
Microclimate and the Zoonotic Cycle of Tick-Borne Encephalitis Virus in Switzerland

Japanese encephalitis virus infection activatescaspase-8 and -9 in a FADD-independent andmitochondrion-dependent manner
Chang-Huei Tsao,1 Hong-Lin Su,2 Yi-Ling Lin,3 Han-Pang Yu,3
Shu-Ming Kuo,2 Ching-I Shen,4 Ching-Wen Chen2 and Ching-Len Liao5
Correspondence
Ching-Len Liao
1Graduate Institute of Life Sciences, National Defense Medical Center, Taiwan, ROC
2The Department of Life Sciences, National Chung-Hsing University, Taiwan, ROC
3Institute of Biomedical Sciences, Academia Sinica, Taiwan, ROC
4The Department of Veterinary Medicine, National Chung-Hsing University, Taiwan, ROC
5Department of Microbiology and Immunology, National Defense Medical Center, Taiwan, ROC
Received 9 January 2008
Accepted 10 April 2008
Japanese encephalitis virus (JEV), a mosquito-borne flavivirus, replicates primarily at the
endoplasmic reticulum and thereby triggers apoptosis of infected cells. This study investigated the
hierarchical activation of the caspase network induced by JEV infection. It was found that JEV
activated the initiators caspase-8 and -9, as well as effector caspase-3, in infected baby hamster
kidney and mouse neuroblastoma (N18) cells. In neuronal N18 cells, JEV infection triggered
cytochrome c release from mitochondria, which in turn activated caspase-9 and -3. Treatment of
JEV-infected N18 cells with cyclosporin A or ruthenium red, which attenuate mitochondrial
injuries, blocked activation of caspase-9 or -3, typifying that, in neuronal cells, this apoptosis
involves the mitochondrial pathway. Alternatively, in caspase-3-deficient MCF-7 cells, JEV
persisted and readily triggered a typical apoptotic response, including cytochrome c release and
full activation of caspase-9 and -8 along with caspase-6, indicating that JEV did not require
caspase-3 to manifest caspase-8 activation and apoptosis. Interestingly, a Fas-associated death-
domain-containing protein (FADD) dominant-negative mutant, which interfered with transmission
of the extracellular death signals into cells through the Fas/tumour necrosis factor (TNF) receptor,
failed to block JEV-induced apoptosis and caspase-8 activation, implying that receptor
oligomerization of the Fas/TNF pathway might not participate in JEV-induced apoptosis. Taken
together, these results illustrate that JEV infection triggers caspase cascades involving the
initiators caspase-8 and -9, probably through FADD-independent but mitochondrion-dependent
pathways.
INTRODUCTION
Caspases are the central executioners for most apoptoticresponses when activated in proteolytic cascades (Cohen,1997; Villa et al., 1997). Apoptosis is implemented byeffector caspases, including caspase-3, -6 and -7, whichcleave a variety of essential cellular targets, and theiractivations are controlled hierarchically by initiator cas-pases such as caspase-8 and -9. Caspase-8 activation istriggered by an extracellular death-receptor signallingpathway involving the Fas/tumour necrosis factor receptor(TNFR), whilst caspase-9 is activated by intracellularapoptotic stimuli through a mitochondrial pathway. Themitochondrial apoptosis pathway is initiated by holocyto-chrome c (Cyto-c) released from mitochondria, whichcauses apoptosome formation and in turn triggers caspase-9 autoactivation (Adams & Cory, 2002; Green & Kroemer,
2004). The death-receptor pathway, via CD95/Fas/Apo1 orTNFR, is activated through oligomerization of membran-ous receptors that recruit procaspase-8 and Fas-associateddeath-domain-containing protein (FADD) to form a
death-inducing signalling complex (DISC). This inducedproximity to DISC formation subsequently stimulatescaspase-8 autoactivation (Chen & Wang, 2002; Muzio etal., 1998). These two pathways function separately in
response to diverse death signals, but may converge to thesame downstream effector caspases, caspase-3, -6 and -7(Cohen, 1997). However, the death-receptor pathway hasbeen reported to potentiate the mitochondrial pathway by
triggering Cyto-c release through caspase-8-cleaved Bidprotein, a pro-apoptotic member of the Bcl-2 family thattargets mitochondria (Li et al., 1998). Caspase-3 and -7may also participate directly in causing Cyto-c release from
Journal of General Virology (2008), 89, 1930–1941 DOI 10.1099/vir.0.2008/000182-0
1930 2008/000182 G 2008 SGM Printed in Great Britain

mitochondria in a feedback-regulation manner (Lakhaniet al., 2006). Crosstalk of this kind between the twopathways further ensures that the given cell will execute theapoptotic process begun by the death stimulus.
Bcl-2 family members are crucial cellular mediators thatmodulate the outer-membrane permeability of mitochon-dria in most apoptotic pathways (Cory & Adams, 2002). Aspro-apoptotic signals to mitochondria, the bcl-2 homo-logue (BH)-3-containing Bid and Bcl-XS lead to release ofapoptogenic factors from the intermembrane space ofmitochondria, including Cyto-c, apoptotic-inducing factor,endonuclease G, Smac/Diablo and Omi/HtrA2 (Green &Kroemer, 2004; Saelens et al., 2004). In contrast, BH1- andBH2-containing anti-apoptotic proteins, such as Bcl-2 andBcl-XL, inhibit mitochondrial permeabilization by formingheterodimers with Bak or Bax and thereby stabilize thepermeability transition (PT) pore complex on mitochon-dria (Green & Kroemer, 2004; Saelens et al., 2004).Vertebrate bcl-2 was the first cellular gene to be recognizedas blocking apoptosis induced by infection of certain RNAviruses (Duncan et al., 1999; Grandgirard et al., 1998; Liaoet al., 1998; Rodgers et al., 1997; Ubol et al., 1994).Moreover, studies involving Sindbis virus (Levine et al.,1993), Semliki Forest virus (Scallan et al., 1997) andinfluenza virus (Hinshaw et al., 1994) have demonstratedthat constitutive bcl-2 expression can prevent infected cellsfrom undergoing apoptosis and help them to becomepersistently infected. These observations suggest that Bcl-2plays a role in forcing an acute cytolytic RNA virus to infectits host cells chronically.
Japanese encephalitis virus (JEV), a mosquito-borneflavivirus, is transmitted to humans through chronicallyinfected mosquitoes. JEV is a positive-sense, single-stranded RNA virus that replicates primarily in thecytoplasm of infected cells. The cytopathic effect (CPE)of this encephalitis may result from the cell death of JEV-infected astrocytes and neuronal cells, or from infiltratedinflammatory cells caused by the cytokines released frominfected astrocytes (Chen et al., 2000, 2004; Liao et al.,2002). JEV infection triggers apoptosis in numerousculture cell lines, such as baby hamster kidney (BHK-21)cells, mouse neuronal N18 cells and human neuralprecursor NT-2 cells (Liao et al., 1997). Overexpressionof bcl-2 can effectively delay JEV-induced cell death andsubsequently convert some target cells into persistentlyinfected cells (Liao et al., 1997, 1998); as the functional Bcl2is primarily located on the mitochondrial membrane, theseobservations suggest that JEV-induced apoptosis mayinvolve the mitochondrial apoptosis pathway. Enforcedbcl-2 expression does not, however, affect JEV replicationand spread during the early lytic infection. Caspase-3-cleaved Bcl-2 products have been found in apoptotic cells,suggesting that caspase-3 activation may participate inJEV-induced apoptosis (Liao et al., 1998). In this study, wefurther characterized caspase activation cascades duringJEV-induced apoptosis, focusing on activation of theinitiators caspase-8 and -9, and the downstream
executioners caspase-3 and -6. We found that JEV infectionappeared to activate caspase-8 by a FADD-independentmanner and to turn on caspase-9 by the mitochondrion-dependent pathway. Although the addition of caspaseinhibitors diminished cell injury, they failed to block JEVreplication, illustrating that completion of the JEV life cycledoes not require caspase activation.
