
Microtubule disassembly induces cytoskeletal remodeling and lung vascular barrier dysfunction: Role...
16
JOURNAL OF CELLULAR PHYSIOLOGY 201:55–70 (2004) Microtubule Disassembly Induces Cytoskeletal Remodeling and Lung Vascular Barrier Dysfunction: Role of Rho-Dependent Mechanisms ANNA A. BIRUKOVA, 1 KSENYA SMUROVA, 1,2 KONSTANTIN G. BIRUKOV, 1 PETER USATYUK, 1 FENG LIU, 1 KOZO KAIBUCHI, 3 ANILA RICKS-CORD, 1 VISWANATHAN NATARAJAN, 1 IRINA ALIEVA, 2 JOE G.N. GARCIA, 1 AND ALEXANDER D. VERIN 1 * 1 Division of Pulmonary and Critical Care Medicine, Johns Hopkins University School of Medicine, Baltimore, Maryland 2 Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Vorobievy Gory, Russia 3 Department of Cell Pharmacology, Nagoya University Graduate School of Medicine, Nagoya, Japan Barrier dysfunction of pulmonary endothelial monolayer is associated with dra- matic cytoskeletal reorganization, activation of actomyosin contractility, and gap formation. The linkage between the microtubule (MT) network and the contractile cytoskeleton has not been fully explored, however, clinical observations suggest that intravenous administration of anti-cancer drugs and MT inhibitors (such as the vinca alkaloids) can lead to the sudden development of pulmonary edema in breast cancer patients. In this study, we investigated the crosstalk between MT and actomyosin cytoskeleton and characterized specific molecular mechanisms of endothelial cells (EC) barrier dysfunction induced by MT inhibitor nocodazole (ND). Our results demonstrate that MT disassembly by ND induced rapid decreases in transendothelial electrical resistance (TER) and actin cytoskeletal remodeling, indicating EC barrier dysfunction. These effects involved ND-induced activation of Rho GTPase. Rho-mediated activation of its downstream target, Rho-kinase, induced phosphorylation of Rho-kinase effector EC MLC phosphatase (MYPT1) at Thr 696 and Thr 850 resulting in MYPT1 inactivation. Phosphatase inhibition leaded to accumulation of diphospho-MLC, which induced acto-myosin polymerization, stress fiber formation and gap formation. Inhibition of Rho-kinase by Y27632 abolished ND-induced MYPT1 phosphorylation, MLC phosphorylation, and stress fiber formation. In addition, MT preservation via the MT stabilizer paclitaxel, Rho inhibition (via C3 exotoxin, or dominant negative (DN)-Rho, or DN-Rho-kinase) attenuated ND-induced TER decreases, stress fiber formation and MLC phosphor- ylation. Collectively, our results demonstrate a leading role for Rho-dependent mechanisms in crosstalk between the MT and actomyosin cytoskeleton, and suggest Rho-kinase and MYPT1 as major Rho effectors mediating pulmonary EC barrier disruption in response to ND-induced MT disassembly. J. Cell. Physiol. 201: 55–70, 2004. ß 2004 Wiley-Liss, Inc. A key vascular endothelial cells (EC) function is to regulate the exchanges across the capillary wall between circulating blood and the interstitial fluid. Reorganization of the endothelial cytoskeleton, which is composed of actin filaments, microtubules (MT), and intermediate filaments, leads to alteration in cell shape and provides a structural basis for increase of vascular permeability, which has been implicated in the patho- genesis of many diseases including asthma, sepsis, and the acute lung injury (Garcia et al., 1995; Lum and Malik, 1996; Dudek and Garcia, 2001). While the importance of MT component in the maintenance of EC barrier function remains largely unexplored, intra- venous administration of anti-cancer drugs and MT ß 2004 WILEY-LISS, INC. Contract grant sponsor: National Heart, Lung, and Blood Institutes; Contract grant numbers: HL67307, HL68062, HL58064; Contract grant sponsor: American Heart Association (to ADV); Contract grant sponsor: Russian Foundation of Biomedical Research (to IBA); Contract grant numbers: RFBR- 03-04-48035, MD-244.2003.04. *Correspondence to: Alexander D. Verin, Division of Pulmonary and Critical Care Medicine, Johns Hopkins University School of Medicine, 5200 Eastern Avenue, MFL Center Tower, Room 676, Baltimore, MD 21224. E-mail: [email protected] Received 1 October 2003; Accepted 12 December 2003 DOI: 10.1002/jcp.20055
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Microtubule disassembly induces cytoskeletal remodeling and lung vascular barrier dysfunction: Role...

JOURNAL OF CELLULAR PHYSIOLOGY 201:55–70 (2004)
Microtubule Disassembly Induces CytoskeletalRemodeling and Lung Vascular Barrier Dysfunction:
Role of Rho-Dependent Mechanisms
ANNA A. BIRUKOVA,1 KSENYA SMUROVA,1,2 KONSTANTIN G. BIRUKOV,1 PETER USATYUK,1
FENG LIU,1 KOZO KAIBUCHI,3 ANILA RICKS-CORD,1 VISWANATHAN NATARAJAN,1 IRINA ALIEVA,2
JOE G.N. GARCIA,1 AND ALEXANDER D. VERIN1*1Division of Pulmonary and Critical Care Medicine,
Johns Hopkins University School of Medicine, Baltimore, Maryland2Belozersky Institute of Physico-Chemical Biology,
Lomonosov Moscow State University, Vorobievy Gory, Russia3Department of Cell Pharmacology,
Nagoya University Graduate School of Medicine, Nagoya, Japan
Barrier dysfunction of pulmonary endothelial monolayer is associated with dra-matic cytoskeletal reorganization, activation of actomyosin contractility, and gapformation. The linkage between the microtubule (MT) network and the contractilecytoskeleton has not been fully explored, however, clinical observations suggestthat intravenous administration of anti-cancer drugs andMT inhibitors (such as thevinca alkaloids) can lead to the sudden development of pulmonary edema in breastcancer patients. In this study, we investigated the crosstalk between MT andactomyosin cytoskeleton and characterized specific molecular mechanisms ofendothelial cells (EC) barrier dysfunction induced by MT inhibitor nocodazole(ND).Our results demonstrate thatMTdisassembly byND induced rapid decreasesin transendothelial electrical resistance (TER) and actin cytoskeletal remodeling,indicating EC barrier dysfunction. These effects involved ND-induced activationof Rho GTPase. Rho-mediated activation of its downstream target, Rho-kinase,induced phosphorylation of Rho-kinase effector EC MLC phosphatase (MYPT1) atThr696 and Thr850 resulting in MYPT1 inactivation. Phosphatase inhibition leadedto accumulation of diphospho-MLC, which induced acto-myosin polymerization,stress fiber formation and gap formation. Inhibition of Rho-kinase by Y27632abolished ND-induced MYPT1 phosphorylation, MLC phosphorylation, and stressfiber formation. In addition, MT preservation via the MT stabilizer paclitaxel, Rhoinhibition (via C3 exotoxin, or dominant negative (DN)-Rho, or DN-Rho-kinase)attenuated ND-induced TER decreases, stress fiber formation and MLC phosphor-ylation. Collectively, our results demonstrate a leading role for Rho-dependentmechanisms in crosstalk between the MT and actomyosin cytoskeleton, andsuggest Rho-kinase and MYPT1 as major Rho effectors mediating pulmonary ECbarrier disruption in response to ND-induced MT disassembly. J. Cell. Physiol.201: 55–70, 2004. � 2004 Wiley-Liss, Inc.
A key vascular endothelial cells (EC) function is toregulate the exchanges across the capillary wallbetween circulating blood and the interstitial fluid.Reorganization of the endothelial cytoskeleton, which iscomposed of actin filaments, microtubules (MT), andintermediate filaments, leads to alteration in cell shapeand provides a structural basis for increase of vascularpermeability, which has been implicated in the patho-genesis of many diseases including asthma, sepsis, andthe acute lung injury (Garcia et al., 1995; Lum andMalik, 1996; Dudek and Garcia, 2001). While theimportance of MT component in the maintenance ofEC barrier function remains largely unexplored, intra-venous administration of anti-cancer drugs and MT
� 2004 WILEY-LISS, INC.
Contract grant sponsor: National Heart, Lung, and BloodInstitutes; Contract grant numbers: HL67307, HL68062,HL58064; Contract grant sponsor: American Heart Association(to ADV); Contract grant sponsor: Russian Foundation ofBiomedical Research (to IBA); Contract grant numbers: RFBR-03-04-48035, MD-244.2003.04.
*Correspondence to: Alexander D. Verin, Division of Pulmonaryand Critical Care Medicine, Johns Hopkins University School ofMedicine, 5200 Eastern Avenue, MFL Center Tower, Room 676,Baltimore, MD 21224. E-mail: [email protected]
Received 1 October 2003; Accepted 12 December 2003
DOI: 10.1002/jcp.20055

