
Microarray analysis of gene expression profiles in human neuroblastoma cells exposed to Ab–Zn and...
15
483 10.2217/FNL.12.35 © 2012 Future Medicine Ltd ISSN 1479-6708 Future Neurol. (2012) 7(4), 483–497 part of Future Neurology Amyloid-b (Ab), the amyloid precursor protein (APP) cleavage product, plays a critical role in the development of Alzheimer’s disease (AD) (BOX 1) [1] . Mechanisms through which Ab exerts its pathogenic actions are not completely under- stood, but a key step in the degenerative cascade leading to AD is associated with an imbalance between Ab production and clearance, ulti- mately causing abnormal peptide accumulation, neuronal death and cognitive decline [2] . Once released at high concentrations, Ab undergoes a well-defined process of aggregation. Ab mono- mers turn into oligomers (the most toxic species) through a self-assembly process that involves nucleation and linear growth [3] ; oligomers then turn into higher ordered structures (protofibrils) and eventually into fibrils, the main constituents of senile plaques (BOX 1) [4] . This pathogenic process is modified by many endogenous and exogenous factors. Within these, metal ions are relevant for at least three reasons: first, they significantly alter Ab aggrega- tion [5] , second, their homeostasis is profoundly altered in AD patients [6] and third, they are found at high concentrations in senile plaques [7,8] . Ab can also act as a potent endogenous pathophysiological metal buffer that hampers the role of brain metals in controlling neuro- transmission, neuronal and neurotrophic sign- aling, thus ultimately affecting cognition. All of these mechanisms contribute to the pillar of the metal hypothesis of AD, which postulates that brain metal dyshomeostasis plays an impor- tant role in the development and progression of AD [5,9,10] . In the present study, we analyzed the gene expression profile changes due to exposure to Ab–zinc (Zn) or Ab–copper (Cu) in a neuronal- like cell line (SH-SY5Y). SH-SY5Y were chosen because as a cell line they offer the advantage of being a homogenous population that does not show the subtype heterogeneity present in primary neuronal cultures, a confounding factor that can make results more difficult to interpret. We employed the same experimental paradigm previously used in a study analyzing gene expression changes triggered by exposure to Ab 1–42 conjugated with Al [11] . Cultures were exposed to Ab 1–42 , Ab 1–42 –Zn, Ab 1–42 –Cu, Zn 2+ Microarray analysis of gene expression profiles in human neuroblastoma cells exposed to Ab–Zn and Ab–Cu complexes Valentina Gatta †1,2 , Alberto Granzotto †3 , Karina Fincati 3 , Denise Drago 4 , Silvia Bolognin 5 , Paolo Zatta 6 & Stefano L Sensi* 3,7,8 1 Department of Oral Health & Biotechnological Sciences, “G. D’Annunzio” University, Chieti-Pescara, Italy 2 Functional Genetics Unit – Center of Excellence in Aging (Ce.S.I.), Chieti, Italy 3 Molecular Neurology Unit - Ce.S.I., Chieti, Italy 4 CNS Repair Unit – INSPE, Biological Mass Spectrometry Unit, San Raffaele Scientific Institute, Milan, Italy 5 Department of Neurological, Neuropsychological, Morphological & Motor Sciences – Physiology & Psychology Unit, Verona, Italy 6 National Research Council, Biomedical Technology Institute (CNR-ITB), Metalloproteins Unit, Department of Biology, University of Padua, Padua, Italy 7 Department of Neuroscience & Imaging, “G. D’Annunzio” University, Chieti, Italy 8 Departments of Neurology & Pharmacology, University of California Irvine, Irvine, California, USA *Author for correspondence: [email protected] † Authors contributed equally to this article Aims: Abnormal metal accumulation is associated with Alzheimer’s disease and plays a relevant role in affecting amyloid- b (Ab) peptide aggregation and neurotoxicity. Material & Methods: In the present study, employing a microarray analysis of 35,129 genes, we analyzed gene expression profile changes due to exposure to Ab 1-42 –Zn or Ab 1-42 –Cu complexes in neuronal-like cells (SH-SY5Y). Results: Microarray data indicated that Ab –Zn or Ab –Cu complexes selectively alter expression of genes mainly related to cell death, inflammatory responses, cytoprotective mechanisms and apoptosis. Conclusions: Taken together, these findings indicate that Ab 1–42 –Zn or Ab 1–42 –Cu show some commonalities in affecting Alzheimer’s disease-related target functions. The overall modulatory activity on these genes supports the idea of a possible net effect resulting in the activation of pathways that counteract toxic effects of Ab –Zn or Ab –Cu. Keywords n amyloid-b 1–42 n Alzheimer’s disease n apoptosis n copper n gene expression n inflammation n microarray n oxidative stress n zinc Research Article For reprint orders, please contact: [email protected]
Transcript of Microarray analysis of gene expression profiles in human neuroblastoma cells exposed to Ab–Zn and...

48310.2217/FNL.12.35 © 2012 Future Medicine Ltd ISSN 1479-6708Future Neurol. (2012) 7(4), 483–497
part of
Futu
re N
eu
rolo
gy
Amyloid-b (Ab), the amyloid precursor protein (APP) cleavage product, plays a critical role in the development of Alzheimer’s disease (AD)(Box 1) [1].
Mechanisms through which Ab exerts its pathogenic actions are not completely under-stood, but a key step in the degenerative cascade leading to AD is associated with an imbalance between Ab production and clearance, ulti-mately causing abnormal peptide accumulation, neuronal death and cognitive decline [2]. Once released at high concentrations, Ab undergoes a well-defined process of aggregation. Ab mono-mers turn into oligomers (the most toxic species) through a self-assembly process that involves nucleation and linear growth [3]; oligomers then turn into higher ordered structures (protofibrils) and eventually into fibrils, the main constituents of senile plaques (Box 1) [4].
This pathogenic process is modified by many endogenous and exogenous factors. Within these, metal ions are relevant for at least three reasons: first, they significantly alter Ab aggrega-tion [5], second, their homeostasis is profoundly altered in AD patients [6] and third, they are
found at high concentrations in senile plaques [7,8]. Ab can also act as a potent endogenous pathophysiological metal buffer that hampers the role of brain metals in controlling neuro-transmission, neuronal and neurotrophic sign-aling, thus ultimately affecting cognition. All of these mechanisms contribute to the pillar of the metal hypothesis of AD, which postulates that brain metal dyshomeostasis plays an impor-tant role in the development and progression of AD [5,9,10].
In the present study, we analyzed the gene expression profile changes due to exposure to Ab–zinc (Zn) or Ab–copper (Cu) in a neuronal-like cell line (SH-SY5Y). SH-SY5Y were chosen because as a cell line they offer the advantage of being a homogenous population that does not show the subtype heterogeneity present in primary neuronal cultures, a confounding factor that can make results more difficult to interpret. We employed the same experimental paradigm previously used in a study analyzing gene expression changes triggered by exposure to Ab
1–42 conjugated with Al [11]. Cultures were
exposed to Ab1–42
, Ab1–42
–Zn, Ab1–42
–Cu, Zn2+
Microarray analysis of gene expression profiles in human neuroblastoma cells exposed to Ab–Zn and Ab–Cu complexes
Valentina Gatta†1,2, Alberto Granzotto†3, Karina Fincati3, Denise Drago4, Silvia Bolognin5, Paolo Zatta6 & Stefano L Sensi*3,7,8
1Department of Oral Health & Biotechnological Sciences, “G. D’Annunzio” University, Chieti-Pescara, Italy2Functional Genetics Unit – Center of Excellence in Aging (Ce.S.I.), Chieti, Italy3Molecular Neurology Unit - Ce.S.I., Chieti, Italy4CNS Repair Unit – INSPE, Biological Mass Spectrometry Unit, San Raffaele Scientific Institute, Milan, Italy5Department of Neurological, Neuropsychological, Morphological & Motor Sciences – Physiology & Psychology Unit, Verona, Italy 6National Research Council, Biomedical Technology Institute (CNR-ITB), Metalloproteins Unit, Department of Biology, University of Padua, Padua, Italy 7Department of Neuroscience & Imaging, “G. D’Annunzio” University, Chieti, Italy8Departments of Neurology & Pharmacology, University of California Irvine, Irvine, California, USA*Author for correspondence: [email protected] †Authors contributed equally to this article
Aims: Abnormal metal accumulation is associated with Alzheimer’s disease and plays a relevant role in affecting amyloid-b (Ab) peptide aggregation and neurotoxicity. Material & Methods: In the present study, employing a microarray analysis of 35,129 genes, we analyzed gene expression profile changes due to exposure to Ab1-42 –Zn or Ab1-42 –Cu complexes in neuronal-like cells (SH-SY5Y). Results: Microarray data indicated that Ab–Zn or Ab–Cu complexes selectively alter expression of genes mainly related to cell death, inflammatory responses, cytoprotective mechanisms and apoptosis. Conclusions: Taken together, these findings indicate that Ab1–42 –Zn or Ab1–42 –Cu show some commonalities in affecting Alzheimer’s disease-related target functions. The overall modulatory activity on these genes supports the idea of a possible net effect resulting in the activation of pathways that counteract toxic effects of Ab–Zn or Ab–Cu.
Keywords
n amyloid-b1–42 n Alzheimer’s disease n apoptosis n copper n gene expression n inflammation n microarray n oxidative stress n zinc
Rese
arc
h A
rticle
For reprint orders, please contact: [email protected]

