
Linkage to Chromosome 1< i> p 36 for Attention-Deficit/Hyperactivity Disorder Traits in School and...
6
Linkage to Chromosome 1p36 for Attention-Deficit/ Hyperactivity Disorder Traits in School and Home Settings Kaixin Zhou, Philip Asherson, Pak Sham, Barbara Franke, Richard J.L. Anney, Jan Buitelaar, Richard Ebstein, Michael Gill, Keeley Brookes, Cathelijne Buschgens, Desmond Campbell, Wai Chen, Hanna Christiansen, Ellen Fliers, Isabel Gabriëls, Lena Johansson, Rafaela Marco, Fernando Mulas, Ueli Müller, Aisling Mulligan, Benjamin M. Neale, Fruhling Rijsdijk, Nanda Rommelse, Henrik Uebel, Lamprini Psychogiou, Xiaohui Xu, Tobias Banaschewski, Edmund Sonuga-Barke, Jacques Eisenberg, Iris Manor, Ana Miranda, Robert D. Oades, Herbert Roeyers, Aribert Rothenberger, Joseph Sergeant, Hans-Christoph Steinhausen, Eric Taylor, Margaret Thompson, and Stephen V. Faraone Background: Limited success has been achieved through previous attention-deficit/hyperactivity disorder (ADHD) linkage scans, which were all designed to map genes underlying the dichotomous phenotype. The International Multi-centre ADHD Genetics (IMAGE) project performed a whole genome linkage scan specifically designed to map ADHD quantitative trait loci (QTL). Methods: A set of 1094 single selected Caucasian ADHD nuclear families was genotyped on a highly accurate and informative single nucleotide polymorphism (SNP) panel. Two quantitative traits measuring the children’s symptoms in home and school settings were collected and standardized according to a population sample of 8000 children to reflect the developmental nature and gender prevalence difference of ADHD. Univariate linkage test was performed on both traits and their mean score. Results: A significant common linkage locus was found at chromosome 1p36 with a locus-specific heritability of 5.1% and a genomewide empirical p .04. Setting-specific suggestive linkage signals were also found: logarithm of odds (LOD) 2.2 at 9p23 for home trait and LOD 2.6 at 11q21 for school trait. Conclusions: These results indicate that given large samples with proper phenotypic measures, searching for ADHD genes with a QTL strategy is an important alternative to using the clinical diagnosis. The fact that our linkage region 1p36 overlaps with the dyslexia QTL DYX8 further suggests it is potentially a pleiotropic locus for ADHD and dyslexia. Key Words: ADHD, linkage, QTL A ttention-deficit/hyperactivity disorder (ADHD [MIM143465]) is a common childhood developmental behavioral disorder that has a prevalence of 4% to 10% in the general popula- tion, depending on the population studied and the precise way the diagnostic criteria are implemented (1). It occurs four to eight times more frequently in boys than in girls (2). Converging evidence from family, twin, and adoption studies suggests a significant genetic contribution to ADHD, with an estimated sib relative risk ( s ) of 4 to 8 and average heritability of .76 (3,4). Both categorical diagnosis and dimensional ratings of ADHD symptoms have been used in molec- ular genetic studies designed to identify the genes that confer risk to the disorder. Convincing evidence of candidate gene association has been obtained for genetic variants within or close to the dopamine D4 and D5 receptor genes from the combined analysis of numerous independent datasets in different populations (4,5). In addition, associations are suggested in several other genes following the meta-analysis of association findings reported in three or more studies, although further independent replications are still required (4). However, the genes identified so far contribute only a small elevated risk to ADHD and a large amount of the population variance is still to be accounted for (6,7). The main method applied during the last decade to localize novel genes that increase the risk for ADHD has been the use of genomewide linkage tests to identify chromosomal regions From the MRC Social Genetic Developmental and Psychiatry Centre (KZ, PA, PS, KB, DC, WC, LJ, BMN, FR, XX, ET, ES-B), King’s College London, London, United Kingdom; Department of Psychiatry (PS), LKS Faculty of Medicine, University of Hong Kong, Pokfulam, Hong Kong; Department of Psychiatry (RJLA, MG, AM), Trinity Centre for Health Sciences, St. James’s Hospital, Dublin, Ireland; Department of Psychiatry (BF, JB, CB, EF), Radboud University Nijmegen Medical Center, Nijmegen, The Netherlands; Department of Human Genetics (BF), Radbound University Nijmegan Medical Centre, Nijmegan, The Netherlands; S. Herzog Memorial Hospital (RE, JE), Jerusalem, Israel; ADHD Clinic (IM), Geha Mental Health Center, Petak Tikvah, Israel; University Clinic for Child and Adolescent Psychiatry (HC, RDO), Essen, Germany; Child and Adolescent Psychiatry (HU, TB, AR), University of Göttingen, Göttingen, Germany; Department of Child and Adolescent Psychiatry and Psychotherapy (TB), Central Institute of Mental Health, Mannheim, Germany; Departments of Experimental Clinical Health Psychology (IG, HR, ES-B), Ghent University, Ghent, Belgium; Department of Developmental and Educational Psychology (RM, AM), University of Valencia, Valencia, Spain; Neuropediatric Service (FM), University Hospital La Fe, Valencia, Spain; Department of Child and Adolescent Psychiatry (UM, H-CS), University of Zurich, Zurich, Switzerland; Department of Clinical Neuropsychology (NR, JS), Vrije Universiteit, Amsterdam, The Netherlands; School of Psychology (LP, ES-B, MT), University of Southampton, Highfield, Southampton, United Kingdom; Departments of Psychiatry and Neuroscience and Physiology (SVF), SUNY Upstate Medical University, Syracuse, New York; and Child Study Center (ES-B), New York University, New York, New York. Address reprint requests to Philip Asherson, M.D., Ph.D., MRC Social Genetic Developmental Psychiatry, Institute of Psychiatry, De Crespigny Park, London SE5 8AF, United Kingdom; E-mail: [email protected]. Received September 27, 2007; revised January 22, 2008; accepted February 22, 2008. BIOL PSYCHIATRY 2008;64:571–576 0006-3223/08/$34.00 doi:10.1016/j.biopsych.2008.02.024 © 2008 Society of Biological Psychiatry
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Linkage to Chromosome 1< i> p 36 for Attention-Deficit/Hyperactivity Disorder Traits in School and...

