
Intracellular parasitism with Toxoplasma gondii stimulates mammalian-target-of-rapamycin-dependent...
18
Intracellular parasitism with Toxoplasma gondii stimulates mammalian-target-of-rapamycin-dependent host cell growth despite impaired signalling to S6K1 and 4E-BP1cmi_1305 983..1000 Yubao Wang, 1 Louis M. Weiss, 1,2 and Amos Orlofsky 1 * Departments of 1 Pathology and 2 Medicine, Albert Einstein College of Medicine, Bronx, New York 10461, USA. Summary The Ser/Thr kinase mammalian-target-of- rapamycin (mTOR) is a central regulator of anabo- lism, growth and proliferation. We investigated the effects of Toxoplasma gondii on host mTOR signal- ling. Toxoplasma invasion of multiple cell types rapidly induced sustained mTOR activation that was restricted to infected cells, as determined by rapamycin-sensitive phosphorylation of ribo- somal protein S6; however, phosphorylation of the growth-associated mTOR substrates 4E-BP1 and S6K1 was not detected. Infected cells still phos- phorylated S6K1 and 4E-BP1 in response to insulin, although the S6K1 response was blunted. Parasite- induced S6 phosphorylation was independent of S6K1 and did not require activation of canonical mTOR-inducing pathways mediated by phospha- tidylinositol 3-kinase–Akt and ERK. Host mTOR was localized in a vesicular pattern surrounding the parasitophorous vacuole, suggesting potential activation by phosphatidic acid in the vacuolar membrane. In spite of a failure to phosphorylate 4E-BP1 and S6K1, intracellular T. gondii triggered host cell cycle progression in an mTOR-dependent manner and progression of infected cells displayed increased sensitivity to rapamycin. Moreover, normal cell growth was maintained during parasite- induced cell cycle progression, as indicated by total cellular S6 levels. The Toxoplasma-infected cell provides a unique example of non-canonical mTOR activation supporting growth that is independent of signalling through either S6K1 or 4E-BP1. Introduction The Ser/Thr kinase mammalian-target-of-rapamycin (mTOR) plays a central role in co-ordinating global cellular metabolic and growth responses to environmental condi- tions with respect to nutritional status, growth factor sig- nalling and stress (Fingar and Blenis, 2004; Hay and Sonenberg, 2004; Wullschleger et al., 2006). A key func- tion of mTOR is the promotion of cellular growth and proliferation. Dysregulation of mTOR is a frequent occur- rence in neoplasms, and excessive stimulation of this pathway leads to defective control of cell survival and proliferation (Petroulakis et al., 2006). Conversely, rapa- mycin, a specific inhibitor of mTOR, prevents or delays mitogenic responses in certain cell types (Jayaraman and Marks, 1993; Slavik et al., 2001; Mourani et al., 2004; Yeager et al., 2008), and is an effective anti-tumour agent in certain settings (Carraway and Hidalgo, 2004; Kurma- sheva et al., 2006; Thomas et al., 2006; LoPiccolo et al., 2008). An even broader role for mTOR as a critical control point for cellular growth is indicated by studies of mTOR- deficient cells that have revealed rapamycin-insensitive growth-promoting functions for this protein (Murakami et al., 2004; Wang et al., 2005). A number of potential downstream effectors of mTOR have been identified, including the S6 kinases (S6K1 and S6K2) as well as several proteins associated with the translational initiation complex (Tee and Blenis, 2005). The latter include the mTOR substrate 4E-BP1, an inhibi- tory factor whose phosphorylation leads to its dissociation from eIF4E and the subsequent recruitment to the complex of eIF4G, which competes with 4E-BP1 for binding to a shared site on eIF4E. The binding of eIF4G, together with its associated RNA helicase, eIF4A, results in the formation of the eIF4F complex, which serves to promote global translation and also modulates the trans- lation of specific mRNAs in a manner that favours anabo- lism, cell growth and proliferation (Dmitriev et al., 2003; Bilanges et al., 2007; Yoon et al., 2007; Ramirez-Valle et al., 2008). An additional potential effector is the riboso- mal protein S6, which is a substrate for S6K1 and S6K2, although its role in translation and cell growth is not well understood (Ruvinsky and Meyuhas, 2006). In addition to these growth-promoting effects on the translational Received 3 November, 2008; revised 19 February, 2009; accepted 22 February, 2009. *For correspondence. E-mail orlofsky@ aecom.yu.edu; Tel. (+1) 718 430 2674; Fax (+1) 718 430 8541. Cellular Microbiology (2009) 11(6), 983–1000 doi:10.1111/j.1462-5822.2009.01305.x First published online 19 March 2009 © 2009 Blackwell Publishing Ltd cellular microbiology
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Intracellular parasitism with Toxoplasma gondii stimulates mammalian-target-of-rapamycin-dependent...

Intracellular parasitism with Toxoplasma gondiistimulates mammalian-target-of-rapamycin-dependenthost cell growth despite impaired signalling to S6K1and 4E-BP1cmi_1305 983..1000
Yubao Wang,1 Louis M. Weiss,1,2 andAmos Orlofsky1*Departments of 1Pathology and 2Medicine, AlbertEinstein College of Medicine, Bronx, New York 10461,USA.
Summary
The Ser/Thr kinase mammalian-target-of-rapamycin (mTOR) is a central regulator of anabo-lism, growth and proliferation. We investigated theeffects of Toxoplasma gondii on host mTOR signal-ling. Toxoplasma invasion of multiple cell typesrapidly induced sustained mTOR activation thatwas restricted to infected cells, as determinedby rapamycin-sensitive phosphorylation of ribo-somal protein S6; however, phosphorylation of thegrowth-associated mTOR substrates 4E-BP1 andS6K1 was not detected. Infected cells still phos-phorylated S6K1 and 4E-BP1 in response to insulin,although the S6K1 response was blunted. Parasite-induced S6 phosphorylation was independent ofS6K1 and did not require activation of canonicalmTOR-inducing pathways mediated by phospha-tidylinositol 3-kinase–Akt and ERK. Host mTORwas localized in a vesicular pattern surrounding theparasitophorous vacuole, suggesting potentialactivation by phosphatidic acid in the vacuolarmembrane. In spite of a failure to phosphorylate4E-BP1 and S6K1, intracellular T. gondii triggeredhost cell cycle progression in an mTOR-dependentmanner and progression of infected cells displayedincreased sensitivity to rapamycin. Moreover,normal cell growth was maintained during parasite-induced cell cycle progression, as indicated by totalcellular S6 levels. The Toxoplasma-infected cellprovides a unique example of non-canonical mTORactivation supporting growth that is independent ofsignalling through either S6K1 or 4E-BP1.
Introduction
The Ser/Thr kinase mammalian-target-of-rapamycin(mTOR) plays a central role in co-ordinating global cellularmetabolic and growth responses to environmental condi-tions with respect to nutritional status, growth factor sig-nalling and stress (Fingar and Blenis, 2004; Hay andSonenberg, 2004; Wullschleger et al., 2006). A key func-tion of mTOR is the promotion of cellular growth andproliferation. Dysregulation of mTOR is a frequent occur-rence in neoplasms, and excessive stimulation of thispathway leads to defective control of cell survival andproliferation (Petroulakis et al., 2006). Conversely, rapa-mycin, a specific inhibitor of mTOR, prevents or delaysmitogenic responses in certain cell types (Jayaraman andMarks, 1993; Slavik et al., 2001; Mourani et al., 2004;Yeager et al., 2008), and is an effective anti-tumour agentin certain settings (Carraway and Hidalgo, 2004; Kurma-sheva et al., 2006; Thomas et al., 2006; LoPiccolo et al.,2008). An even broader role for mTOR as a critical controlpoint for cellular growth is indicated by studies of mTOR-deficient cells that have revealed rapamycin-insensitivegrowth-promoting functions for this protein (Murakamiet al., 2004; Wang et al., 2005).
A number of potential downstream effectors of mTORhave been identified, including the S6 kinases (S6K1 andS6K2) as well as several proteins associated with thetranslational initiation complex (Tee and Blenis, 2005).The latter include the mTOR substrate 4E-BP1, an inhibi-tory factor whose phosphorylation leads to its dissociationfrom eIF4E and the subsequent recruitment to thecomplex of eIF4G, which competes with 4E-BP1 forbinding to a shared site on eIF4E. The binding of eIF4G,together with its associated RNA helicase, eIF4A, resultsin the formation of the eIF4F complex, which serves topromote global translation and also modulates the trans-lation of specific mRNAs in a manner that favours anabo-lism, cell growth and proliferation (Dmitriev et al., 2003;Bilanges et al., 2007; Yoon et al., 2007; Ramirez-Valleet al., 2008). An additional potential effector is the riboso-mal protein S6, which is a substrate for S6K1 and S6K2,although its role in translation and cell growth is not wellunderstood (Ruvinsky and Meyuhas, 2006). In additionto these growth-promoting effects on the translational
Received 3 November, 2008; revised 19 February, 2009; accepted22 February, 2009. *For correspondence. E-mail [email protected]; Tel. (+1) 718 430 2674; Fax (+1) 718 430 8541.
Cellular Microbiology (2009) 11(6), 983–1000 doi:10.1111/j.1462-5822.2009.01305.xFirst published online 19 March 2009
© 2009 Blackwell Publishing Ltd
cellular microbiology

