
Kinetics and energetics of the criegee intermediate in the gas phase. I. The criegee intermediate in...
12
Kinetics and Energetics of the Criegee Intermediate in the Gas Phase. 11. The Criegee Intermediate in the Photooxidation of Formaldehyde, in Alkyldioxy Disproportionation and 0 + Oxoalkane Addition Reactions JOHN T. HERRON, RICHARD I. MARTINEZ,* and ROBERT E. HUIE National Bureau of Standards, Chemical Kinetics Division, Center for Chemical Physics, Washington, I)C 20234, 1J.S.A. Abstract 'The gas-phase kinetics and energetics of the Criegee intermediate, deduced from studies of 0:l-alkene systems, suggest that a hydroxy-substituted Criegee intermediate prohahly participates in the photooxidation of formaldehyde. In contradistinction, the existing evi- dence suggests that the Criegee intermediate and its isomers are prohahly not involved in alkyldioxy disproportionation reactions. In the case of 0 + oxoalkane addition reactions, the Criegee intermediate and its isomers are discussed in terms of a complex equilibrium: 0 + oxoalkane -10 + oxoalkane adduct]. Introduction In the preceding paper [ 11 we discussed the gas-phase kinetics and en- ergetics of the Criegee intermediate in OR-alkene systems. In this paper we discuss its postulated role in reaction systems other than 0:3 + al- kene. The Postulated Intermediacy of Secondary Ozonide Formation in Reaction Systems other than 03 + Alkene The formation and stability of secondary ozonides, as discussed in [l], has some bearing on the question of the formation of Criegee intermediates in alkyldioxy radical disproportionation reactions and in the photooxidation of formaldehyde. * Author to whom correspondence should he addressed International Journal of Chemical Kinetics, Vol. 14,225-236 (1982). Not subject to copyright within the LJnited States. Puhlished by John Wiley & Sons, Inc. CCC 0538-8066/82/030225-12$01.20
Transcript of Kinetics and energetics of the criegee intermediate in the gas phase. I. The criegee intermediate in...

Kinetics and Energetics of the Criegee Intermediate in the Gas Phase.
11. The Criegee Intermediate in the Photooxidation of Formaldehyde, in
Alkyldioxy Disproportionation and 0 + Oxoalkane Addition Reactions
JOHN T . HERRON, RICHARD I. MARTINEZ,* and ROBERT E. HUIE
National Bureau o f Standards, Chemical Kinetics Division, Center for Chemical Physics, Washington, I)C 20234, 1J.S.A.
Abstract
'The gas-phase kinetics and energetics of the Criegee intermediate, deduced from studies of 0:l-alkene systems, suggest that a hydroxy-substituted Criegee intermediate prohahly participates in the photooxidation of formaldehyde. In contradistinction, the existing evi- dence suggests that the Criegee intermediate and its isomers are prohahly not involved in alkyldioxy disproportionation reactions. In the case of 0 + oxoalkane addition reactions, the Criegee intermediate and i t s isomers are discussed in terms of a complex equilibrium: 0 + oxoalkane -10 + oxoalkane adduct].
Introduction
In the preceding paper [ 11 we discussed the gas-phase kinetics and en- ergetics of the Criegee intermediate in OR-alkene systems. In this paper we discuss its postulated role in reaction systems other than 0:3 + al- kene.
The Postulated Intermediacy of Secondary Ozonide Formation in Reaction Systems other than 0 3 + Alkene
The formation and stability of secondary ozonides, as discussed in [l], has some bearing on the question of the formation of Criegee intermediates in alkyldioxy radical disproportionation reactions and in the photooxidation of formaldehyde.
* Author to whom correspondence should he addressed
International Journal of Chemical Kinetics, Vol. 14,225-236 (1982).
Not subject to copyright within the LJnited States. Puhlished by John Wiley & Sons, Inc.
