
MURPESS — MUlti radio pedestrian energy scavenging sensor network

Chemico-Biological Interactions 193 (2011) 129–140
Contents lists available at ScienceDirect
Chemico-Biological Interactions
journal homepage: www.elsevier .com/locate /chembioint
Mangiferin attenuates methylmercury induced cytotoxicity against IMR-32,human neuroblastoma cells by the inhibition of oxidative stress and freeradical scavenging potential
Shubhankar Das, B. Nageshwar Rao, B.S. Satish Rao ⇑Division of Radiobiology and Toxicology, Manipal Life Sciences Centre, Manipal University, Manipal 576 104, Karnataka, India
a r t i c l e i n f o
Article history:Received 23 November 2010Received in revised form 4 June 2011Accepted 6 June 2011Available online 14 June 2011
Keywords:ApoptosisGenotoxicityMangiferinMethylmercuryNeurotoxicity
0009-2797/$ - see front matter � 2011 Elsevier Irelandoi:10.1016/j.cbi.2011.06.002
⇑ Corresponding author. Tel.: +91 820 2922815; faxE-mail addresses: [email protected], satishra
Rao).
a b s t r a c t
Mangiferin (MGN), a C-glucosylxanthone was investigated for its ability to protect against methylmer-cury (MeHg) induced neurotoxicity by employing IMR-32 (human neuroblastoma) cell line. MTT[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] and clonogenic cell survival assays con-firmed the efficacy of MGN supplementation in attenuating MeHg-induced cytotoxicity. Pre-treatmentwith MGN significantly (p < 0.01) inhibited MeHg-induced DNA damage (micronuclei, olive tail momentand % tail DNA) thereby demonstrating MGN’s antigenotoxic potential. Also, pre-treatment with MGNsignificantly reduced MeHg-induced oxidative stress, intra-cellular Ca2+ influx and inhibited depolariza-tion of mitochondrial membrane. MGN pre-treated cells demonstrated a significant (p < 0.05) increase inthe GSH and GST levels followed by a significant (p < 0.05) decrease in malondialdehyde (MDA) forma-tion. In addition, inhibition of MeHg induced apoptotic cell death by MGN was demonstrated by micro-scopic, Annexin-V FITC and DNA fragmentation assays and further confirmed by western blot analysis.The present findings indicated the protective effect of MGN against MeHg induced toxicity, which maybe attributed to its anti-genotoxic, anti-apoptotic and anti-lipid peroxidative potential plausibly becauseof its free radical scavenging ability, which reduced the oxidative stress and in turn facilitated the down-regulation of mitochondrial apoptotic signalling pathways.
� 2011 Elsevier Ireland Ltd. All rights reserved.
1. Introduction
Heavy metals, their inclusion into the environment invariablydepends on various anthropogenic practices that have been associ-ated with adverse health effects and several clinical manifestationsin human beings. Although the toxicities of mercury, lead,cadmium and arsenic have been very well documented [1,2], theirprecise treatment remains elusive. Both mercury and MeHg behaveas potent neurotoxicant and are associated with numerous neuro-logical disorders, such as paresthesia, ataxia, sensory and speechimpairment, and constriction of the visual field [3].
The menacing consequence of MeHg can be personified by thecalamitous outbreaks in Japan, Iraq, Pakistan and Guatemala[4,5]. Major sources of MeHg exposure were by the consumptionof fish from polluted areas, dental amalgams or occupational expo-sures [6–8], and the bioaccumulative nature of MeHg makes it apotent environmental pollutant and has been implicated in neu-ro-development disorders. Prospective studies carried out in FaroeIslands [9], and Seychelles Islands [10] have demonstrated the
d Ltd. All rights reserved.
: +91 820 [email protected] (B.S. Satish
occurrence of neuro-development disorders due to damagingeffects of associated MeHg levels. Though, the precise treatmentmodality for shielding the nervous tissue from being susceptibletoward MeHg assault remains obscure, the human body possessessome internal defence machinery in order to protect by itself. As itis regarded that in case of metal-induced toxicity one of the keyroute towards cellular damage is played by the oxidative stress[1], antioxidants may play a vital role in averting MeHg poisoning.Experimental evidences have shown that plant polyphenols pos-sess diverse health promoting effects [11]. They principally helpby preventing or reducing free radical production, iron chelationand exerting anti-inflammatory action, which has been character-ised in both in vitro and in vivo models [12,13].
Xanthones, in recent times have gained great importance by vir-tue of their numerous pharmacological and biological properties[14]. Mangiferin (MGN, 1,3,6,7-tetrahydroxyxanthone-C2-b-D-glucoside) is a C-glucosylxanthone obtained from the stem bark/leaves of Mangifera indica and has been shown to possess a wide-range of health promoting properties, such as antiviral, antitumor,immunostimulating [15], spasmolytic and antidiabetic [16]. MGNbears a catechol moiety, a pharmacophore with well-establishedanti-oxidant property [16] and also scavenges highly reactive spe-cies such as peroxynitrite and the hydroxyl radical. Besides, MGN is

130 S. Das et al. / Chemico-Biological Interactions 193 (2011) 129–140
reported to have protective efficacy against heavy metal inducedtoxicity in vitro and in vivo model systems [17–19] as well as incase of excitotoxic insults leading to neuronal damage [20]. The po-tential of MGN in crossing the blood-brain-barrier has been estab-lished in an earlier study while demonstrating the ability of MGNagainst ischemia induced oxidative stress and neuronal cell dam-age in an animal model [21].
Therefore, the present study was conducted to evaluate the po-tential role of MGN against MeHg-induced neurotoxicity and toelucidate the underlying mechanism in IMR-32, human neuroblas-toma cell line.
2. Materials and methods
2.1. Chemicals
Methylmercury chloride (MeHgCl, CAS No. 115-09-3), mangif-erin (MGN, CAS No. 4773-96-0), minimum essential medium(MEM), Trypsin, MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide], cytochalasin-B, propidium iodide (PI), ethi-dium bromide (EtBr), RNAse, proteinase K, low and normalmelting agarose; 20,70-dicholorofluorescein diacetate (DCFH2-DA),N-acetyl cysteine (NAC), Triton X-100, Tween-20, Rhodamine123, and fetal bovine serum (FBS) were purchased from SigmaChemical Co. (St. Louis, MO, USA). Acridine orange (AO) was pur-chased from BDH Chemicals Ltd., Poole, England. Fluo-3 was ob-tained from Molecular probes, Invitrogen, (Eugene, OR, USA).Anti-Bcl2, anti-Bax, anti-caspase-3, anti-cytochrome c, anti-Nrf-2and anti-b actin antibodies obtained from Millipore (Temecula,CA, USA). Metallothionein (MT) was purchased from Santa CruzBiotechnology, Inc. (Santa Cruz, CA, USA). Other chemicals men-tioned were procured from Qualigens Fine Chemicals (a divisionof Glaxo SmithKline Pharmaceuticals), Mumbai, India.
2.2. Cell culture conditions
IMR-32 cells originally derived from human neuroblastomawere procured from National Centre for Cell Sciences, Pune, India.The cells were grown in T-25 flasks (Falcon, Becton Dickinson, USA)with loosened caps, containing MEM supplemented with 10% FBS,1% L-glutamine and 50 lg/ml gentamycin sulphate, at 37 �C in anincubator (NuAire, Plymouth, MN, USA) with an atmosphere ofhumidified 5% CO2 in 95% air. For all the assays cells were main-tained at 80–90% confluency in T- 25 cm2 flasks and used as perrequirement.
2.3. Preparation of MeHgCl/MGN solutions
MeHgCl was dissolved in double distilled water at a concentra-tion of 1 mM and further diluted with MEM as per the require-ment. MGN was dissolved in 0.02% DMSO at a concentration of1 mM and diluted with MEM immediately before use.
2.4. Analysis of cell viability by MTT assay
Cell viability after exposure to MeHg or MGN alone or in com-bination was determined by MTT assay as described by Mosmann[22]. The exponentially growing IMR-32 cells were trypsinised andapproximately 104cells/well were plated into 96-well plates andkept in 5% CO2 incubator at 37 �C. Cell cultures were divided intofollowing:
Group-I (MGN alone) – This group of cultures were treated withvarious concentrations of MGN (2.5–100 lM) for 24 and 48 h.Group-II (MeHg alone) – The cell cultures of this group were trea-ted with various concentrations of MeHg (1–50 lM) for 1, 3, 24, 48
and 72 h, respectively. Group-III (MGN + MeHg) – Cell cultures ofthis group were treated with MGN (2.5–100 lM) for 2 h followedby treatment with MeHg 10 lM for 3 h and the cells were allowedto grow for 24 h. The assay was performed with minor modifica-tions as described in the previous paper [17].
2.5. Clonogenic assay
Clonogenic assay was performed according to Puck and Marcus[23]. The experiment was carried out by seeding appropriate num-ber of cells in 6 cm2 petri-dishes in batches. The optimum concen-tration of 10 lM MGN was selected from previous experiment, cellcultures were treated as Group-I (MeHg alone) – the cultures of thisgroup were treated with various concentrations (1–10 lM) ofMeHg for 3 h. Group-II (MGN + MeHg) – the cultures of this groupwere treated with 10 lM of MGN for two h before treating withvarious concentrations (1–10 lM) of MeHg for 3 h.