apparatus, mTOR activation also enhances transcriptionof rRNA, tRNA and ribosomal protein genes (Wullschlegeret al., 2006).
These various effects of mTOR might be envisaged asacting in concert to promote a unified programme of growthand cell cycle progression. Alternatively, distinct sets ofmTOR effectors may act independently to govern distinctfunctions. The latter view is supported by recent studies ofS6K-deficient mice. It was observed that S6K1-deficientmyoblasts are defective with respect to proliferation-associated cell growth; however, this kinase is not requiredfor mTOR-dependent control of either cell proliferation(Ohanna et al., 2005), global protein translation or proteinturnover in these cells (Mieulet et al., 2007). Furthersupport for a specific linkage between S6K1 and cellgrowth comes from the observation that pancreatic b-cellshave a reduced size in S6K1-deficient mice (Pende et al.,2000). Other cell types were unaffected, however, implyingthe likely existence of additional pathways to maintain cellgrowth. The nature of such pathways and their connectionto mTOR are unresolved questions.
The notion that cell growth and cell cycle progressionmay each be governed by distinct mTOR effectors sug-gests that subsets of effectors might be regulated inde-pendently downstream of mTOR. However, there is as yetlittle evidence for such selective regulation of downstreamelements of the pathway.
Intracellular parasites represent a relatively untappedresource for the investigation of regulatory mechanisms inmammalian cells. While cellular responses to parasitismmay involve host defense-related functions, such as TLRsignalling and stress responses, it is becoming increas-ingly clear that they also reflect sophisticated parasiteadaptations that target and modify various aspects of hostcell signalling. In particular, the apicomplexan parasiteToxoplasma gondii, which invades most mammalian cells,has been shown to generate novel modifications ofseveral host cell signalling pathways. For example, infec-tion of fibroblasts by T. gondii leads to initiation of theNF-kB signalling pathway (activation of IKKb and degra-dation of IkB), but the pathway is blocked due to incom-plete phosphorylation and failed nuclear translocation ofp65/RelA (Shapira et al., 2005). In addition, intracelluarToxoplasma can export a secretory kinase termed ROP16to the host cytoplasm and nucleus, resulting in activationof host Stat3, suppression of host IL-12 production andenhanced parasite virulence (Saeij et al., 2007). In addi-tion to ROP16, several other functionally uncharacterizedputative kinases and phosphatases are contained in thespecialized parasite secretory vesicles known as rhop-tries and may potentially modulate host cell signalling(Ravindran and Boothroyd, 2008).
The mTOR pathway represents a potential target formanipulation by T. gondii, as the regulation of the
anabolic/catabolic balance of the host cell may have asubstantial impact on the availability of host-derived nutri-ents to support parasite expansion. We now report thatthe mTOR pathway is functionally activated upon infectionin a manner that leads to an unusual dissociation betweenmTOR and its effectors, resulting in a physiological demon-stration of mTOR-dependent cell growth independent ofsignalling through S6K1 and 4E-BP1.
Results
Stable activation of host mTOR in Toxoplasma-infectedcells
To assess activation of host cell mTOR after infection byT. gondii, we examined the phosphorylation of ribosomalprotein S6 by immunoblot analysis. Initial studies wereconducted in fibroblasts deprived of serum in order tominimize basal mTOR activity. As demonstrated in Fig. 1,an increase in phosphorylated S6 (pS6) was observedwithin 2 h of infection in either BALB/c 3T3 murine fibro-blasts (3T3) (Fig. 1A) or human foreskin fibroblasts (HFF)(Fig. 1B). The induced level of pS6 was maintained for atleast 24 h and was not related to the number of parasitesin the cell (as determined by YFP content using YFP-expressing T. gondii). A similar elevation of pS6 wasobserved in other cultured cells, including HeLa cells andmurine peritoneal exudate macrophages (Fig. 1C). Para-site induction of host pS6 was comparable in intensity tothe response to insulin. Densitometric analysis of bandintensity indicated that the fold increase in pS6 uponinfection was 7.1 � 2.7 for 3T3 (n = 9 independent experi-ments), 3.3 � 0.6 for HeLa (n = 6) and 5.5 � 1.3 for HFFs(n = 4). The increase in pS6 was abolished by treatmentwith 200 ng ml-1 rapamycin (Fig. 1D), confirming that pS6generation reflected activation of host cell mTOR. Rapa-mycin at 20 ng ml-1 was equally effective (Fig. 2D). Theeffect of rapamycin was not due to an inhibition of parasitesurvival or growth (Figs 1D and 7D). No reactivity wasdetected in lysates of extracellular parasites (Fig. 1A).Furthermore, immunostaining of pS6 revealed a general-ized staining of host cytoplasm specific to infected cells,confirming that a host cell pathway had been activated(Fig. 1E). The fact that immunostaining is confined toinfected cells indicates that T. gondii-mediated activationof host cell mTOR is a direct consequence of parasiteinvasion, rather than an indirect result of the secretion ofdiffusible mediators within the culture, or of incidentalcontact between parasites and fibroblasts.
Toxoplasma-induced mTOR is independent of canonicalmTOR-regulatory signals
At least three pathways leading to mTOR activation havebeen described, mediated by either Akt, Erk or amino
984 Y. Wang, L. M. Weiss, and A. Orlofsky
© 2009 Blackwell Publishing Ltd, Cellular Microbiology, 11, 983–1000

acids (principally leucine). Akt activation by class I phos-phatidylinositol 3-kinase (PI3K) results in an inactivatingphosphorylation of tuberin (TSC2), which functions as aGTPase-activating protein suppressing Rheb, a memberof the Ras GTPase superfamily that associates withmTOR and is required for its activation (Li et al., 2004; Baiet al., 2007). In addition to its potential direct activation ofmTOR, Rheb may indirectly stimulate mTOR through itsability to bind and activate phospholipase D (Sun et al.,2008). Phospholipase D is required for mTOR activationin several settings (Foster, 2007) and acts by convertingphosphatidylcholine to phosphatidic acid, which can binddirectly to mTOR (Veverka et al., 2008). The Erk connec-tion to mTOR activation is similar to that of Akt, as itdepends on Erk2-dependent phosphorylation of TSC2(Ma et al., 2005; Rolfe et al., 2005; Arvisais et al., 2006).The amino acid-dependent pathway, less well character-ized, is also dependent on Rheb, as well as the RagGTPase, but it is independent of TSC2 and is mediated byVps34, a class III PI3K (Avruch et al., 2008; Gulati et al.,2008; Sancak et al., 2008).
To investigate these pathways as potential mediatorsof mTOR activation by Toxoplasma, we examinedinfected cells with respect to the activation of thesesignals and their contribution to mTOR activity. Akt phos-phorylation did not correlate with mTOR activation inT. gondii-infected cells. While Akt activation did occurin HFF, it was delayed relative to pS6 accumulation
(Fig. 1B). Furthermore, Akt activation was absent in both3T3 and HeLa cells, and phosphorylation of the Akt sub-strate GSK-3b was not detected (Fig. 2A). An examina-tion of shorter infection times in 3T3 cells failed to reveala transient activation of the kinase (Fig. 2B). Neverthe-less, both basal and infection-induced mTOR activitywere strongly dependent on basal levels of Akt activityin 3T3 cells (Fig. 2C). Inhibitors of PI3k, wortmanninand LY294002 were also inhibitory of both basal andinfection-induced mTOR activity in both 3T3 and HeLacells (Fig. 2C and D), although this result may be lessinformative as these compounds can also inhibitmTOR. Overall, these data indicate that while thePI3K–Akt pathway may play an important permissiverole for mTOR activation, the stimulation of mTOR activ-ity in response to infection is not mediated via thispathway.
With respect to Erk activity, a mild stimulation wasdetected in some experiments at 24 h post infection in3T3 (Fig. 2B) or HeLa cells (data not shown), as well as atransient elevation at 2 h in 3T3 (Fig. 2B). A biphasicresponse of Erk activity to T. gondii infection has beenpreviously reported in HFF (Molestina et al., 2008).However, the kinetics of this variable response did notcorrelate well with the consistent and more uniformincrease in pS6 accumulation (Fig. 2B, lower panel). Inhi-bition of Erk activity with PD98059, an inhibitor of theErk-activating kinase MKK1, resulted in a partial diminu-
Fig. 1. T. gondii induces host cell mTORactivation.A. 3T3 cells were deprived of serum for 1 dayand then infected or not with YFP-RH at anmoi of 8 for the indicated times. Lysates ofhost cells or of free T. gondii tachyzoites (T)were subjected to immunoblotting.B. HFFs were deprived of serum for 1 dayand then infected at the indicated moi for 2, 4,8 or 24 h. Protein extracts were prepared forimmunoblotting.C. Murine peritoneal exudate macrophages orHeLa cells were infected at the indicated moifor 24 h in the absence of serum. UninfectedHeLa cells were stimulated with insulin (Ins)for 30 min prior to harvest. The loadingcontrol is total Akt for macrophages and totalS6 for HeLa.D. 3T3 cells were infected overnight at anmoi. of 4 in the absence of serum. Cells weretreated as indicated with insulin for the final2 h and with rapamycin (Rap, 200 ng ml-1) forthe final 30 min of culture.E. 3T3 cells were infected with YFP-RH(green) overnight in the absence of serum,and then subjected to pS6 (S235/236)immunostaining (red). Brightfield (BF) andfluorescent images of the same field aredisplayed. The data in the figure arerepresentative of either two or threeindependent experiments.
Toxoplasma activates host cell mTOR 985
© 2009 Blackwell Publishing Ltd, Cellular Microbiology, 11, 983–1000
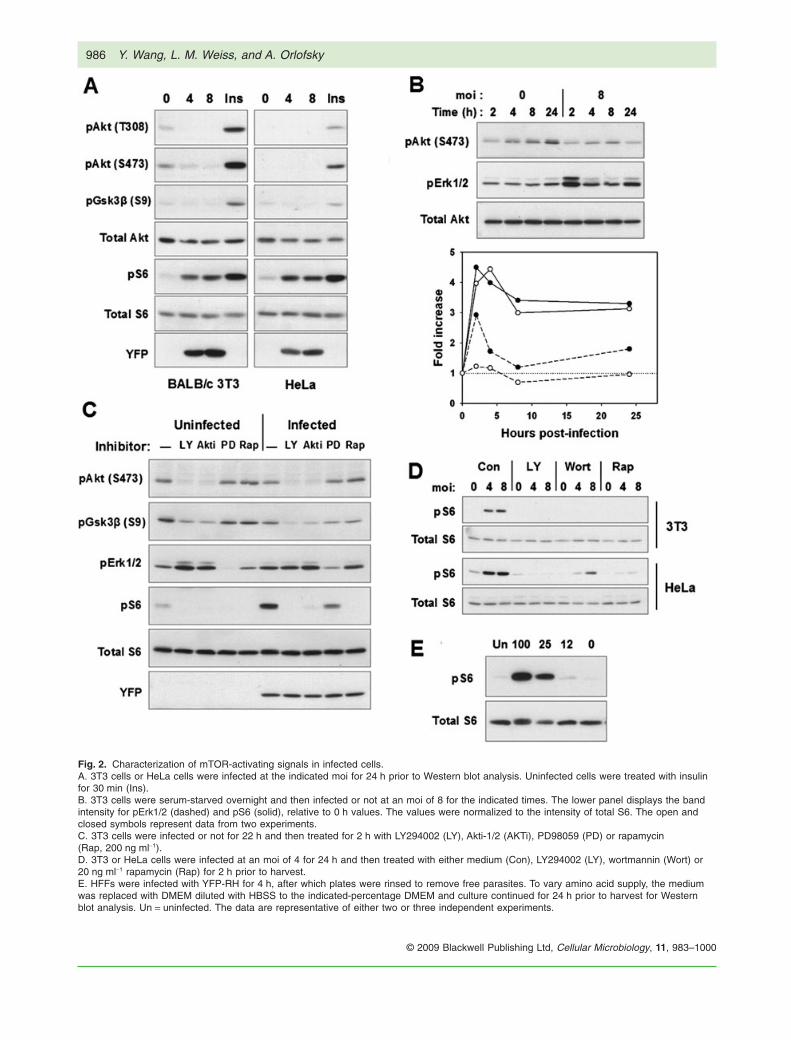
Fig. 2. Characterization of mTOR-activating signals in infected cells.A. 3T3 cells or HeLa cells were infected at the indicated moi for 24 h prior to Western blot analysis. Uninfected cells were treated with insulinfor 30 min (Ins).B. 3T3 cells were serum-starved overnight and then infected or not at an moi of 8 for the indicated times. The lower panel displays the bandintensity for pErk1/2 (dashed) and pS6 (solid), relative to 0 h values. The values were normalized to the intensity of total S6. The open andclosed symbols represent data from two experiments.C. 3T3 cells were infected or not for 22 h and then treated for 2 h with LY294002 (LY), Akti-1/2 (AKTi), PD98059 (PD) or rapamycin(Rap, 200 ng ml-1).D. 3T3 or HeLa cells were infected at an moi of 4 for 24 h and then treated with either medium (Con), LY294002 (LY), wortmannin (Wort) or20 ng ml-1 rapamycin (Rap) for 2 h prior to harvest.E. HFFs were infected with YFP-RH for 4 h, after which plates were rinsed to remove free parasites. To vary amino acid supply, the mediumwas replaced with DMEM diluted with HBSS to the indicated-percentage DMEM and culture continued for 24 h prior to harvest for Westernblot analysis. Un = uninfected. The data are representative of either two or three independent experiments.
986 Y. Wang, L. M. Weiss, and A. Orlofsky
© 2009 Blackwell Publishing Ltd, Cellular Microbiology, 11, 983–1000

