
J Immunol-2014-Tone Gitr En
14
of March 14, 2014. This information is current as Enhancer B and Foxp3 through an κ Regulated by NF- Locus Is Gitr Gene Expression in the Waldmann and Masahide Tone Yamamoto, Masato Tsuda, Christian Peter, Herman Yukiko Tone, Yoko Kidani, Chihiro Ogawa, Kouhei ol.1302174 http://www.jimmunol.org/content/early/2014/03/14/jimmun published online 14 March 2014 J Immunol Material Supplementary 4.DCSupplemental.html http://www.jimmunol.org/content/suppl/2014/03/14/jimmunol.130217 Subscriptions http://jimmunol.org/subscriptions is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/ji/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/cgi/alerts/etoc Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved. Copyright © 2014 by The American Association of 9650 Rockville Pike, Bethesda, MD 20814-3994. The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology at Cedars-Sinai Medical Center on March 14, 2014 http://www.jimmunol.org/ Downloaded from at Cedars-Sinai Medical Center on March 14, 2014 http://www.jimmunol.org/ Downloaded from at Cedars-Sinai Medical Center on March 14, 2014 http://www.jimmunol.org/ Downloaded from at Cedars-Sinai Medical Center on March 14, 2014 http://www.jimmunol.org/ Downloaded from at Cedars-Sinai Medical Center on March 14, 2014 http://www.jimmunol.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of J Immunol-2014-Tone Gitr En

of March 14, 2014.This information is current as
EnhancerB and Foxp3 through anκRegulated by NF-
Locus IsGitrGene Expression in the
Waldmann and Masahide ToneYamamoto, Masato Tsuda, Christian Peter, Herman Yukiko Tone, Yoko Kidani, Chihiro Ogawa, Kouhei
ol.1302174http://www.jimmunol.org/content/early/2014/03/14/jimmun
published online 14 March 2014J Immunol
MaterialSupplementary
4.DCSupplemental.htmlhttp://www.jimmunol.org/content/suppl/2014/03/14/jimmunol.130217
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2014 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
at Cedars-Sinai M
edical Center on M
arch 14, 2014http://w
ww
.jimm
unol.org/D
ownloaded from
at C
edars-Sinai Medical C
enter on March 14, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from
at Cedars-Sinai M
edical Center on M
arch 14, 2014http://w
ww
.jimm
unol.org/D
ownloaded from
at C
edars-Sinai Medical C
enter on March 14, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from
at Cedars-Sinai M
edical Center on M
arch 14, 2014http://w
ww
.jimm
unol.org/D
ownloaded from

The Journal of Immunology
Gene Expression in the Gitr Locus Is Regulated by NF-kB andFoxp3 through an Enhancer
Yukiko Tone,*,1 Yoko Kidani,†,1 Chihiro Ogawa,* Kouhei Yamamoto,* Masato Tsuda,*
Christian Peter,‡ Herman Waldmann,‡ and Masahide Tone*
Glucocorticoid-induced TNFR (Gitr) andOx40, twomembers of the TNFR superfamily, play important roles in regulating activities
of effector and regulatory T cells (Treg). Their gene expression is induced by T cell activation and further upregulated in Foxp3+
Treg. Although the role of Foxp3 as a transcriptional repressor in Treg is well established, the mechanisms underlying Foxp3-
mediated transcriptional upregulation remain poorly understood. This transcription factor seems to upregulate expression not
only of Gitr and Ox40, but also other genes, including Ctla4, Il35, Cd25, all critical to Treg function. To investigate how Foxp3
achieves such upregulation, we analyzed its activity on Gitr and Ox40 genes located within a 15.1-kb region. We identified an
enhancer located downstream of the Gitr gene, and both Gitr and Ox40 promoter activities were shown to be upregulated by the
NF-kB–mediated enhancer activity. We also show, using the Gitr promoter, that the enhancer activity was further upregulated in
conjunction with Foxp3. Foxp3 appears to stabilize NF-kB p50 binding by anchoring it to the enhancer, thereby enabling local
accumulation of transcriptional complexes containing other members of the NF-kB and IkB families. These findings may explain
how Foxp3 can activate expression of certain genes while suppressing others. The Journal of Immunology, 2014, 192: 000–000.
Glucocorticoid-induced TNFR (Gitr; Tnfrsf18) and Ox40(CD134, Tnfrsf4) are members of the TNFR superfamily(1) and are expressed on T cells. Expression of both
receptors is upregulated by activation and, in particular, kept con-stitutively high on CD4+CD25+Foxp3+ regulatory T cells (Treg) (2–4), cells essential to the prevention of autoimmune disease and otherforms of immunopathology (5–7). The development and function ofTreg are regulated by the transcription factor Foxp3, which re-presses many genes by associating with other transcription factors,including NFAT (8), the p65 subunit of NF-kB (9), and Runx1 (10).However, a number of other genes such as Ctla4, Il35, Cd25, Ox40,and Gitr are upregulated in Foxp3+ Treg (2, 11), with all of thesegene products playing essential roles in Treg function (12). Sig-naling through Gitr and Ox40 has been shown to neutralize thesuppressive effects of Treg in vitro (3, 13). Additionally, Ox40signaling inhibits induced Treg (iTreg) development in periphery byblocking TGF-b/TCR signal–mediated induction of Foxp3 (14, 15).The Gitr and Ox40 genes are particularly interesting as models for
Foxp3-mediated transcriptional upregulation, because they are lo-cated within a short 15.1-kb stretch of the mouse genome, sug-gesting control by a common regulatory region. We have previouslyshown a similar clustering of MS4A gene family members, alsoupregulated by Foxp3 (16), where we postulated the existence ofa comparable Foxp3-associated regulatory region at that gene locus.We selected the Gitr/Ox40 rather than the MS4A gene locus forstudy because the latter is far larger, extending over 600-kb, and istherefore more challenging for analysis.Not only are Gitr and Ox40 influential in Treg, but they also act as
costimulatory receptors for effector T cells (Teff) (1, 17–19). Gitrand Ox40 are unique molecules playing seemingly different rolesin Teff and Treg, with the distinct functions possibly determined byexpression levels of these receptors on each T cell subset (low onTeff and high on Treg). In either case, the mechanisms underlyinggene expression remain poorly understood, although we know thatOx40 expression is regulated by constitutively active transcriptionfactors and upregulated by NF-kB in activated Teff (20). To definethe molecular mechanisms responsible for upregulation of Gitr andOx40 in more detail, in this study we sought clues within the Gitr/Ox40 gene locus in both Teff and Treg.We identify an enhancer located downstream of the Gitr gene.
Histone H4 molecules in this region are highly acetylated both inactivated T cells and Foxp3+ Treg. We show that enhancer activity isregulated by NF-kB in activated Teff, and by NF-kB in conjunctionwith Foxp3 in Treg. We propose that Foxp3 stabilizes binding of thep50 subunit of NF-kB to the enhancer. Although p50 does notcontain a transactivation domain, the p50/Foxp3 complex on theenhancer interacts with other members of the NF-kB and IkB family(e.g., p65, c-Rel, B cell lymphoma 3 [Bcl-3]) to supply trans-activation domains to the complex. This may explain how Foxp3 cansuppress expression of many genes while also upregulating others.
Materials and MethodsCell culture
EL4 subclones LAF and BO2 cells were cultured in IMDM with L-glu-tamine and 25 mM HEPES (Cellgro) and 5% FBS. T cells were cultured in
*Research Division of Immunology, Department of Biomedical Sciences, Cedars-Sinai Medical Center, Los Angeles, CA 90048; †Department of Pathology and Lab-oratory Medicine, David Geffen School of Medicine, University of California, LosAngeles, Los Angeles, CA 90095; and ‡Sir William Dunn School of Pathology,University of Oxford, Oxford OX1 3RE, United Kingdom
1Y.T. and Y.K. contributed equally to this work.
Received for publication August 14, 2013. Accepted for publication February 13,2014.
This work was supported by National Institutes of Health Grant R01 A1078987 (toM. Tone), the Cedars-Sinai Medical Center, and a European Research Council ad-vanced investigator grant (to H.W.).
Address correspondence and reprint requests to Dr. Masahide Tone, Department ofBiomedical Sciences, Cedars-Sinai Medical Center, 8700 Beverly Boulevard, LosAngeles, CA 90048. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: Bcl-3, B cell lymphoma 3; ChIP, chromatin im-munoprecipitation; Gitr, glucocorticoid-induced TNFR; HS, hypersensitive site;iTreg, induced regulatory T cell; NAI, NF-kB activation inhibitor; nTreg, naturalregulatory T cell; poly(dI-dC), poly(deoxyinosinic-deoxycytidylic); Treg, regulatoryT cell; WT, wild-type.
Copyright� 2014 by The American Association of Immunologists, Inc. 0022-1767/14/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1302174
Published March 14, 2014, doi:10.4049/jimmunol.1302174 at C
edars-Sinai Medical C
enter on March 14, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from

RPMI 1640 with L-glutamine (Cellgro) and 10% FBS. CD4+ T cells andT cells were isolated from mouse spleen using a CD4+ T cell isolation kit(Miltenyi Biotec) and a Pan T cell isolation kit (Miltenyi Biotec), re-spectively. CD4+CD25+ Treg cells were purified by a cell sorter using anti-CD4 (RM4-5, eBioscience) and anti-CD25 (PC61.5, eBioscience) frompreisolated CD4+ T cells, isolated CD4+CD25+ T cells were analyzed byan internal Foxp3 staining kit (eBioscience), and Foxp3+ T cells were.95%. When required, T cells were stimulated with plate-coated anti-CD3(KT3, 5 mg/ml in PBS). To generate iTreg, CD4+CD252 T cells wereisolated from wild-type (WT) and Foxp3-GFP reporter mice (The JacksonLaboratory) and stimulated with TGF-b (5 ng/ml) (PeproTech), anti-CD28(1 mg/ml), and anti-CD3 (plate-coated). To isolate Foxp3+ iTreg, thestimulated cells were sorted with GFP. Splenocytes and thymocytes wereisolated from Nfkb p50-deficient mice (The Jackson Laboratory) and WTmice.
The preparation of bone marrow–derived dendritic cells has been de-scribed previously (21). Briefly, bone marrow cells from BALB/c micewere cultured for 7 d with GM-CSF (5 ng/ml). When required, bonemarrow–derived dendritic cells were stimulated with LPS (10 mg/ml).