After the various treatments, MeHg or MGN containing mediawere discarded and fresh MEM media was added and the cultureswere left undisturbed for 14 days at 37 �C in 5% CO2 incubator forcolony formation and the colonies were stained, counted to calcu-late the surviving fraction as describe earlier [17].
2.6. Experimental design for assessing DNA damage
A fixed number (5 � 105) of exponentially growing cells wereinoculated into several individual culture flasks, allowed to growfurther 24 h and divided into following groups in triplicates:
Group I (Control): This group of cultures were not treated withMGN/MeHg.Group II (MeHg alone): This group of cultures were treated withdifferent concentrations of MeHg (0.125–2.5 lM) alone for 3 h.Group III: The cultures of this group were treated with MGN (1–25 lM) for 2 h followed by 2.5 lM of MeHg for 3 h.Group IV: This group of cultures were treated with 10 lM MGNfor 2 h followed by MeHg (0.125–2.5 lM) for 3 h.
The cells from the above groups were dislodged by mild trypsinEDTA treatment. Micronucleus (MN) and Comet assay were carriedout from the same stock of cells.
2.6.1. Micronucleus assayThe assay was performed according to the original method of
Fenech and Morley [24] with minor modifications suggested byRao and co-workers [25]. One thousand bi-nucleated cells withwell-preserved cytoplasm were scored from each culture and thefrequency of micronucleated binucleate cells (MNBNC) was deter-mined. The MN identification was done according to the criteria ofFenech and co-workers [26]. The cytokinesis blocked proliferationindex (CBPI) was then calculated according to the method of Sur-ralles and co-workers [27].
2.6.2. Single cell gel electrophoresis (COMET assay)The assay was performed under alkaline conditions according to
the procedure of Singh and co-workers [28] with minor modifica-tions as described in earlier paper [17].
2.7. Cell death by apoptosis
For all the apoptotic assays exponentially growing cells wereseeded at the density of 106 per 6-cm2 plate and divided into fol-lowing groups: Group-I (MeHg alone): the cultures of this groupwere treated with different concentrations of MeHg (1–10 lM)for 24 h. Group-II (MGN + MeHg): the cultures of this group were

S. Das et al. / Chemico-Biological Interactions 193 (2011) 129–140 131
treated with different concentrations (1–25 lM) of MGN 2 h beforetreatment with different concentrations of MeHg (1–10 lM) for24 h.
After the various treatments, cells from the above groups weredislodged by mild trypsin EDTA treatment. Microscopic analysis,Annexin-V FITC and DNA ladder assay were carried out from thesame stock of cells as follows:
2.7.1. Microscopic analysis of apoptotic cellsCyto-morphological evidence for apoptosis/necrosis was differ-
entiated by EtBr/AO staining as described Renvoize and co-workers[29].
2.7.2. Measurement of apoptosis by Annexin V stainingThe externalisation of phosphatidylserine is a reliable early
apoptotic marker which was used to detect apoptotic IMR-32 cellsusing Annexin V-FITC Kit (Sigma–Aldrich, St. Louis, MO, USA)according to the supplier’s instructions which clearly differentiateviable, apoptotic and necrotic cells.
2.7.3. Detection of DNA fragmentation by agarose gel electrophoresisThe extent of DNA fragmentation upon treatment of exponen-
tially growing cells with MeHg alone or in combination withMGN was qualitatively visualized on agarose gel as described byGiri and co-workers [30].
2.8. Experimental design for ROS, intracellular Ca2+ levels,mitochondrial membrane potential (DWm), biochemical estimationsand Western blotting
Exponentially growing cells were seeded at the density of 106
per 6 cm2 plate and treated with different concentrations ofMeHg/MGN after 24 h. Cells were divided into following groups:
Group I (control): Cells were not treated with MeHg or MGN.Group II (MGN alone): Cells were treated with 10 lM MGNalone for 2 h.Group III (MeHg alone): Cells were treated with different con-centrations of MeHg (10 lM) for 1 or 24 h.Group IV (MGN + MeHg): Cells were pre-treated with 10 lMMGN for 2 h and then treated with 10 lM MeHg for 1 or 24 h.
The cells from the above groups were dislodged by mild trypsinEDTA treatment. Following assays were carried out from the samestock of cells.
2.8.1. Evaluation of intracellular reactive oxygen species (ROS)Intracellular ROS levels were estimated using DCF-DA as de-
scribed by Bai and Cederbaum [31]. After various treatments as de-scribed above, MeHg containing medium was removed and freshmedium was added and the cultures were post-incubated for 60,90 and 120 min. After the various post incubation time intervals,the medium was removed and replaced with 5 lM DCFH-DA infor 30 min at 37 �C in 5% CO2 incubator. Cells were then harvestedby trypsinisation, washed with PBS and collected by centrifugationat 1000 rpm for 10 min. Final pellet obtained was re-suspended in3 ml of PBS and the fluorescence was read (excitation at 488 nm;emission at 525 nm) by using fluorescence spectrophotometer(RF-5301PC, Shimadzu). NAC (250 mM) was used as a positive con-trol to ensure validity of the assay.
2.8.2. Estimation of intra-cellular Ca2+ levelsThe assay was performed according to Rijkers and co-workers
[32] with minor modifications. For the above assay, IMR-32 cellswere treated with MGN/MeHg in Ca2+/Mg2+ free media. After the
various treatments, cells from the above groups were post-incu-bated for 60 min, at 37 �C in 5% CO2 incubator. Post-incubated cellswere washed twice with Ca2+/Mg2+ free HBSS and then loaded withFluo-3 (5 lM), a high affinity calcium-indicator and incubated at37 �C, 5% CO2 incubator for 30 min. Cells were finally trypsinisedand re-suspended in 1ml of PBS and were analysed on FACS Calibur(Becton Dickinson, USA) using CellQuest software (excitation at488 nm; emission at 525 nm).
2.8.3. Estimation of mitochondrial membrane potential (DWm)Determination of DWm was done according to the method of
Scaduto and Grotyohann [33]. After the various treatments, cellsfrom the above groups were post-incubated for 60 min, followedby addition of Rhodamine 123 (final concentration 5 lg/ml) andincubated at 37 �C for 20 min in 5% CO2. Cells were then harvestedby trypsinisation, washed twice in chilled PBS and collected bycentrifugation at 1000 rpm for 10 min. The pellet thus obtainedwas finally re-suspended in 1 ml of chilled PBS and the sampleswere acquired on FACS Calibur (Becton Dickinson, USA) using Cell-Quest software (excitation at 488 nm; emission at 525 nm).
2.8.4. Biochemical estimationsFor all the biochemical estimations exponentially growing IMR-
32 cells were seeded at a density of 106cells/ml in T-25 flasks. Cul-tures were divided into different groups as described above. Aftervarious treatments, cells were allowed to grow further by incuba-tion in fresh medium for 24 h. The cells were then lysed in a lysisbuffer appropriate for the requirements of each assay as describedbelow. Concentrations of total protein were measured by themethod of Bradford [34]. The cells were lysed at 4 �C for 2 h using5% w/v metaphosphoric acid (chilled) with mild agitation to ex-tract the cellular GSH by the method of Moron and co-workers[35]. Glutathione-S-transferase (GST) activity was determinedaccording to the procedure of Habig and co-workers [36]. Lipidperoxidation (LPO) indicated by increased malondialdehyde(MDA) level was determined by thiobarbituric reactive substance(TBARS) assay as described by Buege and Aust [37]. LPO was ex-pressed as MDA per milligram of total protein.
2.8.5. Analysis of proteins by Western blottingFor Western blotting exponentially growing IMR-32 cells were
seeded at a density of 106cells/ml in T-25 flasks. Cultures were di-vided as described above. After treatment durations, cells weretrypsinised, washed with PBS and centrifuged at 1000 rpm for10 min to obtain the pellet. Pellets were re-suspended in lysis buf-fer (150 mM sodium chloride, 1.0% Triton X-100, 0.5% sodiumdeoxycholate, 0.1% SDS (sodium dodecyl sulphate), 50 mM Tris,pH 8.0, 1 mM sodium fluoride, 1 mM sodium orthovandate,1:250 protease inhibitor cocktail) and incubated at 4 �C for60 min with mild agitation. Supernatant was collected by centrifu-gation at 12000 rpm for 15 min at 4 �C. Protein content was esti-mated by Bradford assay [34] and the samples were stored at�20 �C and used whenever required. The total proteins (20 lg)were resolved on 12–15% SDS–polyacrylamide gel electrophoresis,transferred onto the nitrocellulose membrane and then immuno-blotted with the corresponding antibodies such as anti-Bcl2, anti-Bax, anti-caspase-3, anti-cytochrome c, anti-Nrf-2, anti-MT andanti-b actin antibodies. The immobilised proteins were incubatedwith goat anti-rabbit IgG (Millipore). Protein expression levelswere detected using enhanced chemiluminescence detection re-agents (Thermo scientific, Rockford, IL, USA) according to the man-ufacturer’s instructions.

