
Protective Role of Nutritional Plants Containing Flavonoids in ...
Upload
independentCategory
view
0download
0
Toxicology 217 (2006) 194–205
Inhibition of human cytochrome CYP1 enzymesby flavonoids of St. John’s wort
Amit Chaudhary, Kristine L. Willett∗
Department of Pharmacology and Environmental Toxicology Research Program, School of Pharmacy,University of Mississippi, 315 Faser Hall, Box 1848, University, MS 38677, USA
Received 2 July 2005; received in revised form 24 August 2005; accepted 22 September 2005Available online 4 November 2005
Abstract
CYP1B1 is involved in metabolizing both polycyclic aromatic hydrocarbons and estradiol to potentially carcinogenic interme-diates, and it is also over-expressed in human cancer cells. In order to investigate whether flavonoids could specifically inhibitCYP1B1, seven flavonoids in St. John’s wort and apigenin were screened for their inhibition of recombinant human CYP1B1 andCYP1A1. While seven flavonoids (myricetin, apigenin, kaempferol, quercetin, amentoflavone, quercitrin and rutin) were slightlymore selective for CYP1B1 EROD inhibition (Kis 0.06–5.96�M) compared to CYP1A1 (Kis 0.20–1.6�M) the difference inKisfor the P450s were not significantly different. Rutin did not inhibit CYP1A1 at concentrations up to 10�M. Kinetic analyses deter-mined that apigenin and amentoflavone were competitive inhibitors of CYP1B1, while quercetin showed mixed type inhibition. Tocharacterize the inhibition potential of these flavonoids, five were studied further for their ability to inhibit TCDD-induced EROD
1 mRNA.in (ICasnzymes
ndpolarfam-1.is
ereashetic
adiol
has
activity in 22Rv1 human prostate cancer cells. 22Rv1 cells express constitutive and TCDD-inducible CYP1A1 and CYP1BIn the cells, the IC50s were similar to those measured for the recombinant CYP1A1 except for amentoflavone. Quercet50:4.1�M), kaempferol (3.8�M), myricetin (3.0�M) and apigenin (3.1�M) caused significant inhibition of EROD activity whereamentoflavone did not cause inhibition. Depending on their bioavailability, flavonoids that can selectively inhibit CYP1 emay be useful as chemoprotective agents in prostate cancer prevention.© 2005 Elsevier Ireland Ltd. All rights reserved.
Keywords: CYP1A1; CYP1B1; Chemoprevention; prostate
1. Introduction
Our research focus is the potential for bioflavonoids toact as therapeutic and/or preventative agents in prostatecancer via inhibition of the CYP1 cytochrome P450enzymes, which have been implicated in carcinogenesis(Chun and Kim, 2003; McFadyen et al., 2004). The P450
Abbreviations: EROD, ethoxyresorufin-O-deethylase; BaP,benzo(a)pyrene; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin
∗ Corresponding author. Tel.: +1 662 915 6691;fax: +1 662 915 5148.
E-mail address: [email protected] (K.L. Willett).
superfamily of genes is involved in the oxidation aexcretion of both endogenous and exogenous noncompounds in the body. The human CYP1 geneily is comprised of CYP1A1, CYP1A2, and CYP1BCYP1A2 is expressed primarily in liver, CYP1A1expressed in both liver and extrahepatic tissues, whCYP1B1 is primarily expressed extrahepatically. TCYP1s are involved in bioactivating polycyclic aromahydrocarbons (PAHs), heterocyclic amines, and estrto mutagenic and carcinogenic intermediates (Murrayet al., 2001). In the prostate, CYP1B1 messagebeen detected in normal (Finnstrom et al., 2001) andbenign prostatic hyperplasia (Luo et al., 2002) and is
0300-483X/$ – see front matter © 2005 Elsevier Ireland Ltd. All rights reserved.doi:10.1016/j.tox.2005.09.010

A. Chaudhary, K.L. Willett / Toxicology 217 (2006) 194–205 195
up-regulated in malignant prostate tissue (Chaib et al.,2001; Carnell et al., 2004). In a Japanese population aCYP1B1 polymorphism at codon 119 has been associ-ated with prostatic carcinogenesis (Tanaka et al., 2002).This polymorphism has also been associated with thehighest catalytic bioactivation of estradiol (Hanna et al.,2000). More recently,Chang et al. (2003)identified afrequent haplotype in CYP1B1 that was associated withan increased risk for prostate cancer. Together these stud-ies, implicate CYP1B1 as a potential therapeutic targetfor reducing development of prostate cancer.
