-2-Hydroxyglutaric aciduria: identiWcation of amutant gene C14orf160, localized on chromosome 14q22.1, Hum.Mol. Genet. 13 (2004) 2803–2811.
[23] R. Rzem, M. Veiga-da-Cunha, G. Noel, S. GoVette, M.C. Nasso-gne, B. Tabarki, C. Scholler, T. Marquardt, M. Vikkula, E. VanSchaftingen, A gene encoding a putative FAD-dependent L-2-hydroxyglutarate dehydrogenase is mutated in L-2-hydroxyglu-taric aciduria, Proc. Natl. Acad. Sci. USA 101 (2004) 16849–16854.
[24] P.G. Barth, R.J. Wanders, H.R. Scholte, N. Abeling, C. Jakobs,R.B. Schutgens, P. Vreken, L-2-Hydroxyglutaric aciduria and lac-tic acidosis, J. Inherit. Metab. Dis. 21 (1998) 251–254.
[25] C.G. da Silva, C.A.J. Ribeiro, G. Leipnitz, C.S. Dutra-Filho, A.T.Wyse, C.M. Wannmacher, J.J. Sarkis, C. Jakobs, M. Wajner, Inhi-bition of cytochrome c oxidase activity in rat cerebral cortex andhuman skeletal muscle by D-2-hydroxyglutaric acid in vitro, Bio-chim. Biophys. Acta 1586 (2002) 81–91.
[26] S. Kölker, V. Pawlak, B. Ahlemeyer, J.G. Okun, F. Horster, E.Mayatepek, J. Krieglstein, G.F. HoVmann, G. Kohr, NMDAreceptor activation and respiratory chain complex V inhibitioncontribute to neurodegeneration in D-2-hydroxyglutaric aciduria,Eur. J. Neurosci. 16 (2002) 21–28.
[27] C.G. da Silva, A.R. Bueno, R.B. Rosa, C.S. Dutra Filho, C.M.Wannmacher, A.T. Wyse, M. Wajner, Inhibition of mitochondrialcreatine kinase activity by D-2-hydroxyglutaric acid in cerebellumof young rats, Neurochem. Res. 28 (2003) 1329–1337.
[28] C.G. da Silva, A.R. Bueno, P.F. Schuck, G. Leipnitz, C.A. Ribeiro,C.M. Wannmacher, A.T. Wyse, M. Wajner, D-2-Hydroxyglutaricacid inhibits creatine kinase activity from cardiac and skeletalmuscle of young rats, Eur. J. Clin. Invest. 33 (2003) 840–847.
[29] R. Anan, M. Nakagawa, M. Miyata, I. Higuchi, S. Nakao, M. Sue-hara, M. Osame, H. Tanaka, Cardiac involvement in mitochon-drial diseases, Circulation 91 (1995) 955–961.
[30] S. Bohlega, K. Tanji, F. Santorelli, M. Hirano, A. al-Jishi, S. DiM-auro, Multiple mitochondrial DNA deletions associated withautosomal recessive ophthalmoplegia and severe cardiomyopathy,Neurology 46 (1996) 1329–1334.
[31] J. Marin-Garcia, R. Ananthakrishnan, M.J. Goldenthal, J.J. Filiano,A. Perez-Atayde, Mitochondrial dysfunction in skeletal muscle ofchildren with cardiomyopathy, Pediatrics 103 (1999) 456–459.
[32] J. Houstek, P. Klement, D. Floryk, H. Antonická, J. Hermanská,M. Kalous, H. Hansíková, H. Houst’ková, S.K.R. Chowdhury, S.Rosipal, S. Kmoch, L. Stratilová, J. Zeman, A novel deWciency ofmitochondrial ATPase of nuclear origin, Hum. Mol. Genet. 8(1999) 1967–1974.
[33] E. Fosslien, Mitochondrial medicine—cardiomyopathy caused bydefective oxidative phosphorylation, Ann. Clin. Lab. Sci. 33 (2003)371–395.