METHODS
Viruses and cell lines. The neurovirulent RP-9 strain of JEV (Chenet al., 1996) was used in this study. Mouse neuroblastoma N18 cellswere grown in RPMI 1640 containing 10 % fetal bovine serum (FBS;Gibco). The human breast epithelial adenocarcinoma cell line MCF-7(ATCC HTB-22) was cultured in high-glucose Dulbecco’s modifiedEagle’s medium (Gibco) supplemented with 10 % FBS. Viruspropagation was carried out in BHK-21 cells and virus titres andthe virus growth curve were determined by plaque assay on BHK-21cells (Chen et al., 1996). Virus titres were determined as p.f.u. ml21.Virus at an m.o.i. of 5 was used to synchronize infection and virustitres correlated linearly with the number of target cells infected byJEV (Liao et al., 1997).
Antibodies, reagents and plasmids. Anti-Cyto-c antibody waspurchased from Pharmingen, anti-CPP32 and anti-PARP antibodieswere purchased from Santa Cruz and anti-actin antibody waspurchased from Chemicon. All cysteine protease inhibitors werepurchased from Clontech and Bachem. Western blotting wasperformed as described previously (Su et al., 2002). Cyclosporin A(CsA) and ruthenium red (RR) were obtained from Sigma. Plasmidscontaining bcl-2 and its mutant D34A were a kind gift from D. E.Griffin and J. M. Hardwick (Cheng et al., 1997; Clem et al., 1998). Theplasmids carrying c-FLIP (Yeh et al., 1998) and FADD dominant-negative mutant (FADD-DN) (Chen & Lai, 2001; Walsh et al., 1998)were obtained from M.-Z. Lai (Academic Sinica, Taipei, Taiwan) andV. Dixit (Genentech, San Francisco, USA), respectively. bcl-2–IRES–DsRed and D34A–IRES–DsRed were constructed by using thebackbone of the pCDNA4-HisMax vector (Invitrogen).
Preparation of subcellular fractions. Preparation of cytosolicfractions from N18 and BHK-21 cells was as described previously(Pastorino et al., 1998) with modifications. Briefly, cells weredisrupted and the nuclei pellets were centrifuged to separate themitochondrial fraction. Supernatants were collected by centrifugationat 100 000 g for 60 min at 4 uC to obtain the cytosolic S-100 fraction.
Apoptotic cell death analysis. Cells grown to approximately 80 %confluence were infected by JEV at an m.o.i. of 5, and flow cytometryusing propidium iodide and Annexin V staining was performed todistinguish apoptosis from necrosis. Only the Annexin V-positivepopulation was recognized as apoptotic cells. In some experiments,BHK-21 cells were transfected with bcl-2–IRES–DsRed, D34A–IRES–DsRed or control DsRed. Alternatively, cells were co-transfected withthe given genes plus the reporter plasmid pEGFP-C1 (Clontech) at aDNA concentration ratio of 5 : 1 and then infected with JEV (m.o.i. of1) at 12 h post-transfection. Apoptotic cells with green fluorescencewere analysed by flow cytometry. In some experiments, the infected,GFP-positive, rounded-up BHK-21 and N18 cells were examinedmicroscopically for the apoptotic hallmarks of cell shrinkage andnuclear condensation (Yu et al., 2002) at the indicated times post-infection (p.i.).
Caspase activity assay. JEV-infected cells (26106) were lysed atvarious times p.i. and the supernatants were incubated withfluorescent caspase substrate (Clontech): 1 mM Ac-DEVD-fac for
Caspase activation in JEV-induced apoptosis
http://vir.sgmjournals.org 1931

caspase-3, 2 mM Ac-IETD-fac for caspase-8 or Ac-LEHD-fac for
caspase-9. Results were obtained using a spectrophotometer
(TopCount; Tecan) with excitation at 400 nm and absorption at
505 nm.
RESULTS
Caspase activation and Cyto-c translocationduring JEV-induced apoptosis
We have established previously that JEV infection cantrigger a typical apoptotic cell death from different types oftarget cell (Liao et al., 1997). Here, we further investigatedhow caspase activation occurs in JEV-infected cells. Usingfluorescent peptide substrates containing individual cas-pase-specific cleavage sites, we found that, in neuronal N18cells, caspase-3, -8 and -9 were first activated after JEVinfection at 24 h p.i. and peaked at 36 h p.i. (Fig. 1a). Theactivation of caspase-9 suggested that JEV infection maydisturb the homeostasis of mitochondria, thereby causingthe target cells to undergo mitochondrion-dependentapoptosis. To test this possibility, we infected N18 cellswith JEV at an m.o.i. of 5 and collected the cytosolicfraction of infected cells to determine the release of Cyto-cfrom mitochondria. Western blot analysis showed thatcytosolic Cyto-c was detected as early as 18 h p.i. in JEV-infected N18 cells (Fig. 1b) and that the levels of Cyto-crelease increased gradually, corresponding well to theseverity of CPE and the number of infected N18 cells thatunderwent cell death (data not shown). These resultssuggested that JEV infection triggers the mitochondrialapoptotic pathway.
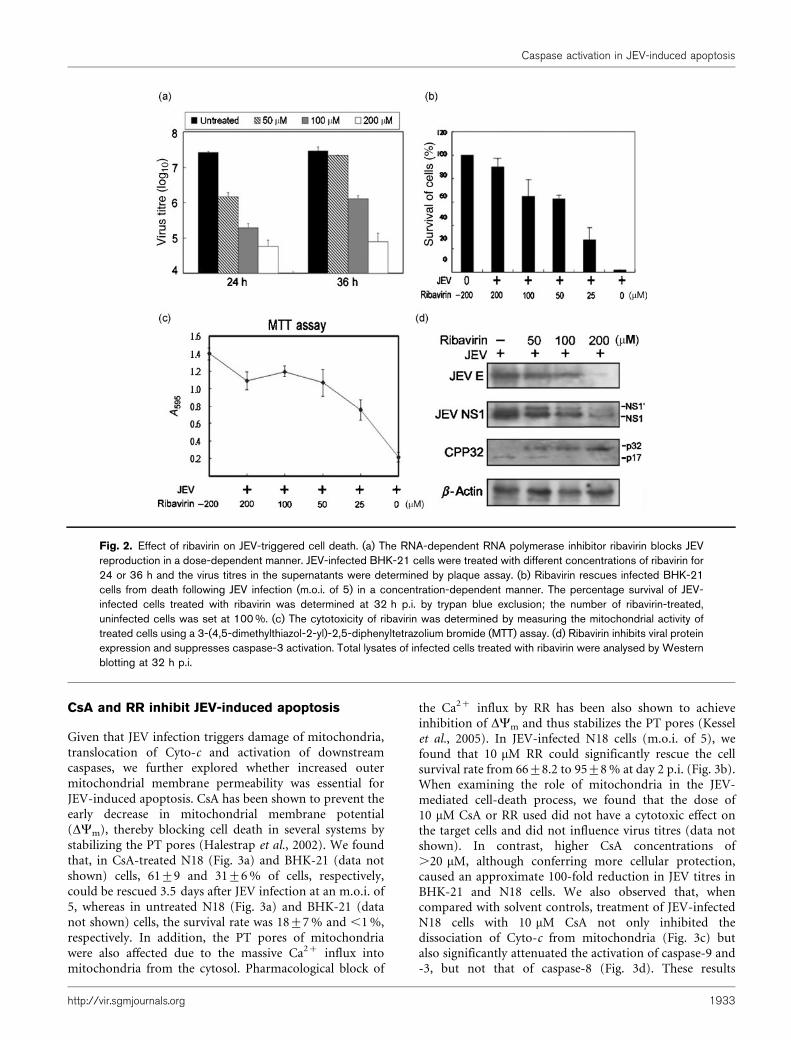
Mitochondrial Cyto-c released into the cytosol causes theformation of apoptosomes, which can in turn activate thedownstream executioner caspase-3 (Adams & Cory, 2002).Activated caspase-3 cleaves several structural and regula-tory proteins, such as poly(ADP-ribose) polymerase(PARP) (Lazebnik et al., 1994), gelsolin (Kothakota etal., 1997) and anti-apoptotic Bcl-2 (Cheng et al., 1997;Grandgirard et al., 1998). We demonstrated that, in JEV-infected N18 cells at 24 h p.i., the levels of p17, thecleavage product and active form of caspase-3 (CPP32),were increased significantly corresponding to a decrease inthe full-length inactive p32, while at the same time, PARPwas cleaved to p85 (Fig. 1b). The gradual increase incaspase-3 activation appeared to correlate well with theseverity of CPE and the number of dead cells seenfollowing JEV infection (data not shown). When wecompared the kinetic changes in the amounts of cytosolicCyto-c with those of p17 cleaved from p32, we found thatCyto-c release appeared to be one of the upstreamapoptotic signals for caspase-3 activation during product-ive JEV replication (Fig. 1c). Similar results for thetranslocation of Cyto-c and caspase activation (Lee et al.,2005) could also be observed in JEV-infected BHK-21 andNT-2 cells (data not shown). To demonstrate that JEVreplication was the direct cause of caspase activation, JEV-infected BHK-21 cells were treated with ribavirin, aspecific RNA-dependent RNA polymerase inhibitor.Ribavirin appeared to inhibit JEV replication (Fig. 2a),to enhance cell-survival rates (Fig. 2b) and to block viralprotein expression (Fig. 2d), as well as concurrentlysuppressing virus-induced caspase-3 activation in a dose-dependent manner (Fig. 2c).
