
LaXp180, a mammalian ActA-binding protein, identified with the yeast two-hybrid system, co-localizes...
14
LaXp180, a mammalian ActA-binding protein, identified with the yeast two-hybrid system, co-localizes with intracellular Listeria monocytogenes Thilo Pfeuffer, 1 Werner Goebel, 1 Julia Laubinger, 2 Michael Bachmann 2² and Michael Kuhn 1 * 1 Lehrstuhl fu ¨ r Mikrobiologie, Theodor-Boveri-Institut fu ¨ r Biowissenschaften der Universita ¨t Wu ¨ rzburg, Am Hubland, 97074 Wu ¨ rzburg, Germany. 2 Institut fu ¨ r Physiologische Chemie der Universita ¨t Mainz, Duesbergweg 6, 55099 Mainz, Germany. Summary The Listeria monocytogenes surface protein ActA is an important virulence factor required for listerial intra- cellular movement by inducing actin polymerization. The only host cell protein known that directly interacts with ActA is the phosphoprotein VASP, which binds to the central proline-rich repeat region of ActA. To identify additional ActA-binding proteins, we applied the yeast two-hybrid system to search for mouse pro- teins that interact with ActA. A mouse cDNA library was screened for ActA-interacting proteins (AIPs) using ActA from strain L. monocytogenes EGD as bait. Three different AIPs were identified, one of which was identical to the human protein LaXp180 (also called CC1). Binding of LaXp180 to ActA was also demonstrated in vitro using recombinant histidine- tagged LaXp180 and recombinant ActA. Using an anti- LaXp180 antibody and fluorescence microscopy, we showed that LaXp180 co-localizes with a subset of intracellular, ActA-expressing L. monocytogenes but was never detected on intracellularly growing but ActA-deficient mutants. Furthermore, LaXp180 bind- ing to intracellular L. monocytogenes was asymmetri- cal and mutually exclusive with F-actin polymerization on the bacterial surface. LaXp180 is a putative bind- ing partner of stathmin, a protein involved in signal transduction pathways and in the regulation of micro- tubule dynamics. Using immunofluorescence, we showed that stathmin co-localizes with intracellular ActA-expressing L. monocytogenes. Introduction The Gram-positive facultative intracellular bacterial patho- gen Listeria monocytogenes encounters different cell types during the course of infection. A number of listerial virulence determinants involved in the intracellular life cycle of L. monocytogenes have been characterized in recent years (reviewed by Kuhn and Goebel, 1995). Intracellular movement of L. monocytogenes inside the host cell cytoplasm, as well as intercellular spread, is mediated by actin polymerization as initially described by Tilney and Portnoy (1989) and Mounier et al . (1990). The cell biology and molecular biology of listerial intracellular movement has been reviewed recently (Ireton and Cossart, 1997; Cossart and Lecuit, 1998). The L. monocytogenes gene actA codes for a proline-rich protein (ActA) of 639 amino acids. Its apparent molecular weight determined by SDS–PAGE is 92 kDa. ActA is a surface protein con- sisting of three distinct parts: the N-terminal part with the transport signal sequence; the central proline-rich repeat region; and the C-terminal region, which includes a mem- brane anchor. Mutations in the actA gene resulted in the loss of virulence in mice, in the lack of intracellular actin polymerization around the bacteria and in the inabil- ity of intracellular movement. Inside the host cells, the actA mutant forms microcolonies that are located near the nucleus (Domann et al ., 1992; Kocks et al ., 1992; Vazquez-Boland et al ., 1992). Whether the ActA protein is distributed asymmetrically on the surface of L. monocytogenes is still under debate. Using immunofluorescence microscopy, data have been presented showing that ActA is not present at the new bacterial pole after cell division, but seems to be concen- trated at the old pole (Kocks et al ., 1993). These results were, however, questioned by independent investigations demonstrating the presence of ActA throughout the sur- face of the bacteria (Niebuhr et al ., 1993). An asymmetric distribution of the ActA protein was shown to be required and sufficient to direct actin-based motility by coating strep- tococci asymmetrically with genetically engineered ActA protein. In a cell-free system, these streptococci, but not uniformly coated ones, moved efficiently in cytoplasmic extracts (Smith et al ., 1995). Deletion of the N-terminal domain of ActA is followed by a total abolishment of actin polymerization and intracellular Cellular Microbiology (2000) 2(2), 101–114 Q 2000 Blackwell Science Ltd Received 5 July, 1999; revised 12 November, 1999; accepted 24 November, 1999. ²Present address: Oklahoma Medical Research Foundation, University of Oklahoma Health Sciences Center, Okla- homa City, USA. *For correspondence. E-mail kuhn@biozentrum. uni-wuerzburg.de; Tel. (49) 931 8884421; Fax ( 49) 931 8884402.
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of LaXp180, a mammalian ActA-binding protein, identified with the yeast two-hybrid system, co-localizes...

LaXp180, a mammalian ActA-binding protein,identi®ed with the yeast two-hybrid system,co-localizes with intracellular Listeria monocytogenes
Thilo Pfeuffer,1 Werner Goebel,1 Julia Laubinger,2
Michael Bachmann2² and Michael Kuhn1*1Lehrstuhl fuÈr Mikrobiologie, Theodor-Boveri-Institut
fuÈr Biowissenschaften der UniversitaÈ t WuÈrzburg,
Am Hubland, 97074 WuÈrzburg, Germany.2Institut fuÈr Physiologische Chemie der UniversitaÈ t Mainz,
Duesbergweg 6, 55099 Mainz, Germany.
Summary
The Listeria monocytogenes surface protein ActA is
an important virulence factor required for listerial intra-
cellular movement by inducing actin polymerization.
The only host cell protein known that directly interacts
with ActA is the phosphoprotein VASP, which binds
to the central proline-rich repeat region of ActA. To
identify additional ActA-binding proteins, we applied
the yeast two-hybrid system to search for mouse pro-
teins that interact with ActA. A mouse cDNA library
was screened for ActA-interacting proteins (AIPs)
using ActA from strain L. monocytogenes EGD as bait.
Three different AIPs were identi®ed, one of which
was identical to the human protein LaXp180 (also
called CC1). Binding of LaXp180 to ActA was also
demonstrated in vitro using recombinant histidine-
tagged LaXp180 and recombinant ActA. Using an anti-
LaXp180 antibody and ¯uorescence microscopy, we
showed that LaXp180 co-localizes with a subset of
intracellular, ActA-expressing L. monocytogenes but
was never detected on intracellularly growing but
ActA-de®cient mutants. Furthermore, LaXp180 bind-
ing to intracellular L. monocytogenes was asymmetri-
cal and mutually exclusive with F-actin polymerization
on the bacterial surface. LaXp180 is a putative bind-
ing partner of stathmin, a protein involved in signal
transduction pathways and in the regulation of micro-
tubule dynamics. Using immuno¯uorescence, we
showed that stathmin co-localizes with intracellular
ActA-expressing L. monocytogenes.
Introduction
The Gram-positive facultative intracellular bacterial patho-
gen Listeria monocytogenes encounters different cell types
during the course of infection. A number of listerial virulence
determinants involved in the intracellular life cycle of L.
monocytogenes have been characterized in recent years
(reviewed by Kuhn and Goebel, 1995).
Intracellular movement of L. monocytogenes inside the
host cell cytoplasm, as well as intercellular spread, is
mediated by actin polymerization as initially described by
Tilney and Portnoy (1989) and Mounier et al. (1990). The
cell biology and molecular biology of listerial intracellular
movement has been reviewed recently (Ireton and Cossart,
1997; Cossart and Lecuit, 1998). The L. monocytogenes
gene actA codes for a proline-rich protein (ActA) of 639
amino acids. Its apparent molecular weight determined
by SDS±PAGE is 92 kDa. ActA is a surface protein con-
sisting of three distinct parts: the N-terminal part with the
transport signal sequence; the central proline-rich repeat
region; and the C-terminal region, which includes a mem-
brane anchor. Mutations in the actA gene resulted in
the loss of virulence in mice, in the lack of intracellular
actin polymerization around the bacteria and in the inabil-
ity of intracellular movement. Inside the host cells, the
actA mutant forms microcolonies that are located near
the nucleus (Domann et al., 1992; Kocks et al., 1992;
Vazquez-Boland et al., 1992).
Whether the ActA protein is distributed asymmetrically
on the surface of L. monocytogenes is still under debate.
Using immuno¯uorescence microscopy, data have been
presented showing that ActA is not present at the new
bacterial pole after cell division, but seems to be concen-
trated at the old pole (Kocks et al., 1993). These results
were, however, questioned by independent investigations
demonstrating the presence of ActA throughout the sur-
face of the bacteria (Niebuhr et al., 1993). An asymmetric
distribution of the ActA protein was shown to be required
and suf®cient to direct actin-based motility by coating strep-
tococci asymmetrically with genetically engineered ActA
protein. In a cell-free system, these streptococci, but not
uniformly coated ones, moved ef®ciently in cytoplasmic
extracts (Smith et al., 1995).
Deletion of the N-terminal domain of ActA is followed by
a total abolishment of actin polymerization and intracellular
Cellular Microbiology (2000) 2(2), 101±114
Q 2000 Blackwell Science Ltd
Received 5 July, 1999; revised 12 November, 1999; accepted 24November, 1999. ²Present address: Oklahoma Medical ResearchFoundation, University of Oklahoma Health Sciences Center, Okla-homa City, USA. *For correspondence. E-mail [email protected]; Tel. (�49) 931 8884421; Fax (�49) 931 8884402.