tion of both basal and parasite-induced mTOR activities in3T3 cells (Fig. 2C). Therefore, while MEK/Erk signallingmay partially contribute to mTOR function in infected cells,the role of this pathway, as with PI3K–Akt, appears to bepermissive and does not provide an explanation forparasite-induced mTOR activation.
We considered the possibility that T. gondii mightamplify signalling through the pathway mediating aminoacid control of mTOR. In this case, infected cells might beexpected to show reduced sensitivity of mTOR activity toamino acid depletion. As shown in Fig. 2E, this is not thecase: parasite-induced pS6 accumulation was stronglydependent on amino acids in a dose-dependent manner.In addition, we have observed no effect on pS6 accumu-lation following siRNA-mediated knockdown of Vps34 ininfected cells (data not shown). Therefore, it is unlikelythat this pathway contributes to parasite-mediated mTORactivation.
Association of mTOR with the parasitophorous vacuole
As our results do not support a major involvement of thecanonical pathways in driving the host mTOR response toToxoplasma infection, it was necessary to search for addi-
tional clues to the mechanism of this response. A notablefeature of Toxoplasma is the ability to export parasite-encoded proteins to the surface of the parasitophorousvacuole, and there is evidence suggesting that such pro-teins can perform a signalling function (Saeij et al., 2007;Ravindran and Boothroyd, 2008). In addition, there isevidence suggesting that the vacuolar membrane isenriched in phosphatidic acid, an important mediator ofmTOR activation (Charron and Sibley, 2002). Several dif-ferent kinds of host vesicles and organelles have beenshown to accumulate in the vicinity of the vacuole (Sinaiet al., 1997; Coppens et al., 2006; Martin et al., 2007). AsmTOR activity, at least when driven by amino acid signal-ling, has recently been shown to involve mTOR associa-tion with vesicular compartments (Sancak et al., 2008),we considered the possibility that vesicular mTOR mightinteract with the vacuole as part of an activating mecha-nism. We therefore investigated the localization of mTORin infected HeLa cells. As shown in Fig. 3A–C, infectedcells uniformly displayed punctate mTOR staining thatwas preferentially localized to a region surrounding theparasitophorous vacuole. In many infected cells, it waspossible to observe a distinct ring of punctate stainapproximately coinciding with the vacuolar membrane
Fig. 3. Localization of mTOR inT. gondii-infected cells. HeLa cells wereinfected overnight with YFP-RH (green) andstained for mTOR (A–D) or pS6 (E) using aCy5-conjugated secondary antibody. (A)–(D)display the Cy5 signal in inverted grayscale.In (E), the red Cy5 signal is merged with agrayscale phase contrast image. (B)represents the boxed region in (A),rotated clockwise. Arrowheads indicatemTOR-stained puncta closely adjoined to theparasitophorous vacuole.C. Illustration of the mTOR-staining pattern ina cell containing two vacuoles. Note thatmTOR-stained puncta are associated withboth vacuoles.D. Uninfected culture.E. Arrowheads indicate infected cellsdisplaying uniform cytoplasmic pS6 stain.Scale bars: 10 mm (A, D and E); 5 mm (B);6 mm (C).
Toxoplasma activates host cell mTOR 987
© 2009 Blackwell Publishing Ltd, Cellular Microbiology, 11, 983–1000

(Fig. 3B). In comparison, uninfected cells displayed a vari-able pattern of punctate mTOR staining that was fre-quently perinuclear (Fig. 3D), as expected for mTOR inamino acid-replete cells (Sancak et al., 2008). In compari-son, pS6 was distributed diffusely through the cytoplasmof infected cells as expected (Fig. 3E). Western blotanalysis showed that infection did not alter the total levelof mTOR (data not shown). These data support the pos-sibility of a functional interaction between mTOR-bearingvesicles and the parasitophorous vacuole.
Activation of mTOR in infected cells is dissociated fromdownstream events
Activation of mTOR by mitogens and growth factors leadsto phosphorylation of S6K1 by mTOR at the hydrophobicmotif site T389 within the catalytic domain, a critical eventin S6K1 activation (Volarevic and Thomas, 2001). Surpris-ingly, infection of 3T3 cells resulted in only a transientphosphorylation of S6K1 at T389 at 2–4 h post infection,even though high levels of pS6 are maintained for24 h (Fig. 4A). Transiently induced S6K1 activity cannotaccount for the sustained elevation of pS6, as the latteris completely reversed by exposure to rapamycin for0.5 h (Fig. 1D). Therefore, the results imply that whileToxoplasma-activated mTOR may initially interact with itscanonical substrate S6K1, this linkage appears to be dis-rupted after the first several hours of infection. We there-fore considered the possibility that at these later time pointsthe infected cell might actively suppress S6K1 activity. Totest this possibility, we asked whether overnight infection of3T3 cells would affect the subsequent activation of S6K1 inresponse to treatment with insulin for 30 min. As shown inFig. 4B, activation of S6K1 was substantially reducedin the infected culture. Very similar results were obtained
with HeLa cells (Fig. 4B) and HFF (data not shown). In cellsinfected at a multiplicity of infection of 8, the insulin-inducedintensity of the pS6K1 band was reduced by 56% for 3T3cells and 51% for HeLa. The inhibition of signalling to S6K1was not the consequence of a general loss of insulinresponsiveness, as insulin-stimulated phosphorylation of4E-BP1 was unimpaired in infected cells (Fig. 6A). There-fore, Toxoplasma infection appears to generate a suppres-sive activity that interrupts signal transduction betweenmTOR and S6K1.
The dissociation of mTOR and S6K1 activation sug-gested that S6 phosphorylation in infected cells is likely tobe S6K1-independent. To test this prediction, we analysedS6 phosphorylation in 3T3 cells treated with S6K1 siRNA.As shown in Fig. 5, treatment with specific siRNA led toa reduction in both total and phosphorylated S6K1, buthad no effect on the Toxoplasma-induced level of pS6.Notably, S6K1 knockdown also had no effect on pS6elevation in response to insulin, in either infected or unin-fected cultures. Therefore, an mTOR-dependent, S6K1-independent mechanism of S6 phosphorylation is presentboth in infected and in insulin-stimulated cells.
In addition to S6K1, a second substrate of mTOR that isphosphorylated in response to growth signals is 4E-BP1.Phosphorylation of 4E-BP1 is hierarchical, with phospho-rylation at two N-terminal sites preceding phosphorylationat Ser64 and Ser69. These late phosphorylations areassociated with the conversion of the faster-migrating aand b isoforms to the slower migrating g isoform in SDS-PAGE, and also with the dissociation of 4E-BP1 fromeIF4E, for which Ser64 phosphorylation appears to beparticularly important (Mothe-Satney et al., 2000a; Wanget al., 2005). We assessed 4E-BP1 phosphorylation inT. gondii-infected 3T3 cells. As shown in Fig. 6A, Toxo-plasma infection failed to generate detectable levels of g
Fig. 4. Absence of sustained S6K1 activation in T. gondii-infected cells.A. 3T3 cells were deprived of serum for 1 day and then infected or not with YFP-RH at an moi of 8 for the indicated times prior to harvest andWestern blot analysis. The asterisk indicates a novel band of unknown identity found consistently and uniquely in infected cells.B. 3T3 or HeLa cells were infected at the indicated moi for 23.5 h followed by treatment with insulin (Ins) for 30 min. The data arerepresentative of two (A) or three (B) independent experiments.
988 Y. Wang, L. M. Weiss, and A. Orlofsky
© 2009 Blackwell Publishing Ltd, Cellular Microbiology, 11, 983–1000

isoform. In contrast to S6K1, no transient phosphorylationwas detected at early time points (data not shown). There-fore, the activation of mTOR in infected cells is disso-ciated from phosphorylation of both S6K1 and 4E-BP1.However, unlike S6K1, whose activation by insulin ispartly suppressed by infection, 4E-BP1 was efficientlyconverted to the g isoform by insulin in both infected anduninfected cells (Fig. 6A). Similar results were obtainedwith HeLa cells (data not shown). Amino acid-dependentphosphorylation of 4E-BP1, which was readily observablein serum-starved MEF cells, was similarly unaffected byToxoplasma infection (Fig. 6B). These results imply aselectivity of the suppressive function of T. gondii withinthe mTOR pathway.
Toxoplasma promotes mTOR-dependent cell cycleprogression and cell growth
The mTOR pathway functions to enhance cell growth andcell cycle progression. We therefore asked whether, inspite of the failure to phosphorylate S6K1 and 4E-BP1,activated mTOR in infected cells could still promote thesefunctions. Quiescent 3T3 cells were infected andassessed for cell cycle progression in the presence orabsence of rapamycin. In order to specifically detect cell-autonomous effects of the parasite, the YFP-transgenicstrain of T. gondii was employed, so that cells of varyinginfection level in the same culture could be compared. Theethanol fixation step required for DNA content analysislimits the sensitivity of YFP detection. By comparison ofethanol-treated samples with aliquots fixed with paraform-aldehyde (PFA) alone, in which the signal from singleparasites is readily detected, it was estimated that the
YFP-positive gate (‘highly infected cells’) in ethanol-treated samples includes cells with eight or more para-sites (Fig. 7A), and should therefore include nearly allcells that were productively infected for the entire cultureinterval. Similar results were obtained in experiments inwhich the effect of ethanol on parasite enumeration wasless pronounced (data not shown). As shown in Fig. 7Band C, infection was a highly efficient inducer of cell cycleentry, leading to the accumulation of 82% YFP-positivecells in S phase by 20 h, compared with < 10% in controluninfected cultures. The YFP-negative fraction of theinfected culture (‘minimally infected cells’, including unin-fected and lightly infected cells) showed only minor pro-gression. The finding of T. gondii-induced S-phase entryin quiescent fibroblasts is confirmatory of similar resultsrecently obtained by two other groups (Brunet et al., 2008;Molestina et al., 2008); however, the data presented heredemonstrate for the first time that this is predominantlydue to a cell-autonomous effect of the parasite on hostcell signalling.
To determine whether activated mTOR in infected cellswas responsible for parasite-induced cell cycle progres-sion, we assessed the effect of rapamycin on cell cyclestatus. As shown in Fig. 7B and C, treatment withrapamycin severely retarded T. gondii-induced cell cycleprogression. At 20 h, rapamycin restored the G1 andS-phase frequencies of the highly infected fraction nearlyto baseline levels. The effect of the drug was even morepronounced when examined with respect to median DNAcontent (Fig. 7C). At 30 h, rapamycin-treated infectedcells had begun to enter S phase, but their progress wasmarkedly inhibited, as revealed by median DNA content(Fig. 7C). The effect of rapamycin was not due to any
Fig. 5. T. gondii-induced S6 phosphorylation is independentof S6K1. 3T3 cells were transfected with the indicatedoligoribonucleotides and then infected with YFP-RH for 23.5 h,followed by stimulation with insulin for 30 min. Lysates wereprepared for Western blot analysis. The data are representative oftwo independent experiments.
Fig. 6. 4E-BP1 phosphorylation is not altered by Toxoplasmainfection.A. 3T3 cells were infected with T. gondii at the indicated moi for23.5 h followed by treatment with insulin for 30 min and Westernblot analysis. The positions of the a, b and g species, representingstates of progressively increased phosphorylation (Khaleghpouret al., 1999), are indicated.B. Mouse embryonic fibroblasts were infected with T. gondii at anmoi of 8 for 24 h. Amino acid concentration ([AA]) was controlled bydilution of DMEM to the indicated percentages in HBSS. The datain the figure are representative of two independent experiments.
Toxoplasma activates host cell mTOR 989
© 2009 Blackwell Publishing Ltd, Cellular Microbiology, 11, 983–1000

