
Nitrite Reduction Mediated by Heme Models. Routes to NO and HNO?

A
(mioHM0©
K
1
ccTch[atCahts
0d
Electrochimica Acta 51 (2006) 6435–6441
Iron(III) protoporphyrin IX—single-wall carbon nanotubes modifiedelectrodes for hydrogen peroxide and nitrite detection
Graziella L. Turdean a,b,∗,1, Ionel Catalin Popescu a, Antonella Curulli c, Giuseppe Palleschi b
a “Babes Bolyai” University, Physical Chemistry Department, Arany Janos 11, 400 028 Cluj-Napoca, Romaniab Dipartimento di Scienze e Technologie Chimiche, Universita di Roma “Tor Vergata”, Via della Ricerca Scientifica 1, 00133 Rome, Italy
c Istituto per lo Studio dei Materiali Nanostrutturati (ISMN) CNR Division 2, Via del Castro Laurenziano 7, 00161 Rome, Italy
Received 14 February 2006; received in revised form 12 April 2006; accepted 18 April 2006Available online 5 June 2006
bstract
Iron(III) protoporphyrin IX (Fe(III)P), adsorbed either on single-walled carbon nanotubes (SWCNT) or on hydroxyl-functionalized SWCNTSWCNT-OH), was incorporated within a Nafion matrix immobilized on the surface of a graphite electrode. From cyclic voltammetric measure-ents, performed under different experimental conditions (pH and potential scan rate), it was established that the Fe(III)P/Fe(II)P redox couple
nvolves 1e−/1H+. The heterogeneous electron transfer process occurred faster when Fe(III)P was adsorbed on SWCNT-OH (∼11 s−1) thann SWCNT (∼4.9 s−1). Both the SWCNT-Fe(III)P- and SWCNT-OH-Fe(III)P-modified graphite electrodes exhibit electrocatalytic activity for
2O2 and nitrite reduction. The modified electrodes sensitivities were found varying in the following sequences: SSWCNT-OH-Fe(III)P = 2.45 mA/≈ SSWCNT-Fe(III)P = 2.95 mA/M > SFe(III)P = 1.34 mA/M for H2O2, and SSWCNT-Fe(III)P = 3.54 mA/M > SFe(III)P = 1.44 mA/M > SSWCNT-OH-Fe(III)P =.81 mA/M for NO2−.
2006 Elsevier Ltd. All rights reserved.
am[
tetcfifb
eywords: Hemin; Single-wall carbon nanotubes; H2O2; Nitrite reduction
. Introduction
The discovery of fullerenes in 1985 and the invention ofarbon nanotubes (CNT) in 1991 opened new perspectives onarbon materials based on flat graphite-like hexagonal layers [1].heir nanometer dimension [2] and their interesting physico-hemical properties, such as excellent electrical conductivity,igh chemical stability and remarkable mechanical strength3,4], recommended them as promising materials for variouspplications. The carbon nanotubes were found to have twoypes of structures: single-wall CNT (SWCNT) and multi-wallNT (MWCNT) [5]. Due to the above mentioned propertiesnd diversity, CNTs of various lengths and internal diameters
ave allowed a wide range of practical applications in elec-roanalytical chemistry and nanotechnology (i.e., probes forcanning microscopy [5], electron field emission sources, actu-∗ Corresponding author. Tel.: +402 64595872; fax: +402 64 590818.E-mail address: [email protected] (G.L. Turdean).
1 ISE member.
tasrtac[
013-4686/$ – see front matter © 2006 Elsevier Ltd. All rights reserved.oi:10.1016/j.electacta.2006.04.028
tors, nanoelectronic devices, batteries, nanotube-reinforcedaterials, hydrogen storage materials [6] and chemical sensors
4,5,7]).In the domain of electroanalytical chemistry, the subtle elec-
ronic behavior of CNT, even if not yet fully characterizedlectrochemically, is associated with several other characteris-ics [8]: (i) CNT are conducting and relatively stable and thusan be used as advanced electrode materials; (ii) CNT can beunctionalized with various chemical groups, facilitating themmobilization of biomolecules; (iii) CNT have a high sur-ace area to weight ratio, favorable to both charge transfer andiomolecule immobilization.
Recently, it has been demonstrated that CNTs can decreasehe electroreduction potential for various redox substrates and,t the same time, increase the reaction rate, improving theelectivity and sensitivity of amperometric detection. For thiseason, CNTs have been used as redox catalysts for electron
ransfer processes involving various electrochemical systems,s for example: calixarene [9], nitrite [10], ferrocene [9],atecholamine [11], H2O2 [12], NADH [13], norepinephrine14], dopamine [15], epinephrine, ascorbic acid [5,9], hemin
6 himic
[c[
poupwa(h[h[cd[d
rinnWdfptg
peowUpttelhit
2
2
AUaaepa
sTJAc0Ktaum
c(w
2
t
saemWenemt
2
P
stal
msit
tiarww
436 G.L. Turdean et al. / Electroc
16], hemoglobin [9,17], myoglobin [18], cytochrome[9,19,20], glucose oxidase [3,21–23] and peroxidases
17,24,25].The study of the direct heterogeneous electron transfer
rocess between electrodes and biomolecules (redox proteins,xidoreductases, etc.) is a convenient and informative means tonderstand the kinetics and thermodynamics of biological redoxrocesses. In this context, hemin (iron(III) protoporphyrin IX),hich has a good stability in solution, a low molecular weight,
nd is relatively inexpensive [26], plays an important role:i) as model molecule for understanding the redox activity ofeme-proteins (e.g. b-cytochromes, peroxidases, catalases)27,28] and oxygen-carrying proteins (myoglobin andemoglobin [29]); (ii) as mimetic compound for peroxidases26] or nitrite reductase [30]); (iii) as mediator, in conditionslose to the native biological environment and for amperometricetection of various species, as for example: O2 [31,32], H2O226], NO/NO2
− [30,33], superoxide [27], tryptophan and itserivatives [34].