132 S. Das et al. / Chemico-Biological Interactions 193 (2011) 129–140
2.9. Isolation of tubulin proteins from goat brain and tubulin assemblyassay
2.9.1. Isolation of tubulin proteinsIsolation of tubulin protein from goat brain was done by succes-
sive cycles of assembly according to the method described by Bori-sy and co-workers [38] with some minor modifications. Briefly,goat brains were procured within 1 h of slaughter and rinsed inice-cold saline in order to remove blood stains. The meningesand superficial blood vessels were carefully removed and the cor-tex was dissected out, homogenised in 1.33 volumes of chilledhomogenisation buffer (0.1 M NaPIPES, 1 mM EGTA, 1 mMMgCl2�6H2O, 0.1 mM GTP; pH 6.9) using a motor driven Potter-El-vehjem tissue grinder. The homogenate was transferred into 45 mlpolycarbonate tubes and centrifuged in Sorvall SS-34 rotor (cooledto 4 �C) at 12,500 rpm for 90 min at 4 �C. Supernatant was col-lected, 1 mM solid GTP was added to it, mixed properly and themixture was incubated in water-bath for 45 min maintained at37 �C with gentle shaking. An increase in turbidity and viscosityis noticed because of polymerisation of tubulin into microtubulesin presence of GTP. This mixture was then centrifuged at18,000 rpm for 45 min at 37 �C (rotor pre-warmed to 37 �C). Thegelatinous pellet obtained was re-suspended in homogenizing buf-fer containing 1 mM GTP (1/8th volume of supernatant obtainedafter first centrifugation step) and incubated on ice for 30 min inorder to depolymerise the microtubules and again centrifuged at18,000 rpm for 40 �C. This constituted one cycle of polymerisa-tion-depolymerisation process. Two more such repeated cycleswere carried out and the protein pellet was collected and storedat �80 �C for further usage. Tubulin protein was checked on 8%SDS-PAGE and quantified by Bradford’s method [34].
Fig. 1. Changes in cell viability of IMR-32 cells treated with various concentrations of MGfor 1 and 3 h; (B) MeHg (1–50 lM) for 12, 24, 48 and 72 h; (C) MGN (2.5–100 lM) for 24 aAll the results are shown as Mean ± SD from the data of a minimum of three separate e
2.9.2. In vitro tubulin assembly assayTubulin assembly assay was performed on 96 well plates as de-
scribed by Fila and co-workers [39]. In brief, the isolated tubulinproteins were diluted to 2.2 mg/ml in 80 mM Na-PIPES, pH 6.8,0.5 mM EGTA, 2 mM MgCl2.6H2O and 5% glycerol. Tubulin poly-merization reaction mixture (100 ll) containing diluted tubulinproteins with 10 lM MeHg, 10 lM MGN were prepared in a 96well plate, and polymerization was started by the addition of1 mM GTP at 37 �C and followed by A340 readings for up to30 min using multi-well spectrophotometer (TECAN, Austria). Col-chicine (10 lM) and taxol (10 lM) served as internal controls forthe experiment.
2.10. Statistical analysis
The significance of the differences between treatments andrespective controls was analysed using the Student’s t-test andone-way analysis of variance and with Bonferroni’s post hoc testusing GraphPAD InStat (La Jolla, CA, USA). All the data were ex-pressed as mean ± SEM or SD.
3. Results
3.1. Cytotoxicity studies using MTT assay
IMR-32 cells treated with different concentrations of MeHg (1–50 lM) showed a concentration as well as time dependant de-crease in cell survival. The inhibitory concentration (IC50) valuesfor MeHg alone were found to be 14.3, 10.1, 7.6, 4.5, 1.9 and1.6 lM for 1, 3, 12, 24, 48 and 72 h respectively (Fig. 1A and B),although the IC50 values for 48 and 72 h did not show a significant
N/MeHg either alone or in combination using MTT assay. (A) MeHg (1–50 lM) alonend 48 h; (D) MGN (2.5–100 lM) for 2 h before treatment with MeHg (10 lM) for 3 h.xperiments (⁄p < 0.05 when compared to MeHg alone group).

Fig. 2. Effect of MGN (10 lM) for 2 h on the cell survival of IMR-32 cells treatedwith different concentrations of MeHg (1–10 lM) for 3 h assessed by clonogenicassay.
Fig. 3. (A) Induction of micronuclei in IMR-32 cells treated with differentconcentrations of MeHg (0.125–2.5 lM) for 3 h. (B) Dose dependant decrease inmicronuclei formation (Y1-axis) and increase in cytokinesis block proliferation
S. Das et al. / Chemico-Biological Interactions 193 (2011) 129–140 133
difference. The various concentrations of MGN (2.5–100 lM) didnot have a significant impact on the cell viability when cells weretreated with it for 24 and 48 h (Fig. 1C). When the cells were trea-ted with increasing concentrations of MGN (2.5–100 lM) for 2 hbefore exposure to IC50 concentration of MeHg (3 h), a gradual in-crease in cell survival was observed. Maximum elevation in cellsurvival was significantly highest (p < 0.01) in 10 lM MGN pre-treated group when compared with other groups (Fig. 1D).
index (CBPI) values (Y2 axis) in IMR-32 cells pre-treated with different doses ofMGN (1–25 lM) for 2 h and then treated with MeHg (2.5 lM) for 3 h. All the resultsare shown as Mean ± SEM from the data of a minimum of three separateexperiments. The significant levels ⁄p < 0.05 and ⁄⁄p < 0.01 when compared withMeHg alone group.
3.2. Clonogenic assay
Clonogenic assay was used to assess the reproductive death ofcells after treatment with a cytotoxic agent and thus it was usedto study the effects of MGN on MeHg induced toxicity. The cyto-protective action of MGN was assessed by pre-treatment withthe optimal dose of MGN at 10 lM for 2 h before exposure to dif-ferent concentrations of MeHg (0.5–10 lM) resulted in increasein cell survival when compared to the MeHg alone treatment witha DRF of 1.37 (Fig. 2).
3.3. Micronucleus assay
The protection against MeHg induced genotoxicity by MGN wasassessed by micronucleus assay. Exposure of IMR-32 cells to vari-ous concentrations of MeHg resulted in a concentration dependentincrease in the frequency of MNBNC (Fig. 3A). Treatment of cellswith 2.5 lM (3 h) of MeHg resulted in a significant (p < 0.001) ele-vation in the MNBNC and this increase in MNBNC was 8.4-foldhigher than that of control MNBNC frequency.
Treatment of the cells to different concentrations of MGN (1–25 lM) for 2 h before treatment with 2.5 lM MeHg (3 h) caused
Table 1Effect of MGN (10 lM) on the micronuclei induction in IMR-32 cells exposed to various c
MeHg (lM) Frequency of micronucleated binucleated cells (MNBNC)/1000 cells
One Two
DDW + MeHg MGN DDW + MeHg MGN
0 38.1 ± 1.21 36.75 ± 1.47 5.9 ± 0.56 2.25 ± 00.125 72.15 ± 1.32 38.12 ± 0.91c 5.85 ± 1.08 4.88 ± 00.25 75.75 ± 1.08 53.49 ± 1.24c 17.5 ± 1.47 7.5 ± 10.5 108.51 ± 1.21 67.75 ± 3.54c 41.5 ± 0.56 12.75 ± 11 115.0 ± 1.47 71.23 ± 1.68c 43.19 ± 1.21 15.02 ± 12.5 142.52 ± 2.23 76.28 ± 1.57c 49.23 ± 2.21 17.47 ± 1
The significant levels a = p < 0.05, b = p < 0.01, c = p < 0.001 and no symbol = non-signific
a decline in the MeHg induced micronuclei formation and a maxi-mum decrease (4.3-fold) in MNBNC frequency was observed for10 lM MGN (Fig. 3B), and this concentration was considered asthe optimum protective concentration as the frequency of micro-nuclei was minimum at this concentration when compared toother concentrations of MGN, therefore, further studies were car-ried out using this MGN concentration.
CBPI was significantly reduced in the MeHg group, whencompared to untreated control group indicating the MeHg-induceddelay in the cell proliferation. Pre-treatment with different concen-trations of MGN normalised CBPI when compared to MeHg 10 lMalone treatment (Fig. 3B). The IMR-32 cells exposed to differentconcentrations of MeHg induced a dose-dependent increase in theMN frequency. Pre-treatment of cells with 10 lM MGN for 2 h be-fore exposure to different concentrations of MeHg resulted in a sig-nificant decline in the frequency of MNBNC when compared withthe concurrent MeHg alone group (Table 1). The frequency of binu-cleate cells bearing one, two and multiple micronuclei increased in
oncentrations of MeHg.
± SEM
Multiple (P3) Total
DDW + MeHg MGN DDW + MeHg MGN
.21 – – 44 ± 2.2 39 ± 1.95
.64b – – 78 ± 3.9 43 ± 2.15b
.04c 8.75 ± 1.21 2.01 ± 0.40b 102 ± 3.4 67 ± 3.14b
.49c 11.24 ± 0.64 3.0 ± 1.21b 161.2 ± 2.8 83.5 ± 3.8c
.84c 15.81 ± 1.29 7.5 ± 0.95b 174 ± 2.54 93.7 ± 2.4c
.89c 20.21 ± 2.56 10.2 ± 1.26a 211.9 ± 2.89 103.9 ± 2.4c
ant, when MGN + MeHg group are compared to corresponding MeHg alone groups.