CCC 0538-8066/82/030225-12$01.20

226 HERRON. MARTINEZ, AND HUIE
Alkyldioxy Disproportionation Reactions
Nangia and Benson [2] have proposed that alkyldioxy radicals containing tu-C-H bonds undergo disproportionation reactions not via the Russell mechanism [3], but rather through a process leading to a hydroperoxide and to a Criegee intermediate RCHOO (which they have identified as the zwitterion’):
(1) ZRCH,OO. - RCH,OOH + RCHTO
followed by a very fast chain sequence:
(2) RCFOO + RCH,OO.- RCHO + RCH,O;
RCH,O. + 0, (3 )
(4 ) RCH,O. + RCH,OO. - RCH,OH + RCHOO
Several chain-terminating reactions were considered, but the most im- portant in terms of this discussion is:
- RC(O)OH ( 8 )
We noted in [l] that kinetic studies [4] do not support a biradical mechanism for reaction (7). Furthermore, as we argued in [l], reaction (8) would lead to a “hot” acid (with up to 100 kcal of excess energy), which would rapidly decompose to molecular and free-radical products. Irre- spective of the intermediacy of reaction (8), however, Niki e t al. [5] have shown that secondary ozonides are not products of the photolysis of azo- methane or azoethane in air in the presence of added CHaCHO, under ex- perimental conditions designed to approximate those used in studying ozone-alkene reactions in which ozonides were readily detected [5,6]. Thus without invoking some unique chemistry for the intermediate formed in reaction ( l ) , the existing evidence does not support the Nangia-Benson mechanism [ 21 .2
Refer to [ I ] for a discussion of their nomenclature. There is some additional indirect evidence that hydroxy-substituted alkyldioxy radicals
also do not conform to the mechanism postulated by Nangia and Renson [2]. Thus from the work of S u e t al. 171 on the photooxidation of HyCO one can infer tha t the reaction sequence (1). (5). (7 ) . (8) does not apply to CH2(0H)OO.. Su e t al. 171 proposed tha t CH2(0H)OO.

CKIEGEE 1NTEHMEI)IATE I N THE: GAS PHASE. I 1 227
T h e Photooxidation of Formaldehyde and I ts Implications for the Reactions of Carbenes with 0 2
The intermediacy of a hydroxy-substituted secondary ozonide [ HO- SOZ]* has been implicitly assumed in the mechanism proposed by Morrison and Heicklen [9] to explain their observations in the photooxidation of formaldehyde:
(9) H,CO + hv - I H + H C O
' T H , + C O
( 1 2 ) I + O , - 10,
(13) 10, + H , C O -[HO-SOZ]*- 2 H C ( O ) O H
In reaction (13) we have included the [HO-SOZ]* which is implicitly as- sumed.
The intermediate I was presumed to be the singlet hydroxymethylene HC(0H) of Sodeau and Lee [lo], and hence 1 0 2 is inferred to be the hy- droxy-substituted Criegee intermediate HC(0H)OO.. However, since we have argued above against the reaction sequence (7) and (8), reaction (13) cannot be the analog of the reaction sequence (5), (7), and (8), but rather it probably represents the analogue of the reaction sequence (14) plus the concwted reaction (15).
Reaction (15) is based on the work of Hull e t al. [4]. The latter was dis- cussed in [ 11 with reference to the concerted thermal decomposition of secondary ozonides. Thus the second molecule of HC(0)OH formed via reaction (13) can be viewed as a hydroxy-substituted analogue of the H2C0 of reaction (15).
radicals were formed in the photooxidation of H2CO via
(&-a ) HOO. + H&O .* [(HOO)CH20.] +-: CH,(OH)OO.
(Independent support for this hypothesis comes from a study of Niki e t al. 181 in which CHr(OH)OONO2 was identified as a product of the C12-photoinitiated oxidation of H2CO in the presence of added NO?.) T h e subsequent fate of the CHz(0H)OO- radical can be de- duced from the studies of S u e t al. 171, who used the isotopically labeled reactants HIW1"O and D&O. T h e Nangia-Henson mechanism 121 would predict the formation of some DC(0)OI); none was found by Su e t al. 171. T h e scheme below incorporates the conclusions of the combined isotopic-labeling experiments of S u e t al. 171 apposite to the mechanism

228 HERRON, MARTINEZ, AND HUIE
Reaction (12) also presents a problem in that the I02 produced in that reaction would probably be extremely "hot," and by analogy with reaction (16),
(16) CH2 + 0 2 - CH202 - products
would be expected to rapidly decompose. We would not expect the ther- mochemistry of reaction (12) to differ greatly from that for reaction (16). Reference to Fig. 1 of [ 1) shows that the Criegee intermediate formed in reaction (16) contains as much as 185 kcal/mol of energy with respect to the ground state of the isomeric acid HC(O)OH, and is expected to have a very short lifetime with respect to decomposition to molecular and free- radical products. This conclusion is supported by the observations that CO, CO2, H2, and probably H are among the products of reaction (16)
There is evidence, however, that Criegee intermediates produced in the reaction of some carbenes with 0 2 can be stabilized. Thus photooxidation of diazo compounds in solution in the presence of aldehyde leads to the formation of ozonides [13], and in the presence of alkenes leads to the for- mation of epoxides [ 14). However, only diaryl-substituted diazo com- pounds lead to ozonide and epoxide formation. For alkyl-substituted diazo compounds there is no evidence for ozonide or epoxide formation. This suggests that diaryl-substituted Criegee intermediates are much more readily stabilized than the corresponding alkyl-substituted Criegee inter- mediates.