Future Neurol. (2012) 7(4)484 future science group
or Cu2+ alone and the transcriptomic profile was investigated with microarray analysis (Box 1). The main goal of the study was to offer a better understanding of biological downstream effects triggered by Ab conjugated with metals, a con-dition that is the pathogenic scenario occurring in the AD brain. To that aim, we took into account only the genes that, compared to the ones changed by exposure to Ab
1–42, Cu or Zn
alone, were selectively modified in cells treated with Ab
1–42–Zn or Ab
1–42–Cu conjugates. These
selectively modified genes were investigated using ingenuity pathway analysis (IPA) to assess their roles and functions (Box 1).
Materials & methodsPreparation of Ab–metal complexesHexafluorisopropanol (HFIP; Sigma-Aldrich, MO, USA) was added to Ab
1–42 (Invitrogen,
CA, USA) for 40 min at room temperature to dissolve possible aggregates, HFIP-dissolved Ab monomers were then divided in aliquots, HFIP was removed under vacuum and lyophilized pep-tides stored at -20°C until used. Ab
1–42 aliquots
(~0.1 mg per vial) were resuspended in dou-ble distilled water (ddH
2O) to reach a 50 µM
concentration and dialyzed for 24 h against a 10 mM ZnCl
2 or CuCl
2 solution using Spectra/
PorR Float-A-LyserR tubes (Spectrum Labs, CA, USA) with 1000 molecular weight cutoffs. Ab
1–42–metal conjugates were then dialyzed
against ddH2O (with three water changes) for
24 h to remove the metal excess. The protein content of the Ab
1–42–metal solutions protein
content was determined, prior to use, by employ-ing the Lowry protein assay. Structure, morphol-ogy and stoichiometry of these Ab
1–42–metal
complexes have been recently described by Bolognin et al. [12] and their toxicity has been previously reported [13].
Cell cultures & treatmentsSH-SY5Y were purchased from the European Collection of Cell Culture (Salisbury, UK) and maintained in a controlled atmosphere (37°C,
5% CO2, 90% humidity). Cell culture medium
contained: DMEM:F12 (1,1) (Gibco, Carlsbad, CA, USA) with l-Glutamine and 15 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, penicillin and streptomycin (100 units/ml and 100 µg/ml, respectively; Gibco, Carlsbad, CA, USA), 15% fetal bovine serum (FBS; Sigma-Aldrich) and MEM supplemented with non-essential amino acids (100×; Sigma-Aldrich). Cells were cultured onto six-well plates at a 3 × 105 cells/well density; once cells reached 80% confluence (~1.2 × 106 cells/well), standard medium was replaced with a 2% FBS medium containing Ab
1–42–Zn and Ab
1–42–Cu at a con-
centration of 0.5 µM. Cells were incubated for 24 h. As a control, sister cultures were treated with media containing only metals at concentra-tions tenfold higher than the one employed for the peptide (5 µM).
RNA isolation, quality control & labelingTotal RNA was extracted from untreated (control) or treated (with Ab
1–42, Ab
1–42–Cu,
Ab1–42
–Zn, Cu or Zn) cultures using the Qiagen® RNA/DNA Mini Kit (Qiagen S.p.A., Milan, Italy) following the manufacturer’s instructions. Purity and quantity of extracted RNA was assessed using the RNA 6000 Nano LabChip® and the Agilent™ Bioanalyzer 2100 (Agilent Technologies, Palo Alto, CA, USA). Then 1 µg of total RNA was amplified using the Superscript™ Indirect RNA Amplification System Kit (Invitrogen, Grand Island, NY) and 10 µg of the obtained aRNA were conjugated with either Alexa Fluor 555 or Alexa Fluor 647 dyes (Invitrogen, NY, USA) following the manu-facturer’s protocol.
MicroarrayModified aRNAs were hybridized on human OpArrays™ DNA microarray slides (con-taining 35,129 total probes corresponding to 29,166 different human genes; Operon Biotechnologies, Inc., Ebersberg, Germany) using a ArrayBooster™ Hybridization Station
Box 1. Key terms.
n Amyloid precursor protein: an integral membrane protein, mainly localized at synapses, whose role is still elusive. Amyloid precursor protein cleavage, is a key step of the amyloid cascade as b- and g-secretase activity on amyloid precursor protein leads to amyloid-b (Ab) formation. n Ab oligomers: small (2.7–4.2 nm), spherical, soluble Ab aggregates. n Ingenuity pathway analysis: a commercial software program used to analyze large amounts of data from gene expression or proteomics
analysis and infer interrelated biological functions, functional networks and pathways. n Microarray: a technique that, by analyzing variations in mRNA levels, allows investigation of gene expression changes. n Ab: a pathogenic peptide containing 36–43 amino acid residues that is involved in Alzheimer’s disease. Ab, aggregated in several
forms, is the main constituent of Alzheimer’s disease plaques.
Research Article Gatta, Granzotto, Fincati et al.

www.futuremedicine.com 485future science group
(Advalytix, Brunnthal, Germany). For each slide, one treated sample (Ab
1–42, Ab
1–42–Cu,
Ab1–42
–Zn, Cu, Zn) was hybridized against one control sample. Hybridization and posthybridi-zation washing were performed following the microarray slide manufacturer’s instructions. A dye-swapping procedure for each experiment was performed in two replicates in which treated and control RNA samples (labeled with either Alexa Fluor 555 or Alexa Fluor 647 fluorochromes) were crossed in both combinations on different slides, for a total of 14 experiments (Ab
1–42: six
slides, Ab1–42
–Cu: two slides, Ab1–42
–Zn: two slides, Cu: two slides, Zn: two slides). For each experiment, a technical replicate was carried out using material from sister cultures. After hybridization, fluorescent signals were scanned using a GenePix® 4000B laser scanner and ana-lyzed with proprietary Genepix® 6.0 software (Molecular Devices Corp., Sunnyvale, CA, USA). Microarray data are stored in the GEO public database (accession number: GSE23000). All the data are ‘minimum information about a microarray experiment’ compliant.
Statistical analysisStatistical analysis of the microarray data was performed using Acuity® 4.0 software (Molecular Devices Corp.). Initial results were normalized using the LOWESS algorithm and filtered to exclude outliers and extreme flags. To reduce the possibility of false-positive/-neg-ative data, spots that did not show a detectable response across all slides were not included in the analysis. The expression value (log ratio) was calculated as base 2 logarithm of ratios of back-ground-corrected intensities that are medians of red dye over green dye fluorescent intensity values. Internal replicates were plotted against technical replicates to obtain single expression values for each gene. The reproducibility of the data was assessed using the X-Y scatter plot and outliers excluded. Finally, using a trimmed mean, spot replicate values within arrays were averaged excluding any spot that had no detect-able response in all three treatment replicates. The two final datasets were composed of 2910 transcripts (Ab
1–42, Ab
1-42–Cu and Cu experi-
ments) and 2814 transcripts (Ab1–42
, Ab1–42
–Zn and Zn experiments).
Statistical testing and fold change criteria were combined to validate significant gene changes. A gene was included in the analysis and considered to be differentially expressed when showing a log ratio higher or equal to 0.5, a value that represents a fold change of 1.4 in
transcript quantity. Student’s t-test was applied and p-values determined for each gene; p-val-ues were then adjusted for false discovery rate (FDR) by employing the q-value method cre-ated by Storey and Dabney (R package q-value 1.0, Bioconductor) [14]. Significance was set at q < 0.015.
Transmission electron microscopy To assess the morphology of Ab
1–42–metal
conjugates, transmission electron microscopy micrographs were collected after dialysis. Briefly, samples at 10 µM protein concentration were absorbed onto glow-discharged carbon-coated butwar films on 400-mesh copper grids; 1% uranyl acetate was used to negatively stain cop-per grids. Samples were observed at 40,000× by transmission electron microscopy (Tecnai G2, FEI, OR, USA).
ResultsSH-SY5Y cells were treated for 24 h with Ab conjugated with Zn or Cu. In parallel control experiments, SH-SY5Y were also treated with Ab alone or metal ions at a concentration tenfold higher than what was used for Ab exposure.
As we have previously reported in the case of Ab–Al complexes, Ab–Zn or Ab–Cu treat-ments significantly affected gene expression profiles [11]. When analyzing these changes, we focused our attention on up- or down-reg-ulated genes strongly related to AD pathology. Unfortunately, technical issues did not allow the validation of microarray data by real-time polymerase chain reaction (RT-PCR); however, to reduce the risk of false-positive/-negative results we employed a conservative statistical analysis that only considered spots to be ‘dif-ferentially expressed’ if they showed a detectable response across all slides and that, on average, had a log ratio higher or equal to 0.5, a value that represents a strong fold change (i.e., 1.4 in transcript quantity).
To increase the clarity and readability of the study, we chose to divide the ‘Results’ section into two sub-sections where we separately describe Ab–Zn or Ab–Cu data. Few gene changes were common to both Ab–metal treatments and are shown as a Venn diagram (Supplementary Figure 1 &
Supplementary taBle 1, see online www.futuremedi-cine.com/doi/suppl/10.2217/FNL.12.35).
Effects of Ab–Zn exposureAb–Zn treatment resulted in selective up- and down-regulation of 88 and 50 transcripts, respec-tively (see Supplementary taBleS 2 & 3). IPA was then
Gene profile of cells exposed to Ab–Zn & Ab–Cu Research Article