EL4 and CD4+ T cells were cultured with NF-kB activation inhibitor(NAI; EMD Biosciences) in DMSO. These cells were pretreated with NAIfor 30 min before stimulation and then cultured with anti-CD3–coated plates.Ox40 and Gitr expression was analyzed by quantitative RT-PCR (EL4), andby FACS (CD4+ T cells, gated by 7-aminoactinomycin D2 and CD4+) withanti-Ox40 (OX-86, eBioscience) and anti-Gitr (DTA-1, eBioscience).
RT-PCR
Total RNAwas isolated from EL4 cells and T cells using an RNeasy Minikit (Qiagen), and cDNA was prepared with an iScript cDNA synthesis kitcontaining random and oligo(dT) primer mixture (Bio-Rad). QuantitativeRT-PCR was performed by using SsoFast EvaGreen supermix (Bio-Rad).PCR primers used were as follows: Gitr, forward, 59-GACCCTCAGT GC-AAGATCTGC-39, reverse, 59-CCTCAGCTGACAACTGCACCTC-39; Ox40,forward, 59-GTAGACCAGGCACCCAACC-39, reverse, 59-GGCCAGACTG-TGGTGGATTGG-39; Gapdh, forward, 59-TGGTGAAGGTCGGTGTGAAC-GGATTT-39, reverse, 59-TGTGCC GTTGAATTTGCCGTGAG-39; 18SrRNA, forward, 59-CTTAGAGGG ACAAGTGGCG-39, reverse, 59-ACG-CTGAGCCAGTCAGTGTA-39. Gitr, Ox40, and Gapdh expression levelswere normalized to 18S rRNA levels.
Construction of luciferase reporter plasmids and luciferaseassay
A DNA fragment (1.22 kb) of the Gitr promoter and its deletion mutantswere amplified by PCR, cloned into the pGL4 basic vector (Promega), andDNA sequences of the all inserted fragments were determined to removedeformed fragments generated by PCR errors. To construct enhancer lu-ciferase reporter plasmids, Pro8 Gitr promoter luciferase reporter plasmidwas used. Potential enhancer DNA fragments (1.6-kb fragment [containingHS1 site], 1.15-kb fragment [containing HS2 site], and 0.28-kb fragment[containing HS3 site]) were PCR amplified and inserted into SalI site lo-cated downstream of the luciferase gene. To construct Ox40 promoter-enhancer plasmid, the enhancer core fragment was inserted into the SalIsite located downstream of the luciferase gene of the Ox40 promoter re-porter plasmid (carrying 1.97-kb DNA fragment as Ox40 promoter) shownin a previous publication (20). The enhancer deletion mutant fragmentswere PCR amplified and integrated in to the SalI site of the Gitr Pro8reporter plasmid. The +39, +136, +183, and WT core enhancer fragmentscontain the same 39 end (+286), and +71, +108, +193, and WT core en-hancer fragments contain the same 59 end (+1). These positions are shownin Supplemental Fig. 3A. Mutations in the enhancer sequence (Fig. 3A)were introduced into the enhancer fragment by PCR assemble procedure,and the mutated fragments were integrated in to the SalI site in the GitrPro8 promoter reporter plasmid. DNA sequences of the all inserted frag-ments were determined to remove defective fragments generated by PCRerrors.
For the luciferase assay, 5 3 106 EL4 LAF (Figs. 2, 3) and EL4 BO2(Fig. 8) cells were transfected by Gene Pulser Xcell (Bio-Rad) with 5 mgluciferase reporter plasmids and 2 mg phRL-TK (Promega) as an internalcontrol plasmid and cultured for 24 h in six-well plates. When required,cells were activated with plate-coated anti-CD3 (5 mg/ml in PBS). Cellswere harvested, and promoter or enhancer activity was analyzed by theDual-Luciferase reporter assay system (Promega). Foxp3, NF-kB p50,p65, and c-Rel expression plasmids were constructed using pMF-neovector (expression is regulated by EF1a promoter). To remove the IkBhomologous from p50, p50 cDNA encoding 1–423 aa was PCR amplifiedand cloned into pMF-neo vector. For cotransfection experiments, 4 mg Gitr
promoter-enhancer plasmid was cotransfected with 4 mg each expressionplasmid indicated in Fig. 8. Total DNA amount was adjusted with theempty pMF-neo vector. EL4 BO2 subclone was used for this assay.
DNase I hypersensitive assay
Isolated nuclei were treated with DNase I (0–25 U/ml) at 25˚C for 5 min in60 mM KCl, 15 mM NaCl, 5 mMMgCl2, 0.1 mM EGTA, 15 mM Tris-HCl(pH 7.4) 0.5 mM DTT, 5% glycerol, and 10% sucrose, and DNAs wereisolated and digested with KpnI, SphI, EcoRI, XbaI, XhoI, or SalI. DNase Ihypersensitive sites were analyzed by Southern blot hybridization, andprobe positions (for KpnI digestion) are indicated in Supplemental Fig. 2.
EMSA
EMSAwas performed with 32P-labeled probes and 2 mg nuclear extracts in20 ml EMSA reaction buffer (2 mg poly(deoxyinosinic-deoxycytidylic)[poly(dI-dC)]·poly(dI-dC) acid, 20 mM HEPES [pH 7.9], 1 mM MgCl2,40 mM KCl, 0.1 mM EDTA, 1 mM DTT, and 12% glycerol). Nuclearextracts were prepared from nonactivated or CD3-activated (for 1.5 and24 h) EL4 cells as described previously (20). To perform competition assays,100-fold excess of unlabeled competitor oligonucleotides was added toEMSA reaction mixture. To perform supershift assay, nuclear extracts inEMSA reaction buffer were incubated for 15 min with anti-Sp1 (Santa CruzBiotechnology, PEP2), anti-Sp3 (Santa Cruz Biotechnology, D-20), anti–NF-kB p50 (Santa Cruz Biotechnology, D-17), and anti–NF-kB p65 (Santa CruzBiotechnology, C-20), and probes were added into the reaction mixture.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assay was performed using PanT cells, CD4+CD25– T cells, TGF-b–induced iTreg (93% purity), andCD4+CD25+Foxp3+ nTreg cells (purity .95%) as described previously(20). These cells were fixed (for 10 min at room temperature in 1%formaldehyde, 4.5 mM HEPES [pH 8.0], 9 mM NaCl, 0.09 mM EDTA,and 0.045 mM EGTA) and sonicated (Bioruptor) in lysis buffer (1% SDS,10 mM EDTA, and 50 mM Tris-HCl [pH 8.0]) with proteinase inhibitor(Sigma-Aldrich, P8340). Precleared lysates were incubated overnight at4˚C with polyclonal anti–acetyl histone H4 (Millipore), anti–NF-kB p50(Abcam, anti–p105/p50-ChIP grade), or control rabbit IgG (Santa CruzBiotechnology). DNA fragments were isolated from the immunoprecipi-tated chromatin and analyzed by real-time PCR with SsoFast EvaGreenSupermix (Bio-Rad). PCR primers for ChIP used were as follows: kB1/kB2, forward, 59-TTACACTGGAAACACCACAGGTGG-39, reverse, 59-TGCTGGCTTCAAGGCAAGGATACA-39; kB3, forward, 59-TGCATTC-CAC TCACGTCCAC-39, reverse, 59-GGGCACTGTCCCTCAGCTAC-39.
Pull-down assay
A 101-bp DNA fragment containing the kB1 and kB2 sites was amplifiedby PCR using biotinylated primers and purified using QIAEX II gel ex-traction system (Qiagen). Purified probes (500 ng) were mixed with nu-clear extract (50 mg) in 400 ml binding buffer (60 mM KCl, 12 mMHEPES [pH 7.9], 4 mM Tris-HCl [pH 8.0], 1 mM EDTA, 1 mM EGTA,and 12% glycerol) containing 1.66 mM DTT, 0.06% BSA, and 20 mg poly(dI-dC) acid at 4˚C for 2 h. When required, 100 pmol Sp1 binding oligofrom CD40 promoter (22) and 500 pmol indicated competitor oligonu-cleotides were added. Then, precleared streptavidin-agarose beads (LifeTechnologies) were mixed with the DNA–nuclear extract mixture for 2 h.The streptavidin-agarose beads were then washed five times with 1 mlbinding buffer, and then 23 SDS sample buffer was added. The sampleswere analyzed by immunoblotting. PCR primers used were as follows: kB,forward, 59-biotin-GAAACACC ACAGGTGGGACA-39, reverse, 59-CACACCCATCAGCCGCCCACA-39; Gitr promoter, forward, 59-biotin-TGGGAGAGGCATGTAGGGGTTAGA-39, reverse, 59-TTTC CGGCAGA-CATCTGAGGT-39.
Immunoprecipitation and immunoblotting
A nuclear extract was prepared from a p50 (1–423 aa)–Foxp3 (FLAG-tagged) transfectant. Nuclear extract (10 mg) was precleared with proteinG (Invitrogen) for 3 h in 5% glycerol, 12 mM HEPES (pH 7.9), 4 mM Tris(pH 8.0) 60 mM KCl, 0.1 mM EDTA, 1 mM DTT, poly(dI-dC)·poly(dI-dC) acid 20 mg, and 4 ml proteinase inhibitor (Sigma-Aldrich, P8340) withor without 92-bp DNA (16–108, Fig. 3). Anti-p50 (2 mg; Santa CruzBiotechnology, C-19) or control goat IgG was added and incubated at 4˚Covernight, and protein G was added and incubated for 2 h at 4˚C. Protein Gwas washed with the binding buffer and analyzed by immunoblotting.
Immunoblotting was performed with anti-p50 (Santa Cruz Biotech-nology, C-19), anti-p65 (Santa Cruz Biotechnology, C-20), anti–c-Rel
2 REGULATION OF Gitr GENE EXPRESSION
at Cedars-Sinai M
edical Center on M
arch 14, 2014http://w
ww
.jimm
unol.org/D
ownloaded from

(Santa Cruz Biotechnology, C), anti-Bcl-3 (Acris, SP7024P), anti-Foxp3(Cell Signaling Technology, D608R), and anti-FLAG (Sigma-Aldrich). Abbinding was detected by using Luminata Forte Western HRP substrate(Millipore).
Statistical analysis
Significance was determined with a Student t test.