The foregoing discussion again suggests that reaction (12) would not be expected to lead to a stabilized HC(0H)OO.. However, the fact remains that the reaction sequence (9)-( 13) appears to successfully explain the limiting quantum yields of 2, 0, and 0, respectively, for the products HC(O)OH, H2, and CO from the direct unsensitized photooxidation of formaldehyde in the presence of a very large excess of 0 2 and HzCO 191. Consequently, if viewed in the context of the discussion of [l] with reference to the enhancement of the quenching and scavenging of hot Criegee in-
[11,12].
proposed by Nangia and Benson [2]:
(a',-a') H i 8 0 1 ' 0 . + D,CO =[(H180 '80)CD,0 . ] ~ C D , ( O H ) ' 8 0 1 8 0 ~
(1') 2CD,(OH)180180~ -CD,(OH)180180D + CD(OH)1801*0.
( 5 ' ) 1 D,CO
( 8 ' )
DC( I80)OD

CRIE(;I.:E: IN‘r‘I.:HME:DIATE IN T H E GAS PHASE. I1 229
termediates by a large excess of added aldehyde, their observations 191 would suggest the following. The nascent very hot I02 formed in reaction (12) may be sufficiently quenched by the HZCO (which is in large excess) so as to form an intermediate, “hot” secondary ozonide in a “quasi-con- certed” process (refer to footnote 10 in [l]), where the quenching molecule (H2CO) can immediately become the scavenger of the quenched 102. If this plausible interpretation is correct, then reactions (12) and (13) would best be replaced by the following scheme, which is consistent with the ex- perimental observations [9] and with our current knowledge of Criegee intermediates in the gas phase.:’
(12’) I + 0,- 10:
decomposi t ion products (CO, CO,, H,O, etc.)
(13’) 10: + H,CO - [IO:(H2CO)]- [HO-SOZ]*- 2HC(O)OH
The Postulated Intermediacy of the Criegee Intermediate and Its Isomers in 0 + Oxoalkane Addition Reactions
Postulated Role of Methylenebis(0xy)
atoms with formaldehyde, In a recent study [15) of the low-pressure (-1.6 torr) reaction of O(:’P)
(17) 0 + H2CO - products
Chang and Barker observed the production of substantial yields of COP (-30%). These yields were unaffected by a significant variation in [02] / [0] . Thus they concluded that a major primary channel of reaction (17) involves addition of the 0 atoms to the carbonyl carbon of H2CO [as distinct from the abstraction of the aldehydic H atom which is usually assumed in reac- tion (17’)4]. Hence they argued that methylenebis(0xy) is the primary
‘’ The “hot” intermediate secondary ozonide of reaction (13‘) clearly needs to have the necessary internal energy in order to decompose in the manner prescribed by Hull et al. 141, and this “quasiconcerted” process provides the means not only to “quench” the nascent Hc(OH)00 . but also to ensure the formation of a “hot” transient secondary ozonide. Pre- sumably, then, it should be possible at very high pressures to quench and trap this intermediate ozonide.
They argued 115) against the reaction sequence (17’) + (18) as the primary source of CO?,
(17‘)
(18) O + H C O -H+CO:!
0 + H2CO + HCO + OH
since a large variation in [O2]/(0] presumably would have significantly affected the CO2 yields because of the competition between reactions (18) and (19).
(19) 0 2 + HCO + HOL, + CO

230 HERRON. MARI'INEZ, AND HUIL.:
adduct of reaction (17)."." This postulate, if correct, could contribute to further understanding of the chemistry of the Criegee intermediate, and will be discussed briefly.