movement. In contrast, deletion of neither the proline-rich
repeat domain nor the C-terminal domain completely inhi-
bits actin assembly. However, the actin tails produced by
L. monocytogenes strains expressing ActA without the pro-
line-rich repeats were signi®cantly shorter, pointing to a
stimulatory function of this region (Lasa et al., 1995; Pistor
et al., 1995; Smith et al., 1996). Among the several actin-
binding proteins localized to the actin tails, a-actinin, the
Arp2/3 complex, capZ, co®lin, coronin, Evl, ezrin/radixin,
®mbrin, Mena, pro®lin, Rac, talin, tropomyosin, VASP, villin
and vinculin (reviewed by Cossart and Lecuit, 1998; David
et al., 1998), only pro®lin, Mena and the vasodilator-stimu-
lated phosphoprotein (VASP) are associated with the sur-
face of the moving bacteria and co-localize with ActA.
VASP, recently shown to bind directly to the proline-rich
repeats of ActA (Chakraborty et al., 1995) and being a nat-
ural ligand of pro®lin (Reinhard et al., 1995), could stimulate
actin assembly by binding to ActA and enhancing the pro®-
lin concentration in the vicinity of the bacterium. VASP also
interacts with ®lamentous actin, thereby linking the bac-
terium to the actin tail (Laurent et al., 1999). The recently
described Arp2/3 complex consisting of seven host cell
proteins found in the actin tails of moving bacteria obviously
represents the host cell actin polymerization machinery
(Welch et al., 1997; 1998). The pure complex is suf®cient
to initiate ActA-dependent actin polymerization on the sur-
face of L. monocytogenes in a cell-free system and is
thought to interact at least transiently with ActA.
The ActA protein is phosphorylated inside the host cells,
which yields three distinct forms of this protein with slightly
different sizes in SDS±PAGE (Brundage et al., 1993).
However, a genetically engineered ActA variant that is
fully functional but lacking the C-terminal region no longer
shows mobility associated with phosphorylation, suggest-
ing that phosphorylation (at least in the C-terminal region)
may not be necessary for movement (Lasa et al., 1995).
ActA functions not only as an inducer of actin polymer-
ization. It was also shown to contribute to the invasion of
epithelial cells and macrophages, probably through bind-
ing to heparan sulphate proteoglycan receptors (Alvarez-
Dominguez et al., 1997). Furthermore, ActA seems to in¯u-
ence host cell signal transduction pathways, as an actA
deletion mutant of L. monocytogenes is partially impaired
in triggering the persistent activation of the mammalian
transcription factor NF-kB (Hauf et al., 1997).
These many different roles of ActA in the intracellular life
cycle of L. monocytogenes are in contrast to our limited
knowledge of putative mammalian proteins interacting with
ActA. We therefore used the yeast two-hybrid system and
ActA as a bait to try to identify new ActA-interacting pro-
teins (AIPs). The yeast two-hybrid system identi®es inter-
acting proteins through the functional restoration of the
yeast GAL4 transcriptional activator in vivo (Fields and
Song, 1989). Although this system has been applied widely
in identifying many types of interacting proteins, it has only
been used once successfully to screen a mammalian cDNA
library for the binding partner of a bacterial protein, namely
the Neisseria gonorrhoeae Opa protein (Williams et al.,
1998).
Results
Isolation of plasmids encoding putative AIPs
The actA gene of L. monocytogenes EGD lacking the
N-terminal signal sequence and the C-terminal membrane
anchor (Domann et al., 1992) was cloned into the GAL4
DNA-binding domain (DB) plasmid pPC97 (Kalmes et al.,
1998) to create pPC97-ActA. The expression of the DB-
ActA fusion protein from pPC97-ActA in the yeast strains
Saccharomyces cerevisiae HF7c and SFY526 was ana-
lysed in a Western blot with protein extracts from both
strains and an anti-GAL4 DB antibody. A protein of the
expected molecular weight is heavily expressed in both
strains harbouring the plasmid pPC97-ActA (Fig. 1). When
pPC97-ActA was transformed into either reporter strain,
it did not result in reporter gene (HIS3 and lacZ ) activation
(data not shown). Thus, pPC97-ActA was used as bait to
screen the mouse embryonic cDNA library pPC67 (Chev-
ray and Nathans, 1992) that was created in the GAL4
transactivation domain (TA) plasmid pPC86.
About 3.3 ´ 106 clones representing <1.9 ´ 106 indepen-
dent cDNAs were screened for ActA-interacting proteins
(AIPs) in S. cerevisiae HF7c. A total of 89 HF7c yeast colo-
nies appeared between 2 and 7 days after transformation.
Of the 89 colonies, 25 (28.1%) had both reporter genes
Q 2000 Blackwell Science Ltd, Cellular Microbiology, 2, 101±114
Fig. 1. Expression of the DB±ActA fusion protein in S. cerevisiae.Protein extracts from S. cerevisiae SFY526 (lanes 1, 3 and 5) orS. cerevisiae HF7c (lanes 2, 4 and 6) harbouring either theDB-ActA expression plasmid pPC97-ActA (lanes 5 and 6) or thecontrol plasmid pVA3 expressing the DB±p53 hybrid protein (lanes1 and 2) or from non-transformed control cells (lanes 3 and 4)were separated on SDS±PAGE, blotted onto nitrocellulose anddeveloped with an anti-GAL4DB antiserum. The DB±p53 fusionprotein is marked with an open arrowhead, and the DB±ActAfusion protein with a ®lled arrowhead.
102 T. Pfeuffer et al.

(HIS3 and lacZ ) activated, possessing mouse embryonic
cDNAs coding for putative AIPs. The 25 pPC67-cDNA plas-
mids were isolated, digested and found to contain inserts
ranging from 30 bp to 2500 bp (data not shown). All plas-
mids were individually retransformed into both yeast strains
HF7c and SFY526 containing pPC97-ActA and, upon this
procedure, only four clones were found to express the lacZ
gene. Such a decrease in positive clones upon retransfor-
mation has also been observed in other yeast two-hybrid
screens. To con®rm that each of the four pPC67 plasmids
(called pPC67-15, pPC67-18, pPC67-37 and pPC67-70)
coded for proteins that interact speci®cally with ActA, they
were retransformed into the yeast reporter strain SFY526
alone and together with the appropriate control plasmid
pPC97. The different pPC67-cDNAs alone or when co-
transformed with pPC97 did not result in the activation of
the lacZ reporter gene (data not shown). Each pPC67-
cDNA resulted in the activation of the lacZ reporter gene
only in combination with pPC97-ActA. These results con-
®rm that each pPC67-cDNA codes for a protein or portion
of a protein that interacts with ActA in vivo.
Characteristics and identity of the AIPs
The inserts from each of the four pPC67-cDNA plasmids
were sequenced either totally (pPC67-15) or partially until
reaching a stop codon (pPC67-18, pPC67-37 and pPC67-
70). The nucleotide and corresponding peptide data are
summarized in Table 1. Each AIP is encoded by an incom-
plete cDNA, coding for the C-terminus of the protein. The
isolation of partial cDNAs is a frequent ®nding in two-hybrid
screens and re¯ects either the initial cloning of incomplete
cDNAs or the fact that only these partial cDNAs result in
GAL4 TA fusions that are in the correct translational read-
ing frame. The deduced fusion proteins encoded by the
inserts of pPC67-18 (2 kb) and pPC67-70 (1.65 kb) are
identical and code for 260 amino acids fused to GAL4 TA
showing homology to mouse-expressed sequence tags
and human clone NH0364H22. pPC67-37 codes for 275
amino acids fused to GAL4 TA, which are 71.9% identical
(on the DNA level) to the human KIAA0646 sequence.
However, there are no functional data available for any of
these mentioned genes or putative proteins.
pPC67-15 harbours a 927 bp insert that codes for 227
amino acids fused to GAL4 TA. The DNA sequence corre-
sponding to the fused peptide is highly homologous (86.2%
identity) with part of a human gene coding for an `unknown
human autoantigen' (LaxP180) (TroÈster et al., 1994; M.
Bachmann and J. Laubinger, unpublished) or KIAA0203
(Nagase et al., 1996) (Fig. 2). No function has yet been
demonstrated for these proteins, but a different part of
Q 2000 Blackwell Science Ltd, Cellular Microbiology, 2, 101±114
Table 1. Summary of AIPs.Size Accession numbers
Clone Insert Peptide Homology AIP Homologues
pPC67-15 927 227 KIAA0203 AJ242720 D86958LaXp180 Z35085
pPC67-37 2500 275 KIAA0646 AJ242721 AB014546pPC67-18 2000 260 EST AJ242722 D19250
EST W62537NH0364H22 AC005036
pPC67-70 1650 260 EST AJ242722 D19250EST W62537NH0364H22 AC005036
Fig. 2. A. Amino acid sequence homology between pPC67-15(middle), LaXp180 (top) and KIAA0203 (bottom).B: Schematic representation of published sequences (KIAA0203and LaXp180) with homology to the insert of pPC67-15. CC1 doesnot overlap with the insert found in pPC67-15, but overlaps withKIAA0203 and LaXp180. Numbers refer to the amino acidnumbering in the KIAA0203 sequence.
LaXp180, a new mammalian ActA-binding protein 103

the sequence was identi®ed earlier in a two-hybrid screen
with the phosphoprotein stathmin as bait (Maucuer et al.,
1995). This protein was called CC1 (accession no. X82318)
and shown to code for a coiled coil-forming protein (Fig.