Inhibiting CYP1B1 selectively could be consideredchemoprotective for several reasons. First,Shimadaet al. (1999)found that recombinant human CYP1B1was more active than CYP1A1 in metabolizingbenzo(a)pyrene (BaP) to the proximate toxicant BaP-7,8-diol. In addition, human CYP1B1 is an estra-diol hydroxylase, acting primarily at the C-4 position.CYP1A1, in contrast, has activity at the C-2, C-6� andC-15� positions of estradiol (Hayes et al., 1996). Inrodent tissues, including the Noble rat prostate, estro-gen induces tumors when estradiol is primarily convertedto the 4-hydroxyestradiol metabolite, yet tumors fail todevelop where 2-hydroxylation predominates (Cavalieriet al., 2002). Lastly, CYP1B1 can detoxify cancer drugs,such as flutamide and possibly docetaxel, which reducestheir cytotoxic potential and effectiveness (Rochat et al.,2001; McFadyen et al., 2001; Bournique and Lemarie,2002). This is particularly significant because CYP1B1is over-expressed in a variety of human tumors comparedtT thata c ifi sa easeC usep andd larlya .,2
lp y form r ofc lud-i pro-c nyl-p unds( ft roleo tinga vi-d 3A,
one of the most important human drug metabolizingenzymes and the cause of adverse drug reactions in somepatients (Vogel, 2001). Other commercially available St.John’s wort extracts and individual components havebeen investigated for their effects on CYP1A1, CYP2D6,CYP2C9, CYP3A4, CYP1A2 and CYP2C19 (Obach,2000). Prior to our work, little has been reported aboutthe effects of flavonoids found in St. John’s wort onCYP1B1 specifically.
In this present study, we characterized eightflavonoids: quercetin, amentoflavone, kaempferol,myricetin, quercitrin, isoquercetin, rutin, and apigenin(Fig. 1) for their inhibition of recombinant CYP1B1and CYP1A1. Apigenin, while not a component of St.John’s wort, is a monomer of amentoflavone and wasincluded in these experiments to determine if the effectsof amentoflavone would reflect its degradation intomonomers. While none of the flavonoids were signif-icantly selective for CYP1B1 compared to CYP1A1,apigenin especially was a relatively potent CYP1B1inhibitor. In addition, we showed that myricetin,apigenin, kaempferol, and quercetin can inhibit ERODactivity, a conventional assay for assessment of CYP1enzyme activity, in the 22Rv1 human prostate cancer cellline. By distinguishing the relative roles of cytochromeP450s (specifically CYP1s) in human prostate cancercell lines, new therapies or chemoprotective strategiesagainst prostate cancer formation may emerge.
2. Methods
taseed-
etinna,
%),rateockthylThe-, fluo-O,7,8-y
antfor
ined
S
o adjacent noncancerous cells (McFadyen et al., 2001).herefore, the hypothesis guiding this study wasbioflavonoid could be potentially anti-carcinogeni
t could inhibit CYP1B1’s ability to metabolize PAHnd estrogen to toxic intermediates and/or decrYP1B1’s ability to detoxify cancer drugs. Becarostate cancer typically has a long latency periodevelops in older men, this disease may be particumenable to chemopreventive approaches (Saleem et al003).
St. John’s wort (Hypericium perforatum) is an herbaroduct widely used as an over the counter remedild to moderate depression. It contains a numbe
lasses of pharmacologically active compounds incng napthodianthrones, flavonoids, phloroglucinols,yanidins, tannins, essential oils, amino acids, pheropanes, xanthones and other water-soluble compoGreeson et al., 2001). It is well known that some ohe flavonoids in St. John’s wort exert a modulatingn the metabolism of drugs and xenobiotics by acs inducers or inhibitors of cytochrome P450s. Eence is prevalent that St. John’s wort induces CYP
Recombinant human CYP1A1 and CYP1B1 + reducSUPERSOMES were obtained from BD Biosciences (Bford, MA). Amentoflavone (purity 99.83%) and isoquerc(99%) were obtained from Chromadex, Inc. (Santa ACA). Quercetin (98%), myricetin (85%), kaempferol (90quercitrin dihydrate (85%), apigenin (95%) and rutin hyd(95%) were obtained from Sigma (St. Louis, MO). Stsolutions (0.1 mM) of flavonoids were prepared in dimesulfoxide (Sigma) and stored at room temperature.ethoxyresorufin (Sigma) solution for ethoxyresorufinO-deethylase assay was prepared in methanol. Resorufinrescamine, NADPH, NADH, bovine serum albumin, MgS4and other chemicals were obtained from Sigma. 2,3Tetrachlorodibenzo-p-dioxin (TCDD) was kindly provided bDr. Stephen Safe, Texas A&M University.
