
Interaction of Serotonin and Fluoxetine: Toward understanding the importance of the chirality of...
7
Interaction of Serotonin and Fluoxetine: Toward Understanding the Importance of the Chirality of Fluoxetine (S form and R form) Prabhat K. Sahu,* ,†,‡ Chun-Hung Wang, ‡ and Shyi-Long Lee* ,‡ Institut fu ¨r Organische Chemie, UniVersita ¨t Wu ¨rzburg, Am Hubland, 97074 Wu ¨rzburg, Germany, and Department of Chemistry and Biochemistry, National Chung Cheng UniVersity, Chia -Yi, 621 Taiwan ReceiVed: July 27, 2009; ReVised Manuscript ReceiVed: August 28, 2009 The present investigation reports the importance of the S and R forms of fluoxetine, as an antidepressant with regards to the chirality taking different types of interactions associated with the neurotransmitter, serotonin. The goal of the present study is to provide predictions and to help experimental and theoretical studies toward understanding the chirality of fluoxetine in drug-ligand interaction, associated with serotonin-reuptake inhibitor drug design studies. Several different conformations for serotonin and fluoxetine complexes have been considered for quantum mechanical calculations. Both the S and R forms of fluoxetine associated with serotonin and fluoxetine complexes are found to be similar in total energy and binding energy values. The present study also supports the conformational effect of the 3-phenyl group of fluoxetine as stereo independent and is consistent with in vitro and in vivo data which indicates that the the eudismic ratio of fluoxetine enantiomers is near unity. The calculated highest stabilization energy values, binding energy values, both in the gas and aqueous phases at MP2/6-31+G*//B3LYP/6-31+G* identify the most possible stable conformer for the serotonin-fluoxetine complex. Introduction Chirality introduces marked selectivity, and often specificity, in drug action. 1 The interaction of enantiomers with pharma- cological receptors can be explained as a hand fits to a glove. Such receptors are often chiral, and the “right-” or “left-handed” drug will only fit to the molecular receptor at the desired site of action. 2 Fluoxetine has been introduced as a potent antide- pressant with a safer treatment alternative to the other two classes of drugs used to treat depressed patients: the monoamine oxidase inhibitors (MAOIs) and the tricyclic antidepressants (TCAs), having an improved side effect profile. 3,4 The phar- macological effect of fluoxetine takes place via inhibition of the presynaptic serotonin-reuptake carrier of neurons. 5 For the last several years, the mechanism to understand the mode of action of these antidepressant drugs on their direct target, the serotonin transport protein, and possible regulatory mechanisms with respect to long-term alleviation of depression, although having been investigated both neurobiologically and clinically, are not yet fully understood. Fluoxetine is used therapeutically as the racemate, and most published pharmacology studies were conducted with the racemate. The two enantiomers have been obtained in optically pure form, and some of the pharmacologi- cal features of these two compounds have also been reported. Wong et al. showed that the (+) isomer was slightly more potent than the (-) isomer as a serotonin-reuptake inhibitor in rat cortical synaptosomes. 6 Fuller and Snoddy demonstrated that the dextrorotatory isomer of fluoxetine was slightly more potent than the levorotatory isomer as an antagonist of p-chloroam- phetamine-induced depletion of whole brain serotonin concen- trations in rats. 7 To date, there appears to be no evidence that either single enantiomer is preferable to racemic fluoxetine. In this report, the importance of the S and R forms of fluoxetine, as an antidepressant with regard to the chirality, has been considered to investigate different type of interactions, associated with the neurotransmitter, serotonin, using different quantum chemical methods. The goal of the present study is to provide predictions and to help experimental and theoretical studies toward understanding the chirality of fluoxetine in drug-ligand interactions, associated with serotonin-reuptake inhibitor drug design studies. To the best of our knowledge, no detailed studies have been found to date which enhance the understanding of the molecular interaction of fluoxetine with serotonin. Serotonin (5-hydroxytryptamine, 5-HT) acts as a neurotransmitter in the central nervous system (CNS) and it influences several physiological processes such as temperature regulation, appetite, sexual behavior, and sleep. Serotonin also plays important role in regulating smooth muscle function in the cardiovascular and gastrointestinal systems. Theoretical investigations for the gas phase and aqueous solution confor- mational analysis for protonated 8 and neutral 9 serotonin have been reported. More recently, LeGreve et al. have reported the resonant two-photon ionization (R2PI), laser-induced fluores- cence (LIF), UV-UV hole-burning, resonant ion-dip infrared (RIDIR), and fluorescence-dip infrared (FDIR) spectra of isolated serotonin cooled in a supersonic expansion. 10 Other than the molecular interaction of fluoxetine with serotonin, previous experimental investigations demonstrate that fluoxetine is a high- affinity ligand and a potent inhibitor of the serotonin transporter found in the human placental brush-border membrane 11 and brain tissue. 12 Computational Details The initial geometries of several different conformations for the interaction of serotonin and fluoxetine are identified in the HyperChem 7.5 package by molecular mechanics method using the AMBER 13 force field. The resulting six different conforma- tions for serotonin and fluoxetine complexes have been con- * E-mail: [email protected] (P.K.S.) and [email protected] (S.-L.L.). † Universita ¨t Wu ¨rzburg. ‡ National Chung Cheng University. J. Phys. Chem. B 2009, 113, 14529–14535 14529 10.1021/jp907151n CCC: $40.75 2009 American Chemical Society Published on Web 09/29/2009
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Interaction of Serotonin and Fluoxetine: Toward understanding the importance of the chirality of...