inhibitors (such as the vinca alkaloids) can lead to thesudden development of pulmonary edema in breastcancer patients suggesting the potential importance ofMT network for regulation of lung permeability (Cattanand Oberg, 1999). Substantial studies, including ourown, have verified that EC barrier maintenance is underclose regulation by competing contractile and tetheringforces generated by the cytoskeletal motor proteins(Lum and Malik, 1996; Dudek and Garcia, 2001;Bogatcheva et al., 2002). The linkage between the MTnetwork and contractile cytoskeleton is not fullyexplored. Recent studies demonstrated the interplaybetween MT and actin cytoskeleton is required during awide variety of processes, including vesicle and orga-nelle transport, tissue cell migration and cytokinesis(Gundersen and Cook, 1999; Waterman-Storer andSalmon, 1999). Disruption of MTs by either colcemid,colchicine, nocodazole (ND), or vinblastine caused for-mation of stress fibers and vinculin-containing focaladhesions (Danowski, 1989; Enomoto, 1996). Subse-quent studies showed that ND-induced isometric con-traction on fibroblasts correlated well with the levelof ND-induced MLC phosphorylation (Kolodney andElson, 1995). A notion supported by further workindicating that MT depolymerization activates Rho/Rho kinase signaling cascade in cultured cell systems(Enomoto, 1996; Zhang et al., 1997; Elbaum et al., 1999;Platts et al., 2002). In addition, colchicine-induced MTdissolution significantly enhanced migration of humanneutrophils accompanied by increase in MLC phosphor-ylaiton, which was attenuated by taxol or Rho kinaseinhibitor, Y-27632 (Niggli, 2003). These results areconsistent with previously described RhoA activationduring cell migration (Worthylake et al., 2001).These data suggest an important role for Rho GTPase-mediated signaling in the cellular responses inducedby MT disruption.
Our previous findings have clearly demonstrated acritical role for MLC phosphorylation in pulmonaryendothelial barrier dysfunction in induced by MTinhibitors, and suggested the involvement of Rho-dependent mechanisms in signaling events initiatedby disruption of MTs (Verin et al., 2001). In this study,we detail the molecular mechanisms of MT-mediatedbarrier regulation with focus on the crosstalk betweenactin filaments and MTs and specifically explored therole of Rho GTP-ase in the link between MTs and ECcontractility.
MATERIALS AND METHODSReagents
Culture medium 199 was obtained from Gibco-BRL(Chagrin Falls, OH). Colustrum-free bovine serum waspurchased from Irvine Scientific (Santa Ana, CA). ECgrowth supplement was from Collaborative Research(Bedford, MA). Antibiotic–antimycotic mixture andnon-essential amino acids were purchased from K.C.Biologicals (Lenexa, KA) and Gibco-BRL, respectively.Unless specified, biochemical reagents were obtainedfrom Sigma (St. Louis, MO). Fura-2, AM (cell perme-able), 4-bromo-A23187, Pluronic acid (F-127), BAPTAAM, Texas Red phalloidin, Alexa 488, and Alexa594 conjugated secondary antibodies were purchasedfrom Molecular Probes (Eugene, OR). Primary anti-
bodies were obtained as follows: MYPT1 polyclonal, b-tubulin, and myosin A antibodies were from Covance,Inc. (Berkeley, CA), site-specific phospho-MYPT1 anti-bodies were from Upstate Biotechnology (Lake Placid,NY), RhoA, HA, and c-Myc rabbit polyclonal antibodieswere from Santa Cruz Biotechnology (Santa Cruz, CA),diphospho-MLC antibodies were from Cell Signalling(Beverly, MA), VE-Cadherin, Rho Kinase antibodieswere from BD Biosciences (Lexington, KY), acetylatedtubulin antibodies were from Accurate Chemical andScientific Corporatoin (Westbury, NY). Rho-kinasespecific inhibitor Y27632, C3 exoenzyme, ML-7, KN-93, PP2, Genistein, LY294002 were obtained fromCalbiochem (La Jolla, CA).
Cell cultures
Bovine pulmonary artery endothelial cells (BPAEC)were obtained frozen at 16 passages from AmericanType Tissue Culture Collection (Rockville, MD; cultureline-CCL 209), and were utilized at passages 19–24.Cells were cultured in M-199 media (Gibco-BRL) sup-plemented with 20% (v/v) colostrums-free bovine serum(Irvine Scientific), 15 mg/ml EC growth supplement(Collaborative Research), 1% antibiotic, and antimyco-tic (10,000 U/ml penicillin, 10 mg/ml streptomycin, and25 mg/ml amphotericin B; K.C. Biologicals, Lenexa, KA),and 0.1 mM non-essential amino acids (Gibco-BRL) andmaintained at 378C in humidified atmosphere of 5%CO2–95% air. The EC grew to contact-inhibited mono-layers with the typical cobblestone morphology. Humanpulmonary artery endothelial cells (HPAEC) wereobtained from Clonetics, BioWhittaker, Inc. (Frederick,MD), propagated in culture medium EGM-2 and used atpassages 6–10.
Immunofluorescent staining
Endothelial cells grown on glass coverslips were fixedafter agonist treatment in 3.7% formaldehyde solutionin PBS for 10 min at 48C, washed three times with PBS,permeabilized with 0.2% triton X-100 in PBS-Tween(PBST) for 30 min at room temperature, and blockedwith 2% BSA in PBST for 30 min. Incubation withantibody of interest was performed in blocking solution(2% BSA in PBST) for 1 h at room temperature followedby staining with either Alexa 488-, or Alexa 594-conjugated secondary antibodies (Molecular Probes).Actin filaments were stained with Texas Red-conju-gated phalloidin (Molecular Probes) for 1 h at roomtemperature. After immunostaining, the glass slideswere analyzed using a Nikon video-imaging systemNikon Instech Co., Tokyo, Japan) consisting of a phasecontrast inverted microscope Nikon Eclipse TE2000connected to Hamamatsu (Hamamatsu Photonics K.K.,Hamamatsu, Japan) digital camera and image proces-sor. The images were recorded and processed usingAdobe Photoshop 6.0 program, using a Pentium III PC.
Image analysis of gap formation andstress fiber formation
Texas Red-stained EC monolayers stimulated witheither ND or vehicle were viewed under microscopeusing 60�/1.40 objective, and images captured asdescribed above. The 16-bit images were analyzed usingMetaVue 4.6 (Universal Imaging, Downington, PA).
56 BIRUKOVA ET AL.