2.1. Recombinant CYP1 EROD assay
To investigate the effect of flavonoids on recombinCYP1A1 and CYP1B1, the EROD assay was adapted96-well plates (white, Costar). The standard curve contaresorufin (500, 200, 100, 50, 20, 15, 10 pmol), 15�g bovineserum albumin, 2�l of DMSO and cofactor solution of HEPE
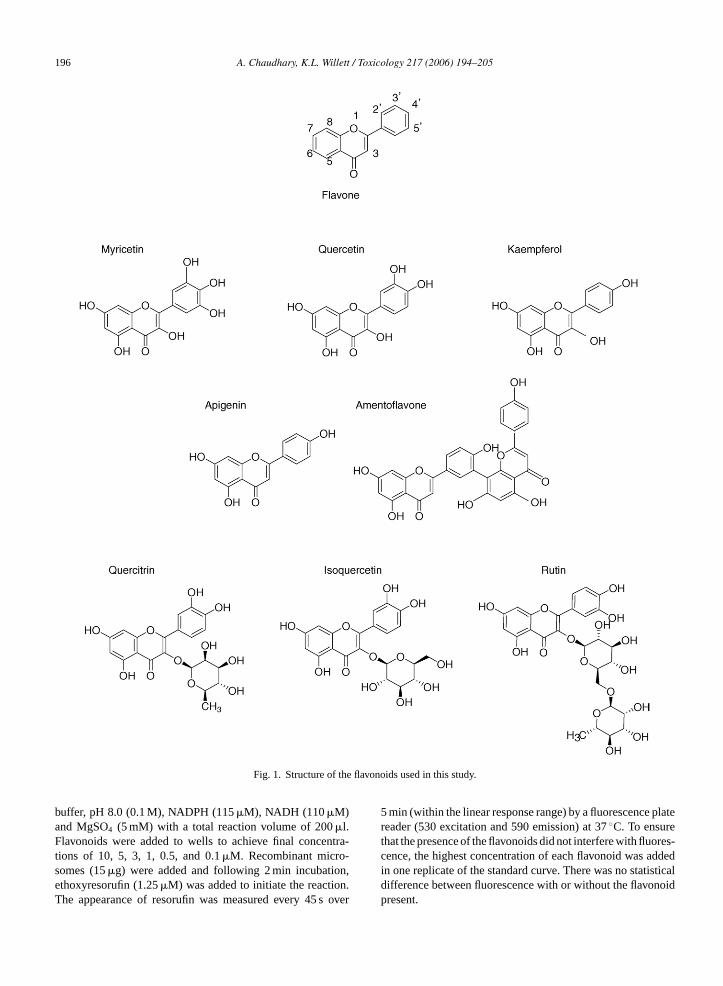
196 A. Chaudhary, K.L. Willett / Toxicology 217 (2006) 194–205
Fig. 1. Structure of the flavonoids used in this study.
buffer, pH 8.0 (0.1 M), NADPH (115�M), NADH (110�M)and MgSO4 (5 mM) with a total reaction volume of 200�l.Flavonoids were added to wells to achieve final concentra-tions of 10, 5, 3, 1, 0.5, and 0.1�M. Recombinant micro-somes (15�g) were added and following 2 min incubation,ethoxyresorufin (1.25�M) was added to initiate the reaction.The appearance of resorufin was measured every 45 s over
5 min (within the linear response range) by a fluorescence platereader (530 excitation and 590 emission) at 37◦C. To ensurethat the presence of the flavonoids did not interfere with fluores-cence, the highest concentration of each flavonoid was addedin one replicate of the standard curve. There was no statisticaldifference between fluorescence with or without the flavonoidpresent.