Fig. 1. JEV infection triggers Cyto-c release and caspase activation. (a) Caspase activation was analysed using fluorescentpeptide substrates (see Methods). Total cell lysates from JEV-infected N18 cells at the indicated times were incubated withcaspase peptide substrates and the amount of cleaved product was measured using a fluorimeter. Results are shown asmeans±SD. (b) Western blot of Cyto-c release and caspase-3 activation after JEV infection. M, Mock infection. p32 indicatesthe intact CPP32/caspase-3, whilst p17 corresponds to its active cleaved product. Full-length PARP (p117) and its cleavageproduct (p85) were also detected. (c) The relative photodensities of the protein bands of Cyto-c and cleavage product p17 ofCPP32 on the Western blot in (b) were measured using a bioimaging analyser system (BAS-1000; Fuji Film) and their banddensities at 36 h p.i. were arbitrarily set at 100 %. Virus titres in the supernatant of JEV-infected N18 cells are shown at theindicated times p.i.
C.-H. Tsao and others
1932 Journal of General Virology 89
CsA and RR inhibit JEV-induced apoptosis
Given that JEV infection triggers damage of mitochondria,translocation of Cyto-c and activation of downstreamcaspases, we further explored whether increased outermitochondrial membrane permeability was essential forJEV-induced apoptosis. CsA has been shown to prevent theearly decrease in mitochondrial membrane potential(DYm), thereby blocking cell death in several systems bystabilizing the PT pores (Halestrap et al., 2002). We foundthat, in CsA-treated N18 (Fig. 3a) and BHK-21 (data notshown) cells, 61±9 and 31±6 % of cells, respectively,could be rescued 3.5 days after JEV infection at an m.o.i. of5, whereas in untreated N18 (Fig. 3a) and BHK-21 (datanot shown) cells, the survival rate was 18±7 % and ,1 %,respectively. In addition, the PT pores of mitochondriawere also affected due to the massive Ca2+ influx intomitochondria from the cytosol. Pharmacological block of
the Ca2+ influx by RR has been also shown to achieveinhibition of DYm and thus stabilizes the PT pores (Kesselet al., 2005). In JEV-infected N18 cells (m.o.i. of 5), wefound that 10 mM RR could significantly rescue the cellsurvival rate from 66±8.2 to 95±8 % at day 2 p.i. (Fig. 3b).When examining the role of mitochondria in the JEV-mediated cell-death process, we found that the dose of10 mM CsA or RR used did not have a cytotoxic effect onthe target cells and did not influence virus titres (data notshown). In contrast, higher CsA concentrations of.20 mM, although conferring more cellular protection,caused an approximate 100-fold reduction in JEV titres inBHK-21 and N18 cells. We also observed that, whencompared with solvent controls, treatment of JEV-infectedN18 cells with 10 mM CsA not only inhibited thedissociation of Cyto-c from mitochondria (Fig. 3c) butalso significantly attenuated the activation of caspase-9 and-3, but not that of caspase-8 (Fig. 3d). These results
Fig. 2. Effect of ribavirin on JEV-triggered cell death. (a) The RNA-dependent RNA polymerase inhibitor ribavirin blocks JEVreproduction in a dose-dependent manner. JEV-infected BHK-21 cells were treated with different concentrations of ribavirin for24 or 36 h and the virus titres in the supernatants were determined by plaque assay. (b) Ribavirin rescues infected BHK-21cells from death following JEV infection (m.o.i. of 5) in a concentration-dependent manner. The percentage survival of JEV-infected cells treated with ribavirin was determined at 32 h p.i. by trypan blue exclusion; the number of ribavirin-treated,uninfected cells was set at 100 %. (c) The cytotoxicity of ribavirin was determined by measuring the mitochondrial activity oftreated cells using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. (d) Ribavirin inhibits viral proteinexpression and suppresses caspase-3 activation. Total lysates of infected cells treated with ribavirin were analysed by Westernblotting at 32 h p.i.
Caspase activation in JEV-induced apoptosis
http://vir.sgmjournals.org 1933

suggested that stabilizing mitochondrial PT pores leadsJEV-infected cells to tolerate the mitochondrial deathsignal, probably involving blockage of the massive Ca2+
influx from the cytosol into the mitochondria.
Caspase-resistant bcl-2 is more potent than wild-type bcl-2 in protecting JEV-infected cells fromapoptosis
Overexpression of bcl-2 has been found to delay the JEV-induced apoptotic process and thereby cause persistent JEVinfections in infected BHK-21 cells that would otherwisehave been killed (Liao et al., 1997, 1998). The protectiveeffect of Bcl-2 in JEV-infected cells also supports thesuggestion that mitochondria are involved in JEV-inducedapoptosis. However, in a previous study (Liao et al., 1998),only approximately 5–10 % of bcl-2-overexpressing BHK-21 cells (B2-5 cells) survived after 3 days of JEV infection,and the cleaved fragment of Bcl-2 was readily observed ininfected cells. It has been demonstrated that activatedcaspase-3 can proteolytically cleave Bcl-2, and, instead, theresulting pro-apoptotic fragmental Bcl-2 further promotesapoptotic processes in the stressed cells (Cheng et al., 1997;Grandgirard et al., 1998). To examine whether the cleavageof Bcl-2 may play a role in the apoptosis caused by JEVinfection, we transiently transfected a caspase-3-resistant
human bcl-2 gene (carrying the D34A mutation) intoBHK-21 cells and infected the transfectants with JEV toexamine their apoptotic response (Grandgirard et al.,1998). As shown in Fig. 4, we found that transientexpression of caspase-resistant D34A indeed inhibitedJEV-induced apoptosis more potently than wild-type Bcl-2 in infected cells, as shown by analysis of apoptoticmorphology (Fig. 4a) and flow cytometry (Fig. 4b). Theseresults illustrated that caspase activation and Bcl-2 playcrucial roles in determining the cell’s fate when respondingto virus attacks.