ResultsA scheme for identifying regulatory regions in the Gitr/Ox40gene locus in Teff and Treg
Because CD4+CD25+Foxp3+ Treg are normally present in rela-tively low abundance, and because primary T cells are not idealfor transfection-based procedures such as luciferase reporter as-says, we adopted the following strategy to identify regulatory re-gions in the Gitr/Ox40 locus in activated Teff and Treg. First, weidentified regulatory regions such as promoters and enhancers inboth activated T cells (using TCR cross-linking by anti-CD3) andthe EL4 T cell line (which is known to retain many features ofT cells), and we characterized the regulatory region in activatedT cells. We then determined whether that regulatory region wasfunctional in both TGF-b iTreg (23, 24) and nTreg and thereafterproceeded to investigate the molecular basis for the upregulationin Foxp3+ Treg. Because Foxp3 gene expression is maintained bytranscription factors induced by TCR-CD3 signaling, these factorsshould translocate into the nuclei of Foxp3+ Treg. Knowing thatFoxp3 can associate with these activated factors in Treg (8–10,25), it was then conceivable that the Foxp3-responsive regulatoryregion coincides with those regions involved in CD3 activation.Ox40 gene expression can be upregulated by CD3 activation in
both T cells and EL4 cells (20), and as shown in Fig. 1A, Gitrgene expression is also upregulated in both cell types, suggestingthat expression of these genes in primary T cells and the EL4 cellline is regulated by the same regulatory mechanisms. Regulationof Foxp3 gene expression in T cells and our EL4 subclone wasalso comparable (26). These data suggest that the EL4 T cell linecan be used to probe Gitr and Ox40 gene expression and that thisinformation is relevant to primary Teff and iTreg.
Identification of an enhancer containing CD3 activationresponse elements
Using this scheme, we analyzed promoter activity in activatedcells. We had previously identified an NF-kB binding site in theOx40 promoter (20) that was involved in the response to CD3. Weused the same approach in the present study to investigate theGitr promoter by using a luciferase reporter assay with a seriesof deletion mutants (Pro1 to Pro8 in Fig. 1B). Gitr promoter ac-tivity was detected in both nonactivated and CD3-activated EL4cells, but no CD3 activation response element was identified withinthe 1.22-kb Gitr promoter. Regardless of activation, promoter ac-tivity was reduced to basal levels by deleting 39 bp from 290(Pro3) to 251 (Pro2). Binding of the transcription factor NFI,which is known to be constitutively and ubiquitously expressed(27), to this region was detected using the EMSA (SupplementalFig. 1). Gitr basal promoter activity seems, therefore, to be con-stitutively regulated through this region, suggesting that this geneexpression is upregulated by other regulatory regions containingCD3 activation response elements in activated T cells.To identify the regulatory regions involved, DNase I hyper-
sensitive assays were performed. Because DNase I hypersensitivesites (HSs) were generated by disruption of nucleosome chromatinstructure by additional factor bindings, some of the HSs are ob-served in promoters and enhancers. Regulatory regions containingCD3 response elements might be located near CD3-activated
T cell–specific HSs. We therefore compared positions of DNaseI HSs in nonactivated and CD3-activated CD4+ T cells, EL4 cells,and bone marrow–derived dendritic cells (Fig. 1C, SupplementalFig. 2). Many common HSs were detected in the Gitr/Ox40 genelocus (Fig. 1D). However, three additional sites were observedonly in CD3-activated cells (Fig. 1C, 1D, Supplemental Fig. 2),which we term HS1, HS2, and HS3 (HS1 was detected only inCD3-activated CD4+ T cells).We next examined enhancer activities in regions containing
HS1, HS2, or HS3 using a luciferase reporter assay. As shown inFig. 2A, DNA fragments containing these HSs were integrateddownstream of the luciferase gene in the Gitr promoter reporterplasmid (Fig. 1B, Pro8). Promoter activity was enhanced with thefragment containing HS3 in both orientations (Fig. 2A, 2B) in CD3-activated cells but not in nonactivated cells. The enhancer activitywas also analyzed using the Ox40 promoter (1.97 kb). Unlikethe Gitr promoter, Ox40 promoter activity is itself increased 2-foldin CD3-activated cells (Fig. 2C, Pro) by NF-kB, as previouslydescribed (20), but this promoter activity was upregulated far moreby this enhancer (Fig. 2C), suggesting strong enhancer activity.Enhancer activity was further analyzed by luciferase assays(Fig. 2A, 2D, 2E). Searching the transcription factor database, wefound three potential NF-kB binding sequences in this enhancerregion (referred to henceforth as kB1, kB2, and kB3) (Fig. 2D,2E, gray boxes). Because the enhancer activity was only detectedin CD3-activated cells but not resting T cells, we hypothesizedthat enhancer activity might be regulated by activated NF-kBthrough these sites. This possibility was investigated by using the59 (Fig. 2D) and 39 (Fig. 2E) deletion mutants of the enhancer.Luciferase activity using the WT enhancer was reduced by dele-tions from +1 to +39 and from +39 to +136 (Fig. 2D). The deletedregions +1/+39 and +39/+136 contain kB1 and kB2 sites, re-spectively (Fig. 2D). Although the 59 deletion mutant (+183) con-tained one potential NF-kB site (kB3), the enhancer activity wassimilar to that of the negative control (Fig. 2D, +183 and no en-hancer). To investigate whether the kB3 site is simply not func-tional, or whether the kB sites cooperate to regulate enhanceractivity, we performed luciferase assays using the 39 deletionmutants (Fig. 2E). Luciferase activity was reduced by the deletioncontaining the kB3 site (Fig. 2E, +193), suggesting that this 39 re-gion containing kB3 cooperatively regulates enhancer activity aswell. Such cooperative regulation was also deduced from the 39deletion mutant +71 containing kB1 but not kB2 and kB3 (Fig. 2E),as the enhancer activity of this deletion mutant was similar to thenegative control. These results strongly suggest that the three NF-kBbinding sequences, identified using the transcription factor database,are strong candidates as regulatory elements within this enhancer.We then analyzed histone H4 acetylation by ChIP as a marker foropen chromatin in the kB1 plus kB2 (kB1 and kB2 cannot be an-alyzed separately by this assay because these two sites are too closeto each other) and kB3 regions (Fig. 2F). H4 molecules in T cellsbecame highly acetylated within 24 h of activation, suggesting thatthe chromatin of these regions opens up after CD3 activation.Histone H4 in these regions was also highly acetylated in nTregbut not in naive CD4+CD252 T cells (Fig. 2G), suggesting that thisregulatory region also functions in nTreg.
NF-kB regulates enhancer activity
As shown in Fig. 2, the 286-bp enhancer sequence (Fig. 3A) en-codes three potential NF-kB binding sequences. Transcription factorbinding to these sites was analyzed by EMSA in Figs. 3B and 4. Toassess the contribution of these potential NF-kB sites and to inves-tigate the existence of other regulatory elements in the enhancer,luciferase reporter assays were performed using mutations in the
The Journal of Immunology 3
at Cedars-Sinai M
edical Center on M
arch 14, 2014http://w
ww
.jimm
unol.org/D
ownloaded from

potential NF-kB binding sequences in the Gitr promoter/enhancerreporter plasmid (Fig. 3A, 3C). No factor binding to the mutatedsequences could be detected by EMSA using a nuclear extract fromCD3-activated cells (Fig. 3B). Mutation of any of the potential NF-kB binding sites reduced enhancer activity, and mutating all threesites eliminated it (Fig. 3C), suggesting that all three sites regulateenhancer activity. The kB1 site was identified as the strongest reg-ulatory element in this enhancer because the enhancer activity wasreduced to ,20% by mutation of the kB1 sequence. However,similar reductions were observed with double mutations at thekB2 and kB3 sites. These results indicate that enhancer activity iscooperatively regulated by the transcription factors binding tokB1, kB2, and kB3 sites.Transcription factor binding to each kB site was detected by
a simple EMSA using a nuclear extract from CD3-activated cells(Fig. 3B). Although all probes encode potential NF-kB bindingsequences, binding patterns were not exactly the same. We there-fore investigated transcription factor binding in more detail (Fig. 4).First, we performed EMSA using nuclear extracts from nonacti-vated and CD3-activated cells (Fig. 4A). We detected both con-stitutive and induced binding to the kB1 and kB2 probes, but onlyinduced binding to the kB3 probe (Fig. 4A). As shown in Sup-plemental Fig. 3, the constitutively active transcription factorsbinding to kB1 and kB2 were Sp1 and Sp3, which bind to se-quences overlapping the NF-kB binding sites. Binding of thesame induced factors to all probes was suggested by inhibition ofbinding to the 32P-labeled kB3 probe with excess of the “cold” kB1and kB2 competitors (Fig. 4B). Because histone H4 acetylation inthis enhancer region was induced by CD3 activation (Fig. 2F),binding of the induced transcription factors is likely to be the keyto regulation. To identify binding of NF-kB to the kB1 and kB2sites, excess cold competitor encoding an Sp1 binding sequence inthe CD40 promoter (22) was added to block Sp1 and Sp3 bindingto the labeled probe. We confirmed NF-kB p50 binding to these
sites by supershift EMSA using anti–NF-kB p50. All complexes oftranscription factors and the probes were supershifted with anti–NF-kB p50, and some of complexes were supershifted with anti–NF-kBp65, but unmoving complexes were still detected with this anti-p65(Fig. 4C). This suggests that all complexes contain p50; in otherwords, all kB sites were bound by p50 homodimers, p50/p65 het-erodimers, and/or p50 heterodimers with other transcription factorsincluding other NF-kB members (as shown later, binding of c-Reland Bcl-3 was detected by a DNA pull-down assay).Any contribution of Sp1 and Sp3 to this enhancer activity is
likely to be minimal, as they are present constitutively. However, itcould be that they are bound in nonactivated cells but then replacedby NF-kB on activation, as described for Foxo1 and Foxp3 bind-ing to Foxp3 gene (28).