[34] N.S. Baker, H.B. Sarnat, R.M. Jack, K. Patterson, D.W. Shaw, S.P.Herndon, D-2-Hydroxyglutaric aciduria: hypotonia, corticalblindness, seizures, cardiomyophathy and cylindrical spirals inskeletal muscle, J. Child Neurol. 12 (1997) 31–36.
[35] A. Cassina, R. Radi, DiVerential inhibitory action of nitric oxideand peroxynitrite on mitochondrial electron transport, Arch. Bio-chem. Biophys. 328 (1996) 309–316.
[36] J.C. Fischer, W. Ruitenbeek, J.A. Berden, J.M. Trijbels, J.H. Veerk-amp, A.M. Stadhouders, R.C. Sengers, A.J. Janssen, DiVerentialinvestigation of the capacity of succinate oxidation in human skel-etal muscle, Clin. Chim. Acta 153 (1985) 23–36.
[37] P. Rustin, D. Chretien, T. Bourgeron, B. Gerard, A. Rotig, J.M.Saudubray, A. Munnich, Biochemical and molecular investiga-tions in respiratory chain deWciencies, Clin. Chim. Acta 228 (1994)35–51.
[38] A.H.V. Schapira, J.M. Cooper, D. Dexter, J.B. Clark, P. Jenner,C.D. Marsden, Mitochondrial complex I deWciency in Parkinson’sdisease, J. Neurochem. 54 (1990) 823–827.
[39] R.W. Browne, D. Armstrong, Reduced glutathione and glutathi-one disulWde, Methods Mol. Biol. 108 (1998) 347–352.
[40] A.J. Kowaltowski, A.E. Vercesi, R.F. Castilho, Mitochondrialmembrane protein thiol reactivity with N-ethylmaleimide or mer-salyl is modiWed by Ca2+: correlation with mitochondrial perme-ability transition, Biochim. Biophys. Acta 1318 (1997) 395–402.
[41] C.S. Dutra-Filho, M. Wajner, E. Gassen, R. Candiago, A. Wihl-elms, H. Malfussi, C.M.D. Wannmacher, EVect of organic acids onin vitro glucose oxidation by cerebral cortex of young rats, Med.Sci. Res. 23 (1995) 25–26.
[42] T. Mosmann, Rapid colorimetric assay for cellular growth andsurvival: application to proliferation and cytotoxicity assays, J.Immunol. Methods 65 (1983) 55–63.
[43] O.H. Lowry, N.J. Rosebrough, A.L. Farr, R.J. Randall, Proteinmeasurement with the Folin phenol reagent, J. Biol. Chem. 193(1951) 265–267.
[44] M. Dixon, E.C. Webb, Enzymes, second ed., Longmans, London,1964.
[45] A. Cornish-Bowden, A simple graphical method for determiningthe inhibition constants of mixed, uncompetitive and non-compet-itive inhibitors, Biochem. J. 137 (1974) 143–144.
[46] S.Y. Wong, A. Matsuno-Yagi, Y. HateW, Kinetics of ATP hydroly-sis by F1-ATPase and the eVects of anion activation, removal oftightly bound nucleotides, and partial inhibition of the ATPase bycovalent modiWcation, Biochemistry 23 (1984) 5004–5009.
[47] Y.M. Milgrom, M.B. Murataliev, P.D. Boyer, Bi-site activationoccurs with the native and nucleotide-depleted mitochondrial F1-ATPase, Biochem. J. 330 (1998) 1037–1043.
[48] F. Antunes, A. Boveris, E. Cadenas, On the mechanism and biol-ogy of cytochrome oxidase inhibition by nitric oxide, Proc. Natl.Acad. Sci. USA 101 (2004) 16774–16779.
[49] R. Radi, A. Cassina, R. Hodara, Nitric oxide and peroxynitriteinteractions with mitochondria, Biol. Chem. 383 (2002) 401–409.