Images were differentially segmented between gaps(black) and cells (highest gray value) based on imagegrayscale levels. The gap formation was expressed as aratio of the gap area to the area of the whole image.Similarly, actin fibers were marked out, and the ratio tothe cell area covered by stress fibers to the whole cellarea was determined. The same technique was used formeasurements of focal adhesions, assembled MTs,and amount of phosphorylated MLC. The values werestatistically processed using Sigma Plot 7.1 (SPSSScience, Chicago, IL) software.
Myosin light chain (MLC) phosphorylation assay
Assays were performed as it was previously describedin detail (Garcia et al., 1995; Verin et al., 2001). Briefly,after incubation with the agonist of interest or vehiclecontrols EC monolayers were lysed in 10% trichloroace-tic acid, and the precipitates homogenated and sub-jected to urea polyacrylamide gel electrophosphoresisfollowed by immunoblotting with anti-MLC antibody.Immuno-reactive proteins were detected using en-hanced chemiluminescent detection system (ECL,Amersham, Little Chalfront, Buckinghamshire, UK)and the separated unphosphorylated, monophosphory-lated and diphosphorylated forms of MLC quantified bylaser scanning densitometry.
Western immunoblotting
Protein extracts were separated by SDS–PAGE on10% gels, transferred to nitrocellulose membrane (30 Vfor 18 h, or 90 V for 2 h), and probed with specificantibodies as previously described (Verin et al., 2001).Immunoreactive proteins were detected using enhancedchemiluminescent detection system (ECL) according tothe manufacturer’s protocol (Amersham).
Rho kinase phosphorylation assay
Phosphorylation of Rho kinase induced by MTdisruption was measured in Rho kinase immunopreci-pitates obtained from 32P-labeled cells. EC monolayersin 60 mm dishes were serum starved in PO4
� -freeDMEM for 16 h followed by radioactive labelingwith g-32P-orthophosphate for 4 h. After rinsing withPO4
�-free DMEM, cells were preincubated with taxol(10 mM), or vehicle controls for 30 min, and then treatedwith ND (1 mM) for 30 min. Following a brief rinse withPBS, cells were lysed in boiling lysis buffer containing10 mM Tris pH 7.4, 1 mM sodium ortho-vanadate and1% SDS. Cell lysates were transferred into microcen-trifuge tubes and boiled for 5 min. Cell debris wasremoved by centrifugation at 16,000g for 5 min. Rhokinase was immunoprecipitated from supernatantsusing 20 mg of monoclonal anti-Rho kinase antibodyfollowed by 1-h incubation with pre-equilibrated ProteinG 4 Fast Flow Sepharose (Amersham) at 48C with gentleagitation. After four-time washing with immunopreci-pitation buffer (20 mM Tris pH 7.4, 300 mM NaCl, 2 mMEDTA, 2 mM EGTA, 0.4 mM sodium ortho-vanadate,2% Triton X-100, 1% NP-40), the immunoprecipitatedcomplex was resuspended in 2� SDS sample buffer andboiled for 5 min. The supernatant was subjected toelectrophoresis on 7% SDS–PAGE, protein transferredto nitrocellulose, and exposed to phosphoimager plateovernight. The intensity of phosphorylated Rho kinase
bands were quantified using Molecular DynamicsPhosphoimager 445 SI and the total Rho kinase proteinfrom the same blot was detected by Western blottingusing monoclonal anti-Rho kinase antibody.
Measurement of [Ca2þ]i
Human or bovine PAECs, grown on glass cover slips(�95% confluence) were loaded with 5 mM Fura-2 AM(Grynkiewicz et al., 1985) in 1 ml of basic medium (116mM NaCl, 5.37 mM KCl, 26.2 mM NaHCO3, 1.8 mMCaCl2, 0.81 mM MgSO4, 1.02 mM NaHPO4, 5.5 mMglucose, 10 mM HEPES/HCl, pH 7.4) supplementedwith 0.1% BSA and 0.03% Pluronic F-217. Cells wereincubated at 378C for 15 min in 95% O2 and 5% CO2,rinsed twice and inserted diagonally in the 1.0 cm acryliccuvettes filled with 3 ml basic medium at 378C. Fura-2fluorescence was measured with an Aminco-BowmanSeries 2 luminescence spectrometer (SLM/Aminco,Urbana, IL) at excitation wavelengths of 340 and380 nm and emission wavelength of 510 nm. Intracel-lular [Ca2þ]i in nM was calculated from the 340/380 ratiousing calibration curves and software.
Expression plasmids and transfection protocol
Plasmids encoding dominant negative Rho (N19Rho)and dominant negative Rho-kinase (a RB-PH(TT)mutant, which is the C-terminal fragment of Rho-kinasemutated at Rho-binding sites) have been previouslydescribed (Amano et al., 1998). EC grown in D35 petridishes at 70% confluence were incubated with 2 ml ofOPTI-MEM medium containing 2 mg DNA and 20 ml ofFugene 6 (Boehringer Mannheim-Roche, Indianapolis,IN) for 6 h in CO2 incubator at 378C. Following washing(DMEMþ10% FCS) cells were incubated for additional24 h, and used for experiments with ND stimulation.
Rho activation assay
Rho activation in EC culture was analyzed using Rhoassay kit available from Upstate Biotechnology. Briefly,pulmonary EC grown in 100 mm petri dishes wereserum-starved for 2 h followed by stimulation with ND(0.5 mM) for the indicated periods of time. At the end ofexperiment, cells were rinsed with ice-cold PBS, and500 ml of lysate buffer was added to each dish. Cells werescraped, and cell lysates were prepared according tomanufacturer’s protocol. After centrifugation at 10,000gfor 10 min, supernatants were incubated with rhotekinRho-binding peptide immobilized on agarose, and acti-vated GTP-Rho bound to rhotekin-agarose was detectedby Western blot with anti-Rho antibody.
Measurement of transendothelial electricalresistance (TER)
The cellular barrier properties were measured usingthe highly sensitive biophysical assay with an electricalcell–substrate impedance sensing system (ECIS)(Applied Biophysics, Troy, NY) described previously(Garcia et al., 1997; Verin et al., 2001).
Neutrophil migration assay
Human neutrophils (PMNs) were purified, loaded intotranswell chambers, and migration was initiated byLTB4 as previously described (Garcia et al., 1998). Tostudy the effect of ND on transendothelial migration
MICROTUBULES AND ENDOTHELIAL BARRIER DYSFUNCTION 57

of neutrophils, EC were stimulated with 0.5 mM NDalone, or in combination with LTB4. Measurementswere performed as previously described (Garcia et al.,1998).
Statistical Analysis
Results are expressed as means�SD of three to fiveindependent experiments. Stimulated samples werecompared with controls by unpaired Student’s t-test.For multiple-group comparisons, one-way analysis ofvariance (ANOVA) followed by the post-hoc Fisher’stest were used. P<0.05 was considered statisticallysignificant.
RESULTSEffect of MT disruption on human pulmonary
endothelial cell barrier function
Our previous studies have demonstrated that disrup-tion of MT caused increased permeability of the bovineendothelial monolayer (Verin et al., 2001). In thepresent study, we examined the effect of MT inhibitorson barrier function in human lung endothelium andexplored cytoskeletal and signaling mechanisms of ECbarrier dysfunction induced by MT disassembly. First,we monitored electrical resistance across human ECmonolayers after treatment with two structurallydifferent MT inhibitors, ND and vinblastine (VB). BothND (Fig. 1, part A) and VB (Fig. 1, part B) induced dose-dependent and sustained decreases in HPAEC trans-monolayer electrical resistance (TER) with maximaldecline by 30 min with 0.5 mM ND and 0.1 mM VB. Thesedata demonstrate greater sensitivity of human pulmon-ary EC to MT inhibitors, as compared to bovinepulmonary EC described in our previous study (Verinet al., 2001). MT disassembly not only increased HPAECpermeability for micromolecules, but also facilitatedtransendothelial neutrophil migration. In Figure 1,parts C and D depicts the enhanced neutrophil migra-tion across HPAEC monolayers pretreated with ND inresponse to potent neutrophil chemoattractant leuko-triene B4 (LTB4), as compared to HPAEC treated withvehicle.
Effect of MT alteration on MT structure andcytoskeletal rearrangement
Using bovine pulmonary EC, we have previouslydemonstrated direct crosstalk between MT disassemblyand actin cytoskeletal remodeling (Verin et al., 2001;Petrache et al., 2003). To confirm that the effect of MTinhibitors on human pulmonary EC barrier propertiescan be attributed to MT disassembly, we next examinedthe effect of ND on MT structure. In Figure 2, part Ademonstrates that in control HPAEC MT organize intofaint lattice network, and ND treatment completelydisrupted MTs. MT destabilization and disassemblyafter ND treatment was further confirmed by thestaining of acetylated tubulin representing the stableMT pool (Fig. 2, part B). Treatment of HPAEC with NDcaused complete disappearance of acetylated microtu-bules (Fig. 2, part C) reflected by quantitative analysis ofMT pool in control HPAEC and in cells treated withdifferent doses of ND. Thus, dose-dependent MTdisassembly in response to ND treatment accompaniedby decreased pool of stable MT is highly consistent with
the dose-dependent increase in HPAEC permeabilityinduced by ND (Fig. 1, parts A, B).
We have previously demonstrated that MT disassem-bly induces stress fiber formation (Verin et al., 2001). Wenext examined the direct relationship between rapid MTdynamics and microfilament reorganization and againobserved that ND treatment of EC monolayers causedMT dissolution and stress fiber formation (Fig. 3,parts A, B). Removal of MT inhibitors caused rapidrestoration of MT structure (evident at 5 min after NDwithdrawal and completed by 30 min) (Fig. 3, parts A,B), which was accompanied by rapid disappearance ofstress fibers, restoration of cortical actin ring by 30 min,and disappearance of paracellular gaps characteristic ofnon-contractile EC phenotype and indicating barrierrestoration. Direct TER measurements confirmed rapidEC barrier restoration after ND withdrawal (data notshown). Reversibility of ND-induced changes in cytos-keleton and permeability suggests that barrier dysfunc-tion is not due to cytotoxic effect of ND. These data areconsistent with our previous findings that ND at con-centrations, which were used in this study did not affectcell viability (Verin et al., 2001). Using MT stabilizers,taxol (Fig. 3, part D) and epothilon B (data not shown),we next demonstrated, that stabilization of MT networkby both reagents abrogated ND-induced MT disassem-bly and significantly attenuated ND-induced F-actinrearrangement (Fig. 3, part C). Quantitative analysisof gap formation (Fig. 3, part D) demonstrates thatstabilization of MT by taxol preserved EC monolayersagainst ND-induced gap formation. These effects wereassociated with abrogation of ND-induced TER decline(Verin et al., 2001). Thus, our data demonstrate a tightlinkage between MT dynamics and precise arrangementof the actin cytoskeleton.
Rearrangements of actin cytoskeleton and cellcontacts induced by MT disruption
Effects of MT disruption on actomyosin remodelingwere next examined by immunofluorescent staining.ND-induced formation of massive stress fibers wasaccompanied by robust myofilament assembly (Fig. 4,parts A, B). Microfilaments were co-localizad with actinstress fibers reflecting contractile phenotype (mergedimages on Fig. 4, part C). We and others have previouslyshown the important role of MLC phosphorylation inthe agonist-induced actomyosin contraction leading tobarrier compromise (Wysolmerski and Lagunoff, 1990;Garcia et al., 1995; Verin et al., 2001). Consistent withthese data increased actomyosin interaction was accom-panied by increases in MLC phosphorylation andaccumulation of diphosphorylated MLC on the acto-myosin bundles (Fig. 4, part D). Using MetaVue 4.6software, we performed quantitative analysis of phos-phorylated MLC and have demonstrated that NDstimulation induced 2.6-fold increases in MLC phos-phorylation compared to untreated cells (22.03% forvehicle and 56.89% for ND treatment). These im-munofluorescent images clearly indicate intracellularmorphological changes associated with activation ofactomyosin contraction, cell retraction, and barrierdysfunction induced by MT disruption.
Several studies suggest the important role of cell–cellcontacts (adherens junctions) and cell–substrate con-
58 BIRUKOVA ET AL.