A. Chaudhary, K.L. Willett / Toxicology 217 (2006) 194–205 197
2.2. Inhibition kinetics
The flavonoid inhibition kinetics of CYP1B1-mediatedEROD activity was determined with methods similar tothose described above with slight modifications. Recombinantmicrosomes (15�g) were incubated in buffer with DMSOor one of the two concentrations of flavonoid, which wereselected to be in the vicinity of IC50. Quercetin (0.5 and 1�M),apigenin (0.1 and 0.5�M) and amentoflavone (1 and 3�M)were investigated. Following 2 min incubation, increasing con-centrations of ethoxyresorufin (substrate = 0.1, 0.2, 0.3, 0.4,0.6, 1.0, 2.0�M) were added to initiate the reaction. After7–20 min, 100�l of methanol was added to stop the reaction.Plates were read in fluorescence plate reader at 530 nm/590 nmfor resorufin production.
2.3. Data analyses and statistics
GraphPad Prism (San Diego, CA) was used for statisti-cal analysis of data. The nonlinear regression curve fit withsigmoidal dose response (variable slope) function was usedto calculate IC50 and EROD inhibition for the CYP1A1and CYP1B1 recombinant microsome experiments.Ki val-ues were calculated according to the Cheng–Prusoff equation:Ki = IC50/(1 + [S]/Km), where [S] is the substrate concentra-tion (1.25�M) and Km (affinity constant) was calculated forCYP1A1 (0.054�M) and CYP1B1 (0.35�M) by GraphPadPrism 4.Ki values for each compound were statistically com-pared by Student’st-test a significance level ofp < 0.05.
For the inhibition kinetics studies,Vmax and Km valueswere determined by the nonlinear regression curve fit usingthe Michaelis–Menten equation. TheKis were calculated withG(w ancel wasu
2
an-a ed)s 5%a ereg x-i y 4–5d
2(
P1a ctedi ately1 red f
incubation, media was removed and new media (500�l) wasadded to each well. Flavonoids were added in triplicate toachieve final concentration of 0.5, 1.5, 2.5 and 5�M and eachexperiment was repeated on three different plates. After 30 minof incubation with flavonoid, media was removed from thecells, and rinsed with 100�l phosphate buffered saline (PBS),then an additional 185�l of PBS was added to each well. Aprotein standard curve was prepared by adding increasing con-centrations of BSA (stock, 2 mg/ml) and resorufin in triplicatein the first three columns of each plate (final concentrationrange 20–160�g/well and 7.5–75 pmol/well, respectively).The plate was incubated at 37◦C, and the reaction wasinitiated by addition of ethoxyresorufin (final concentration1.25�M). The reaction was stopped after 15 min by the adding100�l of fluorescamine. Plates were read in fluorescent platereader (HTS 7000 bioassay plate reader, Perkin-Elmer,Norwalk, CT) at 530 nm/590 nm for resorufin productionand 405 nm/535 nm for fluorescamine protein determinationas described byWillett et al. (1998). To calculate IC50s ofEROD inhibition in the prostate cancer cell experiments datawas probit transformed prior to linear regression and IC50
calculation.
2.6. Quantitative reverse transcription real time PCR forCYP1A1 and CYP1B1
Cells were seeded in six-well plates with 5× 106 cells perwell in 2 ml of media. After 24 h, the media was changed,and the cells were exposed to 0.001% DMSO or TCDD(10 nM). After 6 h, the media was removed and the wellswere rinsed with 2 ml of PBS. The total RNA was extractedusing RNeasy® Plant Mini Kit (Qiagen, Valencia, CA)according to the manufacturer’s protocol. The RNA quality
lyseralong
ntsers,NTP
1,nedI1B1
nds forCRons
to
ingmedem.inP1A1
raphPad using the equation for competitive inhibitionv =Vmax[S])/(Km(1 + [I]/Ki ) + [S]). Global one-way ANOVAsere used to determine treatment effects with a signific
evel of p < 0.05. The Student–Newman–Keuls methodsed for pair-wise multiple comparisons.
.4. Cell and culture conditions
The human prostate cancer cell line, 22Rv1 (ATCC, Mssas, VA), was cultured in RPMI-1640 medium (modifiupplemented with 10% (v/v) fetal bovine serum andntibiotics as suggested by ATCC. Stock culture cells wrown in 75 cm2 flasks at 37◦C in a humidified air/carbon dio
de (95%/5%) atmosphere and subcultured (1:5 ratio) everays using 0.25% trypsin and 0.05% EDTA (ATCC).