A t room temperature the specific rate of 0 + CH&HO is approximately three times that of 0 + H&O [16-181, while that of 0 + acetone could not be measured [19a] by the standard competitive technique [19b] and was found to be about 700 times slower than that of 0 + CH&HO [20]. This trend in reactivity can be readily rationalized in terms of abstraction of a hydrogen atom from the labile aldehydic C-H bond (cf. discussion in [all). On the other hand, however, if addition were a major process and were to occur as postulated by Chang and Barker [15], one would expect that the relative ordering of reactivity of 0 atoms toward carbonyl compounds would be R&O > RCHO > H2CO. Hence addition of 0 atoms to oxalkanes is, in general, not consistent with experiment [15-201, a t least not in the manner postulated by Chang and Barker [15].
Apart from the possibility that the 0 + H&O reaction is exceptional, an explanation for the experimentally determined trend in reactivity (ko+KcHo > ko+H2C0 >> ko+R2CO) [15-211 could conceivably be proposed in terms of an addition process if all of the following three conditions could be satisfied simultaneously: (i) if a reversible reaction were involved in which the equilibrium strongly favors the reactants 0 + oxoalkane (i.e., no reaction),
(20,-20) 0 + oxoalkane ?z- [0 + oxoalkane adduct]
(i i) if a fast reaction, which is presumably available only to the adducts of 0 + aldehydes and not to the adducts of 0 + ketones, were to shift the equilibrium (20,-20) to the right by removing the adduct as i t is formed, and (iii) if the reaction involved in (i i) were to lead to the same experi- mentally observed product distributions which are now attributed to ab- straction of the aldehydic H. These conditions are discussed next.
Conditions Required for the Occurrence of 0 + Oxoalkane Addi t ion Reactions
Condition (i i i): Products expected from 0 + oxoulkane addition re- uctions. In Fig. 1 of [ 11, the energetics of @ suggest that the decomposition
Reference to Fig. 1 of [ 11 indicates that this newly formed biradical would possess some- what less internal energy than the same biradical formed in the 03-ethene reaction in the absence of collisional stabilization. Chang and Barker [15] also estimated that the activation energy for isomerization of methylenebis(0xy) to the "hot" excited acid is E 5 10.5 kcalimol (this value, which was incorporated into Fig. 1 of [l], is too large to be consistent with the observed stability of dioxirane (cf. [ 11). The kinetics implications for methylenebis~oxy) of having such a high activation barrier for isomerization to the acid were discussed in [l].
Measurement of the initial free-radical product distribution arising from their postulated methylenebis(oxy) adduct of 0 + HzCO would be most useful in resolving the question of the origin of their observed stable decomposition products.
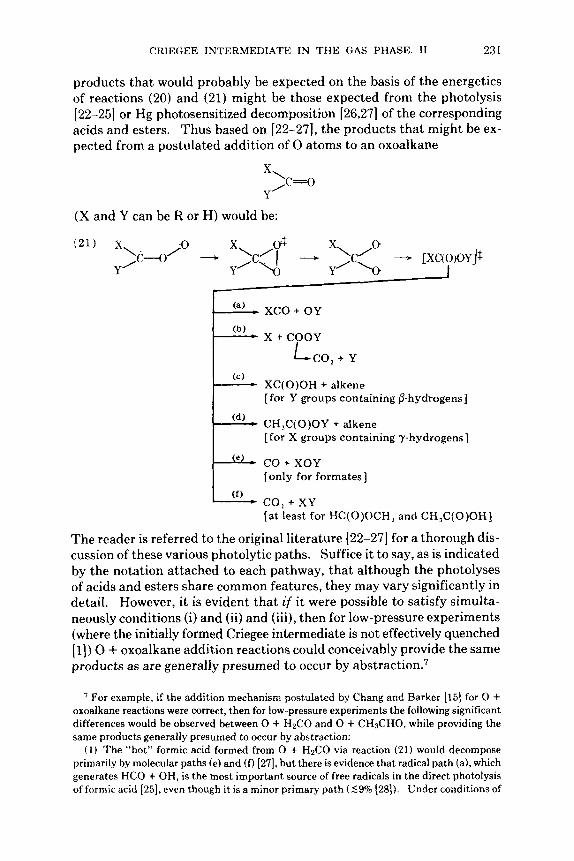
CRIEGEE INTERMEDIATE I N T H E GAS PHASE. I1 231
products that would probably be expected on the basis of the energetics of reactions (20) and (21) might be those expected from the photolysis [22-251 or Hg photosensitized decomposition [26,27] of the corresponding acids and esters. Thus based on [22-271, the products that might be ex- pected from a postulated addition of 0 atoms to an oxoalkane
x\ ,c=o Y
(X and Y can be R or H) would be:
xco + OY
x + COOY
L e o , + Y
XC(0)OH + alkene [for Y groups containing @-hydrogens]
CH,C(O)OY + alkene [ fo r X groups containing 7-hydrogens]
co f XOY [only for formates]
co, + XY [at least for HC(O)OCH, and CH,C(O)OH]