Interaction of Serotonin and Fluoxetine: Toward Understanding the Importance of theChirality of Fluoxetine (S form and R form)
Prabhat K. Sahu,*,†,‡ Chun-Hung Wang,‡ and Shyi-Long Lee*,‡
Institut fur Organische Chemie, UniVersitat Wurzburg, Am Hubland, 97074 Wurzburg, Germany, andDepartment of Chemistry and Biochemistry, National Chung Cheng UniVersity, Chia -Yi, 621 Taiwan
ReceiVed: July 27, 2009; ReVised Manuscript ReceiVed: August 28, 2009
The present investigation reports the importance of the S and R forms of fluoxetine, as an antidepressant withregards to the chirality taking different types of interactions associated with the neurotransmitter, serotonin.The goal of the present study is to provide predictions and to help experimental and theoretical studies towardunderstanding the chirality of fluoxetine in drug-ligand interaction, associated with serotonin-reuptake inhibitordrug design studies. Several different conformations for serotonin and fluoxetine complexes have beenconsidered for quantum mechanical calculations. Both the S and R forms of fluoxetine associated with serotoninand fluoxetine complexes are found to be similar in total energy and binding energy values. The presentstudy also supports the conformational effect of the 3-phenyl group of fluoxetine as stereo independent andis consistent with in vitro and in vivo data which indicates that the the eudismic ratio of fluoxetine enantiomersis near unity. The calculated highest stabilization energy values, binding energy values, both in the gas andaqueous phases at MP2/6-31+G*//B3LYP/6-31+G* identify the most possible stable conformer for theserotonin-fluoxetine complex.
Introduction
Chirality introduces marked selectivity, and often specificity,in drug action.1 The interaction of enantiomers with pharma-cological receptors can be explained as a hand fits to a glove.Such receptors are often chiral, and the “right-” or “left-handed”drug will only fit to the molecular receptor at the desired siteof action.2 Fluoxetine has been introduced as a potent antide-pressant with a safer treatment alternative to the other twoclasses of drugs used to treat depressed patients: the monoamineoxidase inhibitors (MAOIs) and the tricyclic antidepressants(TCAs), having an improved side effect profile.3,4 The phar-macological effect of fluoxetine takes place via inhibition ofthe presynaptic serotonin-reuptake carrier of neurons.5 For thelast several years, the mechanism to understand the mode ofaction of these antidepressant drugs on their direct target, theserotonin transport protein, and possible regulatory mechanismswith respect to long-term alleviation of depression, althoughhaving been investigated both neurobiologically and clinically,are not yet fully understood. Fluoxetine is used therapeuticallyas the racemate, and most published pharmacology studies wereconducted with the racemate. The two enantiomers have beenobtained in optically pure form, and some of the pharmacologi-cal features of these two compounds have also been reported.Wong et al. showed that the (+) isomer was slightly more potentthan the (-) isomer as a serotonin-reuptake inhibitor in ratcortical synaptosomes.6 Fuller and Snoddy demonstrated thatthe dextrorotatory isomer of fluoxetine was slightly more potentthan the levorotatory isomer as an antagonist of p-chloroam-phetamine-induced depletion of whole brain serotonin concen-trations in rats.7 To date, there appears to be no evidence thateither single enantiomer is preferable to racemic fluoxetine.
In this report, the importance of the S and R forms offluoxetine, as an antidepressant with regard to the chirality, hasbeen considered to investigate different type of interactions,associated with the neurotransmitter, serotonin, using differentquantum chemical methods. The goal of the present study is toprovide predictions and to help experimental and theoreticalstudies toward understanding the chirality of fluoxetine indrug-ligand interactions, associated with serotonin-reuptakeinhibitor drug design studies. To the best of our knowledge, nodetailed studies have been found to date which enhance theunderstanding of the molecular interaction of fluoxetine withserotonin. Serotonin (5-hydroxytryptamine, 5-HT) acts as aneurotransmitter in the central nervous system (CNS) and itinfluences several physiological processes such as temperatureregulation, appetite, sexual behavior, and sleep. Serotonin alsoplays important role in regulating smooth muscle function inthe cardiovascular and gastrointestinal systems. Theoreticalinvestigations for the gas phase and aqueous solution confor-mational analysis for protonated8 and neutral9 serotonin havebeen reported. More recently, LeGreve et al. have reported theresonant two-photon ionization (R2PI), laser-induced fluores-cence (LIF), UV-UV hole-burning, resonant ion-dip infrared(RIDIR), and fluorescence-dip infrared (FDIR) spectra ofisolated serotonin cooled in a supersonic expansion.10 Other thanthe molecular interaction of fluoxetine with serotonin, previousexperimental investigations demonstrate that fluoxetine is a high-affinity ligand and a potent inhibitor of the serotonin transporterfound in the human placental brush-border membrane11 andbrain tissue.12
Computational Details
The initial geometries of several different conformations forthe interaction of serotonin and fluoxetine are identified in theHyperChem 7.5 package by molecular mechanics method usingthe AMBER13 force field. The resulting six different conforma-tions for serotonin and fluoxetine complexes have been con-
* E-mail: [email protected] (P.K.S.) and [email protected](S.-L.L.).