Fig. 4. (A) Genotoxic effect of various concentrations of MeHg (0.125–2.5 lM) for3 h treatment on IMR-32 cells as assessed by DNA damage using comet assay. (B)Effect of different concentration of MGN (1–25 lM) for 2 h on MeHg (2.5 lM) for3 h induced DNA damage in IMR-32 cells. (C) Protective effect of optimalconcentration of MGN (10 lM) against various concentrations of MeHg (0.125–2.5 lM) for 3 h induced DNA damage. The significant levels compared to therespective MeHg group and the other symbols are as in Fig 3.
134 S. Das et al. / Chemico-Biological Interactions 193 (2011) 129–140
a concentration dependent manner and the highest number ofmicronuclei was observed for 2.5 lM of MeHg. MGN pre-treatmentresulted in a significant decline in the induction of cells with one,two and multiple micronuclei. The frequencies of two and multipleMNBNC were always lower in MGN + MeHg group when comparedwith concurrent MeHg alone group (Table 1).
3.4. Comet assay
Alkaline single cell gel electrophoresis (comet assay) was used todetect the cellular DNA damage induced by MeHg. Exposure of IMR-32 cells to MeHg (0.125–2.5 lM) resulted in an increase in the per-centage of tail DNA (% TDNA) and olive tail moment (OTM) in a con-centration dependent manner indicating DNA damage (Fig. 4A).Pre-treatment of IMR-32 cells with different concentrations (1–25 lM) of MGN before exposure to 10 lM MeHg caused declinein the MeHg induced percentage of tail DNA and OTM and a maxi-mum decrease was observed for 10 lM MGN (Fig. 4B), and was con-sidered as the optimum protective concentration.
A separate experiment was conducted to study the effect of10 lM MGN on the DNA damage induced by increasing doses ofMeHg (0.125–2.5 lM). Treatment of IMR-32 cells with differentconcentrations of MeHg alone caused a concentration dependentincrease in the DNA damage. Pre-treatment with 10 lM MGNshowed significant (p < 0.001) decrease in the DNA damage whencompared with respective MeHg alone treated group (Fig. 4C).
3.5. Microscopic analysis of Apoptotic cells
To assess the degree of protection by MGN against MeHg in-duced apoptosis and necrosis, microscopic AO/EtBr dual stainingwas used. Cells were differentiated into live, early apoptotic, lateapoptotic and necrotic from the uptake of AO/EtBr stain. Live cellswere seen as bright green coloured nuclei with intact and uniformcell membrane. Early apoptotic cells have green nuclei, but perinu-clear chromatin condensation was visible as bright green patchesor fragments; late apoptotic cells have orange to red nuclei withcondensed or fragmented chromatin; necrotic cells have uniformorange to red nuclei.
A dose dependant increase in apoptosis and necrosis was ob-served in cells treated with MeHg (1–10 lM) alone for 24 h, necro-sis percent being maximum in case of 10 lM of MeHg (Fig. 5A).Treatment of cells with optimum dose of MGN (10 lM) for 2 hprior to different concentrations of MeHg (1–10 lM) for 24 hshowed a dose dependant decrease in percent apoptosis and necro-sis when compared with respective MeHg groups (Fig. 5A).
3.6. FITC–Annexin V staining
The type of cell death induced by MeHg, cells treated with orwithout MGN/MeHg was assessed with flow cytometry by a doublestaining with Annexin V and PI. The Annexin V-FITC-positive pop-ulation of the cells (apoptotic cells; lower right quadrant) was notsignificantly increased in the control and MGN-alone-treated cells[Fig. 5B (a and b)]. However, the apoptotic cell population was in-creased to 24.72%, and also the Annexin V-FITC-positive/PI-positivepopulation (necrosis and late apoptotic cells; upper right quadrant)was increased to 9.37% in the MeHg-alone group [Fig. 5B (c)]. Asshown in Fig. 5B (d), treatment with optimal concentrations ofMGN (10 lM) prior to MeHg reduced both apoptotic and necroticcells.
3.7. Detection of DNA fragmentation by Ladder assay
To confirm further, the effect of MGN on MeHg induced apopto-sis, DNA was isolated from the treated and untreated IMR-32 cells
and electrophoresed on 1.5% agarose gel (Fig. 5C). Nucleosomalladder formation in cells pre-treated with MGN (1, 2.5, 5, 10 and25 lM) showed decrease in the intensity of the ladder indicatingthe inhibition of apoptosis when compared to the MeHg (10 lM)alone. Further, it was observed that the 10 lM of MGN pre-treat-ment significantly inhibited the MeHg induced apoptosis as indi-

Fig. 5. (A) Protective effect of MGN (10 lM) for 2 h on various concentrations of MeHg (1–10 lM) for 24 h induced apoptotic and necrotic damage in IMR-32 cells assessed byacridine orange/ethidium bromide staining. (B) Effect of MGN (10 lM) for 2 h on MeHg (10 lM) for 24 h induced apoptosis assessed by Annexin V-FITC flow cytometrymethod. Early apoptotic cells, which are the Annexin V-FITC-positive/PI-negative population of cells, are reported in the lower right quadrant. Necrotic and late apoptoticcells, which are the Annexin V-FITC-positive/PI-positive population of cells, are reported in the upper right quadrant. (a) Control cells; (b) MGN (10 lM) alone; (c) MeHg(10 lM) alone; (d) MGN (10 lM) + MeHg (10 lM). (C) Effect of MGN (2 h) on MeHg (24 h) induced DNA fragmentation assessed by gel electrophoresis. Lane 1 and 7 – 3kbmarker; Lane 2 – Control; Lane 3 – MeHg (1 lM); Lane 4 – MeHg (2.5 lM); Lane 5 – MeHg (5 lM); Lane 6 and 8 – MeHg (10 lM); Lane 9 – MGN (1 lM) + MeHg (10 lM); Lane10 – MGN (2.5 lM) + MeHg (10 lM); Lane 11 – MGN (5 lM) + MeHg (10 lM); Lane 12 – MGN (10 lM) + MeHg (10 lM); Lane 13 – MGN (25 lM) + MeHg (10 lM); Lane 14 –MGN (10 lM) alone; Lane 15 – MGN (10 lM) + MeHg (1 lM); Lane 16 – MGN (10 lM) + MeHg (2.5 lM); Lane 17 – MGN (10 lM) + MeHg (5 lM); Lane 18 – MGN(10 lM) + MeHg (10 lM). The significant levels compared to the respective MeHg group and the other symbols are as in Fig 3.
S. Das et al. / Chemico-Biological Interactions 193 (2011) 129–140 135
cated by the lesser intensity when compared with other pre-trea-ted groups (Fig. 5C, Lane 12).
A separate experiment was conducted to study the influence ofoptimal dose of MGN (10 lM) on the DNA fragmentation inducedby different concentrations of MeHg (1–10 lM). Treatment ofIMR-32 cells with different concentrations of MeHg showed in-crease in intensity of ladder formation with the increase in MeHgconcentrations (Fig. 5C, Lane 3–6). Treatment of IMR-32 cells withMGN (10 lM) before treating with various concentrations of MeHgshowed a dose-dependent reduction in the intensity of ladderwhen compared with respective MeHg alone groups (Fig. 5C, Lane15–18).
3.8. Estimation of ROS generation
Estimation of intra-cellular ROS was done using DCFH-DA. Thecells treated with various concentrations of MeHg (2.5–10 lM)showed dose dependent increase in DCF fluorescence levels (indic-ative of intracellular ROS levels) (Fig. 6A). However, addition ofMGN inhibited the increase in DCHF oxidation as measured at60, 90 and 120 min of post-treatment when compared to MeHgalone levels (Fig. 6B). A known reactive oxygen intermediate scav-enger, N-acetyl cysteine (250 mM) was used as a positive control,shown to be capable of resulting in significantly (p < 0.05) time-
dependent decrease in ROS generation in the IMR-32 cells whencompared with other treatment groups.
3.9. Determination of intracellular Ca2+ levels and mitochondrialmembrane potential (DWm)
Fluo-3, a fluorescein chromophore-derived Ca2+ probe was usedto estimate the intra-cellular Ca2+ levels. Once bound to Ca2+, Fluo-3 exhibits a proportional increase in the emitted spectrum whichin turn suggests whether there is an increase or decrease in intra-cellular Ca2+ levels. Flow-cytometric analysis of IMR-32 cells trea-ted with MeHg (10 lM) alone showed a marked increase in intra-cellular Ca2+ levels as compared to control (untreated) and MGN(10 lM) alone treated groups. Cells pre-treated with MGN(10 lM) for 2 h prior to MeHg exposure showed a significant de-crease in Ca2+ concentrations as represented in histogram(Fig. 6C). This clearly shows that the rise in cytoplasmic Ca2+ levelsduring MeHg exposure mainly occur due to release of Ca2+ fromintracellular reservoirs (endoplasmic reticulum; ER) as the role ofextracellular Ca2+ was omitted by performing the assay in Ca2+ freemedia. From the above findings, it is obvious that this rise in intra-cellular Ca2+ levels facilitate in the process of depletion of DWm.