Fig. 7. Infection promotes mTOR-dependent cell cycle progression. 3T3 cells were deprived of serum beginning 1 day prior to infection withYFP-RH at an moi of 2. The cells were cultured for 20 or 30 h in the presence or absence of 200 ng ml-1 rapamycin (Rap) and then fixed with0.5% PFA for 1 h. PFA-only aliquots were reserved for parasite enumeration (A, upper panel; D) and the remainder of the samples weretreated with ethanol, stained with propidium iodide (PI) and analysed by flow cytometry.A. Estimation of parasite content in PI-stained samples. The upper panel displays a YFP histogram for ethanol-treated, PI-stained cells from aculture infected for 30 h (solid line). The shaded area represents the gate defined as YFP-positive (‘highly infected cells’, 57% of the total). Anuninfected culture is shown for comparison (dashed line). The lower panel displays a PFA-only aliquot of the same sample. The upper 57% ofthe distribution is shown to contain cells with at least eight parasites per cell. Bars indicate fractions containing uninfected cells (U), singlyinfected cells (1) and cells with 1–8 parasites (1–8).B. Histograms display DNA content for samples cultured for the indicated times. Cells were either from uninfected cultures (shaded), theYFP-negative (‘minimally infected’) fraction of infected cultures (dashed) or the YFP-positive (‘highly infected’) fraction (solid, unshaded).Rap, rapamycin-treated.C. Data from (B) was analysed with respect to cell cycle phase distribution and median DNA content (normalized to the mean PI intensity ofcells in G1, with G1 DNA content = 2n).D. The parasite content of total infected cells in PFA-only aliquots was analysed. YFP intensity was normalized to the mean intensity of freeparasites in the same sample. Data in the figure represent the mean � SEM of triplicates and are representative of two independentexperiments.
990 Y. Wang, L. M. Weiss, and A. Orlofsky
© 2009 Blackwell Publishing Ltd, Cellular Microbiology, 11, 983–1000

alteration of parasite proliferation by the drug (Fig. 7D).These data imply that the level of mTOR activation ininfected cells is sufficient to support S phase entry, andthat mTOR-mediated promotion of cell cycle progressioncan occur even in the absence of elevated levels of S6K1and 4E-BP1 phosphorylation.
We next asked whether the mTOR-dependent cellcycle-promoting effect of T. gondii was redundant with theeffects of serum. Quiescent serum-deprived 3T3 cellswere briefly infected and then replated at subconfluentdensity in the presence of serum to initiate cell cycle entry.As shown in Fig. 8, cell cycle progression in the highlyinfected fraction was enhanced relative to the minimallyinfected fraction in the same culture. After 24 h, 19% ofminimally infected cells were in G1 and 48% in G2,whereas only 5% of highly infected cells were in G1 and68% were in G2 (Fig. 8B). Treatment with rapamycinreduced the G2 frequency to 12% for minimally infectedand 10% for highly infected cells. Therefore, as evidentfrom Fig. 8B, the effect of the drug was more pronouncedin the highly infected relative to the minimally infectedfraction, and even more so in comparison with the unin-fected culture. We draw two inferences from these find-ings. First, T. gondii-induced cell cycle progression isnon-redundant with serum-induced signals. Second, the
heightened rapamycin sensitivity of infected cells sug-gests that the elevation of mTOR activity in these cellscontributes to the observed acceleration of cell cycleprogression.
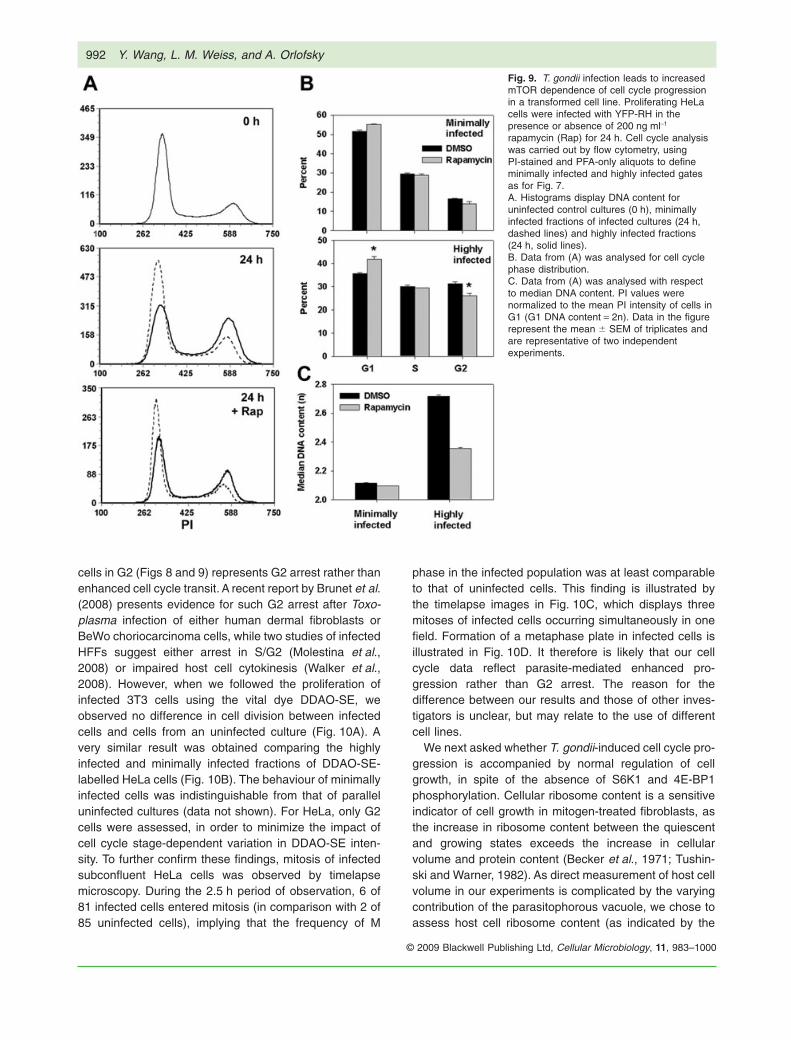
We next asked whether T. gondii could generaterapamycin-sensitive cell cycle progression in transformedcells whose growth was normally less mTOR-dependentthan that of 3T3. The cell cycle status of asynchronouslyproliferating HeLa cells displayed minimal sensitivity torapamycin (Fig. 9) The behaviour of uninfected cultureswas indistinguishable from the minimally infected fractionof infected cultures (data not shown). After 24 h, highlyinfected HeLa cells displayed significantly reduced G1frequency (36 � 0.6% versus 52 � 0.7% for minimallyinfected) and increased G2 frequency (31 � 0.6% versus16 � 0.3%). This effect of the parasite was sensitive torapamycin: the gain in median DNA content in the highlyinfected compared with minimally infected fraction wasreduced by 60% in the presence of the drug (Fig. 9C).Therefore, in both primary and transformed cells, infectionled to an alteration of cell cycle progression, and theparasite effect was more mTOR-dependent than the pro-gression of uninfected cells.
For both 3T3 and HeLa cells, an alternative interpreta-tion of our data is that the accumulation of highly infected
Fig. 8. T. gondii-induced cell cycleprogression is non-redundant with serum. 3T3cells were deprived of serum beginning 1 dayprior to infection with YFP-RH at an moi of 4.After 4 h cells were trypsinized and replatedat low density in DMEM containing 10%serum with or without 200 ng ml-1 rapamycin(Rap). Cell cycle analysis was carried out byflow cytometry, using PI-stained and PFA-onlyaliquots to define minimally infected andhighly infected gates as for Fig. 7.A. Histograms display DNA content foruninfected control cultures (thin lines),minimally infected fractions of infectedcultures (dashed lines) and highly infectedfractions (thick lines).B. Data from (A) was analysed for cell cyclephase distribution. Data represent themean � SEM of triplicates and arerepresentative of two independentexperiments.
Toxoplasma activates host cell mTOR 991
© 2009 Blackwell Publishing Ltd, Cellular Microbiology, 11, 983–1000
cells in G2 (Figs 8 and 9) represents G2 arrest rather thanenhanced cell cycle transit. A recent report by Brunet et al.(2008) presents evidence for such G2 arrest after Toxo-plasma infection of either human dermal fibroblasts orBeWo choriocarcinoma cells, while two studies of infectedHFFs suggest either arrest in S/G2 (Molestina et al.,2008) or impaired host cell cytokinesis (Walker et al.,2008). However, when we followed the proliferation ofinfected 3T3 cells using the vital dye DDAO-SE, weobserved no difference in cell division between infectedcells and cells from an uninfected culture (Fig. 10A). Avery similar result was obtained comparing the highlyinfected and minimally infected fractions of DDAO-SE-labelled HeLa cells (Fig. 10B). The behaviour of minimallyinfected cells was indistinguishable from that of paralleluninfected cultures (data not shown). For HeLa, only G2cells were assessed, in order to minimize the impact ofcell cycle stage-dependent variation in DDAO-SE inten-sity. To further confirm these findings, mitosis of infectedsubconfluent HeLa cells was observed by timelapsemicroscopy. During the 2.5 h period of observation, 6 of81 infected cells entered mitosis (in comparison with 2 of85 uninfected cells), implying that the frequency of M
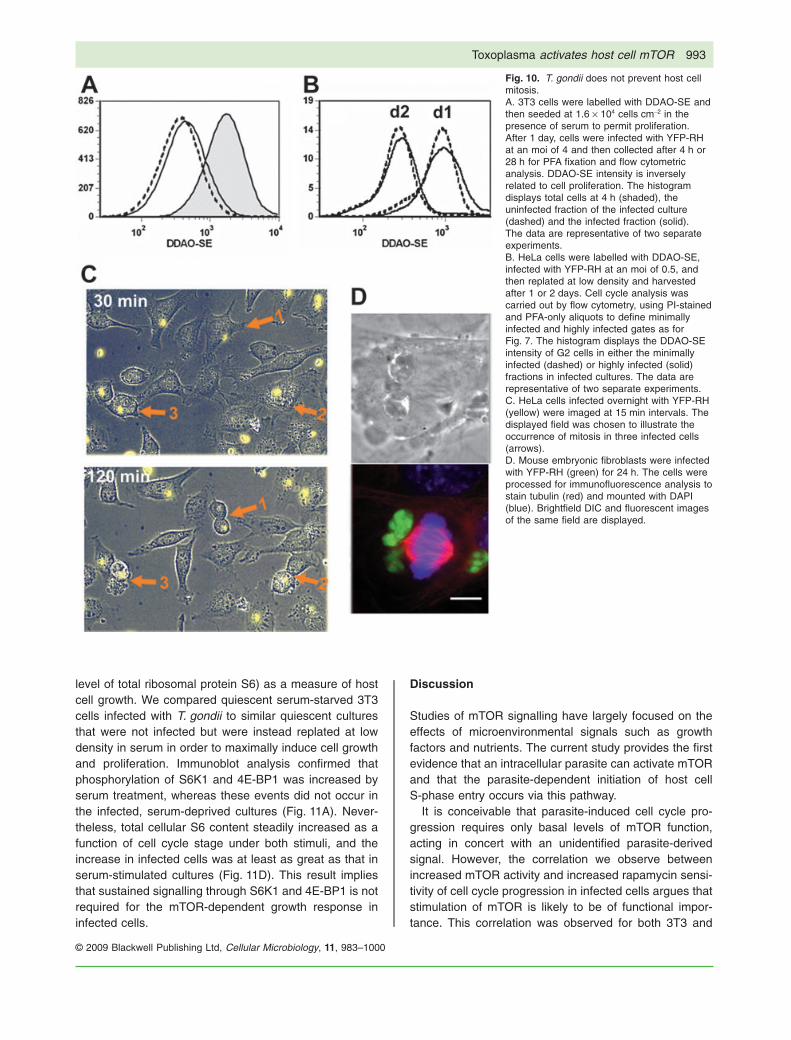
phase in the infected population was at least comparableto that of uninfected cells. This finding is illustrated bythe timelapse images in Fig. 10C, which displays threemitoses of infected cells occurring simultaneously in onefield. Formation of a metaphase plate in infected cells isillustrated in Fig. 10D. It therefore is likely that our cellcycle data reflect parasite-mediated enhanced pro-gression rather than G2 arrest. The reason for thedifference between our results and those of other inves-tigators is unclear, but may relate to the use of differentcell lines.
We next asked whether T. gondii-induced cell cycle pro-gression is accompanied by normal regulation of cellgrowth, in spite of the absence of S6K1 and 4E-BP1phosphorylation. Cellular ribosome content is a sensitiveindicator of cell growth in mitogen-treated fibroblasts, asthe increase in ribosome content between the quiescentand growing states exceeds the increase in cellularvolume and protein content (Becker et al., 1971; Tushin-ski and Warner, 1982). As direct measurement of host cellvolume in our experiments is complicated by the varyingcontribution of the parasitophorous vacuole, we chose toassess host cell ribosome content (as indicated by the
Fig. 9. T. gondii infection leads to increasedmTOR dependence of cell cycle progressionin a transformed cell line. Proliferating HeLacells were infected with YFP-RH in thepresence or absence of 200 ng ml-1
rapamycin (Rap) for 24 h. Cell cycle analysiswas carried out by flow cytometry, usingPI-stained and PFA-only aliquots to defineminimally infected and highly infected gatesas for Fig. 7.A. Histograms display DNA content foruninfected control cultures (0 h), minimallyinfected fractions of infected cultures (24 h,dashed lines) and highly infected fractions(24 h, solid lines).B. Data from (A) was analysed for cell cyclephase distribution.C. Data from (A) was analysed with respectto median DNA content. PI values werenormalized to the mean PI intensity of cells inG1 (G1 DNA content = 2n). Data in the figurerepresent the mean � SEM of triplicates andare representative of two independentexperiments.
992 Y. Wang, L. M. Weiss, and A. Orlofsky
© 2009 Blackwell Publishing Ltd, Cellular Microbiology, 11, 983–1000
level of total ribosomal protein S6) as a measure of hostcell growth. We compared quiescent serum-starved 3T3cells infected with T. gondii to similar quiescent culturesthat were not infected but were instead replated at lowdensity in serum in order to maximally induce cell growthand proliferation. Immunoblot analysis confirmed thatphosphorylation of S6K1 and 4E-BP1 was increased byserum treatment, whereas these events did not occur inthe infected, serum-deprived cultures (Fig. 11A). Never-theless, total cellular S6 content steadily increased as afunction of cell cycle stage under both stimuli, and theincrease in infected cells was at least as great as that inserum-stimulated cultures (Fig. 11D). This result impliesthat sustained signalling through S6K1 and 4E-BP1 is notrequired for the mTOR-dependent growth response ininfected cells.
Discussion
Studies of mTOR signalling have largely focused on theeffects of microenvironmental signals such as growthfactors and nutrients. The current study provides the firstevidence that an intracellular parasite can activate mTORand that the parasite-dependent initiation of host cellS-phase entry occurs via this pathway.
It is conceivable that parasite-induced cell cycle pro-gression requires only basal levels of mTOR function,acting in concert with an unidentified parasite-derivedsignal. However, the correlation we observe betweenincreased mTOR activity and increased rapamycin sensi-tivity of cell cycle progression in infected cells argues thatstimulation of mTOR is likely to be of functional impor-tance. This correlation was observed for both 3T3 and
Fig. 10. T. gondii does not prevent host cellmitosis.A. 3T3 cells were labelled with DDAO-SE andthen seeded at 1.6 ¥ 104 cells cm-2 in thepresence of serum to permit proliferation.After 1 day, cells were infected with YFP-RHat an moi of 4 and then collected after 4 h or28 h for PFA fixation and flow cytometricanalysis. DDAO-SE intensity is inverselyrelated to cell proliferation. The histogramdisplays total cells at 4 h (shaded), theuninfected fraction of the infected culture(dashed) and the infected fraction (solid).The data are representative of two separateexperiments.B. HeLa cells were labelled with DDAO-SE,infected with YFP-RH at an moi of 0.5, andthen replated at low density and harvestedafter 1 or 2 days. Cell cycle analysis wascarried out by flow cytometry, using PI-stainedand PFA-only aliquots to define minimallyinfected and highly infected gates as forFig. 7. The histogram displays the DDAO-SEintensity of G2 cells in either the minimallyinfected (dashed) or highly infected (solid)fractions in infected cultures. The data arerepresentative of two separate experiments.C. HeLa cells infected overnight with YFP-RH(yellow) were imaged at 15 min intervals. Thedisplayed field was chosen to illustrate theoccurrence of mitosis in three infected cells(arrows).D. Mouse embryonic fibroblasts were infectedwith YFP-RH (green) for 24 h. The cells wereprocessed for immunofluorescence analysis tostain tubulin (red) and mounted with DAPI(blue). Brightfield DIC and fluorescent imagesof the same field are displayed.
Toxoplasma activates host cell mTOR 993
© 2009 Blackwell Publishing Ltd, Cellular Microbiology, 11, 983–1000

