
Kinetics and mechanism of the reduction of the molybdatopentaamminecobalt(III) ion by aqueous...
11
Inorganica Chimica Acta 331 (2002) 279 – 289 www.elsevier.com/locate/ica Kinetics and mechanism of the reduction of the molybdatopentaamminecobalt(III) ion by aqueous sulfite and aqueous potassium hexacyanoferrate(II) Alvin A. Holder a , Tara P. Dasgupta b, * a Department of Biological and Chemical Sciences, Uniersity of the West Indies, Cae Hill Campus, PO Box 64, Bridgetown, St. Michael, Barbados b Department of Chemistry, Uniersity of the West Indies, Mona Campus, Mona, Kingston 7, Jamaica Received 22 June 2001; accepted 12 November 2001 Dedicated to Professor A.G. Sykes Abstract A detailed investigation on the oxidation of aqueous sulfite and aqueous potassium hexacyanoferrate(II) by the title complex ion has been carried out using the stopped-flow technique over the ranges, 0.01 [S(IV)] T 0.05 mol dm −3 , 4.47 pH 5.12, and 24.9 37.6 °C and at ionic strength 1.0 mol dm −3 (NaNO 3 ) for aqueous sulfite and 0.01 [Fe(CN) 6 4 − ] 0.11 mol dm −3 , 4.54 pH 5.63, and 25.0 35.3 °C and at ionic strength 1.0 or 3.0 mol dm −3 (NaNO 3 ) for the hexacyanoferrate(II) ion. Both redox processes are dependent on pH and reductant concentration in a complex manner, that is, for the reaction with aqueous sulfite, k obs ={(k 1 K 1 K 2 K 3 +k 2 K 1 K 4 [H + ])[S(IV)] T ]/([H + ] 2 +K 1 [H + ] +K 1 K 2 ) and for the hexacyanoferrate(II) ion, k obs = {(k 1 K 3 K 4 K 5 +k 2 K 3 K 6 [H + ])[Fe(CN) 6 4 − ] T )/([H + ] 2 +K 3 [H + ] +K 3 K 4 ). At 25.0 °C, the value of k 1 (the composite of k 1 K 3 ) is 0.77 0.07 mol −1 dm 3 s −1 , while the value of k 2 (the composite of k 2 K 4 ) is (3.78 0.17) ×10 −2 mol −1 dm 3 s −1 for aqueous sulfite. For the hexacyanoferrate(II) ion, k 1 (the composite of k 1 K 5 ) is 1.13 0.01 mol −1 dm 3 s −1 , while the value of k 2 (the composite of k 2 K 6 ) is 2.36 0.05 mol −1 dm 3 s −1 at 25.0 °C. In both cases there was reduction of the cobalt(III) centre to cobalt(II), but there was no reduction of the molybdenum(VI) centre. k 22 , the self-exchange rate constant, for aqueous sulfite (as SO 3 2 − ) was calculated to be 5.37 ×10 −12 mol −1 dm 3 s −1 , while for Fe(CN) 6 4 − , it was calculated to be 1.10 ×10 9 mol −1 dm 3 s −1 from the Marcus equations. © 2002 Elsevier Science B.V. All rights reserved. Keywords: Redox; Ferrocyanide; Molybdenum(VI); Aqueous sulfite; Cobalt(III) 1. Introduction Over the past 2 decades a number of systems have been studied in our laboratory and by collaborators elsewhere that involve the reactions of various aqua transition metal complex ions with ‘free’ sulfite in aqueous solution. The studies reported include the reac- tions of the aquapentaammine complexes of cobalt(III) [1 – 3], rhodium(III) [4], platinum(IV) [5], and chromiu- m(III) [4], along with (tren)Co(OH 2 ) 2 3 + (tren =2, 2, 2-triaminotriethylamine) [3]. The kinetic data show that in each instance the most significant first step in the overall process is very rapid nucleophilic attack by ligand hydroxide on dissolved SO 2 to form an O- bonded sulfito complex, a reaction which is readily reversed by immediate acidification [1 – 5]. Metal-to- oxygen bonding is not involved in this reversible pro- cess as confirmed by NMR measurements [6]. Subsequent reactions in the cobalt(III) systems com- prise O-bonded to S-bonded isomerisation, internal re- dox, sulfite ion addition, or a combination of these processes depending on the pH and the nature of the N 4 or N 5 ligand grouping [1]. The hexacyanoferrate(II) anion on the other hand can reduce some pentaamminecobalt(III) complexes to Co(II) via an outer-sphere electron transfer step [7]. In the past, the reductions of a series of substituted (pyri- dine)pentaamminecobalt(III) complexes by hexacyano- ferrate(II) proceed via the formation of an ion-pair, * Corresponding author. Fax: +1-876-977 1835. E-mail address: [email protected] (T.P. Dasgupta). 0020-1693/02/$ - see front matter © 2002 Elsevier Science B.V. All rights reserved. PII:S0020-1693(02)00686-2
Transcript of Kinetics and mechanism of the reduction of the molybdatopentaamminecobalt(III) ion by aqueous...

Inorganica Chimica Acta 331 (2002) 279–289
www.elsevier.com/locate/ica
Kinetics and mechanism of the reduction of themolybdatopentaamminecobalt(III) ion by aqueous sulfite and
aqueous potassium hexacyanoferrate(II)
Alvin A. Holder a, Tara P. Dasgupta b,*a Department of Biological and Chemical Sciences, Uni�ersity of the West Indies, Ca�e Hill Campus, PO Box 64, Bridgetown,
St. Michael, Barbadosb Department of Chemistry, Uni�ersity of the West Indies, Mona Campus, Mona, Kingston 7, Jamaica
Received 22 June 2001; accepted 12 November 2001
Dedicated to Professor A.G. Sykes
Abstract
A detailed investigation on the oxidation of aqueous sulfite and aqueous potassium hexacyanoferrate(II) by the title complexion has been carried out using the stopped-flow technique over the ranges, 0.01� [S(IV)]T�0.05 mol dm−3, 4.47�pH�5.12,and 24.9���37.6 °C and at ionic strength 1.0 mol dm−3 (NaNO3) for aqueous sulfite and 0.01� [Fe(CN)6
4−]�0.11 moldm−3, 4.54�pH�5.63, and 25.0���35.3 °C and at ionic strength 1.0 or 3.0 mol dm−3 (NaNO3) for the hexacyanoferrate(II)ion. Both redox processes are dependent on pH and reductant concentration in a complex manner, that is, for the reaction withaqueous sulfite, kobs={(k1K1K2K3+k2K1K4[H+])[S(IV)]T]/([H+]2+K1[H+]+K1K2) and for the hexacyanoferrate(II) ion, kobs={(k1K3K4K5+k2K3K6[H+])[Fe(CN)6
4−]T)/([H+]2+K3[H+]+K3K4). At 25.0 °C, the value of k1� (the composite of k1K3) is0.77�0.07 mol−1 dm3 s−1, while the value of k2� (the composite of k2K4) is (3.78�0.17)×10−2 mol−1 dm3 s−1 for aqueoussulfite. For the hexacyanoferrate(II) ion, k1� (the composite of k1K5) is 1.13�0.01 mol−1 dm3 s−1, while the value of k2� (thecomposite of k2K6) is 2.36�0.05 mol−1 dm3 s−1 at 25.0 °C. In both cases there was reduction of the cobalt(III) centre tocobalt(II), but there was no reduction of the molybdenum(VI) centre. k22, the self-exchange rate constant, for aqueous sulfite (asSO3
2−) was calculated to be 5.37×10−12 mol−1 dm3 s−1, while for Fe(CN)64−, it was calculated to be 1.10×109 mol−1 dm3
s−1 from the Marcus equations. © 2002 Elsevier Science B.V. All rights reserved.