Rhodamine 123 is a lipophilic cationic dye which enters themitochondria and gets retained within the mitochondria due to
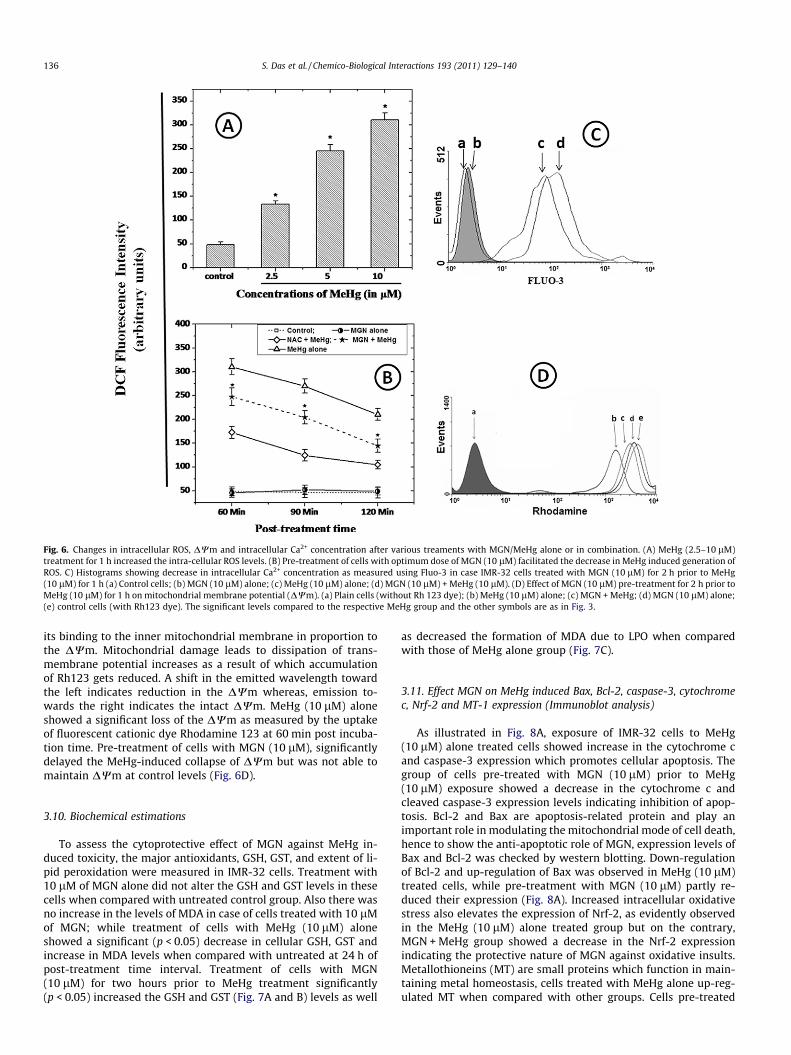
Fig. 6. Changes in intracellular ROS, DWm and intracellular Ca2+ concentration after various treaments with MGN/MeHg alone or in combination. (A) MeHg (2.5–10 lM)treatment for 1 h increased the intra-cellular ROS levels. (B) Pre-treatment of cells with optimum dose of MGN (10 lM) facilitated the decrease in MeHg induced generation ofROS. C) Histograms showing decrease in intracellular Ca2+ concentration as measured using Fluo-3 in case IMR-32 cells treated with MGN (10 lM) for 2 h prior to MeHg(10 lM) for 1 h (a) Control cells; (b) MGN (10 lM) alone; (c) MeHg (10 lM) alone; (d) MGN (10 lM) + MeHg (10 lM). (D) Effect of MGN (10 lM) pre-treatment for 2 h prior toMeHg (10 lM) for 1 h on mitochondrial membrane potential (DWm). (a) Plain cells (without Rh 123 dye); (b) MeHg (10 lM) alone; (c) MGN + MeHg; (d) MGN (10 lM) alone;(e) control cells (with Rh123 dye). The significant levels compared to the respective MeHg group and the other symbols are as in Fig. 3.
136 S. Das et al. / Chemico-Biological Interactions 193 (2011) 129–140
its binding to the inner mitochondrial membrane in proportion tothe DWm. Mitochondrial damage leads to dissipation of trans-membrane potential increases as a result of which accumulationof Rh123 gets reduced. A shift in the emitted wavelength towardthe left indicates reduction in the DWm whereas, emission to-wards the right indicates the intact DWm. MeHg (10 lM) aloneshowed a significant loss of the DWm as measured by the uptakeof fluorescent cationic dye Rhodamine 123 at 60 min post incuba-tion time. Pre-treatment of cells with MGN (10 lM), significantlydelayed the MeHg-induced collapse of DWm but was not able tomaintain DWm at control levels (Fig. 6D).
3.10. Biochemical estimations
To assess the cytoprotective effect of MGN against MeHg in-duced toxicity, the major antioxidants, GSH, GST, and extent of li-pid peroxidation were measured in IMR-32 cells. Treatment with10 lM of MGN alone did not alter the GSH and GST levels in thesecells when compared with untreated control group. Also there wasno increase in the levels of MDA in case of cells treated with 10 lMof MGN; while treatment of cells with MeHg (10 lM) aloneshowed a significant (p < 0.05) decrease in cellular GSH, GST andincrease in MDA levels when compared with untreated at 24 h ofpost-treatment time interval. Treatment of cells with MGN(10 lM) for two hours prior to MeHg treatment significantly(p < 0.05) increased the GSH and GST (Fig. 7A and B) levels as well
as decreased the formation of MDA due to LPO when comparedwith those of MeHg alone group (Fig. 7C).
3.11. Effect MGN on MeHg induced Bax, Bcl-2, caspase-3, cytochromec, Nrf-2 and MT-1 expression (Immunoblot analysis)
As illustrated in Fig. 8A, exposure of IMR-32 cells to MeHg(10 lM) alone treated cells showed increase in the cytochrome cand caspase-3 expression which promotes cellular apoptosis. Thegroup of cells pre-treated with MGN (10 lM) prior to MeHg(10 lM) exposure showed a decrease in the cytochrome c andcleaved caspase-3 expression levels indicating inhibition of apop-tosis. Bcl-2 and Bax are apoptosis-related protein and play animportant role in modulating the mitochondrial mode of cell death,hence to show the anti-apoptotic role of MGN, expression levels ofBax and Bcl-2 was checked by western blotting. Down-regulationof Bcl-2 and up-regulation of Bax was observed in MeHg (10 lM)treated cells, while pre-treatment with MGN (10 lM) partly re-duced their expression (Fig. 8A). Increased intracellular oxidativestress also elevates the expression of Nrf-2, as evidently observedin the MeHg (10 lM) alone treated group but on the contrary,MGN + MeHg group showed a decrease in the Nrf-2 expressionindicating the protective nature of MGN against oxidative insults.Metallothioneins (MT) are small proteins which function in main-taining metal homeostasis, cells treated with MeHg alone up-reg-ulated MT when compared with other groups. Cells pre-treated
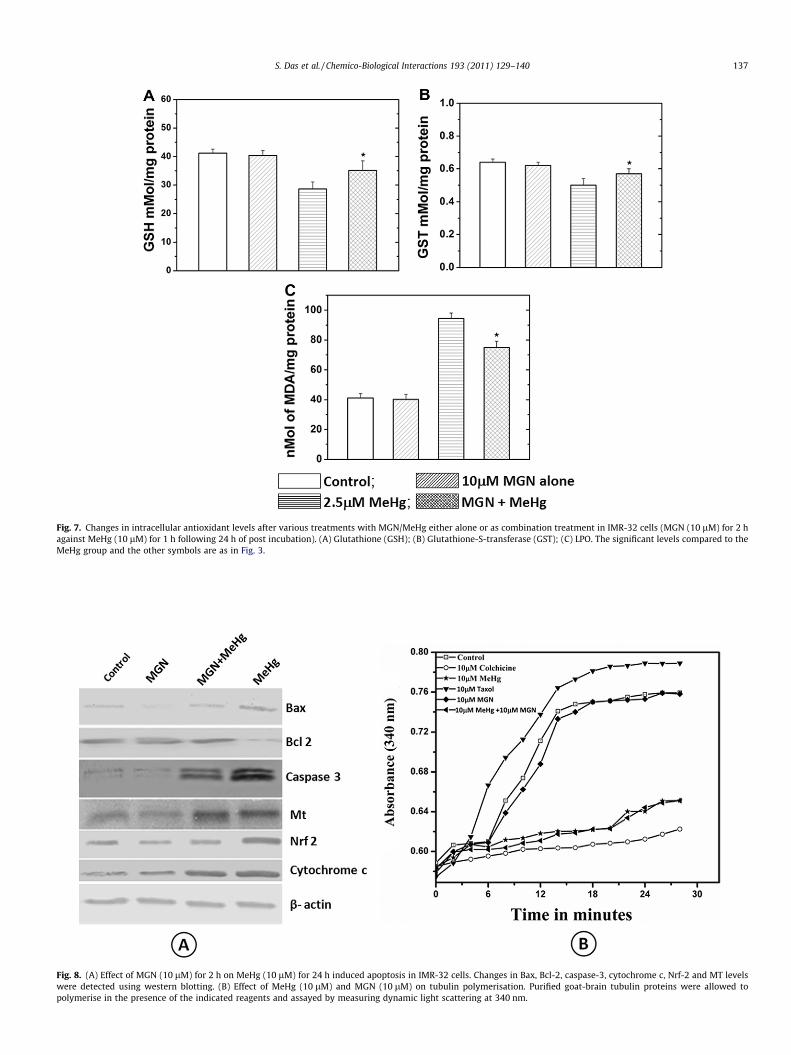
Fig. 7. Changes in intracellular antioxidant levels after various treatments with MGN/MeHg either alone or as combination treatment in IMR-32 cells (MGN (10 lM) for 2 hagainst MeHg (10 lM) for 1 h following 24 h of post incubation). (A) Glutathione (GSH); (B) Glutathione-S-transferase (GST); (C) LPO. The significant levels compared to theMeHg group and the other symbols are as in Fig. 3.