The reader is referred to the original literature [22-271 for a thorough dis- cussion of these various photolytic paths. Suffice it to say, as is indicated by the notation attached to each pathway, that although the photolyses of acids and esters share common features, they may vary significantly in detail. However, it is evident that if it were possible to satisfy simulta- neously conditions (i) and (ii) and (iii), then for low-pressure experiments (where the initially formed Criegee intermediate is not effectively quenched [I]) 0 + oxoalkane addition reactions could conceivably provide the same products as are generally presumed to occur by ab~tract ion.~
For example, if the addition mechanism postulated by Chang and Barker (151 for 0 + oxoalkane reactions were correct, then for low-pressure experiments the following significant differences would be observed between 0 + HPCO and 0 + CHsCHO, while providing the same products generally presumed to occur by abstraction:
( 1 ) The “hot” formic acid formed from 0 + H2CO via reaction (21) would decompose primarily by molecular paths (e) and (f) (271, but there is evidence that radical path (a), which generates HCO + OH, is the most important source of free radicals in the direct photolysis of formic acid (251, even though it is a minor primary path (59% [28]). Under conditions of

232 HERRON, MARTINEZ. A N D HUIE
Condition (i): Equilibrium (20,-20) favors the reactants. If addition via reaction (20) were a viable process, then the adduct of 0 + H2CO would have to be one of the three isomers of the Criegee intermediate: dioxy- methylene, dioxirane, or methylenebis(oxy) [ 11. Based on the energetics of Fig. 1 of [l], one can readily show that only for dioxymethylene (i.e., the “stabilized” Criegee intermediate) would the equilibrium favor the reac- tants.8
Since condition (i), together with conditions (ii) and (iii), must be sat- isfied in order for addition to be a viable process for the 0 + H2CO reaction,
excess [O] (with or without 0 2 ) . the HCO would be rapidly converted to OH via fast radical reactions such as 0 + HCO - OH + CO, 0 2 + HCO + HO2 + CO, and H 0 2 + 0 - OH + 0 2
[29]. Then both the OH formed directly in (a) and indirectly from HCO could react with the HzCO in the fast abstraction reaction OH + H2CO - H20 + HCO (291. It is interesting to note that path (e) could possibly account for the unexplained amount of H20 formed in low- pressure experiments under conditions [O] > [HzCO] [refer to discussion in [30]1 if 0 + ox- oalkane addition were possible.
(2) The “hot” acetic acid or methyl formate which might be formed from 0 + CH:%CHO via reaction (21) would decompose predominantly by the formation of free radicals (23,271; paths (a) and (b) together account for approximately 50% of the primary decomposition of CH&(O)OH and approximately 7@%0 of that from HC(0)OCHs [27]. Hence the 0 + CH:jCHO addition reaction would provide a much greater free-radical activity than that provided by the 0 + HzCO addition reaction, which was discussed above in (1). Methane is also a primary product of the photolysis of both acetic acid and methyl formate [27]. This could possibly account for the unexplained source of the CH4 found in low-pressure discharge-flow experi- ments (refer to discussion in [30]) if 0 + oxoalkane addition were possible. Moreover, the CHsCO formed via path (a) might also contribute to the yield of the diacetyl observed in the low-pressure 0 + CH3CHO system [30], especially since the OH formed in (a) would react with CHsCHO to produce H2O and generate additional CH&O 1301.