LHHKRHULIH
Bwp
Mncd
Rea
Csf
K
Atdmfc8du
F
A
R
0d
inkage to Chromosome 1p36 for Attention-Deficit/yperactivity Disorder Traits in School andome Settings
aixin Zhou, Philip Asherson, Pak Sham, Barbara Franke, Richard J.L. Anney, Jan Buitelaar,ichard Ebstein, Michael Gill, Keeley Brookes, Cathelijne Buschgens, Desmond Campbell, Wai Chen,anna Christiansen, Ellen Fliers, Isabel Gabriëls, Lena Johansson, Rafaela Marco, Fernando Mulas,eli Müller, Aisling Mulligan, Benjamin M. Neale, Fruhling Rijsdijk, Nanda Rommelse, Henrik Uebel,amprini Psychogiou, Xiaohui Xu, Tobias Banaschewski, Edmund Sonuga-Barke, Jacques Eisenberg,
ris Manor, Ana Miranda, Robert D. Oades, Herbert Roeyers, Aribert Rothenberger, Joseph Sergeant,ans-Christoph Steinhausen, Eric Taylor, Margaret Thompson, and Stephen V. Faraone
ackground: Limited success has been achieved through previous attention-deficit/hyperactivity disorder (ADHD) linkage scans, whichere all designed to map genes underlying the dichotomous phenotype. The International Multi-centre ADHD Genetics (IMAGE) projecterformed a whole genome linkage scan specifically designed to map ADHD quantitative trait loci (QTL).
ethods: A set of 1094 single selected Caucasian ADHD nuclear families was genotyped on a highly accurate and informative singleucleotide polymorphism (SNP) panel. Two quantitative traits measuring the children’s symptoms in home and school settings wereollected and standardized according to a population sample of 8000 children to reflect the developmental nature and gender prevalenceifference of ADHD. Univariate linkage test was performed on both traits and their mean score.
esults: A significant common linkage locus was found at chromosome 1p36 with a locus-specific heritability of 5.1% and a genomewidempirical p � .04. Setting-specific suggestive linkage signals were also found: logarithm of odds (LOD) � 2.2 at 9p23 for home trait and LOD � 2.6t 11q21 for school trait.
onclusions: These results indicate that given large samples with proper phenotypic measures, searching for ADHD genes with a QTLtrategy is an important alternative to using the clinical diagnosis. The fact that our linkage region 1p36 overlaps with the dyslexia QTL DYX8
urther suggests it is potentially a pleiotropic locus for ADHD and dyslexia.PS, Krsity
elandGeneD Clindole
TB), C, GheversityNeuroton, U
dy CeDevel
ey Words: ADHD, linkage, QTL
ttention-deficit/hyperactivity disorder (ADHD [MIM143465])is a common childhood developmental behavioral disorderthat has a prevalence of 4% to 10% in the general popula-
ion, depending on the population studied and the precise way theiagnostic criteria are implemented (1). It occurs four to eight timesore frequently in boys than in girls (2). Converging evidence from
amily, twin, and adoption studies suggests a significant geneticontribution to ADHD, with an estimated sib relative risk (�s) of 4 to and average heritability of .76 (3,4). Both categorical diagnosis andimensional ratings of ADHD symptoms have been used in molec-lar genetic studies designed to identify the genes that confer risk to
rom the MRC Social Genetic Developmental and Psychiatry Centre (KZ, PA,Kingdom; Department of Psychiatry (PS), LKS Faculty of Medicine, UniveAM), Trinity Centre for Health Sciences, St. James’s Hospital, Dublin, IrMedical Center, Nijmegen, The Netherlands; Department of Human Netherlands; S. Herzog Memorial Hospital (RE, JE), Jerusalem, Israel; ADHChild and Adolescent Psychiatry (HC, RDO), Essen, Germany; Child and ADepartment of Child and Adolescent Psychiatry and Psychotherapy (Experimental Clinical Health Psychology (IG, HR, ES-B), Ghent UniversityAM), University of Valencia, Valencia, Spain; Neuropediatric Service (FM), Uni(UM, H-CS), University of Zurich, Zurich, Switzerland; Department of ClinicalPsychology (LP, ES-B, MT), University of Southampton, Highfield, Southamp(SVF), SUNY Upstate Medical University, Syracuse, New York; and Child Stu
ddress reprint requests to Philip Asherson, M.D., Ph.D., MRC Social Genetic8AF, United Kingdom; E-mail: [email protected].
eceived September 27, 2007; revised January 22, 2008; accepted February 22, 2
006-3223/08/$34.00oi:10.1016/j.biopsych.2008.02.024
the disorder. Convincing evidence of candidate gene associationhas been obtained for genetic variants within or close to thedopamine D4 and D5 receptor genes from the combined analysis ofnumerous independent datasets in different populations (4,5). Inaddition, associations are suggested in several other genes followingthe meta-analysis of association findings reported in three or morestudies, although further independent replications are still required(4). However, the genes identified so far contribute only a smallelevated risk to ADHD and a large amount of the populationvariance is still to be accounted for (6,7).
The main method applied during the last decade to localizenovel genes that increase the risk for ADHD has been the use ofgenomewide linkage tests to identify chromosomal regions
B, DC, WC, LJ, BMN, FR, XX, ET, ES-B), King’s College London, London, Unitedof Hong Kong, Pokfulam, Hong Kong; Department of Psychiatry (RJLA, MG,; Department of Psychiatry (BF, JB, CB, EF), Radboud University Nijmegentics (BF), Radbound University Nijmegan Medical Centre, Nijmegan, Theic (IM), Geha Mental Health Center, Petak Tikvah, Israel; University Clinic for
scent Psychiatry (HU, TB, AR), University of Göttingen, Göttingen, Germany;entral Institute of Mental Health, Mannheim, Germany; Departments of
nt, Belgium; Department of Developmental and Educational Psychology (RM, Hospital La Fe, Valencia, Spain; Department of Child and Adolescent Psychiatrypsychology (NR, JS), Vrije Universiteit, Amsterdam, The Netherlands; School ofnited Kingdom; Departments of Psychiatry and Neuroscience and Physiology
nter (ES-B), New York University, New York, New York.opmental Psychiatry, Institute of Psychiatry, De Crespigny Park, London SE5
008.
BIOL PSYCHIATRY 2008;64:571–576© 2008 Society of Biological Psychiatry

cwwtrmdcpt
(uc(smsaFlswtpcdm
plarhstfafp
M
(fpsalce(dKsabcpscT
572 BIOL PSYCHIATRY 2008;64:571–576 K. Zhou et al.
w
ontaining putative risk loci. To date, six independent genome-ide ADHD linkage scans have been conducted, all of whichere designed to adopt the categorical diagnosis as the pheno-
ype (8–14). These scans nominated several novel genomicegions that might harbor ADHD susceptibility genes, with theost prominent findings on chromosomes 5p and 17p beingetected in several studies. However, none of these loci wereonsistently detected and no genes that can explain these linkageeaks have been clearly identified, indicating the complexity ofhe genetic risk factors underlying ADHD.