Artificial caspase inhibitors block JEV-inducedapoptosis without modulating virus titres
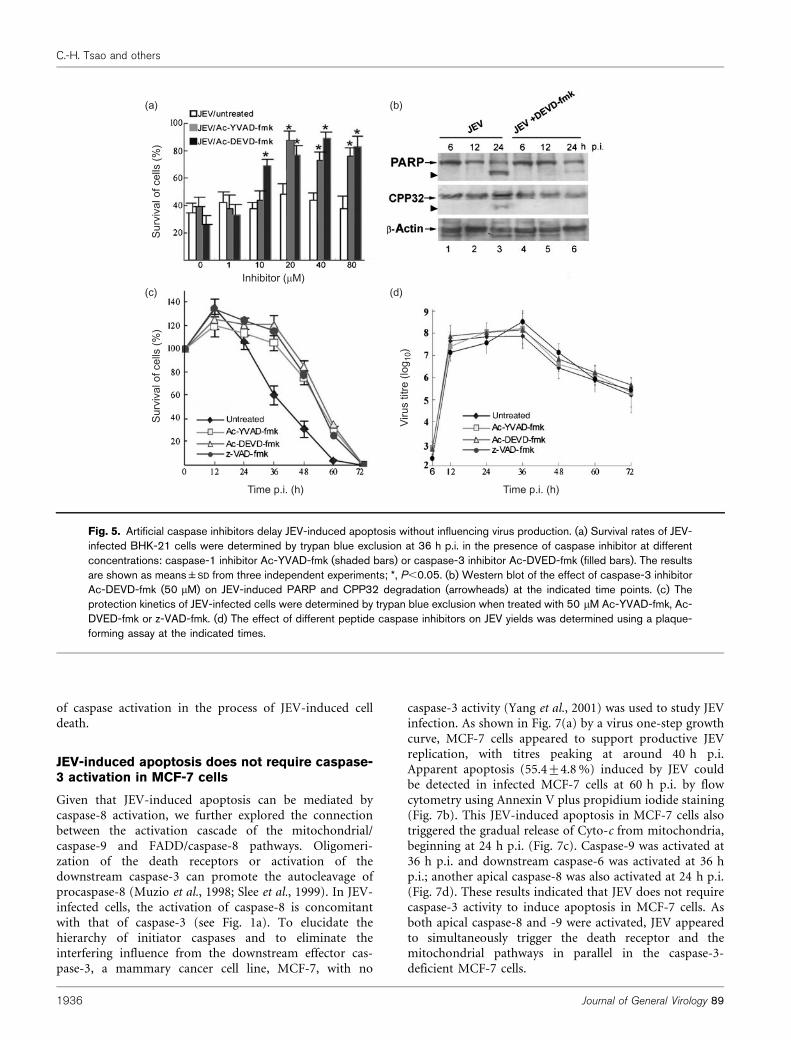
To examine further the relationship between the caspaseactivation and JEV-induced apoptosis, artificial cell-permeable caspase inhibitors were utilized in the followingexperiments. Ac-YVAD-fmk and Ac-DEVD-fmk are clas-sified as caspase-1- and caspase-3-specific inhibitors,respectively (Villa et al., 1997). Ac-VAD-fmk, a pan-caspase inhibitor, blocks activation of caspase-1, -3, -6 and-7 (Villa et al., 1997). Addition of Ac-YVAD-fmk or Ac-DEVD-fmk showed a dose-dependent pattern of rescuefrom JEV-induced cell death (Fig. 5a), and Western blotanalysis further confirmed that the addition of 20 mM Ac-
Fig. 3. CsA and RR delay the JEV-induced death process. (a, b) JEV-infected N18 cells were treated with 10 mM CsA (a) or10 mM RR (b) and survival rates were measured by trypan blue exclusion at the indicated times. (c) The effect of CsA on Cyto-crelease in infected N18 cells was analysed by Western blotting at the indicated times. (d) The effect of CsA treatment on JEV-induced caspase activation was measured at 36 h p.i. using the substrates described in Fig. 1(a). Statistical data represent themeans±SD derived from three independent experiments. *, Statistical significance using Student’s t-test (P,0.05).
C.-H. Tsao and others
1934 Journal of General Virology 89

DEVD-fmk effectively blocked degradation of full-lengthCPP32 and PARP in JEV-infected cells (Fig. 5b). We foundthat, despite these peptide caspase inhibitors havingdifferent target specificities, their suppression of JEV-induced cell death appeared to vary only slightly (Fig. 5c).Notably, JEV-infected cells treated with artificial caspaseinhibitors did not interfere with virus yields at any of thetime points examined (Fig. 5d), suggesting that, althoughthese inhibitors delay the JEV-triggered apoptotic proces-sing, completion of the JEV life cycle does not requirecaspase activation. Nevertheless, these synthetic inhibitorsfailed to induce persistent JEV infection when the culturedperiod was extended (data not shown).
Viral proteins p35 and CrmA and cellular FLIPattenuate JEV-induced apoptosis
The baculovirus p35 protein and poxvirus CrmA proteinhave been used as virus caspase inhibitors in severalapoptotic models. Overexpression of p35 blocks activationof caspase-1 and -3, whilst CrmA inhibits the activity ofcaspase-1 and -8 in apoptotic cells (Dorstyn & Kumar,1997; Tschopp et al., 1998). We investigated the effect ofp35 or CrmA on JEV-induced apoptosis in N18 and BHK-21 cells. We found that both p35 (Fig. 6a, d) and CrmA(Fig. 6b) showed a comparable block in JEV-induced
apoptosis in a dose-dependent manner in infected N18 orBHK-21 cells. Quantitatively, p35 seemed to provide aslightly more potent anti-apoptotic effect than CrmAfollowing JEV-induced apoptosis (Fig. 6a, b), similar toprevious observations for reactive oxygen species (ROS)-induced apoptosis (Datta et al., 1997). In addition, wenoticed that stable p35 expression helped JEV to establish apersistent infection in the target cells (Fig. 6f), suggestingthat, in addition to being a well-known caspase inhibitor,p35 may also confer an additional as-yet-unidentified anti-apoptotic function to rescue doomed JEV-infected cells.
To characterize further the role of caspase-8 in JEV-infected cells, a cellular version of the caspase-8 inhibitor,c-FLIP, was transiently overexpressed in N18 cells. Higherheterodimer affinity between c-FLIP and procaspase-8inhibits caspase-8 activation by sequestering the dormantprocaspase-8 from the DISC formation (Peter, 2004). Wefound that overexpression of c-FLIP in N18 cells indeedenhanced the cell survival rates in response to JEV infection(Fig. 6c). Similarly, the enforced expression of p35 (Fig. 6e),crmA or c-FLIP (data not shown) did not significantlyaffect virus titres, at least during primary JEV infection.Taking these results together, despite not affecting virustitres, inhibition of apoptotic responses by both artificialand natural caspase inhibitors established the crucial role
Fig. 4. Caspase-resistant bcl-2 is more potent than wild-type bcl-2 in inhibiting JEV-induced apoptosis. (a) BHK-21 cells wereco-transfected with human wild-type bcl-2, caspase-resistant bcl-2 (D34A) or pcDNA3 as a control together with pEGFP-C1(DNA concentration ratio of 5 : 1); pcDNA3 was used if necessary to adjust the final amount of DNA transfected to 2 mg. At12 h post-transfection, cells were infected with JEV (m.o.i. of 5) and EGFP+-infected cells with apoptotic characteristics werecounted microscopically at 36 h p.i. as described in Methods. The results are shown as means±SD derived from threeindependent experiments; *, P,0.05 using Student’s t test. (b) The anti-apoptotic effect of bcl-2 or D34A on JEV-infected cells.BHK-21 cells (5�105) were transiently transfected with DsRed control, bcl-2–IRES–DsRed or D34A–IRES–DsRed; after12 h, the resulting cells were infected with JEV at an m.o.i. of 1 and analysed by FACS at 36 h p.i. to determine the percentageof Annexin V+ cells among the DsRed+ population. The data presented were derived from two independent FACS analyses.