It is implicitly assumed in this discussion of condition (i) that reaction (20,-20) represents a true equilibrium wherein reaction (-20) is the only loss process available to the adduct- clearly a gross oversimplification intended to facilitate comprehension of the probable con- sequences of condition (i) with reference to the three isomers of the Criegee intermediate. Thus, e.g., if the activation energy for reaction (-20’)
0 + H,CO = H’
(20’ ,-20’)
is E-20 = AH-20, + E20, N 11.9 kcal/mol (where E20 has been identified with the experi- mentally determined activation energy of -3 kcal/mol for 0 + HzCO [15], and from Fig. 1 of [l], AH-20 = -AH20 N 8.9 kcal/mol), one can estimate the equilibrium constant a t 300 K for reaction (20’,-20’) by using the known k 2 ( ~ N 10” cm3/mol-s [15] and by assuming some value for the preexponential factor for reaction (-209, A-20. Hence in the absence of other loss processes for
\C/O- /
if A-20, were about 10I6 s-l, reaction (-20’) would be highly favored over reaction (20’) a t 300 K for pressures between 1 and lo00 torr; i.e., under these conditions the degree of dissociation a of the o---o
\n /

CRIEGEE IN'I'ERMEIIIA'I'E IN T H E GAS PHASE. I1 233
and since the above discussion suggests that only dioxymethylene might satisfy condition ( i ) , it is important to consider the kinetics implications of having dioxymethylene (and its substituted derivatives) be the primary adduct for 0 + H&O (and its substituted analogues).
The obvious and most important implication of the above discussion of condition (i) is that the Criegee intermediate formed in 03-alkene reactions would have two available decomposition channels: the one to molecular and free-radical products and the one to 0 + oxoalkane. This warrants further study to uniquely determine whether or not this latter possibility is important, especially in view of the fact that the Criegee intermediate formed in the 03 + 2,3-dimethyl-2-butene reaction [ (CH3)&OO.] corre- sponds to that which would supposedly arise from 0 + (CH3)2CO if such an addition reaction were in fact possible. Hence if the reason 0 + (CH:<)&O is very much slower than 0 + H&O [19,20] is because the equilibrium (20,-20) strongly favors the reactants and because there is no exit channel available for the adduct other than reaction (-20), then this would imply that the (CH:i)&OO- formed via 0:j + (CH:,)&=C(CH:j)? would promptly decompose to 0 + (CH:J&O, thus converting the O:< + (CH:{)&=C(CH:Jz system into a very reactive 0 + (CH:3)&=C(CH:J:! system. Hence one would expect to find in an 0s-alkene system the products characteristic of 0 + alkene reactions [20,30]. There is no evi- dence that such is the case [31,32].
A second implication of the above discussion of condition ( i ) is the fol- lowing. If dioxymethylenes were the primary adducts, then one might conclude that the reactions of 0 + oxoalkanes were exact analogues of 0 + alkene reactionsg inasmuch as electrophilic addition of 0 atoms to the electron-rich 0 atom of the C=O bond would have to be involved in for- mation of the dioxymethylenes.'O Hence one might anticipate that the number of alkyl substituents on the C of the C=O bond of oxoalkanes would increasingly favor such an electrophilic addition of 0 atoms to the 0 of the C=O bond both because of the very favorable inductive effect of alkyl substituents'O (not borne out by the experimentally observed trend in aldehydic C-H bond dissociation energies [21]) and because of the in- creased number of degrees of freedom which would tend to promote sta- in favor o f 0 + H2CO would range between about 1 and 0.9 for pressures between 1 and 1000 torr, respectively. Even if A-2" were as low a s 10" s-I, (r would range between about 0.5 and 0.02 for pressures between 1 and lo00 torr, respectively. In contradistinction, similar cal- culations show that for 0 + H2CO 2 [dioxirane or methylenebis(oxy)] the equilibrium would instead highly favor the adduct.
Cf.: 0.1 + aldehyde reactions appear to be exact analogues of 0 3 + alkene reactions
' O Cvetanovic 120,301 showed tha t the primary s tep in 0 + alkene reactions involves pref- erential electrophilic addition of the 0 atom to the less substituted carbon atom of the C=C hond; one might argue that this is the location where one has the highest electron density due to the "electron-donating" inductive effect of the alkyl substituents located on the more highly substituted C of the C=C bond.
[M].

234 HERRON, MARTINEZ, AND HUIE
bihation of the primary adduct. Thus based on the preceding discussion, one would probably expect the following ordering for 1220: R2CO > RCHO. This is not borne out by experiment [15-201.
Speculation concerning condition (ii) is clearly unwarranted until the above conflicts have been resolved. However, the observations of Chang and Barker [ 151 warrant additional experiments to determine whether or not 0 + oxoalkane reactions might possibly proceed via addition instead of (or in addition to) abstraction,ll especially in view of the very recent observations of Diem and Lee [34]12 and of the plausibility of condition (iii), as discussed above.