2). As stathmin is a putative regulator in many mammalian
signal transduction pathways (Ozon et al., 1997), interac-
tion of LaXp180 (CC1) with ActA and stathmin might link
intracellular L. monocytogenes to host cell signal trans-
duction pathways. The amino acid sequences of the pro-
teins fused to GAL4 TA in the plasmids pPC67-15,
pPC67-18 and pPC67-37 did not contain any obvious
motifs or any similarity with each other.
Interaction of ActA variants with the C-terminal part
of LaXp180 encoded in pPC67-15
To identify the region of ActA needed for the interaction
with LaXp180, we performed YTH experiments with yeast
strains expressing different ActA variants (Fig. 3) and the
C-terminal part of LaXp180 encoded as a TA fusion protein
in pPC67-15. As shown in Table 2, neither the N-terminal
domain (NActA) nor the C-terminal domain (CActA) of
ActA interacted with LaXp180. In contrast, fusion of the
proline-rich repeat region (PActA) to the DB domain of
GAL4 also resulted in LacZ expression, even in the absence
of LaXp180. Such a false-positive result with the proline-
rich region has also been reported by others (Mourrain
et al., 1997). However, expression of either the N-terminal
domain plus the proline-rich region (NPActA) or the C-ter-
minal domain plus the proline-rich region (PCActA) gave
positive results (Table 2), showing that these ActA variants
interacted with LaXp180 in a highly speci®c (PCActA)
or reasonably speci®c (NPActA) manner. These results
underline the importance of the proline-rich region for
ef®cient interaction, but ¯anking sequences are required
for speci®city. However, even upon deletion of the middle
region of ActA harbouring the proline-rich repeats
(DPActA), a weak but reproducible and speci®c expression
of lacZ was observed in the yeast strains, pointing to a
weak interaction of the DPActA variant with LaXp180.
ActA binds LaXp180 in vitro
To con®rm the results of yeast two-hybrid assays that lister-
ial ActA binds mammalian LaXp180, we investigated the
ability of ActA to bind the C-terminal part of LaXp180 in
vitro. A crude cell extract from Escherichia coli pQHS30,
containing large amounts of (His)6-tagged LaXp180, was
loaded onto a nickel-agarose column and washed exten-
sively. The column was then loaded with a crude extract
from E. coli pCYB4-ActA, washed extensively, and the
proteins were eluted with an imidazole gradient. Protein
fractions were separated by SDS±PAGE, and ActA and
LaXp180 were detected with the respective antisera (Fig.
4). ActA eluted from the column in large quantities in the
same fractions as LaXp180, indicating that ActA was
bound to LaXp180 (Fig. 4, top left). The listerial extracellu-
lar protein InlC (Engelbrecht et al., 1996) does not interact
with ActA in a yeast two-hybrid assay (data not shown).
We therefore used an N-terminal histidine-tagged InlC pro-
tein (F. Engelbrecht, S. Gholami and W. Goebel, unpub-
lished results) as a control. It was loaded onto the column
before adding recombinant ActA. As shown in Fig. 4 (top
right), elution with imidazole yielded only traces of ActA
in fractions separated from those harbouring the InlC pro-
tein. The addition of ActA to the column without prior load-
ing with any His-tagged protein resulted in binding of some
ActA. However, the ActA protein eluted from the column in
a strikingly different pattern and in much lower amounts
than from columns preloaded with His-tagged LaXp180
(data not shown). Taken together, these data suggest
that recombinant LaXp180 speci®cally binds recombinant
ActA and therefore support the ®ndings with the yeast
two-hybrid system.
LaXp180 is expressed in different mammalian cell lines
and co-localizes with L. monocytogenes in infected cells
LaXp180 expression in different human (Caco-2 epithelial
cells), monkey (Cos-1 ®broblasts) and mouse (P388D1-
macrophage-like cells, bone marrow-derived macrophages)
Q 2000 Blackwell Science Ltd, Cellular Microbiology, 2, 101±114
30 613263 423
190 470
ActA
NActA
PActA
CActA
NPActA
PCActA
∆PActA
264 429
∆
Fig. 3. Schematic representation of the ActA protein and thevariants fused to the GAL4 DB domain in pPC97.
Table 2. Results of b-galactosidase assaystesting the interaction of variants of ActAexpressed as DB fusion proteins with LaXp180encoded as a TA fusion protein by pPC67-15 inS. cerevisiae SFY526 (third row).
ActA NActA PActA CActA NPActA PCActA DPActA
ÿ ÿ ÿ ÿ ÿ ÿ ÿ ÿ
pPC86 ÿ ÿ ��� ÿ � ÿ ÿ
pPC67-15 ��� ÿ ��� ÿ ��� ��� �
As controls, strains harbouring only the vector (second row) or without plasmid (first row) areincluded.
104 T. Pfeuffer et al.

cell lines was investigated by reverse transcriptase±poly-
merase chain reaction (RT±PCR). RNA was isolated from
L. monocytogenes-infected and non-infected cells, reverse
transcribed and ampli®ed with appropriate primer pairs as
described (Kuhn and Goebel, 1994). As shown in Fig. 5,
LaXp180-speci®c mRNA was detected in all cell lines ana-
lysed. As a control, b-actin-speci®c RT±PCRs were also
included. Infection of these cells with L. monocytogenes
for 4 h did not result in any measurable change in the
LaXp180 mRNA concentrations (Fig. 5). Furthermore, a
single protein of 194 kDa was immunoprecipitated from
Cos-1 cells with the anti-LaXp180 antiserum, but not with
the preimmune serum (Fig. 6). The amount of LaXp180
protein present in Cos-1 cells is obviously low, as the pro-
tein was not detected in immunoblots with total-cell lysates
without prior immunoprecipitation. The size of the immuno-
precipitated protein is in good agreement with the theore-
tical molecular weight (182.8 kDa) of the gene product
predicted for the ORF KIAA0203 (Nagase et al., 1996).
The intracellular distribution of LaXp180 in Cos-1 and
Caco-2 cells was analysed using immuno¯uorescence
with the preadsorbed polyclonal anti-LaXp180 antiserum
and LRSC-labelled secondary antibodies. At 4 after infec-
tion, the cells were ®xed and stained as described
Q 2000 Blackwell Science Ltd, Cellular Microbiology, 2, 101±114
Fig. 4. In vitro binding of ActA to LaXp180.Left. Elution pro®les of the C-terminal, His-tagged fragment of LaXp180 (middle) and recombinant ActA (top) from a nickel-agarose columnloaded ®rst with crude protein extracts containing LaXp180 and, second with ActA. Both proteins elute simultaneously upon adding increasingconcentrations of imidazole.Right. Elution pro®les of the His-tagged InlC (middle) and recombinant ActA (top) from a nickel-agarose column loaded ®rst with crude proteinextracts containing InlC and second with ActA. ActA does not bind ef®ciently to the column and does not co-elute with InlC.Bottom left. Quantitative analysis of the elution pro®les: (ÐÐ) ActA; (± ±) LaXp180.Bottom right. (ÐÐ) ActA; (± ±) InlC. Left scale: arti®cial units for eluted ActA (absolute values). Right scale: relative arti®cial units forLaXp180 (left) or InlC (right) given as a percentage of the highest intensity.
LaXp180, a new mammalian ActA-binding protein 105
previously (Kuhn et al., 1990). The antiserum labels the
nucleus, but only moderately labels the cytoplasm of the
cells, suggesting that the protein recognized is mainly
found in association with the nucleus. Upon infection, a
varying number of intracellular L. monocytogenes are
labelled with the anti-LaXp180 antiserum, demonstrating
that LaXp180 co-localizes with intracellular L. monocyto-
genes, most probably through binding to ActA (Fig. 7A
and B). In contrast, direct labelling of broth-grown L. mono-
cytogenes could not be achieved (data not shown). It is,
however, important to note that all the intracellular bac-
teria were never labelled by the anti-LaXp180 antiserum.
The co-localization of LaXp180 with intracellular L. mono-
cytogenes is, however, highly speci®c for the expression
of the actA gene by L. monocytogenes, as a mutant lack-
ing the entire actA gene grows intracellularly but was
never found to be labelled by the treatment with anti-
LaXp180 antiserum (Fig. 7E and F) and, as expected, was
never associated with ®lamentous actin (Fig. 7G and H).