Caspase activation in JEV-induced apoptosis
http://vir.sgmjournals.org 1935
of caspase activation in the process of JEV-induced celldeath.
JEV-induced apoptosis does not require caspase-3 activation in MCF-7 cells
Given that JEV-induced apoptosis can be mediated bycaspase-8 activation, we further explored the connectionbetween the activation cascade of the mitochondrial/caspase-9 and FADD/caspase-8 pathways. Oligomeri-zation of the death receptors or activation of thedownstream caspase-3 can promote the autocleavage ofprocaspase-8 (Muzio et al., 1998; Slee et al., 1999). In JEV-infected cells, the activation of caspase-8 is concomitantwith that of caspase-3 (see Fig. 1a). To elucidate thehierarchy of initiator caspases and to eliminate theinterfering influence from the downstream effector cas-pase-3, a mammary cancer cell line, MCF-7, with no
caspase-3 activity (Yang et al., 2001) was used to study JEVinfection. As shown in Fig. 7(a) by a virus one-step growthcurve, MCF-7 cells appeared to support productive JEVreplication, with titres peaking at around 40 h p.i.Apparent apoptosis (55.4±4.8 %) induced by JEV couldbe detected in infected MCF-7 cells at 60 h p.i. by flowcytometry using Annexin V plus propidium iodide staining(Fig. 7b). This JEV-induced apoptosis in MCF-7 cells alsotriggered the gradual release of Cyto-c from mitochondria,beginning at 24 h p.i. (Fig. 7c). Caspase-9 was activated at36 h p.i. and downstream caspase-6 was activated at 36 hp.i.; another apical caspase-8 was also activated at 24 h p.i.(Fig. 7d). These results indicated that JEV does not requirecaspase-3 activity to induce apoptosis in MCF-7 cells. Asboth apical caspase-8 and -9 were activated, JEV appearedto simultaneously trigger the death receptor and themitochondrial pathways in parallel in the caspase-3-deficient MCF-7 cells.
(a) (b)
(c) (d)
Surv
ival
of c
ells
(%)
Surv
ival
of c
ells
(%)
Viru
s tit
re (l
og10
)
Inhibitor (mM)
Time p.i. (h) Time p.i. (h)
Fig. 5. Artificial caspase inhibitors delay JEV-induced apoptosis without influencing virus production. (a) Survival rates of JEV-infected BHK-21 cells were determined by trypan blue exclusion at 36 h p.i. in the presence of caspase inhibitor at differentconcentrations: caspase-1 inhibitor Ac-YVAD-fmk (shaded bars) or caspase-3 inhibitor Ac-DVED-fmk (filled bars). The resultsare shown as means±SD from three independent experiments; *, P,0.05. (b) Western blot of the effect of caspase-3 inhibitorAc-DEVD-fmk (50 mM) on JEV-induced PARP and CPP32 degradation (arrowheads) at the indicated time points. (c) Theprotection kinetics of JEV-infected cells were determined by trypan blue exclusion when treated with 50 mM Ac-YVAD-fmk, Ac-DVED-fmk or z-VAD-fmk. (d) The effect of different peptide caspase inhibitors on JEV yields was determined using a plaque-forming assay at the indicated times.
C.-H. Tsao and others
1936 Journal of General Virology 89

Caspase-8 activation in JEV-induced apoptosis isnot mediated through an FADD-dependentpathway
To clarify whether the death signal to trigger caspase-8activation was extrinsic during JEV-induced apoptoticprocessing, we stably expressed a FADD-DN gene (Chen &Lai, 2001; Newton et al., 2001) in MCF-7 and BHK-21 cells(Fig. 8a), in which FADD-DN can interrupt the externalFas/TNFR sending apoptotic signals inside the cell totrigger the internal caspase-8 activation. We found thatFADD-DN expression in MCF-7 clones, despite success-fully reducing TNF-a-induced apoptosis, failed to blockJEV-induced apoptosis (Fig. 8b); similar results were alsoseen for BHK-21 cells expressing FADD-DN (data notshown). Treatment of JEV-infected MCF-7 cells with 50 or100 mM z-IETD-fmk (a caspase-8-specific inhibitor) sig-nificantly enhanced the survival rate compared with theuntreated control (Fig. 8c), indicating that caspase-8 playsa role, at least in part, in JEV-induced apoptosis in MCF-7cells. In addition, FADD-DN expression did not appear tointerfere with activation of caspase-9, -8 or -3 in eitherJEV-infected BHK-21 (Fig. 8d) or MCF-7 cells (Fig. 8e).
Taken together, these observations suggest that the initiatorcaspase-8 activation induced by JEV infection may bemediated through the death-receptor pathway in anFADD-independent and as-yet-unknown manner.
DISCUSSION
Although JEV infection appears to cause both apoptosisand necrosis of target cells, we failed to distinguish thehierarchy between the apoptotic and necrotic pathwaysfollowing JEV infection in this study. The interplaybetween the two processes is of interest and remains tobe studied further. Despite the exact upstream death signalsremaining unclear, the results from infected neural N18(Fig. 1) and fibroblast BHK-21 (Fig. 4) cells, together withour previous findings (Liao et al., 1997), clearly indicatethat JEV is not only an inducer of apoptotic death but canalso activate caspase cascades in infected cells (Figs 1, 7 and8). The simultaneous activation of caspase-8 and -9 (Fig. 7)together with the specific suppression of caspase-9, but notcaspase-8, by CsA (Fig. 3d) seem to suggest that JEV cantrigger the death-receptor pathway and the mitochondrial
Apop
totic
cel
ls (%
)
Viru
s tit
re (l
og10
)Ap
opto
tic c
ells
(%)
Apop
totic
cel
ls (%
)
(a) (b)
(d) (e) (f)
(c)
Fig. 6. The natural caspase inhibitors CrmA, p35 and FLIP suppress JEV-induced apoptosis. BHK-21 (open bars) or N18(filled bars) cells (5�105) were transiently transfected with 0.4 mg reporter pEGFP-C1 plus the indicated amounts of individualplasmid carrying p35 (a), CrmA (b) or FLIP (c); pcDNA3 was used to adjust the amount of DNA transfected to 2 mg (for p35and FLIP) or 4 mg (for CrmA). After 24 h, the transfected cells were infected with JEV (m.o.i. of 5) and the apoptotic EGFP+
cells were counted as described in Fig. 4. The results are shown as means±SD from three independent experiments; *, P,0.05.(d) DNA ladder formation induced by JEV infection. Low-molecular-mass DNA was extracted from uninfected BHK-21 (lanes 1and 2), JEV-infected BHK-21 cells stably expressing p35 (lanes 3 and 4) and JEV-infected BHK-21 cells (lanes 5 and 6), andanalysed by 2 % agarose gel electrophoresis. (e) The effect of p35 expression on JEV replication. Virus titres from supernatantsof p35- or pcDNA3-transfected BHK-21 cells were determined at 18 or 24 h p.i. (f) p35 transient expression results in BHK-21cells persistently infected with JEV. After infection, some p35-transfected BHK-21 cells became persistently infected by JEV,indicated by staining with anti-JEV NS1 monoclonal antibody at 72 h p.i.
Caspase activation in JEV-induced apoptosis
http://vir.sgmjournals.org 1937

pathway separately and in parallel. It will be of interest toreveal the upstream signals that trigger these two pathwaysfollowing JEV infection.