[50] J. Chen, D.R. Petersen, S. Schenker, G.I. Henderson, Formation ofmalondialdehyde adducts in livers of rats exposed to ethanol: rolein ethanol-mediated inhibition of cytochrome c oxidase, AlcoholClin. Exp. Res. 24 (2000) 544–552.
[51] J. Chen, G.I. Henderson, G.L. Freeman, Role of 4-hydroxynone-nal in modiWcation of cytochrome c oxidase in ischemia/reper-fused rat heart, J. Mol. Cell. Cardiol. 33 (2001) 1919–1927.
[52] A.M. Cassina, R. Hodara, J.M. Souza, L. Thomson, L. Castro, H.Ischiropoulos, B.A. Freeman, R. Radi, Cytochrome c nitration byperoxynitrite, J. Biol. Chem. 275 (2000) 21409–21415.
[53] A. Latini, K. Scussiato, R.B. Rosa, S. Llesuy, A. Bello-Klein, C.S.Dutra-Filho, M. Wajner, D-2-Hydroxyglutaric acid induces oxida-tive stress in cerebral cortex of young rats, Eur. J. Neurosci. 17(2003) 2017–2022.
[54] A. Latini, K. Scussiato, R.B. Rosa, S. Llesuy, A. Bello-Klein, C.S.Dutra-Filho, M. Wajner, Induction of oxidative stress by

A. Latini et al. / Molecular Genetics and Metabolism 86 (2005) 188–199 199
L-2-hydroxyglutaric acid in rat brain, J. Neurosci. Res. 74 (2003)103–110.
[55] P.M. Elias, J. Goerke, D.S. Friend, B.E. Brown, Freeze-fractureidentiWcation of sterol-digitonin complexes in cell and liposomemembranes, J. Cell Biol. 78 (1978) 577–596.
[56] A. Lombes, H. Nakase, H.J. Tritschler, B. Kadenbach, E. Bonilla,D.C. DeVivo, E.A. Schon, S. DiMauro, Biochemical and molecu-lar analysis of cytochrome c oxidase deWciency in Leigh’s syn-drome, Neurology 41 (1991) 491–498.
[57] R. Van Coster, A. Lombes, D.C. DeVivo, T.L. Chi, W.E. Dodson,S. Rothman, E.J. Orrechio, W. Grover, G.T. Berry, J.F. Schwartz,et al., Cytochrome c oxidase-associated Leigh syndrome: pheno-typic features and pathogenic speculations, J. Neurol. Sci. 104(1991) 97–111.
[58] B. Kadenbach, M. Hüttemann, S. Arnold, I. Lee, E. Bender, Mito-chondrial energy metabolism is regulated via nuclear-coded sub-units of cytochrome c oxidase, Free Radic. Biol. Med. 29 (2000)211–221.
[59] S. DiMauro, J.R. Mendell, Z. Sahenk, D. Bachman, A. Scarpa,R.M. ScoWeld, C. Reiner, Fatal infantile mitochondrial myophatyand renal dysfunction due to cytochrome c oxidase deWciency,Neurology 30 (1980) 795–804.
[60] P.E. Minchom, R.L. Dormer, I.A. Hughes, D. Stansbie, A.R.Cross, G.A. Hendry, O.T. Jones, M.A. Johnson, H.S. Sherratt,D.M. Turnbull, Fatal infantile mitochondrial myopathy due tocytochrome c oxidase deWciency, J. Neurol. Sci. 60 (1983) 453–463.
[61] D.M. Glerum, B.H. Robinson, R.A. Capaldi, Fibroblasts andcytochrome c oxidase deWciency, in: A. Azzi, Z. Drahota, S. Papa(Eds.), Molecular Basis of Membrane Associated Diseases,Springer-Verlag, Berlin, 1989, pp. 228–238.
[62] B.H. Robinson, Lacticacidemia: biochemical, clinical, and geneticconsiderations, in: H. Harris, K. Hirschhorn (Eds.), Advances inHuman Genetics, Plenum Press, New York, 1989, pp. 151–179.