Careful analysis of the ¯uorescence images also revealed
that the anti-LaXp180 antiserum labelled the bacteria
asymmetrically, with heavy labelling at only one pole
(Fig. 7B and Fig. 8, second row). Furthermore, double
immuno¯uorescence with the anti-LaXp180 antibody
(LRSC-labelled secondary antibody) and with ¯uorescein
isothiocyanate (FITC)-phalloidin to label ®lamentous actin
(Fig. 7C and D) showed that LaXp180 binding and ®lamen-
tous actin accumulation on the bacterial surface are largely
mutually exclusive: in most cases, one pole appeared red
as a result of the anti-LaXp180 label and the opposite pole
green because of the FITC-phalloidin label, as shown in
Fig. 7C and D and Fig. 8 (third and fourth rows). Some-
times, both labels overlap in the middle region of the bac-
teria, resulting in a yellow appearance. Dividing bacteria
Q 2000 Blackwell Science Ltd, Cellular Microbiology, 2, 101±114
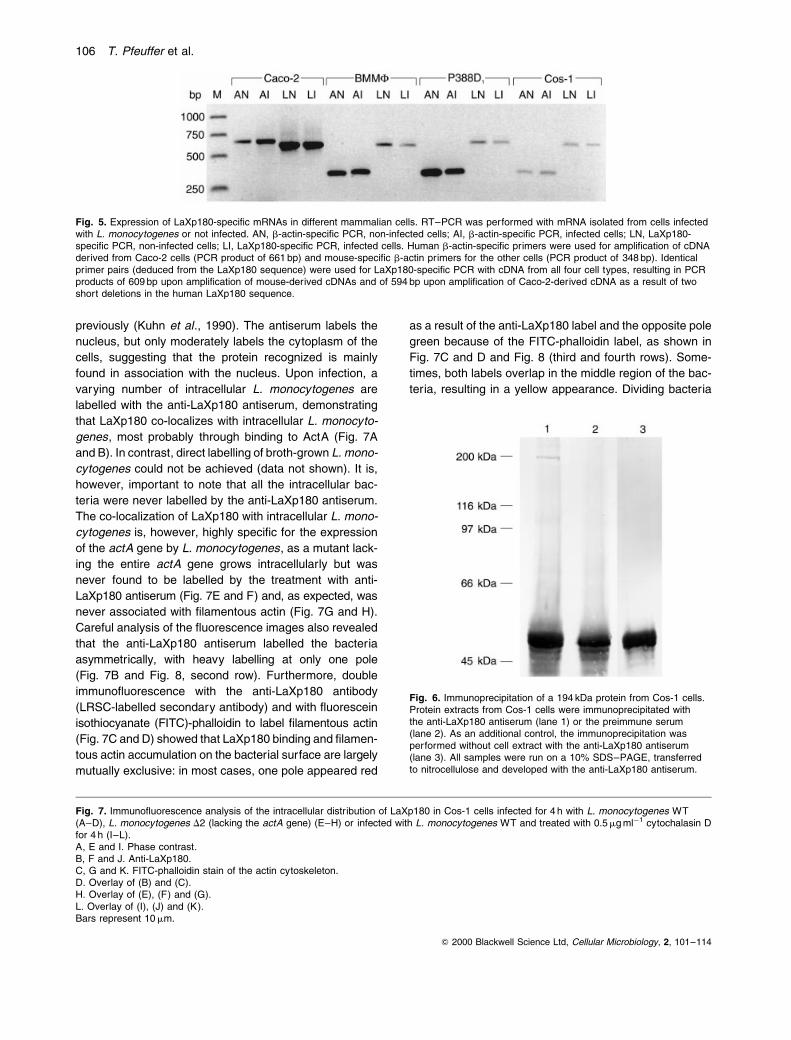
Fig. 5. Expression of LaXp180-speci®c mRNAs in different mammalian cells. RT±PCR was performed with mRNA isolated from cells infectedwith L. monocytogenes or not infected. AN, b-actin-speci®c PCR, non-infected cells; AI, b-actin-speci®c PCR, infected cells; LN, LaXp180-speci®c PCR, non-infected cells; LI, LaXp180-speci®c PCR, infected cells. Human b-actin-speci®c primers were used for ampli®cation of cDNAderived from Caco-2 cells (PCR product of 661 bp) and mouse-speci®c b-actin primers for the other cells (PCR product of 348 bp). Identicalprimer pairs (deduced from the LaXp180 sequence) were used for LaXp180-speci®c PCR with cDNA from all four cell types, resulting in PCRproducts of 609 bp upon ampli®cation of mouse-derived cDNAs and of 594 bp upon ampli®cation of Caco-2-derived cDNA as a result of twoshort deletions in the human LaXp180 sequence.
Fig. 6. Immunoprecipitation of a 194 kDa protein from Cos-1 cells.Protein extracts from Cos-1 cells were immunoprecipitated withthe anti-LaXp180 antiserum (lane 1) or the preimmune serum(lane 2). As an additional control, the immunoprecipitation wasperformed without cell extract with the anti-LaXp180 antiserum(lane 3). All samples were run on a 10% SDS±PAGE, transferredto nitrocellulose and developed with the anti-LaXp180 antiserum.
Fig. 7. Immuno¯uorescence analysis of the intracellular distribution of LaXp180 in Cos-1 cells infected for 4 h with L. monocytogenes WT(A±D), L. monocytogenes D2 (lacking the actA gene) (E±H) or infected with L. monocytogenes WT and treated with 0.5 mg mlÿ1 cytochalasin Dfor 4 h (I±L).A, E and I. Phase contrast.B, F and J. Anti-LaXp180.C, G and K. FITC-phalloidin stain of the actin cytoskeleton.D. Overlay of (B) and (C).H. Overlay of (E), (F) and (G).L. Overlay of (I), (J) and (K).Bars represent 10 mm.
106 T. Pfeuffer et al.

are labelled red in the middle region where the cells sepa-
rate and have actin ®laments (green label) present at the
old poles (Fig. 8). To exclude the possibility that the polar
recruitment of LaXp180 results from steric exclusion by
the actin cloud, we analysed LaXp180 recruitment after
cytochalasin D treatment. As shown in Fig. 7I±L, cyto-
chalasin treatment of L. monocytogenes-infected cells
resulted in microcolony formation and inhibition of ®lamen-
tous actin accumulation, as expected. Nevertheless,
LaXp180 recruitment was still clearly asymmetrical, as
Q 2000 Blackwell Science Ltd, Cellular Microbiology, 2, 101±114
LaXp180, a new mammalian ActA-binding protein 107

only one pole of the bacteria was labelled by the anti-
LaXp180 antiserum.
Stathmin co-localizes with L. monocytogenes in
infected cells
The recent identi®cation of a part of LaXp180 (called CC1)
as a stathmin-interacting protein (Maucuer et al., 1995)
prompted us to investigate whether stathmin also co-local-
izes with intracellular L. monocytogenes. At 4 h after infec-
tion, infected Cos-1 cells were stained with af®nity-puri®ed
polyclonal antistathmin antibodies (Koppel et al., 1990).
In non-infected cells, the antiserum mainly labelled the
nucleus and partially the cytoplasm with bright small
spots (data not shown), as described earlier (Gavet et
al., 1998). Upon infection, intracellular L. monocytogenes
were labelled with the antiserum, demonstrating that stath-
min co-localizes with intracellular listeriae, most probably
through interaction with surface-bound LaXp180 (Fig.
9A±C). As already described for the LaXp180 staining,
not all intracellular bacteria were labelled but, again, the
Q 2000 Blackwell Science Ltd, Cellular Microbiology, 2, 101±114
Fig. 8. Details from Fig. 7A±D showing the mutual exclusion ofLaXp180 binding to L. monocytogenes and F-actin polymerizationon the surface of the bacteria.First row. Phase contrast.Second row. Anti-LaXp180.Third row. FITC-phalloidin.Fourth row. Overlays.
Fig. 9. Immuno¯uorescence analysis of the intracellular distribution of stathmin in Cos-1 cells infected for 4 h with L. monocytogenes WT(A±C) or L. monocytogenes D2 (lacking the actA gene) (D and E).A and D. Phase contrast.B and E. Antistathmin.C. Overlay of (A) and (B). The arrowhead in (C) points to a bacterium that was not labelled by the antistathmin antiserum. Inserts showindividual bacteria at higher magni®cation.F and G. Phase contrast (F) and immuno¯uorescence (G) of L. monocytogenes WT-infected Cos-1 cells stained with antistathmin antiserum,which was preincubated with the antigenic peptide. Bars represent 10 mm.
108 T. Pfeuffer et al.

co-localization of stathmin with intracellular bacteria was
dependent on the expression of the actA gene, as actA
deletion mutants growing in microcolonies were never
labelled by the antistathmin antiserum (Fig. 9D and E).
As an additional control, Cos-1 cells infected with the wild-
type L. monocytogenes strain were stained with antiserum,
which was blocked by the addition of the antigenic peptide
(Gavet et al., 1998). As shown in Fig. 9F and G, this
blocked antiserum did not label the bacteria, proving the
speci®city of the labelling shown in Fig. 9B and C. In con-
trast to the highly asymmetrical labelling found with the
anti-LaXp180 antiserum, the stathmin labelling pattern
was different: only some of the bacteria were labelled
asymmetrically (predominantly bacteria with actin tails;
see upper insert in Fig. 9A±C), and most were found asso-
ciated with stathmin around the bacteria appearing in bright
distinct spots (lower insert in Fig. 9A±C). The reason for
this spotted stathmin staining is currently unknown.
Discussion
Using the yeast two-hybrid system, we identi®ed four plas-
mids containing mouse cDNAs coding for three different
proteins interacting with ActA, demonstrating that the
yeast two-hybrid system can be used to screen for host±
pathogen interacting proteins with a bacterial protein as
bait. Although we do not know whether all these interac-
tions identi®ed in our screen do occur during the natural
course of an L. monocytogenes infection, we provide evi-
dence for in vivo interaction of ActA with LaXp180, one of
the proteins identi®ed.
The DNA sequence of one of the protein fragments
(pPC67-15) identi®ed was nearly identical to parts of two
recently identi®ed human cDNA clones. The ®rst one was
sequenced in the course of a genomic sequencing project
(Nagase et al., 1996). The second sequence encodes part
of a protein identi®ed as an `unknown human autoantigen',
which is reactive with serum from a patient with primary
SjoÈgrens' syndrome (TroÈster et al., 1994; M. Bachmann
and J. Laubinger, unpublished). Recent sequence data
show that both cDNA clones mentioned above belong to
a coding region of 4775 bp, which could give rise to a
large protein of <183 kDa. The portion of the protein we
identi®ed is homologous to the extreme C-terminal end of
this coding region. Additionally, a central part of this protein
was recently identi®ed in a yeast two-hybrid screen with
the mammalian phosphoprotein stathmin as bait and
was called coiled coil-forming protein 1 (CC1) (in the pre-
sent study, we used the term LaXp180 for the whole puta-
tive protein).