HeLa cells. These data, as well as the many studiesdocumenting the cell-dependence of rapamycin sen-sitivity (Slavik et al., 2001; Noh et al., 2004; Zeiseret al., 2008) imply the existence in host cells of a re-gulated balance between mTOR-dependent and mTOR-independent proliferative pathways, and suggest that thepresence of intracellular Toxoplasma tips this balancetowards mTOR dependence. While several mechanismscould produce such an effect, including downregulation ofan mTOR-independent pathway, the simplest explanationof our results is that T. gondii-induced mTOR activity isresponsible for the increased mTOR dependence of hostcell cycle transit.
A remarkable aspect of our data is that parasite-induced cell cycle progression and cell growth, whilemTOR-dependent, take place in the absence of eithersustained activation of S6K1 or phosphorylation of4E-BP1. Ohanna et al. (2005) have described indepen-
dence of mTOR-dependent cell cycle progression fromS6K1 for cultured myoblasts from S6K1-deficient mice.While mTOR-mediated S6 phosphorylation remainedintact in S6K1-deficient myoblasts, as in our studies withToxoplasma-infected cells, this phosphorylation did notaccount for the mTOR-dependent proliferation, whichremained unchanged in S6K1/S6K2 double knockoutcells that no longer phosphorylated S6 (Ohanna et al.,2005). It therefore seems likely that mTOR-dependent cellcycle progression can proceed via a pathway, as yetuncharacterized, that is independent of both S6K1 andS6. Our study of parasite-infected cells demonstrates, inaddition, that such S6K1-independent progression cantake place through mTOR signalling that does not involvephosphorylation of 4E-BP1. Consistent with this finding,transfection with non-phosphorylatable mutants of4E-BP1 had no effect on proliferation in S6K-deficientmyoblasts (Ohanna et al., 2005). However, the roles of
Fig. 11. T. gondii-induced cell cycleprogression is accompanied by normal hostcell growth. 3T3 cells were deprived of serumfor 1 day. Cells were then either trypsinizedand replated at one half original cell density inthe presence of serum, or infected withYFP-RH at an moi of 1 in the absence ofserum (without replating), or harvested (G0).Cells treated with serum or infected werecultured for 30 h and then harvested. Aliquotsof harvested cells were processed for eitherWestern blot analysis or cell cycle analysis,using PI-stained and PFA-only aliquots todefine minimally infected and highly infectedgates as for Fig. 6. Prior to PI staining, cellswere stained with antitotal S6 antibody.A. Western blot analysis of duplicate samplesof either G0, serum-treated or infected cells.B. Definition of gates used for S6 contentanalysis. The lower panel represents thehighly infected fraction of an infected culture.Shaded segments of the DNA histogramsrepresent the gates used to define G0/G1,early S, mid S and late S fractions.C. Representative staining of total S6 (solidline) compared with a control with secondaryantibody alone (dashed line). The datarepresent total cells from a G0 sample.D. The intensity of total S6 staining isdisplayed for the indicated cell cycle fractions,as defined in (B). Data representmean � SEM for three to six replicates. Thetwo panels represent two independentexperiments (the left panel corresponds to thedata shown in A–C).
994 Y. Wang, L. M. Weiss, and A. Orlofsky
© 2009 Blackwell Publishing Ltd, Cellular Microbiology, 11, 983–1000