Although not originally designed to map quantitative trait lociQTL) for ADHD symptoms, three of these linkage studies alsosed QTL linkage methods by using the total DSM-IV symptomount (DSM-TOT) as a quantitative measure of ADHD severity11–13). No clear QTL linkage evidence was discovered in thesetudies and there was hardly any overlap between the dichoto-ous and quantitative trait linkage test results. There are a few
tudy design issues that could explain these unimpressive resultspart from the complexity of the genetic influences on ADHD.irst, the power to detect QTLs of small to moderate effect wasimited in these studies, which consisted of relatively smallamples of 100 to 300 sib pairs. Second, the DSM-TOT measuresere not adjusted for age and gender in these studies to reflect
he developmental course of ADHD symptoms and the genderrevalence difference. Third, the variance of DSM-TOT in thelinical samples is less than that from the standardized scoreserived from standard ratings scales of ADHD symptoms that inost cases adopt 4-point scales of severity for each symptom.Another concern for QTL mapping of ADHD is that only
arental DSM-TOT but no teacher data were applied in previousinkage scans. Three twin studies have examined this questionnd while all show that parent and teacher rated ADHD dataeflecting behavior in the home and school settings are highlyeritable, there are substantial unique genetic effects (setting-pecific loci) in addition to shared genetic effects between thewo measures (common loci) (15–18). Here, we report resultsrom the first QTL linkage scan designed specifically to use agend gender standardized quantitative scores of ADHD symptomsrom both home and school settings, using a large single-roband ascertained sample.
ethods and Materials
For this study, the International Multi-centre ADHD GeneticsIMAGE) project recruited 1094 European Caucasian nuclearamilies ascertained through DSM-IV combined-subtype ADHDrobands. The study design was originally established with thepecific aim of completing genomewide linkage using QTLpproaches. Detailed recruitment procedures have been pub-ished previously (19). Families were identified at 12 specialistlinical centers across eight European countries: Belgium (Gh-nt), Germany (Essen and Goettingen), Ireland (Dublin), IsraelTel-Aviv and Jerusalem), Netherlands (Nijmegan and Amster-am), Spain (Valencia), Switzerland (Zurich), and the Unitedingdom (London and Southampton). The probands and theiriblings were aged between 5 and 17 years at the time ofssessment, and at least one, and in most cases two, of theiriological parents were also available for DNA collection. Clini-al research assessments were administered by qualified childsychiatrists or trained interviewers, after which blood or buccalamples were collected for DNA extraction either directly or fromell lines that were established at Rutgers University, New Jersey.
he following exclusion criteria were applied for the probandsww.sobp.org/journal
and their sibs: 1) IQ � 70; 2) a diagnosis of schizophrenia orautism or neurological disorders such as epilepsy and braininjury; 3) any genetic or medical disorder associated with exter-nalizing behaviors that might mimic ADHD; 4) ethnicity otherthan white European origin, and 5) not living at home with atleast one biological parent.
The final sample of 4529 genotyped subjects reported in thislinkage study included 2545 children and their parents. Ethicalapproval for the study was obtained from National Institute ofHealth registered ethical review boards for each center. All thefamilies gave informed consent for the collection of clinical data,DNA and cell line storage at Rutgers, and sharing of anonymousclinical and genotype data with the scientific community.
Two quantitative traits measuring the children’s ADHD symp-toms at home and at school were obtained from their parents andteachers, respectively, by using the revised long versions of theConners’ Rating Scales (20). The DSM-IV symptom subscales(N-subscales), consisting of four-point scales for each of the 18DSM-IV ADHD symptom items, were used to rate the children’sbehavior. The raw scores from both informants were adjusted forage and gender according to a normative profile, which has adistribution of mean � 50 and SD � 10 derived from apopulation sample of 8000 children (20). After the transforma-tion, the standardized scores from parents (PNscore) and teach-ers (TNscore) were used as the quantitative trait measures in theQTL linkage analysis.
Descriptive statistics for the two quantitative traits are listed inTable 1. The distribution of both traits is shifted toward the moresevere end in the ADHD children and their unaffected sibscompared with the normal population mean of 50. As expectedfrom the bivariate distribution of the PNscore and TNscore in thepopulation, both proband and sibling scores are distributedcloser to the extreme, and the correlation in children with ADHDis lower than that in the unaffected group. To detect QTLs thatcould account for the overlap between parent and teacherscores, we tested for QTL linkage using both the PNscore andTNscore independently, in addition to the mean of the twoscores. The use of the mean score for the parent and teacherrated ADHD symptoms is expected to reduce the variancespecific to each informant and improve the power to detectcommon genetic factors.
Genotyping was completed by the Center for Inherited Dis-ease Research (CIDR) using Illumina’s BeadArray technology(http://www.illumina.com) on a BeadLab system (Illumina). Atotal number of 5545 autosomal single nucleotide polymor-phisms (SNPs) from the Illumina Linkage IVb SNP panel weresuccessfully assayed with a call rate of 99.6% and reproduction
Table 1. Phenotypic Features of the 2545 Children from 1094 NuclearFamilies
ADHDa (n � 1141) Non-ADHD (n � 1404)
Male : Female 975 : 166 714 : 690Age 10.7 � 2.8 11 � 3.3PNscore 77.3 � 9.4 54.8 � 13TNscore 73.8 � 11.6 55.6 � 13.3Mean Score 75.5 � 8.2 55.1 � 11.2Correlationb .21 .44
ADHD, attention-deficit/hyperactivity disorder; PNscore, standardizedscores from parents; TNscore, standardized scores from teachers.
aA total number of 1141 children (967 probands and 174 sibs) made theresearch diagnosis criteria of DSM-IV combined type ADHD.
bPearson’s correlation coefficient between PNscore and TNscore.
rpBt(f
pROtP(ieMflwdeSa
Trenpmpdrc
R
rwK9Tsi
K. Zhou et al. BIOL PSYCHIATRY 2008;64:571–576 573
ate of 99.994%. The markers were ordered and placed on thehysical map according to Genome Build 35 (National Center foriotechnology Information, http://www.ncbi.nlm.nih.gov/). In-
erpolated genetic distances from the deCODE genetic mapSturlugotu 8, IS-101, Reykjavik, Iceland) were used in theollowing linkage analysis (21).