Keywords: Redox; Ferrocyanide; Molybdenum(VI); Aqueous sulfite; Cobalt(III)
1. Introduction
Over the past 2 decades a number of systems havebeen studied in our laboratory and by collaboratorselsewhere that involve the reactions of various aquatransition metal complex ions with ‘free’ sulfite inaqueous solution. The studies reported include the reac-tions of the aquapentaammine complexes of cobalt(III)[1–3], rhodium(III) [4], platinum(IV) [5], and chromiu-m(III) [4], along with (tren)Co(OH2)2
3+ (tren=2, 2�,2�-triaminotriethylamine) [3]. The kinetic data showthat in each instance the most significant first step inthe overall process is very rapid nucleophilic attack by
ligand hydroxide on dissolved SO2 to form an O-bonded sulfito complex, a reaction which is readilyreversed by immediate acidification [1–5]. Metal-to-oxygen bonding is not involved in this reversible pro-cess as confirmed by NMR measurements [6].Subsequent reactions in the cobalt(III) systems com-prise O-bonded to S-bonded isomerisation, internal re-dox, sulfite ion addition, or a combination of theseprocesses depending on the pH and the nature of theN4 or N5 ligand grouping [1].
The hexacyanoferrate(II) anion on the other handcan reduce some pentaamminecobalt(III) complexes toCo(II) via an outer-sphere electron transfer step [7]. Inthe past, the reductions of a series of substituted (pyri-dine)pentaamminecobalt(III) complexes by hexacyano-ferrate(II) proceed via the formation of an ion-pair,
* Corresponding author. Fax: +1-876-977 1835.E-mail address: [email protected] (T.P. Dasgupta).
0020-1693/02/$ - see front matter © 2002 Elsevier Science B.V. All rights reserved.
PII: S0 0 2 0 -1693 (02 )00686 -2

A.A. Holder, T.P. Dasgupta / Inorganica Chimica Acta 331 (2002) 279–289280
followed by internal electron transfer within the ion-pair [8]. It is postulated that the ion-pairs featureapproach by Fe(CN)6
4− on the ammonia side of thecobalt(III) complexes [8]. The electron transfer processis assumed to be adiabatic and the variations in rate areassociated with changes in the reduction potentialsand/or rate constants for self-exchange of thecobalt(III) complexes [8].
The title complex {[(H3N)5CoOMoO3]+} is binuclearwith a cobalt(III) centre and a molybdenum(VI) centre,so it is possible that one or both metal centres can bereduced by both reductants, that is, Co(III) to Co(II),Mo(VI) to Mo(V) (or even lower oxidation states). Alsoit is possible that there could be anation by the aqueoussulfite followed by internal redox depending on the pH.Clearly both reactions with aqueous sulfite and thehexacyanoferrate(II) (ferrocyanide) anion are of vitalimportance.
2. Experimental
2.1. Materials
All reagents were of analytical grade (BDH). Ultra-pure water, obtained by deionising distilled water usinga Milli-Q Reagent Grade water system, was used forpreparative work and to make up solutions for allphysical measurements. Solid Na2S2O5 (BDH) was thesource of aqueous sulfite; this salt is very stable in thesolid form but hydrates rapidly and completely whendissolved in water to yield aqueous sulfite [1].
2.2. Preparation of the complex
[(H3N)5CoOMoO3]ClO4 was prepared by the methodoutlined previously [9]. Visible spectrum: �max (nm (0.1mol dm−3 Na2MoO4) 360 and 526 (� (dm3 mol−1
cm−1) 61 and 92). Lit. [9]: �max (nm (0.1 mol dm−3
Na2MoO4) 360 and 526 (� (dm3 mol−1 cm−1) 61.3 and91.7).
2.3. Kinetic measurements
All visible and ultraviolet spectra were recorded witheither a Pye Unicam Model PU 8800 or a Hewlett–Packard 8452A diode array spectrophotometer. All ki-netic measurements were made using a Hi-Tech SF-51stopped-flow spectrophotometer interfaced with a com-puter. The spectrophotometer syringes were immersedin a water-bath linked to a thermostat system (HaakeD8) capable of maintaining temperatures within �0.02 °C.
For the electron transfer reactions involving aqueoussulfite solution or K4Fe(CN)6 (potassium hexacyanofer-rate(II)) the following was carried out: Stock solutions
of the complex of known concentration were preparedin volumetric flasks (10 cm3). The ionic strength of eachsolution was adjusted to the required value with appro-priate amounts of NaNO3 and the flasks were ther-mostatted for 10 min before introducing the solutionsinto the syringes of the stopped-flow apparatus. Theapparatus was equilibrated at the reaction temperaturefor at least 0.5 h prior to use and the solutions werekept in one of the thermostatted drive syringes for 5min before each experiment.
The other syringe contained the respective reducingagent, buffer (potassium hydrogen phthalate–NaOH orCH3CO2Na–CH3CO2H), and supporting electrolyte(NaNO3). This syringe with its contents was also pre-thermostatted at the desired temperature. Both reac-tants were then mixed in the stopped-flowspectrophotometer after triggering. Na2H2EDTA (withthe reductant, supporting electrolyte, and buffer) wasused in the reaction involving Fe(CN)6
4− so as tosequester the Co(aq)
2+ formed [7]. Constant ionic strengthwas maintained at 1.0 mol dm−3 (NaNO3) in all cases(or 3.0 mol dm−3 in one case for the reaction with theferrocyanide anion). The NaNO3 solution was stan-dardised by an ion-exchange method using Dowex 50W-X8 (50 mesh, H+ form) resin. The redox processeswere followed at 524 nm, where the largest absorbancechange occurred. An excess of reductant was used in allcases to ensure pseudo-first order conditions.