† Universitat Wurzburg.‡ National Chung Cheng University.
J. Phys. Chem. B 2009, 113, 14529–14535 14529
10.1021/jp907151n CCC: $40.75 2009 American Chemical SocietyPublished on Web 09/29/2009

sidered for quantum mechanical calculations via an ab initioand density functional theory (DFT) method. All these differentconformations have been optimized at the B3LYP/6-31+G* 14
level. Single point calculations at MP2/6-31+G*//B3LYP/6-31+G* 15 have also been carried out to better estimate thehydrogen bonding strengths. Harmonic vibrational frequencyhas also been analyzed for serotonin, fluoxetine monomers (withboth racemic forms) at the B3LYP/cc-pVTZ16 level of theory.The vibrational analysis obtained for the serotonin monomerhas been compared to previously reported van Mourik andEmson’s calculated results9 and with recently reported experi-mental results.10 The vibrational frequency analysis for all thesix stable serotonin-fluoxetin complexes have also been carriedout at B3LYP/6-31+G*. To analyze the solvent polarity effect,the COSMO approach17 has also been taken into account at theMP2/6-31+G*//B3LYP/6-31+G* level. All the electronic struc-ture calculations are performed by using the Gaussian 03package.18
Results and Discussion
1. Serotonin and Fluoxetine Monomers. The geometryparameters of serotonin monomer (5-hydroxytryptamine, 5-HT)and fluoxetine monomers with both racemic forms have beenobtained at the B3LYP/6-31+G* and B3LYP/cc-pVTZ levelsof theory. Figure 1.1 (cf the Supporting Information) shows thegeometry optimized structure of serotonin monomer using theB3LYP/6-31+G* method. Figure 1.2 (cf the Supporting Infor-mation) shows the geometry optimized structure of the fluoxetineS form using the B3LYP/6-31+G* method. Compared with vanMourik and Emson’s computed result, Gpy(out) and Gph(out)are found as the two most stable minima, the lone pair in theethylamine group is oriented away from the indole ring andone of the ethylamine hydrogens is directed toward the indoleπ-cloud. Our obtained results are similar to van Mourik andEmson’s computed result,9 but the orientation of the hydroxylgroup is directed away from the ethylamine group, which isalso supported by Alagona et al.8 and LeGreve et al.10 It hasalso been found that the relative energy difference between theanti and syn conformation is within the range 0.1-0.4 kcal/mol.10 We also note that the results obtained at B3LYP/cc-pVTZand B3LYP/6-31+G* are found to be very similar, and hence,we have chosen the B3LYP/6-31+G* method for our furthercalculations and discussions, as compared to the larger B3LYP/cc-pVTZ to meet with the high computational cost.
Figure 1.3 (cf the Supporting Information) shows thecalculated IR spectra of serotonin and fluoxetine monomers withboth racemic forms at the B3LYP/cc-pVTZ level of theory.There are other theoretical studies8 about the IR spectra ofvarious conformers of serotonin monomer. Table 1.1 (cf theSupporting Information) lists the higher stretching frequenciesscaled with a factor of 0.9603 as for the B3LYP method forthe serotonin monomer. From Table 1.1, our results are in goodagreement with the experimental Fourier transform infrared(FTIR) spectrum.9,10 In serotonin monomer, alkyl chain CHstretch bands are predicted to lie between 2840 and 2950 cm-1,the aromatic CH stretches between 3020 and 3110 cm-1, andthe NH stretches associated with the amine group between 3340and 3420 cm-1. The OH and indole NH stretch are close toeach other, and the aromatic CH stretches are rather similar.
The calculated harmonic vibrational frequencies and intensi-ties of the fluoxetine monomers with both racemic forms at theB3LYP/cc-pVTZ level of theory are listed in Table 1.2 (cf theSupporting Information). From Table 1.2, in both forms offluoxetine, alkyl chain CH stretch bands lie between 2810 and
2980, the aromatic CH stretches between 3020 and 3110 cm-1,and the NH stretches associated with the ethylamine group near3380 ( 15 cm-1. Because of the lack of experimental evidence,we note the vibrational frequencies as a point of reference forthe future experimental work.