S6K1 and 4E-BP1 in growth may be cell-dependent.S6K1-deficient mice display a selective defect in pancre-atic b-cell size (Pende et al., 2000), and forced expressionof hypophosphorylated 4E-BP1 leads to marked inhibitionof global translation in HEK293 cells, in a manner directlyrelated to the ability of the mutant proteins to bind eIF4E(Mothe-Satney et al., 2000b). Ongoing studies in ourlaboratory are directed to examining the ability of mTORto enhance global translation in parasitized cells in spite ofthe persistence of hypophosphorylated forms of 4E-BP1.Broad upregulation of host cell translation is indicated bypreliminary results showing recruitment of host ribosomalproteins to polysomes in infected cells (data not shown).Alternatively, mTOR function in these cells may befocused to translational enhancement of specific mRNAs.A recent comparison of the alterations in host cell tran-scriptome and proteome after T. gondii-parasitized cellsdetected many instances of potential translational regula-tion in response to infection (Nelson et al., 2008).
Dissociation between mTOR and its effectors, S6K1and eIF4E, is a highly unusual occurrence. In myotubecultures, amino acid starvation is reported to partly reduceS6K1 activity, while leaving intact mTOR-dependent S6phosphorylation (Talvas et al., 2006). In our experiments,mTOR-mediated S6 phosphorylation is strictly aminoacid-dependent. We are not aware of any previous studyin which upregulation of pS6 levels via mTOR is notaccompanied by phosphorylation of S6K1 and 4E-BP1. Itis possible that this unique pattern represents an adapta-tion of the parasite. A recent study indicates thatphosphorylation of S6 may serve to protect cells fromapoptosis mediated by TRAIL, a death receptor ligandexpressed by cytotoxic T cells and of potential importanceto the control of T. gondii infection (Jeon et al., 2008). Onthe other hand, both S6K1 and 4E-BP1 have recentlybeen implicated in the mediation of mTOR-dependenteffects on cytoskeletal organization (Liu et al., 2008).There is evidence that Toxoplasma actively modifies thehost cell cytoskeleton (Coppens et al., 2006) and maytherefore need to limit interfering effects derived fromhost mTOR. The pattern of mTOR-dependent signallingwe have observed might represent a parasite-initiatedmechanism to optimize host cell survival and structuralremodelling.
How might Toxoplasma achieve uncoupling of mTORactivation from phosphorylation of S6K1 and 4E-BP1?Several investigators have demonstrated that these sub-strates share an N-terminal TOS (TOR signalling) motifthat is essential for efficient phosphorylation by mTOR,and that this motif does not mediate interaction directlywith mTOR but rather with the mTOR-binding protein,Raptor (Schalm and Blenis, 2002). One mechanism thatcould explain our findings is that T. gondii invasion gen-erates a factor that reduces the availability of the TOS-
interacting site in the N-terminal portion of Raptor. Underthis mechanism, other sites on the Raptor scaffold wouldremain available for interaction with other potential mTORsubstrates, including the kinase responsible for the pS6generation in infected cells. An obvious candidate for sucha kinase is S6K2. Our attempts to assess S6K2 activityhave so far been inconclusive (data not shown). A poten-tial difficulty with this mechanism is that S6K2 also con-tains a TOS motif (Schalm and Blenis, 2002); however, arole for this motif, or of interaction with Raptor, has notbeen established for this kinase. In the case of at leastone other Raptor-binding (and TOS-motif-containing)protein, phospholipase D2, it has been demonstrated thatbinding can occur to the C-terminal region of Raptor, evenwhen the N-terminal region is simultaneously occupiedwith S6K1 or 4E-BP1 (Ha et al., 2006). A second difficultywith this mechanism is that it does not account for theability of insulin to efficiently induce 4E-BP1 phosphory-lation in infected cells. However, insulin or serum-inducedphosphorylation of 4E-BP1, in contrast to that of S6K1,has been reported to occur in some instances via arapamycin-insensitive mechanism (Wang et al., 2005;Choo et al., 2008), and we have noted a partial rapamycinresistance for the generation of the g band of 4E-BP1 byinsulin in 3T3 cells (data not shown). Finally, the possibilitymust be considered that the absence of phosphorylatedS6K1 and 4E-BP1 reflects the parasite-induced activationof specific phosphatase activity rather than a restriction ofmTOR function. The phosphatase PP2A has been pro-posed to act on both S6K1 and 4E-BP1 (Lin andLawrence, 1997; Peterson et al., 1999).
A further question raised by our results is how Toxo-plasma invasion induces S6 phosphorylation. Based onthe exquisite sensitivity both to rapamycin and to aminoacid withdrawal, as well as dependence on basal Aktactivity, we have concluded that the immediate cause ofpS6 generation is mTOR activation, although we cannotabsolutely rule out the contribution of unknown kinaseswith similar regulatory features. Our data do not clearlyidentify a mechanism for host mTOR activation. Canonicalpathways that act through Akt or Erk activation, or alter-natively via amino acids and Vps34, do not sufficientlyaccount for mTOR activity in infected cells. While basalAkt function was required for mTOR activity, we observedno correlation between mTOR and Akt activation andindeed could detect no elevation of Akt phosphorylation ateither Thr308 or Ser473 in infected 3T3 cells. Similarly, weobserved a partial dependence of S6 phosphorylationon basal Erk function, but only a modest and variableelevation of Erk activity that did not correlate with pS6accumulation.
A possible explanation for these negative findings isthat T. gondii initiates host mTOR signalling at a pointdownstream of Akt and Erk. A candidate for this point of
Toxoplasma activates host cell mTOR 995
© 2009 Blackwell Publishing Ltd, Cellular Microbiology, 11, 983–1000

intervention is suggested by the observation that hostphosphatidic acid, an important downstream mediator ofmTOR signalling from several stimuli, becomes highlyenriched in a membrane that surrounds the parasite andlikely represents the parasitophorous vacuolar membrane(Charron and Sibley, 2002). It is plausible that phospha-tidic acid concentrated at this membrane could directlyassociate with host mTOR and facilitate its activation.Consistent with this hypothesis, we observed that hostmTOR was localized to a region surrounding the vacuole,and often to puncta that appeared to line the vacuolarmembrane. The hypothesis accounts not only for the inde-pendence of the parasite-mediated mTOR response fromAkt and Erk stimulation, but also for the dependence ofthis response on basal Akt and Erk. Activation of mTOR byeither exogenous phosphatidic acid or overexpression ofphospholipase D1 requires PI3K activity, although Aktactivation is not elevated, implying a dependence onbasal Akt function (Foster, 2007). Basal Akt signals mightenhance mTOR activation by maintaining adequate GTPloading of mTOR-associated Rheb. Amino acid signallingmay contribute to the localization of mTOR to Rheb-bearing vesicles (Sancak et al., 2008). Parasite-mediatedcolocalization of these vesicles to phosphatidic acid at thevacuolar surface may then lead to the induction of mTORactivity.
Alternative mechanisms may link mTOR activation tothe trafficking of mTOR-bearing vesicles to the parasito-phorous vacuole. One possibility is that the concentrationof these vesicles in the vicinity of the vacuole is sufficientto increase mTOR activity, perhaps by facilitating inter-action between mTOR-bearing vesicles with vesiclesbearing an mTOR-activating factor. Alternatively, parasite-imposed trafficking of these vesicles might alter theirmaturation in a manner facilitating mTOR activation.Finally, it is possible that vesicular mTOR is activated bysignals initiated by a parasite-encoded factor. Toxoplasmais capable of exporting parasite-encoded proteins eitherto the surface of the parasitophorous vacuole, wherecontact with host cytosol is possible, or alternatively intohost cytosol and nuclei (Ravindran and Boothroyd, 2008).The parasite secretory organelles known as rhoptriescontain a number of proteins that can potentially functionas kinases or phosphatases that might initiate signaltransduction cascades in the host cell, and there is evi-dence for the export of signalling kinases to the vacuolarsurface (Molestina and Sinai, 2005). The rhoptries arethought to be capable of releasing their contents at a veryearly stage of parasite invasion; nevertheless, at least onerhoptry product, ROP16, is able to generate effects onhost cell signalling at least 18 h post infection (Saeij et al.,2007), consistent with the kinetics we have observed forS6 phosphorylation. Alternatively, the initiating factor maybe a parasite moiety that engages a host cell surface
receptor. During the process of attachment and invasionof host cells, Toxoplasma secretes a number of proteinsthat can bind the host cell membrane, and whose effectson host cells are yet to be characterized (Soldati et al.,2001).
The potential role of the host mTOR pathway in Toxo-plasma pathogenesis will require further investigation. Wehave already alluded to a potential function of pS6 underapoptotic stress, and also to the possibility that the para-site might modify the translational regulation of specificmRNA, for example, encoding mediators of host defense.An additional possibility arises from our recent finding thathost cell autophagy becomes supportive of parasitegrowth when ambient amino acid concentration isreduced to physiological levels (Wang et al., 2009). Thisobservation gave rise to the hypothesis that Toxoplasmaderives nutritive benefit by driving host amino acidsthrough a ‘futile cycle’ of protein synthesis and degrada-tion (Orlofsky, 2009), resulting in increased amino acidflux through host lysosomes that can be captured by theparasitophorous vacuole (Coppens et al., 2006). It will beof interest to determine the impact of host mTOR impair-ment under conditions of nutritional stress. The nutritionaldemand from the parasitophorous vacuole is expected toincrease exponentially with time, and therefore the criticalpathogenetic role of mTOR under this hypothesismight only occur during the late stages of intracellularresidence.
Another noteworthy aspect of our results is the demon-stration of the ability of T. gondii-infected cells to progressthrough an apparently normal complete cell cycle, includ-ing cytokinesis. This has not been observed previously,and several recent studies, using cell lines different fromthe ones studied here, suggest that the cell cycle ofinfected cells is impaired or incomplete (Brunet et al.,2008; Molestina et al., 2008; Walker et al., 2008). It ispossible that parasite interference in the late stages ofhost cell mitosis is a cell-dependent phenomenon. Itwill be interesting to determine whether the parasite-dependent reorganization of host microtubules is similarin cell lines that either do or do not progress throughmitosis after infection. It may also be significant that thecell lines in which we observed mitosis (3T3 and HeLa)failed to activate Akt upon infection; in comparison, Aktwas activated in infected HFFs, in which defective mitosishas been reported (Walker et al., 2008). Further investi-gation will be needed to determine the basis for alterna-tive outcomes with respect to host cell cycle completion inToxoplasma-infected cells.
In summary, this study identifies several novel featuresof Toxoplasma–host cell interaction: the stimulation ofhost mTOR; the uncoupling of mTOR signalling from thegrowth-promoting effectors, S6K1 and eIF4E; and theability of mTOR to support both cell growth and cell cycle
996 Y. Wang, L. M. Weiss, and A. Orlofsky
© 2009 Blackwell Publishing Ltd, Cellular Microbiology, 11, 983–1000