The kinetic data were collected and processed with aHi-Tech Scientific IS-1 software suite V 1.0a. Thepseudo-first order rate constants (kobs) were determinedby a non-linear least squares regression to fit the curveof the photomultiplier voltage versus time. The re-ported rate constants are an average of at least threekinetic runs. The standard deviation for each kobs is�5%. The pH of each solution was measured on anOrion Research EA 920 Expandable Ion Analyser,fitted with a Cole Partner combination electrode.
2.4. Stoichiometry
The redox stoichiometry [reductant: complex] wasdetermined by measuring the absorbance at 416 nm (forFe(CN)6
4−) and 524 nm (for aqueous sulfite) ofbuffered solutions containing the complex at a fixedconcentration while varying added reductant, then de-termining the break in the plot of absorbance versus theratio, [complex]: [reductant].
3. Results and discussions
3.1. Reduction with aqueous sulfite
3.1.1. Nature of the reactionOn reacting [(H3N)5CoOMoO3]+ in aqueous sulfite
solutions at pH 5.29 (potassium hydrogen phthalate–

A.A. Holder, T.P. Dasgupta / Inorganica Chimica Acta 331 (2002) 279–289 281
Fig. 1. Repetitive scan for the reduction of [(H3N5)CoOMoO3]+ by aqueous sulfite at 23.8 °C. [complex]=1×10−3 mol dm−3, [S(IV)]=0.05mol dm−3, I=1.0 mol dm−3 (NaNO3), pH 5.66 (buffer=CH3CO2H–CH3CO2Na), cycle time=25 s.
sodium hydroxide buffer) there was an immediate yel-low coloration, which slowly got paler with time. At theend of the reaction a pale pink solution left behind. Thepale pink solution was due to the formation of Co(aq)
2+,which was identified by the thiocyanate–acetone test[10]. Molybdate(VI) was tested in the solution by usingthe molybdyl ‘oxinate’ method [11]; this proved positivefor molybdate(VI). Fig. 1 shows a repetitive scan of thereaction over the wavelength range. Preliminary studiesof the reaction of aqueous sulfite with the title complexion indicate that there is a uniphasic reaction at 524nm, resulting in the reduction of [(H3N)5CoOMoO3]+
to Co(aq)2+.
3.2. Stoichiometry of the reaction
The stoichiometric data in Table 1 are consistentwith the reaction summarised in the followingequations:
2[(H3N)5CoOMoO3]+ +2H2O+2SO2
� 2[(H3N)5CoOSO2]++4H++2MoO42− (1)
[(H3N)5CoOSO2]+�Co(aq)2+ +5NH3+SO3
−� (2)
[(H3N)5CoOSO2]++SO3−�
�Co(aq)2+ +5NH3+SO2+SO4
2− (3)
Overall:
2[(H3N)5CoOMoO3]++2H2O+SO2�2Co(aq)2+
+6NH3+4NH4++2MoO4
2− +SO42− (4)
We favour direct reduction by the SO32− ligand (Eq.
(2)), with the formation of the radical anion [13], SO3�−
and Co(aq)2+. The SO3
�− radical anion is then scavengedby the [(H3N)5CoOSO2]+ ion. Analyses of the finalreaction mixture confirmed the presence [10] of Co(aq)
2+
at concentrations exactly equal to the initial concentra-tions of the Co(III) complex, and this finding wasindependent of reaction conditions. The identity of thesulfur-containing reaction product with aqueous BaCl2
Table 1Stoichiometry of the reaction between [(H3N)5CoOMoO3]+ andaqueous sulphite at 25.0 °C, pH 5.64 (CH3CO2Na–CH3CO2Hbuffer), I=1.0 mol dm−3 (NaNO3), [complex]=2×10−3 mol dm−3,�=524 nm, cell path length=1.00 cm
Absorbance[S(IV)]T/[complex]
0.0625 0.1480.1230.125
0.250 0.1000.0650.375
0.500 a 0.0251.000 0.024
0.0251.5002.000 0.0232.500 0.024
0.0233.0003.500 0.0284.000 0.026
0.0264.5000.0245.000
5.500 0.030
a Break point occurs at [S(IV)]T/[complex]=0.50.

A.A. Holder, T.P. Dasgupta / Inorganica Chimica Acta 331 (2002) 279–289282
Table 2Pseudo-first order rate constants for the reaction between[(H3N)5CoOMoO3]+ and aqueous sulfite
�=24.9 °C[S(IV)]T (mol �=31.8 °C �=37.6 °Cdm−3)
103 kobs (s−1)103 kobs (s−1) 102 kobs (s−1)
3.25– 0.9150.01–0.02 1.601.76 (2.93) a
–– (3.17) –0.0258.220.03 2.112.39 (3.47)–– (3.60) –0.035
2.84 (3.78)0.04 9.31 2.34–– (3.98) –0.045
3.35 (4.13)0.05 10.3 2.68
Variation in [S(IV)]T, �=524 nm, [complex]=0.5×10−3 mol dm−3,I=1.0 mol dm−3 (NaNO3), pH 4.88 (potassium hydrogen phthalate–NaOH buffer).
a Values in parentheses are at pH 5.34.
Scheme 1.
Fig. 2. A plot of kobs vs. [S(IV)] for the reduction of [(H3N)5-CoOMoO3]+ by aqueous sulfite at 37.6 °C.
was inconclusive as Ba(aq)2+ precipitates SO4
2−, SO32−
and also MoO42− [12].
It is quite interesting to note that only the cobalt(III)centre of the binuclear title complex ion was reduced tocobalt(II) by aqueous sulfite during the redox process.