We identified and corrected 40 pedigree errors by testingairwise subject relationships throughout the sample withELPAIR (http://csg.sph.umich.edu/boehnke/relpair.php) (22).ne hundred five SNPs were dropped due to significant depar-
ure (p � .01) from Hardy-Weinberg equilibrium reported byEDSTATS (http://www.sph.umich.edu/csg/abecasis/PedStats/)23). Another 33 SNPs were dropped due to excessive Mendeliannconsistencies found by PedCheck (http://watson.hgen.pitt.du/register/docs/pedcheck.html) (24). The remaining sporadicendelian inconsistencies were removed by dropping all the
amily members’ genotypes on the erroneous SNP. Genotypeseading to unlikely recombinants were removed by Merlin (http://ww.sph.umich.edu/csg/abecasis/Merlin/) (25). After the aboveata cleaning process, a total number of 5407 autosomal SNPsntered into our linkage analysis with an average resolution of 1.66NPs/cM. The average entropy information content was 95.2%cross the genome and never dropped below 81% (25).
Univariate QTL linkage was examined for the PNscore, theNscore, and their mean score by Merlin, which implements aegression-based procedure using trait-squared sums and differ-nces to predict identity by descent (IBD) sharing between anyoninbred relative pairs (25,26). With the population distributionarameters of mean, variance, and heritability specified, thisethod can be applied to selected samples with similar statisticalower to the variance component linkage tests. Since linkageisequilibrium between adjacent SNPs can lead to inflated loga-ithm of odds (LOD) scores, we applied the criteria of r2 � .05 toluster correlated SNPs into combined markers (23).
esults
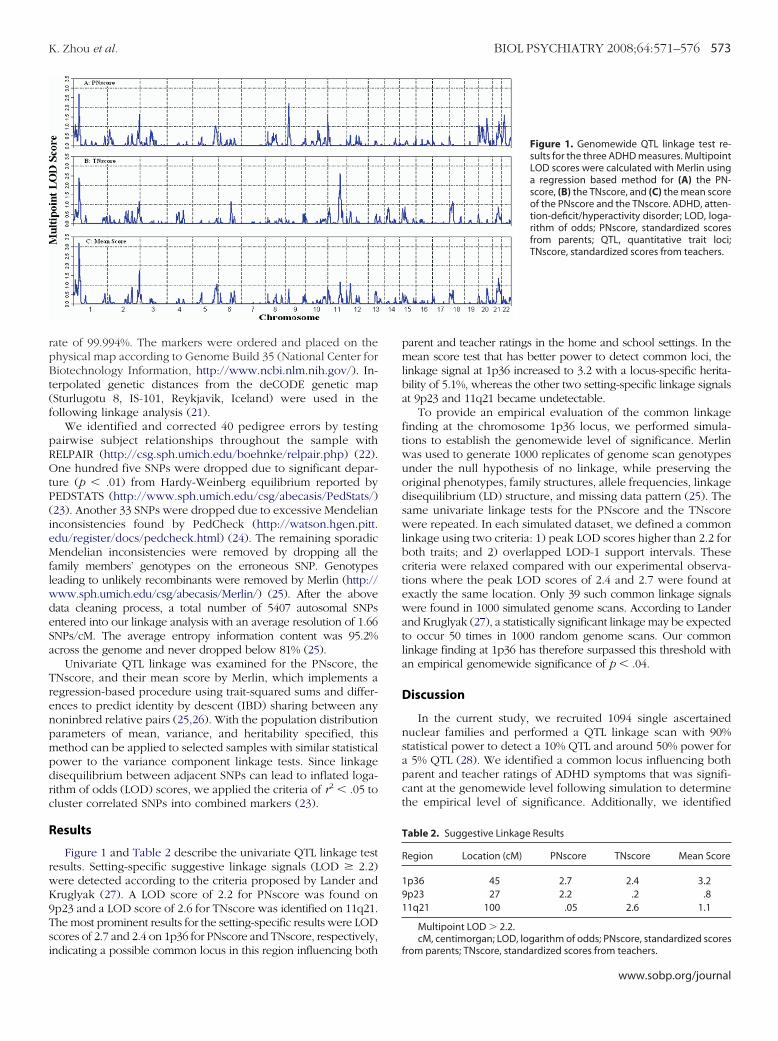
Figure 1 and Table 2 describe the univariate QTL linkage testesults. Setting-specific suggestive linkage signals (LOD � 2.2)ere detected according to the criteria proposed by Lander andruglyak (27). A LOD score of 2.2 for PNscore was found onp23 and a LOD score of 2.6 for TNscore was identified on 11q21.he most prominent results for the setting-specific results were LODcores of 2.7 and 2.4 on 1p36 for PNscore and TNscore, respectively,
ndicating a possible common locus in this region influencing bothparent and teacher ratings in the home and school settings. In themean score test that has better power to detect common loci, thelinkage signal at 1p36 increased to 3.2 with a locus-specific herita-bility of 5.1%, whereas the other two setting-specific linkage signalsat 9p23 and 11q21 became undetectable.
To provide an empirical evaluation of the common linkagefinding at the chromosome 1p36 locus, we performed simula-tions to establish the genomewide level of significance. Merlinwas used to generate 1000 replicates of genome scan genotypesunder the null hypothesis of no linkage, while preserving theoriginal phenotypes, family structures, allele frequencies, linkagedisequilibrium (LD) structure, and missing data pattern (25). Thesame univariate linkage tests for the PNscore and the TNscorewere repeated. In each simulated dataset, we defined a commonlinkage using two criteria: 1) peak LOD scores higher than 2.2 forboth traits; and 2) overlapped LOD-1 support intervals. Thesecriteria were relaxed compared with our experimental observa-tions where the peak LOD scores of 2.4 and 2.7 were found atexactly the same location. Only 39 such common linkage signalswere found in 1000 simulated genome scans. According to Landerand Kruglyak (27), a statistically significant linkage may be expectedto occur 50 times in 1000 random genome scans. Our commonlinkage finding at 1p36 has therefore surpassed this threshold withan empirical genomewide significance of p � .04.