NF-kB regulates gene expression in the Gitr/Ox40 locus
As shown above, enhancer activity is regulated by NF-kB. Becausethe EMSA result (Fig. 4C) suggests that NF-kB p50 is a maincomponent of the binding factors (p50 homodimer and hetero-dimer with other transcription factors), we further analyzed p50binding in T cells by ChIP. p50 binding to the kB1 plus kB2 andkB3 regions was increased in T cells 24 h after CD3 activation(Fig. 5A). This information, together with previous evidence thatNF-kB p50 and p65 bind to the Ox40 promoter (20), indicates thatgene expression in the Gitr/Ox40 locus is predominantly regulatedby NF-kB p50. To test this, we analyzed Gitr and Ox40 expressionin CD3-activated CD4+ T cells isolated from the spleens of micelacking the Nf-kb p50 gene (Fig. 5B). Gitr and Ox40 expressionwas upregulated by CD3 activation on CD4+ T cells from bothmice, but the Gitrhigh (36.8%) and Ox40high (39.3%) cell pop-ulations from WT mice were less than half (Gitrhigh, 15.2%;Ox40high, 18.7%) of values observed from p50-deficient mice. Thisresult suggests that both Gitr and Ox40 expression are regulated byNF-kB p50. This conclusion was not matched by the observation
FIGURE 1. Regulation of Gitr gene expression in nonactivated (Non) and CD3-activated (CD3) T cells. (A) Gitr RNA expression levels were analyzed
by quantitative RT-PCR using RNA from nonactivated and CD3-activated (activated with anti-CD3) EL4 cells and T cells. Expression levels of Gitr RNA
were normalized relative to 18S rRNA levels. (B) Gitr promoter activity was analyzed by luciferase reporter assay using Gitr promoter deletion mutants. All
promoter fragments contain the same 39 ends (+46), and positions of the 59 ends are indicated in the right side of the graph. The promoter activity was
analyzed using EL4 under indicated conditions. (C) DNase I hypersensitive assay was performed. Nuclei from indicated cells were treated by DNase I under
different concentrations (low [left] to high [right], indicated by the triangle). DNA isolated from the nuclei was digested with KpnI and analyzed by
Southern blot hybridization using the probe shown in (D). CD3-activated T cell specific DNA fragments are indicated by an arrow (HS3). (D) The activated
T cell (including EL4)–specific DNase I hypersensitive sites are indicated by solid arrows (HS1, HS2, and HS3) in the upper the map of the Ox40 and Gitr
gene loci, and common DNase I hypersensitive sites in nonactivated and CD3-activated cells are indicated by arrows with dotted lines below the map.
Exons are indicated by gray boxes, and position of the probe for the Southern blot hybridization is indicated by a solid line. Data are representative of three
(A) or four (B) independent experiments (error bars indicate the SD of triplicate samples) or three to four independent experiments with different restriction
enzymes (C, D).
4 REGULATION OF Gitr GENE EXPRESSION
at Cedars-Sinai M
edical Center on M
arch 14, 2014http://w
ww
.jimm
unol.org/D
ownloaded from

that a significant number of Gitrhigh and Ox40high cells could bedetected in p50-deficient mice, despite NF-kB p50 preferablybinding to these NF-kB sites in the enhancer (Fig. 4C) and in theOx40 promoter (20). We propose that the lack of NF-kB p50 iscompensated by binding of a very similar family member, NF-kBp52, as previously suggested using NF-kB p50-, p52-, and p50/p52-deficient mice (29, 30). Unfortunately, the p50/p52 double-deficient mice would not be suitable to test this hypothesis, asthese mice have a major problem in bone development and exhibita profound immunodeficiency (29, 30). To overcome this problem,we analyzed Gitr and Ox40 expression by using NAI. Expressionof Gitr and Ox40 RNAs was strongly inhibited by NAI in CD3-activated EL4 cells in a dose-dependent manner (Fig. 5C, 5D), incontrast to the housekeeping gene control (Gapdh) (Fig. 5E). Gitrand Ox40 cell surface expression on CD3-activated T cells wasalso inhibited by NAI (Fig. 5F). These results indeed suggest thatGitr/Ox40 expression depends on NF-kB.
NF-kB p50 regulates enhancer activity in Treg
Upregulation of Gitr and Ox40 expression in Foxp3+ Treg hasbeen previously demonstrated (2–4). The enhancer we have identi-
fied in the present study might be involved in this process. Toassess whether this enhancer functions in iTreg and nTreg, weexamined NF-kB p50 binding to this region by ChIP assay inthese cells. p50 binding to the enhancer (kB1 plus kB2 and kB3regions) was detected in nTreg, but not in naive CD4+CD25–
T cells (Fig. 6A). iTreg were generated using CD4+CD252 T cellsfrom Foxp3-GFP reporter mice by stimulation with TGF-b plusanti-CD3 plus anti-CD28; 37% cells were Foxp3+ iTreg after 48 h,and these were isolated (93% purity) using cell sorting and GFPfluorescence (Fig. 6B). p50 was bound to the enhancer in theseFoxp3+ iTreg, but not in CD4+CD252 T cells (Fig. 6B).We also examined Gitr expression in CD4+Foxp3+ Treg isolated
from p50-deficient mice. Because Ox40 promoter activity is reg-ulated by NF-kB, and NF-kB p50 binding to the promoter is de-tectable by ChIP assay (20), we investigated only Gitr expressionto avoid any confusion. Splenocytes and thymocytes from thesemice had a larger proportion of CD4+Foxp3+ T cells with low Gitrexpression than did those from control WT mice (Fig. 6C). Thissupports the argument that p50 is involved in upregulating Gitr ex-pression in Treg. We speculate, therefore, that those GitrhighCD4+
Foxp3+ T cells in p50-deficient mice are generated by com-
FIGURE 2. Identification of an enhancer in CD3-activated T cells and Treg. Assays were performed with nonactivated (Non) and CD3-activated (CD3)
cells. (A) Structures of the promoter/enhancer reporter plasmids used in (B)–(E) are illustrated. DNA fragments containing HS1, HS2, or HS3 sites were
inserted downstream of the luciferase gene in both orientations (AS, antisense; S, sense) and these plasmids were used in (B). The reporter plasmid
containing the enhancer in sense orientation was used in (C)–(E). Luciferase assays were performed using EL4 cells under indicated conditions. (B)
Luciferase reporter plasmids were constructed using the Gitr promoter as shown in (A). Luciferase activities were compared with that given by the negative
control vector (Basic, no promoter and no enhancer). (C) The enhancer activity was detected with both the Ox40 and the Gitr promoter. The promoter in the
Gitr promoter/enhancer with HS3 (sense) plasmid [shown in (A)] was swapped with the Ox40 promoter, and the luciferase assay was performed using the
Ox40 and Gitr promoter (Pro) and the Ox40 and Gitr promoter/ enhancer (Pro plus HS3) reporters. (D and E) Luciferase assays were performed using
the WT enhancer and 59 deletion mutants (D) and 39 deletion mutants (E). These deletion mutants are illustrated with the possible NF-kB sites indicated by
gray boxes (kB1, kB2, and kB3). (F) Acetylation of histone H4 molecules in the chromatin region containing the kB1 plus kB2 and kB3 sites in CD3-
activated T cells (T cells were activated by anti-CD3 for indicated times) was analyzed by ChIP using anti–acetyl-histone H4 (AcH4) or control IgG. (G)
Acetylation of histone H4 was also analyzed using CD4+CD252 T cells (CD252) and nTreg as described in (F). Data are representative of more than three
(B–G) independent experiments (error bars indicate the SD of triplicate samples).
The Journal of Immunology 5
at Cedars-Sinai M
edical Center on M
arch 14, 2014http://w
ww
.jimm
unol.org/D
ownloaded from

pensatory NF-kB p52. Taken together, we conclude that Gitr ex-pression is upregulated by the enhancer in conjunction with p50molecules in both nTreg and iTreg.
Foxp3 binds both to p50 and to the enhancer DNA
Because the enhancer seems to be the key regulatory region in thelocus, it is likely that Gitr and Ox40 expression is further upreg-ulated in Treg by Foxp3-mediated mechanisms operating throughthe enhancer. We noticed that within the enhancer the kB1 bind-ing sequence is followed by a potential Foxp3 binding sequence,analogous to the NFAT-Foxp3 binding site in the IL-2 promoter(8) (Fig. 7A, Supplemental Fig. 3D). p50 binding to the kB2 sitewas weak, yet this site also possesses a similar arrangement(Supplemental Fig. 3A). We therefore analyzed Foxp3 binding tothe kB1 plus kB2 region using a DNA pull-down assay and nu-clear extracts from CD4+CD25– T cells stimulated for 72 h withTGF-b plus anti-CD3 plus anti-CD28. Foxp3 was coprecipitatedwith the biotinylated kB probe but not a control probe of the samelength from the Gitr promoter (Fig. 7B). To confirm Foxp3binding to the potential site (39 side of the p50 binding site) (Fig.7A), we performed a competition assay with kB1 and Foxp3 com-petitors (i.e., nonbiotinylated double-stranded oligonucleotides shownin Fig. 7A). Stimulation-specific NF-kB p50 and Foxp3 binding
to the kB probe was detected (Fig. 7C), and both binding to theenhancer probe was completely inhibited by the kB1 competitor(containing the kB1 binding site and the potential Foxp3 bindingsite, Fig. 7A; Fig. 7C, kB1), suggesting that p50 and Foxp3 bindto the competitor. Foxp3 binding was also inhibited by a Foxp3competitor (lacking the kB1 binding site but containing the po-tential Foxp3-binding site, Fig. 7A; Fig. 7C, Foxp3); this indi-cates that Foxp3 binds to the overlapping region containing thepotential Foxp3 binding site (Fig. 7A). Because Foxp3 binds toboth NFAT and the IL-2 promoter (Supplemental Fig. 3D), weexamined whether Foxp3 binds directly to enhancer DNA and/orin association with p50. To examine this, p50 binding to the en-hancer was inhibited in the presence of a competitor containingonly a p50 binding sequence from the CD40 promoter (CD40–NF-kBcompetitor) (22) (Fig. 7A, 7D), but Foxp3 binding was minimallyaffected (Fig. 7D); this suggests that Foxp3 binds to DNA in theenhancer. However, p50 binding to the enhancer was not com-pletely inhibited by a 500-fold excess of the CD40–NF-kB com-petitor (containing only NF-kB site) (Fig. 7D), but was completelyinhibited by the kB1 competitor (containing NF-kB plus Foxp3binding sites) (Fig. 7C). This suggests that a p50/CD40–NF-kBcompetitor complex binds to Foxp3 on the probe (i.e., a protein–protein interaction), as shown in Supplemental Fig. 3E. Thispossibility was supported by a coimmunoprecipitation experimentwhere FLAG-tagged Foxp3 was coprecipitated with anti-p50 with-out DNA (Fig. 7E). Foxp3 seems therefore to bind to both p50 andDNA in the enhancer (Fig. 8D).
Foxp3 upregulates gene expression in conjunction with p50
We confirmed that p50/Foxp3 binds to the enhancer in a manneranalogous to NFAT/Foxp3 binding to the IL-2 promoter (but note
FIGURE 3. Three NF-kB binding sites in the enhancer core sequence.