3.3. Aqueous sulfite, temperature and pH dependence ofthe reaction
Kinetic runs were carried out over the ranges, 0.01�[S(IV)]T�0.05 mol dm−3, 4.47�pH�5.12, and24.9���37.6 °C ranges, and at ionic strength 1.0 moldm−3 (NaNO3). Individual kinetic runs showed first-order kinetics in [complex] for the uniphasic reaction.The pseudo-first order rate constants (kobs) for thereaction at the various [S(IV)]T, temperatures, and pHare shown in Tables 2 and 3. The rate of the reactionincreases with increasing [S(IV)]T and tends to reach asaturation point at high [S(IV)]T. A behaviour such asthis, is indicative of an ion-pair formation as shown inScheme 1, where
kobs=k2K4[S(IV)]/(1+K4[S(IV)]) (5)
A plot showing the saturation behaviour is shown inFig. 2, from which k2= (5.29�0.32)×10−2 s−1 andK4=21.1�1.8 mol−1 dm3 s−1 are calculated (data inTable 2 at 37.6 °C) after carrying out linear regressionon the inverse of Eq. (5), where the slope, 1/(k2K4) andthe intercept, 1/k2. Over the pH range the main existing
Table 3Pseudo-first order rate constants for the reaction between [(H3N)5CoOMoO3]+ and aqueous sulfite
�=25.0 °C pHpHpH �=31.2 °C �=36.4 °C
103 kobs (s−1) 102 kobs (s−1)103 kobs (s−1)
–– – 4.47 1.28–2.08 –4.50 – 4.53 1.392.25 4.654.60 5.56 4.64 1.582.54 4.744.71 6.14 4.75 1.72
4.834.82 1.874.826.832.794.92 4.922.95 7.93 4.94 1.995.03 3.12 5.02 8.73 5.01 2.15
15.12 2.349.7215.1013.325.07
Variation in pH and temperature, �=524 nm, [complex]=0.5×10−3 mol dm3, I=1.0 mol dm−3 (NaNO3), [S(IV)]T=0.045 mol dm−3,Buffer=potassium hydrogen phthalate–NaOH.

A.A. Holder, T.P. Dasgupta / Inorganica Chimica Acta 331 (2002) 279–289 283
sulfite species are the HSO3− and SO3
2− ions, while[(H3N)5CoOMoO3]+ and [(H3N)5CoOMoO2(OH)]2+,exist in equilibrium [9], but since the pK1 of[(H3N)5CoOMoO2(OH)]2+ is 2.09; the deprotonatedspecies, [(H3N)5CoOMoO3]+, is the reactive species inthe reaction as over the pH range and it exists in�99% as the predominant cobalt(III) species. The ob-served pseudo-first order rate constants, kobs, increasewith an increase in pH (Table 3), and this observationmay be interpreted in terms of the reactive species viathe proposed mechanism in Scheme 1 where K1=1.26×10−2 mol dm−3 and K2=5.01×10−7 moldm−3 at 25 °C [2]. Based on the increase of kobs withpH it can be deduced that SO3
2− is the more reactivespecies in the electron transfer reaction; so the k1 pathis the main contributing path over the pH range.
The proposed mechanism given in Scheme 1 leads tothe expression:
kobs={(k1K1K2K3+k2K1K4[H+])[S(IV)]T}
/([H+]2+K1[H+]+K1K2) (6)
where [S(IV)]T represents the total sulfite concentration.The expression can be rearranged to give:
{kobs([H+]2+K1[H+]+K1K2)[S(IV)]T−1}
=k1K1K2K3+k2K1K4[H+] (7)
A plot of the left hand side of Eq. (7) versus [H+] islinear in the pH range 4.47�pH�5.12, the slope beingk2K1K4 and intercept being k1K1K2K3. A plot at 25.0 °Cshown in Fig. 3 is linear over the pH range. A linearregressional analysis was carried out and due to theinability to separate k1 from K3 and k2 from K4, it wasnecessary to combine k1 and K3 to give k1� , and tocombine k2 and K4 to give k2� . The rate and activationparameters, along with the pK1 and pK2 values are [2]shown in Table 4.
In our work the separation of the equilibrium con-stant and the rate constant for the electron transfer hasproven to be difficult. This is due to the equilibrium ofthe two reactive species, namely, HSO3
− and SO32−. It
would not be correct to neglect the contribution fromHSO3
− in the k2 path (see Scheme 1).At 25.0 °C the value of k1� is 0.77�0.07 mol−1 dm3
s−1, while the value of k2� is (3.60�0.18)×10−2
mol−1 dm3 s−1; so one can see that SO32− reacts about
21 times faster than HSO3−. The transition state in-
volving HSO3− is more ordered than that involving
SO32− as can be seen from the respective values for
entropy of activation, the values being �S2� =116�88
J mol−1 K−1 and �S1� =204�27 J mol−1 K−1,
respectively.We can compare our studies with electron transfer
reactions involving sulfite and other Co(III) complexes.What is noticeable is that after the formation of theion-pair in Scheme 1 there is intermolecular electrontransfer. The rate constant for the intramolecular elec-tron transfer [1] of the complex, [(H3N)5CoOSO2]+,after SO2 addition to [(H3N)5CoOH2]3+, within the pHrange 3–7, is 1.4×10−2 s−1. The activation parame-ters are �H� =112.5 kJ mol−1 and �S� is 96.7 Jmol−1 K−1 at 25 °C. For [(H3N)5CoSO3]+, the rate [5]constant is �1×10−4 s−1. Our rate of electron trans-fer by SO3
2− occurs at a rate of 0.77 mol−1 dm3 s−1 at25.0 °C, which is faster than either of the electrontransfer reaction involving either Co(III) complex. Theactivation parameters (�H� and �S�) for the SO3
2−,term (k1� ) during the electron transfer process is com-parable with those for the [(H3N)5CoSO3]+ system.
The [(tren)Co(OH2)(OSO2)]+ complex undergoes[2,5,13] only a redox reaction for pH�5 but at higherpH values shows evidence for the disappearance of theredox reaction, while addition of a second sulfite anionyields a stable complex of stoichiometry [(tren)Co-(SO3)2]−, which predominates at a pH of 7.2 or higher[13].Similarly, the bis(ethylenediamine) species, cis-[(en)2Co(OSO2)]+ exhibits [14] redox over the pHranges 3–7 as the only observable secondary process.
Fig. 3. A plot of [kobs([H+]2+K1[H+]+K1K2][S(IV)]T
−1] vs. [H+]for the reduction of [(H3N)5CoOMoO3]+ by aqueous sulfite at25.0 °C.
Table 4Rate parameters and pK values a for the reaction between[(H3N)5CoOMoO3]+ and aqueous sulfite
pK2� (°C) k1� (mol−1 dm3 pK1102 k2� (mol−1 dm3
s−1) s−1)
3.60�0.180.77�0.0725.0 6.301.901.9831.2 6.302.63�0.10 6.85�0.33
6.3036.4 5.85�0.52 21.54�1.28 2.03
�H1� =134�8 kJ mol−1; �H2
� =116�27 kJ mol−1; �S1� =204�
27 J mol−1 K−1; �S2� =116�88 J mol−1 K−1.
a Ref. [2].