tacts (focal adhesions) (Dejana, 1997; Schaphorstet al., 1997) in the regulation of agonist-inducedhyperpermeability. Immunofluorescent staining of con-trol and ND-stimulated EC showed dramatic remodel-ing of both adherens junctions and focal adhesions inresponse to MT disassembly. In Figure 5, part A depictsstaining of control and ND-treated EC for VE-Cadherin,the major protein of adherens junctions, and demon-strates that MT disassembly caused a loss of adherensjunction integrity. In Figure 5, part B shows immuno-fluorescent staining for vinculin, an essential compo-nent of focal adhesions. Using MetaVue 4.6 software,
we performed quantitative analysis of focal adhesionsas described in Materials and Methods. Our resultsindicate that ND-induced MT disruption caused for-mation of enlarged focal adhesions (0.32�0.10 mm2 forunstimulated cells, and 0.63�0.17 for ND-treatedcells) that serve as anchoring sites for actomyosin.Taken together, these data demonstrate that MTdisruption induces dramatic barrier dysfunction inpulmonary endothelium, which involves actomyosin-driven contractile response, activation of focal adhe-sions, and partial disassembly of adherens junctioncomplexes.
Fig. 1. Effect of microtubule (MT) inhibitors on endothelial cells (EC)barrier function. Parts A, B: EC were plated on gold microelectrodesand were cultured to confluence. Growth medium was replaced withserum-free Opti-MEM, and after equilibration and stabilization,transendothelial electrical resistance (TER) was monitored for severalhours. Results are representative of three independent experiments.Part A: At the time indicated by the arrow, human pulmonaryendothelial cells (HPAEC) were treated with either vehicle (0.1%DMSO) or nocodazole (ND) at different concentrations, and TER wasmonitored for 2 h. Part B: At the time indicated by the arrow, HPAECwere treated with either vehicle (Opti-MEM) or vinblastine at
different concentrations, and TER was monitored for 2 h. Parts C,D: Neutrophil migration assay was performed as previously described(Garcia et al., 1998). Part C shows results of quantitative analysis ofbasal and LTB4-induced (5 mM) neutrophil transmigration throughthe EC monolayers pretreated with either vehicle, or ND (0.5 mM).Results are representative of five independent experiments. Part Ddepicts images of neutrophils migrated through EC monolayers withand without ND treatment, in response to LTB4 as chemoattractant.[Color figure can be viewed in the online issue, which is available atwww.interscience.wiley.com.]
MICROTUBULES AND ENDOTHELIAL BARRIER DYSFUNCTION 59

Fig. 2. Effect ND on MT structure. HPAEC grown on glass coverslipswere challenged either with vehicle (0.1% DMSO) or ND (0.5 mM) for30 min. After stimulation, cells were fixed and stained for b-tubulin(part A), or acetylated tubulin (part B) with specific antibodies.Results are representative of three independent experiments. Part C:MT dissolution induced by ND was assessed by morphometric analysisof b-tubulin-stained HPAEC performed using MetaVue software, as
described in Materials and Methods. MTs were marked out, and theratio of the area covered by MTs to the whole cell area was calculated.Data are expressed as % of control corresponding to non-stimulatedcells and represent results of three independent experiments. [Colorfigure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
60 BIRUKOVA ET AL.

Fig. 3. Effect of MT alteration on EC cytoskeletal structure. Parts A,B: Confluent bovine pulmonary artery endothelial cells (BPAEC)grown on glass coverslips were challenged with either vehicle (0.1%DMSO) or ND (1 mM) for 30 min. To remove ND medium was replacedwith growth medium for indicated periods of time. Cells were fixed andstained with b-tubulin antibody (part A), or Texas Red phalloidin forF-actin (part B). Results are representative of three independentexperiments. Part C: Confluent BPAEC grown on glass coverslipswere pretreated with either vehicle (0.1% DMSO) or taxol (5 mM) for30 min, and then treated with either vehicle (0.1% DMSO) or ND
(1 mM) for 30 min. After stimulation cells were fixed and stained withTexas Red phalloidin for F-actin. Shown are representative results offive independent experiments. Part D: Gap formation was assessed byincreases in the ratio of the integrated gap area to the area of thewhole image. Actin fibers were marked out, and the ratio of the areacovered by stress fibers to the whole cell area was calculated. Data areexpressed as % of control corresponding to non-stimulated cells andrepresent results of three independent experiments. [Color figure canbe viewed in the online issue, which is available at www.interscience.wiley.com.]
MICROTUBULES AND ENDOTHELIAL BARRIER DYSFUNCTION 61

Fig. 4. Effect of MT inhibitors on EC cytoskeletal organization.BPAEC grown on glass coverslips were challenged either with vehicle(0.1% DMSO) or ND (1 mM) for 30 min. After stimulation, cells werefixed and stained with Texas Red phalloidin for F-actin (part A), ornon-muscle myosin A (part B). Part C represents merged images fromparts A and B and demonstrates increase in association of myosin andF-actin stress fibers, correlating with activation of acto-myosin driven
contraction. Results are representative of three independent experi-ments. Part D: Immunofluorescent images of EC stained with anti-diphospho-MLC antibody. Inset shows Western blot detection ofdiphospho-MLC in EC treated with either vehicle or ND (1 mM) for30 min. Results are representative of three independent experiments.[Color figure can be viewed in the online issue, which is available atwww.interscience.wiley.com.]
62 BIRUKOVA ET AL.