Future Neurol. (2012) 7(4)486 future science group
carried out to identify biological functions of the genes. For upregulated transcripts we found that the main functions involved were: protein synthesis, carbohydrate metabolism, cellular assembly and organization, cellular function and maintenance, lipid metabolism, nervous system development and function, neurologi-cal diseases, cell death and energy production (Figure 1). As for downregulated genes, functions
included: cell morphology, cellular development, cell death, cellular assembly and organization, nervous system development and function, lipid metabolism and free radical scavenging (Figure 2).
AD-related upregulated genesCAST: this gene encodes for calpastatin, an endogenous calpain inhibitor. Calpains are cal-cium-activated intracellular cysteine proteases
0
1
2
3
4
5
Pro
tein
syn
thes
is
Car
bohy
drat
e m
etab
olis
m
Cel
lula
r as
sem
byan
d or
gani
zatio
n
Cel
lula
r fu
nctio
nan
d m
aint
enan
ce
Lipi
d m
etab
olis
m
Ner
vous
sys
tem
dev
and
func
Neu
rolo
gica
l dis
ease
Cel
l dea
th
Ene
rgy
prod
uctio
n
-lo
g (
p-v
alu
e)
Threshold
Category
Protein synthesis
Carbohydrate metabolismCellular assembly and organizationCellular function and maintenanceLipid metabolismNervous system development and functionNeurological disease
Cell death
Energy production
p-value
4.06E-05-2.53E-02
3.38E-03-4.31E-02
3.38E-03-4.96E-02
3.38E-03-4.31E-02
3.38E-03-4.63E-023.38E-03-4.96E-02
3.38E-03-4.63E-02
1.01E-02-3.98E-02
1.01E-02-1.01E-02
Molecules
IL1RN, BIN3, RPL17, USP11, RPL21, CUTA, RPL26, CAST, RPS3, EEF1GARF1, SCARB1, CHGA, IL1RN, INPP5K
ARF1, WNT3A, MYL6, SCARB1, MT2A, ACTB, KTPN,CAST, MSTO1FTL, ARF1, WNT3A, SCARB1, MT2A, IL1RN,IL21R, KPTN, PFDN5, CAST, MECOMARF1, SCARB1, IL1RN, SULT1A1, INPP5KARF1, WNT3A, CHGA, MT2A, IL1RN, CAST
UPF2, FTL, CHGA, SLC15A1, IL1RN, PQBP1, ACTB, RPL17, RPL21, TUFT1, CAST, nullACTB, NAA35, CBX5, PSIP1, MECOM, HSPA4,TADA3, WNT3A, SCARB1, ENO1, IL1RN, MT2A, PQBP1, EIF5A, CAST, RPS3IL1RN
Figure 1. Ingenuity pathway analysis functional analysis of upregulated genes modulated by exposure to amyloid-b–Zn. (A) Bar chart shows key modulated functions. (B) Table shows genes upregulated by amyloid-b–Zn treatment related to functions shown in (A).Dev: Development; func: Function.
Research Article Gatta, Granzotto, Fincati et al.

www.futuremedicine.com 487future science group
involved in physiological and neurotoxic proc-esses. The role of calpains in AD is still elusive; however, important activities are described as far as involvement in BACE1 expression, APP processing and Ab deposition in an AD mouse model [15].
EIF5A: the protein encoded by this gene, called eukaryotic translation initiator factor 5A, modulates protein synthesis. Interestingly, EIF5A affects the activity of NGF in the modulation of neurotrophism and neuroprotection [16].
ENO1: this gene encodes for enolase-a (also known as phosphopyruvate hydratase), a key enzyme for glycidic metabolism. A study has shown that oxidative stress mediates ENO1 inactivation and that can have a role in AD development in subjects showing signs of mild cognitive impairment [17].
HSPA4 (also known as HSP70): this gene encodes for HSP4 (70 kDa isoform), a chap-erone that is involved in protein assembly. As recently reported [18], HSPA4 upregulation coun-teracts intracellular Ab toxicity; however, some data also indicate that the protein can promote neuroprotection in AD [19].
IL1RN: this gene encodes for the IL-1 receptor antagonist. Levels of the protein are increased in AD patients [20]. Furthermore, IL1RN overex-pression resulted in marked reduction of brain volume without upregulation of compensatory proinflammatory cytokines in an AD mouse model [21].
MT2A: this gene encodes for metal ion bind-ing metallothionein (MT) 2A. MT2A shows high affinity for heavy metals such as Zn2+ and Cu2+. MT2A, as all MTs, is an antioxidant and cytoprotective [22].
PQBP1: the protein encoded by this gene is a nuclear polyglutamine-binding protein involved in transcription activation. Knockout models for this gene show learning impairment [23]; PQBP1 dysfunction causes mitochondrial-induced cell death [24] while mutations are involved in X-linked mental retardation [25].
PSIP1: this gene encodes for a transcription co-activator. Data from fetal and adult human brains indicate a possible role of the protein in the regulation of neurogenesis and neuroepithelial stem cell differentiation [26].
RPS3: this gene encodes for the 40S ribos-omal protein S3. RPS3 overexpression has been reported to occur in ischemia-damaged hippocampal CA1 neurons likely exerting neuroprotective effects [27].
SCARB1: this gene encodes for the high density lipoproteins receptor. An involvement
of the receptor has been reported in an AD mouse model; SCARB1 downregulation has been shown to enhance amyloid pathology and exacerbate memory deficits [28].
WNT3A : this gene encodes for WNT3, a protein involved in transcription signaling and cell development. WNT3 expression appears to counteract Ab-mediated blockade of neuro-nal differentiation as well as damage of neural progenitor cells [29].
AD-related underexpressed genesACE: this gene encodes for the ACE, a protein involved in catalyzing conversion of angio-tensin I to the physiologically active peptide angiotensin II. Interestingly, recent data sug-gest that the enzyme also participates in Ab degradation [30].
ACIN1 (also known as Acinus): the protein encoded by this gene induces apoptotic chro-matin condensation after cleavage induced by caspase-3 activity [31].
C5: this gene encodes for C5, a protein of the complement family. C5 plays a critical role in inflammation and induction of apoptosis. The role of C5 in AD is controversial. Some studies indicate an increase in C5 expression in post-mortem AD brains; however, in a transgenic AD model (APP23) no changes have been found [32].
CCNG1: this gene encodes for cyclin G1, a protein participating in the regulation of the cell cycle and in negative regulation of apoptosis. CCNG1 has been shown to be overexpressed in a zebrafish AD model carrying human presenilin point mutations [33].
CITED2 : the protein encoded by the CITED2 gene plays several roles in proliferation, differentiation, migration, development, and apoptosis. Upregulation of CITED2 promotes cellular death, whereas its deficiency is cytopro-tective [34]. A recent study also suggested that CITED2 can affect neuronal differentiation [35].
CLIC4: this gene encodes for the chloride intracellular channel, CLIC4. The channel is ubiquitously distributed in cells. In neu-rons, CLIC4 is present in the plasma mem-brane, in intracellular compartments [36] and its translocation to the nucleus involved in stress-induced apoptosis [37]. Furthermore, the CLIC4 homolog, CLIC1 has been reported to promote Ab-induced reactive oxygen species (ROS) generation in microglia [38].
NFE2L1: the role played by the protein encoded by this gene is still obscure, although some findings speculate involvement in the modulation of redox responses. Supporting this
Gene profile of cells exposed to Ab–Zn & Ab–Cu Research Article