A.A. Holder, T.P. Dasgupta / Inorganica Chimica Acta 331 (2002) 279–289284
Table 5Summary of the activation parameters for various redox reactionsinvolving some cobalt(III) complexes and sulfite (‘free’ and co-ordi-nated) in aqueous solution
Complex a �H� �S� Reference(J mol−1 K−1)(kJ mol−1)
[(H3N)5CoOSoO2]+ (1) 96.7�117.6112.5�0.8 [2]34.3�27.6[(tren)Co(OH2)(OSO2)]+ (2) [5]101.3�34.3
4.6�29.3104.6�9.6 [16][(phen)2Co(SO3H)SO3] (3)132.6�2.1[(bpy)2Co(SO3H)SO3] (4) 91.6�6.7 [16]118.4�7.9 51.5�24.3[(phen)2Co(SO3H)- [16]
(OH2)]2+ (5)204�27[(H3N)5CoOMoO3]+ (6) this work134�8
[(H3N)5CoOMoO3]+ (6) 116�27 116�88 this workA (7) 117.6�0.4117.6�5.9 [17]
A, [(H3N)3Co(�-OH)2(�-SO3)Co(NH3)3]2+.a Each complex is numbered in Fig. 4.
transfer between the cobalt(III) centre and the sulfitoligand.
The activation parameters in Table 5 are very similarand can be used in an isokinetic plot of �H� versus�S� since
�H� =�Go� +�o�S� (8)
where �o is the isokinetic temperature and �Go� is the
intrinsic free energy of activation. Fig. 4 shows such aplot where the linearity proves that a common mecha-nism exists 15 for the sulfito cobalt(III) complexes andthe reaction involving aqueous sulfite and the titlecomplex ion, with �Go
� =104�5 kJ mol−1 and �o=144�51 K.
3.4. Reduction with hexacyanoferrate(II)
3.4.1. Nature of the reactionOn mixing Fe(CN)6
4− and [(H3N)5CoOMoO3]+ inaqueous solution a precipitate (presumably cobalt(II)hexacyanoferrate(II) and/or cobalt(II) hexacyanofer-rate(III)) was formed, and, therefore, homogeneouskinetic measurements were precluded. However, whendisodium dihydrogen ethylenediaminetetraacetate(Na2H2EDTA) was added to the solution to complexthe cobalt(II) formed, no precipitate was produced [7].A repetitive scan for the reaction at 25.0 °C is shown inFig. 5, where an increase in absorbance is observed.
3.5. Stoichiometry of the reaction
The stoichiometric data in Table 6 are consistentwith the reaction summarised in the following equation:
Fe(CN)64− + [(H3N)5CoOMoO3]++H2EDTA2−
+3H+�Fe(CN)63− +CoEDTA2− +5NH4
+
+MoO42− (9)
Eventually a reddish purple colour is produced overa period of hours. The formation of the reddish purplecolour is straightforward. The Fe(CN)6
3− andCoEDTA2− produced in Eq. (9) undergo an electrontransfer reaction [8] that produces purple CoEDTA−.This reaction is very slow and does not interfere withthe reaction of interest, Eq. (9).
Fe(CN)62− +CoEDTA2−
�CoEDTA−+Fe(CN)64− (10)
The purple product of CoEDTA− was identifiedspectrophotometrically.
3.6. Aqueous potassium hexacyanoferrate,temperature and pH dependence of the reaction
Kinetic runs were carried out over the ranges, 0.01�[Fe(CN)6
4−]�0.11 mol dm−3, 4.54�pH�5.63, and
Fig. 4. An isokinetic plot of the activation parameters for variousredox reactions involving some cobalt(III) complexes and sulfite(‘free’ and co-ordinated) in aqueous solution.
Meanwhile, the long chain tetraethylenepentaammine(tetren) analogue of �� S-[(tetren)Co(OSO2)]+, under-goes a slow internal rearrangement to the S-bondedform with no evidence for redox even at elevatedtemperatures [3]. There is evidence of a complex ana-tion reaction by aqueous sulfite at much higher pH withthe title complex ion, with the production of yellowcobalt(III) complex [15].
A summary of data concerning the redox reactions ofa selection of sulfito cobalt(III) complexes is shown inTable 5. As depicted in Table 5 the activation parame-ters found for the �-sulfito species compares reasonablywell with those of the mononuclear systems implying asimilarity in nature of the mechanisms. However, it isbelieved that the �-sulfito species undergoes a twoelectron transfer process with the reduction of the twocobalt(III) centres and the simultaneous oxidation ofS(IV) to S(VI). This contrasts with the mononuclearcobalt(III) sulfito species which involves one-electron
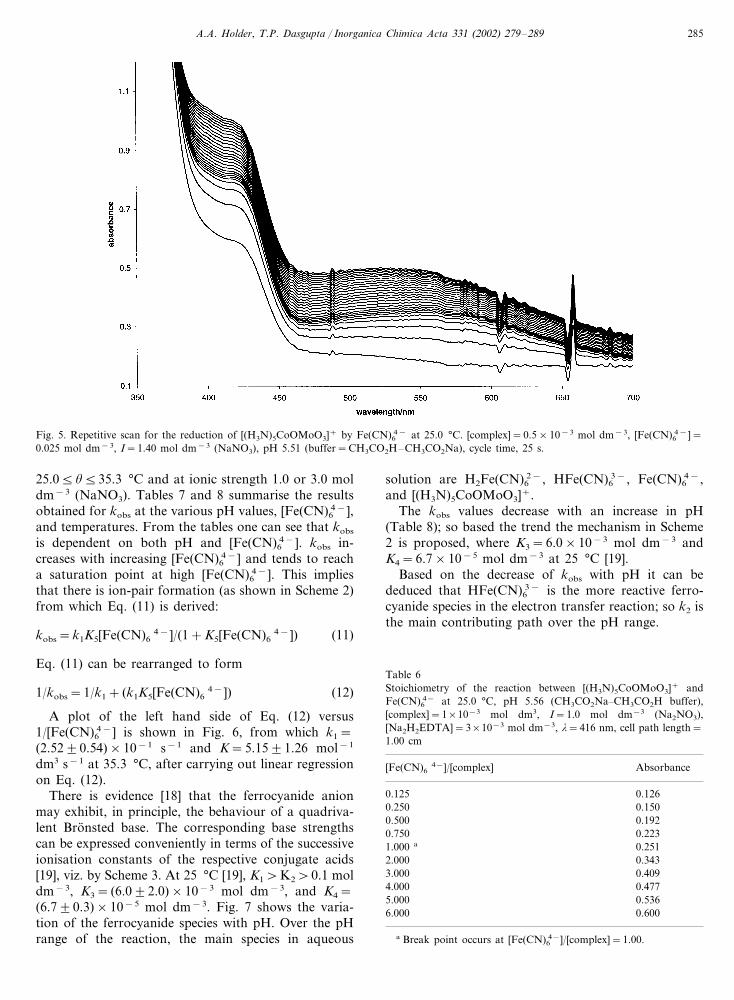
A.A. Holder, T.P. Dasgupta / Inorganica Chimica Acta 331 (2002) 279–289 285
Fig. 5. Repetitive scan for the reduction of [(H3N)5CoOMoO3]+ by Fe(CN)64− at 25.0 °C. [complex]=0.5×10−3 mol dm−3, [Fe(CN)6
4−]=0.025 mol dm−3, I=1.40 mol dm−3 (NaNO3), pH 5.51 (buffer=CH3CO2H–CH3CO2Na), cycle time, 25 s.