Taking advantage of the fact that hemin has a porphyrining, promoting its adsorption on carbonaceous materials, it wasmmobilized on a CNT surface for O2 detection [35]. Unfortu-ately, it was reported that when dispersed in aqueous mediaanotubes aggregate easily because of the substantial van deraals attractions [36]. Consequently, in order to improve the
ispersion of CNTs in water, which is an important conditionor their use in biomedical application, several approaches wereroposed: polymer wrapping [37,38]; CNT coating with surfac-ants [37,38]; CNT functionalization with –OH [39] or –COOHroups [19,24].
Taking into account all these considerations, the aim of thisaper was to obtain modified electrodes for H2O2 and NO2
−lectrocatalytic reduction, based on the immobilization of heminn SWCNT and its further incorporation within a Nafion matrix,hich was deposited on the surface of a graphite electrode.sing the rate constants for the heterogeneous electron transferrocess, estimated with the Laviron approach [40], a quantita-ive evaluation of the influence of SWCNT functionalization onhe electron transfer process between hemin and the graphitelectrode was performed. For the first time, to our best know-edge, the electrocatalytic activities of the hemin-SWCNT- andemin-SWCNT-OH-modified graphite electrodes towards twomportant biological analytes, H2O2 and nitrite, have been quan-itatively evaluated.
. Materials
.1. Chemicals
Single-walled carbon nanotubes (SWCNT) (CarboLexP-grade, 1.2–1.5 nm diameter) were acquired from (Aldrich,SA). The iron(III) protoporphyrin IX chloride (Fe(III)P), 2-
mino-2-hydroxymethyl-1,3-propanediol(tris-(hydroxymethyl)
mino methane) (TRIS), Nafion solution (5% in methanol withquivalent weight of about 1100) and potassium bromide wereurchased from Fluka, Switzerland. KOH for CNT function-lization was from Carlo Erba (Italy). The sodium dodecylmtt
a Acta 51 (2006) 6435–6441
ulphate (SDS) and the K3[Fe(CN)6] were from Sigma, Italy.he hydrogen peroxide stock solution (30%, w/v) was from.T. Baker, Italy and the sodium nitrite from Merck, Germany.
phosphate buffer solution (PBS; pH 7, ionic strength 0.2 M)ontaining 0.05 M KBr was prepared from 0.05 M KH2PO4,.05 M K2HPO4. Also, a 0.05 M Tris–HCl containing 0.05 MBr solution was employed as supporting electrolyte. In order
o perform the pH-dependent experiments, the pH of PBS wasdjusted with HCl or KOH. The buffer solutions were preparedsing distilled-deionized water and were kept refrigerated toinimize bacterial growth.The 5 mM Fe(III)P solution was prepared by dissolving its
hloride salt in a 0.05 M Tris–HCl buffer, containing 0.05 M KBrpH 8). All chemicals were of analytical grade and were usedithout further purification.
.2. Apparatus
Cyclic voltammetric investigations were carried out using arace analyzer (model AMEL 433, Amel, Milan, Italy).
All electrochemical measurements were done using atandard single-compartment three electrode cell equipped with
platinum counter electrode, an Ag|AgCl, KClsat referencelectrode (Amel, 805/CPG/6) and a disc working electrodeade from spectral graphite (3 mm diameter) (Ringsdorff-erke, Gmbh, Bonn-Bad Godesberg, Germany). Prior to
xperiments the buffer solutions were purged with high-purityitrogen (Linde, Italy) for at least 15 min and a nitrogennvironment was then kept over the solution in the cell duringeasurements. All experiments were performed at room
emperature.
.3. Preparation of G/SWCNT–Fe(III)P-Nafion electrode
The graphite electrode was wet polished with Carbimet paperSA (600 grit) (Buehler, USA) and rinsed with distilled water.
The 0.010 g of SWCNT were dispersed in 50 ml aqueousolution of 1 wt% SDS. After homogenization and ultrasonica-ion, the suspension of SWCNT obtained was ultracentrifugatedt 120,000 G for 4 h. For all further experiments only the upperayer of the stable SWCNT suspension was used.
Functionalized SWCNT (SWCNT-OH) were obtained byixing 0.01 g SWCNT with 50 ml of 10 M KOH for 1 h. The
uspension obtained was heated for 1 h at 100 ◦C and then driedn a stove. After washing with water, the dried SWCNT-OH werereated as described above.
SWCNT of 500 �l or SWCNT-OH suspensions were mixedhoroughly with 5 mM Fe(III)P solution. Then, 2 �l of the result-ng mixture were cast onto the surface of the graphite electrodend allowed to dry at ambient temperature. This operation wasepeated successively three times. Finally, 2 �l of 5% Nafionas dropped on the modified electrode surface. The electrodesere stored at 4 ◦C.