Fig. 8. (A) Effect of MGN (10 lM) for 2 h on MeHg (10 lM) for 24 h induced apoptosis in IMR-32 cells. Changes in Bax, Bcl-2, caspase-3, cytochrome c, Nrf-2 and MT levelswere detected using western blotting. (B) Effect of MeHg (10 lM) and MGN (10 lM) on tubulin polymerisation. Purified goat-brain tubulin proteins were allowed topolymerise in the presence of the indicated reagents and assayed by measuring dynamic light scattering at 340 nm.
S. Das et al. / Chemico-Biological Interactions 193 (2011) 129–140 137

138 S. Das et al. / Chemico-Biological Interactions 193 (2011) 129–140
with MGN (10 lM) did not significantly inhibit the activation ofMT (Fig. 8A).
3.12. Tubulin assay
MeHg has the ability to modify the free –SH groups of aminoacids present in the tubulin subunits rendering them inactive.For tubulin-polymerisation depolymerisation assay, optimum doseof MeHg (10 lM) as well as MGN (10 lM) was chosen from previ-ous experiments. MGN alone at a concentration of 10 lM did notaffect the polymerisation process and it was similar to control.Ten micromolar MeHg alone when compared to control had a sig-nificant effect on the polymerisation of tubulin subunits. Incombination treatment, it was ineffective in inhibiting the depoly-merisation of tubulin proteins. Colchicine (10 lM) and taxol(10 lM) served as internal controls for the experiment (Fig. 8B).
4. Discussion
A number of toxic heavy metals, such as mercury, lead, cad-mium etc., find various industrial applications owing to whichtheir entry into the environment has become inevitable. Earlierstudies have elucidated that MeHg (the organic form) acts as a po-tent neurotoxicant by breaching the blood-brain-barrier [40] andaffecting the central nervous system [3,7,8,41]. Although, severalchelating agents and antagonists, such as sodium 2,3-dimercaptopropane-1-sulphonate, 2,3-dimercapto succinic acid [42], mineralssuch as selenium and zinc and small thiol molecules have beenfound to be promising [43], their practical applicability is limiteddue to their inherent toxicity at effective doses. Due to the lowacceptability of these chemical protectors, the search has beenshifted towards natural products as nutraceuticals possess betterhealth promoting effects when compared to synthetic protectors.
MeHg intoxication therapy may include the identification andcharacterisation of novel therapeutic agents, especially from phy-to-chemicals. Thus the importance of natural antioxidants has in-creased during the recent years owing to their numerouspharmacological properties. It is well known that nutritional sup-plements from extracts of medicinal plants and herbs can modu-late physiological functions and thereby provide beneficial effectsagainst various human diseases [44,45]. Besides, plant-derivedcompounds have shown their protective role against heavy metaltoxicity [17,46]. As MGN has been well established for its variouspharmacological properties [16] current study was designed to testthe hypothesis that MGN, at micromolar concentrations, is capableof minimising the toxic effects of MeHg imposed on IMR-32 cells,considering these cells as a suitable model for studying heavy me-tal induced-neuronal abnormality.
As expected, MeHg inhibited the growth of IMR-32 cells in aconcentration and time dependent manner. However, MGN alonedid not decrease the cell viability even when IMR-32 cells were ex-posed to 100 lM for 48 h indicating its non-toxic nature. Interest-ingly, increase in cell viability was observed in cells pre-treatedwith MGN before treating with MeHg. These findings corroborateearlier studies demonstrating the protective potential of aminoacids and antioxidants such as NAC, cysteine, GSH against MeHg-induced cytotoxicty in various cell lines and primary cultures[43]. Exposure to toxicants alters the cellular proliferation and alsoaffects the reproductive potential; hence in order to assess the cel-lular toxicity of MeHg the clonogenic assay was performed. MeHgresulted in a dose-dependent decline in the surviving fraction ofIMR-32 cells, while pre-treatment with MGN afforded significantincrease in cell survival with a dose modification factor of 1.37, fur-ther supporting our MTT results.
Several lines of evidence indicate that oxidative stress and ROSformed in the presence of MeHg could be responsible for its toxiceffects in cells or organs. However, the mechanism of tissue dam-age by MeHg is not completely known, but many studies confirmMeHg as a lipophilic compound, which passes through cell mem-branes relatively easily and initiate chain of events which damagethe intoxicated cell or tissue [47]. MeHg can also act as a poison forthe mitotic apparatus as it inactivates the tubulin proteins [48,49]resulting in microtubule inhibition which is followed by mitotic ar-rest of cells. Our present findings confirmed the ability of MeHg forinhibiting microtubule formation as shown by the in vitro tubulinpolymerisation–depolymerisation assay, which is a vital cytoskele-tal element for the formation of spindle fibres. Presence of MGNwas ineffective in inhibiting the depolymerisation of tubulin pro-teins in vitro. However, the role of MGN in vivo needs to be studiedfurther to elucidate whether MGN by itself or its metabolites haveany influence on the MeHg induced tubulin depolymerisation.There are reports related to the occurrence of cytogenetic damageobserved as chromatid breaks in peripheral lymphocytes from theindividuals exposed to MeHg [50]. Therefore, we used alkaline sin-gle cell gel electrophoresis (comet) and micronucleus assay togauge whether non-genotoxic doses of MGN (10 lM) was potentenough to prevent the genotoxic effects of MeHg on IMR-32 cells.However, our results from the micronucleus study did not give anyinformation on the anuegenic effect as we did not carry out FISHassay for discriminating the centromere positive (anuegenic) andcentromere negative (clastogenic) micronuclei. The exact mecha-nism by which MGN amends the genotoxic nature of MeHg is stillunspecified but our finding confirms the anti-genotoxic nature ofMGN as shown in earlier findings by the inhibition of heavy metalinduced free radicals and oxidative damage [17].
Cells intoxicated with MeHg show an increase in ROS levels,which in turn results in peroxidation of membrane lipids causingfurther damage. Also, MeHg has a high binding affinity for thiolrich molecules, which results in the depletion of intracellularGSH and other antioxidants leading to accumulation of ROS [51].ROS accumulation inside the cells increase the mitochondrialhydrogen peroxide production and LPO of mitochondrial mem-brane, resulting in loss of membrane integrity and finally leadingto cell death [52]. Results from our study showed that treatmentwith MeHg resulted in increase in ROS levels in a concentrationas well as time dependent manner and MGN at an optimal concen-tration (10 lM) was potent enough to shield the cells from oxida-tive damage. MGN acts as an effective anti-oxidant, mainly onaccount of its catechol moiety with a 6,7-dihydroxylated structure,which provides it the ability to neutralise the ROS generated. It isevident that increase in ROS causes depletion of GSH, GST andother thiol-rich enzymes that actively participate in the cellularantioxidant defence mechanism and peroxidation of membranelipids, resulting in MDA. Along with ROS, the byproducts of LPOmay confer cellular damage by interacting with cellular macromol-ecules, including the genetic material [53] consequently leading tocell death. We observed a significant (p < 0.05) diminution inMeHg induced ROS levels associated with an increase in the in-tra-cellular GSH and GST levels, along with decrease in MDA, whencells were preconditioned with MGN. Also, it has been proved thatMGN possesses the ability to inhibit Fenton’s-type reaction by che-lating Fe2+ ions which diminish ROS and consequently there is acutback in the cellular MDA levels [54]. This observation clearlysuggests that MGN facilitates the reduction in ROS-induced oxida-tive damage thereby maintaining homeostasis of the enzymaticantioxidant system and circumventing LPO.