Formation of central stress fibers and actomyosincontraction resulting in cell retraction and barrierdysfunction is opposed by tethering forces applied byintercellular junctions and cortical actin structures(Ingber, 1997). We have previously demonstrated thatplatelet-derived phospholipid, sphingosine 1-phosphate,attenuates thrombin and LPS-induced permeabilityin vitro and in vivo and exerts its barrier-protective effecton endothelial monolayers via rac-dependent peripheralactin remodeling and enhancement of peripheral actinring (Garcia et al., 2001; Shikata et al., 2003). Our resultssuggest, that HPAEC pretreatment with sphingosine 1-phosphate prior to ND challenge significantly attenu-ated ND-induced maximal drop in TER and completelyreversed ND-induced TER decline (Fig. 6) indicatingpotentially important role of the interplay betweencortical actin and central stress fibers in regulation ofendothelial permeability.
Role of intracellular [Ca2þ] on ND-induced stressfiber formation and MLC phosphorylation
We and others have previously shown the importantrole of MLC phosphorylation in the agonist-inducedactomyosin contraction leading to barrier compromise(Garcia et al., 1995; Goeckeler and Wysolmerski, 1995;Verin et al., 2001). Our previous studies have demon-strated that MT disassembly increased MLC phosphor-ylation in a time-dependent manner, which correlatedwith EC permeability increases, however, ND-inducedMT disruption did not affect MLCK activity (Verin et al.,2001). To further elucidate mechanisms underlying
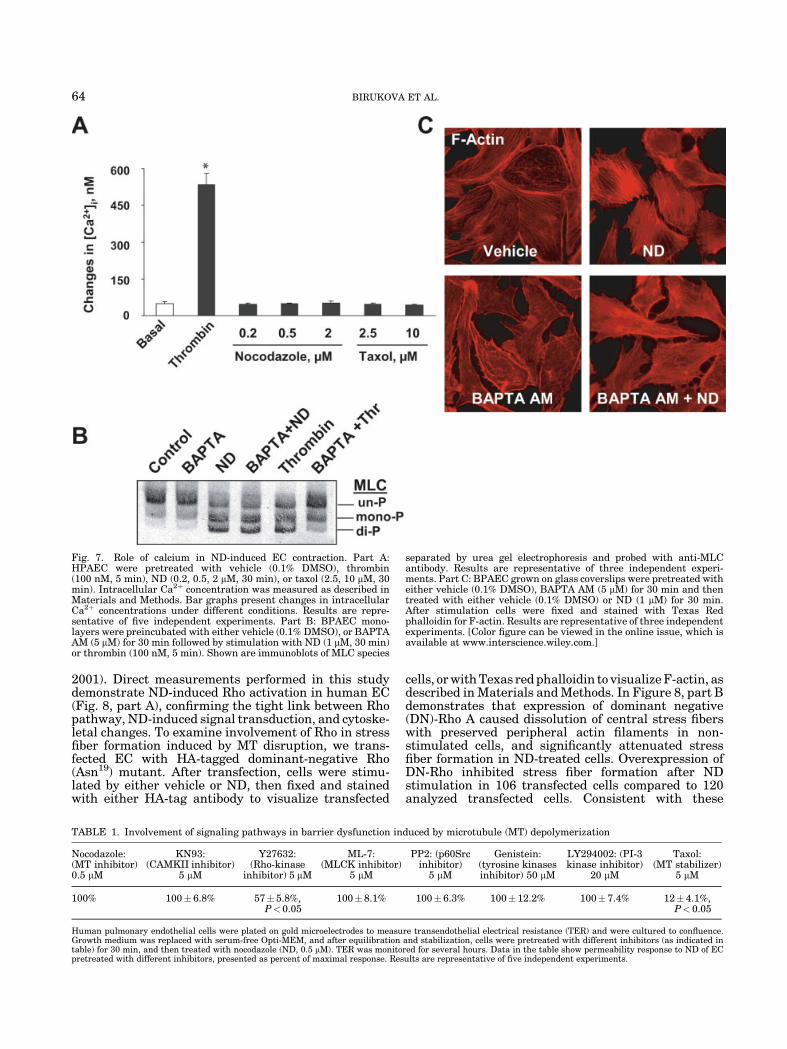
ND-induced MLC phosphorylation, we examined poten-tial effect of ND on intracellular [Ca2þ] levels. Measure-ments of intracellular [Ca2þ] in HPAEC (Fig. 7) andBPAEC (data not shown) stimulated with ND demon-strated that neither MT disruption by ND, nor MTstabilization by taxol affected intracellular [Ca2þ]levels. In contrast, thrombin stimulation induced dra-matic intracellular [Ca2þ] elevation consistent withpreviously published data (Garcia et al., 1995). Next,we examined effects of cell permeable Ca2þ chelator,BAPTA AM, on ND-induced stress fiber formation andMLC phosphorylation. Preincubation with BAPTA AM(5 mM) abolished thrombin-induced MLC phosphoryla-tion, but had no effect on ND-induced MLC phosphor-ylation (Fig. 7, part B). In addition, BAPTA AMpretreatment decreased the basal levels of polymerizedactin, however, it did not prevent stress fiber formationinduced by MT dissolution (Fig. 7, part C). Thus, ourpublished data and results of this study suggest thatMLCK- and Ca2þ-mediated pathways are not involvedin the ND-induced contractile response and EC barrierdysfunction.
To further determine the leading mechanisms of ECbarrier dysfunction induced by MT disassembly, weused inhibitory analysis. HPAEC monolayers werepretreated with inhibitors of key signal transductionpathways involved in EC barrier regulation, then cellswere stimulated with ND, and TER changes weremonitored thereafter. Table 1 demonstrates that amongsignaling pathways tested, only the MT stabilizer taxoland Rho-kinase inhibitor Y27632 significantly attenu-ated ND-induced TER decreases suggesting involve-ment of Rho-mediated pathways in ND-induced barrierdisruption.
Role of Rho pathway in EC barrier compromisecaused by MT inhibitors
We have previously demonstrated involvement ofsmall GTPase Rho in the ND-induced barrier dysfunc-tion in bovine pulmonary endothelium (Verin et al.,
Fig. 5. Effect of MT inhibitors on adherens junctions and focaladhesion complexes. Parts A, B: BPAEC grown on glass coverslipswere challenged either with vehicle (0.1% DMSO) or ND (1 mM) for30 min. After stimulation, cells were fixed and stained for specificmarker of endothelial adherens junctions, VE-cadherin (part A), andfocal adhesion protein, vinculin (part B). Results are representative ofthree independent experiments. [Color figure can be viewed in theonline issue, which is available at www.interscience.wiley.com.]
Fig. 6. Effect of S1P on ND-induced increases in EC permeability. Atthe time indicated by first arrow HPAEC were pretreated with eithervehicle (Opti-MEM) or S1P 1 mM for 10 min. At the time indicated bysecond arrow HPAEC were treated with either vehicle (0.1% DMSO)or ND (0.5 mM), and TER monitored for 10 h. Shown are representa-tive results of three independent experiments. [Color figure can beviewed in the online issue, which is available at www.interscience.wiley.com.]
MICROTUBULES AND ENDOTHELIAL BARRIER DYSFUNCTION 63
2001). Direct measurements performed in this studydemonstrate ND-induced Rho activation in human EC(Fig. 8, part A), confirming the tight link between Rhopathway, ND-induced signal transduction, and cytoske-letal changes. To examine involvement of Rho in stressfiber formation induced by MT disruption, we trans-fected EC with HA-tagged dominant-negative Rho(Asn19) mutant. After transfection, cells were stimu-lated by either vehicle or ND, then fixed and stainedwith either HA-tag antibody to visualize transfected
cells, or with Texas red phalloidin to visualize F-actin, asdescribed in Materials and Methods. In Figure 8, part Bdemonstrates that expression of dominant negative(DN)-Rho A caused dissolution of central stress fiberswith preserved peripheral actin filaments in non-stimulated cells, and significantly attenuated stressfiber formation in ND-treated cells. Overexpression ofDN-Rho inhibited stress fiber formation after NDstimulation in 106 transfected cells compared to 120analyzed transfected cells. Consistent with these
Fig. 7. Role of calcium in ND-induced EC contraction. Part A:HPAEC were pretreated with vehicle (0.1% DMSO), thrombin(100 nM, 5 min), ND (0.2, 0.5, 2 mM, 30 min), or taxol (2.5, 10 mM, 30min). Intracellular Ca2þ concentration was measured as described inMaterials and Methods. Bar graphs present changes in intracellularCa2þ concentrations under different conditions. Results are repre-sentative of five independent experiments. Part B: BPAEC mono-layers were preincubated with either vehicle (0.1% DMSO), or BAPTAAM (5 mM) for 30 min followed by stimulation with ND (1 mM, 30 min)or thrombin (100 nM, 5 min). Shown are immunoblots of MLC species
separated by urea gel electrophoresis and probed with anti-MLCantibody. Results are representative of three independent experi-ments. Part C: BPAEC grown on glass coverslips were pretreated witheither vehicle (0.1% DMSO), BAPTA AM (5 mM) for 30 min and thentreated with either vehicle (0.1% DMSO) or ND (1 mM) for 30 min.After stimulation cells were fixed and stained with Texas Redphalloidin for F-actin. Results are representative of three independentexperiments. [Color figure can be viewed in the online issue, which isavailable at www.interscience.wiley.com.]
TABLE 1. Involvement of signaling pathways in barrier dysfunction induced by microtubule (MT) depolymerization
Nocodazole:(MT inhibitor)0.5 mM
KN93:(CAMKII inhibitor)
5 mM
Y27632:(Rho-kinase
inhibitor) 5 mM
ML-7:(MLCK inhibitor)
5 mM
PP2: (p60Srcinhibitor)
5 mM
Genistein:(tyrosine kinasesinhibitor) 50 mM
LY294002: (PI-3kinase inhibitor)
20 mM
Taxol:(MT stabilizer)
5 mM
100% 100� 6.8% 57� 5.8%,P<0.05
100�8.1% 100� 6.3% 100� 12.2% 100� 7.4% 12� 4.1%,P< 0.05
Human pulmonary endothelial cells were plated on gold microelectrodes to measure transendothelial electrical resistance (TER) and were cultured to confluence.Growth medium was replaced with serum-free Opti-MEM, and after equilibration and stabilization, cells were pretreated with different inhibitors (as indicated intable) for 30 min, and then treated with nocodazole (ND, 0.5 mM). TER was monitored for several hours. Data in the table show permeability response to ND of ECpretreated with different inhibitors, presented as percent of maximal response. Results are representative of five independent experiments.
64 BIRUKOVA ET AL.