Discussion
In the current study, we recruited 1094 single ascertainednuclear families and performed a QTL linkage scan with 90%statistical power to detect a 10% QTL and around 50% power fora 5% QTL (28). We identified a common locus influencing bothparent and teacher ratings of ADHD symptoms that was signifi-cant at the genomewide level following simulation to determinethe empirical level of significance. Additionally, we identified
Figure 1. Genomewide QTL linkage test re-sults for the three ADHD measures. MultipointLOD scores were calculated with Merlin usinga regression based method for (A) the PN-score, (B) the TNscore, and (C) the mean scoreof the PNscore and the TNscore. ADHD, atten-tion-deficit/hyperactivity disorder; LOD, loga-rithm of odds; PNscore, standardized scoresfrom parents; QTL, quantitative trait loci;TNscore, standardized scores from teachers.
Table 2. Suggestive Linkage Results
Region Location (cM) PNscore TNscore Mean Score
1p36 45 2.7 2.4 3.29p23 27 2.2 .2 .811q21 100 .05 2.6 1.1
Multipoint LOD � 2.2.cM, centimorgan; LOD, logarithm of odds; PNscore, standardized scores
from parents; TNscore, standardized scores from teachers.
www.sobp.org/journal

tn
dactapaIbttsnFnbtqGAncags
ltattOmwossftstr
Qcctsmsfcisdtvged
574 BIOL PSYCHIATRY 2008;64:571–576 K. Zhou et al.
w
wo setting-specific loci that were suggestive of linkage but didot pass genomewide levels of significance.
The use of QTL approaches to map genes for ADHD has beeniscussed in the literature with no firm conclusions drawn on thedded value of this approach over the analysis of diagnosedases alone. We have previously discussed the potential value ofhis approach that is supported by available family and twin datand for the following reasons consider that QTL strategiesrovide an important complementary strategy (29). First, thepproach is supported by our quantitative genetic analyses of theMAGE sample family data, which shows a familial associationetween ADHD and quantitative trait measures of ADHD symp-om scores in siblings, with estimated proband-sibling correla-ions in the region of .3 (29). Furthermore, the sibling distributionshow no evidence of bimodality that might suggest some disconti-uity of the genetic influences between the probands and siblings.urthermore, since the diagnosis can only be made when theumber of symptoms exceeds a threshold, the dichotomous trait isy its very nature dimensional (2). Second, nearly all twin studieshat demonstrate high heritability for ADHD symptoms have useduantitative measurements in general population twin samples.roup heritability approaches that estimated the heritability ofDHD using various thresholds for the extreme group also provideo evidence of discontinuity of the genetic influences (29,30). Inontrast, however, the analysis of twin data using latent classpproaches has suggested that the genetic influences on extremeroups may not be the same as those that influence levels of ADHDymptoms throughout the general population (31).
Quantitative trait loci studies of ADHD therefore add to theiterature in two important ways. First, if the QTL model holdsrue, which states that different frequencies of the same risklleles will influence the level of ADHD symptoms throughouthe population, then we can expect a greatly increased power forhe detection of risk loci from the adoption of QTL strategies (26).n the other hand, the alternative finding that some genetic risksay be specific to the extreme group would also be importantith different predictions for the severity, course, and outcomef ADHD symptoms related, at least in part, to presence ofpecific genetic risk factors. The relative unpopularity of QTLtudies for molecular genetic studies of ADHD has arisen mainlyrom concerns over the lack of direct clinical relevance and uncer-ainty that the QTL model holds true (4). Furthermore, in the linkagetudies to date, little additional information has been obtained fromhe small number of QTL studies, which were mostly designed toeplicate the findings from dichotomous trait studies (3).
Our study was specifically designed to increase the power forTL approaches through the selection of extreme cases (ADHD
ombined type was recently estimated to occur in 2.34% of malehildren) (32). In the original design, we intended to select onlyhe most informative subset of extreme high- and low-scoringiblings for QTL linkage (33); however, the study design wasodified to include probands plus all of their siblings, since the
election process would have excluded many suitable familiesor association studies and made the ascertainment processonsiderably more complex. Furthermore our power calculationsndicated that we would need to screen many more proband-ibling pairs to achieve greater power using the selected siblingesign. The most significant common linkage locus detected inhis study on chromosome 1p36 explains 5.1% of the totalariance. Since the overall phenotypic variance from additiveenetic factors is around 76%, this suggests that many more QTLsxist of far smaller effect that were not detectable using the study
esign and sample size implemented in this study. In fact, this isww.sobp.org/journal
expected, since linkage has far less power to detect small tomoderate genetic effects than association, and recent studiesusing whole genome association approaches suggest that formost common complex disorders only small genetic effects willexist (34). The genes identified as being associated with ADHDso far confer odds ratios of around 2.2 to 1.4, which is equivalentto QTL effects of �1% assuming a simple additive genetic model(6). This partially explains why the three previous ADHD linkagescans that used between 100 to 300 sib pairs failed to detect anyof the QTLs reported in this study. It also suggests that only verylarge samples will have the required power to replicate a QTLfinding that contributes 5% to the total phenotypic variance usinglinkage approaches. However, this would not be the case if thelinkage finding were explained by a single common variant. Forexample, to replicate a 5% QTL in a threshold-selected case-control study, assuming a liability threshold model with casesselected two standard deviations above the mean and risk-increas-ing allele frequency of 50%, only 740 cases and control subjectswould be needed to obtain 80% statistical power at � � 10�7 (35).However, if the 5% QTL detected in our linkage scan is a cumulatedeffect of five variants, each accounting for only 1% of the pheno-typic variance, then the sample size needed to detect each of themwould be 3400 cases and control subjects (35).
A major question that we had to address in this study was thebest way to select suitable quantitative traits for QTL studies ofADHD. Although a large number of instruments are available,assessing ADHD as a quantitative trait for molecular geneticstudies remains challenging due to its developmental nature, sexdifferences, and rater specificity. The DSM-TOT score that hasbeen used in previous ADHD QTL analyses is not an idealmeasurement despite being the most comparable quantitativetrait to the DSM-IV categorical diagnosis. The symptom count isgenerally less informative than rating scales that show increasedvariance in the general population, and the proper adjustmentfor age and gender is not clearly established. In contrast, thestandardized scores of the Conners’ Rating Scales were devel-oped from a large population survey and are well-adjusted forthe covariates of age and gender to have uniform populationdistributions. These rating scales are therefore suitable for QTLstudies, since they can be informative when applied to eitherclinical or population samples or both.