In order to demonstrate LaXp180±ActA interaction in
vitro, we cloned the 467 C-terminal amino acids of
LaXp180 in E. coli as a histidine-tagged fusion protein
and were able to show speci®c LaXp180±ActA interaction
by a biochemical approach. The observed retardation of
ActA on a nickel-agarose column was highly dependent
on prior loading with LaXp180, con®rming the data of the
yeast-based assays.
The expression of the isolated N-, P- and C-domains of
ActA was used to try to identify the LaXp180-binding
region of ActA. It became obvious that the proline-rich
repeat region is necessary for ef®cient ActA±LaXp180
interaction. However, the ¯anking regions on either side
of the proline-rich repeats are needed to confer speci®city
to the interaction, as the proline-rich repeats alone, when
fused to the DB domain of GAL4, gave false-positive
results, as also described by others (Mourrain et al.,
1997). The way in which the N-terminal or the C-terminal
domain of ActA contributes to the ActA±LaXp180 inter-
action and confers speci®city is not currently understood.
One might speculate that the ¯anking regions mask the
`sticky' proline-rich repeats in order to allow protein±
protein interaction only with LaXp180.
The generation of a polyclonal anti-LaXp180 antiserum
allowed us to investigate the intracellular localization of
LaXp180 in L. monocytogenes-infected Cos-1 cells. These
cells were shown to express signi®cant levels of LaXp180-
speci®c mRNA (which did not change upon infection)
as well as a single 194 kDa protein reacting speci®cally
with the anti-LaXp180 antiserum. The immunoprecipita-
tion experiment also demonstrated for the ®rst time the
expression of a full-length LaXp180 protein.
As shown by immuno¯uorescence analysis, the anti-
LaXp180 antiserum labelled intracellularly but not extra-
cellularly growing L. monocytogenes. However, only a
subfraction of the bacteria were labelled, suggesting that
LaXp180 only binds to some of the intracellular listeriae.
The reason for this observation is not known, but might
result from either differential expression of the actA gene
or limited cytoplasmic pools of free LaXp180. Limited avail-
ability of LaXp180 to cytoplasmic L. monocytogenes could
be explained by the preferential presence of LaXp180 in
the nucleus, as shown by immuno¯uorescence labelling.
The at least partial nuclear localization of LaXp180 was
not unexpected, as a nuclear localization sequence is pre-
sent in the N-terminal part of the protein. Speci®city of the
observed intracellular co-localization of L. monocytogenes
with LaXp180 is demonstrated by the fact that LaXp180
does not co-localize with the bacteria upon infection
with a L. monocytogenes strain lacking the entire actA
gene. Surprisingly, most of the bacteria were not labelled
uniformly but, rather, asymmetrically with one pole being
labelled and the opposite one being without label, a ®nding
resembling the distribution of the ActA-binding cellular
protein VASP (Chakraborty et al., 1995). However, in con-
trast to VASP, which co-localizes with F-actin on the bac-
terial surface, the LaXp180 binding site and F-actin
polymerization site on the bacterial surface are mutually
Q 2000 Blackwell Science Ltd, Cellular Microbiology, 2, 101±114
LaXp180, a new mammalian ActA-binding protein 109

exclusive. In most cases, one pole of the bacteria (the new
one upon division) is covered with LaXp180, and the other
(the old one) is covered with F-actin. The reason for this
asymmetrical distribution of LaXp180 and VASP is not
known. However, we could exclude the possibility that
the presence of the actin cloud simply results in steric
exclusion of LaXp180 recruitment, as cytochalasin D treat-
ment, which abolished ®lamentous actin accumulation,
does not interfere with the polar distribution of LaXp180
on the bacterial surface. It was argued that ActA itself is
asymmetrically distributed on the bacterial surface, being
accumulated at the site of F-actin assembly (Kocks et al.,
1993). However, work by others (Niebuhr et al., 1993)
has demonstrated that ActA is indeed found equally
throughout the surface of L. monocytogenes, and it has
been suggested that F-actin polymerization might be speci-
®cally connected with a modi®ed form of ActA (Chakraborty
et al., 1995). It is known that ActA becomes phosphory-
lated inside mammalian cells (Brundage et al., 1993),
but phosphorylation is obviously not necessary for actin
polymerization (Lasa et al., 1995). Our ®nding could be
explained by the hypothesis that ActA found at the two
poles of the bacterium is functionally different as a result
of modi®cation (e.g. phosphorylation) and that the one
form speci®cally interacts with LaXp180 and the other
with VASP and potential other factors, resulting in F-actin
polymerization. Whether LaXp180 itself could be the cellu-
lar factor modifying ActA is a matter of speculation, and
awaits further investigations.
As mentioned above, a portion of LaXp180 was identi-
®ed as a protein interacting with the phosphoprotein stath-
min, which is the prototype of a growing family of closely
related proteins. Stathmin is a ubiquitous protein of
about 19 kDa, originally proposed to be a relay integrating
various intracellular signaling pathways. The members of
this family all contain an N-terminal regulatory domain
with four serine residues and a C-terminal a-helical
domain responsible for coiled coil±protein interactions
(Gavet et al., 1998). The reported LaXp180±stathmin
interaction (Maucuer et al., 1995), together with the results
on LaXp180±ActA interaction and the ActA-dependent
stathmin labelling of L. monocytogenes described here,
obviously link the listerial surface protein ActA directly to
cellular signaling pathways. We recently reported on the
failure of a L. monocytogenes actA deletion mutant to sus-
tain fully persistent activation of the mammalian transcrip-
tion factor NF-kB in P388D1 macrophages upon infection
(Hauf et al., 1997). The now reported link of ActA to the
regulatory phosphoprotein stathmin and, hence, cellular
signal transduction pathways might open a way of under-
standing this unexpected ®nding. Depending on their
phosphorylation state, all members of the stathmin family
act on the microtubule network (Gavet et al., 1998). How-
ever, the role of microtubules in the intracellular life cycle
of L. monocytogenes has not been investigated in detail,
and it is only known that microtubule-depolymerizing
drugs, such as nocodazole, inhibit L. monocytogenes inva-
sion of endothelial cells (Greiffenberg et al., 1998), macro-
phages (Kuhn, 1998) and dendritic cells (Guzman et al.,
1995). Furthermore, tubulin was found in the F-actin tails
of intracellularly moving bacteria (Buchwalow et al.,
1997). Whether stathmin plays a role in any of these
processes is currently unknown.
In our hands, the yeast two-hybrid system was found to
be a useful method of identifying cellular-binding proteins
of a listerial virulence factor, which might open a new way
to study host±pathogen interactions. The system is, how-
ever, not free of limitations, as exempli®ed by the fact that
the well-known ActA-binding protein VASP (Chakraborty
et al., 1995) was not identi®ed in our screen. The reason
for this bias is not known but might result from either the
yeast two-hybrid system itself, or VASP being under-
represented in the gene bank used.
Experimental procedures
GAL4 DNA-binding domain±actA fusions in pPC97
The two-hybrid vectors pPC97 (Kalmes et al., 1998), pPC86and the cDNA library pPC67 (Chevray and Nathans, 1992)were kindly provided by T. Schuster (MSZ, WuÈrzburg, Ger-many) with the permission of D. Nathans (Baltimore, MD,USA). The PCR was used to amplify the actA gene from L.monocytogenes EGD cell lysate (Bubert et al., 1997). The58 PCR primers (see Table 3) all contain Sal I sites and the38 PCR primers all contain Bgl II sites. The PCR fragmentsgenerated with these primers were digested with Sal I andBgl II, which created fragments encoding either amino acids30±613 of the ActA protein (ActA) lacking the signal peptideand the membrane anchor (Domann et al., 1992) or encodingonly parts of ActA (see Table 3 and Fig. 3). These fragmentswere unidirectionally cloned into the multiple cloning site of theGAL4 DNA-binding domain plasmid pPC97 to create the plas-mids pPC97-ActA, pPC97-NActA, pPC97-PActA, pPC97-CActA, pPC97-NPActA, pPC97-PCActA and pPC97-DPActA.
GAL4 activation domain±cDNA fusions in pPC86
The GAL4 activation domain±cDNA fusion library pPC67created by Chevray and Nathans (1992) was used in thisstudy. cDNA synthesized from poly(A) RNA derived from14.5-day-old CD-1 mouse embryos was cloned into the multi-ple cloning site of the GAL4 activation domain plasmid pPC86.Plasmid DNA was then isolated from 2 ´ 106 colonies.
Expression of the DB±ActA fusion proteins
Protein extract was prepared from Saccharomyces cerevisiaepPC97-ActA (leu2) by suspending the pellet from 1.5 ml of aculture grown for 48 h in SD medium lacking leucine (Match-maker protocol PT 1265-1) in 30 ml of sample buffer
Q 2000 Blackwell Science Ltd, Cellular Microbiology, 2, 101±114
110 T. Pfeuffer et al.

[0.125 M Tris, pH 6.8, 4% SDS, 10% (v/v) 2-mercaptoethanol,20% glycerol, 0.04% bromophenol blue], boiling for 3 min, vor-texing for 10 min with the same amount of glass beads(0.45 mm), the addition of 100 ml of sample buffer, vortexingagain for 10 min and a short centrifugation. For Westernblot analysis, 15 ml of the sample was separated by SDS±PAGE (10%) according to the method of Laemmli (1970)and transferred onto a nitrocellulose membrane (Hybond-C,Amersham). The membrane was blocked with PBS (pH 7.4,2% BSA, 0.05% Tween 20) overnight and incubated with arabbit anti-GAL4DB antibody (sc-577, Santa Cruz) diluted1:1000 in PBS (0.05% Tween 20) for 2 h. After washingthree times (5 min each) in PBS (0.05% Tween 20), the mem-brane was incubated with a goat anti-rabbit immunoglobulinantibody conjugated with horseradish peroxidase (sc-2004,Santa Cruz) diluted 1:1000 for 1 h. Immunodetection was per-formed using the enhanced chemoluminescence system(Amersham), according to the manufacturer's protocol.