Fig. 8. Role of Rho GTPase in ND-induced EC barrier dysfunction.Part A: HPAEC were incubated with ND (0.5 mM) for 15 min, and Rhoactivation assay was performed as described in Materials andMethods. ND stimulation increased the levels of Rho-GTP indicatingactivation of Rho activity. Results are representative of threeindependent experiments. Part B: BPAEC were transiently trans-fected with dominant-negative Rho A bearing HA-tag as described inMaterials and Methods, treated with either vehicle with (0.1% DMSO)or ND (1 mM, 30 min), then fixed and stained with either Texas Redphalloidin to visualize F-Actin, or with anti-HA antibody to detect
overexpressing cells. Part C: BPAEC were plated on gold microelec-trodes to measure TER and were cultured to confluence. ECmonolayers were first permeabilized by lipofectamine treatmentin the presence or absence of specific Rho inhibitor, C3 exoenzyme(2.5 mg/ml) as previously described (Borbiev et al., 2000) followed bystimulation with ND (1 mM). TER was monitored for 15 h. Results arerepresentative of three independent experiments. [Color figure can beviewed in the online issue, which is available at www.interscience.wiley.com.]
MICROTUBULES AND ENDOTHELIAL BARRIER DYSFUNCTION 65

results, inhibition of Rho activity by specific inhibitor,C3 exoenzyme (Fig. 8, part C), significantly attenuatedthe increase in EC permeability after ND stimulation,further confirming involvement of Rho in EC contrac-tion induced by MT inhibitors.
Effects of Rho-kinase inhibitionon ND-induced EC barrier dysfunction
The status of MLC phosphorylation is criticallydependent upon the balance between MLCK and MLCP
(MLC phosphatase) activities. The key downstreameffector of Rho is Rho-kinase, which inactivates myosin-specific phosphatase via phosphorylation of myosinphosphatase 130-kDa regulatory subunit (MYPT1) atThr696 and Thr850 (Fukata et al., 2001). We nextexamined the involvement of Rho-kinase in ND-inducedbarrier dysfunction. In Figure 9, part A demonstratesautoradiography of Rho-kinase immunoprecipitatedfrom P32-labeled cells. EC treatment with ND inducedphosphorylation of Rho-kinase, which was inhibited by
Fig. 9. Role of Rho-kinase in ND-induced barrier dysfunction. PartA: HPAEC were pretreated with either vehicle (0.1% DMSO), or taxol(5 mM) for 30 min followed by treatment with either vehicle (0.1%DMSO) or ND (0.5 mM) for 30 min. Measurement of Rho kinasephosphorylation was performed as described in Materials andMethods. Results are representative of three independent experi-ments. Part B: BPAEC were transiently transfected with dominant
negative Rho-kinase construct bearing myc-tag. Shown are immuno-fluorescent images of EC treated with either vehicle (0.1% DMSO) orND (1 mM, 30 min), then fixed and stained for F-Actin or with anti-mycantibody to visualize dominant negative Rho-kinase-overexpressingcells. Results are representative of three independent experiments.[Color figure can be viewed in the online issue, which is available atwww.interscience.wiley.com.]
66 BIRUKOVA ET AL.

taxol. To investigate the effect of Rho-kinase inhibitionon cytoskeletal changes induced by MT disassembly, wetransiently transfected EC with the RB-PH (TT) frag-ment of Rho-kinase, which functions as Rho-kinasedominant negative (DN) mutant (Amano et al., 1999).After transfection, cells were fixed and stained witheither myc-tag antibody to visualize transfected cells, orwith Texas red phalloidin to visualize F-actin. As shownin Figure 9, part B overexpression of DN-Rho-kinasesignificantly decreased the amount of actin filaments inunstimulated cells, and completely prevented stressfiber formation upon ND stimulation. Consistentwith these data, pharmacological inhibition of Rho-kinase activity by Y27632 completely blocked ND-induced MYPT1 phosphorylation (Fig. 10, part A), and
MLC phosphorylation (Fig. 10, part B) after NDtreatment. Next, we performed quantitative analysisof intracellular actin filaments after different treatmentconditions, as described in Materials and Methods. ECwere preincubated with Y27632 or vehicle, followedby treatment with ND (0.2 or 0.5 mM) or vehicle. InFigure 10, part C shows that Rho-kinase inhibitionsignificantly attenuated stress fiber formation in ND-treated HPAEC, thus further confirming the directinvolvement of Rho-kinase pathway in stress fiberformation and MLC phosphorylation in EC induced byMT disassembly. Taken together, our data demonstratethe key role for Rho-dependent mechanisms in ND-mediated barrier disruption in the human pulmonaryendothelium.
Fig. 10. Role of Rho-kinase inhibition in ND-induced barrier dysfunction. Part A: ConfluentHPAECs were preincubated with Y-27632 (Y, 5 mM)for 1 h followed by ND stimulation (0.5 mM, 30 min),or treated with thrombin (100 nM, 5 min). Cells werelysed and probed on Western blotting with eitherphospho-Thr696 MYPT1 antibody, or pan-MYPT1antibody. Results are representative of three inde-pendent experiments. Part B: BPAEC grown oncoverslips were preincubated with either vehicle(0.1% DMSO) or Y-27632 (Y, 5 mM) for 1 h, and thentreated with either vehicle vehicle (0.1% DMSO) orND (1 mM, 30 min). Cells were stained with diphos-pho-MLC antibody. Shown are representative resultsfrom three independent experiments. Part C: Effectof Rho kinase inhibition on ND-induced stress fiberformation was assessed by morphometric analysis ofTexas Red phalloidin-stained HPAEC performedusing MetaVue software, as described in Materialsand Methods. Actin fibers were marked out, and theratio of the area covered by stress fibers to the wholecell area was calculated. Data are expressed as % ofcontrol corresponding to non-stimulated cells andrepresent results of three independent experiments.[Color figure can be viewed in the online issue, whichis available at www.interscience.wiley.com.]
MICROTUBULES AND ENDOTHELIAL BARRIER DYSFUNCTION 67