Another question we needed to address was the number ofdifferent quantitative phenotypes to include in this analysis.There is evidence of both unique and common genetic effects onparent and teacher scores but also on hyperactivity versusinattention scores. Since our main objective was to determinewhether the QTL approach could identify loci related to thecombined type of ADHD, we decided to focus on the overlapbetween the measures, both in terms of symptom subtype andsituational pervasiveness. Furthermore, we did not wish toreduce our power further by inclusion of multiple differentphenotypic measures, so we decided to analyze inattention andhyperactivity as a single combined measure since estimates ofthe genetic correlation of around .6 suggest that the majority ofgenetic effects are shared between the two domains (36).
In contrast, when considering the combined inattention-hyperactivity/impulsivity scores from parent and teacher ratings,both the phenotypic and genetic correlations between the twosituation-specific measures were relatively low, suggesting thatthere may be substantial differences in the genes underlyinggenetic risk for the two situations (16). For this reason, weincluded the two scales as separate dimensions in addition to
taking the mean. By testing PNscore and TNscore individually,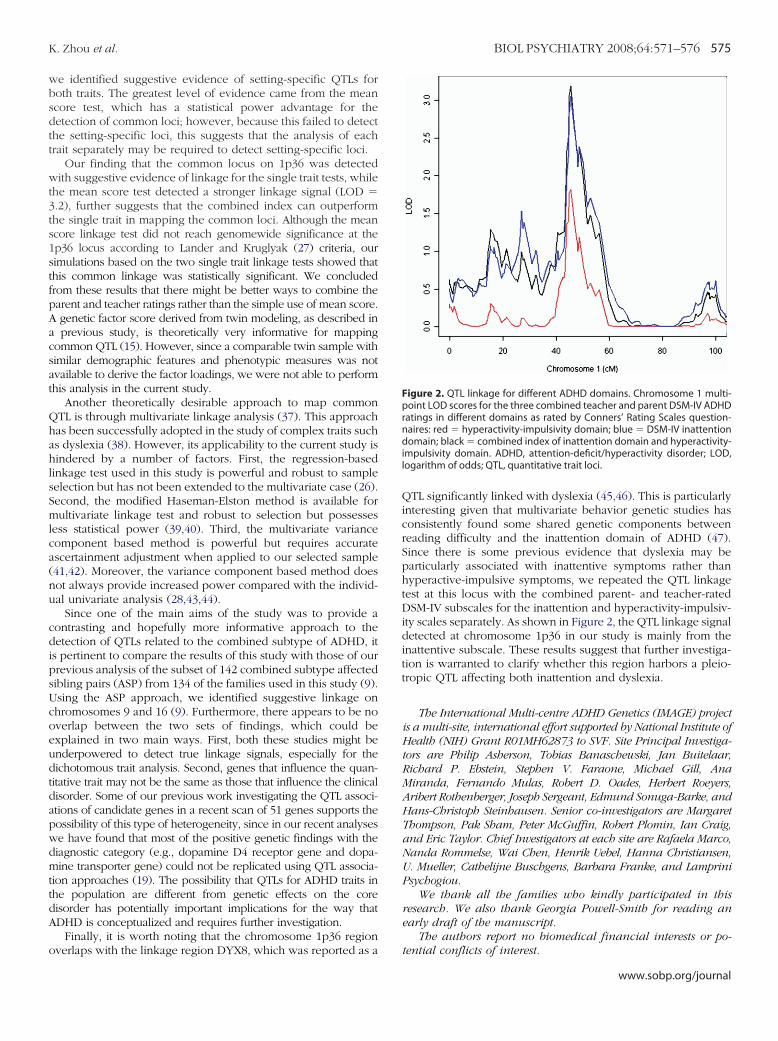
wbsdtt
wt3ts1stfpAacsat
QhahlsSmlca(nu
cdipsUcoeudtdapwdmttdA
o
K. Zhou et al. BIOL PSYCHIATRY 2008;64:571–576 575
e identified suggestive evidence of setting-specific QTLs foroth traits. The greatest level of evidence came from the meancore test, which has a statistical power advantage for theetection of common loci; however, because this failed to detecthe setting-specific loci, this suggests that the analysis of eachrait separately may be required to detect setting-specific loci.
Our finding that the common locus on 1p36 was detectedith suggestive evidence of linkage for the single trait tests, while
he mean score test detected a stronger linkage signal (LOD �.2), further suggests that the combined index can outperformhe single trait in mapping the common loci. Although the meancore linkage test did not reach genomewide significance at thep36 locus according to Lander and Kruglyak (27) criteria, ourimulations based on the two single trait linkage tests showed thathis common linkage was statistically significant. We concludedrom these results that there might be better ways to combine thearent and teacher ratings rather than the simple use of mean score.genetic factor score derived from twin modeling, as described inprevious study, is theoretically very informative for mapping
ommon QTL (15). However, since a comparable twin sample withimilar demographic features and phenotypic measures was notvailable to derive the factor loadings, we were not able to performhis analysis in the current study.
Another theoretically desirable approach to map commonTL is through multivariate linkage analysis (37). This approachas been successfully adopted in the study of complex traits suchs dyslexia (38). However, its applicability to the current study isindered by a number of factors. First, the regression-basedinkage test used in this study is powerful and robust to sampleelection but has not been extended to the multivariate case (26).econd, the modified Haseman-Elston method is available forultivariate linkage test and robust to selection but possesses
ess statistical power (39,40). Third, the multivariate varianceomponent based method is powerful but requires accuratescertainment adjustment when applied to our selected sample41,42). Moreover, the variance component based method doesot always provide increased power compared with the individ-al univariate analysis (28,43,44).
Since one of the main aims of the study was to provide aontrasting and hopefully more informative approach to theetection of QTLs related to the combined subtype of ADHD, its pertinent to compare the results of this study with those of ourrevious analysis of the subset of 142 combined subtype affectedibling pairs (ASP) from 134 of the families used in this study (9).sing the ASP approach, we identified suggestive linkage onhromosomes 9 and 16 (9). Furthermore, there appears to be noverlap between the two sets of findings, which could bexplained in two main ways. First, both these studies might benderpowered to detect true linkage signals, especially for theichotomous trait analysis. Second, genes that influence the quan-itative trait may not be the same as those that influence the clinicalisorder. Some of our previous work investigating the QTL associ-tions of candidate genes in a recent scan of 51 genes supports theossibility of this type of heterogeneity, since in our recent analysese have found that most of the positive genetic findings with theiagnostic category (e.g., dopamine D4 receptor gene and dopa-ine transporter gene) could not be replicated using QTL associa-
ion approaches (19). The possibility that QTLs for ADHD traits inhe population are different from genetic effects on the coreisorder has potentially important implications for the way thatDHD is conceptualized and requires further investigation.