Screening for AIPs in S. cerevisiae HF7c
This entire procedure was performed according to the Match-maker protocol (PT 1265-1). The yeast host strain HF7c(trp1±, leu2±, his3±; reporter HIS3 and lacZ ) was ®rst trans-formed with pPC97-ActA (leu2 ) to isolate HF7c containing thepPC97-ActA plasmid on SD plates lacking leucine. This yeaststrain was then transformed with 1250 mg of the pPC67 library(trp1 ) and plated on SD plates lacking tryptophan, leucineand histidine and containing 20 mM 3-amino triazole (to sup-press leaky HIS3 activation in HF7c). To determine the num-ber of cDNAs screened, an aliquot of the transformationmixture was plated on SD plates lacking tryptophan and leu-cine. The HF7c colonies that grew on medium lacking trypto-phan, leucine and histidine, resulting from HIS3 reporter geneactivation, were assayed for the second reporter gene (lacZ )by performing b-galactosidase ®lter assays.
pPC67 plasmids (trp1, ampr) encoding putative AIPs wereextracted from HF7c according to the method of Robzyk andKassir (1992), transformed into E. coli DH5a via electro-poration and selected for on LB medium plates containing100 mg mlÿ1 ampicillin. Restriction analysis was performedon the plasmids reisolated from DH5a using Sal I and Bgl II.
Each of the different pPC67 plasmids was retransformedinto both yeast reporter strains HF7c and SFY526 (trp1±,leu2±; reporter lacZ ) alone or in combination with pPC97 orpPC97-ActA. b-Galactosidase ®lter assays were performedin duplicate on the yeast transformants.
Determining the identities of AIPs
The cDNA inserts of the plasmids encoding putative AIPs(pPC67-15, pPC67-18, pPC67-37, pPC67-70) were sequ-enced using a commercial T7 sequencing kit (Pharmacia).Synthetic oligonucleotide primers were annealed to thepPC67 plasmids serving as templates. Radiolabelling wascarried out with [a-32P]-dATP (3000 Ci mmolÿ1; Amersham-Buchler). Both strands of the templates were determined.Database searches using the nucleotide and correspondingpeptide sequences were performed using the Genetics Com-puter Group (GCG) software package. Percentage identity/similarity were determined with BESTFIT. Generally, defaultvalues for selectable parameters were accepted.
Cloning and expression of ActA and a C-terminal
fragment of LaXp180 in E. coli and generation of
antisera against both proteins
The nucleotide sequence encoding the C-terminal 467amino acids of LaXp180 was cloned into the (His)6-fusionplasmid pQE30 (Qiagen) in two steps. First, a 178 bp frag-ment was PCR ampli®ed from plasmid pKS30, which was co-isolated during screening a peripheral blood lymphocyte±cDNA expression library (TroÈster et al., 1994), using theprimers 58-CTAAGAGGATCCATGACAATTGAA-38 (BamHIsite underlined) and 58-CCAATTTACTCTGCAGTTC-38 andligated into pGEM-T (Promega), resulting in plasmid pGEM-T1. A 160 bp BamHI±Pst I fragment from pGEM-T1 wasthen cloned into pQE30 (Qiagen) to produce an N-terminal(His)6-fusion in plasmid pQE30-1. In a second step, a1299 bp Pst I fragment from pKS30 was ligated into Pst I-cutpQE30-1, resulting in plasmid pQHS30. The recombinantHis-tagged LaXp180 fusion protein was expressed in E. coliM15 [pREP4] and af®nity puri®ed over Ni-NTA columns (Qia-gen), as described in the QIAexpressionist manual (Qiagen).
Q 2000 Blackwell Science Ltd, Cellular Microbiology, 2, 101±114
Table 3. Primer pairs used for the generationof the DB±ActA fusion proteins. ActA 58-CGAGGTCGACAGCGACAGATAGCGAAGAT-38 aa 30±613
58-TAGTAGATCTGCGTCGTATGGTTCCCTGGTTCTTC-38NActA 58-CGAGGTCGACAGCGACAGATAGCGAAGAT-38 aa 30±263
58-GGTGGCGAGATCTCCGAAGCATTTACCTCTTC-38PActA 58-AGGTAAAGTCGACGGACTTCCCGCCACCACCT-38 aa 264±429
58-TCCTCTAGATCTTAAATCAGCTAGGCGATCAAT-38CActA 58-CAGAAGAGTCGACTGATCGCCTAGCTGATTTAAG-38 aa 423±613
58-TAGTAGATCTGCGTCGTATGGTTCCCTGGTTCTTC-38NPActA 58-CGAGGTCGACAGCGACAGATAGCGAAGAT-38 aa 30±429
58-TCCTCTAGATCTTAAATCAGCTAGGCGATCAAT-38PCActA 58-AGGTAAAGTCGACGGACTTCCCGCCACCACCT-38 aa 264±613
58-TAGTAGATCTGCGTCGTATGGTTCCCTGGTTCTTC-38DPActAa 58-CGAGGTCGACAGCGACAGATAGCGAAGAT-38 aa 30±190 and 470±613
58-TAGTAGATCTGCGTCGTATGGTTCCCTGGTTCTTC-38
a. Cell lysate from L. monocytogenes strain A49 (Hauf et al., 1997) was used for PCR, whichharbours a deletion in the actA gene removing the whole proline-rich repeat region.The numbering of the amino acids present in the fusion proteins refer to Domann et al. (1992).The Sal I and Bgl II sites introduced are underlined.
LaXp180, a new mammalian ActA-binding protein 111

Fractions containing the (His)6-tagged LaXp180 were pooledand additionally puri®ed by SDS±PAGE, Coomassie bluestaining, subsequent cutting of the corresponding proteinband and electroelution using the Biotrap equipment (Schlei-cher & Schuell). Recombinant (His)6-tagged LaXp180 wasused to immunize a New Zealand White rabbit (CharlesRiver) subcutaneously with 200 mg of protein and an equalvolume of Freund's complete adjuvant (Sigma). Six and 11weeks after the initial immunization, a booster injection of150 mg of (His)6-tagged LaXp180 with an equal volume ofFreund's incomplete adjuvant (Sigma) was given. The anti-LaXp180 serum was obtained 16 weeks after the ®rst immuni-zation. Preadsorption of the antiserum was performed asfollows: 1 ml of anti-LaXp180 antiserum was added to a bacter-ial pellet from 2 ml of logarithmically grown L. monocytogenesEGD, vortexed, incubated for 20 min at room temperature andcentrifuged. This procedure was repeated twice.
For the puri®cation of recombinant ActA, we used theImpact I system (New England Biolabs) according to themanufacturer's protocol. The nucleotide sequence encodingthe ActA protein (amino acids 30±607), lacking the N-terminalsignal sequence and the C-terminal membrane, anchor wasampli®ed using PCR with the 58 primer (58-GGTGGGT-CCATGGCGACAGATAGCGAAGATTCTAG-38) including aNco I site (underlined) and the 38primer (58-GTATGGTTC-CCGGGTTCTTCCTTCGCTTTTTCGTCTTC-38) containinga Xma I site (underlined) from L. monocytogenes EGD celllysate. The PCR fragment was digested with Nco I andXma I and cloned unidirectionally into the multiple cloningsite of plasmid pCYB4 to create the plasmid pCYB4-ActAencoding an ActA±intein±CBD (chitin-binding domain) fusionprotein. The recombinant protein was expressed in E. coliDH5a and passed through a chitin column (New England Bio-labs). After extensive washing, the self-cleavage of the fusionprotein still remaining on the chitin matrix was induced withdithiothreitol (DTT), and the recombinant ActA was eluted.Fractions containing ActA were pooled and additionally puri-®ed by cutting out the corresponding protein band from pre-parative SDS±PAGE gels after Coomassie blue staining.Electroelution was performed using the Biotrap equipment(Schleicher & Schuell). Immunization of a New ZealandWhite rabbit (Charles River) with puri®ed ActA was performedessentially as described above for the LaXp180 protein.