2. Serotonin-Fluoxetine Complex. 2.1. Geometry Param-eters.Sixdifferentstableconformationsfortheserotonin-fluoxetinecomplex have been investigated. According to the moleculardynamics (MD) simulation of serotonin and 5-HT2A receptor,the NH2 group of serotonin can form two strong hydrogen bondswithD155andS159of5-HT2Areceptor,N-H(serotonin) · · ·O(aspartate)and O-H(serine) · · ·N(serotonin),19,20 and it also reveals that theethylamine group of serotonin does not form hydrogen bondswith fluoxetine. Figures 1-6 show the six different stable
Figure 1. Conformer 1 of the serotonin-fluoxetine complex usingthe B3LYP/6-31+G* method. (a) S form of fluoxetine. (b) R form offluoxetine. Atom color: carbon (grey), hydrogen (white), oxygen (red),nitrogen (blue), and fluorine (cyan).
14530 J. Phys. Chem. B, Vol. 113, No. 43, 2009 Sahu et al.

conformations for the serotonin-fluoxetine complex at theB3LYP/6-31+G* level of theory. Table 1.3 (cf the SupportingInformation) shows the geometrical parameters of serotoninmonomer and serotonin in the serotonin-fluoxetine complexusing the B3LYP/6-31+G* method. Because of the chiralityof fluoxetine, the stable structures of serotonin in each form offluoxetine differ. The differences of the dihedral angles γC, γN,C47-C44-N42-H41, and C47-C44-N42-H43 in the six conform-ers are very similar, as compared to either the S or R forms offluoxetine. Table 1.4 (cf the Supporting Information) shows thegeometrical parameters of fluoxetine monomer and fluoxetinein the serotonin-fluoxetine complex using the B3LYP/6-31+G*method. Because of the chirality of fluoxetine, the dihedral
angles C32-C29-C16-O15, C32-C29-C16-O15, C32-C29-C16-C18, and C29-C16-O15-C3 have nearly opposite signs in the Sand R forms for all six conformers.
2.2. Hydrogen Bonding Parameters. Table 1 lists thehydrogen bonding parameters for all six different conformationsof the serotonin-fluoxetine complex at the B3LYP/6-31+G*level. It has been observed that the interaction of serotonin andfluoxetine are mainly due to hydrogen bonding, which followstwo different approaches. One is through the hydroxyl groupof serotonin (N · · ·H-O or F · · ·H-O) and the other throughthe indole NH (F · · ·H-N or N · · ·H-N). Both of these twokinds of hydrogen bond interactions are observed for conformers1-4, whereas only N · · ·H-N type interactions were observedfor conformers 5 and 6. In conformers 1 and 2, the hydrogen
Figure 2. Conformer 2 of the serotonin-fluoxetine complex usingthe B3LYP/6-31+G* method. (a) S form of fluoxetine. (b) R form offluoxetine. Atom color: carbon (grey), hydrogen (white), oxygen (red),nitrogen (blue), and fluorine (cyan).
Figure 3. Conformer 3 of the serotonin-fluoxetine complex usingthe B3LYP/6-31+G* method. (a) S form of fluoxetine. (b) R form offluoxetine. Atom color: carbon (grey), hydrogen (white), oxygen (red),nitrogen (blue), and fluorine (cyan).
Interaction of Serotonin and Fluoxetine J. Phys. Chem. B, Vol. 113, No. 43, 2009 14531

bond involved through the hydroxyl group of serotonin,N35 · · ·H65 (N · · ·H-O type) are less than 2.0 Å, and the bondangle ∠N35H65O64 are more linear as compared to thosehydrogen bond involved through the indole NH, F14 · · ·H56
(F · · ·H-N type). In conformer 3 and 4, it has been observedthat the hydrogen bonds involved through the indole NH,N35 · · ·H56 (N · · ·H-N type), are around 2.0 Å and the bondangles ∠N35H56N55 are more linear as compared to thosehydrogen bonds involved through the hydroxyl group ofserotonin, F14 · · ·H65 (F · · ·H-O type). In conformers 5 and 6,hydrogen bonds only involved through the indole NH, N35 · · ·H56
(N · · ·H-N type), are observed, the bond length N35 · · ·H56 isaround 2.0 Å, and the bond angles ∠N35H56N55 are all nearlylinear.
Rotational constants for the six stable conformers ofserotonin-fluoxetine complexes using B3LYP/6-31+G* are
listed in Table 1.5 (cf the Supporting Information) for futurereference and experimental support.
2.3. Energetics. Table 1.6 (cf the Supporting Information)shows the total energy for the six stable conformers ofserotonin-fluoxetine complexes calculated both at the gas andaqueous phases with the B3LYP/6-31+G* and MP2/6-31+G*//B3LYP/6-31+G* methods, using the COSMO approach. It isinteresting to note that for all these six different stableconformers, the stabilization energies are found to be similar(within 3 kcal/mol) both in the gas phase and even in theaqueous phase. However, the MP2 method is of course, well-
Figure 4. Conformer 4 of the serotonin-fluoxetine complex usingthe B3LYP/6-31+G* method. (a) S form of fluoxetine. (b) R form offluoxetine. Atom color: carbon (grey), hydrogen (white), oxygen (red),nitrogen (blue), and fluorine (cyan).