Virus replication can cause mitochondrial injury by thegeneration of secondary toxic metabolites, such as ROS,nitric oxide and superphysiological Ca2+ concentrations,which may serve as the upstream signals to modulate themitochondrial PT pores, leading progressively to DYm
collapse, Cyto-c release and caspase-9 activation (Cai &Jones, 1998; Green & Kroemer, 2004; Tan et al., 1998). Inan early stage of JEV-induced apoptosis, we found a changein mitochondrial potential and the generation of ROS inN18 cells (Lin et al., 2004), and even cells treated with UV-inactivated JEV caused ROS-dependent mitochondrialinjury and a decrease in DYm (Lin et al., 2004).Alternatively, we have shown previously that a high levelof JEV replication on the endoplasmic reticulum (ER)membrane triggers classical ER stress and an unfoldingprotein response (Su et al., 2002), which may lead todepletion of the storage of Ca2+ in the ER lumen, causingan elevation of Ca2+ concentrations to abnormal levels andactivating ER-associated caspase-12, which in turn triggersthe mitochondrial pathway. It was therefore not unexpec-ted to see in this study that stabilizing the mitochondrial
PT pores by CsA or blocking the influx of Ca2+ into themitochondria by RR could delay the progression of JEV-induced apoptosis. The ectodomain of the M protein ofJEV has been shown to be an apoptotic inducer (Catteau etal., 2003a), probably by triggering DYm collapse, but notby generating ROS or activating caspase-9 (Catteau et al.,2003b). Conceivably, JEV infection may also causedisruption of DYm by producing certain toxic secondarymetabolites, such as ROS, nitric oxide or abnormal Ca2+
concentrations in the target cells.
Several RNA viruses have been shown to readily activate thecaspase-8-associated apoptotic pathway (Bitzer et al., 1999;Clarke & Tyler, 2003; Liu et al., 2006). In the case of reovirus,the induced apoptosis involves the activation of deathreceptors DR5 or DR4 with TNF-related apoptosis-inducingligand (TRAIL, also called Apo2L), which depends mainlyon the intracellular FADD association with procaspase-8(Clarke & Tyler, 2003). On the other hand, althoughcaspase-8 is often activated through the death-receptorpathway in many systems, Sendai virus can activate apicalcaspase-8 without involvement of the upstream deathreceptors during the apoptotic process (Bitzer et al., 1999),but whether this caspase-8 activation is dependent or not onassociation with FADD to form DISC remains unclear. Our
(a)
(d)
Caspase-9
(b) (c)Vi
rus
titre
(log
10)
Apop
totic
cel
ls (%
)
Caspase-8 Caspase-6 Caspase-3
Fig. 7. JEV infection triggers apoptosis and caspase activation in the absence of caspase-3 in MCF-7 cells. (a) JEV replicationlevels were determined in MCF-7 cells by a virus one-step growth curve. (b) Apoptotic cells in MCF-7 cells infected by JEV(m.o.i. of 1) were analysed by flow cytometry using Annexin V staining at the indicated times p.i. M, Mock infection at 60 h p.i. (c)Cyto-c release in the cytosolic fraction was detected by Western blotting of JEV-infected MCF-7 cells at the times indicated p.i.(d) Activation of different caspases after JEV infection was determined using specific fluorescent peptide substrates (seeMethods) at the indicated times p.i. Results are shown as means±SD derived from three independent experiments. *, P,0.05.
C.-H. Tsao and others
1938 Journal of General Virology 89

results provide a similar example in that JEV infection wasshown to induce apical caspase-8 activation in a FADD-independent manner, leading to apoptosis, even withoutcaspase-3 (Figs 7 and 8). Suppression of the mitochondrialapoptotic pathway by CsA treatment failed to inhibit JEV-induced caspase-8 activation (Fig. 3d), indicating these weretwo separate apoptotic pathways occurring in infected cells.This observation also differs from a previous study ofrhinovirus-induced apoptosis via the mitochondrial path-way without caspase-8 activation (Deszcz et al., 2005). Asoverexpression of NS3 protein derived from West Nile virusor dengue virus has been shown to initiate caspase-8-dependent apoptosis in neural cells (Ramanathan et al.,2006; Shafee & AbuBakar, 2003), it is of interest to identifywhich of the protein(s) of JEV may be responsible fortriggering apoptosis in infected cells in a caspase-8-dependent manner.
FADD is the final common link between the death-domain-containing receptors and caspase-8. How canprocaspase-8 undergo autocleavage without the involve-ment of FADD in an apoptotic process? Anticancer drug-mediated caspase-8 activation has been documented as anFADD-independent process (Wesselborg et al., 1999), butits activation mechanism remains largely unidentified.Although the activation of caspase-8 by caspase-3 has beenreported occasionally (Slee et al., 1999), we have shown inthe present study that JEV-induced caspase-8 activationwas able to occur without caspase-3 in infected MCF-7 cells(Fig. 7), indicating that effector caspase-3 is not alwaysessential for the caspase-8-mediated apoptotic pathway inJEV-infected cells. Using the same FADD-DN approach aswe did in this study, it has been shown previously(Schrantz et al., 2001) that transforming growth factor-b
induces FADD-independent activation of caspase-8 in theapoptotic pathway regulated by p38 MAP kinase. In ourprevious study (Su et al., 2002), we demonstrated that JEVreplication triggers the unfolding protein response ininfected cells as indicated by induction of certainchaperones, resulting in apoptotic cell death. Moreover,JEV infection also activates the expression of transcriptionfactor CHOP/GADD153 and triggers the activation of p38MAP kinase. Ectopic expression of CHOP could enhanceJEV-induced apoptosis, whereas treatment with a p38-specific inhibitor, SB203580, appeared to attenuate JEV-induced apoptosis. Conceivably, it could be the activatedp38 MAP kinase that triggers FADD-independent activa-tion of caspase-8 in JEV-induced apoptosis. Alternatively,the possibility that the TRAIL pathway might alsoparticipate in JEV-induced cell death cannot be excluded,as the cytokine TRAIL has previously been reported toactivate the caspase cascade by an FADD-independentmechanism (Pan et al., 1997; Sheridan et al., 1997).
In summary, we found that JEV replication induced acaspase-mediated apoptosis and that caspase inhibitors,although able to alleviate the apoptotic process, had noeffect on virus replication, consistent with a previousobservation from West Nile virus-induced apoptosis(Kleinschmidt et al., 2007). These observations thereforesuggest that flavivirus replication does not require caspaseactivation to complete the virus life cycle. JEV infection notonly triggers caspase-9 activation via the mitochondrialpathway but also induces caspase-8 activation separatelythrough a FADD-independent death-receptor pathway.Taken together, our results suggest that caspase cascadestriggered by JEV infection may start from activation of theapical caspase-8 and -9, probably through a FADD-independent but mitochondrial-dependent pathway.
ACKNOWLEDGEMENTS
This research work was supported by the National Science Council
(NSC96-2320-B-016- 027-My3 and NSC96-3112-B-016-002) and the
Department of Defense (DOD96-07-03), Taipei, Taiwan, ROC.