In vitro LaXp180±ActA interaction
A 125 ml culture of E. coli M15 pQHS30 grown in 2YT medium,containing 100 mg mlÿ1 ampicillin and 30 mg mlÿ1 kanamycinwas induced at an OD600 of 1.0 by the addition of 1 mMIPTG for 4 h. After centrifugation (6000 r.p.m. for 15 min at48C), the pellet was washed once and resuspended in 10 mlof binding buffer [50 mM NaH2PO4, 300 mM NaCl, 10 mM imi-dazole, 10 mM 2-mercaptoethanol, 1 mM phenylmethylsul-phonyl ¯uoride (PMSF), pH 7.2]. This solution was Frenchpressed, centrifuged (16000 r.p.m. at 48C for 30 min) andthe supernatant containing (His)6-tagged LaXp180 was passedthrough a column containing 12 ml of Ni-NTA agarose (Qia-gen), equilibrated with binding buffer. After extensive washingwith 100 ml of binding buffer, the crude cell extract containingrecombinant ActA was loaded on the column, which wasobtained by growing a 125 ml culture of E. coli DH5a
pCYBActA in 2YT medium containing 100 mg mlÿ1 ampicillin,induced at an OD600 of 0.9 with 1 mM IPTG for 3.5 h at 308C,centrifuged, washed once, resuspended in 10 ml of bindingbuffer, French pressed and centrifuged (16000 r.p.m. at 48Cfor 30 min). 2-Mercaptoethanol was added to the supernatantat a ®nal concentration of 20 mM and incubated for 2 h at 48Cto induce cleavage of the intein±ActA fusion. After adding fourvolumes of binding buffer, the solution was loaded on thecolumn. After thorough washing with 300 ml of binding buffer,the (His)6-tagged LaXp180 was eluted with binding buffer con-taining a gradient of 10±250 mM imidazole. Fractions of 1.5 mleach were collected, the proteins precipitated with 10% TCAand analysed on Western blots using the anti-LaXp180 andanti-ActA antisera in a dilution of 1:1000.
Bacteria, mammalian cell culture and infection
L. monocytogenes strain EGD and strain D2 (lacking theentire actA gene) (Hauf et al., 1997) used for cell culture infec-tions were grown in brain±heart infusion broth (BHI; Difco) at378C with aeration. Mid-log phase cultures were washed twicein phosphate-buffered saline (PBS) and stored at ÿ808C.Caco-2 epithelial cells, Cos-1 ®broblasts and P388D1 macro-phages were grown in RPMI-1640 medium supplementedwith 10% heat-inactivated FCS (all from Gibco). Cells wereseeded 48 h before infection in 60-mm-diameter tissue cultureplates (Greiner) at 5 ´ 106 cells/plate for RNA isolation or in12-well tissue culture plates containing round cover slides at2 ´ 105 cells wellÿ1. The cells were infected with Listeria togive a multiplicity of infection (MOI) of 12.5 (strain EGD) or50 (strain D2) bacteria per eukaryotic cell, centrifuged ontothe monolayer (1200 r.p.m. for 10 min at room temperature)and incubated for 50 min. They were washed with PBS andincubated further in medium containing gentamicin (50 mgmlÿ1) to kill extracellular bacteria and prevent reinfection.
RNA isolation, cDNA synthesis and RT±PCR
At 4 h after infection, total RNA was extracted from the cellsusing the guanidinium thiocyanate procedure, as describedby Chomczynski and Sacchi (1987). Complementary DNAwas synthesized from 10 mg of total RNA with the Stratagene®rst-strand synthesis kit, and the PCR ampli®cation was per-formed as described in detail earlier (Kuhn and Goebel,1994). The following primers were used for LaXp180-speci®cPCR: 58-CGGGATAAAGATTTGATAGAGTC-38 and 58-GC-CTTTTTGGCTTGACAGTATTC-38. Appropriate low volumesof cDNA were ampli®ed essentially as described to ensurethat possible differences in mRNA expression levels betweeninfected and non-infected cells were detectable. The reactionproducts were visualized by electrophoresis, and the speci®ci-ties of the ampli®ed bands were con®rmed by their predictedsizes. As an internal control, PCR ampli®cations with mouse(Kuhn and Goebel, 1994) and human (Arnold et al., 1993)b-actin-speci®c primers were always included. RNA samplesnot treated with reverse transcriptase were also included inthe ampli®cation reactions to check for DNA contamination.
Immunoprecipitation of LaXp180 from Cos-1 cells
Cells grown in a 90-mm-diameter dish (cooled to 08C) were
Q 2000 Blackwell Science Ltd, Cellular Microbiology, 2, 101±114
112 T. Pfeuffer et al.

lysed by the addition of 1 ml of ice-cold lysis buffer (50 mMTris-HCl, pH 8.0, 150 mM NaCl, 0.02% sodium azide, 0.1%SDS, 100 mg mlÿ1 PMSF, 1 mg mlÿ1 aprotinin, 1% Igepal,0.5% sodium deoxycholate) and scraped from the dish after20 min. After centrifugation (12 000 g for 2 min at 48C), thesupernatant was added to 5 ml of the antiserum and 0.2 mlof NET-gel buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl,0.1% Igepal, 1 mM EDTA pH 8.0, 0.25% gelatin, 0.02% sodiumazide). After 1 h incubation at 48C with shaking, 2 mg of pro-tein A±Sepharose (Sigma) was added, and the mixture wasincubated further for 1 h. The pellet was washed twice withNET-gel buffer and once with 10 mM Tris-HCl, pH 8.0, 0.1%Igepal, with 20 min shaking between each centrifugationstep. Sample buffer (20 ml) was then added to the pellet,heated to 1008C for 5 min and again centrifuged. The super-natant (15 ml) was run on a 10% SDS±polyacrylamide geland Western blotted.
Immuno¯uorescence assays
Cell monolayers grown on glass cover slides were infectedwith L. monocytogenes, as described above. At 4 h after infec-tion, the cells were processed for immuno¯uorescence asdescribed earlier in detail (Kuhn et al., 1990; Mounier et al.,1990). Brie¯y, the cells were permeabilized by treatment with0.5% Triton X-100 for 1 min, ®xed with acetone for 3 min, incu-bated with the anti-LaXp180 antiserum diluted 1:40 in PBS(30 min at room temperature) and washed with PBS threetimes. They were then incubated with a lissamine rhodamine(LRSC)-conjugated goat anti-rabbit IgG (Dianova) diluted 1:40in PBS with 2% BSA for 30 min, washed three times andmounted on a slide in PBS/50% glycerol. For stathmin label-ling, the cells were ®xed with methanol for 6 min, blocked withPBS/3% BSA for 1 h at room temperature and incubated over-night at 48C with the antistathmin antiserum C (Koppel et al.,1990) diluted 1:200 in PBS, washed three times in PBS/0.1%Tween 20 and then incubated with the LRSC-conjugated sec-ondary antiserum diluted 1:100 for 2 h. After three washes,the cover slides were mounted in PBS/50% glycerol. Forvisualization of ®lamentous actin, the cells were additionallyincubated in FITC-labelled phalloidin (Sigma), as describedpreviously. Pictures were taken with a ¯uorescence-equippedmicroscope (Leitz Dialux 20) and an electronic camera (Prince-ton Instruments). Digital images were processed using META-
MORPH software (Universal Imaging Corporation).
Acknowledgements
We thank J. Daniels and A. Demuth for critical reading of themanuscript, F. Engelbrecht for providing plasmid pQE30-InlCand for many discussions, and A. Sobel for the kind gift of theantistathmin antiserum, the blocked antiserum and for helpfulcomments on our staining results. This work was supportedby grant SFB479-B1 to W.G.
References
Alvarez-Dominguez, C., Vazquez-Boland, J.A., Carrasco-Marin,
E., Lopez-Mato, P., and Leyva-Cobian, F. (1997) Host cell
heparan sulfate proteoglycans mediate attachment and entryof Listeria monocytogenes, and the listerial surface protein
ActA is involved in heparan sulfate receptor recognition. Infect
Immun 65: 78±88.
Arnold, R., Scheffer, J., KoÈnig, B., and KoÈnig, W. (1993) Effectsof Listeria monocytogenes and Yersinia enterocolitica on cyto-
kine gene expression and release from human polymorpho-
nuclear granulocytes and epithelial (HEp-2) cells. InfectImmun 61: 2545±1552.
Brundage, R.A., Smith, G.A., Camilli, A., Theriot, J.A., and
Portnoy, D.A. (1993) Expression and phosphorylation of the
Listeria monocytogenes ActA protein in mammalian cells.Proc Natl Acad Sci USA 90: 11890±11894.
Bubert, A., Kestler, H., GoÈtz, M., BoÈckmann, R., and Goebel, W.
(1997) The Listeria monocytogenes iap gene as an indicator
gene for the study of PrfA-dependent regulation. Mol GenGenet 256: 54±62.
Buchwalow, I.B., Emoto, M., Brich, M., and Kaufmann, S.H.E.
(1997) Involvement of tubulin and inhibitory G proteins in the
interaction of Listeria monocytogenes with mouse hepato-cytes. Infect Immun 65: 1095±1097.
Chakraborty, T., Ebel, F., Domann, E., Niebuhr, K., Gerstel, B.,
Pistor, S., et al. (1995) A focal adhesion factor directly linkingintracellularly motile Listeria monocytogenes and Listeria iva-
novii to the actin-based cytoskeleton of mammalian cells.
EMBO J 14: 1314±1321.
Chevray, P.M., and Nathans, D. (1992) Protein interaction clon-ing in yeast: identi®cation of mammalian proteins that react
with the leucine zipper of Jun. Proc Natl Acad Sci USA 89:5789±5793.
Chomczynski, P., and Sacchi, N. (1987) Single-step method ofRNA isolation by acid guanidinium thiocyanate-phenol-chloro-
form extraction. Anal Biochem 162: 156±159.
Cossart, P., and Lecuit, M. (1998) Interactions of Listeria mono-cytogenes with mammalian cells during entry and actin-based
movement: bacterial factors, cellular ligands and signaling.
EMBO J 17: 3797±3806.
David, V., Gouin, E., Troys, M.V., Grogan, A., Segal, A.W.,Ampe, C., et al. (1998) Identi®cation of co®lin, coronin, Rac
and capZ in actin tails using a Listeria af®nity approach. J Cell
Sci 111: 2877±2884.