Figure 5. Conformer 5 of the serotonin-fluoxetine complex usingthe B3LYP/6-31+G* method. (a) S form of fluoxetine. (b) R form offluoxetine. Atom color: carbon (grey), hydrogen (white), oxygen (red),nitrogen (blue), and fluorine (cyan).
14532 J. Phys. Chem. B, Vol. 113, No. 43, 2009 Sahu et al.

known to account for the correlation effect and to better estimatethe energy values as compared to B3LYP method, as reflectedin the listed values.
Table 2 shows the gas phase binding energy for the six stableconformers of serotonin-fluoxetine complexes using B3LYP/6-31+G* and MP2/6-31+G*//B3LYP/6-31+G* methods. Thecalculated binding energy values for both the S and R forms ofeach conformer are found to be very similar, which supportsthe finding that eudismic ratio21 of fluoxetine enantiomers isnear unity. However, in the B3LYP/6-31+G* method, the orderof the stability of binding energies of the six conformers is 1 >2 > 5-6 > 3 > 4. From this stability ordering, it is clear thatthe nature of hydrogen bond interactions for each conformerdescribed in the earlier section is playing an important role,
whereas the conformational effect of the 3-phenyl group offluoxetine is stereo independent. For conformers 1 and 2, thetwo hydrogen bonds involved through the hydroxyl group ofserotonin, N35 · · ·H65 (N · · ·H-O type), and the other hydrogenbond involved through the indole NH, F14 · · ·H56 (F · · ·H-Ntype), are found to be associated with higher interaction ascompared to those of conformers 3 and 4, the hydrogen bondinvolved through the indole NH, N35 · · ·H56 (N · · ·H-N type),and the other hydrogen bond involved through hydroxyl groupof serotonin, F14 · · ·H65 (F · · ·H-O type). Though in conformers5 and 6, hydrogen bonds are only involved through the indoleNH, N35 · · ·H56 (N · · ·H-N type), the more linear hydrogen bondangles ∠N35H56N55 for conformers 5 and 6 provide evidence insupport of their higher stability ordering, as compared toconformers 3 and 4. To better estimate the binding energy valueswith correlation effect and dispersion contribution taken intoaccount, in the MP2/6-31+G*//B3LYP/6-31+G* method, the
Figure 6. Conformer 6 of the serotonin-fluoxetine complex usingthe B3LYP/6-31+G* method. (a) S form of fluoxetine. (b) R form offluoxetine. Atom color: carbon (grey), hydrogen (white), oxygen (red),nitrogen (blue), and fluorine (cyan).
TABLE 1: Hydrogen Bonding Parameters for the Six StableConformers of Serotonin-Fluoxetine Complexesa
conformersN35 · · ·H65s
O64
F14 · · ·H56sN55
N35 · · ·H56sN55
F14 · · ·H65sO64
1S form 1.90/155.7 3.05/137.2 -/- -/-R form 1.90/155.7 3.05/137.1 -/- -/-2S form 1.89/166.8 2.98/140.9 -/- -/-R form 1.89/167.7 3.01/140.8 -/- -/-3S form -/- -/- 2.15/154.1 2.46/150.7R form -/- -/- 2.15/154.2 2.45/150.84S form -/- -/- 2.11/166.0 2.40/153.3R form -/- -/- 2.10/167.2 2.44/152.45S form -/- -/- 2.02/178.2 -/-R form -/- -/- 2.02/178.1 -/-6S form -/- -/- 2.02/176.4 -/-R form -/- -/- 2.02/176.7 -/-a All the H-bond distances are in angstroms; the corresponding
bond angles are in degrees.
TABLE 2: Binding Energies for the Six Stable Conformersof Serotonin-Fluoxetine Complexesa
conformersB3LYP/
6-31+G* bMP2/6-31+G*//
B3LYP/6-31+G* b,dMP2/6-31+G*//
B3LYP/6-31+G* c,d
1S form -7.78 -21.94 (3.80) -11.50 (4.15)R form -7.78 -21.93 (3.80) -11.50 (4.15)2S form -7.49 -20.65 (5.19) -11.70 (6.14)R form -7.53 -20.88 (5.21) -11.82 (6.20)3S form -5.28 -19.94 (7.36) -8.21 (8.77)R form -5.28 -19.95 (7.36) -8.20 (8.78)4S form -4.97 -17.98 (8.24) -7.55 (9.88)R form -4.98 -17.91 (8.25) -7.54 (9.90)5S form -6.26 -11.81 (8.09) -7.02 (9.62)R form -6.27 -11.74 (8.09) -6.99 (9.62)6S form -6.28 -11.66 (7.85) -7.19 (9.15)R form -6.28 -11.58 (7.86) -7.14 (9.17)
a All the energy values are in kilocalories per mole. b Gas phase.c Aqueous phase. d The corresponding dipole moment values (inDebye) are given in parentheses.