Summary and Conclusions
The gas-phase kinetics and energetics of the Criegee intermediate, de- duced from studies of 03-alkene systems [l], suggest that the observations of Morrison and Heicklen [9] and of Diem and Lee [34]12 on the photoox- idation of formaldehyde are consistent with the probable involvement of a hydroxy-substituted Criegee intermediate in this system. In contrad- istinction, the existing evidence suggests that the Criegee intermediate or its isomers are probably not involved in alkyldioxy disproportionation re- actions. Additional work is warranted to further explore the possibility that a complex equilibrium may be involved in the 0 + oxoalkane sys- tems
0 + oxoalkane Y [O + oxoalkane adduct] - products
especially in view of the observation by Diem and Lee [34] of 0 3 production in the 02-matrix photooxidation of formaldehyde in the absence of 0 2
photolysis. 12,13 Such a complex equilibrium might possibly provide an
l1 For example, under conditions of high total pressure and with a large excess of aldehyde, observation of secondary ozonide and/or hydroxyalkyl alkanoate formation in an 0 + aldehyde system may provide evidence for the formation of a primary dioxymethylene and/or methy- lenebis(oxy) adduct, respectively. Moreover, reaction (21) would predict some fairly unique products in low-pressure experiments depending on whether or not the rearrangement acid or ester isomer of the primary adduct contains P-H’s on the Y or y-H’s on the X substitu- ents.
Diem and Lee [34] reported that the major products observed in the 02-matrix photo- oxidation (X > 240 nm) of formaldehyde were CO2 and HzO. They also observed as minor products CO, HOz, Hp02, CH30H, HCOOH, and 03. Observation of HCOOH and 0 3 pro- duction in the absence of 0 2 photolysis (since X > 240 nm) may possibly provide the first ev- idence in support of the hypothesis that the Criegee intermediate involved in the equilibrium (20,-20) may have a decomposition channel leading to the formation of oxygen atoms, and thence 0 3 . The implications of their [34] observations will be discussed elsewhere [35].
l3 Note added i n proof: Hatakeyama et al. (HBOA) [J. Phys. Chem., 85,2249 (198l)l re- cently reported that HpCO and 0 3 are also products of the reaction of 3CHz with 0 2 . To ac- count for these products, they suggested that i n addition to their reaction (13) [their reaction numbers are designated here as (13HBOA), etc.],
(13HBOA) 3CH2 + 0 2 - HpCO + O(3P)

CRIEGEE INTERMEDIATE IN THE GAS PHASE. I1 235
explanation for the difference in the CO2 yields YcoZ reported by Chang and Barker ( Yco2 - 0.3) [ 151 and by Morrison and Heicklen ( Yco2 < 0.16) [36] for the 0 + H2CO reaction, especially since in the complex chemical system used by Morrison and Heicklen [36] any dioxymethylene formed via reaction (20) would probably be rapidly removed by its reaction with NOz, etc. [I], in competition with its decomposition to COZ, etc.
Acknowledgment
The authors wish to acknowledge very helpful discussions with Dr. R. J. Cvetanovic, and the information kindly provided by Dr. H. Niki on his research results prior to their publication.
Bibliography
J. T. Herron, R. I. Martinez, and R. E. Huie, Int. J . Chem. Kinet., 14,201 (1982) (the preceding paper). P. S. Nangia and S. W. Benson, Int. J. Chem. Kinet., 12,43 (1980). G. A. Russell, J. Am. Chem. SOC., 79,3871 (1957). I,. A. Hull, I. C. Hisatsune, and J. Heicklen, J. Phys. Chem., 76,2659 (1972). H. Niki, P. D. Maker, C. M. Savage, and L. P. Breitenbach, private communication; J . Phys. Chem., 85,877 (1981). H. Niki, P. D. Maker, C. M. Savage, and L. P. Breitenbach, Chem. Phys. Lett . , 46,327 (1977). F. Su, J. G. Calvert, J. H. Shaw, H. Niki, P. 1). Maker, C. M. Savage, and L. P. Breiten- bach, Chem. Phys. Lett., 65,221 (1979). H. Niki, P. D. Maker, C. M. Savage, and L. P. Breitenbach, Chem. Phys. Lett . , 72,71 (1980).