DISCUSSION
Our results directly demonstrate rapid activation ofsmall GTPase Rho in human and bovine pulmonaryartery EC in response to ND-induced MT disassembly,and link this event to actin cytoskeletal remodeling,MLC phosphorylation, actomyosin assembly, EC con-traction, and disruption of the endothelial barrier. Thevaried signaling mechanisms triggered by MT depoly-merization have been previously studied in various celltypes with clear tissue- and cell-specific responses(Danowski, 1989; Enomoto, 1996; Cook et al., 1998;Platts et al., 2002; Niggli, 2003). For example, disas-sembly of MTs attenuates polarity and migration infibroblasts (Waterman-Storer et al., 1999), but inducesdevelopment of polarity and migration in neutrophils(Niggli, 2003). Interestingly, MT organization itself maybe regulated by Rho, and mechanisms vary in differentcell types. In fibroblasts, RhoA activation increases thepool of stable MT (Cook et al., 1998). In contrast, inneuroblastoma cells, RhoA activation induces phosphor-ylation of the MT-associated protein Tau, dissociation ofTau from MT and subsequent MT destabilization (Sayaset al., 1999). Overexpression of the wild-type RhoAproduced disruption of MT in UTA6 cells (Song et al.,2000). Thus, published data emphasize the importanceof characterization of cell-specific mechanisms of Rho-MT interactions.
In our studies, stress fiber formation and gap forma-tion strongly correlated with MT polymerization state,as EC monolayer integrity and F-actin organizationtypical for uncompromised EC barrier was completelyrestored by 30 min after ND withdrawal. Instant Rhoactivation by MT disassembly may be explained by therelease of Rho activating factors bound to MTs. Thecycling of Rho GTPases between inactive GDP-boundform and activated GTP-bound form is controlled byguanosine nucleotide exchange factors (GEF) andGTPase-activating proteins (Bishop and Hall, 2000).GEFs mediate receptor-dependent Rac, Cdc42, and Rhoregulation by external stimuli. A number of GEFs targetmultiple GTPases, whereas others specifically targetRac (Tiam1, Vav2, bPIX), Cdc42 (Vav2, intersectin) orRho (p115RhoGEF, GEF-H1, Lfc) (Zheng, 2001;Schmidt and Hall, 2002). Regulation of GEF activitiesinvolves several mechanisms including phosphorylationby protein kinase A (O’Connor and Mercurio, 2001), Srcfamily tyrosine kinases (Bustelo, 2000), Ca2þ/Calmodu-lin-dependent kinase II (Fleming et al., 1999), or phos-phoinositides (Zheng, 2001). Recent studies haveidentified two GEFs, Lfc and GEF-H1, which existbound to MT (Glaven et al., 1996; Ren et al., 1998). InMT-bound state, the guanine-exchange activity of GEF-H1 is suppressed, whereas GEF-H1 release, caused byMT disassembly, stimulates Rho-specific GEF activity(Krendel et al., 2002). Further studies in our laboratoryare underway to explore this potential mechanism ofRho activation.
Another example of heterogeneity of cellular re-sponses to MT depolymerization is the effect of MTinhibitors on intracellular Ca2þ concentrations. Plattset al. (1999) found that MT inhibitors do not produce anincrease Ca2þ in association with the enhancing vaso-constriction in rat arterioles. In contrast, Gomez et al.
(2000) found that intracellular Ca2þ levels wereincreased in rat cardiac myocytes after treatment withMT inhibitors. Our results suggest that ND-induced MTdisruption in human pulmonary EC did not affectintracellular Ca2þ levels. We have previously demon-strated that pharmacological inhibitor of Ca2þ/calmo-dulin-dependent MLC kinase, ML-7, did not abolishMT-induced stress fiber formation and MLC phosphor-ylation (Verin et al., 2001). Thus, our novel data showinglack of ND effect on intracellular Ca2þ concentration inhuman pulmonary EC further support MLCK-indepen-dent mechanism of MLC phosphorylation and barrierdysfunction in ND-treated HPAEC, thus emphasizing aleading role of the Rho-mediated pathway.
Among several Rho targets, Rho-kinase is directlyinvolved in actin stress fiber formation and regulation ofMLC phosphorylation (Amano et al., 1996; Essler et al.,1998; Verin et al., 2001). Binding of GTP-Rho facilitatesinteractions of Rho-kinase with target proteins, andtranslocation of the protein complex to the target celllocation (Leung et al., 1996; Ishizaki et al., 1997). In thisstudy, we demonstrated ND-induced phosphorylation ofRho-kinase that correlated with increased Rho-kinaseactivity towards MLC. To the best of our knowledge thispotential regulation of Rho-kinase by phosphorylationhas not been previously described. Thus, our strikingfinding suggest a potential novel mechanism for mod-ulation of Rho-dependent regulation of Rho-kinase.Further studies are underway to characterize a proteinkinase involved in Rho-kinase phosphorylation.
Rho-kinase increases MLC phosphorylation by twopotential mechanisms: direct phosphorylation of MLCSer19, Thr18, and indirectly via phosphorylation of theregulatory subunit of myosin phosphatase (MYPT1) atSer696 and Ser850 (Kawano et al., 1999), which sup-presses MYPT1 activity (Amano et al., 1996). Using site-specific antibodies to MYPT1 phospho-Ser696 and phos-pho-Ser850, we demonstrate that MT disassemblycaused MYPT1 phosphorylation by Rho-kinase, whichwas linked to increased phospho-MLC accumulation inhuman pulmonary EC. Direct involvement of Rho–Rho-kinase–MYPT1 pathway in ND-induced MLC phos-phorylation and cytoskeletal remodeling was furtherdemonstrated in our study, as pharmacological inhibi-tion of Rho-kinase by Y27632 abolished MYPT1 phos-phorylation at Ser696, decreased ND-induced MLCphosphorylation (Fig. 10), and Rho downregulation byC3 exotoxin attenuated ND-induced decreases inTER (Fig. 8). Consistent with these data, overexpressionof dominant negative Rho-kinase abolished ND-inducedstress fiber formation and accumulation of diphospho-MLC in pulmonary EC. Figure 11 summarizes theresults of our study and previous reports and depicts apotential mechanism by which Rho-mediated path-way may be involved in barrier dysfunction induced byMT inhibitors. MT disassembly by MT inhibitorsstimulates accumulation of Rho-GTP, possibly viarelease from MTs and activation of guanine nucleotideexchange factor(s) including Rho-specific factor GEF-H1, and causes rapid activation of Rho-kinase, andinactivation of MYPT1, that culminates in robust MLCphosphorylation, stress fiber formation, actomyosincontraction, cell retraction, and disruption of EC mono-layer integrity.
68 BIRUKOVA ET AL.

In this study, we show that a number of signalingmolecules such as CaMKII, tyrosin kinases includingp60Src, MLC kinase, and PI-3 kinase are not involved inND-induced barrier disruption (Table 1 and page 19).Recent studies have shown that different PKC isoformsand PKA could be involved in the regulation of MTstability (Manie et al., 1993; Banan et al., 2002) anddisruption of MT network may lead to activation of MAPkinases (Erk 1/2, p38 MAPK, JNK) (Stone and Cham-bers, 2000). These signaling events may contribute tocytoskeletal remodeling and permeability changes.Ongoing studies in our laboratory are designed toinvestigate the potential role of the MAP kinases, PKAand PKC in ND-induced barrier dysfunction.
Thus, our results suggest a leading role of Rho-dependent mechanisms in development of vascularleak and pathogenesis of pulmonary edema that maydevelop as a complication in the course of cytostatictherapy utilizing MT-disrupting agents. Therefore,pharmacological inhibition or gene targeting of smallGTPase Rho and its effector Rho-kinase in pulmonaryvasculature, as well as modulation of myosin phospha-
tase activity in pulmonary EC may represent importantstrategies for the prevention of pulmonary edema underthese conditions.
LITERATURE CITED
Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, et al.1996. Phosphorylation and activation of myosin by Rho-associatedkinase (Rho-kinase). J Biol Chem 271(34):20246–20249.
Amano M, Chihara K, et al. 1998. Myosin II activation promotesneurite retraction during the action of Rho and Rho-kinase. GenesCells 3(3):177–188.
Amano M, Chihara K, et al. 1999. The COOH terminus of Rho-kinasenegatively regulates rho-kinase activity. J Biol Chem 274(45):32418–32424.
Banan A, Fields JZ, et al. 2002. PKC-zeta is required in EGFprotection of microtubules and intestinal barrier integrity againstoxidant injury. Am J Physiol Gastrointest Liver Physiol 282(5):G794–G808.
Bishop AL, Hall A. 2000. Rho GTPases and their effector proteins.Biochem J 348(Pt 2):241–255.
Bogatcheva NV, Garcia JG, et al. 2002. Molecular mechanismsof thrombin-induced endothelial cell permeability. Biochemistry(Mosc) 67(1):75–84.
Borbiev T, Nurmukhambetova S, et al. 2000. Introduction of C3exoenzyme into cultured endothelium by lipofectamine. AnalBiochem 285(2):260–264.
Fig. 11. Proposed schema of Rho-mediated regulation of EC cytoskeletal rearrangement and barrierfunction induced by MT depolymerization. See Discussion for details. [Color figure can be viewed in theonline issue, which is available at www.interscience.wiley.com.]
MICROTUBULES AND ENDOTHELIAL BARRIER DYSFUNCTION 69