The surface coverage with hemin (Γ , mol cm−2) was esti-ated by integrating the peak surface area (Q) under its reduc-ion peak recorded at low potential scan rate, and assuming thathe redox process involves one electron.

himic
3
3
ptSKsnsan–w
s[–ait
3
pFa
Fpspvc
htdptfFd
�
cccaompt–tf
obKs
G.L. Turdean et al. / Electroc
. Results and discussions
.1. The SWCNT functionalization
Among the techniques known to improve the SWCNT dis-ersion in aqueous solution, two approaches were compared inhe present work: (i) SWCNT coating with a surfactant; (ii) theWCNT functionalization with –OH groups by treatment withOH at room temperature and the subsequent coating with a
urfactant. The first method was chosen because the thermody-amic tendency toward bundling is overcome in the presence ofurfactants [16,36,41]. The second method is supposed to inducesimple solid-phase mechano-chemical exfoliation of carbon
anotubes, producing bundles of SWCNT functionalized withOH groups to various degrees and having a higher solubility inater [39].FTIR spectra, recorded for SWCNT treated by 10 M KOH,
howed a broad band centered between 3100 and 3400 cm−1
39,42], which is characteristic to the stretching vibration ofOH groups (Fig. 4 in reference [43]). It was supposed that thelkali treatment introduced the –OH groups on the already exist-ng defects, without breaking the graphite structure nor cuttinghe tube [43].
.2. Electrochemical behavior of hemin-modified electrodes
Fig. 1 shows the cyclic voltammograms recorded in phos-hate buffer (pH 7) at G/SWCNT-Nafion and at G/SWCNT-e(III)P-Nafion electrodes, prepared with non-functionalizednd functionalized SWCNT. As expected, in the absence of
ig. 1. Cyclic voltammograms of hemin-modified graphite electrodes incor-orated in SWCNT (. . .) and SWCNT–OH (– – –). Experimental conditions:tarting potential, 0 V vs. Ag/AgCl, KClsat; potential scan rate, 50 mV s−1; sup-orting electrolyte, 0.1 M phosphate buffer (pH 7); N2 saturated solution. Theoltammetric response of G/SWCNT-Nafion electrode (—) was included foromparison.
ifb3ih
stctcTevi
wrcvpcipcil
pt
a Acta 51 (2006) 6435–6441 6437
emin, no redox peaks were observed for either electrode inhe potential range investigated. By contrast, a pair of well-efined redox peaks was clearly seen when the hemin wasresent on the electrode surface. The pair peaks was attributedo a quasi-reversible monoelectron transfer (�EFWHM = 197 mVor G/SWCNT-Fe(III)P-Nafion and 177 mV for G/SWCNT-OH-e(III)P-Nafion), involving the Fe(III)/Fe(II) redox couple coor-inated in the porphyrinic ring [44].
For both modified electrodes investigated, the value ofEFWHM was higher than the theoretical value (90.5/n mV)
orresponding to a surface confined redox couple [40,42], indi-ating either a certain non-uniformity of the adsorbed redoxouples, or the existence of repulsive interactions between thective redox centers [45]. The slight decrease of the peak splitbserved for G/SWCNT-OH-Fe(III)P-Nafion (�Ep = 35 mV)odified electrode in comparison with the peak split corres-
onding to G/SWCNT-Fe(III)P-Nafion (�Ep = 55 mV) elec-rodes was attributed to a beneficial effect due to the presence ofOH groups. It can be suggested that the –OH groups increasehe van der Waals interactions between hemin and SWCNT,avoring its adsorption on the electrode material [37,39].
The formal potential (E◦′; defined as the average valuef the anodic and cathodic peak potentials) was not affectedy SWCNT functionalization: −0.500 V versus Ag/AgCl,Clsat for G/SWCNT-OH-Fe(III)P-Nafion and −0.515 V ver-
us Ag/AgCl, KClsat for G/SWCNT-Fe(III)P-Nafion. However,n both cases the E◦′ value was more negative than that observedor hemin immobilized on MWCNT deposited on a glassy car-on electrode covered with Ta (E◦′ = −0.34 V versus Ag/Ag,M KCl) [35]. This difference probably reflects the specific
nfluence of the immobilization matrix on the redox behavior ofemin.
Over a relatively wide range of scan potential (5–2000 mV−1) the anodic and cathodic peak currents increase linearly withhe potential scan rate (v), indicating a surface confined redoxouple [44]. This conclusion is also supported by the slope ofhe log I versus log v dependence, which was found to be verylose to 1, for both electrodes investigated (results not shown).he Fe(IIIP/Fe(II)P redox couple bound in the porphyrinic ringxhibits a quasi-reversible behavior as shown by a Ipa/Ipc ratioery close to 1 [46] and by the slight increase of �Ep withncrease in the potential scan rate.