Interaction of Serotonin and Fluoxetine J. Phys. Chem. B, Vol. 113, No. 43, 2009 14533
trend for the calculated binding energy for all six differentconformers is found to be 1 > 2 > 3 > 4 > 5 > 6 in contrast tothose obtained with B3LYP/6-31+G*. The trend reversal forthe binding energy values of conformers 3 and 4 is observeddue to the contribution of the electron correlation and dispersioneffect. However, the highest binding energy values are obtainedfor conformer 1 (21.94 kcal/mol for the S form and 21.93 kcal/mol for the R form).
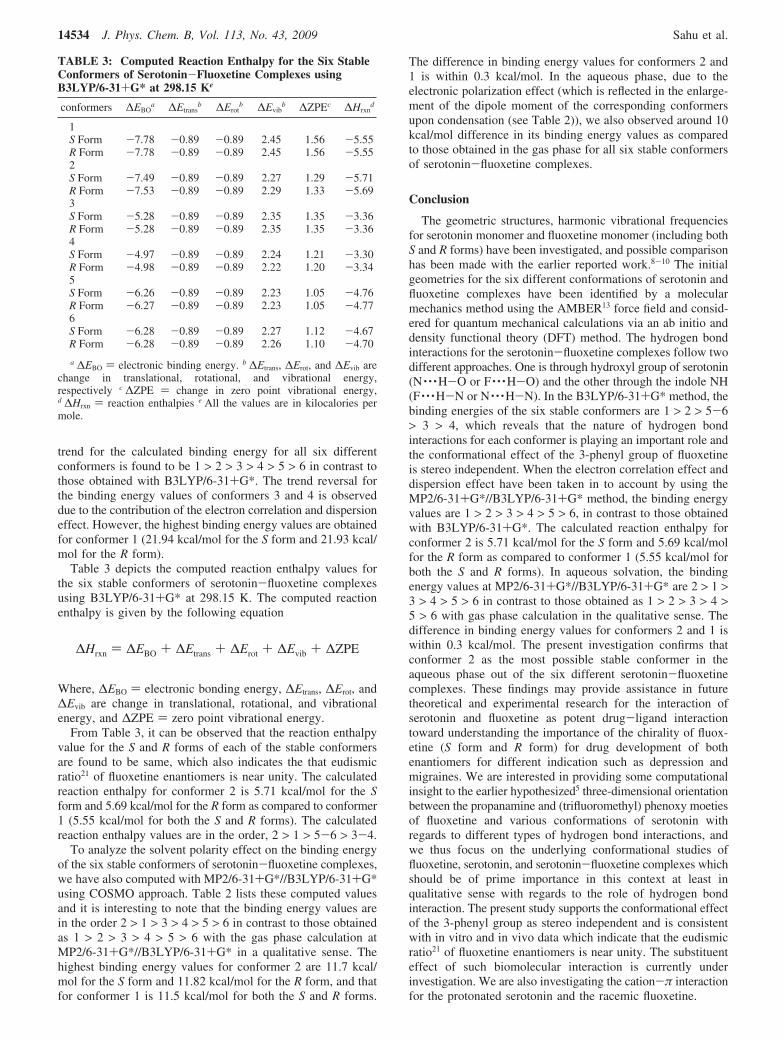
Table 3 depicts the computed reaction enthalpy values forthe six stable conformers of serotonin-fluoxetine complexesusing B3LYP/6-31+G* at 298.15 K. The computed reactionenthalpy is given by the following equation
Where, ∆EBO ) electronic bonding energy, ∆Etrans, ∆Erot, and∆Evib are change in translational, rotational, and vibrationalenergy, and ∆ZPE ) zero point vibrational energy.
From Table 3, it can be observed that the reaction enthalpyvalue for the S and R forms of each of the stable conformersare found to be same, which also indicates the that eudismicratio21 of fluoxetine enantiomers is near unity. The calculatedreaction enthalpy for conformer 2 is 5.71 kcal/mol for the Sform and 5.69 kcal/mol for the R form as compared to conformer1 (5.55 kcal/mol for both the S and R forms). The calculatedreaction enthalpy values are in the order, 2 > 1 > 5-6 > 3-4.
To analyze the solvent polarity effect on the binding energyof the six stable conformers of serotonin-fluoxetine complexes,we have also computed with MP2/6-31+G*//B3LYP/6-31+G*using COSMO approach. Table 2 lists these computed valuesand it is interesting to note that the binding energy values arein the order 2 > 1 > 3 > 4 > 5 > 6 in contrast to those obtainedas 1 > 2 > 3 > 4 > 5 > 6 with the gas phase calculation atMP2/6-31+G*//B3LYP/6-31+G* in a qualitative sense. Thehighest binding energy values for conformer 2 are 11.7 kcal/mol for the S form and 11.82 kcal/mol for the R form, and thatfor conformer 1 is 11.5 kcal/mol for both the S and R forms.
The difference in binding energy values for conformers 2 and1 is within 0.3 kcal/mol. In the aqueous phase, due to theelectronic polarization effect (which is reflected in the enlarge-ment of the dipole moment of the corresponding conformersupon condensation (see Table 2)), we also observed around 10kcal/mol difference in its binding energy values as comparedto those obtained in the gas phase for all six stable conformersof serotonin-fluoxetine complexes.