[63] E.A. Shoubridge, Cytochrome c oxidase deWciency, Am. J. Med.Genet. 106 (2001) 46–52.
[64] S. Arnold, B. Kadenbach, Cell respiration is controlled by ATP,an allosteric inhibitor of cytochrome c oxidase, Eur. J. Biochem.249 (1997) 350–354.
[65] S. Arnold, B. Kadenbach, Intramitochondrial ATP/ADP ratioscontrol cytochrome c oxidase activity allosterically, FEBS Lett.443 (1999) 105–108.
[66] B. Kadenbach, S. Arnold, A second mechanism of respiratorycontrol, FEBS Lett. 447 (1999) 131–134.
[67] V. Geromel, B. Parfait, J.C. von Kleist-Retzow, D. Chretien, A.Munnich, A. Rotig, P. Rustin, The consequences of a mild respira-tory chain deWciency on substrate competitive oxidation in humanmitochondria, Biochim. Biophys. Res. Commum 236 (1997) 643–646.
[68] J. Müller-Hocker, Cytochrome c oxidase deWciency cardiomyo-cytes in the human heart—an age-related phenomenon. Ahistochemical ultracytochemical study, Am. J. Pathol. 134 (1989)1167–1173.
[69] R. Carrozzo, T. Rizza, S. Lucioli, R. Pierini, E. Bertini, F.M. San-torelli, A mitochondrial ATPase6 mutation is associated withLeigh syndrome in a family and aVects proton Xow and adenosinetriphosphate output when modeled in Escherichia coli, Acta Pae-diatr. 93 (2004) 65–67.
[70] E. Ciafaloni, F.M. Santorelli, S. Shanske, T. Deonna, E. Roulet, C.Janzer, G. Pescia, S. DiMauro, Maternally inherited Leigh syn-drome, J. Pediatr. 122 (1993) 419–422.
[71] Y. Tatuch, R.A. Pagon, B. Vlcek, R. Roberts, M. Korson, B.H.Robinson, The 8993 mtDNA mutation: heteroplasmy and clinicalpresentation in three families, Eur. J. Hum. Genet. 2 (1994) 35–43.
[72] G. Manfredi, J. Fu, J. Ojaimi, J.E. Sadlock, J.Q. Kwong, J. Guy,E.A. Schon, Rescue of a deWciency in ATP synthesis by transfer ofMTATP6, a mitochondrial DNA-encoded gene, to the nucleus,Nat. Genet. 30 (2002) 394–399.
[73] D. Zhang, J.L. Mott, P. Farrar, J.S. Ryerse, S.W. Chang, M. Ste-vens, G. Denniger, H.P. Zassenhaus, Mitochondrial DNA muta-tions activate the mitochondrial apoptotic pathway and causedilated cardiomyopathy, Cardiovasc. Res. 57 (2003) 147–157.
[74] I.J. Holt, A.E. Harding, R.K.H. Petty, J.A. Morgan-Hughes, A newmitochondrial disease associated with mitochondrial DNA het-eroplasmy, Am. J. Hum. Genet. 46 (1990) 428–433.
[75] J. Houstek, P. Klement, D. Floryk, H. Antonická, J. Hermanská,M. Kalous, H. Hansíková, H. Houst’ková, S.K.R. Chowdhury, S.Rosipal, S. Kmoch, L. Stratilová, Z. Zeman, A novel deWciency ofmitochondrial ATPase of nuclear origin, Hum. Mol. Gen. 8 (1999)1967–1974.
[76] M. Erecinska, S. Cherian, I.A. Silver, Energy metabolism in mamma-lian brain during development, Prog. Neurobiol. 73 (2004) 397–445.
[77] V.A. Saks, A.V. Kuznetsov, V.V. Kuprianov, M.V. Miceli, W.E.Jacobus, Creatine kinase of rat heart mitochondria. The demon-stration of functional coupling to oxidative phosphorylation in aninner membrane-matrix preparation, J. Biol. Chem. 260 (1985)7757–7764.




