Future Neurol. (2012) 7(4)488 future science group
idea, NFE2L1 has been shown to mediate cellu-lar adaptation to redox stress in the endoplasmic reticulum [39] and to be critical to favor redox balance in liver cells during development [40].
NR4A1 (as known as TR3): this gene encodes for a protein of the so-called ‘death receptor fam-ily’. A close correlation between NR4A1 expres-sion and AD-affected brain regions has been shown in patients [41].
ZNF148 : this gene encodes for a DNA-binding protein. ZNF148 acts as a proapoptotic factor, enhancing Bak expression and promoting cell death in hepatocellular cancer cells [42].
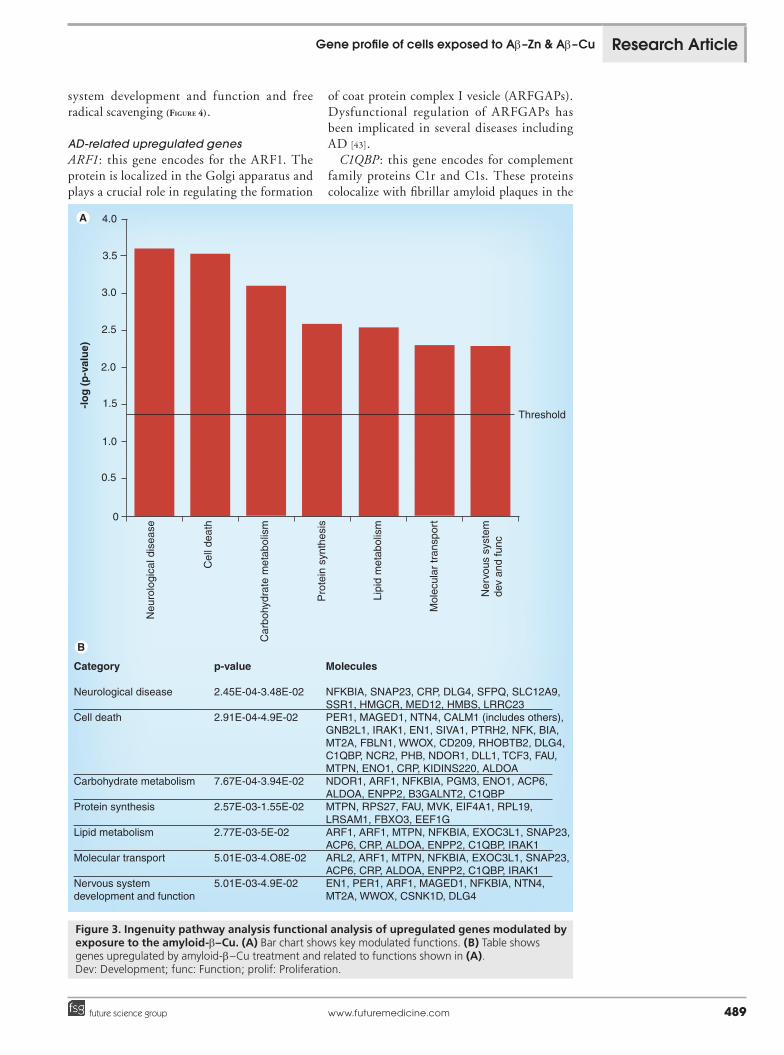
Effects of Ab–Cu exposureAb–Cu exposure resulted in selective upregula-tion and downregulation of 189 and 210 genes, respectively (Supplementary taBleS 4 & 5). IPA of the upregulated gene dataset showed transcripts involved in: neurological diseases, cell death, carbohydrate metabolism, protein synthesis, lipid metabolism, molecular transport and nerv-ous system development and function (Figure 3). Functions related to downregulated genes were: cell death, neurological disease, lipid metabo-lism, cell morphology, cellular growth and proliferation, inflammatory response, nervous
Cel
lula
r as
sem
byan
d or
gani
zatio
n
Cel
lula
r de
velo
pmen
t
Lipi
d m
etab
olis
m
Ner
vous
sys
tem
dev
and
func
-lo
g (
p-v
alu
e)
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
Threshold
Category
Cell morphologyCellular developmentCell death
Cellular assemblyand organizationNervous systemdevelopment and functionLipid metabolismFree radicalscavenging
p-value
1.06E-03-4.87E-021.28E-03-3.69E-021.57E-03-4.95E-02
1.57E-03-4.6E-02
1.57E-03-2.78E-02
3.13E-03-4.15E-022,17E-02-3.95E-02
Molecules
CCNG1, NR4A1, NEGH, C5, CLIC4, ACE, CITED2LAMA1, C5, CLIC4, CITED2, NFE2L1, ACECCNG1, ZNF148, ACIN1, NR4A1, NEFH, C5,CLIC4, CITED2, ACE, NFE2L1HAUS5, NEFH, C5, PIK3R4, C11orf73, ACE
KCNJ1, NEFH, C5, CITED2, ACE
NR4A1, C5, PIK3R4, CITED2, ACENR4A1, C5
Cel
l dea
th
Cel
l mor
phol
ogy
Free
rad
ical
scav
engi
ng
Figure 2. Ingenuity pathway analysis functional analysis of downregulated genes modulated by exposure to the amyloid-b–Zn. (A) Bar chart shows key modulated functions. (B) Table shows genes downregulated by amyloid-b–Zn treatment and related to functions shown in (A).Dev: Development; func: Function.
Research Article Gatta, Granzotto, Fincati et al.
www.futuremedicine.com 489future science group
system development and function and free radical scavenging (Figure 4).
AD-related upregulated genesARF1: this gene encodes for the ARF1. The protein is localized in the Golgi apparatus and plays a crucial role in regulating the formation
of coat protein complex I vesicle (ARFGAPs). Dysfunctional regulation of ARFGAPs has been implicated in several diseases including AD [43].
C1QBP: this gene encodes for complement family proteins C1r and C1s. These proteins colocalize with fibrillar amyloid plaques in the
Neu
rolo
gica
l dis
ease
Ner
vous
sys
tem
dev
and
func
-lo
g (
p-v
alu
e)
Threshold
Category
Neurological disease
Cell death
Carbohydrate metabolism
Protein synthesis
Lipid metabolism
Molecular transport
Nervous system development and function
p-value
2.45E-04-3.48E-02
2.91E-04-4.9E-02
7.67E-04-3.94E-02
2.57E-03-1.55E-02
2.77E-03-5E-02
5.01E-03-4.O8E-02
5.01E-03-4.9E-02
Molecules
NFKBIA, SNAP23, CRP, DLG4, SFPQ, SLC12A9, SSR1, HMGCR, MED12, HMBS, LRRC23PER1, MAGED1, NTN4, CALM1 (includes others), GNB2L1, IRAK1, EN1, SIVA1, PTRH2, NFK, BIA, MT2A, FBLN1, WWOX, CD209, RHOBTB2, DLG4, C1QBP, NCR2, PHB, NDOR1, DLL1, TCF3, FAU, MTPN, ENO1, CRP, KIDINS220, ALDOANDOR1, ARF1, NFKBIA, PGM3, ENO1, ACP6, ALDOA, ENPP2, B3GALNT2, C1QBPMTPN, RPS27, FAU, MVK, EIF4A1, RPL19, LRSAM1, FBXO3, EEF1GARF1, ARF1, MTPN, NFKBIA, EXOC3L1, SNAP23, ACP6, CRP, ALDOA, ENPP2, C1QBP, IRAK1ARL2, ARF1, MTPN, NFKBIA, EXOC3L1, SNAP23, ACP6, CRP, ALDOA, ENPP2, C1QBP, IRAK1EN1, PER1, ARF1, MAGED1, NFKBIA, NTN4, MT2A, WWOX, CSNK1D, DLG4
0
0.5
1.0
1.5
2.5
3.0
4.0
3.5
2.0
Cel
l dea
th
Car
bohy
drat
e m
etab
olis
m
Pro
tein
syn
thes
is
Lipi
d m
etab
olis
m
Mol
ecul
ar tr
ansp
ort
Figure 3. Ingenuity pathway analysis functional analysis of upregulated genes modulated by exposure to the amyloid-b–Cu. (A) Bar chart shows key modulated functions. (B) Table shows genes upregulated by amyloid-b–Cu treatment and related to functions shown in (A). Dev: Development; func: Function; prolif: Proliferation.
Gene profile of cells exposed to Ab–Zn & Ab–Cu Research Article

Future Neurol. (2012) 7(4)490 future science group
-lo
g (
p-v
alu
e)
Category
Cell death
Neurological disease
Lipid metabolism
Cell morphology
Cellular growth and proliferation
Inflammatory response
Nervous system development and functionFree radical scavenging
p-value
6.05E-04-4.8E-02
1.9E-03-3.6E-02
3.7E-03-4.8E-02
5.45E-03-3.69E-02
5.45E-03-4.8E-02
5.45E-03-4.59E-02
5.45E-03-2.69E-02
8.11E-03-8.11E-03
Molecules
SAT1, GAPDH, USP2, ACIN1, PTK2 (includes EG:14083), TACR1, PRDM2, TXK, BMI1, LPAR2, PPP3CB, INPP5B, HSPBAP1, SORT1, HIPK2, PARVA, DUT, SRA1, IRF4, HIPK3, MAD2L1, CDC42BPA, YARS, MDK, BCL2A1, FOXC2, FANCAAPBA2, AGAP1, SAT1, GAPDH, BEST3, PDE1A, USP2, TOR3A, PTK2 (inludes EG:14083), TACR1, HTR1B, BMI1, PRDM2, PPP3CB, HECW1, LAMA1, GSTM4, DARS, SORT1, NR2F6, HIPK2, CHRNA3, PARVA, IRF4, HIPK3, FXYD1, CXorf40A/CXorf40B, TTC23L, RXRG, CDC42BPA, PEX5, YARS, C21orf91PTK2 (includes EG:14083), LTA4H, ABCD3, PEX5, HSPA1L, INPP5B, PCYT2, TSHR, SAT1, ECH1PTK2 (includes EG:14083), MAD2L1, COL5A3, PARVA, HTR1B, SYNE2, LAMA1IRF4, RUVBL1, PTK2 (includes EG:14083), TACR1, MAD2L1, BMI1, TXK, LPAR2, LAMA1, TSHR, MDK, HIPK2, BCL2A1, FANCAPTK2 (includes EG:14083), TACR1, IRF4, INPP5B, TSHR, MDK, BCL2A1PTK2 (includes EG:!4083), APBA2, COL5A3, BMI1, HTR1B, PEX5, LAMA1, MDK, CHRNA3, FOXC2ME3
Cel
l dea
th
0.0
0.5
1.0
1.5
2.0
2.5
3.5
3.0
4.0
Neu
rolo
gica
l dis
ease
Lipi
d m
etab
olis
m
Cel
l mor
phol
ogy
Cel
lula
r gr
owth
and
pro
lif
Infla
mm
ator
y re
spon
se
Ner
vous
sys
tem
de
v an
d fu
nc
Free
rad
ical
sca
veng
ing
Threshold
Figure 4. Ingenuity pathway analysis functional analysis of downregulated genes modulated by exposure to the amyloid-b–Cu. (A) Bar chart shows key modulated functions. (B) Table shows genes downregulated by amyloid-b–Cu treatment and related to functions shown in (A).Dev: Development; func: Function; prolif: Proliferation.
Research Article Gatta, Granzotto, Fincati et al.