Conclusion
The geometric structures, harmonic vibrational frequenciesfor serotonin monomer and fluoxetine monomer (including bothS and R forms) have been investigated, and possible comparisonhas been made with the earlier reported work.8-10 The initialgeometries for the six different conformations of serotonin andfluoxetine complexes have been identified by a molecularmechanics method using the AMBER13 force field and consid-ered for quantum mechanical calculations via an ab initio anddensity functional theory (DFT) method. The hydrogen bondinteractions for the serotonin-fluoxetine complexes follow twodifferent approaches. One is through hydroxyl group of serotonin(N · · ·H-O or F · · ·H-O) and the other through the indole NH(F · · ·H-N or N · · ·H-N). In the B3LYP/6-31+G* method, thebinding energies of the six stable conformers are 1 > 2 > 5-6> 3 > 4, which reveals that the nature of hydrogen bondinteractions for each conformer is playing an important role andthe conformational effect of the 3-phenyl group of fluoxetineis stereo independent. When the electron correlation effect anddispersion effect have been taken in to account by using theMP2/6-31+G*//B3LYP/6-31+G* method, the binding energyvalues are 1 > 2 > 3 > 4 > 5 > 6, in contrast to those obtainedwith B3LYP/6-31+G*. The calculated reaction enthalpy forconformer 2 is 5.71 kcal/mol for the S form and 5.69 kcal/molfor the R form as compared to conformer 1 (5.55 kcal/mol forboth the S and R forms). In aqueous solvation, the bindingenergy values at MP2/6-31+G*//B3LYP/6-31+G* are 2 > 1 >3 > 4 > 5 > 6 in contrast to those obtained as 1 > 2 > 3 > 4 >5 > 6 with gas phase calculation in the qualitative sense. Thedifference in binding energy values for conformers 2 and 1 iswithin 0.3 kcal/mol. The present investigation confirms thatconformer 2 as the most possible stable conformer in theaqueous phase out of the six different serotonin-fluoxetinecomplexes. These findings may provide assistance in futuretheoretical and experimental research for the interaction ofserotonin and fluoxetine as potent drug-ligand interactiontoward understanding the importance of the chirality of fluox-etine (S form and R form) for drug development of bothenantiomers for different indication such as depression andmigraines. We are interested in providing some computationalinsight to the earlier hypothesized5 three-dimensional orientationbetween the propanamine and (trifluoromethyl) phenoxy moetiesof fluoxetine and various conformations of serotonin withregards to different types of hydrogen bond interactions, andwe thus focus on the underlying conformational studies offluoxetine, serotonin, and serotonin-fluoxetine complexes whichshould be of prime importance in this context at least inqualitative sense with regards to the role of hydrogen bondinteraction. The present study supports the conformational effectof the 3-phenyl group as stereo independent and is consistentwith in vitro and in vivo data which indicate that the eudismicratio21 of fluoxetine enantiomers is near unity. The substituenteffect of such biomolecular interaction is currently underinvestigation. We are also investigating the cation-π interactionfor the protonated serotonin and the racemic fluoxetine.
TABLE 3: Computed Reaction Enthalpy for the Six StableConformers of Serotonin-Fluoxetine Complexes usingB3LYP/6-31+G* at 298.15 Ke
conformers ∆EBOa ∆Etrans
b ∆Erotb ∆Evib
b ∆ZPEc ∆Hrxnd
1S Form -7.78 -0.89 -0.89 2.45 1.56 -5.55R Form -7.78 -0.89 -0.89 2.45 1.56 -5.552S Form -7.49 -0.89 -0.89 2.27 1.29 -5.71R Form -7.53 -0.89 -0.89 2.29 1.33 -5.693S Form -5.28 -0.89 -0.89 2.35 1.35 -3.36R Form -5.28 -0.89 -0.89 2.35 1.35 -3.364S Form -4.97 -0.89 -0.89 2.24 1.21 -3.30R Form -4.98 -0.89 -0.89 2.22 1.20 -3.345S Form -6.26 -0.89 -0.89 2.23 1.05 -4.76R Form -6.27 -0.89 -0.89 2.23 1.05 -4.776S Form -6.28 -0.89 -0.89 2.27 1.12 -4.67R Form -6.28 -0.89 -0.89 2.26 1.10 -4.70
a ∆EBO ) electronic binding energy. b ∆Etrans, ∆Erot, and ∆Evib arechange in translational, rotational, and vibrational energy,respectively c ∆ZPE ) change in zero point vibrational energy,d ∆Hrxn ) reaction enthalpies e All the values are in kilocalories permole.
∆Hrxn ) ∆EBO + ∆Etrans + ∆Erot + ∆Evib + ∆ZPE
14534 J. Phys. Chem. B, Vol. 113, No. 43, 2009 Sahu et al.

Acknowledgment. This research is supported by the NationalScience Council (NSC) of Taiwan and the computationalresource is partially supported by National Center for High-Performance Computing (NCHC), Hsin-Chu, Taiwan.