:WH2 reacts with 0 2 also via their reaction (12),
and that (13HBOA) is followed by (14HBOA). ( 12HBOA) ‘CH2 + 0 2 + M + H2COO. + M
(14HROA) O(’P) + 0 2 + M - 0 3 + M We believe instead that reaction (13HBOA) might proceed not in addition to (12HBOA), but rather oia “(12HBOA)”; that is:
“(12HBOA)” ’CH, + 0, +H,COO.
I “(13HBOA)” H,CO + o ( 3 ~ )
{:her :e.c;;m;!tion products discussed in refs. [ 1 ] and [ 321 .]
Note that “(13HBOA)” corresponds to the reaction (-20) of this paper, and, more specifically, to the reaction (-20’) which is discussed in footnote 8 of this paper. Hence, we believe that the observations of Hatakeyama et al. provide additional support for the possible involvement of the complex equilibrium postulated in this paper.

236 HERRON, MARI’INEZ, A N D H U l E
191 B. M. Morrison, Jr., and J . Heick1en.J. Photochem., 11, 183 (1979). 1101 J. R. Sodeau and E. K. C. Lee, Chem. Phys. Let t . , 57,71 (1978). Ill] R. L. Russell and F. S. Rowland, J . Am. Chem. Soc., 90,1671 (1968). 1121 D. S. Y. Hsu and M. C. Lin, Int. J . Chem. Kinet . , 9,507 (1977). [13] R. W. Murray and A. Suzui, J . Am. Chem. Soc., 93,4963 (1971). 1141 T. A. Hinrichs, V. Ramachandran, and R. W. Murray, J . A m . Chem. Soc., 101, 1282
[ 151 J. S. Chang and .J. R. Barker, J . Phys. Chem., 83,3059 (1979). [16] J. T. Herron and R. E. Huie, J . Phys. Chem. Ref . D a t a , 2,467 (1973). 1171 G. P. R. Mack and R. A. Thrush, J . Chem. Soc., Faraday Trans. 1,69, 208 (1973). [l8] G. P. R. Mack and B. A. Thrush, J . Chem. Soc., Faraday Trans. I , 70,178 (1974). 1191 (a) K. tJ. Cvetanovic, private communication; (b) Adu. Photochem., 1, 115 (1963). 1201 J. H. Lee and R. R. ‘rimmons, Int. J . Chem. Kinet . , 9,133 (1977). 121) D. I,. Singleton, R. 1. Irwin, and R. <J. Cvetanovic, Can. J . Chem., 55,3321 (1977). [22] P. Ausloos and E. W. R. Steacie, Can. J . C‘hem., 33, 1530 (1955). 1231 P. Ausloos, Can. J . Chem., 36, 383 (1958). 1241 R. Borkowski and P. Ausloos, J . A m . Chem. Soc., 83, 1053 (1961 ). 1251 R. Gorden, Jr . , and P. Ausloos, J . f h y s . Chem., 65,1033 (1961). I261 R. F. Pottie and F. P. Lossing, Can. J . Chem., 39, 1900 (1961). [27] P. Kebarle and F. P. Lossing, Can. J . Chem., 37,389 (1959). 1281 ,J. T . Herron and R. E. Huie, J . Am. Chem. Soc., 99,5430 (1977). 1291 R. F. Hampson, Jr., and D. Garvin, “Reaction Rate and Photochemical Data for Atmo-
spheric Chemistry-1977,” National Bureau of Standards, Tech. Note 513, U.S. Gov- ernment Printing Office, Washington, DC, 1978.
1301 R. E. Huie and J. T. Herron, “Reactions of Atomic Oxygen O(“P) with Organic Com- pounds,” in “Progress in Reaction Kinetics,” vol. 8, K. R. Jennings and R. B. Cundall, Eds., Pergamon, Oxford, 1975, pp. 1-80.
(1979).
[31] T. Vrhaski and H. J. Cvetanovic, Can. J . Chem., 38,1063 (1960). 1321 R. I. Martinez, d. T. Herron, and R. E. Huie, J . Am. Chem. SOC. , 103,3807 (1981). [33] R. I. Martinez, Int. J . Chem. Kinet., 14,237 (1982) (the following paper). 1341 M. Diem and E. K. C. Lee, presented a t 15th Intl. Symp. on Free Radicals, Ingonish
1351 R. I. Martinez and J . T. Herron, to be published. [:El R. M. Morrison, Jr., and J. Heicklen, J . Photochem., 15, 131 (1981).
Reach, Canada, June 2-7, 1981.
Received February 19,1981 Accepted June 26,1981




