www.futuremedicine.com 491future science group
hippocampus and cerebral cortex of AD patients. Activated complement components are found in association with AD lesions and can contribute to local inflammation [44,45].
CRP: this gene encodes for C-reactive pro-tein. C-reactive protein plasma levels increase as a result of tissue injury, infection or inflam-matory stimuli, and have been reported in AD patients [46].
DLG4: this gene encodes for the postsynaptic scaffold protein PSD-95, which is involved in synaptic clustering and trafficking of ionotropic glutamate receptors. A correlation between AD and increased PSD-95 protein levels has been reported by some authors [47]; however, others have shown different results [48].
DLL1: this gene encodes for DLL1, a pro-tein that is a downstream effector involved in Notch signaling, a key step in the cascade lead-ing to Ab production. Upregulation of DLL1 has been shown in Down’s syndrome and AD patients [49].
ENO1: (described above in the ‘Ab–Zn’ section).
ENPP2: this gene encodes for a protein that has a role in active myelination and/or late stages of oligodendrocyte differentiation. Intriguingly, microarray analysis of rat brains revealed that mRNA levels of ENPP2 are increased by estro-gens in the hippocampus, an interesting link as estrogens have a role in AD and promote neuronal survival against Ab-mediated neurotoxicity [50–52].
GNB2L1 (als known as RACK-1): this gene encodes for the guanine nucleotide binding protein (G protein). GNB2L1 has been suggested to pro-mote Ab-driven cognitive impairment by favoring blockade of muscarinic receptor signaling [53] and metabotropic glutamate receptor activation [54]. Furthermore, GNB2L1 overexpression is effec-tive in rescuing GABAergic neurotransmission in Ab-treated cortical neurons [55].
HMGCR: this gene encodes for HMG-CoA reductase that is the rate-limiting enzyme controlling cholesterol synthesis in the brain. Overexpression of HMGCR results in increased cholesterol plasma levels, induction of Ab production and increased AD risk [56].
IRAK-1: the protein encoded by this gene is the IL-1 receptor-associated kinase 1, a serine/threo-nine kinase associated with IL-1 receptor upon stimulation. The protein modulates a plethora of biological processes, including activation of MAPK activity, activation of NF-kB-inducing kinase activity and Toll-like/IL-1 receptor sig-naling. Interestingly, unlike what we find here, the protein has been found to be decreased in
human astroglial cells exposed to Ab and in the hippocampus and neocortex of AD patients [57].
KIDINS220 (also known as ARMS): this gene encodes for a transmembrane scaffold protein that plays a critical role in BDNF-TrkB signal-ing [58]. Downregulation of this gene results in aberrant neuronal development [59].
MAGED1: this gene encodes for a protein member of the melanoma antigen gene fam-ily. Although this protein is not expressed in normal adult tissue it should be noted that the molecule is a cell-death inducer involved in neu-ronal development. Pharmacological inhibition of MAGED1 activity has been shown to reduce Ab-mediated toxicity in cortical neurons [60].
MED12: the protein encoded by this gene is involved in neuronal development. MED12 is a new component of APP-dependent nuclear-signaling pathway [61] and MED12 mutations are associated with cognitive and behavioral dysfunction in humans [62].
MT2A : (described above in the ‘Ab–Zn’ section).NDOR1-NR1: this gene encodes for a NADPH-dependent dif lavin reductase. NR1 levels decrease in the frontal cortex and hippocampus of AD patients [63].
NFKBIA: the molecule encoded by this gene is involved in the inhibition of NF-kB-mediated inflammatory responses. NF-kB is one of the major pathways activated during Ab-driven inflammation occurring in AD [64].
PER1/CSNK1A1: these are two genes involved in circadian rhythms. Circadian rhythms are altered in dementia. Thus, changes in PER1 mRNA expression fit with the alterations of the sleep/wake cycle found in AD; however, signifi-cant differences in PER1 expression have not been observed in a recent study with AD patients [65,66].
PHB: this gene encodes for prohibitin, a pro-tein involved in a variety of functions in many cell types. The functions of prohibitin in neu-rons remain largely unknown; however, recent findings suggest a possible protective role against ROS-mediated neuronal death [67].
PTRH2 (also known as BIT1): this gene encodes for Bit1, an effector of anoikis [68]. Overexpression of Bit1 has been reported to mediate cell survival in adherent cells through activation of NF-kB pathways [69].
SIVA1: the protein encoded by the SIVA1 gene plays a role in the activation of apoptotic path-ways. SIVA1 also negatively regulates NF-kB activity [70].
SNAP23: the protein encoded by this gene is an important regulator of transport vesicle
Gene profile of cells exposed to Ab–Zn & Ab–Cu Research Article

Future Neurol. (2012) 7(4)492 future science group
docking and fusion. SNAP-23 is relevant for the functional regulation of postsynaptic glutamate receptors. Loss of SNAP-23 in transgenic mice leads to a marked decrease in NMDA receptor surface expression and NMDA receptor-mediated currents [71–73].
WWOX: the molecule encoded by this gene, the WW domain-containing oxidoreductase, is a proapoptotic protein. A correlation between downregulation of WWOX and increased tau phosphorylation has been observed in AD hippocampi [74].
AD-related downregulated genesABCD3 : the protein encoded by this gene belongs to the superfamily of ATP-binding cas-sette transporters. A recent report indicates a significant increase in ABCD3 expression upon inflammatory conditions [75].
APBA2: this gene encodes for a member of the X11 family that interacts with APP and inhib-its production of proteolytic APP fragments, thereby suppressing Ab production [76].
BCL2A1: this gene encodes for a member of the BCL-2 protein family. BCL2A1 overexpres-sion reduces release of cytocrome C, thereby exerting potent antiapoptotic activity. Bcl-2 is crucial to reduce caspase activation.
CHRNA3: this gene encodes for a member of the nicotinic acetylcholine receptor family pro-teins. An association between polymorphisms of this gene and AD development has been reported, suggesting a role in the pathogenesis of sporadic AD [77].
FOXC2: the role of this gene is still elusive. The encoded protein, FOXC2, belongs to the forkhead family of transcription factors. Data indicate possible involvement of this gene in the NF-kB-activating signals as downstream effector of inflammatory or stress-like stimuli [78].
GSTP1: glutathione-S-transferase P1 is encoded by this gene. A recent study indicates a protective role of the molecule against Cdk5-induced neuronal toxicity. GSTP1 is effective in inhibiting Cdk5 and reducing oxidative stress [79].
HIPK2: this gene encodes for a serine/threonine kinase crucial to maintaining p53 wild-type activ-ity. Homodomain interacting protein kinase 2 is involved in Ab-driven toxicity although a study found no interaction between Ab treatment and HIPK2 mRNA expression in HEK-293 cells as well as in fibroblasts from AD patients [80].
HSPA1L: this gene encodes for a 70 kDa HSP. Similarly to HSPA4, Hsp70 has been reported to exert a protective role against intracellular Ab-driven toxicity [18].
HTR1B: this gene encodes for the serotonin-ergic receptor subtype 5-HT1B. 5-HT1B is a putative target for antidepressant activity [81].
MDK: this gene encodes for a hepatin-bind-ing growth factor. This factor is involved in the repair and development of several tissues including neurons [82].
PMAIP1: the molecule encoded by this gene is involved in proapoptotic mechanisms by interfer-ing with p53/p73 signaling. As far as AD, altera-tions in APP function induce expression of several p53/p73 target genes including PMAIP1 [83].
PTK2 (also known as FAK ): this gene encodes for a tyrosine kinase involved in cell adhesion. Ab-driven PTK2 activation has been reported as one of the earliest biochemical responses trig-gered by Ab exposure in human and rat cortical cultures [84].
DiscussionIn recent years, great attention has been focused on how changes in Ab conformation can affect the neurotoxic properties of the peptide and it is now established that oligomers are the most toxic species. Thus, factors promoting Ab capa-bility to retain its oligomeric structure strongly influence the pathogenic weight of the peptide. Several endogenous factors alter Ab folding and, among these, metals appear to have a primary role in the process. We have previously shown that, when conjugated with Ab, several metals modified cell viability most likely by changing peptide folding [12,13]. However, it must be noted that oligomerization is not the only pathogenic factor in AD as Ab (in any aggregation form) favors the detrimental destabilization of brain metal homeostasis [5,9,85,86].
As with the results, the discussion is divided into two subsections.
Ab–ZnZn greatly affects brain physiology and pathol-ogy [87]. In AD, loss of Zn homeostasis triggers a series of downstream harmful events [88]. We have previously shown that Ab–Zn promotes rapid formation of amorphous aggregates [12] (a phenomenon confirmed in this set of experiments; see Supplementary Figure 2) and induces some levels of SH-SY5Y toxicity [13]. Microarray analysis shows that exposure to Ab–Zn evokes a complex net-work of responses that can exert compensatory and protective actions. Unfortunately, the number of transcripts we have found to be changed is too low and heterogeneous to obtain an IPA-generated pathway. Nevertheless, it is possible to speculate that Ab–Zn promotes gene expression changes
Research Article Gatta, Granzotto, Fincati et al.

www.futuremedicine.com 493future science group
that belong to a potential pathway centered on cell death modulation. This hypothesis is supported by the coexistence of downregulation of proapop-totic transcripts (ACIN1, CAST, CITED2 and ZNF148). Moreover, the metal peptide complex is likely to promote an additional pathway centered on compensatory and protective responses as we observed the downregulation of AD-related genes that are usually overexpressed in the disease (ACE, C5, IL1RN and NR4A1) along with upregulation of cytoprotective genes (EIF5A, ENO1, HSPA4, PSIP1, RPS3 and WNT3A). Interestingly, two of the upregulated transcripts (HSPA4 and WNT3A) are reported to counteract Ab toxic-ity. MT2A overexpression is also important and potentially a key component of this hypothesized protective pathway as MTs are crucial in provid-ing Zn2+ and Cu2+ homeostasis, reducing oxidative stress and regulating authophagy [89].
Taken together, microarray results along with previous data support the hypothesis that Ab–Zn (when not forming oligomeric struc-tures) is unable to induce the expression of proa-poptotic pathways but, on the contrary, activates compensatory and protective mechanisms, thus counteracting its toxicity [90].
Ab–CuBrain Cu levels undergo significant changes with aging [91]. In AD, data indicate that cortical Cu levels are decreased [92], a process resulting from sequestration of the cation in amyloid plaques [8]. Cu can promote oxidative stress [5,6,86]; however, the metal is also involved in synaptic inhibition of glutamatergic neurotransmission [93] and its decreased availability can promote excitotoxic-ity. Furthermore, decreased intracellular lev-els of Cu favor the internalization of Ab and Cu in cholesterol-rich lipid rafts of the plasma membrane, thereby facilitating the formation of Ab–Cu complexes [94]. Thus, the issue of age- and AD-related loss of Cu homeostasis is critical in the theoretical framework of the metal hypothesis of AD [5,10,85,95,96].
Ab–Cu shows many commonalities with Ab–Zn. This is probably due to high biophysi-cal similarities of the two metal ions. They have the same ionic charge (2+), and a similar ionic radius (~87 pm), which likely promotes simi-lar changes in structural Ab conformation. As with Ab–Zn, Cu conjugated with Ab generates amorphous aggregates of high molecular weight [12,97]. However, Ab–Cu has a selective feature: the complex shows less pronounced exposure of hydrophobic clusters and is more soluble. It is important to highlight that Cu2+, even when still
bound to Ab, can be reduced to Cu+ and promote oxidative stress [5,12,98] by generating hydrogen peroxide [99–102]. This Cu-mediated oxidative stress has been shown to be significant in neu-ronal cultures and has an important role in AD [5,10]. Reduced Cu also generates hydroxyl radicals by Fenton reaction, an upstream event leading to harmful molecular modifications (i.e., protein carbonyl modifications, DNA damage and lipid peroxidation) found in AD [5,10].
We have previously shown that Ab–Cu con-jugates exert moderate levels of SH-SY5Y toxic-ity most likely by promoting ROS production [103]. Our microarray data show that Ab–Cu interferes with the NF-kB pathway. NF-kB is a transcription factor involved in cellular response against several stressors, including ROS, and in AD-related inflammatory responses [64,104]. Our findings indicate an indirect action on NF-kB. In fact, the complex promotes upregulation of two genes that negatively control its activity (NFKBIA and SIVA1). It must be noted that this negative modulation is potentially coun-teracted by simultaneous overexpression of PTRH2, a gene that is cytoprotective through activation of NF-kB-related pathways [69].
As with Ab–Zn, the number of changed tran-scripts triggered by Ab–Cu exposure is too low and heterogeneous to obtain an IPA-generated pathway. However, some speculative pathways can be hypothesized.
Contrary to what we have found in the case of Ab–Zn (where antiapoptotic and compen-satory/protective pathways seem to prevail), Ab–Cu appears to produce an overall homeo-static response in which expression of pro- and antiapoptotic genes reaches a balance. Indeed, along with upregulation of proapoptotic genes (MAGED1 and WWOX ), we found a simultane-ous underexpression of antiapoptotic transcripts (BCL2A1, GSTP1 and MDK ).
A speculative pathway that can be proposed is centered on proteins that counteract oxida-tive stress. Supporting that hypothesis we report overexpression of transcripts like PHB and MT2A that encode for scavenger molecules. In the case of MT2A, it is possible that free radicals generated by Ab–Cu promote intra cellular Zn and Cu release from MTs, a process that further induces MT2A expression (as the gene is con-trolled by the Cu/Zn-sensitive metal-responsive transcription factor-1) [105].
ConclusionHow the described up- or down-regulated tran-scripts modulate AD pathogenic pathways is still
Gene profile of cells exposed to Ab–Zn & Ab–Cu Research Article