progression in the absence of signalling through thesetwo effectors. Each of these effects points to an area offurther investigation that may enhance our understandingof both parasite–host cell co-adaptation and the mecha-nisms underlying the co-ordination of growth and prolif-eration in mammalian cells. In light of these findings, it willbe of particular interest to assess the impact of T. gondiion host cell translational regulation, and this area is afocus of ongoing studies.
Experimental procedures
Parasite and mammalian cell culture
YFP-expressing T. gondii RH strain (YFP-RH; a kind gift of Dr B.Striepen, University of Georgia) was maintained in primary HFF.After cell lysis, parasites were recovered by centrifugation andresuspended in Dulbecco’s minimal essential medium (DMEM,Invitrogen). All cell culture was performed in DMEM containing(except as indicated) 10% foetal calf serum (HyClone) in theabsence of antibiotics at 5% CO2. BALB/c 3T3 cells wereobtained from Dr R. Baserga (Thomas Jefferson University).HeLa cells were obtained from the late Dr M. Horwitz (AlbertEinstein College of Medical). Peritoneal exudate macrophageswere obtained as peritoneal lavage from mice injected intraperi-toneally 4 days earlier with 1 ml of 3% thioglycolate broth. Cellswere pooled from at least three mice and stored as frozen ali-quots. Cells were infected 24 h after plating in multi-well dishes.Prior to infection, one well was harvested for cell counting todetermine multiplicity of infection (moi). Initial experiments wereconducted using cells deprived of serum beginning 1 day prior toinfection. Subsequently, it was observed that similar parasite-dependent signals were obtained when serum was removed atthe time of infection. This procedure was routinely adopted(except as indicated), as it was less stressful to host cells. Whereindicated, cells were treated with insulin (200 nM; Invitrogen),LY294002 (10 mM; LC Laboratories), Akti-1/2 (10 mM; EMDBiosciences), PD98059 (20 mM; LC Laboratories), wortmannin(200 nM; EMD Biosciences) or the indicated concentrations ofrapamycin (LC labs).
siRNA transfection
Cells were transfected with either non-specific oligoribonucleo-tides or S6K1 RNAi (ON-TARGETplus SMARTpool, L-040893-00, Dharmacon), using lipofectamine 2000 (Invitrogen) accordingto the manufacturer’s protocol. Cells were replated 24 h aftertransfection, infected for 24 h and then harvested for Westernblot analysis.
Immunoblot analysis
Cells were lysed in RIPA buffer (1% NP-40, 0.5% sodium deoxy-cholate, 0.1% SDS in PBS) supplemented with protease inhibitorand phosphatase inhibitor cocktails (Sigma). Protein concentra-tion was determined by the microbicinchoninic acid assay(Pierce). Ten or twenty micrograms of protein extracts wasresolved by SDS-PAGE, followed by transfer to PVDF mem-branes (Millipore). Membranes were blocked with 5% non-fat milk
in TPBS (0.5% Tween-20 in PBS) and subsequently probed withprimary antibody in TPBS containing 1% bovine serum albumin(BSA). After overnight incubation at 4°C, blots were washed withTPBS, incubated with horseradish peroxidase-conjugated sec-ondary antibody in 5% milk in TPBS for 1 h at room temperature,and imaged using enhanced chemiluminescence (Pierce) fol-lowed by exposure to X-ray film. Primary antibodies from CellSignalling include: anti-pAkt (S473) (9271), anti-total Akt (9272),anti-pGsk3b(S9) (9336), anti-pERK1/2 (T202/Y204) (9101),anti-pS6k1 (T389) (9234), anti-total S6K1 (9202), anti-pS6(S235/236) 2211, anti-total S6 (2217), anti-4E-BP1 (9644). Otherprimary antibodies include anti-actin (Abcam) and anti-YFP(Becton Dickinson). Band intensities were analysed usingImageJ.
Immunofluorescence and timelapse imaging
Cells were seeded onto coverslips (Fisher scientific) in a 24-wellplate. For analysis, cells were fixed with 4% buffered PFA andpermeabilized with 0.1% Triton X-100. After blocking with 10%foetal bovine serum in PBS, the samples were incubated at 4°Covernight with anti-pS6 (S235/236) diluted in 1% BSA in PBS.Cells were then washed and incubated with Cy5-conjugated anti-rabbit IgG (Jackson ImmunoResearch) for 1 h at room tempera-ture. After extensive washing, the samples were dried andmounted with GOLDEN anti-fade reagent (Invitrogen). Imageswere collected on a fluorescence microscope (Olympus 1 ¥ 81)in the Albert Einstein College of Medicine Analytical ImagingFacility. For timelapse imaging, cells were grown in coverslip-thickness chamber slides (Nunc) and infected overnight withYFP-RH at an moi of 4. The medium was replaced with mediumcontaining 20 mM HEPES (pH 7.5) and timelapse imageswere collected at 15 min intervals on a Zeiss AxioObserverfluorescence microscope.
Flow cytometry
For vital dye labelling, plates were rinsed with Hank’s balancedsalt solution (HBSS) and the cells were incubated with 1 mMDDAO-SE (CellTrace Far Red, Invitrogen) in HBSS for 15 minand then washed with medium. Cells were harvested with trypsinand single-cell suspensions were prepared by repeated tritura-tion on ice. After centrifugation, cells were suspended in 500 mlcold PBS, followed by addition of 500 ml 1% PFA in PBS. After a1 h incubation on ice, cells were washed with PBS containing0.5% BSA. Aliquots (‘PFA-only’) were reserved for assessment ofparasite content in the absence of ethanol treatment. The remain-der of the sample was centrifuged and the pellet suspended bythe addition of 70% ethanol (-20°C) in a dropwise manner whilevortexing. For determination of S6 content, cells were thenwashed several times with 0.5% BSA in PBS and incubated with100 ml anti-S6 (1:100 in BSA-PBS) for 1 h at room temperature.The cells were then washed, incubated with Cy5-conjugatedanti-rabbit IgG for 30 min, and washed extensively. Prior to flowcytometry, all samples (except the ‘PFA only’ aliquots) werewashed with PBS containing 0.5% BSA, and incubated with50 mg ml-1 propidium iodide and 50 mg ml-1 RNase A for 30 min at37°C. Flow cytometry was performed on a FacsCalibur (BectonDickinson). Data were analysed using FCSExpress (De NovoSoftware), except for cell cycle analysis, which was performed in
Toxoplasma activates host cell mTOR 997
© 2009 Blackwell Publishing Ltd, Cellular Microbiology, 11, 983–1000

FlowJo (TreeStar) using a Watson pragmatic model in which theG2/G1 ratio, as well as the G1 and G2 coefficients of variation,were constrained. Prior to cell cycle analysis, the contribution ofparasite DNA to PI intensity was corrected by using compensa-tion software in FCSExpress to remove the dependence of PIintensity on YFP intensity in G1 phase cells. Compensationsamples were used to correct for minor spillover from the PIsignal (FL3) to Cy5 (FL4).
Acknowledgements
The authors wish to acknowledge Drs Jonathan Backer, RenatoBaserga, Umadas Maitra, Boris Striepen and Jonathan Warnerfor valuable suggestions and reagents. We also thank Ms YanfenMa for assistance with parasite preparation. This study was sup-ported by funds from National Institutes of Health Grant AI-55358to A.O. and AI-39454 to L.M.W., and by the Flow Cytometry Coreof the Center for AIDS Research (AI-51519).
References
Arvisais, E.W., Romanelli, A., Hou, X.Y., and Davis, J.S.(2006) AKT-independent phosphorylation of TSC2 and acti-vation of mTOR and ribosomal protein S6 kinase signalingby prostaglandin F2 alpha. J Biol Chem 281: 26904–26913.
Avruch, J., Long, X., Ortiz-Vega, S., Rapley, J., Papageor-giou, A., and Dai, N. (2008) Amino acid regulation of TORcomplex 1. Am J Physiol Endocrinol Metab (epub ahead ofprint) doi: 10.1152/ajpendo.90645.2008.
Bai, X.C., Ma, D.Z., Liu, A.L., Shen, X.Y., Wang, Q.J.M., Liu,Y.J., and Jiang, Y. (2007) Rheb activates mTOR byantagonizing its endogenous inhibitor, FKBP38. Science318: 977–980.
Becker, H., Stanners, C.P., and Kudlow, J.E. (1971) Controlof macromolecular synthesis in proliferating and restingSyrian hamster cells in monolayer culture. II. Ribosomecomplement in resting and early G1 cells. J Cell Physiol 77:43–50.
Bilanges, B., Argonza-Barrett, R., Kolesnichenko, M.,Skinner, C., Nair, M., Chen, M., and Stokoe, D. (2007)Tuberous sclerosis complex proteins 1 and 2 controlserum-dependent translation in a TOP-dependent and –independent manner. Mol Cell Biol 27: 5746–5764.
Brunet, J., Pfaff, A.W., Abidi, A., Unoki, M., Nakamura, Y.,Guinard, M., et al. (2008) Toxoplasma gondii exploitsUHRF1 and induces host cell cycle arrest at G2 to enableits proliferation. Cell Microbiol 10: 908–920.
Carraway, H., and Hidalgo, M. (2004) New targets for therapyin breast cancer – mammalian target of rapamycin (mTOR)antagonists. Breast Cancer Res 6: 219–224.
Charron, A.J., and Sibley, L.D. (2002) Host cells: mobilizablelipid resources for the intracellular parasite Toxoplasmagondii. J Cell Sci 115: 3049–3059.
Choo, A.Y., Yoon, S.O., Kim, S.G., Roux, P.P., and Blenis, J.(2008) Rapamycin differentially inhibits S6Ks and 4E-BP1to mediate cell-type-specific repression of mRNA transla-tion. Proc Natl Acad Sci USA 105: 17414–17419.
Coppens, I., Dunn, J.D., Romano, J.D., Pypaert, M., Zhang,H., Boothroyd, J.C., and Joiner, K.A. (2006) Toxoplasma
gondii sequesters lysosomes from mammalian hosts in thevacuolar space. Cell 125: 261–274.
Dmitriev, S.E., Terenin, I.M., Dunaevsky, Y.E., Merrick, W.C.,and Shatsky, I.N. (2003) Assembly of 48S translation ini-tiation complexes from purified components with mRNAsthat have some base pairing within their 5 ‘’untranslatedregions. Mol Cell Biol 23: 8925–8933.
Fingar, D.C., and Blenis, J. (2004) Target of rapamycin(TOR): an integrator of nutrient and growth factor signalsand coordinator of cell growth and cell cycle progression.Oncogene 23: 3151–3171.
Foster, D.A. (2007) Regulation of mTOR by phosphatidicacid? Cancer Res 67: 1–4.
Gulati, P., Gaspers, L.D., Dann, S.G., Joaquin, M., Nobukuni,T., Natt, F., et al. (2008) Amino acids activate mTORComplex 1 via Ca2+/CaM signaling to hVps34. Cell Metab7: 456–465.
Ha, S.H., Kim, D.H., Kim, I.S., Kim, J.H., Lee, M.N., Lee, H.J.,et al. (2006) PLD2 forms a functional complex withmTOR/raptor to transduce mitogenic signals. Cell Signal18: 2283–2291.
Hay, N., and Sonenberg, N. (2004) Upstream and down-stream of mTOR. Genes Dev 18: 1926–1945.
Jayaraman, T., and Marks, A.R. (1993) Rapamycin-Fkbp12blocks proliferation, induces differentiation, and inhibitsCdc2 kinase-activity in a myogenic cell-line. J Biol Chem268: 25385–25388.
Jeon, Y.J., Kim, I.K., Hong, S.H., Nan, H., Kim, H.J., Lee, H.J.,et al. (2008) Ribosomal protein S6 is a selective mediator ofTRAIL-apoptotic signaling. Oncogene 27: 4344–4352.
Khaleghpour, K., Pyronnet, S., Gingras, A.C., and Sonenberg,N. (1999) Translational homeostasis: eukaryotic translationinitiation factor 4E control of 4E-binding protein 1 and p70,S6 kinase activities. Mol Cell Biol 19: 4302–4310.
Kurmasheva, R.T., Huang, S., and Houghton, P.J. (2006)Predicted mechanisms of resistance to mTOR inhibitors.Br J Cancer 95: 955–960.
Li, Y., Corradetti, M.N., Inoki, K., and Guan, K.L. (2004)TSC2: filling the GAP in the mTOR signaling pathway.Trends Biochem Sci 29: 32–38.
Lin, T.A., and Lawrence, J.C. (1997) Control of PHAS-Iphosphorylation in 3T3-L1 adipocytes: effects of inhibitingprotein phosphatases and the p70 (S6K) signallingpathway. Diabetologia 40: S18–S24.
Liu, L., Chen, L., Chung, J., and Huang, S. (2008) Rapamycininhibits F-actin reorganization and phosphorylation of focaladhesion proteins. Oncogene 27: 4998–5010.
LoPiccolo, J., Blumenthal, G.M., Bernstein, W.B., andDennis, P.A. (2008) Targeting the PI3K/Akt/mTOR path-way: effective combinations and clinical considerations.Drug Resist Updat 11: 32–50.
Ma, L., Chen, Z.B., Erdjument-Bromage, H., Tempst, P., andPandolfi, P.P. (2005) Phosphorylation and functional inac-tivation of TSC2 by Erk: Implications for tuberous sclerosisand cancer pathogenesis. Cell 121: 179–193.
Martin, A.M., Liu, T., Lynn, B.C., and Sinai, A.P. (2007) TheToxoplasma gondii parasitophorous vacuole membrane:transactions across the border. J Eukaryot Microbiol 54:25–28.
Mieulet, V., Roceri, M., Espeillac, C., Sotiropoulos, A.,Ohanna, M., Oorschot, V., et al. (2007) S6 kinase inacti-
998 Y. Wang, L. M. Weiss, and A. Orlofsky
© 2009 Blackwell Publishing Ltd, Cellular Microbiology, 11, 983–1000