The decrease of the peak intensities observed when heminas immobilized on SWCNT-OH, in comparison with those
ecorded for hemin immobilized on SWCNT (see Figs. 1 and 2),ould be the result of a decrease of the hemin redox acti-ity induced by the increase of the matrix tightness due to theresence of –OH groups in the local microenvironment. This isonsistent with the deviation from the linear correlation, whichn the case of the SWCNT-OH based matrix, appears at higherotential scan rate (Fig. 2). This behavior indicates the lesserontribution of diffusion to the global process of charge transfern the more compact matrix (SWCNT-OH) in comparison with
oose matrix (SWCNT) [47].In order to perform a quantitative evaluation of the matrixermeability variation as a function of SWCNT functionaliza-ion, cyclic voltammograms were recorded for different potential
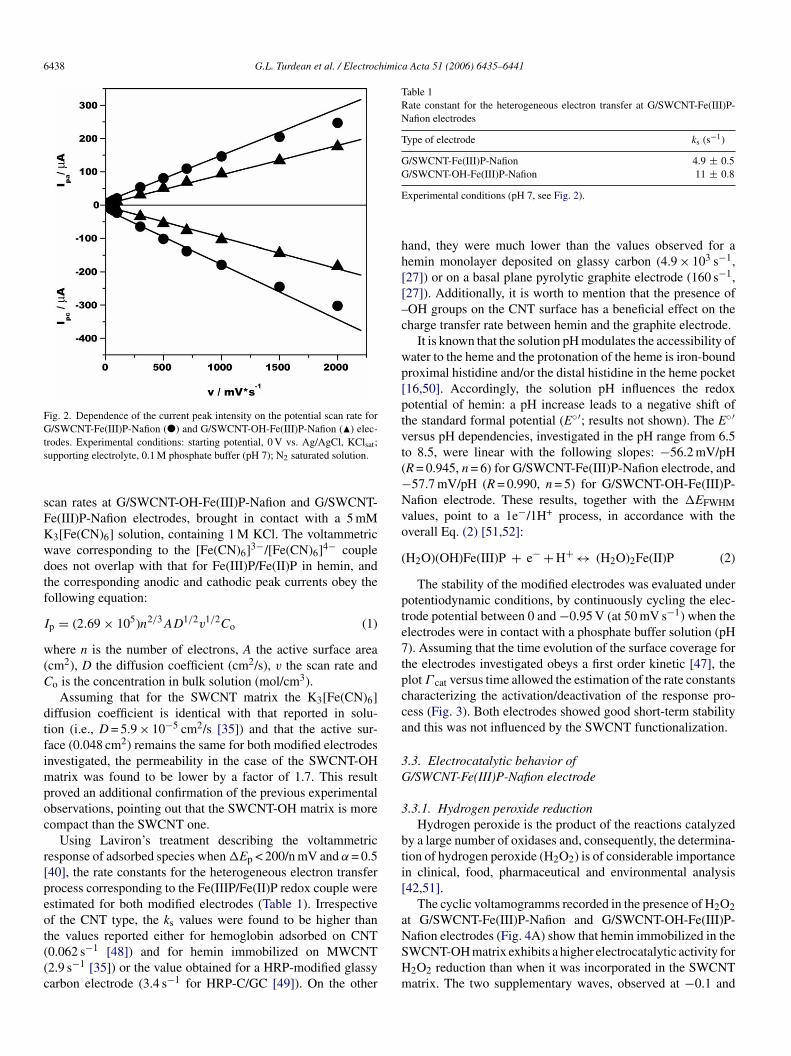
6438 G.L. Turdean et al. / Electrochimica Acta 51 (2006) 6435–6441
Fig. 2. Dependence of the current peak intensity on the potential scan rate forG/SWCNT-Fe(III)P-Nafion (�) and G/SWCNT-OH-Fe(III)P-Nafion (�) elec-ts
sFKwdtf
I
w(C
dtfimpoc
r[peot((c
Table 1Rate constant for the heterogeneous electron transfer at G/SWCNT-Fe(III)P-Nafion electrodes
Type of electrode ks (s−1)
G/SWCNT-Fe(III)P-Nafion 4.9 ± 0.5G/SWCNT-OH-Fe(III)P-Nafion 11 ± 0.8
E
hh[[–c
wp[ptvt(−Nvo
(
pte7tpcca
3G
3
bti[
a
rodes. Experimental conditions: starting potential, 0 V vs. Ag/AgCl, KClsat;upporting electrolyte, 0.1 M phosphate buffer (pH 7); N2 saturated solution.
can rates at G/SWCNT-OH-Fe(III)P-Nafion and G/SWCNT-e(III)P-Nafion electrodes, brought in contact with a 5 mM3[Fe(CN)6] solution, containing 1 M KCl. The voltammetricave corresponding to the [Fe(CN)6]3−/[Fe(CN)6]4− coupleoes not overlap with that for Fe(III)P/Fe(II)P in hemin, andhe corresponding anodic and cathodic peak currents obey theollowing equation:
p = (2.69 × 105)n2/3AD1/2v1/2Co (1)
here n is the number of electrons, A the active surface areacm2), D the diffusion coefficient (cm2/s), v the scan rate ando is the concentration in bulk solution (mol/cm3).
Assuming that for the SWCNT matrix the K3[Fe(CN)6]iffusion coefficient is identical with that reported in solu-ion (i.e., D = 5.9 × 10−5 cm2/s [35]) and that the active sur-ace (0.048 cm2) remains the same for both modified electrodesnvestigated, the permeability in the case of the SWCNT-OH
atrix was found to be lower by a factor of 1.7. This resultroved an additional confirmation of the previous experimentalbservations, pointing out that the SWCNT-OH matrix is moreompact than the SWCNT one.