Future Neurol. (2012) 7(4)494 future science group
ReferencesPapers of special note have been highlighted as:n of interestnn of considerable interest
1. Querfurth HW, LaFerla FM. Alzheimer’s disease. N. Engl. J. Med. 362(4), 329–344 (2010).
2. De Strooper B. Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol. Rev. 90(2), 465–494 (2010).
3. Knowles TPJ, Waudby CA, Devlin GL et al. An analytical solution to the kinetics of breakable filament assembly. Science 326(5959), 1533–1537 (2009).
4. Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Ann. Rev. Biochem. 75(1), 333–366 (2006).
5. Roberts BR, Ryan TM, Bush AI, Masters CL, Duce JA. The role of metallobiology and amyloid-b peptides in Alzheimer’s disease. J. Neurochem. 120(Suppl. 1), 149–166 (2011).
not completely understood and warrants further investigation. However, present data along with previous findings lend support to the idea that metal ions play a critical role in shaping Ab con-formation, a process that in the case of Zn or Cu activates complex biological responses that appear to modulate apoptotic cell death and the handling of oxidative stress [11,12,106]. It should be stressed that according to the metal hypoth-esis of AD, Ab can also exert negative actions that impinge on neuronal and synaptic viability, neurotrophic signaling and cognitive functions. In that respect the role of Ab in sequestering synaptic Zn, thereby reducing its availability, appears critically important as the ion modulates long-term potentiation, the inhibition of NMDA receptors and promotes the maturation of BDNF from biologically inactive pro-BDNF (reviewed in [107]). Substantiating this idea, genetic deple-tion of synaptic Zn in a transgenic mouse model (ZnT3KO mice) reproduces the cognitive defi-cits found in AD mice [108], while dietary Zn supplementation counteracts these deficits and promotes significantly increased levels of BDNF in transgenic-AD mice [109]. The role of synaptic Cu has been investigated less but appears equally important at least for the modulation of NMDA receptors and related excitotoxicity as well as for synaptic viability, correct synaptic signaling and mitochondrial functioning [93,110].
Further unraveling of the pathogenic implica-tions of these Ab/metal-driven changes offers a challenge and an opportunity in AD-related research.
Future perspectiveOur results are in line with a large body of data indicating that metal dyshomeostasis (for Zn, Cu and Fe in particular) is gaining great
attention as a key factor in AD development and progression [5,9,10,85,95]. Further investiga-tion is required to clarify the molecular deter-minants of this dyshomeostasis. The imple-mentation of novel metal-sensitive fluorescent probes, along with sophisticated in vivo multi-photon brain imaging, single-molecule track-ing and optogenetic approaches, will definitely help to unravel mechanisms that promote opti-mal functioning of metal transporters, metal buffering systems as well as the sequestering role of intracellular organelles (endoplasmic reticulum, mitochondria, Golgi among oth-ers) in maintaining ion levels within a physi-ological range. These tools will also provide key information on how the disease affects the homeostatic systems, notions that are essential to design novel disease-modifying drugs for AD treatment.
Financial & competing interests disclosureSL Sensi is supported by funds from the Italian Department of Education (FIRB 2003, PRIN 2006).The authors have no other relevant affiliations or finan-cial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research The authors state that they have obtained appropriate insti tutional review board approval or have followed the princi ples outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investi gations involving human sub-jects, informed consent has been obtained from the participants involved.
Executive summary
n Amyloid-b (Ab) dysmetabolism has been shown to critically participate in the Alzheimer’s disease-related neurotoxic cascade. n In the Alzheimer’s disease brain, Ab misfolding and aggregation promote formation of senile plaques and is greatly influenced by the
alteration of metal levels and availability. n Ab–Zn and Ab–Cu complexes selectively alter gene expression in neuron-like cell lines. n Ingenuity pathway analysis reveals that most of the genes differentially expressed after Ab–Zn or Ab–Cu exposure are related to ‘cell
death’, ‘free radical scavenging’, ‘inflammatory response’, ‘cell morphology’ and ‘lipid metabolism’ functions.
Research Article Gatta, Granzotto, Fincati et al.

www.futuremedicine.com 495future science group
nn Extensive review on the role of metals in Alzheimer’s disease (AD).
6. Bonda DJ, Lee H-gon, Blair JA et al. Role of metal dyshomeostasis in Alzheimer’s disease. Metallomics 3(3), 267–270 (2011).
7. Lovell M. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 158(1), 47–52 (1998).
8. Miller LM, Wang Q, Telivala TP et al. Synchrotron-based infrared and x-ray imaging shows focalized accumulation of Cu and Zn co-localized with b-amyloid deposits in Alzheimer’s disease. J. Struct. Biol. 155(1), 30–37 (2006).
9. Duce JA, Bush AI. Biological metals and Alzheimer’s disease: implications for therapeutics and diagnostics. Progr. Neurobiol. 92(1), 1–18 (2010).
10. Milardi D, Rizzarelli E. Preface. In: Neurodegeneration: Metallostasis and Proteostasis. Milardi D, Rizzarelli E (Eds). Royal Society of Chemistry, Cambridge, UK, 5–8 (2011).
11. Gatta V, Drago D, Fincati K et al. Microarray analysis on human neuroblastoma cells exposed to aluminum, b(1–42)-amyloid or the b(1–42)-amyloid aluminum complex. PloS ONE 6(1), E15965 (2011).
n Study showing the modulation of gene expression triggered by b-amyloid (Ab)
1–42–Al.
12. Bolognin S, Messori L, Drago D et al. Aluminum, copper, iron and zinc differentially alter amyloid-Ab (1-42) aggregation and toxicity. Int. J. Biochem. Cell Biol. 43(6), 877–885 (2011).
13. Drago D, Bettella M, Bolognin S et al. Potential pathogenic role of b-amyloid(1-42)-aluminum complex in Alzheimer’s disease. Int. J. Biochem. Cell. Biol. 40(4), 731–746 (2008).
14. Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. USA 100(16), 9440–9445 (2003).
15. Liang B, Duan BY, Zhou XP, Gong JX, Luo ZG. Calpain activation promotes BACE1 expression, amyloid precursor protein processing, and amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J. Biol. Chem. 285(36), 27737–27744 (2010).
16. Huang Y, Higginson DS, Hester L, Park MH, Snyder SH. Neuronal growth and survival mediated by eIF5A, a polyamine-modified translation initiation factor. Proc. Natl Acad. Sci. USA 104(10), 4194–4199 (2007).
17. Butterfield DA, Poon HF, St Clair D et al. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the
development of Alzheimer’s disease. Neurobiol. Dis. 22(2), 223–232 (2006).
18. Cui J, Wang Y, Dong Q et al. Morphine protects against intracellular amyloid toxicity by inducing estradiol release and upregulation of Hsp70. J. Neurosci. 31(45), 16227–16240 (2011).
19. Turturici G, Sconzo G, Geraci F. Hsp70 and its molecular role in nervous system diseases. Biochem. Res. Int. 2011, 618127 (2011).
20. Yasuhara O, Matsuo A, Terai K et al. Expression of interleukin-1 receptor antagonist protein in post-mortem human brain tissues of Alzheimer’s disease and control cases. Acta Neuropathol. 93(4), 414–420 (1997).
21. Oprica M, Hjorth E, Spulber S et al. Studies on brain volume, Alzheimer-related proteins and cytokines in mice with chronic overexpression of IL-1 receptor antagonist. J. Cell. Mol. Med. 11(4), 810–825 (2007).
22. Mocchegiani E, Costarelli L, Giacconi R et al. Zinc-binding proteins (metallothionein and a-2 macroglobulin) and immunosenescence. Exp. Gerontol. 41(11), 1094–1107 (2006).
23. Tamura T, Horiuchi D, Chen YC et al. Drosophila PQBP1 regulates learning acquisition at projection neurons in aversive olfactory conditioning. J. Neurosci. 30(42), 14091–14101 (2010).
24. Marubuchi S, Wada YI, Okuda T et al. Polyglutamine tract-binding protein-1 dysfunction induces cell death of neurons through mitochondrial stress. J. Neurochem. 95(3), 858–870 (2005).
25. Kalscheuer VM, Freude K, Musante L et al. Mutations in the polyglutamine binding protein 1 gene cause X-linked mental retardation. Nat. Genet. 35(4), 313–315 (2003).
26. Chylack LT, Fu L, Mancini R et al. Lens epithelium-derived growth factor (LEDGF/p75) expression in fetal and adult human brain. Exp. Eye Res. 79(6), 941–948 (2004).
27. Hwang IK, Yoo KY, Kim DW et al. Ischemia-induced ribosomal protein S3 expressional changes and the neuroprotective effect against experimental cerebral ischemic damage. J. Neurosci. Res. 86(8), 1823–1835 (2008).
28. Thanopoulou K, Fragkouli A, Stylianopoulou F, Georgopoulos S. Scavenger receptor class B type I (SR-BI) regulates perivascular macrophages and modifies amyloid pathology in an Alzheimer mouse model. Proc. Natl Acad. Sci. USA 107(48), 20816–20821 (2010).
29. Shruster A, Eldar-Finkelman H, Melamed E, Offen D. Wnt signaling pathway overcomes
the disruption of neuronal differentiation of neural progenitor cells induced by oligomeric amyloid b-peptide. J. Neurochem. 116(4), 522–529 (2011).
30. Akatsu H, Ogawa N, Kanesaka T et al. Higher activity of peripheral blood angiotensin-converting enzyme is associated with later-onset of Alzheimer’s disease. J. Neurol. Sci. 300(1–2), 67–73 (2011).
31. Sahara S, Aoto M, Eguchi Y et al. Acinus is a caspase-3-activated protein required for apoptotic chromatin condensation. Nature 401(6749), 168–173 (1999).
32. Reichwald J, Danner S, Wiederhold KH, Staufenbiel M. Expression of complement system components during aging and amyloid deposition in APP transgenic mice. J. Neuroinflamm. 6, 35 (2009).
33. Newman M, Tucker B, Nornes S, Ward A, Lardelli M. Altering presenilin gene activity in zebrafish embryos causes changes in expression of genes with potential involvement in Alzheimer’s disease pathogenesis. J. Alzheimers Dis. 16(1), 133–147 (2009).
34. Gonzalez YR, Zhang Y, Behzadpoor D et al. CITED2 signals through peroxisome proliferator-activated receptor-g to regulate death of cortical neurons after DNA damage. J. Neurosci. 28(21), 5559–5569 (2008).
35. Dijkmans TF, van Hooijdonk LWA, Schouten TG et al. Identification of new nerve growth factor-responsive immediate-early genes. Brain Res. 1249, 19–33 (2009).
36. Suginta W, Karoulias N, Aitken A, Ashley RH. Chloride intracellular channel protein CLIC4 (p64H1) binds directly to brain dynamin I in a complex containing actin, tubulin and 14-3-3 isoforms. Biochem. J. 359(Pt 1), 55–64 (2001).
37. Suh KS, Mutoh M, Nagashima K et al. The organellular chloride channel protein CLIC4/mtCLIC translocates to the nucleus in response to cellular stress and accelerates apoptosis. J. Biol. Chem. 279(6), 4632–4641 (2004).
38. Milton RH, Abeti R, Averaimo S et al. CLIC1 function is required for b-amyloid-induced generation of reactive oxygen species by microglia. J. Neurosci. 28(45), 11488–11499 (2008).
39. Zhang Y, Crouch DH, Yamamoto M, Hayes JD. Negative regulation of the Nrf1 transcription factor by its N-terminal domain is independent of Keap1: Nrf1, but not Nrf2, is targeted to the endoplasmic reticulum. Biochem. J. 399(3), 373–385 (2006).
40. Chen L, Kwong M, Lu R et al. Nrf1 is critical for redox balance and survival of liver cells during development. Mol. Cell. Biol. 23(13), 4673–4686 (2003).
Gene profile of cells exposed to Ab–Zn & Ab–Cu Research Article