Finally, it is worth noting that the chromosome 1p36 region
verlaps with the linkage region DYX8, which was reported as aQTL significantly linked with dyslexia (45,46). This is particularlyinteresting given that multivariate behavior genetic studies hasconsistently found some shared genetic components betweenreading difficulty and the inattention domain of ADHD (47).Since there is some previous evidence that dyslexia may beparticularly associated with inattentive symptoms rather thanhyperactive-impulsive symptoms, we repeated the QTL linkagetest at this locus with the combined parent- and teacher-ratedDSM-IV subscales for the inattention and hyperactivity-impulsiv-ity scales separately. As shown in Figure 2, the QTL linkage signaldetected at chromosome 1p36 in our study is mainly from theinattentive subscale. These results suggest that further investiga-tion is warranted to clarify whether this region harbors a pleio-tropic QTL affecting both inattention and dyslexia.
The International Multi-centre ADHD Genetics (IMAGE) projectis a multi-site, international effort supported by National Institute ofHealth (NIH) Grant R01MH62873 to SVF. Site Principal Investiga-tors are Philip Asherson, Tobias Banaschewski, Jan Buitelaar,Richard P. Ebstein, Stephen V. Faraone, Michael Gill, AnaMiranda, Fernando Mulas, Robert D. Oades, Herbert Roeyers,Aribert Rothenberger, Joseph Sergeant, Edmund Sonuga-Barke, andHans-Christoph Steinhausen. Senior co-investigators are MargaretThompson, Pak Sham, Peter McGuffin, Robert Plomin, Ian Craig,and Eric Taylor. Chief Investigators at each site are Rafaela Marco,Nanda Rommelse, Wai Chen, Henrik Uebel, Hanna Christiansen,U. Mueller, Cathelijne Buschgens, Barbara Franke, and LampriniPsychogiou.
We thank all the families who kindly participated in thisresearch. We also thank Georgia Powell-Smith for reading anearly draft of the manuscript.
The authors report no biomedical financial interests or po-
Figure 2. QTL linkage for different ADHD domains. Chromosome 1 multi-point LOD scores for the three combined teacher and parent DSM-IV ADHDratings in different domains as rated by Conners’ Rating Scales question-naires: red � hyperactivity-impulsivity domain; blue � DSM-IV inattentiondomain; black � combined index of inattention domain and hyperactivity-impulsivity domain. ADHD, attention-deficit/hyperactivity disorder; LOD,logarithm of odds; QTL, quantitative trait loci.
tential conflicts of interest.
www.sobp.org/journal

1
1
1
1
1
1
1
1
1
1
22
2
2
576 BIOL PSYCHIATRY 2008;64:571–576 K. Zhou et al.
w
1. Faraone SV, Sergeant J, Gillberg C, Biederman J (2003): The worldwideprevalence of ADHD: Is it an American condition? World Psychiatry2:104 –113.
2. Amercian Psychiatric Association (1994): Diagnostic and Statistical Man-ual of Mental Disorders, 4th ed. Washington, DC: American PsychiatricAssociation.
3. Asherson P (2004): Attention-deficit hyperactivity disorder in the post-genomic era. Eur Child Adolesc Psychiatry 13(suppl 1):I50 –I70.
4. Faraone SV, Perlis RH, Doyle AE, Smoller JW, Goralnick JJ, Holmgren MA,Sklar P (2005): Molecular genetics of attention-deficit/hyperactivity dis-order. Biol Psychiatry 57:1313–1323.
5. Li D, Sham PC, Owen MJ, He L (2006): Meta-analysis shows significantassociation between dopamine system genes and attention deficit hy-peractivity disorder (ADHD). Hum Mol Genet 15:2276 –2284.
6. Kuntsi J, Neale BM, Chen W, Faraone SV, Asherson P (2006): The IMAGEproject: Methodological issues for the molecular genetic analysis ofADHD. Behav Brain Funct 2:27.
7. Waldman ID, Gizer IR (2006): The genetics of attention deficit hyperac-tivity disorder. Clin Psychol Rev 26:396 – 432.
8. Arcos-Burgos M, Castellanos FX, Pineda D, Lopera F, Palacio JD, PalacioLG, et al. (2004): Attention-deficit/hyperactivity disorder in a populationisolate: Linkage to loci at 4q13.2, 5q33.3, 11q22, and 17p11. Am J HumGenet 75:998 –1014.
9. Asherson P, Zhou K, Anney RJ, Franke B, Buitelaar J, Ebstein R, et al.(2008): A high density SNP linkage scan with 142 combined subtypeADHD sib pairs identifies linkage regions on chromosomes 9 and 16[published online ahead of print January 8]. Mol Psychiatry.
0. Bakker SC, van der Meulen EM, Buitelaar JK, Sandkuijl LA, Pauls DL,Monsuur AJ, et al. (2003): A whole-genome scan in 164 Dutch sib pairswith attention-deficit/hyperactivity disorder: Suggestive evidence forlinkage on chromosomes 7p and 15q. Am J Hum Genet 72:1251–1260.
1. Faraone SV, Doyle AE, Lasky-Su J, Sklar PB, D’Angelo E, Gonzalez-Hey-drich J, et al. (2007): Linkage analysis of attention deficit hyperactivitydisorder [published online ahead of print December 14]. Am J Med GenetB Neuropsychiatr Genet.
2. Fisher SE, Francks C, McCracken JT, McGough JJ, Marlow AJ, MacPhie IL,et al. (2002): A genomewide scan for loci involved in attention-deficit/hyperactivity disorder. Am J Hum Genet 70:1183–1196.
3. Hebebrand J, Dempfle A, Saar K, Thiele H, Herpertz-Dahlmann B, LinderM, et al. (2006): A genome-wide scan for attention-deficit/hyperactivitydisorder in 155 German sib-pairs. Mol Psychiatry 11:196 –205.
4. Ogdie MN, Macphie IL, Minassian SL, Yang M, Fisher SE, Francks C, et al.(2003): A genomewide scan for attention-deficit/hyperactivity disorderin an extended sample: Suggestive linkage on 17p11. Am J Hum Genet72:1268 –1279.
5. Curran S, Rijsdijk F, Martin N, Marusic K, Asherson P, Taylor E, Sham P(2003): CHIP: Defining a dimension of the vulnerability to attentiondeficit hyperactivity disorder (ADHD) using sibling and individual dataof children in a community-based sample. Am J Med Genet B Neuropsy-chiatr Genet 119:86 –97.