(A) DNA sequence of the enhancer core (286 bp) is shown. NF-kB bind-
ing sites (kB1, kB2, and kB3) are indicated in bold. Transcription factor
binding to kB1, kB2, and kB3 probes (probe positions are underlined) with
or without mutations (indicated by italic capital letters for the kB sites) were
used for EMSA in (B) and Fig. 4, and enhancer activity with mutations of the
kB sites were analyzed in (C). (B) Transcription factor binding to the WTand
mutant kB probes [shown in (A)] were analyzed using the indicated probes
and a nuclear extract from CD3-activated EL4 cells with anti-CD3 for 1.5 h.
(C) Enhancer activity was analyzed using the enhancer fragment with or
without mutations shown in (A). Luciferase assays were performed in non-
activated (Non) and CD3-activated (CD3) EL4 cells using the indicated
plasmids, and activities were compared with the negative control plasmid (no
promoter and no enhancer; Basic). Positions of the potential NF-kB binding
sites (kB1, kB2, and kB3) are indicated by gray boxes. The mutated sites are
indicated by3. Data are representative of more than three (B, C) independent
experiments (error bars indicate the SD of triplicate samples).
FIGURE 4. NF-kB binding to the enhancer. Binding of transcription
factors to the enhancer region was analyzed by EMSA performed using 32P-
labeled indicated probes (Fig. 3A). (A) EMSA using nuclear extracts from
nonstimulated (Non) and CD3-activated (CD3) (24 h activation) EL4 cells.
(B) Competition EMSA using the kB3 probe with a nuclear extract prepared
from CD3-activated (1.5 h) EL4 cells. Added cold competitors (100-fold
excess) are indicated by +. (C) Supershift EMSA using a nuclear extract
prepared from CD3-activated (1.5 h) EL4 cells, indicated probes, and anti–
NF-kB p50 (anti-p50) and anti–NF-kB p65 (anti-p65). To block Sp1 and
Sp3 binding to the kB1 and kB2 probes, cold Sp1 competitor (100-fold)
was added to the reaction mixture. Added Abs are indicated by +. Data are
representative of more than three independent experiments (A–C).
6 REGULATION OF Gitr GENE EXPRESSION
at Cedars-Sinai M
edical Center on M
arch 14, 2014http://w
ww
.jimm
unol.org/D
ownloaded from

that Foxp3 represses IL-2 expression). We investigated the func-tional relevance of this binding using a luciferase reporter assay.Hori et al. (31) have demonstrated that Gitr expression is upreg-ulated by ectopically expressed Foxp3 in CD3-activated CD4+
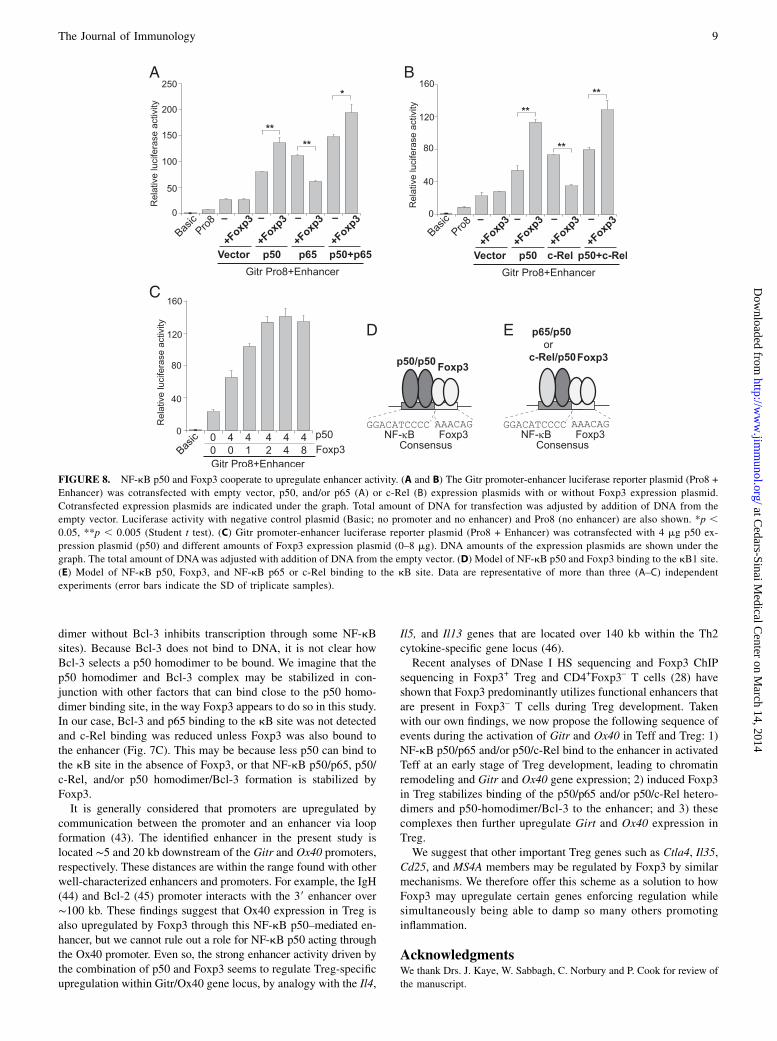
CD252 T cells. We therefore coexpressed Foxp3 in CD3-activatedEL4 cells and performed the luciferase assay with the Gitr pro-moter/enhancer reporter plasmid. Because NF-kB p50 and p65bind to the enhancer (Fig. 4C) and NF-kB c-Rel regulates Tregdevelopment (32–36), we additionally coexpressed p50 and/or p65(Fig. 8A) and p50 and/or c-Rel (Fig. 8B). Enhancer activity wasincreased by coexpressed p50, p65, or p50 plus p65 (Fig. 8A) andby coexpressed p50, c-Rel, or p50 plus c-Rel (Fig. 8B). Importantly,any p50-mediated upregulation was further increased by Foxp3
coexpression. In contrast, p65- (Fig. 8A) and c-Rel–mediated(Fig. 8B) upregulation was reduced by Foxp3, and indeed re-pression of p65 and c-Rel by Foxp3 was previously described(9, 37). This inhibitory effect of Foxp3 was neutralized by p50coexpression (p50 plus p65 and p50 plus c-Rel expression).Taken together, these findings suggest that p50 and Foxp3 co-operate to upregulate the Gitr promoter by binding to the en-hancer. To confirm this, the Gitr promoter/enhancer reporterwas cotransfectedwith a fixed amount of the p50 expression plasmid,but with different amounts of the Foxp3 expression plasmid. En-hancer activity was increased by Foxp3 coexpression in a dose-de-pendent manner (Fig. 8C). A cooperative effect of p50 and Foxp3 onthe enhancer was also observed using the Ox40 promoter/enhancerreporter plasmid (Supplemental Fig. 3G) operating through theenhancer only, or through both the promoter and enhancer.How might Foxp3 upregulate gene expression using a p50 that
possesses no transactivation domain? Usually, NF-kB p50 posi-tively regulates gene expression by associating with other NF-kBand IkB family members that contain transactivation domains(e.g., through a p50/p65 heterodimer, p50/c-Rel heterodimer, orp50 homodimer/Bcl-3). In support of this, we detected p65, c-Rel,and Bcl-3 binding to the kB probe (Fig. 7C). We then asked whatadditional necessary part Foxp3 might play. We analyzed p50binding to the kB probe with or without Foxp3. We consideredusing a nuclear extract from CD3-activated T cells (no Foxp3), butthe amount of p50 in this nuclear extract is different from that inFoxp3+ T cells (stimulated with TGF-b). We therefore trappedFoxp3 protein in the same nuclear extract by adding a Foxp3 com-petitor (Fig. 7C) and performed a DNA pull-down assay using thekB probe. p50 binding to the enhancer probe was reduced as aresult of inhibition of Foxp3 binding to the probe (Fig. 7C), sug-gesting that Foxp3 stabilizes p50 binding by itself binding to bothp50 and DNA (Fig. 8D), thereby enabling accumulation of p50/p65and p50/c-Rel heterodimer (Fig. 8E) and/or p50/p50 homodimer/Blc-3 (Supplemental Fig. 3F) to the enhancer. To be able to un-derstand the whole picture of the Foxp3-mediated transcriptionalupregulation, further investigation is obviously required. However,it is clear that the activation function of the transactivation domainsof p65 and c-Rel do not seem to be inhibited by such indirectbinding of Foxp3 mediated via p50. This would be consistent withthe enhanced luciferase activity seen when all components (Fig.8A, p50, p65, and Foxp3; Fig. 8B, p50, c-Rel, and Foxp3) werecoexpressed.
DiscussionWe have identified a strong enhancer within the Gitr/Ox40 locusthat is regulated by NF-kB and Foxp3. Gitr gene expression isupregulated by NF-kB through this enhancer in activated Teff, andfurther upregulated by NF-kB in conjunction with Foxp3 in Treg.It is well established that Foxp3 can bind to other transcriptionfactors and inhibit activities of these factors (as shown in Fig. 8A,8B, the activity of the p65 and c-Rel subunits of NF-kB wasinhibited by coexpression of Foxp3). However, the enhancer ac-tivity was upregulated by coexpression of the p50 subunit of NF-kB with Foxp3. Foxp3 seems to stabilize binding of the NF-kBp50 to this enhancer. To explain how Foxp3 might stimulate p50-mediated transcription while lacking a transactivation domain (38),we suggest that p50 associates with other family members (e.g.,p65 and c-Rel) to exploit their activation domains (38). Indeed,we have also shown that p65 and c-Rel bind to kB sites with p50in the enhancer (Figs. 4C, 7C). Foxp3 could bind to the p50molecule and DNA without altering the p50/p65 and p50/c-Relcomplex formation (Fig. 8E) or inhibiting the transactivation do-main of p65 and c-Rel. Although nuclear NF-kB levels are tightly
FIGURE 5. NF-kB regulates Gitr and Ox40 gene expression. (A) p50
binding to kB1 plus kB2 and kB3 regions in CD3-activated T cells
(activated by anti-CD3 for indicated times) were analyzed by ChIP using
anti-p50 (p50) and control IgG. (B) Gitr and Ox40 expression on CD4+
T cells from p50-deficient (p50 KO) and WT mice. Nonstimulated (Non)
and CD3-activated (CD3) CD4+ T cells from indicated mice were ana-
lyzed by FACS using anti-Gitr and anti-Ox40. The percentages of CD4+
Gitrhigh or CD4+Ox40high population are indicated. (C–E) EL4 cells were
activated with anti-CD3 and cultured with or without NAI concentrations
(nanomolars) are indicated. (C) Gitr, (D) Ox40, or (E) Gapdh expression
were analyzed by quantitative RT-PCR (expression levels normalized
relative to 18S rRNA levels). (F) Gitr and Ox40 expression in nonacti-
vated (gray) and CD3-activated (solid line) T cells with or without NAI
(100 nM) analyzed by FACS. Data are representative of more than three
(A–F) independent experiments (error bars indicate the SD of triplicate
samples).