Using Laviron’s treatment describing the voltammetricesponse of adsorbed species when �Ep < 200/n mV and α = 0.540], the rate constants for the heterogeneous electron transferrocess corresponding to the Fe(IIIP/Fe(II)P redox couple werestimated for both modified electrodes (Table 1). Irrespectivef the CNT type, the ks values were found to be higher than
he values reported either for hemoglobin adsorbed on CNT0.062 s−1 [48]) and for hemin immobilized on MWCNT2.9 s−1 [35]) or the value obtained for a HRP-modified glassyarbon electrode (3.4 s−1 for HRP-C/GC [49]). On the otherNSHm
xperimental conditions (pH 7, see Fig. 2).
and, they were much lower than the values observed for aemin monolayer deposited on glassy carbon (4.9 × 103 s−1,27]) or on a basal plane pyrolytic graphite electrode (160 s−1,27]). Additionally, it is worth to mention that the presence ofOH groups on the CNT surface has a beneficial effect on theharge transfer rate between hemin and the graphite electrode.
It is known that the solution pH modulates the accessibility ofater to the heme and the protonation of the heme is iron-boundroximal histidine and/or the distal histidine in the heme pocket16,50]. Accordingly, the solution pH influences the redoxotential of hemin: a pH increase leads to a negative shift ofhe standard formal potential (E◦′; results not shown). The E◦′ersus pH dependencies, investigated in the pH range from 6.5o 8.5, were linear with the following slopes: −56.2 mV/pHR = 0.945, n = 6) for G/SWCNT-Fe(III)P-Nafion electrode, and57.7 mV/pH (R = 0.990, n = 5) for G/SWCNT-OH-Fe(III)P-afion electrode. These results, together with the �EFWHMalues, point to a 1e−/1H+ process, in accordance with theverall Eq. (2) [51,52]:
H2O)(OH)Fe(III)P + e− + H+ ↔ (H2O)2Fe(II)P (2)
The stability of the modified electrodes was evaluated underotentiodynamic conditions, by continuously cycling the elec-rode potential between 0 and −0.95 V (at 50 mV s−1) when thelectrodes were in contact with a phosphate buffer solution (pH). Assuming that the time evolution of the surface coverage forhe electrodes investigated obeys a first order kinetic [47], thelot Γ cat versus time allowed the estimation of the rate constantsharacterizing the activation/deactivation of the response pro-ess (Fig. 3). Both electrodes showed good short-term stabilitynd this was not influenced by the SWCNT functionalization.
.3. Electrocatalytic behavior of/SWCNT-Fe(III)P-Nafion electrode
.3.1. Hydrogen peroxide reductionHydrogen peroxide is the product of the reactions catalyzed
y a large number of oxidases and, consequently, the determina-ion of hydrogen peroxide (H2O2) is of considerable importancen clinical, food, pharmaceutical and environmental analysis42,51].
The cyclic voltamogramms recorded in the presence of H2O2t G/SWCNT-Fe(III)P-Nafion and G/SWCNT-OH-Fe(III)P-
afion electrodes (Fig. 4A) show that hemin immobilized in theWCNT-OH matrix exhibits a higher electrocatalytic activity for2O2 reduction than when it was incorporated in the SWCNTatrix. The two supplementary waves, observed at −0.1 and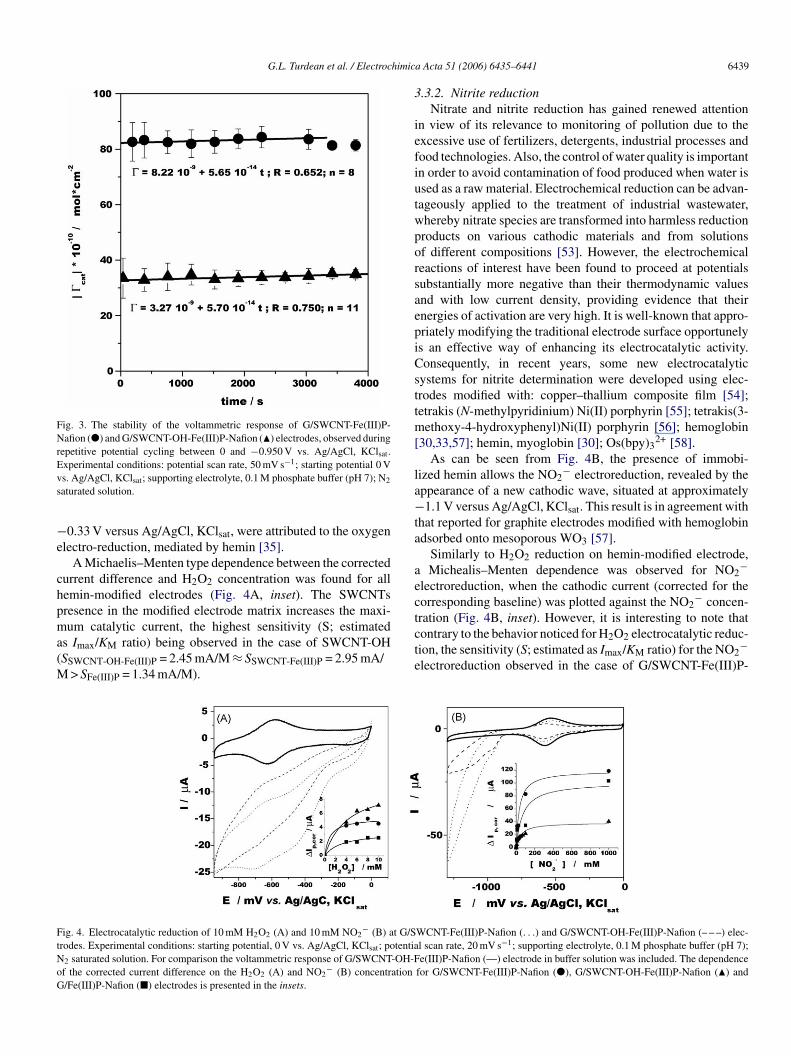
G.L. Turdean et al. / Electrochimic
Fig. 3. The stability of the voltammetric response of G/SWCNT-Fe(III)P-Nafion (�) and G/SWCNT-OH-Fe(III)P-Nafion (�) electrodes, observed duringrepetitive potential cycling between 0 and −0.950 V vs. Ag/AgCl, KClsat.E −1
vs
−e
chpma(M
3
iefiutwporsaepiCsttm[
la−ta
aect
FtNoG
xperimental conditions: potential scan rate, 50 mV s ; starting potential 0 Vs. Ag/AgCl, KClsat; supporting electrolyte, 0.1 M phosphate buffer (pH 7); N2
aturated solution.