6. Martin N, Scourfield J, McGuffin P (2002): Observer effects and heritabil-ity of childhood attention-deficit hyperactivity disorder symptoms. Br JPsychiatry 180:260 –265.
7. Hartman CA, Rhee SH, Willcutt EG, Pennington BF (2007): Modeling raterdisagreement for ADHD: Are parents or teachers biased? J Abnorm ChildPsychol 35:536 –542.
8. Thapar A, Harrington R, Ross K, McGuffin P (2000): Does the definition ofADHD affect heritability? J Am Acad Child Adolesc Psychiatry 39:1528–1536.
9. Brookes K, Xu X, Chen W, Zhou K, Neale B, Lowe N, et al. (2006): Theanalysis of 51 genes in DSM-IV combined type attention deficit hyper-activity disorder: Association signals in DRD4, DAT1 and 16 other genes.Mol Psychiatry 11:934 –953.
0. Conners CK (2003): Conners’ Rating Scales-Revised, 6th ed. Toronto: MHS.1. Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Rich-
ardsson B, et al. (2002): A high-resolution recombination map of thehuman genome. Nat Genet 31:241–247.
2. Epstein MP, Duren WL, Boehnke M (2000): Improved inference of rela-tionship for pairs of individuals. Am J Hum Genet 67:1219 –1231.
3. Abecasis GR, Wigginton JE (2005): Handling marker-marker linkage dis-
equilibrium: Pedigree analysis with clustered markers. Am J Hum Genet77:754 –767.ww.sobp.org/journal
24. O’Connell JR, Weeks DE (1998): PedCheck: A program for identificationof genotype incompatibilities in linkage analysis. Am J Hum Genet 63:259 –266.
25. Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2002): Merlin–rapidanalysis of dense genetic maps using sparse gene flow trees. Nat Genet30:97–101.
26. Sham PC, Purcell S, Cherny SS, and Abecasis GR (2002): Powerful regres-sion-based quantitative-trait linkage analysis of general pedigrees. Am JHum Genet 71:238 –253.
27. Lander E, Kruglyak L (1995): Genetic dissection of complex traits: Guidelinesfor interpreting and reporting linkage results. Nat Genet 11:241–247.
28. Sham PC, Zhao JH, Cherny SS, Hewitt JK (2000): Variance-componentsQTL linkage analysis of selected and non-normal samples: Conditioningon trait values. Genet Epidemiol 19:S22–S28.
29. Chen W, Zhou K, Sham P, Franke B, Kuntsi J, Campbell D, et al. (2008):DSM-IV combined type ADHD shows familial association with siblingtrait scores: A sampling strategy for QTL linkage [published onlineahead of print January 11]. Am J Med Genet B Neuropsychiatr Genet.
30. Thapar A, Holmes J, Poulton K, Harrington R (1999): Genetic basis ofattention deficit and hyperactivity. Br J Psychiatry 174:105–111.
31. Todd RD, Rasmussen ER, Neuman RJ, Reich W, Hudziak JJ, Bucholz KK,et al. (2001): Familiality and heritability of subtypes of attention deficithyperactivity disorder in a population sample of adolescent femaletwins. Am J Psychiatry 158:1891–1898.
32. Ford T, Goodman R, Meltzer H (2003): The British Child and AdolescentMental Health Survey 1999: The prevalence of DSM-IV disorders. J AmAcad Child Adolesc Psychiatry 42:1203–1211.
33. Purcell S, Cherny SS, Hewitt JK, Sham PC (2001): Optimal sibship selec-tion for genotyping in quantitative trait locus linkage analysis. HumHered 52:1–13.
34. Altshuler D, Daly M (2007): Guilt beyond a reasonable doubt. Nat Genet39:813– 815.
35. Purcell S, Cherny SS, Sham PC (2003): Genetic power calculator: Designof linkage and association genetic mapping studies of complex traits.Bioinformatics 19:149 –150.
36. McLoughlin G, Ronald A, Kuntsi J, Asherson P, Plomin R (2007): Geneticsupport for the dual nature of attention deficit hyperactivity disorder:Substantial genetic overlap between the inattentive and hyperactive-impulsive components. J Abnorm Child Psychol 35:999 –1008.
37. Boomsma DI, Dolan CV (1998): A comparison of power to detect a QTL insib-pair data using multivariate phenotypes, mean phenotypes, andfactor scores. Behav Genet 28:329 –340.
38. Marlow AJ, Fisher SE, Francks C, MacPhie IL, Cherny SS, RichardsonAJ, et al. (2003): Use of multivariate linkage analysis for dissection of acomplex cognitive trait. Am J Hum Genet 72:561–570.
39. Allison DB, Thiel B, St Jean P, Elston RC, Infante MC, Schork NJ (1998):Multiple phenotype modeling in gene-mapping studies of quantitativetraits: Power advantages. Am J Hum Genet 63:1190 –1201.
40. Amos CI, Elston RC, Bonney GE, Keats BJ, Berenson GS (1990): A multivariatemethod for detecting genetic linkage, with application to a pedigree withan adverse lipoprotein phenotype. Am J Hum Genet 47:247–254.
41. Amos C, de Andrade M, Zhu D (2001): Comparison of multivariate testsfor genetic linkage. Hum Hered 51:133–144.
42. Blangero J, Almasy L (1997): Multipoint oligogenic linkage analysis ofquantitative traits. Genet Epidemiol 14:959 –964.
43. Evans DM, Duffy DL (2004): A simulation study concerning the effect ofvarying the residual phenotypic correlation on the power of bivariatequantitative trait loci linkage analysis. Behav Genet 34:135–141.
44. Martin N, Boomsma D, Machin G (1997): A twin-pronged attack oncomplex traits. Nat Genet 17:387–392.
45. Tzenova J, Kaplan BJ, Petryshen TL, Field LL (2004): Confirmation of adyslexia susceptibility locus on chromosome 1p34-p36 in a set of 100Canadian families. Am J Med Genet B Neuropsychiatr Genet 127:117–124.
46. de Kovel C, Franke B, Hol FA, Lebrec J, Maassen B, Brunner H, et al. (2007):Confirmation of dyslexia susceptibility loci on chromosomes 1p and 2p,but not 6p in a Dutch sib-pair collection [published online ahead of printSeptember 20]. Am J Med Genet B Neuropsychiatr Genet.
47. Willcutt EG, Pennington BF, Olson RK, DeFries JC (2007): Understandingcomorbidity: A twin study of reading disability and attention-deficit/
hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet 144:709 –714.



