.5. 22Rv1 prostate cell ethoxyresorufin-O-deethylaseEROD) assay
In order to determine the effects of flavonoids on CYctivity in the 22Rv1 cells, the EROD assay was condu
n 48-well plates. Plates were seeded with approxim05 cells per well in 500�l media. After 24 h, the cells weosed with 0.01�M TCDD in DMSO (0.1%). After 24 h o
and concentration was determined using Agilent Bioana2100 (RNA 6000 Nano Assay; Agilent Technologies, PAlto, CA). The synthesis of cDNA was conducted with 250total RNA using Taqman® Reverse Transcription Reage(Applied Biosystems, Foster City, CA): random hexammultiscribe reverse transcriptase, RNase inhibitor, deoxymix, 25 mM MgCl2, 10× RT buffer in a 25�l reaction. Thetemperature program used was 25◦C for 10 min, 37◦C for60 min and 95◦C for 5 min. Transcript abundance of CYP1ACYP1B1 and�-actin (endogenous control) was determiby quantitative real time RT-PCR based on SYBR® Greenassay. Gene specific primers (100 nM) for CYP1A1, CYPand�-actin were previously published (Lin et al., 2003). ThecDNA corresponding to 10 ng for CYP1A1, CYP1B1 a�-actin of reverse-transcribed RNA served as templateeach of triplicate 25�l PCR reactions using SYBR Green PMaster Mix (Applied Biosystems). The PCR amplificatiusing universal thermal cycling conditions accordingthe manufacturer (10 min hold at 95◦C followed by 40cycles of denaturing (95◦C/15 s) and annealing/extend(60◦C/1 min)) and fluorescence detection were perforwith the GeneAmp® 5700 Sequence Detection SystCYP1A1, CYP1B1 and�-actin were amplified in triplicateseparate reactions on the same plate. The amount of CY
198 A. Chaudhary, K.L. Willett / Toxicology 217 (2006) 194–205
and CYP1B1 mRNA, normalized to�-actin were given by theformula 2−��CT , whereCT was the threshold cycle indicatingthe fractional cycle number at which the amount of amplifiedCYP1A1 or CYP1B1 reached a threshold, fixed at 0.5 in theseexperiments. The�CT value was determined by subtractingthe average�-actin CT-value from the average CYP1A1 orCYP1B1CT-value. Then, the calculation of��CT involvedsubtraction of the�CT-value of the calibrator (in our case thecalibrator was average�CT-value of DMSO treated cells) fromthe �CT-value of each sample (ABI PRISM 7700 SequenceDetection System User’s Manual). Accordingly, CYP1A1and CYP1B1 mRNA levels were reported as fold change inabundance relative to DMSO control. Statistical differencesbetween the control and treated samples were determinedusing one-way ANOVA followed by Student–Newman–Keulspost hoc (p < 0.05) using GraphPad Prism.
3. Results
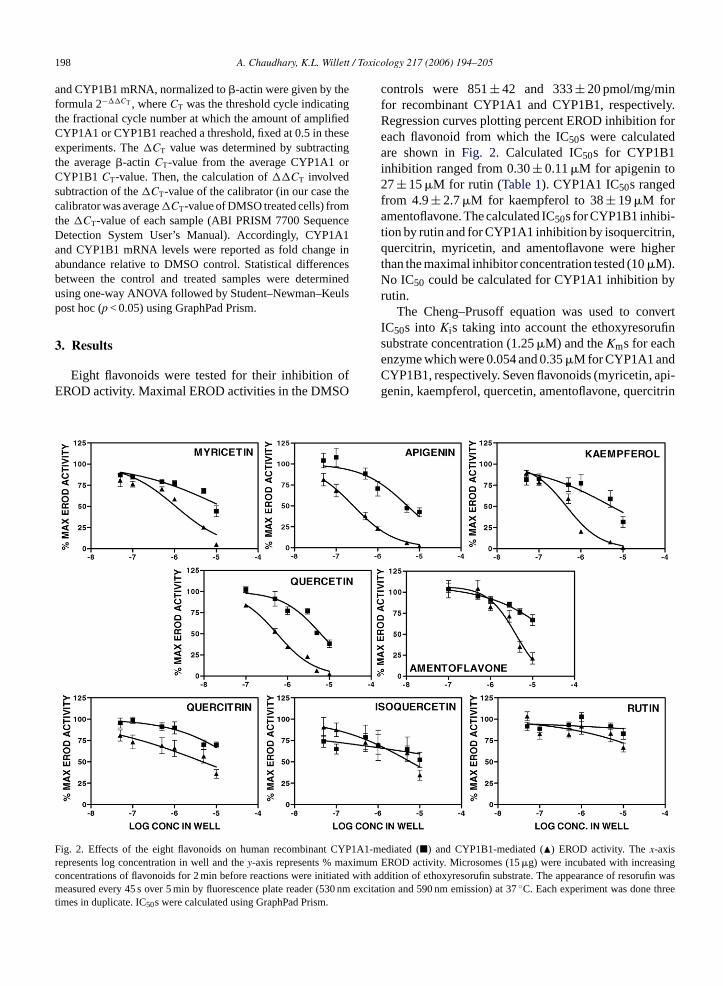
Eight flavonoids were tested for their inhibition ofEROD activity. Maximal EROD activities in the DMSO