There is a strong relationship between mitochondrial dysfunc-tioning, ROS generation and disruption of intra-cellular Ca2+
homeostasis in the process of MeHg intoxication. Elevated intracel-lular Ca2+ acts as a secondary messenger for numerous signalling

S. Das et al. / Chemico-Biological Interactions 193 (2011) 129–140 139
processes which in turn help in cellular demise [55]. In case of dis-ruption of Ca2+ homeostasis, the cytoplasm gets flooded with Ca2+
as a result of either outpouring from intracellular Ca2+ reservoir(mainly endoplasmic reticulum) or an incursion from the externalenvironment. Increased cytosolic Ca2+ levels cause the progressiveuptake of Ca2+ by the mitochondria. Increase in mitochondrial Ca2+
levels initiates cascades of reactions within the mitochondria thatdisturb its normal functioning as well as creates permeability tran-sition pores (PTP), which in turn dissipates the DWm. As obviousfrom the present study, IMR-32 cells treated with MeHg showeddepletion in DWm whereas cells preconditioned with MGN priorto MeHg exposure exhibited restoration of DWm to a certain ex-tent. Also, incubation of cells with 10 lM of MeHg (in Ca2+-freegrowth media) showed an increase in intracellular Ca2+ suggestingthat due to increased oxidative stress there is a surge of Ca2+ intothe cytoplasm from the endoplasmic reticulum or sarcoplasmicreticulum (ER/SR) through the ER/SR channels, which in turn leadsto depletion of DWm. Conversely, MGN pre-treatment (10 lM) ofIMR-32 cells before MeHg (10 lM) exposure showed a marked de-crease in cytosolic Ca2+ concentrations.
Exposure to toxicants may result in either partial damage of cel-lular functions or cell death, depending on the type of assault, thetype of cells and their internal defence mechanisms. The level ofexposure determines whether a cell has to be eliminated by apop-tosis, which is an active process requiring energy (ATP), or by pas-sive cell death via necrosis [56]. Apoptotic cell death induced byMeHg has been very well elucidated using in vitro and in vivo mod-els [57]. In agreement with previous works, our study showed thatMeHg induced apoptosis in IMR-32 cells in a dose-dependent man-ner. There was a significant increase in the DNA fragmentation, in-creased Annexin V-FITC-positive (apoptotic) cell population andapoptotic and necrotic indices in cells treated with MeHg(10 lM) alone. MGN (10 lM) conferred protection to the cellsagainst oxidative damage thereby preventing much of DNA dam-age. There was a considerable increase in percentage of live cellswhen stained with AO/EtBr with a significant decrease in the per-cent of apoptotic cell population in cells treated with optimal doseof MGN (10 lM).
Apoptotic cell death requires a series of events that need to oc-cur in a sequential manner. Loss of Ca2+ homeostasis, depletion ofDWm, and elevated ROS levels are conducive enough to force a cellto undergo apoptosis. During the loss of DWm there is a persistentopening of PTP leading to rupturing of the outer mitochondrialmembrane that eventually releases cytochrome c from the trans-mitochondrial membrane space. Western blot analysis of proteinsisolated from IMR-32 cells treated with MeHg (10 lM) aloneshowed an elevation in Bax, cleaved caspase-3, cytochrome c levelsand a decrease in Bcl-2 expression levels clearly indicating thatMeHg induces cell death via the mitochondrial pathway. Also, anelevation in the expression of cellular Nrf-2 levels was observedin case of MeHg alone treated group, which is indicative ofincreased oxidative stress as Nrf-2 helps in the process of amelio-ration of oxidative stress and helps in the co-ordinated transcrip-tional activation of genes coding for antioxidant repertoire bybinding to the antioxidant response element [58]. Test groupspre-treated with MGN before exposing to MeHg partly reducedthe expression of Nrf-2 indicating that MGN interfered with thegeneration of ROS and helped in maintenance of intracellularhomeostasis. MT is a small cysteine-rich protein recognised asthe most abundant metal-binding proteins and is known to playa critical role in cellular detoxification of inorganic species bysequestering metal ions and by scavenging ROS [59]. It is wellknown that the expression of MT proteins is brought about by awide of range of stimuli, including metals, hormones, cytokines,inflammation and oxidative stress. In the present study, MT wasevaluated by western blotting and no significant differences with
respect to MT levels of the cells treated with or without MGN/MeHg were found. It is therefore likely that the cytoprotective ef-fects of MGN observed clearly preclude the role of MT.
To conclude, the present findings indicated the protective effectof MGN against MeHg- induced toxicity, which may be attributedto its anti-genotoxic, anti-apoptotic and anti-lipid peroxidative po-tential plausibly because of its free radical scavenging ability,which reduced the oxidative stress and in turn facilitated thedown-regulation of mitochondrial apoptotic signalling pathways.Although, several earlier studies have implicated the usefulnessof MGN against conditions such as oxidative stress, excitotoxic in-sults, heavy metal toxicity etc., further studies need to be realisedin order to understand whether MGN would show a similar healthpromoting effects in case of mercury intoxicated normal neuronsor by using animal models of neurodegenerative disorders.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Acknowledgement
The authors are thankful to Dr. K. Satyamoorthy, Director, Man-ipal Life Sciences Centre, Manipal University, Manipal for his helpand for all the facilities offered by TIFAC-CORE in Pharmacogenom-ics, MLSC.
References
[1] K. Jomova, M. Valko, Advances in metal-induced oxidative stress and humandisease, Toxicology 283 (2011) 65–87.
[2] S. Yoon, S.S. Han, S.V. Rana, Molecular markers of heavy metal toxicity – a newparadigm for health risk assessment, J. Environ. Biol. 29 (2008) 1–14.
[3] K.H. Taber, R.A. Hurley, Mercury exposure: effects across the lifespan, J.Neuropsychiatry Clin. Neurosci. 20 (2008) 384–389.
[4] S.B. Elhassani, The many faces of methylmercury poisoning, J. Toxicol. Clin.Toxicol. 19 (1982) 875–906.
[5] F. Bakir, H. Rustam, S. Tikriti, S.F. Al-Damluji, H. Shihristani, Clinical andepidemiological aspects of methylmercury poisoning, Postgrad. Med. J. 56(1980) 1–10.
[6] G. Guzzi, C. Minoia, P.D. Pigatto, G. Severi, Methylmercury, amalgams, andchildren’s health, Environ. Health Perspect. 114 (2006) 149–150.
[7] D. Mergler, H.A. Anderson, L.H. Chan, K.R. Mahaffey, M. Murray, M. Sakamoto,A.H. Stern, Methylmercury exposure and health effects in humans: aworldwide concern, Ambio 36 (2007) 3–11.
[8] S. Diez, Human health effects of methylmercury exposure, Rev. Environ.Contam. Toxicol. 198 (2009) 111–132.
[9] D.C. Rice, Identification of functional domains affected by developmentalexposure to methylmercury: Faroe islands and related studies,Neurotoxicology 21 (2000) 1039–1044.
[10] E. van Wijngaarden, C. Beck, C.F. Shamlaye, E. Cernichiari, P.W. Davidson, G.J.Myers, T.W. Clarkson, Benchmark concentrations for methyl mercury obtainedfrom the 9-year follow-up of the Seychelles Child Development Study,Neurotoxicology 27 (2006) 702–709.
[11] N. Khan, V.M. Adhami, H. Mukhtar, Tea polyphenols in chemoprevention ofprostate cancer: preclinical and clinical studies, Nutr. Cancer 61 (2009) 836–841.
[12] N.A. Kelsey, H.M. Wilkins, D.A. Linseman, Nutraceutical antioxidants as novelneuroprotective agents, Molecules 15 (2010) 7792–7814.
[13] J. Dai, R.J. Mumper, Plant phenolics: extraction, analysis and their antioxidantand anticancer properties, Molecules 15 (2010) 7313–7352.
[14] M.M. Pinto, M.E. Sousa, M.S. Nascimento, Xanthone derivatives: new insightsin biological activities, Curr. Med. Chem. 12 (2005) 2517–2538.
[15] S. Guha, S. Ghosal, U. Chattopadhyay, Antitumor, immunomodulatory andanti-HIV effect of mangiferin, a naturally occurring glucosylxanthone,Chemotherapy 42 (1996) 443–451.
[16] N. Wauthoz, A. Balde, E.S. Balde, M. Van Damme, P. Duez, Ethnopharmacologyof mangifera indica l. Bark and pharmacological studies of its main C-glucosylxanthone, mangiferin, Int. J. Biomed. Pharm. Sci. 1 (2007) 112–119.
[17] B.S. Satish Rao, M.V. Sreedevi, B. Nageshwar Rao, Cytoprotective andantigenotoxic potential of Mangiferin, a glucosylxanthone against cadmiumchloride induced toxicity in HepG2 cells, Food Chem. Toxicol. 47 (2009) 592–600.
[18] E.K. Viswanadh, B.N. Rao, B.S. Rao, Antigenotoxic effect of mangiferin andchanges in antioxidant enzyme levels of Swiss albino mice treated withcadmium chloride, Hum. Exp. Toxicol. 29 (2010) 409–418.

140 S. Das et al. / Chemico-Biological Interactions 193 (2011) 129–140
[19] S. Agarwala, B. Nageshwar Rao, K. Mudholkar, R. Bhuwania, B.S.S. Rao,Mangiferin, a dietary xanthone protects against mercury-induced toxicity inHepG2 cells, Environ. Toxicol. doi:10.1002/tox.20620.
[20] M.R. Campos-Esparza, M.V. Sanchez-Gomez, C. Matute, Molecular mechanismsof neuroprotection by two natural antioxidant polyphenols, Cell Calcium 45(2009) 358–368.