MCI-7(a)
(b)
Surv
ival
of c
ells
(%)
Surv
ival
of c
ells
(%)
(c)
(d)
Caspa
se-9
Caspa
se-8
Caspa
se-9
Caspa
se-8
Caspa
se-3
(e)
FADD-DN
BHK-21
Fig. 8. JEV induces caspase-8 activation in a FADD-independentmanner. (a) Transgene (Tg) expression of FADD-DN in MCF-7(lane 1) and BHK-21 (lane 3) cells was examined by Westernblotting using an anti-Flag antibody; Wt (lanes 2 and 4) indicatesnon-transfected parental cells. (b) Survival rates of MCF-7/FADD-DN-transfected (open bars) and MCF-7 (filled bars) cells inresponse to treatment with 1 ng TNF-a ml”1 or infection with JEVat an m.o.i. of 5 were determined at 48 h p.i. by trypan blueexclusion. (c) The rescue from cell death of JEV-infected MCF-7cells treated with 50 or 100 mM z-IETD-fmk (caspase-8 inhibitor)was determined at 48 h p.i. (d, e) The effect of FADD-DNexpression on JEV-triggered caspase activation was determined inBHK-21 (d) and MCF-7 (e) cells using fluorescent peptidesubstrates (see Methods). Results are shown as means±SD
derived from three independent experiments; *, P,0.05.
Caspase activation in JEV-induced apoptosis
http://vir.sgmjournals.org 1939

REFERENCES
Adams, J. M. & Cory, S. (2002). Apoptosomes: engines for caspaseactivation. Curr Opin Cell Biol 14, 715–720.
Bitzer, M., Prinz, F., Bauer, M., Spiegel, M., Neubert, W. J., Gregor, M.,Schulze-Osthoff, K. & Lauer, U. (1999). Sendai virus infectioninduces apoptosis through activation of caspase-8 (FLICE) andcaspase-3 (CPP32). J Virol 73, 702–708.
Cai, J. & Jones, D. P. (1998). Superoxide in apoptosis. Mitochondrialgeneration triggered by cytochrome c loss. J Biol Chem 273,11401–11404.
Catteau, A., Kalinina, O., Wagner, M. C., Deubel, V., Courageot, M. P.& Despres, P. (2003a). Dengue virus M protein contains aproapoptotic sequence referred to as ApoptoM. J Gen Virol 84,2781–2793.
Catteau, A., Roue, G., Yuste, V. J., Susin, S. A. & Despres, P. (2003b).Expression of dengue ApoptoM sequence results in disruptionof mitochondrial potential and caspase activation. Biochimie 85,789–793.
Chen, Y. & Lai, M. Z. (2001). c-Jun NH2-terminal kinase activationleads to a FADD-dependent but Fas ligand-independent cell death inJurkat T cells. J Biol Chem 276, 8350–8357.
Chen, M. & Wang, J. (2002). Initiator caspases in apoptosis signalingpathways. Apoptosis 7, 313–319.
Chen, L. K., Lin, Y. L., Liao, C. L., Lin, C. G., Huang, Y. L., Yeh, C. T., Lai,S. C., Jan, J. T. & Chin, C. (1996). Generation and characterization oforgan-tropism mutants of Japanese encephalitis virus in vivo and invitro. Virology 223, 79–88.
Chen, C. J., Liao, S. L., Kuo, M. D. & Wang, Y. M. (2000). Astrocyticalteration induced by Japanese encephalitis virus infection.Neuroreport 11, 1933–1937.
Chen, C. J., Chen, J. H., Chen, S. Y., Liao, S. L. & Raung, S. L. (2004).Upregulation of RANTES gene expression in neuroglia by Japaneseencephalitis virus infection. J Virol 78, 12107–12119.
Cheng, E. H., Kirsch, D. G., Clem, R. J., Ravi, R., Kastan, M. B.,Bedi, A., Ueno, K. & Hardwick, J. M. (1997). Conversion of Bcl-2 to aBax-like death effector by caspases. Science 278, 1966–1968.
Clarke, P. & Tyler, K. L. (2003). Reovirus-induced apoptosis: aminireview. Apoptosis 8, 141–150.
Clem, R. J., Cheng, E. H., Karp, C. L., Kirsch, D. G., Ueno, K.,Takahashi, A., Kastan, M. B., Griffin, D. E., Earnshaw, W. C. & otherauthors (1998). Modulation of cell death by Bcl-xL through caspaseinteraction. Proc Natl Acad Sci U S A 95, 554–559.
Cohen, G. M. (1997). Caspases: the executioners of apoptosis. BiochemJ 326, 1–16.
Cory, S. & Adams, J. M. (2002). The Bcl2 family: regulators of thecellular life-or-death switch. Nat Rev Cancer 2, 647–656.
Datta, R., Kojima, H., Banach, D., Bump, N. J., Talanian, R. V.,Alnemri, E. S., Weichselbaum, R. R., Wong, W. W. & Kufe, D. W.(1997). Activation of a CrmA-insensitive, p35-sensitive pathway inionizing radiation-induced apoptosis. J Biol Chem 272, 1965–1969.
Deszcz, L., Gaudernak, E., Kuechler, E. & Seipelt, J. (2005).Apoptotic events induced by human rhinovirus infection. J GenVirol 86, 1379–1389.
Dorstyn, L. & Kumar, S. (1997). Differential inhibitory effects ofCrmA, P35, IAP and three mammalian IAP homologues on apoptosisin NIH3T3 cells following various death stimuli. Cell Death Differ 4,570–579.
Duncan, R., Muller, J., Lee, N., Esmaili, A. & Nakhasi, H. L. (1999).Rubella virus-induced apoptosis varies among cell lines and ismodulated by Bcl-XL and caspase inhibitors. Virology 255, 117–128.
Grandgirard, D., Studer, E., Monney, L., Belser, T., Fellay, I.,Borner, C. & Michel, M. R. (1998). Alphaviruses induce apoptosis in
Bcl-2-overexpressing cells: evidence for a caspase-mediated, proteo-
lytic inactivation of Bcl-2. EMBO J 17, 1268–1278.
Green, D. R. & Kroemer, G. (2004). The pathophysiology of
mitochondrial cell death. Science 305, 626–629.
Halestrap, A. P., McStay, G. P. & Clarke, S. J. (2002). The
permeability transition pore complex: another view. Biochimie 84,
153–166.
Hinshaw, V. S., Olsen, C. W., Dybdahl-Sissoko, N. & Evans, D.(1994). Apoptosis: a mechanism of cell killing by influenza A and B
viruses. J Virol 68, 3667–3673.
Kessel, D., Castelli, M. & Reiners, J. J. (2005). Ruthenium red-
mediated suppression of Bcl-2 loss and Ca2+ release initiated by
photodamage to the endoplasmic reticulum: scavenging of reactive
oxygen species. Cell Death Differ 12, 502–511.
Kleinschmidt, M. C., Michaelis, M., Ogbomo, H., Doerr, H. W. &Cinatl, J., Jr (2007). Inhibition of apoptosis prevents West Nile virus
induced cell death. BMC Microbiol 7, 49.
Kothakota, S., Azuma, T., Reinhard, C., Klippel, A., Tang, J., Chu, K.,McGarry, T. J., Kirschner, M. W., Koths, K. & other authors (1997).Caspase-3-generated fragment of gelsolin: effector of morphological
change in apoptosis. Science 278, 294–298.
Lakhani, S. A., Masud, A., Kuida, K., Porter, G. A., Jr, Booth, C. J.,Mehal, W. Z., Inayat, I. & Flavell, R. A. (2006). Caspases 3 and 7: key
mediators of mitochondrial events of apoptosis. Science 311, 847–851.
Lazebnik, Y. A., Kaufmann, S. H., Desnoyers, S., Poirier, G. G. &Earnshaw, W. C. (1994). Cleavage of poly(ADP-ribose) polymerase by
a proteinase with properties like ICE. Nature 371, 346–347.
Lee, C. J., Liao, C. L. & Lin, Y. L. (2005). Flavivirus activates
phosphatidylinositol 3-kinase signaling to block caspase-dependent
apoptotic cell death at the early stage of virus infection. J Virol 79,
8388–8399.
Levine, B., Huang, Q., Isaacs, J. T., Reed, J. C., Griffin, D. E. &Hardwick, J. M. (1993). Conversion of lytic to persistent alphavirus
infection by the bcl-2 cellular oncogene. Nature 361, 739–742.