Domann, E., Wehland, J., Rohde, M., Pistor, S., Hartl, M., Goebel,W., et al. (1992) A novel bacterial virulence gene in Listeria
monocytogenes required for host cell micro®lament interaction
with homology to the proline-rich region of vinculin. EMBO J11: 1981±1990.
Engelbrecht, F., Chun, S.K., Ochs, C., Hess, J., Lottspeich, F.,
Goebel, W., et al. (1996) A new PrfA-regulated gene of Listeria
monocytogenes encoding a small, secreted protein whichbelongs to the family of internalins. Mol Microbiol 21: 823±
837.
Fields, S., and Song, O. (1989) A novel genetic system to detect
protein±protein interactions. Nature 340: 245±246.Gavet, O., Ozon, S., Manceau, V., Lawler, S., Curmi, P., and
Sobel, A. (1998) The stathmin phosphoprotein family: intracel-
lular localization and effects on the microtubule network. J Cell
Sci 111: 3333±3346.Greiffenberg, L., Goebel, W., Kim, K.S., Weiglein, I., Bubert, A.,
Engelbrecht, F., et al. (1998) Interaction of Listeria monocyto-
genes with human brain microvascular endothelial cells: InlB-dependent invasion, long-term intracellular growth, and spread
from macrophages to endothelial cells. Infect Immun 66: 5260±
5267.
Guzman, C.A., Rohde, M., Chakraborty, T., Domann, E., Hudel,
Q 2000 Blackwell Science Ltd, Cellular Microbiology, 2, 101±114
LaXp180, a new mammalian ActA-binding protein 113

M., Wehland, J., et al. (1995) Interaction of Listeria monocyto-genes with mouse dendritic cells. Infect Immun 63: 3665±
3673.
Hauf, N., Goebel, W., Fiedler, F., Sokolovic, Z., and Kuhn, M.
(1997) Listeria monocytogenes infection of P388D1 macro-phages results in a biphasic NF-kB (RelA/p50) activation
induced by lipoteichoic acid and bacterial phospholipases
and mediated by IkBa and IkBb degradation. Proc Natl AcadSci USA 94: 9394±9399.
Ireton, K., and Cossart, P. (1997) Host±pathogen interactions dur-
ing entry and actin-based movement of Listeria monocyto-
genes. Annu Rev Genet 31: 113±138.Kalmes, A., Hagemann, C., Weber, C.K., Wixler, L., Schuster, T.,
and Rapp, U.R. (1998) Interaction between the protein kinase
B-Raf and the a-subunit of the 11S proteasome regulator. Can-
cer Res 58: 2986±2990.Kocks, C., Gouin, E., Tabouret, M., Berche, P., Ohayon, H., and
Cossart, P. (1992) L. monocytogenes-induced actin assembly
requires the actA gene product, a surface protein. Cell 68:521±531.
Kocks, C., Hellio, R., Gounon, P., Ohayon, H., and Cossart, P.
(1993) Polarized distribution of Listeria monocytogenes sur-
face protein ActA at the site of directional actin assembly. JCell Sci 105: 699±710.
Koppel, J., Boutterin, M.C., Doye, V., Peyro-Saint-Paul, H., and
Sobel, A. (1990) Developmental tissue expression and phylo-
genetic conservation of stathmin, a phosphoprotein associatedwith cell regulations. J Biol Chem 265: 3703±3707.
Kuhn, M. (1998) The microtubule depolymerizing drugs nocoda-
zole and colchicine inhibit the uptake of Listeria monocytogenes
by P388D1 macrophages. FEMS Microbiol Lett 160: 87±90.Kuhn, M., and Goebel, W. (1994) Induction of cytokines in phago-
cytic mammalian cells infected with virulent and avirulent Lis-
teria strains. Infect Immun 62: 348±356.Kuhn, M., and Goebel, W. (1995) Molecular studies on the viru-
lence of Listeria monocytogenes. Genet Eng 17: 31±51.
Kuhn, M., Prevost, M.C., Mounier, J., and Sansonetti, P.J. (1990)
A nonvirulent mutant of Listeria monocytogenes does notmove intracellularly but still induces polymerization of actin.
Infect Immun 58: 3477±3486.
Laemmli, U.K. (1970) Cleavage of structural proteins during the
assembly of the head of bacteriophage T4. Nature 227:680±685.
Lasa, I., David, V., Gouin, E., Marchand, J.B., and Cossart, P.
(1995) The amino-terminal part of ActA is critical for the actin-based motility of Listeria monocytogenes ; the central proline-
rich region acts as a stimulator. Mol Microbiol 18: 425±436.
Laurent, V., Loisel, T.P., Harbeck, B., Wehman, A., Grobe, L.,
Jockusch, B.M., et al. (1999) Role of proteins of the Ena/VASP family in actin-based motility of Listeria monocytogenes.
J Cell Biol 144: 1245±1258.
Maucuer, A., Camonis, J.H., and Sobel, A. (1995) Stathmin inter-
action with a putative kinase and coiled-coil-forming proteindomains. Proc Natl Acad Sci USA 92: 3100±3104.
Mounier, J., Ryter, A., Coquis-Rondon, M., and Sansonetti, P.J.
(1990) Intracellular and cell-to-cell spread of Listeria monocy-
togenes involves interaction with F-actin in the enterocytelikecell line Caco-2. Infect Immun 58: 1048±1058.
Mourrain, P., Lasa, I., Gautreau, A., Gouin, E., Pugsley, A., and
Cossart, P. (1997) ActA is a dimer. Proc Natl Acad Sci USA94: 10034±10039.
Nagase, T., Seki, N., Ishikawa, K., Ohira, M., Kawarabayasi, Y.,Ohara, O., et al. (1996) Prediction of the coding sequences of
unidenti®ed human genes. VI. The coding sequences of 80
new genes (KIAA0201±KIAA0280) deduced by analysis of
cDNA clones from cell line KG-1 and brain. DNA Res 3:321±329.
Niebuhr, K., Chakraborty, T., Rohde, M., Gazlig, T., Jansen, B.,
Kollner, P., et al. (1993) Localization of the ActA polypeptide ofListeria monocytogenes in infected tissue culture cell lines:
ActA is not associated with actin `comets'. Infect Immun 61:2793±2802.
Ozon, S., Maucuer, A., and Sobel, A. (1997) The stathmin family± molecular and biological characterization of novel mammalian
proteins expressed in the nervous system. Eur J Biochem 248:794±806.
Pistor, S., Chakraborty, T., Walter, U., and Wehland, J. (1995)The bacterial actin nucleator protein ActA of Listeria monocy-
togenes contains multiple binding sites for host micro®lament
proteins. Curr Biol 5: 517±525.
Reinhard, M., Giehl, K., Abel, K., Haffner, C., Jarchau, T., Hoppe,V., et al. (1995) The proline-rich focal adhesion and micro®la-
ment protein VASP is a ligand for pro®lins. EMBO J 14: 1583±
1589.Robzyk, K., and Kassir, Y. (1992) A simple and highly ef®cient
procedure for rescuing autonomous plasmids from yeast.
Nucleic Acids Res 20: 3790.
Smith, G.A., Portnoy, D.A., and Theriot, J.A. (1995) Asymmetricdistribution of the Listeria monocytogenes ActA protein is
required and suf®cient to direct actin-based motility. Mol Micro-
biol 17: 945±951.
Smith, G.A., Theriot, J.A., and Portnoy, D.A. (1996) The tandemrepeat domain in the Listeria monocytogenes ActA protein
controls the rate of actin-based motility, the percentage of
moving bacteria, and the localization of vasodilator-stimulatedphosphoprotein and pro®lin. J Cell Biol 135: 647±660.
Tilney, L.G., and Portnoy, D.A. (1989) Actin ®laments and the
growth, movement, and spread of the intracellular bacterial
parasite, Listeria monocytogenes. J Cell Biol 109: 1597±1608.TroÈster, H., Metzger, T.E., Semsei, I., Schwemmle, M.,
Winterpacht, A., Zabel, B., et al. (1994) One gene, two tran-
scripts: isolation of an alternative transcript encoding for the
autoantigen La/SS-B from a cDNA library of a patient with pri-mary SjoÈgrens' syndrome. J Exp Med 180: 2059±2067.
Vazquez-Boland, J.A., Kocks, C., Dramsi, S., Ohayon, H., Geof-
froy, C., Mengaud, J., et al. (1992) Nucleotide sequence of thelecithinase operon of Listeria monocytogenes and possible
role of lecithinase in cell-to-cell spread. Infect Immun 60:219±230.
Welch, M.D., Iwamatsu, A., and Mitchison, T.J. (1997) Actin poly-merization is induced by Arp2/3 protein complex at the surface
of Listeria monocytogenes. Nature 385: 265±269.
Welch, M.D., Rosenblatt, J., Skoble, J., Portnoy, D.A., and Mitch-
ison, T.J. (1998) Interaction of human Arp2/3 complex and theListeria monocytogenes ActA protein in actin ®lament nuclea-
tion. Science 281: 105±108.
Williams, J.M., Chen, G.C., Zhu, L., and Rest, R.F. (1998) Using
the yeast two-hybrid system to identify human epithelial cellproteins that bind gonococcal Opa proteins: intracellular gono-
cocci bind pyruvate kinase via their Opa proteins and require
host pyruvate for growth. Mol Microbiol 27: 171±186.
Q 2000 Blackwell Science Ltd, Cellular Microbiology, 2, 101±114
114 T. Pfeuffer et al.




