0.33 V versus Ag/AgCl, KClsat, were attributed to the oxygenlectro-reduction, mediated by hemin [35].
A Michaelis–Menten type dependence between the correctedurrent difference and H2O2 concentration was found for allemin-modified electrodes (Fig. 4A, inset). The SWCNTsresence in the modified electrode matrix increases the maxi-um catalytic current, the highest sensitivity (S; estimated
s Imax/KM ratio) being observed in the case of SWCNT-OHSSWCNT-OH-Fe(III)P = 2.45 mA/M ≈ SSWCNT-Fe(III)P = 2.95 mA/
> SFe(III)P = 1.34 mA/M).
cte
ig. 4. Electrocatalytic reduction of 10 mM H2O2 (A) and 10 mM NO2− (B) at G/S
rodes. Experimental conditions: starting potential, 0 V vs. Ag/AgCl, KClsat; potentia
2 saturated solution. For comparison the voltammetric response of G/SWCNT-OH-Ff the corrected current difference on the H2O2 (A) and NO2
− (B) concentration/Fe(III)P-Nafion (�) electrodes is presented in the insets.
a Acta 51 (2006) 6435–6441 6439
.3.2. Nitrite reductionNitrate and nitrite reduction has gained renewed attention
n view of its relevance to monitoring of pollution due to thexcessive use of fertilizers, detergents, industrial processes andood technologies. Also, the control of water quality is importantn order to avoid contamination of food produced when water issed as a raw material. Electrochemical reduction can be advan-ageously applied to the treatment of industrial wastewater,hereby nitrate species are transformed into harmless reductionroducts on various cathodic materials and from solutionsf different compositions [53]. However, the electrochemicaleactions of interest have been found to proceed at potentialsubstantially more negative than their thermodynamic valuesnd with low current density, providing evidence that theirnergies of activation are very high. It is well-known that appro-riately modifying the traditional electrode surface opportunelys an effective way of enhancing its electrocatalytic activity.onsequently, in recent years, some new electrocatalytic
ystems for nitrite determination were developed using elec-rodes modified with: copper–thallium composite film [54];etrakis (N-methylpyridinium) Ni(II) porphyrin [55]; tetrakis(3-ethoxy-4-hydroxyphenyl)Ni(II) porphyrin [56]; hemoglobin
30,33,57]; hemin, myoglobin [30]; Os(bpy)32+ [58].
As can be seen from Fig. 4B, the presence of immobi-ized hemin allows the NO2
− electroreduction, revealed by theppearance of a new cathodic wave, situated at approximately1.1 V versus Ag/AgCl, KClsat. This result is in agreement with
hat reported for graphite electrodes modified with hemoglobindsorbed onto mesoporous WO3 [57].
Similarly to H2O2 reduction on hemin-modified electrode,Michealis–Menten dependence was observed for NO2
−lectroreduction, when the cathodic current (corrected for theorresponding baseline) was plotted against the NO2
− concen-ration (Fig. 4B, inset). However, it is interesting to note that
ontrary to the behavior noticed for H2O2 electrocatalytic reduc-ion, the sensitivity (S; estimated as Imax/KM ratio) for the NO2−lectroreduction observed in the case of G/SWCNT-Fe(III)P-
WCNT-Fe(III)P-Nafion (. . .) and G/SWCNT-OH-Fe(III)P-Nafion (– – –) elec-l scan rate, 20 mV s−1; supporting electrolyte, 0.1 M phosphate buffer (pH 7);e(III)P-Nafion (—) electrode in buffer solution was included. The dependence
for G/SWCNT-Fe(III)P-Nafion (�), G/SWCNT-OH-Fe(III)P-Nafion (�) and

6 himic
No[tS
4
twtsbst–ic
vgwHO
mratb
A
dpCtptPF
R
[[[[
[[
[[
[
[[[
[
[[
[
[
[
[
[
[
[
[
[
[
[
[
[[[
[[
[
[
[
[[
440 G.L. Turdean et al. / Electroc
afion electrodes [SSWCNT-Fe(III)P] was higher than thatbserved for G/SWCNT-OH-Fe(III)P-Nafion electrodesSSWCNT-OH-Fe(III)P]. Thus, the following sequence of sensi-ivity decreasing was obtained: SSWCNT-Fe(III)P = 3.54 mA/M >Fe(III)P = 1.44 mA/M > SSWCNT-OH-Fe(III)P = 0.81 mA/M.