Future Neurol. (2012) 7(4)496 future science group
41. Newman SJ, Bond B, Crook B et al. Neuron-specific localisation of the TR3 death receptor in Alzheimer’s disease. Brain Res. 857(1–2), 131–140 (2000).
42. To AKY, Chen GG, Chan UPF et al. ZBP-89 enhances Bak expression and causes apoptosis in hepatocellular carcinoma cells. Biochim. Biophys. Acta 1813(1), 222–230 (2011).
43. Scheper W, Zwart R, Sluijs P et al. Alzheimer’s presenilin 1 is a putative membrane receptor for rab GDP dissociation inhibitor. Hum. Mol. Genet. 9(2), 303–310 (2000).
44. Shen Y, Li R, McGeer EG, McGeer PL Neuronal expression of mRNAs for complement proteins of the classical pathway in Alzheimer brain. Brain Res. 769(2), 391–395 (1997).
45. Zhou J, Fonseca MI, Pisalyaput K, Tenner AJ. Complement C3 and C4 expression in C1q sufficient and deficient mouse models of Alzheimer’s disease. J. Neurochem. 106(5), 2080–2092 (2008).
46. Mulder SD, Hack CE, van der Flier WM et al. Evaluation of intrathecal serum amyloid P (SAP) and C-reactive protein (CRP) synthesis in Alzheimer’s disease with the use of index values. J. Alzheimers Dis. 22(4), 1073–1079 (2010).
47. Leuba G, Savioz A, Vernay A et al. Differential changes in synaptic proteins in the Alzheimer frontal cortex with marked increase in PSD-95 postsynaptic protein. J. Alzheimers Dis. 15(1), 139–151 (2008).
48. Pham E, Crews L, Ubhi K et al. Progressive accumulation of amyloid-b oligomers in Alzheimer’s disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J. 277(14), 3051–3067 (2010).
49. Fischer DF, van Dijk R, Sluijs JA et al. Activation of the Notch pathway in Down syndrome: cross-talk of Notch and APP. FASEB J. 19(11), 1451–1458 (2005).
50. Aston C, Jiang L, Sokolov BP. Transcriptional profiling reveals evidence for signaling and oligodendroglial abnormalities in the temporal cortex from patients with major depressive disorder. Mol. Psych. 10(3), 309–322 (2005).
51. Mateos L, Persson T, Kathozi S, Gil-Bea FJ, Cedazo-Minguez A. Estrogen protects against amyloid-b toxicity by estrogen receptor a-mediated inhibition of Daxx translocation. Neurosci. Lett. 506(2), 245–250 (2012).
52. Takeo C, Ikeda K, Horie-Inoue K, Inoue S. Identification of Igf2, Igfbp2 and Enpp2 as estrogen-responsive genes in rat hippocampus. Endocrin. J. 56(1), 113–120 (2009).
53. Zhong P, Gu Z, Wang X et al. Impaired modulation of GABAergic transmission by muscarinic receptors in a mouse transgenic model of Alzheimer’s disease. J. Biol. Chem. 278(29), 26888–26896 (2003).
54. Tyszkiewicz JP, Yan Z. b-Amyloid peptides impair PKC-dependent functions of metabotropic glutamate receptors in prefrontal cortical neurons. J. Neurophys. 93(6), 3102–3111 (2005).
55. Liu W, Dou F, Feng J, Yan Z. RACK1 is involved in b-amyloid impairment of muscarinic regulation of GABAergic transmission. Neurobiol. Aging 32(10), 1818–1826 (2011).
56. Rodríguez-Rodríguez E, Mateo I, Infante J et al. Interaction between HMGCR and ABCA1 cholesterol-related genes modulates Alzheimer’s disease risk. Brain Res. 1280, 166–171 (2009).
57. Cui JG, Li YY, Zhao Y, Bhattacharjee S, Lukiw WJ. Differential regulation of interleukin-1 receptor-associated kinase-1 (IRAK-1) and IRAK-2 by microRNA-146a and NF-kB in stressed human astroglial cells and in Alzheimer disease. J. Biol. Chem. 285(50), 38951–38960 (2010).
58. Wu SH, Arévalo JC, Neubrand VE et al. The ankyrin repeat-rich membrane spanning (ARMS)/Kidins220 scaffold protein is regulated by activity-dependent calpain proteolysis and modulates synaptic plasticity. J. Biol. Chem. 285(52), 40472–40478 (2010).
59. Higuero AM, Sánchez-Ruiloba L, Doglio LE et al. Kidins220/ARMS modulates the activity of microtubule-regulating proteins and controls neuronal polarity and development. J. Biol. Chem. 285(2), 1343–1357 (2010).
60. Di Certo MG, Corbi N, Bruno T et al. NRAGE associates with the anti-apoptotic factor Che-1 and regulates its degradation to induce cell death. J. Cell. Sci. 120(Pt 11), 1852–1858 (2007).
61. Xu X, Zhou H, Boyer TG. Mediator is a transducer of amyloid-precursor-protein-dependent nuclear signalling. EMBO Rep. 12(3), 216–222 (2011).
62. Turner AJ, Belyaev ND, Nalivaeva NN. Mediator: the missing link in amyloid precursor protein nuclear signalling. EMBO Rep. 12(3), 180–181 (2011).
63. Amada N, Aihara K, Ravid R, Horie M. Reduction of NR1 and phosphorylated Ca2+/calmodulin-dependent protein kinase II levels in Alzheimer’s disease. Neuroreport 16(16), 1809–1813 (2005).
64. Couturier J, Paccalin M, Morel M et al. Prevention of the b-amyloid peptide-induced inflammatory process by inhibition of
double-stranded RNA-dependent protein kinase in primary murine mixed co-cultures. J. Neuroinflamm. 8, 72 (2011).
65. Tseng IJ, Liu HC, Yuan RY et al. Expression of inducible nitric oxide synthase (iNOS) and period 1 (PER1) clock gene products in different sleep stages of patients with cognitive impairment. J. Clin. Neurosci. 17(9), 1140–1143 (2010).
66. Etchegaray JP, Machida KK, Noton E et al. Casein kinase 1 d regulates the pace of the mammalian circadian clock. Mol. Cell. Biol. 29(14), 3853–3866 (2009).
67. Zhou P, Qian L, D’Aurelio M et al. Prohibitin reduces mitochondrial free radical production and protects brain cells from different injury modalities. J. Neurosci. 32(2), 583–592 (2012).
68. Jan Y, Matter M, Pai J-tung et al. A mitochondrial protein, Bit1, mediates apoptosis regulated by integrins and Groucho/TLE corepressors. Cell 116(5), 751–762 (2004).
69. Griffiths GS, Grundl M, Leychenko A et al. Bit-1 mediates integrin-dependent cell survival through activation of the NFkB pathway. J. Biol. Chem. 286(16), 14713–14723 (2011).
70. Gudi R, Barkinge J, Hawkins S et al. Siva-1 negatively regulates NF-kB activity: effect on T-cell receptor-mediated activation-induced cell death (AICD). Oncogene 25(24), 3458–3462 (2006).
71. Suh YH, Terashima A, Petralia RS et al. A neuronal role for SNAP-23 in postsynaptic glutamate receptor trafficking. Nat. Neurosci. 13(3), 338–343 (2010).
72. Mukaetova-Ladinska EB, Xuereb JH, Garcia-Sierra F et al. Lewy body variant of Alzheimer’s disease: selective neocortical loss of t-SNARE proteins and loss of MAP2 and a-synuclein in medial temporal lobe. Sci. World J. 9, 1463–1475 (2009).
73. Bragina L, Candiracci C, Barbaresi P et al. Heterogeneity of glutamatergic and GABAergic release machinery in cerebral cortex. Neuroscience 146(4), 1829–1840 (2007).
74. Sze CI, Su M, Pugazhenthi S et al. Down-regulation of WW domain-containing oxidoreductase induces tau phosphorylation in vitro. A potential role in Alzheimer’s disease. J. Biol. Chem. 279(29), 30498–30506 (2004).
75. Gray E, Ginty M, Kemp K, Scolding N, Wilkins A. Peroxisome proliferator-activated receptor-a agonists protect cortical neurons from inflammatory mediators and improve peroxisomal function. Eur. J. Neurosci. 33(8), 1421–1432 (2011).
Research Article Gatta, Granzotto, Fincati et al.