Bustelo XR. 2000. Regulatory and signaling properties of the Vavfamily. Mol Cell Biol 20(5):1461–1477.
Cattan CE, Oberg KC. 1999. Vinorelbine tartrate-induced pul-monary edema confirmed on rechallenge. Pharmacotherapy 19(8):992–994.
Cook TA, Nagasaki T, et al. 1998. Rho guanosine triphosphatasemediates the selective stabilization of microtubules induced bylysophosphatidic acid. J Cell Biol 141(1):175–185.
Danowski BA. 1989. Fibroblast contractility and actin organizationare stimulated by microtubule inhibitors. J Cell Sci 93(Pt 2):255–266.
Dejana E. 1997. Endothelial adherens junctions: Implications in thecontrol of vascular permeability and angiogenesis. J Clin Invest100(11 Suppl):S7–S10.
Dudek SM, Garcia JG. 2001. Cytoskeletal regulation of pulmonaryvascular permeability. J Appl Physiol 91(4):1487–1500.
Elbaum M, Chausovsky A, et al. 1999. Microtubule involvement inregulating cell contractility and adhesion-dependent signalling: Apossible mechanism for polarization of cell motility. Biochem SocSymp 65:147–172.
Enomoto T. 1996. Microtubule disruption induces the formation ofactin stress fibers and focal adhesions in cultured cells: Possibleinvolvement of the rho signal cascade. Cell Struct Funct 21(5):317–326.
Essler M, Amano M, et al. 1998. Thrombin inactivates myosin lightchain phosphatase via Rho and its target Rho kinase in humanendothelial cells. J Biol Chem 273(34):21867–21874.
Fleming IN, Elliott CM, et al. 1999. Ca2þ/calmodulin-dependentprotein kinase II regulates Tiam1 by reversible protein phosphor-ylation. J Biol Chem 274(18):12753–12758.
Fukata Y, Amano M, et al. 2001. Rho–Rho-kinase pathway in smoothmuscle contraction and cytoskeletal reorganization of non-musclecells. Trends Pharmacol Sci 22(1):32–39.
Garcia JG, Davis HW, et al. 1995. Regulation of endothelial cell gapformation and barrier dysfunction: Role of myosin light chainphosphorylation. J Cell Physiol 163(3):510–522.
Garcia JG, Schaphorst KL, et al. 1997. Mechanisms of ionomycin-induced endothelial cell barrier dysfunction. Am J Physiol 273(1 Pt1):L172–L184.
Garcia JGN, Verin AD, et al. 1998. Adherent neutrophils activateendothelial myosin light chain kinase: Role in transendothelialmigration. J Appl Physiol 84(5):1817–1821.
Garcia JG, Liu F, et al. 2001. Sphingosine 1-phosphate promotesendothelial cell barrier integrity by Edg-dependent cytoskeletalrearrangement. J Clin Invest 108(5):689–701.
Glaven JA, Whitehead IP, et al. 1996. Lfc and Lsc oncoproteinsrepresent two new guanine nucleotide exchange factors for the RhoGTP-binding protein. J Biol Chem 271(44):27374–27381.
Goeckeler ZM, Wysolmerski RB. 1995. Myosin light chain kinase-regulated endothelial cell contraction: The relationship betweenisometric tension, actin polymerization, and myosin phosphoryla-tion. J Cell Biol 130(3):613–627.
Gomez AM, Kerfant BG, et al. 2000. Microtubule disruption modulatesCa(2þ) signaling in rat cardiac myocytes. Circ Res 86(1):30–36.
Grynkiewicz G, Poenie M, et al. 1985. A new generation of Ca2þ
indicators with greatly improved fluorescence properties. J BiolChem 260(6):3440–3450.
Gundersen GG, Cook TA. 1999. Microtubules and signal transduction.Curr Opin Cell Biol 11(1):81–94.
Ingber DE. 1997. Tensegrity: The architectural basis of cellularmechanotransduction. Annu Rev Physiol 59:575–599.
Ishizaki T, Naito M, et al. 1997. p160ROCK: A Rho-associated coiled-coil forming protein kinase, works downstream of Rho and inducesfocal adhesions. FEBS Lett 404(2–3):118–124.
Kawano Y, Fukata Y, et al. 1999. Phosphorylation of myosin-bindingsubunit (MBS) of myosin phosphatase by Rho-kinase in vivo. J CellBiol 147(5):1023–1038.
Kolodney MS, Elson EL. 1995. Contraction due to microtubuledisruption is associated with increased phosphorylation of myosinregulatory light chain. Proc Natl Acad Sci USA 92(22):10252–10256.
Krendel M, Zenke FT, et al. 2002. Nucleotide exchange factor GEF-H1mediates cross-talk between microtubules and the actin cytoskele-ton. Nat Cell Biol 4(4):294–301.
Leung T, Chen XQ, et al. 1996. The p160 RhoA-binding kinaseROK alpha is a member of a kinase family and is involved inthe reorganization of the cytoskeleton. Mol Cell Biol 16(10):5313–5327.
Lum H, Malik AB. 1996. Mechanisms of increased endothelialpermeability. Can J Physiol Pharmacol 74(7):787–800.
Manie S, Schmid-Alliana A, et al. 1993. Disruption of microtubulenetwork in human monocytes induces expression of interleukin-1but not that of interleukin-6 nor tumor necrosis factor-alpha.Involvement of protein kinase A stimulation. J Biol Chem 268(18):13675–13681.
Niggli V. 2003. Microtubule-disruption-induced and chemotactic-peptide-induced migration of human neutrophils: Implications fordifferential sets of signalling pathways. J Cell Sci 116(Pt 5):813–822.
O’Connor KL, Mercurio AM. 2001. Protein kinase A regulates Rac andis required for the growth factor-stimulated migration of carcinomacells. J Biol Chem 276(51):47895–47900.
Petrache I, Birukova A, et al. 2003. The role of the microtubules intumor necrosis factor-alpha-induced endothelial cell permeability.Am J Respir Cell Mol Biol 28(5):574–581.
Platts SH, Falcone JC, et al. 1999. Alteration of microtubulepolymerization modulates arteriolar vasomotor tone. Am J Physiol277(1 Pt 2):H100–H106.
Platts SH, Martinez-Lemus LA, et al. 2002. Microtubule-dependentregulation of vasomotor tone requires Rho-kinase. J Vasc Res 39(2):173–182.
Ren Y, Li R, et al. 1998. Cloning and characterization of GEF-H1: Amicrotubule-associated guanine nucleotide exchange factor for Racand Rho GTPases. J Biol Chem 273(52):34954–34960.
Sayas CL, Moreno-Flores MT, et al. 1999. The neurite retractioninduced by lysophosphatidic acid increases Alzheimer’s disease-likeTau phosphorylation. J Biol Chem 274(52):37046–37052.
Schaphorst KL, Pavalko FM, et al. 1997. Thrombin-mediated focaladhesion plaque reorganization in endothelium: Role of proteinphosphorylation. Am J Respir Cell Mol Biol 17(4):443–455.
Schmidt A, Hall A. 2002. Guanine nucleotide exchange factors for RhoGTPases: Turning on the switch. Genes Dev 16(13):1587–1609.
Shikata Y, Birukov KG, et al. 2003. S1P induces FA remodeling inhuman pulmonary endothelial cells: Role of Rac, GIT1, FAK, andpaxillin. J Appl Physiol 94(3):1193–1203.
Song Y, Wong C, et al. 2000. Overexpression of wild-type RhoAproduces growth arrest by disrupting actin cytoskeleton andmicrotubules. J Cell Biochem 80(2):229–240.
Stone AA, Chambers TC. 2000. Microtubule inhibitors elicit differ-ential effects on MAP kinase (JNK, ERK, and p38) signaling path-ways in human KB-3 carcinoma cells. Exp Cell Res 254(1):110–119.
Verin AD, Birukova A, et al. 2001. Microtubule disassembly increasesendothelial cell barrier dysfunction: Role of MLC phosphorylation.Am J Physiol Lung Cell Mol Physiol 281(3):L565–L574.
Waterman-Storer CM, Salmon E. 1999. Positive feedback interactionsbetween microtubule and actin dynamics during cell motility. CurrOpin Cell Biol 11(1):61–67.
Waterman-Storer CM, Worthylake RA, et al. 1999. Microtubulegrowth activates Rac1 to promote lamellipodial protrusion infibroblasts. Nat Cell Biol 1(1):45–50.
Worthylake RA, Lemoine S, et al. 2001. RhoA is required for monocytetail retraction during transendothelial migration. J Cell Biol154(1):147–160.
Wysolmerski RB, Lagunoff D. 1990. Involvement of myosin light-chain kinase in endothelial cell retraction. Proc Natl Acad Sci USA87(1):16–20.
Zhang Q, Magnusson MK, et al. 1997. Lysophosphatidic acid andmicrotubule-destabilizing agents stimulate fibronectin matrixassembly through Rho-dependent actin stress fiber formation andcell contraction. Mol Biol Cell 8(8):1415–1425.
Zheng Y. 2001. Dbl family guanine nucleotide exchange factors.Trends Biochem Sci 26(12):724–732.
70 BIRUKOVA ET AL.




