[21] G. Martinez Sanchez, E. Candelario-Jalil, A. Giuliani, O.S. Leon, S. Sam, R.Delgado, A.J. Nunez Selles, Mangifera indica L. extract (QF808) reducesischaemia-induced neuronal loss and oxidative damage in the gerbil brain,Free Radic. Res. 35 (2001) 465–473.
[22] T. Mosmann, Rapid colorimetric assay for cellular growth and survival:application to proliferation and cytotoxicity assays, J. Immunol. Methods 65(1983) 55–63.
[23] T.T. Puck, P.I. Marcus, A rapid method for viable cell titration, clone productionwith hela cells in tissue culture: the use of X-irradiated cells to supplyconditioning factors, Proc. Natl. Acad. Sci. U S A 41 (1955) 432–437.
[24] M. Fenech, A.A. Morley, Measurement of micronuclei in lymphocytes, Mutat.Res. 147 (1985) 29–36.
[25] B.S.S. Rao, R. Shanbhoge, D. Upadhya, G.C. Jagetia, S.K. Adiga, P. Kumar, K.Guruprasad, P. Gayathri, Antioxidant, anticlastogenic and radioprotectiveeffect of Coleus aromaticus on Chinese hamster fibroblast cells (V79) exposedto gamma radiation, Mutagenesis 21 (2006) 237–242.
[26] M. Fenech, W.P. Chang, M. Kirsch-Volders, N. Holland, S. Bonassi, E. Zeiger,HUMN project: detailed description of the scoring criteria for the cytokinesis-block micronucleus assay using isolated human lymphocyte cultures, Mutat.Res. 534 (2003) 65–75.
[27] J. Surralles, N. Xamena, A. Creus, J. Catalan, H. Norppa, R. Marcos, Induction ofmicronuclei by five pyrethroid insecticides in whole-blood and isolatedhuman lymphocyte cultures, Mutat. Res. 341 (1995) 169–184.
[28] N.P. Singh, M.T. McCoy, R.R. Tice, E.L. Schneider, A simple technique forquantitation of low levels of DNA damage in individual cells, Exp. Cell Res. 175(1988) 184–191.
[29] C. Renvoize, A. Biola, M. Pallardy, J. Breard, Apoptosis: identification of dyingcells, Cell Biol.Toxicol. 14 (1998) 111–120.
[30] M. Herrmann, H.M. Lorenz, R. Voll, M. Grunke, W. Woith, J.R. Kalden, A rapidand simple method for the isolation of apoptotic DNA fragments, Nucleic AcidsRes. 22 (1994) 5506–5507.
[31] J. Bai, A.I. Cederbaum, Catalase protects HepG2 cells from apoptosis induced byDNA-damaging agents by accelerating the degradation of p53, J. Biol. Chem.278 (2003) 4660–4667.
[32] G.T. Rijkers, L.B. Justement, A.W. Griffioen, J.C. Cambier, Improved method formeasuring intracellular Ca++ with fluo-3, Cytometry 11 (1990) 923–927.
[33] R.C. Scaduto Jr., L.W. Grotyohann, Measurement of mitochondrial membranepotential using fluorescent rhodamine derivatives, Biophys. J. 76 (1999) 469–477.
[34] M.M. Bradford, A rapid and sensitive method for the quantitation ofmicrogram quantities of protein utilizing the principle of protein-dyebinding, Anal. Biochem. 72 (1976) 248–254.
[35] M.S. Moron, J.W. Depierre, B. Mannervik, Levels of glutathione, glutathionereductase and glutathione S-transferase activities in rat lung and liver,Biochim. Biophys. Acta. 582 (1979) 67–78.
[36] W.H. Habig, M.J. Pabst, W.B. Jakoby, Glutathione S-transferases. The firstenzymatic step in mercapturic acid formation, J. Biol. Chem. 249 (1974) 7130–7139.
[37] J.A. Buege, S.D. Aust, Microsomal lipid peroxidation, Methods Enzymol. 52(1978) 302–310.
[38] G.G. Borisy, J.M. Marcum, J.B. Olmsted, D.B. Murphy, K.A. Johnson, Purificationof tubulin and associated high molecular weight proteins from porcine brainand characterization of microtubule assembly in vitro, Ann. NY Acad. Sci. 253(1975) 107–132.
[39] C. Fila, C. Metz, P. van der Sluijs, Juglone inactivates cysteine-rich proteinsrequired for progression through mitosis, J. Biol. Chem. 283 (2008) 21714–21724.
[40] L.E. Kerper, N. Ballatori, T.W. Clarkson, Methylmercury transport across theblood-brain barrier by an amino acid carrier, Am. J. Physiol. 262 (1992) 761–765.
[41] W. Zheng, M. Aschner, J.F. Ghersi-Egea, Brain barrier systems: a new frontier inmetal neurotoxicological research, Toxicol. Appl. Pharmacol. 192 (2003) 1–11.
[42] G.N. George, R.C. Prince, J. Gailer, G.A. Buttigieg, M.B. Denton, H.H. Harris, I.J.Pickering, Mercury binding to the chelation therapy agents DMSA and DMPSand the rational design of custom chelators for mercury, Chem. Res. Toxicol. 17(2004) 999–1006.
[43] J.P. Rooney, The role of thiols, dithiols, nutritional factors and interactingligands in the toxicology of mercury, Toxicology 234 (2007) 145–156.
[44] M. Modak, P. Dixit, J. Londhe, S. Ghaskadbi, A.D.T. Paul, Indian herbs and herbaldrugs used for the treatment of diabetes, J. Clin. Biochem. Nutr. 40 (2007) 163–173.
[45] H. Marona, N. Szkaradek, A. Rapacz, B. Filipek, M. Dybala, A. Siwek, M. Cegla, E.Szneler, Preliminary evaluation of pharmacological properties of somexanthone derivatives, Bioorg. Med. Chem. 17 (2009) 1345–1352.
[46] J.L. Franco, T. Posser, F. Missau, M.G. Pizzolatti, A.R. Dos Santos, D.O. Souza, M.Aschner, J.B. Rocha, A.L. Dafre, M. Farina, Structure-activity relationship offlavonoids derived from medicinal plants in preventing methylmercury-induced mitochondrial dysfunction, Environ. Toxicol. Pharmacol. 30 (2010)272–278.
[47] C.C. Bridges, R.K. Zalups, Molecular and ionic mimicry and the transport oftoxic metals, Toxicol. Appl. Pharmacol. 204 (2005) 274–308.
[48] K. Miura, N. Koide, S. Himeno, I. Nakagawa, N. Imura, The involvement ofmicrotubular disruption in methylmercury-induced apoptosis in neuronal andnonneuronal cell lines, Toxicol. Appl. Pharmacol. 160 (1999) 279–288.
[49] A.F. Castoldi, S. Barni, I. Turin, C. Gandini, L. Manzo, Early acute necrosis,delayed apoptosis and cytoskeletal breakdown in cultured cerebellar granuleneurons exposed to methylmercury, J. Neurosci. Res. 59 (2000) 775–787.
[50] M.I. Amorim, D. Mergler, M.O. Bahia, H. Dubeau, D. Miranda, J. Lebel, R.R.Burbano, M. Lucotte, Cytogenetic damage related to low levels of methylmercury contamination in the Brazilian Amazon, An. Acad. Bras. Cienc. 72(2000) 497–507.
[51] A.F. Castoldi, T. Coccini, S. Ceccatelli, L. Manzo, Neurotoxicity and moleculareffects of methylmercury, Brain Res. Bull. 55 (2001) 197–203.
[52] W.D. Atchison, M.F. Hare, Mechanisms of methylmercury-inducedneurotoxicity, FASEB J. 8 (1994) 622–629.
[53] L.J. Marnett, Lipid peroxidation-DNA damage by malondialdehyde, Mutat. Res.424 (1999) 83–95.
[54] G. Pardo Andreu, R. Delgado, J. Velho, N.M. Inada, C. Curti, A.E. Vercesi,Mangifera indica L. extract (Vimang) inhibits Fe2+-citrate-inducedlipoperoxidation in isolated rat liver mitochondria, Pharmacol. Res. 51(2005) 427–435.
[55] T.L. Limke, S.R. Heidemann, W.D. Atchison, Disruption of intraneuronaldivalent cation regulation by methylmercury: are specific targets involved inaltered neuronal development and cytotoxicity in methylmercury poisoning?,Neurotoxicology 25 (2004) 741–760
[56] S. Orrenius, B. Zhivotovsky, P. Nicotera, Regulation of cell death: the calcium-apoptosis link, Nat. Rev. Mol. Cell. Biol. 4 (2003) 552–565.
[57] S. Ceccatelli, E. Dare, M. Moors, Methylmercury-induced neurotoxicity andapoptosis, Chem. Biol. Interact. 188 (2010) 301–308.
[58] J.W. Kaspar, S.K. Niture, A.K. Jaiswal, Nrf2:INrf2 (Keap1) signaling in oxidativestress, Free Radic. Biol. Med. 47 (2009) 1304–1309.
[59] G.K. Andrews, Regulation of metallothionein gene expression by oxidativestress and metal ions, Biochem. Pharmacol. 59 (2000) 95–104.
Copyright © 2022 FDOKUMEN




