The Journal of Immunology 7
at Cedars-Sinai M
edical Center on M
arch 14, 2014http://w
ww
.jimm
unol.org/D
ownloaded from

regulated by negative feedback in activated T cells, we detectedNF-kB p50, p65, and c-Rel in 72 h–stimulated T cells with TGF-bplus anti-CD3 plus anti-CD28. Nuclear translocation of thesetranscription factors sustained in Treg through TGF-b signalingand/or a Foxp3-mediated pathway. In CD3-activated T cells,nuclear p65 levels peak 2–6 h after activation (39, 40). Althoughwe detected p65 binding to the kB probe using a nuclear extractfrom 72 h–stimulated T cells, p65 levels may have diminishedfrom those found earlier after activation. c-Rel levels seem to
remain high even after p65 decreased (40). The Foxp3 promoteractivity is regulated by c-Rel (34), and nTreg development isalso regulated by c-Rel through CNS3 in the Foxp3 gene (36).Taken together, p50/c-Rel heterodimers may be a key componentin upregulating Gitr gene expression. Alternatively, transactivationdomains might also be supplied by an IkB family member Bcl-3,which can bind to the p50 homodimer (41, 42) (SupplementalFig. 3F). Gene expression upregulated by the p50 homodimer/Bcl-3 complex seems to depend on NF-kB sites (the p50 homo-
FIGURE 6. NF-kB p50 regulates Gitr expression in
Treg. (A) p50 binding to the kB1 plus kB2 and kB3
regions in nTreg and CD4+CD252 T cells were ana-
lyzed by ChIP using anti-p50 (p50) and control IgG. (B)
CD4+CD252 T cells from Foxp3-GFP reporter mice
were cultured with TGF-b plus anti-CD3 plus anti-CD28
for 48 h, and Foxp3+ iTreg were isolated by sorting with
GFP (93% were Foxp3+ T cells). p50 binding to the kB1
plus kB2 and kB3 regions in iTreg and CD4+CD252
T cells was analyzed by ChIP using anti-p50 (p50) and
control IgG. (C) Splenocytes and thymocytes were iso-
lated from p50-deficient (p50 KO) and WT mice. These
cells were stained with anti-CD4, anti-Foxp3, and anti-
Gitr. Gitr expression in CD4+Foxp3+ population was
analyzed. Data are representative of more than three
(A–C) independent experiments (error bars indicate
the SD of triplicate samples).
FIGURE 7. Binding of NF-kB p50 and Foxp3 to kB sites. (A) DNA sequences of the kB1, Foxp3, and CD40 NF-kB (from CD40 promoter) (CD40)
competitors. NF-kB binding sequences are indicated by bold, and a potential Foxp3 binding sequence is indicated by bold italic. (B) DNA pull-down assays
were performed using a biotinylated enhancer probe containing the kB1 plus kB2 sites (enhancer) or the same length of the Gitr promoter probe and
a nuclear extract prepared from stimulated CD4+CD252 T cells with TGF-b plus anti-CD3 plus anti-CD28 for 72 h. The coprecipitated proteins were
analyzed by immunoblotting with anti-p50 and anti-Foxp3. (C) DNA pull-down assays were performed using the kB probe and nuclear extracts prepared
from nonstimulated (Non) and TGF-b plus anti-CD3 plus anti-CD28–stimulated (72 h) CD4+CD25– T cells (left panel). Competition DNA pull-down assay
was performed using the same probe and nuclear extract (from stimulated cells) and using nonbiotinylated kB1 or Foxp3 competitor [shown in (A), right
panel]. Coprecipitated proteins were analyzed by immunoblotting with anti-p50, anti-Foxp3, anti-p65, anti–c-Rel, and anti–Bcl-3. (D) DNA pull-down
assays were performed with a biotinylated kB probe and the nonbiotinylated CD40 NF-kB competitor [shown in (A)]. Coprecipitated proteins were an-
alyzed by immunoblotting with anti-p50 and anti-Foxp3. (E) p50 was precipitated by anti-p50 or control IgG (without DNA), and coprecipitated FLAG-
tagged Foxp3 was detected using an anti-FLAG. Data are representative of more than three (B–E) independent experiments.
8 REGULATION OF Gitr GENE EXPRESSION
at Cedars-Sinai M
edical Center on M
arch 14, 2014http://w
ww
.jimm
unol.org/D
ownloaded from
dimer without Bcl-3 inhibits transcription through some NF-kBsites). Because Bcl-3 does not bind to DNA, it is not clear howBcl-3 selects a p50 homodimer to be bound. We imagine that thep50 homodimer and Bcl-3 complex may be stabilized in con-junction with other factors that can bind close to the p50 homo-dimer binding site, in the way Foxp3 appears to do so in this study.In our case, Bcl-3 and p65 binding to the kB site was not detectedand c-Rel binding was reduced unless Foxp3 was also bound tothe enhancer (Fig. 7C). This may be because less p50 can bind tothe kB site in the absence of Foxp3, or that NF-kB p50/p65, p50/c-Rel, and/or p50 homodimer/Bcl-3 formation is stabilized byFoxp3.It is generally considered that promoters are upregulated by
communication between the promoter and an enhancer via loopformation (43). The identified enhancer in the present study islocated ∼5 and 20 kb downstream of the Gitr and Ox40 promoters,respectively. These distances are within the range found with otherwell-characterized enhancers and promoters. For example, the IgH(44) and Bcl-2 (45) promoter interacts with the 39 enhancer over∼100 kb. These findings suggest that Ox40 expression in Treg isalso upregulated by Foxp3 through this NF-kB p50–mediated en-hancer, but we cannot rule out a role for NF-kB p50 acting throughthe Ox40 promoter. Even so, the strong enhancer activity driven bythe combination of p50 and Foxp3 seems to regulate Treg-specificupregulation within Gitr/Ox40 gene locus, by analogy with the Il4,
Il5, and Il13 genes that are located over 140 kb within the Th2cytokine-specific gene locus (46).Recent analyses of DNase I HS sequencing and Foxp3 ChIP
sequencing in Foxp3+ Treg and CD4+Foxp3– T cells (28) haveshown that Foxp3 predominantly utilizes functional enhancers thatare present in Foxp3– T cells during Treg development. Takenwith our own findings, we now propose the following sequence ofevents during the activation of Gitr and Ox40 in Teff and Treg: 1)NF-kB p50/p65 and/or p50/c-Rel bind to the enhancer in activatedTeff at an early stage of Treg development, leading to chromatinremodeling and Gitr and Ox40 gene expression; 2) induced Foxp3in Treg stabilizes binding of the p50/p65 and/or p50/c-Rel hetero-dimers and p50-homodimer/Bcl-3 to the enhancer; and 3) thesecomplexes then further upregulate Girt and Ox40 expression inTreg.We suggest that other important Treg genes such as Ctla4, Il35,
Cd25, and MS4A members may be regulated by Foxp3 by similarmechanisms. We therefore offer this scheme as a solution to howFoxp3 may upregulate certain genes enforcing regulation whilesimultaneously being able to damp so many others promotinginflammation.
AcknowledgmentsWe thank Drs. J. Kaye, W. Sabbagh, C. Norbury and P. Cook for review of
the manuscript.
FIGURE 8. NF-kB p50 and Foxp3 cooperate to upregulate enhancer activity. (A and B) The Gitr promoter-enhancer luciferase reporter plasmid (Pro8 +
Enhancer) was cotransfected with empty vector, p50, and/or p65 (A) or c-Rel (B) expression plasmids with or without Foxp3 expression plasmid.
Cotransfected expression plasmids are indicated under the graph. Total amount of DNA for transfection was adjusted by addition of DNA from the
empty vector. Luciferase activity with negative control plasmid (Basic; no promoter and no enhancer) and Pro8 (no enhancer) are also shown. *p ,0.05, **p , 0.005 (Student t test). (C) Gitr promoter-enhancer luciferase reporter plasmid (Pro8 + Enhancer) was cotransfected with 4 mg p50 ex-
pression plasmid (p50) and different amounts of Foxp3 expression plasmid (0–8 mg). DNA amounts of the expression plasmids are shown under the
graph. The total amount of DNAwas adjusted with addition of DNA from the empty vector. (D) Model of NF-kB p50 and Foxp3 binding to the kB1 site.
(E) Model of NF-kB p50, Foxp3, and NF-kB p65 or c-Rel binding to the kB site. Data are representative of more than three (A–C) independent
experiments (error bars indicate the SD of triplicate samples).
The Journal of Immunology 9
at Cedars-Sinai M
edical Center on M
arch 14, 2014http://w
ww
.jimm
unol.org/D
ownloaded from

DisclosuresThe authors have no financial conflicts of interest.
References1. Watts, T. H. 2005. TNF/TNFR family members in costimulation of T cell
responses. Annu. Rev. Immunol. 23: 23–68.2. McHugh, R. S., M. J. Whitters, C. A. Piccirillo, D. A. Young, E. M. Shevach,
M. Collins, and M. C. Byrne. 2002. CD4+CD25+ immunoregulatory T cells:gene expression analysis reveals a functional role for the glucocorticoid-inducedTNF receptor. Immunity 16: 311–323.
3. Shimizu, J., S. Yamazaki, T. Takahashi, Y. Ishida, and S. Sakaguchi. 2002.Stimulation of CD25+CD4+ regulatory T cells through GITR breaks immuno-logical self-tolerance. Nat. Immunol. 3: 135–142.
4. Takeda, I., S. Ine, N. Killeen, L. C. Ndhlovu, K. Murata, S. Satomi,K. Sugamura, and N. Ishii. 2004. Distinct roles for the OX40-OX40 ligand in-teraction in regulatory and nonregulatory T cells. J. Immunol. 172: 3580–3589.
5. Josefowicz, S. Z., L. F. Lu, and A. Y. Rudensky. 2012. Regulatory T cells:mechanisms of differentiation and function. Annu. Rev. Immunol. 30: 531–564.
6. Sakaguchi, S. 2004. Naturally arising CD4+ regulatory T cells for immunologicself-tolerance and negative control of immune responses. Annu. Rev. Immunol.22: 531–562.