25.0���35.3 °C and at ionic strength 1.0 or 3.0 moldm−3 (NaNO3). Tables 7 and 8 summarise the resultsobtained for kobs at the various pH values, [Fe(CN)6
4−],and temperatures. From the tables one can see that kobs
is dependent on both pH and [Fe(CN)64−]. kobs in-
creases with increasing [Fe(CN)64−] and tends to reach
a saturation point at high [Fe(CN)64−]. This implies
that there is ion-pair formation (as shown in Scheme 2)from which Eq. (11) is derived:
kobs=k1K5[Fe(CN)64−]/(1+K5[Fe(CN)6
4−]) (11)
Eq. (11) can be rearranged to form
1/kobs=1/k1+ (k1K5[Fe(CN)64−]) (12)
A plot of the left hand side of Eq. (12) versus1/[Fe(CN)6
4−] is shown in Fig. 6, from which k1=(2.52�0.54)×10−1 s−1 and K=5.15�1.26 mol−1
dm3 s−1 at 35.3 °C, after carrying out linear regressionon Eq. (12).
There is evidence [18] that the ferrocyanide anionmay exhibit, in principle, the behaviour of a quadriva-lent Bronsted base. The corresponding base strengthscan be expressed conveniently in terms of the successiveionisation constants of the respective conjugate acids[19], viz. by Scheme 3. At 25 °C [19], K1�K2�0.1 moldm−3, K3= (6.0�2.0)×10−3 mol dm−3, and K4=(6.7�0.3)×10−5 mol dm−3. Fig. 7 shows the varia-tion of the ferrocyanide species with pH. Over the pHrange of the reaction, the main species in aqueous
solution are H2Fe(CN)62−, HFe(CN)6
3−, Fe(CN)64−,
and [(H3N)5CoOMoO3]+.The kobs values decrease with an increase in pH
(Table 8); so based the trend the mechanism in Scheme2 is proposed, where K3=6.0×10−3 mol dm−3 andK4=6.7×10−5 mol dm−3 at 25 °C [19].
Based on the decrease of kobs with pH it can bededuced that HFe(CN)6
3− is the more reactive ferro-cyanide species in the electron transfer reaction; so k2 isthe main contributing path over the pH range.
Table 6Stoichiometry of the reaction between [(H3N)5CoOMoO3]+ andFe(CN)6
4− at 25.0 °C, pH 5.56 (CH3CO2Na–CH3CO2H buffer),[complex]=1×10−3 mol dm3, I=1.0 mol dm−3 (Na2NO3),[Na2H2EDTA]=3×10−3 mol dm−3, �=416 nm, cell path length=1.00 cm
[Fe(CN)64−]/[complex] Absorbance
0.125 0.1260.250 0.1500.500 0.1920.750 0.223
0.2511.000 a
0.3432.0000.4093.0000.4774.0000.5365.000
6.000 0.600
a Break point occurs at [Fe(CN)64−]/[complex]=1.00.

A.A. Holder, T.P. Dasgupta / Inorganica Chimica Acta 331 (2002) 279–289286
Table 7Pseudo-first order rate constants for the reaction between[(H3N)5CoOMoO3]+ and Fe(CN)6
4−
[Fe(CN)64−] (mol dm−3) 102 kobs (s−1)
0.01 1.272.100.023.300.03
0.04 4.315.170.056.080.066.980.077.600.08
0.09 8.329.070.10
0.11 9.57
Variation in [Fe(CN)64−], [complex]=0.5×10−3 mol dm−3,
[Na2H2EDTA]=1.5×10−3 mol dm−3, �=524 nm, I=3.0 moldm−3 (NaNO3), pH 5.59 (CH3CO2Na–CH3CO2H buffer), �=35.3 °C.
The proposed mechanism given in Scheme 2 leads tothe expression:
kobs={(k1K3K4K5+k2K3K6[H+])[Fe(CN)64−]T}
/([H+]2+K3[H+]+K3K4) (13)
where [Fe(CN)64−]T represents the total ferrocyanide
concentration.The expression can be rearranged to give:
{kobs([H+]2+K3[H+]+K3K4[Fe(CN)64−]T−1}
=k1K3K4K5+k2K3K6[H+] (14)
A plot of the left hand side of Eq. (14) versus [H+] islinear in the range 4.53�pH�5.63, the slope beingk2K3K6 and the intercept being k1K3K4K5 A plot at25.0 °C shown in Fig. 8 is linear over the pH range. Alinear regressional analysis was carried out and due tothe inability to separate k1 from K5 and k2 from K6, itwas necessary to combine k1 and K5 to give k1� , and tocombine k2 and K6 to give k2� . K3 and K4 are indepen-dent of temperature as deduced from literature [19].The rate and activation parameters are shown in Table9.
At 25.0 °C the value of k1� is 1.13�0.01 mol−1 dm3
s−1, while the value of k2� is 2.36�0.05 mol−1 dm3; soone can see that HFe(CN)6
3− reacts with
Table 8Pseudo-first order rate constants for the reaction between[(H3N)5CoOMoO3]+ and Fe(CN)6
4−
�=30.4 °C pH �=35.3 °C�=25.0 °CpH pH
102 kobs (s−1) 102 kobs (s−1) 101 kobs(s−1)
3.79 4.544.54 8.73 4.53 1.594.62 8.473.61 4.63 1.544.63
4.723.46 1.474.73 8.294.724.82 3.36 7.93 1.444.834.83
4.943.13 7.715.04 4.94 1.385.05 1.335.16 7.55 5.053.10
1.305.175.27 7.353.06 5.165.38 5.273.01 1.245.277.165.62 5.416.965.402.98 1.21
–6.865.63 –––
Variation in pH and temperature, �=524 nm, [complex]=0.5×10−3
mol dm−3, I=1.0 mol dm−3 (NaNO3), [Fe(CN)64−]=0.025 mol
dm−3, Buffer=CH3CO2Na–CH3CO2H, [Na2H2EDTA]=1.5×10−3
mol dm−3.