www.futuremedicine.com 497future science group
76. Saito Y, Sano Y, Vassar R et al. X11 proteins regulate the translocation of amyloid b-protein precursor (APP) into detergent-resistant membrane and suppress the amyloidogenic cleavage of APP by b-site-cleaving enzyme in brain. J. Biol. Chem. 283(51), 35763–35771 (2008).
77. Kawamata J, Shimohama S. Association of novel and established polymorphisms in neuronal nicotinic acetylcholine receptors with sporadic Alzheimer’s disease. J. Alzheimers Dis. 4(2), 71–76 (2002).
78. Li X, Massa PE, Hanidu A et al. IKKa, IKKb, and NEMO/IKKg are each required for the NF-k B-mediated inflammatory response program. J. Biol. Chem. 277(47), 45129–45140 (2002).
79. Sun KH, Chang KH, Clawson S et al. Glutathione-S-transferase P1 is a critical regulator of Cdk5 kinase activity. J. Neurochem. 118(5), 902–914 (2011).
80. Lanni C, Nardinocchi L, Puca R et al. Homeodomain interacting protein kinase 2: a target for Alzheimer’s b amyloid leading to misfolded p53 and inappropriate cell survival. PloS ONE 5(4), E10171 (2010).
81. Clark MS, Neumaier JF. The 5-HT1B receptor: behavioral implications. Psychopharmacol. Bull. 35(4), 170–185 (2001).
82. Sakakima H, Yoshida Y, Yamazaki Y et al. Disruption of the midkine gene (Mdk) delays degeneration and regeneration in injured peripheral nerve. J. Neurosci. Res. 87(13), 2908–2915 (2009).
83. Benosman S, Meng X, Von Grabowiecki Y et al. Complex regulation of p73 isoforms after alteration of amyloid precursor polypeptide (APP) function and DNA damage in neurons. J. Biol. Chem. 286(50), 43013–43025 (2011).
84. Williamson R, Scales T, Clark BR et al. Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-b peptide exposure: involvement of Src family protein kinases. J. Neurosci. 22(1), 10–20 (2002).
85. Faller P, Hureau C. Metal ions in neurodegenerative diseases. Coord. Chem. Rev. (2012) (In Press).
86. Kozlowski H, Luczkowski M, Remelli M, Valensin D. Copper, zinc and iron in neurodegenerative diseases (Alzheimer’s, Parkinson’s and prion diseases). Coord. Chem. Rev. (2012) (In Press).
87. Sensi SL, Paoletti P, Bush AI, Sekler I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 10(11), 780–791 (2009).
88. Corona C, Pensalfini A, Frazzini V, Sensi SL. New therapeutic targets in Alzheimer’s disease: brain deregulation of calcium and zinc. Cell Death Dis. 2, E176 (2011).
nn Extensive review on the role of Ca and Zn in AD.
89. Lee SJ, Koh JY. Roles of zinc and metallothionein-3 in oxidative stress-induced lysosomal dysfunction, cell death, and autophagy in neurons and astrocytes. Mol. Brain 3(1), 30 (2010).
90. Duce JA, Tsatsanis A, Cater MA et al. Iron-export ferroxidase activity of b-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 142(6), 857–867 (2010).
nn Key study indicating the interplay between Fe and Zn in AD.
91. Squitti R, Salustri C. Agents complexing copper as a therapeutic strategy for the treatment of Alzheimer’s disease. Curr. Alzheimer Res. 6(6), 476–487 (2009).
92. Deibel MA, Ehmann WD, Markesbery WR. Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer’s disease: possible relation to oxidative stress. J. Neurol. Sci. 143(1), 137–142 (1996).
93. Schlief ML, Gitlin JD. Copper homeostasis in the CNS: a novel link between the NMDA receptor and copper homeostasis in the hippocampus. Mol. Neurobiol. 33(2), 81–90 (2006).
94. Hung YH, Robb EL, Volitakis I et al. Paradoxical condensation of copper with elevated b-amyloid in lipid rafts under cellular copper deficiency conditions: implications for Alzheimer disease. J. Biol. Chem. 284(33), 21899–21907 (2009).
95. Zatta P, Drago D, Bolognin S, Sensi SL. Alzheimer’s disease, metal ions and metal homeostatic therapy. Trends Pharmacol. Sci. 30(7), 346–355 (2009).
nn Comprehensive review on the role of metals in AD.
96. Hung Y, Bush A, Cherny R. Copper in the brain and Alzheimer’s disease. J. Biol. Chem. 15(1), 61–76 (2010).
97. Chen WT, Liao YH, Yu HM, Cheng IH, Chen YR. Distinct effects of Zn2+, Cu2+, Fe3+, and Al3+ on amyloid-b stability, oligomerization, and aggregation. J. Biol. Chem. 286(11), 9646–9656 (2011).
98. Tõugu V, Tiiman A, Palumaa P. Interactions of Zn(II) and Cu(II) ions with Alzheimer’s amyloid-b peptide. Metal ion binding, contribution to fibrillization and toxicity. Metallomics 3(3), 250–261 (2011).
99. Huang X. Cu(II) Potentiation of Alzheimer Ab Neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J. Biol. Chem. 274(52), 37111–37116 (1999).
100. Opazo C, Huang X, Cherny RA et al. Metalloenzyme-like activity of Alzheimer’s disease b-amyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H
2O
2.
J. Biol. Chem. 277(43), 40302–40308 (2002).
101. Tabner BJ, Turnbull S, El-Agnaf OMA, Allsop D. Formation of hydrogen peroxide and hydroxyl radicals from Ab and a-synuclein as a possible mechanism of cell death in Alzheimer’s disease and Parkinson’s disease. Free Rad. Biol. Med. 32(11), 1076–1083 (2002).
102. Nelson TJ. Oxidation of cholesterol by amyloid precursor protein and b-amyloid peptide. J. Biol. Chem. 280(8), 7377–7387 (2005).
103. Granzotto A, Zatta P. Resveratrol acts not through anti-aggregative pathways but mainly via its scavenging properties against Ab and Ab–metal complexes toxicity. PloS ONE 6(6), E21565 (2011).
104. Camandola S, Mattson MP. NF-kB as a therapeutic target in neurodegenerative diseases. Expert Opin. Therap. Targets 11(2), 123–132 (2007).
105. Heuchel R, Radtke F, Georgiev O et al. The transcription factor MTF-1 is essential for basal and heavy metal-induced metallothionein gene expression. EMBO J. 13(12), 2870–2875 (1994).
106. Drago D, Bolognin S, Zatta P. Role of metal ions in the Ab oligomerization in Alzheimer’s disease and in other neurological disorders. Curr. Alzheimer Res. 5(6), 500–507 (2008).
107. Sensi SL, Paoletti P, Koh JY, Aizenman E, Bush AI, Hershfinkel M. The neurophysiology and pathology of brain zinc. J. Neurosci. 31(45), 16076–16085 (2011).
108. Adlard PA, Parncutt JM, Finkelstein DI, Bush AI. Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 30(5), 1631–1636 (2010).
109. Corona C, Masciopinto F, Silvestri E et al. Dietary zinc supplementation of 3xTg-AD mice increases BDNF levels and prevents cognitive deficits as well as mitochondrial dysfunction. Cell Death Dis. 1, E91 (2010).
110. Uranga RM, Giusto NM, Salvador GA. Effect of transition metals in synaptic damage induced by amyloid b peptide. Neuroscience 170(2), 381–389 (2010).
Gene profile of cells exposed to Ab–Zn & Ab–Cu Research Article


















![^_`]ab - DPU – Direitos Humanos](https://static.fdokumen.com/doc/165x107/6334d004d2b728420307a0c7/ab-dpu-direitos-humanos.jpg)