. Conclusions
This study provides a new example of hemin immobiliza-ion on simple and –OH functionalized SWCNTs incorporatedithin a Nafion matrix, which was deposited on a graphite elec-
rode by a simple and reproducible drop-coating method. Ashown by potential scan rate and pH experiments, the immo-ilized hemin retain its normal voltammetric response, corre-ponding to a quasi-reversible, 1e−/1H+ process in both inves-igated matrices. Greater reversibility was noticed in the case ofOH functionalized SWCNT, expressed as a significant increasen the rate constant for the heterogeneous electron transfer pro-ess and by a slight decrease in the peak potential split.
For the first time, to our knowledge, the electrocatalytic acti-ities of the hemin-SWCNT- and hemin-SWCNT-OH-modifiedraphite electrodes towards H2O2 and nitrite electroreduction,ere quantitatively evaluated. It was found that only for the2O2 electrocatalytic reduction was the presence of SWCNT-H beneficial for the hemin catalytic activity.Thus, the use of –OH functionalized SWCNTs as electrode
aterial opens a simple and elegant way to construct new ampe-ometric biosensors for detection the small signal of moleculesnd to investigate the direct electron transfer for redox pro-eins incorporating a hemin-like active center (as for example,-cytochromes, peroxidases, myoglobin and hemoglobin).
cknowledgements
G.L.T. acknowledges the financial support for the post-octoral fellowship from NATO-CNR Italy. The work was sup-orted by Grants Program A-55/375/2004, A-85/375/2005 fromNCSIS—Romanian Ministry of National Education. A special
hanks to Adriana Coppe (University “La Sapienza”) for thereparation of the –OH functionalised SWCNT. A.C. and G.P.hank the Consiglio Nazionale delle Ricerche (C.N.R.) Targetroject MADESS II subproject: sensors and the MIUR projectIRB 2001 for financial support.
eferences
[1] M. Inagaki, K. Kaneko, T. Nishizawa, Carbon 42 (2004) 1401.[2] Z. He, J. Chen, D. Liu, H. Tang, W. Deng, Y. Kuang, Mater. Chem. Phys.
85 (2004) 396.[3] W. Liang, Y. Zhuobin, Sensors 3 (2003) 544.[4] W. Huang, W. Hu, J. Song, Talanta 61 (2003) 411.[5] H. Luo, Z. Shi, N. Li, Z. Gu, Q. Zhuang, Anal. Chem. 73 (2001) 915.[6] P.P. Prosini, A. Pozio, S. Botti, R. Ciardi, J. Power Sources 118 (2003) 265.[7] H. Tang, J. Chen, S. Yao, L. Nie, G. Deng, Y. Kuang, Anal. Biochem. 331
(2004) 89.[8] J.N. Wohlstadter, J.L. Wilbur, G.B. Sigal, H.A. Biebuyck, M.A. Billadeau,
L. Dong, A.B. Fischer, S.R. Gudibande, S.H. Jameison, J.H. Kenten, J.Leginus, J.K. Leland, R.J. Massey, S.J. Wohlstadter, Adv. Mater. 15 (2003)1184.
[[[
[
a Acta 51 (2006) 6435–6441
[9] B.S. Sherigara, W. Kutner, F. D’Souza, Electroanalysis 15 (2003) 753.10] P. Liu, J. Hu, Sens. Actuators B 84 (2002) 194.11] Z. Xu, X. Chen, X. Qu, S. Dong, Electroanalysis 16 (2004) 684.12] J. Wang, M. Musameh, Y. Lin, J. Am. Chem. Soc. 125 (2003) 2408.13] R. Antiochia, I. Lavagnini, P. Pastore, F. Magno, Bioelectrochemistry 64
(2004) 157.14] J. Wang, M. Li, Z. Shi, N. Li, Z. Gu, Electroanalysis 14 (2002) 225.15] X.-X. Yan, D.-W. Pang, Z.-X. Lu, J.-Q. Lu, H. Tong, J. Electroanal. Chem.
569 (2004) 47.16] C. Cai, J. Chen, Anal. Biochem. 325 (2004) 285.17] Y.-D. Zhao, Y.-H. Bi, W.-D. Zhang, Q.-M. Luo, Talanta 65 (2005)
489.18] L. Zhang, G.-C. Zhao, X.-W. Wei, Z.-S. Yang, Chem. Letts. 33 (2004)
86.19] J. Wang, M. Li, Z. Shi, N. Li, Z. Gu, Anal. Chem. 74 (2002) 1993.20] Z.-Z. Yin, G.-C. Zhao, X.-W. Wei, Chem. Letts. 34 (2005) 992.21] S. Sotiropoulou, V. Gavalas, V. Vamvakaki, N.A. Chaniotakis, Biosens.