Supporting Information Available: Figures 1.1-1.3 andTables 1.1-1.6. This material is available free of charge viaInternet at http://pubs.acs.org.
References and Notes
(1) Tucker, G. T. Lancet 2000, 355, 1085–1087.(2) Ther. Lett. 2002; 45.(3) Neal, M. J. Medical Pharmacology at a Glance; Blackwell
Publishing: New York, 2005.(4) Medicinal Chemistry and Drug DiscoVery; Burger, A., Ed.; John
Wiley & Sons, Inc.: New York, 2006.(5) (a) Wong, D. T.; Horng, J. S.; Bymaster, F. P.; Hauser, K. L.;
Molloy, B. B. Life Sci. 1974, 15, 471. (b) Wong, D. T.; Bymaster, F. P.;Horng, J. S.; Molloy, B. B. J. Pharmacol. Exp. Ther. 1975, 193, 804. (c)Robertson, D. W.; Jones, N. D.; Swartzendruber, J. K.; Yang, K. S.; Wong,D. T. J. Med. Chem. 1988, 31, 185–189.
(6) Wong, D. T.; Bymaster, F. P.; Reid, L. R.; Fuller, R. W.; Perry,K. W. Drug DeV. Res. 1985, 6, 397.
(7) Fuller, R. W.; Snoddy, H. D. Pharmacol., Biochem. BehaV. 1986,24, 281.
(8) (a) Alagona, G.; Ghio, C.; Nagy, P. I. J. Chem. Theory Comput.2005, 1, 801–806. (b) Pratuangdejkul, J.; Jaudon, P.; Ducrocq, C.;Nosoongnoen, W.; Guerin, G.; Conti, M.; Loric, S.; Launay, J.; Manivet,P. J. Chem. Theory Comput. 2006, 2, 746–760. (c) Pisterzi, L. F.; Almeida,D. R. P.; Chass, G. A.; Torday, L. L.; Papp, J. G.; Varro, A.; Csizmadia,I. G. Chem. Phys. Lett. 2002, 365, 542–551. (d) Alagona, G.; Ghio, C. J.Mol. Structure: THEOCHEM 2006, 769, 123–134.
(9) van Mourik, T.; Emson, L. E. V. Phys. Chem. Chem. Phys. 2002,4, 5863–5871. Bayari, S.; Saglam, S.; Ustundag, H. F. J. Mol. Strcuture:THEOCHEM 2005, 726, 225–232 (the FTIR spectrum recorded with theKBr technique was obtained from Patir’s result).
(10) LeGreve, T. A.; Baquero, E. E.; Zwier, T. S. J. Am. Chem. Soc.2007, 129, 4028–4038.
(11) Cool, D. R.; Liebach, F. H.; Ganapathy, V. Biochem. Pharmacol.1990, 40, 2161–2167.
(12) Backstrom, I.; Bergstrom, M.; Marcusson, J. Brain Res. 1989, 486,261–268.
(13) Cornell, W. D.; Cieplak, P.; Bayly, C. I.; Gould, I. R.; Merz, K. M.,Jr.; Ferguson, D. M.; Spellmeyer, D. C.; Fox, T.; Caldwell, J. W.; Kollman,P. A. J. Am. Chem. Soc. 1995, 117, 5179–5197.
(14) Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652.(15) (a) Head-Gordon, M.; Pople, J. A.; Frisch, M. J. Chem. Phys. Lett.
1988, 153, 503. (b) Frisch, M. J.; Head-Gordon, M.; Pople, J. A. Chem.Phys. Lett. 1990, 166, 275.
(16) (a) Dunning, T. H., Jr. J. Chem. Phys. 1989, 90, 1007–1023. (b)Davidson, E. R. Chem. Phys. Lett. 1996, 260, 514–518.
(17) Barone, V.; Cossi, M. J. Phys. Chem. A 1998, 102, 1995.(18) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb,
M. A.; Cheeseman, J. R. ; Montgomery J. A., Jr.; Vreven, T.; Kudin, K. N.;Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.;Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.;Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.;Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li,X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.;Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.;Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.;Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich,S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.;Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.;Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz,P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al.Laham, M. A.;Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson,B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian 03,revision C.02; Gaussian, Inc.: Wallingford, CT, 2004.
(19) Weinstein, H.; Zhang, D. Receptor models and ligand-inducedresponses: new insights for structure-activity relations. In QSAR andMolecular Modeling: Concepts; Sanz, F., Giraldo, J., Manaut, F., Eds.; ProusScience publishers: Barcelona, 1995; pp 497-507.
(20) Roth, B. L.; Willins, D. L.; Kristiansen, K.; Kroeze, W. K.Pharmacol. Ther. 1998, 79, 231–257.
(21) Lehmann, P. A. Trends Pharmacol. Sci. 1982, 3, 103 (eudismicratio ) ratio of affinities of two enantiomers).
JP907151N
Interaction of Serotonin and Fluoxetine J. Phys. Chem. B, Vol. 113, No. 43, 2009 14535




