Fig. 6. A plot of kobs−1 vs. [Fe(CN)6
4−]−1 for the reduction of[(H3N)5CoOMoO3]+ by Fe(CN)6
4− at 35.3 °C.
Scheme 2. Scheme 3.

A.A. Holder, T.P. Dasgupta / Inorganica Chimica Acta 331 (2002) 279–289 287
Fig. 7. A plot showing the percent speciation of Fe(CN)64− (A),
HFe(CN)63− (B), and H2Fe(CN)6
2− (C), with variation in pH.
k1= (2.52�0.54)×10−1 s−1 and that K5=5.15�1.26mol−1 dm3 at 35.3 °, I=3.0 mol dm−3 (NaNO3), andpH 5.59; from Eq. (12), k1� =1.30�0.60 mol−1 dm3
s−1. This value of k1� =1.30�0.60 mol−1 dm3 s−1 isless than the value k1� =4.60�0.06 mol−1 dm3 s−1 at35.3 °C and I=1.0 mol dm−3 (NaNO3) from Table 9.This shows that k1� decreases with an increase in ionicstrength, a clear indication that the effect of ionicstrength on rates of ionic reactions is well established[20]. For species of like charges, the rate increases withan increase in ionic strength, while for species of oppo-site charges, the rate decreases as the ionic strengthincreases. The rate is unaffected by ionic strength foruncharged species. From our decrease in k1� with anincrease in ionic strength from 1.0 to 3.0 mol dm−3, itshows that species of unlike charges due to[(H3N)5CoOMoO3]+ and Fe(CN)6
4− reacting to formthe ion-pair in Scheme 2.
We now compare our reaction between[(H3N)5CoOMoO3]+ and Fe(CN)6
4− with other reac-tions involving Fe(CN)6
4− and other cobalt(III) com-plexes. Our value of K5=5.15�1.26 mol−1 dm3 is less(1/55 of the value) than that of 3.0×102 mol−1 dm3
from the reaction [21] of [(H3N)5Co(OAc)]2+ andFe(CN)6
4−, while k1 for the acetate complex is 3.7×10−4 s−1. For the reaction involving[(H3N)5CoOH2)]3+ and Fe(CN)6
4− [7], K=1500�100mol−1 dm3 and k1= (1.9�0.1)×10−1 s−1, whichshows that the two values of k1 are very similar underexperimental errors. The value of K5 for our system ismuch lower than for [(H3N)5Co(OAc)]2+ and[(H3N)5CoOH2]3+.
The value of k1 for our system is slightly higher thanthe system reported by Miralles et al. [8] for reductionsof various substituted (pyridine)pentaamminecobalt(III)complexes by Fe(CN)6
4−. Their k1 values ranged from1.51×10−2 to 76×10−2 s−1, while their K5 valueswere much higher than ours, clearly due to the +3charge on the cobalt(III) complexes. Miralles et al.[8] concluded that internal, outer-sphere electrontransfer process occur within the ion-pair containing apyridine bound to the cobalt(III) centre. Based on thisfact, we can conclude that an internal, outer-sphereelectron transfer process has occurred within the ion-
Fig. 8. A plot of {kobs([H+]2+K3[H+]+K3K4][Fe(CN)6
4−]T−1} vs.
[H+] for the reduction of [(H3N)5CoOMoO3]+ by Fe(CN)64− at
25.0 °C. Table 9Rate parameters for the reduction of [(H3N)5CoOMoO3]+ byFe(CN)6
4−
k2� (mol−1 dm3 s−1)� (°C) k1� (mol−1 dm3 s−1)
2.36�0.051.13�0.0125.02.66�0.0130.4 5.55�0.07
35.3 4.60�0.06 10.5�0.2
�H1� =102�8 kJ mol−1; �H2
� =108�4 kJ mol−1; �S1� =99�31
J mol−1 K−1; �S2� =126�17 J mol−1 K−1.
[(H3N)5CoOMoO3]+ about two times faster thanFe(CN)6
4−. The transition state involving Fe(CN)64− is
more ordered than that involving HFe(CN)63− as can
be seen from the respective values for the entropy ofactivation, the values being �S1
� =99�31 J mol−1
K−1 and �S2� =126�17 J mol−1 K−1, respectively.
k1� =4.60�0.06 mol−1 dm3 s−1 at 35.3 °C fromTable 9 and given the fact that k1� =k1K5 and that

A.A. Holder, T.P. Dasgupta / Inorganica Chimica Acta 331 (2002) 279–289288
Table 10Summary of the activation parameters for the reduction of [(H3N)5CoOMoO3]+ by some reducing agents
�H1� (kJ mol−1)Reductant a �S1
� (J mol−1 K−1) �H2� (kJ mol−1) �S2
� (J mol−1 K−1)
−124�30Ascorbate (1) b 99�929�7 67�3599�31Fe(CN)6
4− (2) c 108�4102�8 126�17204�27 116�27 116�88134�8Aqueous sulfite (3) c
a Each reductant is numbered in Fig. 9.b [22].c This work.