Bioelectron. 18 (2003) 211.22] A. Guiseppi-Elie, C. Lei, R.H. Baughman, Nanotechnology 13 (2002)
559.23] J. Wang, Electroanalysis 17 (2005) 7.24] X. Yu, D. Chattopadhyay, I. Galeska, F. Papadimitrakopoulos, J.F. Rusling,
Electrochem. Commun. 5 (2003) 408.25] J.-Z. Xu, J.-J. Zhu, Q. Wu, Z. Hu, H.-Y. Chen, Electroanalysis 15 (2003)
219.26] Y.-L. Zhang, C.-X. Zhang, H.-X. Shen, Electroanalysis 13 (2001)
1431.27] J. Chen, U. Wollenberger, F. Lisdat, B. Ge, F.W. Scheller, Sens. Actuators
B 70 (2000) 115.28] T. Lotzbeyer, W. Schuhmann, H.-L. Schmidt, J. Electroanal. Chem. 395
(1995) 341.29] T. Sagara, S. Takeuchi, K.-i. Kumazaki, N. Nakashima, J. Electroanal.
Chem. 396 (1995) 525.30] D. Mimica, J.H. Zagal, F. Bedioui, J. Electroanal. Chem. 497 (2001)
106.31] N. Zheng, Y. Zeng, P.G. Osborne, Y. Li, W. Chang, Z. Wang, J. Appl.
Electrochem. 32 (2002) 129.32] S.L.P. Dias, Y. Gushikem, E.S. Ribeiro, E.V. Benvenutti, J. Electroanal.
Chem. 523 (2002) 64.33] D. Mimica, J.H. Zagal, F. Bedioui, Electrochem. Commun. 3 (2001)
435.34] C.G. Nan, Z.Z. Fena, W.X. Li, D.J. Ping, C.H. Qin, Anal. Chim. Acta 452
(2002) 245.35] J.-S. Ye, Y. Wen, W.D. Zhang, H.-F. Cui, L.M. Gan, G.Q. Xu, F.-S. Sheu,
J. Electroanal. Chem. 562 (2004) 241.36] M.F. Islam, E. Rojas, D.M. Bergey, A.T. Johnson, A.G. Yodh, Nano Lett.
3 (2003) 269.37] C.A. Dyke, J.M. Tour, Chem. Eur. J. 10 (2004) 812.38] K. Chattopadhyay, S. Mazumdar, Bioelectrochemistry 53 (2000) 17.39] H. Pan, L. Liu, Z.-X. Guo, L. Dai, F. Hang, D. Zhu, R. Czerw, D.L. Caroll,
Nano Lett. 3 (2003) 29.40] E. Laviron, J. Electroanal. Chem. 101 (1979) 19.41] V. Georgakilas, K. Kordatos, M. Prato, D.M. Guldi, M. Holzinger, A.
Hirsch, J. Am. Chem. Soc. 124 (2002) 760.42] B. Wang, J. Zhang, G. Cheng, S. Dong, Anal. Chim. Acta 407 (2000)
111.43] A. Curulli, S. Nunziante Cesaro, A. Coppe, C. Silvestri, G. Palleschi,
Microchim. Acta 152 (2006) 225.44] R.W. Murray, in: A.J. Bard (Ed.), Electroanalytical Chemistry, vol. 13,
Marcel Dekker, New York, 1983, p. 191.45] M.J. Honeychourch, G.A. Rechnitz, Electroanalysis 5 (1998) 285.46] A.J. Bard, L.R. Faulkner, Electrochemical Methods, Wiley-VCH, New
York, 1980, p. 522.
47] A. Ciszewski, G. Milczarek, Anal. Chem. 72 (2000) 3203.48] Y.-D. Zhao, Y.-H. Bi, W.-D. Zhang, Q.-M. Luo, Talanta 65 (2005) 489.49] D. Sun, C. Cai, X. Li, W. Xing, T. Lu, J. Electroanal. Chem. 566 (2004)415.50] T.G. Spiro, A.A. Jarzecki, Curr. Opin. Chem. Biol. 5 (2001) 715.

himic
[[
[[
G.L. Turdean et al. / Electroc
51] Q. Wang, G. Lu, B. Yang, Sens. Actuators B 99 (2004) 50.52] D.L. Pilloud, X. Chen, P.L. Dutton, C.C. Moser, J. Phys. Chem. B 104
(2000) 2868.53] M.J. Moorcroft, J. Davis, R.G. Compton, Talanta 54 (2001) 785.54] I.G. Casella, M. Gatta, J. Electroanal. Chem. 568 (2004) 183.
[
[[[
a Acta 51 (2006) 6435–6441 6441
55] S. Trevin, F. Bedioui, J. Devynck, J. Electroanal. Chem. 408 (1996)261.
56] S. Trevin, F. Bedioui, J. Devynck, Talanta 43 (1996) 303.57] J.-J. Feng, J.-J. Xu, H.Y. Chen, Electrochem. Commun. 8 (2006) 77.58] P. Liu, J. Hu, Sens. Actuators B 84 (2002) 194.
Copyright © 2022 FDOKUMEN




