vation impairs growth and translational target phosphory-lation in muscle cells maintaining proper regulation ofprotein turnover. Am J Physiol 293: C712–C722.
Molestina, R.E., and Sinai, A.P. (2005) Detection of a novelparasite kinase activity at the Toxoplasma gondii parasito-phorous vacuole membrane capable of phosphorylatinghost I kappa B alpha. Cell Microbiol 7: 351–362.
Molestina, R.E., El Guendy, N., and Sinai, A.P. (2008) Infec-tion with Toxoplasma gondii results in dysregulation of thehost cell cycle. Cell Microbiol 10: 1153–1165.
Mothe-Satney, I., Brunn, G.J., McMahon, L.P., Capaldo,C.T., Abrahams, R.T., and Lawrence, J.C. (2000a) Mam-malian target of rapamycin-dependent phosphorylation ofPHAS-I in four (S/T) P sites detected by phospho-specificantibodies. J Biol Chem 275: 33836–33843.
Mothe-Satney, I., Yang, D.Q., Fadden, P., Haystead, T.A.J.,and Lawrence, J.C. (2000b) Multiple mechanisms controlphosphorylation of PHAS-I in five (S/T) P sites that governtranslational repression. Mol Cell Biol 20: 3558–3567.
Mourani, P.M., Garl, P.J., Wenzlau, J.M., Carpenter, T.C.,Stenmark, K.R., and Weiser-Evans, M.C.M. (2004) Unique,highly proliferative growth phenotype expressed byembryonic and neointimal smooth muscle cells is drivenby constitutive Akt, mTOR, and p70S6K signaling and isactively repressed by PTEN. Circulation 109: 1299–1306.
Murakami, M., Ichisaka, T., Maeda, M., Oshiro, N., Hara, K.,Edenhofer, F., et al. (2004) mTOR is essential for growthand proliferation in early mouse embryos and embryonicstem cells. Mol Cell Biol 24: 6710–6718.
Nelson, M.M., Jones, A.R., Carmen, J.C., Sinai, A.P., Burch-more, R., and Wastling, J.A. (2008) Modulation of the hostcell proteome by the intracellular apicomplexan parasiteToxoplasma gondii. Infect Immun 76: 828–844.
Noh, W.C., Mondesire, W.H., Peng, J.Y., Jian, W.G., Zhang,H.X., Dong, J.J., et al. (2004) Determinants of rapamycinsensitivity in breast cancer cells. Clin Cancer Res 10:1013–1023.
Ohanna, M., Sobering, A.K., Lapointe, T., Lorenzo, L., Praud,C., Petroulakis, E., et al. (2005) Atrophy of S6K1(-/-) skel-etal muscle cells reveals distinct mTOR effectors for cellcycle and size control. Nature Cell Biol 7: 286–294.
Orlofsky, A. (2009) Toxoplasma-induced autophagy: awindow into nutritional futile cycles in mammalian cells?Autophagy, in press.
Pende, M., Kozma, S.C., Jaquet, M., Oorschot, V., Burcelin,R., Marchand-Brustel, Y., et al. (2000) Hypoinsulinaemia,glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature 408: 994–997.
Peterson, R.T., Desai, B.N., Hardwick, J.S., and Schreiber,S.L. (1999) Protein phosphatase 2A interacts with the70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycin-associated protein. Proc Natl Acad Sci USA 96:4438–4442.
Petroulakis, E., Mamane, Y., Le Bacquer, O., Shahbazian,D., and Sonenberg, N. (2006) mTOR signaling: implica-tions for cancer and anticancer therapy. Br J Cancer 94:195–199.
Ramirez-Valle, F., Braunstein, S., Zavadil, J., Formenti, S.C.,and Schneider, R.J. (2008) eIF4GI links nutrient sensing bymTOR to cell proliferation and inhibition of autophagy.J Cell Biol 181: 293–307.
Ravindran, S., and Boothroyd, J.C. (2008) Secretion of pro-teins into host cells by Apicomplexan parasites. Traffic 9:647–656.
Rolfe, M., Mcleod, L.E., Pratt, P.F., and Proud, C.G. (2005)Activation of protein synthesis in cardiomyocytes by thehypertrophic agent phenylephrine requires the activation ofERK and involves phosphorylation of tuberous sclerosiscomplex 2 (TSC2). Biochem J 388: 973–984.
Ruvinsky, I., and Meyuhas, O. (2006) Ribosomal protein S6phosphorylation: from protein synthesis to cell size. TrendsBiochem Sci 31: 342–348.
Saeij, J.P.J., Coller, S., Boyle, J.P., Jerome, M.E., White,M.W., and Boothroyd, J.C. (2007) Toxoplasma co-optshost gene expression by injection of a polymorphic kinasehomologue. Nature 445: 324–327.
Sancak, Y., Peterson, T.R., Shaul, Y.D., Lindquist, R.A.,Thoreen, C.C., Bar-Peled, L., and Sabatini, D.M. (2008)The Rag GTPases bind raptor and mediate amino acidsignaling to mTORC1. Science 320: 1496–1501.
Schalm, S.S., and Blenis, J. (2002) Identification of a con-served motif required for mTOR signaling. Curr Biol 12:632–639.
Shapira, S., Harb, O.S., Margarit, J., Matrajt, M., Han, J.,Hoffmann, A., et al. (2005) Initiation and termination ofNF-kappa B signaling by the intracellular protozoan para-site Toxoplasma gondii. J Cell Sci 118: 3501–3508.
Sinai, A.P., Webster, P., and Joiner, K.A. (1997) Associationof host cell endoplasmic reticulum and mitochondria withthe Toxoplasma gondii parasitophorous vacuole mem-brane: a high affinity interaction. J Cell Sci 110: 2117–2128.
Slavik, J.M., Lim, D.G., Burakoff, S.J., and Hafler, D.A. (2001)Uncoupling p70 (s6) kinase activation and proliferation:rapamycin-resistant proliferation of human CD8 (+) T lym-phocytes. J Immunol 166: 3201–3209.
Soldati, D., Dubremetz, J.F., and Lebrun, M. (2001) Micro-neme proteins: structural and functional requirements topromote adhesion and invasion by the apicomplexanparasite Toxoplasma gondii. Int J Parasitol 31: 1293–1302.
Sun, Y., Fang, Y., Yoon, M.S., Zhang, C., Roccio, M., Zwart-kruis, F.J., et al. (2008) Phospholipase D1 is an effector ofRheb in the mTOR pathway. Proc Natl Acad Sci USA 105:8286–8291.
Talvas, J., Obled, A., Fafournoux, P., and Mordier, S. (2006)Regulation of protein synthesis by leucine starvationinvolves distinct mechanisms in mouse C2C12 myoblastsand myotubes. J Nutr 136: 1466–1471.
Tee, A.R., and Blenis, J. (2005) mTOR, translational controland human disease. Sem Cell Dev Biol 16: 29–37.
Thomas, G.V., Tran, C., Mellinghoff, I.K., Welsbie, D.S.,Chan, E., Fueger, B., et al. (2006) Hypoxia-inducible factordetermines sensitivity to inhibitors of mTOR in kidneycancer. Nat Med 12: 122–127.
Tushinski, R.J., and Warner, J.R. (1982) Ribosomal proteinsare synthesized preferentially in cells commencing growth.J Cell Physiol 112: 128–135.
Veverka, V., Crabbe, T., Bird, I., Lennie, G., Muskett, F.W.,Taylor, R.J., and Carr, M.D. (2008) Structural characteriza-tion of the interaction of mTOR with phosphatidic acid anda novel class of inhibitor: compelling evidence for a central
Toxoplasma activates host cell mTOR 999
© 2009 Blackwell Publishing Ltd, Cellular Microbiology, 11, 983–1000

role of the FRB domain in small molecule-mediated regu-lation of mTOR. Oncogene 27: 585–595.
Volarevic, S., and Thomas, G. (2001) Role of S6 phosphory-lation and S6 kinase in cell growth. Prog Nucleic Acid ResMol Biol 65: 101–127.
Walker, M.E., Hjort, E.E., Smith, S.S., Tripathi, A., Hornick,J.E., Hinchcliffe, E.H., et al. (2008) Toxoplasma gondiiactively remodels the microtubule network in host cells.Microbes Infect 10: 1440–1449.
Wang, X.M., Beugnet, A., Murakami, M., Yamanaka, S., andProud, C.G. (2005) Distinct signaling events downstreamof mTOR cooperate to mediate the effects of amino acidsand insulin on initiation factor 4E-binding proteins. Mol CellBiol 25: 2558–2572.
Wang, Y., Weiss, L.M., and Orlofsky, A. (2009) Host cellautophagy is induced by Toxoplasma gondii and contrib-utes to parasite growth. J Biol Chem 284: 1694–1701.
Wullschleger, S., Loewith, R., and Hall, M.N. (2006) TORsignaling in growth and metabolism. Cell 124: 471–484.
Yeager, N., Brewer, C., Cai, K.Q., Xu, X.X., and Di Cristofano,A. (2008) Mammalian target of rapamycin is the keyeffector of phosphatidylinositol-3-OH-initiated proliferativesignals in the thyroid follicular epithelium. Cancer Res 68:444–449.
Yoon, S., Lee, M.Y., Park, S.W., Moon, J.S., Koh, Y.K., Ahn,Y.H., et al. (2007) Up-regulation of Acetyl-CoA carboxylasealpha and fatty acid synthase by human epidermal growthfactor receptor 2 at the translational level in breast cancercells. J Biol Chem 282: 26122–26131.
Zeiser, R., Leveson-Gower, D.B., Zambricki, E.A.,Kambham, N., Beilhack, A., Loh, J., et al. (2008) Differen-tial impact of mammalian target of rapamycin inhibition onCD4(+) CD25(+) Foxp3(+) regulatory T cells compared withconventional CD4(+) T cells. Blood 111: 453–462.
1000 Y. Wang, L. M. Weiss, and A. Orlofsky
© 2009 Blackwell Publishing Ltd, Cellular Microbiology, 11, 983–1000




