7. Shevach, E. M. 2000. Regulatory T cells in autoimmmunity. Annu. Rev.Immunol. 18: 423–449.
8. Wu, Y., M. Borde, V. Heissmeyer, M. Feuerer, A. D. Lapan, J. C. Stroud,D. L. Bates, L. Guo, A. Han, S. F. Ziegler, et al. 2006. FOXP3 controls regu-latory T cell function through cooperation with NFAT. Cell 126: 375–387.
9. Bettelli, E., M. Dastrange, and M. Oukka. 2005. Foxp3 interacts with nuclearfactor of activated T cells and NF-kB to repress cytokine gene expression andeffector functions of T helper cells. Proc. Natl. Acad. Sci. USA 102: 5138–5143.
10. Ono, M., H. Yaguchi, N. Ohkura, I. Kitabayashi, Y. Nagamura, T. Nomura,Y. Miyachi, T. Tsukada, and S. Sakaguchi. 2007. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature 446: 685–689.
11. Collison, L. W., C. J. Workman, T. T. Kuo, K. Boyd, Y. Wang, K. M. Vignali,R. Cross, D. Sehy, R. S. Blumberg, and D. A. Vignali. 2007. The inhibitorycytokine IL-35 contributes to regulatory T-cell function. Nature 450: 566–569.
12. Vignali, D. A., L. W. Collison, and C. J. Workman. 2008. How regulatory T cellswork. Nat. Rev. Immunol. 8: 523–532.
13. Valzasina, B., C. Guiducci, H. Dislich, N. Killeen, A. D. Weinberg, andM. P. Colombo. 2005. Triggering of OX40 (CD134) on CD4+CD25+ T cellsblocks their inhibitory activity: a novel regulatory role for OX40 and its com-parison with GITR. Blood 105: 2845–2851.
14. So, T., and M. Croft. 2007. Cutting edge: OX40 inhibits TGF-b- and antigen-driven conversion of naive CD4 T cells into CD25+Foxp3+ T cells. J. Immunol.179: 1427–1430.
15. Vu, M. D., X. Xiao, W. Gao, N. Degauque, M. Chen, A. Kroemer, N. Killeen,N. Ishii, and X. C. Li. 2007. OX40 costimulation turns off Foxp3+ Tregs. Blood110: 2501–2510.
16. Howie, D., K. F. Nolan, S. Daley, E. Butterfield, E. Adams, H. Garcia-Rueda,C. Thompson, N. J. Saunders, S. P. Cobbold, Y. Tone, et al. 2009. MS4A4B isa GITR-associated membrane adapter, expressed by regulatory T cells, whichmodulates T cell activation. J. Immunol. 183: 4197–4204.
17. Rogers, P. R., J. Song, I. Gramaglia, N. Killeen, and M. Croft. 2001. OX40promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival ofCD4 T cells. Immunity 15: 445–455.
18. Song, J., S. Salek-Ardakani, P. R. Rogers, M. Cheng, L. Van Parijs, and M. Croft.2004. The costimulation-regulated duration of PKB activation controls T celllongevity. Nat. Immunol. 5: 150–158.
19. Tone, M., Y. Tone, E. Adams, S. F. Yates, M. R. Frewin, S. P. Cobbold, andH. Waldmann. 2003. Mouse glucocorticoid-induced tumor necrosis factor receptorligand is costimulatory for T cells. Proc. Natl. Acad. Sci. USA 100: 15059–15064.
20. Tone, Y., Y. Kojima, K. Furuuchi, M. Brady, Y. Yashiro-Ohtani,M. L. Tykocinski, and M. Tone. 2007. OX40 gene expression is up-regulated bychromatin remodeling in its promoter region containing Sp1/Sp3, YY1, and NF-kB binding sites. J. Immunol. 179: 1760–1767.
21. Tone, M., Y. Tone, P. J. Fairchild, M. Wykes, and H. Waldmann. 2001. Regu-lation of CD40 function by its isoforms generated through alternative splicing.Proc. Natl. Acad. Sci. USA 98: 1751–1756.
22. Tone, M., Y. Tone, J. M. Babik, C. Y. Lin, and H. Waldmann. 2002. The role of Sp1and NF-kB in regulating CD40 gene expression. J. Biol. Chem. 277: 8890–8897.
23. Chen, W., W. Jin, N. Hardegen, K. J. Lei, L. Li, N. Marinos, G. McGrady, andS. M. Wahl. 2003. Conversion of peripheral CD4+CD252 naive T cells to CD4+
CD25+ regulatory T cells by TGF-b induction of transcription factor Foxp3. J.Exp. Med. 198: 1875–1886.
24. Fantini, M. C., C. Becker, G. Monteleone, F. Pallone, P. R. Galle, andM. F. Neurath. 2004. Cutting edge: TGF-b induces a regulatory phenotype inCD4+CD252 T cells through Foxp3 induction and down-regulation of Smad7. J.Immunol. 172: 5149–5153.
25. Rudra, D., P. deRoos, A. Chaudhry, R. E. Niec, A. Arvey, R. M. Samstein,C. Leslie, S. A. Shaffer, D. R. Goodlett, and A. Y. Rudensky. 2012. Transcriptionfactor Foxp3 and its protein partners form a complex regulatory network. Nat.Immunol. 13: 1010–1019.
26. Tone, Y., K. Furuuchi, Y. Kojima, M. L. Tykocinski, M. I. Greene, and M. Tone.2008. Smad3 and NFAT cooperate to induce Foxp3 expression through its en-hancer. Nat. Immunol. 9: 194–202.
27. Gronostajski, R. M. 2000. Roles of the NFI/CTF gene family in transcription anddevelopment. Gene 249: 31–45.
28. Samstein, R. M., A. Arvey, S. Z. Josefowicz, X. Peng, A. Reynolds,R. Sandstrom, S. Neph, P. Sabo, J. M. Kim, W. Liao, et al. 2012. Foxp3 exploitsa pre-existent enhancer landscape for regulatory T cell lineage specification. Cell151: 153–166.
29. Franzoso, G., L. Carlson, L. Xing, L. Poljak, E. W. Shores, K. D. Brown,A. Leonardi, T. Tran, B. F. Boyce, and U. Siebenlist. 1997. Requirement for NF-kB in osteoclast and B-cell development. Genes Dev. 11: 3482–3496.
30. Iotsova, V., J. Caamano, J. Loy, Y. Yang, A. Lewin, and R. Bravo. 1997.Osteopetrosis in mice lacking NF-kB1 and NF-kB2. Nat. Med. 3: 1285–1289.
31. Hori, S., T. Nomura, and S. Sakaguchi. 2003. Control of regulatory T cell de-velopment by the transcription factor Foxp3. Science 299: 1057–1061.
32. Deenick, E. K., A. R. Elford, M. Pellegrini, H. Hall, T. W. Mak, and P. S. Ohashi.2010. c-Rel but not NF-kB1 is important for T regulatory cell development. Eur.J. Immunol. 40: 677–681.
33. Long, M., S. G. Park, I. Strickland, M. S. Hayden, and S. Ghosh. 2009. Nuclearfactor-kB modulates regulatory T cell development by directly regulating ex-pression of Foxp3 transcription factor. Immunity 31: 921–931.
34. Ruan, Q., V. Kameswaran, Y. Tone, L. Li, H. C. Liou, M. I. Greene, M. Tone, andY. H. Chen. 2009. Development of Foxp3+ regulatory t cells is driven by thec-Rel enhanceosome. Immunity 31: 932–940.
35. Vang, K. B., J. Yang, A. J. Pagan, L. X. Li, J. Wang, J. M. Green, A. A. Beg, andM. A. Farrar. 2010. Cutting edge: CD28 and c-Rel-dependent pathways initiateregulatory T cell development. J. Immunol. 184: 4074–4077.
36. Zheng, Y., S. Josefowicz, A. Chaudhry, X. P. Peng, K. Forbush, andA. Y. Rudensky. 2010. Role of conserved non-coding DNA elements in theFoxp3 gene in regulatory T-cell fate. Nature 463: 808–812.
37. Loizou, L., K. G. Andersen, and A. G. Betz. 2011. Foxp3 interacts with c-Rel tomediate NF-kB repression. PLoS ONE 6: e18670.
38. Li, Q., and I. M. Verma. 2002. NF-kB regulation in the immune system. Nat.Rev. Immunol. 2: 725–734.
39. Kingeter, L. M., S. Paul, S. K. Maynard, N. G. Cartwright, and B. C. Schaefer.2010. Cutting edge: TCR ligation triggers digital activation of NF-kB. J.Immunol. 185: 4520–4524.
40. Mittal, A., S. Papa, G. Franzoso, and R. Sen. 2006. NF-kB-dependent regulationof the timing of activation-induced cell death of T lymphocytes. J. Immunol.176: 2183–2189.
41. Franzoso, G., V. Bours, V. Azarenko, S. Park, M. Tomita-Yamaguchi, T. Kanno,K. Brown, and U. Siebenlist. 1993. The oncoprotein Bcl-3 can facilitate NF-kB-mediated transactivation by removing inhibiting p50 homodimers from select kBsites. EMBO J. 12: 3893–3901.
42. Fujita, T., G. P. Nolan, H. C. Liou, M. L. Scott, and D. Baltimore. 1993. Thecandidate proto-oncogene bcl-3 encodes a transcriptional coactivator that acti-vates through NF-kB p50 homodimers. Genes Dev. 7: 1354–1363.
43. Bulger, M., and M. Groudine. 1999. Looping versus linking: toward a model forlong-distance gene activation. Genes Dev. 13: 2465–2477.
44. Ren, X., R. Siegel, U. Kim, and R. G. Roeder. 2011. Direct interactions ofOCA-B and TFII-I regulate immunoglobulin heavy-chain gene transcription byfacilitating enhancer-promoter communication. Mol. Cell 42: 342–355.
45. Gong, F., L. Sun, Z. Wang, J. Shi, W. Li, S. Wang, X. Han, and Y. Sun. 2011. TheBCL2 gene is regulated by a special AT-rich sequence binding protein 1-medi-ated long range chromosomal interaction between the promoter and the distalelement located within the 39-UTR. Nucleic Acids Res. 39: 4640–4652.
46. Spilianakis, C. G., and R. A. Flavell. 2004. Long-range intrachromosomalinteractions in the T helper type 2 cytokine locus. Nat. Immunol. 5: 1017–1027.