Li, H., Zhu, H., Xu, C. J. & Yuan, J. (1998). Cleavage of BID by caspase 8
mediates the mitochondrial damage in the Fas pathway of apoptosis.
Cell 94, 491–501.
Liao, C. L., Lin, Y. L., Wang, J. J., Huang, Y. L., Yeh, C. T., Ma, S. H. &
Chen, L. K. (1997). Effect of enforced expression of human bcl-2 on
Japanese encephalitis virus-induced apoptosis in cultured cells. J Virol
71, 5963–5971.
Liao, C. L., Lin, Y. L., Shen, S. C., Shen, J. Y., Su, H. L., Huang, Y. L.,
Ma, S. H., Sun, Y. C., Chen, K. P. & Chen, L. K. (1998). Antiapoptotic
but not antiviral function of human bcl-2 assists establishment of
Japanese encephalitis virus persistence in cultured cells. J Virol 72,
9844–9854.
Liao, S. L., Raung, S. L. & Chen, C. J. (2002). Japanese encephalitis
virus stimulates superoxide dismutase activity in rat glial cultures.
Neurosci Lett 324, 133–136.
Lin, R. J., Liao, C. L. & Lin, Y. L. (2004). Replication-incompetent
virions of Japanese encephalitis virus trigger neuronal cell death by
oxidative stress in a culture system. J Gen Virol 85, 521–533.
Liu, Y., Pu, Y. & Zhang, X. (2006). Role of the mitochondrial signaling
pathway in murine coronavirus-induced oligodendrocyte apoptosis.
J Virol 80, 395–403.
Muzio, M., Stockwell, B. R., Stennicke, H. R., Salvesen, G. S. & Dixit,V. M. (1998). An induced proximity model for caspase-8 activation.
J Biol Chem 273, 2926–2930.
C.-H. Tsao and others
1940 Journal of General Virology 89

Newton, K., Kurts, C., Harris, A. W. & Strasser, A. (2001). Effects of adominant interfering mutant of FADD on signal transduction inactivated T cells. Curr Biol 11, 273–276.
Pan, G., O’Rourke, K., Chinnaiyan, A. M., Gentz, R., Ebner, R., Ni, J. &Dixit, V. M. (1997). The receptor for the cytotoxic ligand TRAIL.Science 276, 111–113.
Pastorino, J. G., Chen, S. T., Tafani, M., Snyder, J. W. & Farber, J. L.(1998). The overexpression of Bax produces cell death uponinduction of the mitochondrial permeability transition. J Biol Chem273, 7770–7775.
Peter, M. E. (2004). The flip side of FLIP. Biochem J 382, e1–e3.
Ramanathan, M. P., Chambers, J. A., Pankhong, P., Chattergoon, M.,Attatippaholkun, W., Dang, K., Shah, N. & Weiner, D. B. (2006). Hostcell killing by the West Nile virus NS2B–NS3 proteolytic complex:NS3 alone is sufficient to recruit caspase-8-based apoptotic pathway.Virology 345, 56–72.
Rodgers, S. E., Barton, E. S., Oberhaus, S. M., Pike, B., Gibson, C. A.,Tyler, K. L. & Dermody, T. S. (1997). Reovirus-induced apoptosis ofMDCK cells is not linked to viral yield and is blocked by Bcl-2. J Virol71, 2540–2546.
Saelens, X., Festjens, N., Vande Walle, L., van Gurp, M., van Loo, G.& Vandenabeele, P. (2004). Toxic proteins released from mitochon-dria in cell death. Oncogene 23, 2861–2874.
Scallan, M. F., Allsopp, T. E. & Fazakerley, J. K. (1997). bcl-2 acts earlyto restrict Semliki Forest virus replication and delays virus-inducedprogrammed cell death. J Virol 71, 1583–1590.
Schrantz, N., Bourgeade, M. F., Mouhamad, S., Leca, G., Sharma, S.& Vazquez, A. (2001). p38-mediated regulation of an Fas-associateddeath domain protein-independent pathway leading to caspase-8activation during TGFb-induced apoptosis in human Burkittlymphoma B cells BL41. Mol Biol Cell 12, 3139–3151.
Shafee, N. & AbuBakar, S. (2003). Dengue virus type 2 NS3 proteaseand NS2B–NS3 protease precursor induce apoptosis. J Gen Virol 84,2191–2195.
Sheridan, J. P., Marsters, S. A., Pitti, R. M., Gurney, A., Skubatch, M.,Baldwin, D., Ramakrishnan, L., Gray, C. L., Baker, K. & other authors(1997). Control of TRAIL-induced apoptosis by a family of signalingand decoy receptors. Science 277, 818–821.
Slee, E. A., Harte, M. T., Kluck, R. M., Wolf, B. B., Casiano, C. A.,Newmeyer, D. D., Wang, H. G., Reed, J. C., Nicholson, D. W. & otherauthors (1999). Ordering the cytochrome c-initiated caspase cascade:hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in acaspase-9-dependent manner. J Cell Biol 144, 281–292.
Su, H. L., Liao, C. L. & Lin, Y. L. (2002). Japanese encephalitis virusinfection initiates endoplasmic reticulum stress and an unfoldedprotein response. J Virol 76, 4162–4171.
Tan, S., Sagara, Y., Liu, Y., Maher, P. & Schubert, D. (1998). Theregulation of reactive oxygen species production during programmedcell death. J Cell Biol 141, 1423–1432.
Tschopp, J., Thome, M., Hofmann, K. & Meinl, E. (1998). The fight ofviruses against apoptosis. Curr Opin Genet Dev 8, 82–87.
Ubol, S., Tucker, P. C., Griffin, D. E. & Hardwick, J. M. (1994).Neurovirulent strains of alphavirus induce apoptosis in bcl-2-expressing cells: role of a single amino acid change in the E2glycoprotein. Proc Natl Acad Sci U S A 91, 5202–5206.
Villa, P., Kaufmann, S. H. & Earnshaw, W. C. (1997). Caspases andcaspase inhibitors. Trends Biochem Sci 22, 388–393.
Walsh, C. M., Wen, B. G., Chinnaiyan, A. M., O’Rourke, K., Dixit, V. M.& Hedrick, S. M. (1998). A role for FADD in T cell activation anddevelopment. Immunity 8, 439–449.
Wesselborg, S., Engels, I. H., Rossmann, E., Los, M. & Schulze-Osthoff, K. (1999). Anticancer drugs induce caspase-8/FLICEactivation and apoptosis in the absence of CD95 receptor/ligandinteraction. Blood 93, 3053–3063.
Yang, X. H., Sladek, T. L., Liu, X., Butler, B. R., Froelich, C. J. & Thor,A. D. (2001). Reconstitution of caspase 3 sensitizes MCF-7 breastcancer cells to doxorubicin- and etoposide-induced apoptosis. CancerRes 61, 348–354.
Yeh, J. H., Hsu, S. C., Han, S. H. & Lai, M. Z. (1998). Mitogen-activatedprotein kinase kinase antagonized Fas-associated death domainprotein-mediated apoptosis by induced FLICE-inhibitory proteinexpression. J Exp Med 188, 1795–1802.
Yu, S. W., Wang, H., Poitras, M. F., Coombs, C., Bowers, W. J.,Federoff, H. J., Poirier, G. G., Dawson, T. M. & Dawson, V. L. (2002).Mediation of poly(ADP-ribose) polymerase-1-dependent cell deathby apoptosis-inducing factor. Science 297, 259–263.
Caspase activation in JEV-induced apoptosis
http://vir.sgmjournals.org 1941
Copyright © 2022 FDOKUMEN




