pair formed between [(H3N)5CoOMoO3]+ andFe(CN)6
4−.The rate constants and thermodynamic parameters
for the reduction of [(H3N)5CoOMoO3]+ by some re-ductants, viz. L-ascorbic acid [22], Fe(CN)6
4−, andaqueous sulfite, are summarised in Table 10. The acti-vation parameters for the reactions are very similar andcan be used in an isokinetic plot of �H� versus �S�.The isokinetic plot is shown in Fig. 9. It is concludedthat a common mechanism exists, with �Go
� =72�3kJ mol−1 and �o=324�22 K. Below this temperaturethe reactions are controlled by �H�, that is, the lowerthe �H� value, the higher the rate constant; and abovethis temperature by �S� values. It was evident thatfrom the studies carried out on this work, that anouter-sphere electron transfer took place with the re-duction of the Co(III) centre to Co(aq)
2+ when reductantssuch as L-ascorbic acid [22], Fe(CN)6
4−, and aqueoussulfite are used. The molybdenum(VI) centre, however,remains unaffected.3.7. Calculation of self-exchange rate constants forboth aqueous sulfite (as SO3
2−) and Fe(CN)64−
Redox reactions involving cobalt(III) complexes areoften viewed as outer-sphere electron transfer reactions[22]. The equations from the Marcus theory [23–25] areused in predicting rate constants for heteronuclearouter-sphere redox reactions from the self-exchangerate constants for each partner and the overall equi-librium constant:
k12= (k11k22k12 f12)1/2 (15)
log f12=(log K12)2
4 log�k11k22
1022
� (16)
ln K12=nF(E11° −E22° )
RT(17)
where k22, the self-exchange rate constant for eitherreductant, k11, the self-exchange rate constant for theoxidant, E11° , redox potential for the oxidant{[(H3N)5CoOMoO3]+}, E22° , redox potential for thereductant, K12, the equilibrium constant for the crossreaction, and k12, the second order rate constant for thecross reaction. Normally one would use the Marcus
equations [23–25] on knowing the self-exchange ratesof both reductants and oxidants, in order to calculatek12 and compare it with the observed k12, but in ourcase, k22 for aqueous sulfite (as SO3
2−) is not known sowe need to calculate it. The self-exchange rate constant,k22, for Fe(CN)6
4− has been reported as (0.1–9)×104
mol−1 dm3 s−1 in water at various ionic strengths[26,27]. The self-exchange rate constant, k11, for[(H3N)5CoOMoO3]+, is 2.32×10−5 mol−1 dm3 s−1
[22], k12 (k1� ) (0.77 mol−1 dm3 s−1 at 25.0 °C for thecross reaction involving SO3
2−) is the cross-reactionrate constant, E22° , −0.93 V [28] for the SO4
2−, H2O/SO3
2−,OH− couple, while E11° for [(H3N)5CoOMoO3]+,+0.12 V [22]. K12 is the equilibrium constant (5.67×1017), and k22 is the self-exchange rate constant foraqueous sulfite as SO3
2−, which is unknown. From therate constant for the aqueous sulfite (as SO3
2−) reduc-tion of [(H3N)5CoOMoO3]+ (k12=0.77 mol−1 dm3
s−1), the self-exchange rate constant, k22, for aqueoussulfite (as SO3
2−) was calculated to be 5.37×10−12
mol−1 dm3 s−1.For the reaction involving Fe(CN)6
4−, where k12 (k1� )(1.13 mol−1 dm3 s−1 at 25.0 °C) is the cross-reactionrate constant, k11, the self-exchange rate constant for[(H3N)5CoOMoO3]+, is 2.32×10−5 mol−1 dm3 s−1
[22], k22 is the self-exchange rate constant for
Fig. 9. An isokinetic plot of the activation parameters for thereduction of [(H3N)5CoOMoO3]+by some reducing agents.

A.A. Holder, T.P. Dasgupta / Inorganica Chimica Acta 331 (2002) 279–289 289
Fe(CN)64−, E22° , +0.36 V at zero ionic strength [29] for
the Fe(CN)63−/Fe(CN)6
4− couple, while E11° for[(H3N)5CoOMoO3]+, +0.12 V [22]. We calculated k22
for Fe(CN)64− as 1.10×109 mol−1 dm3 s−1 from the
Marcus equations [23–25], which is much higher thanthe reported value of (0.1–9)×104 mol−1 dm3 s−1 inwater at various ionic strengths [26]. This discrepancyprobably is due to the method carried out and theapproximations used in the calculations by the authors[27] in determining k22 for Fe(CN)6
4−. We believe thatour k22 value is correct under the controlled conditionscarried out for the cross reaction.
In summary, k22, the self-exchange rate constant, foraqueous sulfite (as SO3
2−) was calculated to be 5.37×10−12 mol−1 dm3 s−1, while for Fe(CN)6
4−, it wascalculated to be 1.10×109 mol−1 dm3 s−1 from theMarcus equations [23–25].
Acknowledgements
Funding for this work was provided by the Depart-ment of Chemistry, and a Postgraduate Award by theBoard of Graduate Studies, University of the WestIndies, Mona Campus, Jamaica (to A.A.H.) is grate-fully acknowledged.
References
[1] R. van Eldik, G.M. Harris, Inorg. Chem. 19 (1980) 880.[2] A.A. El-Awady, G.M. Harris, Inorg. Chem. 20 (1981)
1660.
[3] A.C. Dash, A.A. El-Awady, G.M. Harris, Inorg. Chem. 20(1981) 3160.
[4] R. van Eldik, Inorg. Chim. Acta 42 (1980) 49.[5] K.C. Koshy, G.M. Harris, Inorg. Chem. 22 (1983) 2947.[6] R. van Eldik, J. von Jouanne, H. Kelm, Inorg. Chem. 21 (1982)
2818.[7] D. Gaswick, A. Haim, J. Am. Chem. Soc. 93 (1971) 7347.[8] A.J. Miralles, A.P. Szecsy, A. Haim, Inorg. Chem. 21 (1982) 697.[9] A.A. Holder, T.P. Dasgupta, J. Chem. Soc., Dalton Trans.
(1996) 2637.[10] R.G. Hughes, J.F. Endicott, M.Z. Hoffman, D.A. House, J.
Chem. Edu. 46 (1969) 440.[11] Vogel’s Textbook of Quantitative Inorganic Analysis, ELBS, 4th
ed., p. 472.[12] Vogel’s Textbook of Quantitative Inorganic Analysis, ELBS, 4th
ed., p. 452.[13] A.A. El-Awady, G.M. Harris, Inorg. Chem. 20 (1981) 4251.[14] T.P. Dasgupta, G.M. Harris, Inorg. Chem. 23 (1984) 4399.[15] A.A. Holder, T.P. Dasgupta, unpublished work.[16] V.K. Joshi, R. van Eldik, G.M. Harris, Inorg. Chem. 25 (1986)
2229.[17] G. Powell, M.Ph. Thesis, University of the West Indies, Jamaica,
1988.[18] L.M. Kolthoff, Z. Anorg. Allg. Chem. 110 (1920) 143.[19] J. Jordan, G.J. Ewing, Inorg. Chem. 1 (1962) 586.[20] J.N. Bronsted, Z. Phys. Chem. 102 (1922) 169.[21] A. Miralles, A. Haim, R.E. Armstrong, J. Am. Chem. Soc. 99
(1977) 1416.[22] A.A. Holder, T.P. Dasgupta, S.-C. Im, Transition Met. Chem.
22 (1997) 135.[23] R.A. Marcus, Discuss. Faraday Soc. 29 (1960) 21.[24] R.A. Marcus, Can. J. Chem. 55 (1959) 37.[25] R.A. Marcus, J. Phys. Chem. 853 (1963) 67.[26] K.S. Alleman, K. Weber, S.E. Creager, J. Phys. Chem. 100
(1996) 17050.[27] M. Shpoer, G. Ron, A. Lowenstein, Inorg. Chem. 4 (1965) 361.[28] F.A. Cotton, G. Wilkinson, Advanced Inorganic Chemistry, 4th
ed., Wiley, USA, p. 532.[29] A.G. Sharpe, The Chemistry of Cyano Complexes of the Transi-
tion Metals, Academic Press, 1976, p. 106.




