10 REGULATION OF Gitr GENE EXPRESSION
at Cedars-Sinai M
edical Center on M
arch 14, 2014http://w
ww
.jimm
unol.org/D
ownloaded from

P3 AGCTGGCCTGAAGCCCAGTCTGAGG M1 AAGATCTCTGAAGCCCAGTCTGAGG M2 AGCTAGATCTAAGCCCAGTCTGAGG M3 AGCTGGCAGATCTCCCAGTCTGAGG M4 AGCTGGCCTGAGATCTAGTCTGAGG M5 AGCTGGCCTGAAGAGATCTCTGAGG M6 AGCTGGCCTGAAGCCCAGATCTAGG M7 AGCTGGCCTGAAGCCCAGTAGATCT GCTGGC CCCAG
M1 M2 M3 M4 M5 M6 M7 P3
CCTCAGATGTCTGCCGGAAACTGGAGGTGGAGCTGGCCTGAAGCCCAGTCTGAGGGGT P1
Pro2 (–51 to +46) Pro3 (–90 to +46)
P2 P3
P1 P2 P3
–
Probe: GL NFI
Competitor Probe
A
B C D
Supplemental Fig. 1. Transcription factor NFI binding to the sequence between –90 and –51 (A) The deleted sequence from Pro3 to Pro2 are shown, and the position of the EMSA probes (P1, P2 and P3) are indicated by underlines. (B) EMSA was performed using 32P-labeled P1, P2 and P3 probes and a nuclear extract from EL4 cells. (C) EMSA was performed with P3 wild type and mutant probes. DNA sequences of the P3 and its mutant probes are shown under the EMSA result. The transcription factor binding sequences were determined by EMSA and shown under the probe sequence list. This sequence is similar to the consensus sequence of a transcription factor NFI (TTGGCNNNNNGCCAA). (D) Previously, we showed NFI binding to the Gitr ligand (GL) promoter [Tone et al. (2003) PNAS 100, 15059–15064]. EMSA was performed with the GL probe (–). The NFI binding to the GL probe was disappeared with 100-fold excess the GL and the P3 competitors, indicating NFI binding to the P3 sequence. NFI is indeed binding to the Gitr promoter region.
Consensus:

DNase I Conc.
Non CD3
Probe 1
Non CD3
Probe 2
Non CD3
Probe 3
Non CD3 CD4+ T cell
Non LPS bmDC
Probe 1 Probe 3 DNase I Conc.
EL4
Gitr gene
1.0 kb
Kpn I sites HS1 HS2
Probe 1
Ox40 gene HS3
Probe 2 Probe 3
Non CD3 CD4+ T cell
Non LPS bmDC
A
B
C
Supplemental Fig. 2. DNase I hypersensitive sites in the Ox40/Gitr gene locus. (A) Positions of Ox40 and Gitr genes in this locus are illustrated. Exons are indicated by gray boxes. The activation specific DNase I hypersensitive (HS) sites HS1 (green arrow), HS2 (orange arrow) and HS3 (blue arrow) are indicated with common HS sites (black arrows with dotted lines). Positions of the probes used in (B) and (C) are indicated by red bars. (B) DNase I HS assay was performed using non-activated (Non) and anti-CD3 activated (CD3) EL4 cells. Isolated nuclei were partially digested with different concentrations of DNase I (DNase I Conc., indicated by yellow triangles). DNA isolated from these nuclei was digested with Kpn I and analyzed by Southern blot hybridization using the indicated probes. Positions of the probes are shown in (A). Activation specific and common bands generated with the HS sites are indicated by arrows with the same colors shown in (A). (C) DNase I HS assay was performed as described in (B). Non-stimulated (Non) and LPS-stimulated (LPS) bone marrow derived dendritic cells (bmDC), and Non-activated (Non) and anti-CD3 activated (CD3) CD4+ T cells were used. Activated T cell specific bands generated with the HS sites are indicated by arrows with the same colors shown in (A).
AGTTACACTGGAAACACCACAGGTGGGACATCCCCAAACAGATGACGTGTGTGCAAATACCCACAGGCTGTATGTGCTGGAAACCCCTCCTGTGGGCGGC
TGATGGGTGTGATAGTGGGATGGCAGGGGATTCTGCAGGGTGGGGTGAGGGGTGTCGTCTTTGCTGTATCCTTGCCTTGAAGCCAGCAGCCTGCATTCCA
CTCACGTCCACCCCAGGGGCATGAGATGAGCCGTGCCAGCCTGTGAGTTAGATCTGGGGATTTCCACTAAGAACTGTGAGCTGTGC
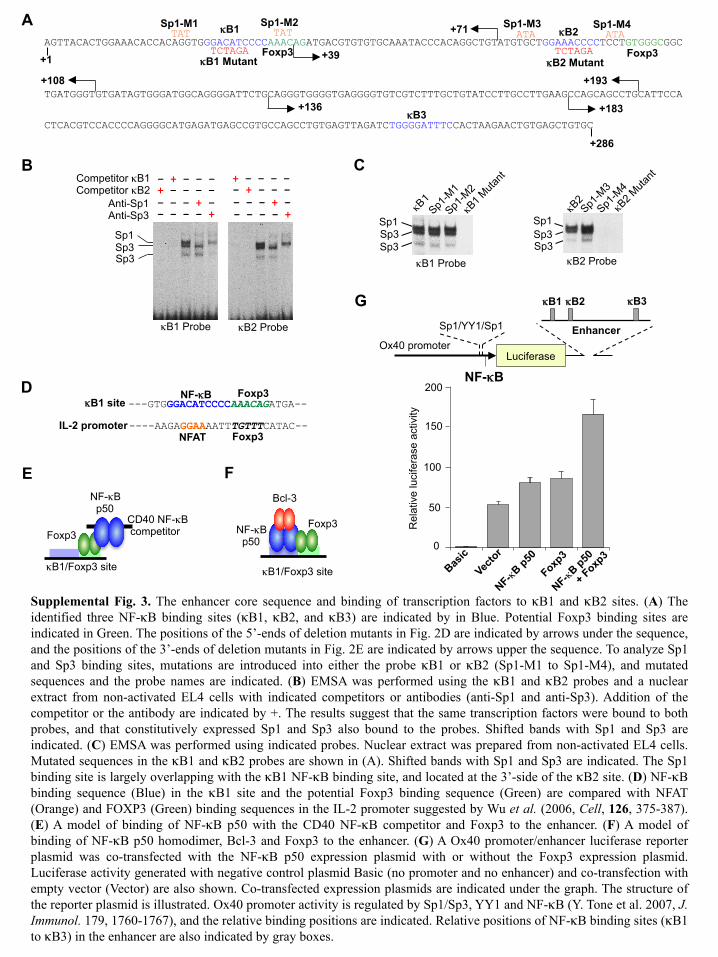
TAT TAT ATA ATA κB1 κB2
κB3
Sp1-M1 Sp1-M2 Sp1-M3 Sp1-M4 A
+39
+71
B
Sp3 Sp1
Sp3 κB1 Probe
TCTAGA κB1 Mutant κB2 Mutant
TCTAGA Foxp3 Foxp3
– + – – –
– – – – + – – – + – + – – – – – + – – –
– – – – + – – – + –
+ – – – – Competitor κB1 Competitor κB2
Anti-Sp1 Anti-Sp3
Sp1 Sp3 Sp3
C
+108
+136 +183
+193
+286
+1
κB1 Probe κB2 Probe
Sp3 Sp1
Sp3 κB2 Probe
---GTGGGACATCCCCAAACAGATGA--
----AAGAGGAAAATTTGTTTCATAC--
NF-κB Foxp3 κB1 site
Foxp3 NFAT IL-2 promoter
κB1/Foxp3 site
CD40 NF-κB competitor
NF-κB p50
Foxp3 NF-κB p50
Foxp3
κB1/Foxp3 site
Bcl-3
F E
Supplemental Fig. 3. The enhancer core sequence and binding of transcription factors to κB1 and κB2 sites. (A) The identified three NF-κB binding sites (κB1, κB2, and κB3) are indicated by in Blue. Potential Foxp3 binding sites are indicated in Green. The positions of the 5’-ends of deletion mutants in Fig. 2D are indicated by arrows under the sequence, and the positions of the 3’-ends of deletion mutants in Fig. 2E are indicated by arrows upper the sequence. To analyze Sp1 and Sp3 binding sites, mutations are introduced into either the probe κB1 or κB2 (Sp1-M1 to Sp1-M4), and mutated sequences and the probe names are indicated. (B) EMSA was performed using the κB1 and κB2 probes and a nuclear extract from non-activated EL4 cells with indicated competitors or antibodies (anti-Sp1 and anti-Sp3). Addition of the competitor or the antibody are indicated by +. The results suggest that the same transcription factors were bound to both probes, and that constitutively expressed Sp1 and Sp3 also bound to the probes. Shifted bands with Sp1 and Sp3 are indicated. (C) EMSA was performed using indicated probes. Nuclear extract was prepared from non-activated EL4 cells. Mutated sequences in the κB1 and κB2 probes are shown in (A). Shifted bands with Sp1 and Sp3 are indicated. The Sp1 binding site is largely overlapping with the κB1 NF-κB binding site, and located at the 3’-side of the κB2 site. (D) NF-κB binding sequence (Blue) in the κB1 site and the potential Foxp3 binding sequence (Green) are compared with NFAT (Orange) and FOXP3 (Green) binding sequences in the IL-2 promoter suggested by Wu et al. (2006, Cell, 126, 375-387). (E) A model of binding of NF-κB p50 with the CD40 NF-κB competitor and Foxp3 to the enhancer. (F) A model of binding of NF-κB p50 homodimer, Bcl-3 and Foxp3 to the enhancer. (G) A Ox40 promoter/enhancer luciferase reporter plasmid was co-transfected with the NF-κB p50 expression plasmid with or without the Foxp3 expression plasmid. Luciferase activity generated with negative control plasmid Basic (no promoter and no enhancer) and co-transfection with empty vector (Vector) are also shown. Co-transfected expression plasmids are indicated under the graph. The structure of the reporter plasmid is illustrated. Ox40 promoter activity is regulated by Sp1/Sp3, YY1 and NF-κB (Y. Tone et al. 2007, J. Immunol. 179, 1760-1767), and the relative binding positions are indicated. Relative positions of NF-κB binding sites (κB1 to κB3) in the enhancer are also indicated by gray boxes.
D 200
150
100
50
0
Rel
ativ
e lu
cife
rase
act
ivity
Luciferase
κB1 κB2 κB3
NF-κB
Sp1/YY1/Sp1
Ox40 promoter Enhancer
G




