controls were 851± 42 and 333± 20 pmol/mg/minfor recombinant CYP1A1 and CYP1B1, respectively.Regression curves plotting percent EROD inhibition foreach flavonoid from which the IC50s were calculatedare shown inFig. 2. Calculated IC50s for CYP1B1inhibition ranged from 0.30± 0.11�M for apigenin to27± 15�M for rutin (Table 1). CYP1A1 IC50s rangedfrom 4.9± 2.7�M for kaempferol to 38± 19�M foramentoflavone. The calculated IC50s for CYP1B1 inhibi-tion by rutin and for CYP1A1 inhibition by isoquercitrin,quercitrin, myricetin, and amentoflavone were higherthan the maximal inhibitor concentration tested (10�M).No IC50 could be calculated for CYP1A1 inhibition byrutin.
The Cheng–Prusoff equation was used to convertIC50s into Kis taking into account the ethoxyresorufinsubstrate concentration (1.25�M) and theKms for eachenzyme which were 0.054 and 0.35�M for CYP1A1 andCYP1B1, respectively. Seven flavonoids (myricetin, api-genin, kaempferol, quercetin, amentoflavone, quercitrin
CYP1Aaximu ng
Fig. 2. Effects of the eight flavonoids on human recombinantrepresents log concentration in well and they-axis represents % m
concentrations of flavonoids for 2 min before reactions were initiated wmeasured every 45 s over 5 min by fluorescence plate reader (530 nmtimes in duplicate. IC50s were calculated using GraphPad Prism.1-mediated (�) and CYP1B1-mediated (�) EROD activity. Thex-axism EROD activity. Microsomes (15�g) were incubated with increasi
ith addition of ethoxyresorufin substrate. The appearance of resorufin wasexcitation and 590 nm emission) at 37◦C. Each experiment was done three
A. Chaudhary, K.L. Willett / Toxicology 217 (2006) 194–205 199
Table 1IC50 andKi values (±S.E.) and ratio of CYP1A1 to CYP1B1Ki for flavonoid-mediated inhibition of EROD activity using recombinant humanCYP1B1 and CYP1A1 enzymes
CYP1A1 CYP1B1 Ki (CYP1A1:CYP1B1)
IC50 (�M) Ki (�M) IC50 (�M) Ki (�M)
Myricetin 34 ± 26 1.4± 1.1 1.1± 0.27 0.23± 0.06 6.2Apigenin 5.1± 1.8 0.21± 0.02 0.30± 0.11 0.06± 0.02 3.5Kaempferol 4.9± 2.7 0.20± 0.11 0.48± 0.12 0.10± 0.02 2.0Quercetin 6.0± 0.88 0.25± 0.04 0.55± 0.10 0.12± 0.02 2.1Amentoflavone 38± 19 1.6± 0.78 4.6± 1.4 0.99± 0.31 1.6Quercitrin 30± 6.2 1.2± 0.26 4.7± 3.2 1.0± 0.69 1.2Isoquercetin 14± 7.1 0.58± 0.30 4.1± 2.0 0.89± 0.43 0.7Rutin N.I. N.I. 27± 15 6.0± 3.20* –
Ki values were calculated according to the Cheng–Prusoff equation:Ki = IC50/(1 + [S]/Km), where [S] is the substrate concentration (1.25�M) andKm (affinity constant) was calculated for CYP1A1 (0.054�M) and CYP1B1 (0.35�M) by GraphPad Prism 4. N.I. indicates no inhibition.
* TheKi for rutin is statistically different than all otherKi s (p < 0.05, ANOVA,n = 3).
and rutin) were more selective for CYP1B1 ERODinhibition compared to CYP1A1 (Table 1). While ourgoal was to identify flavonoids that were more selectivefor CYP1B1 inhibition, none of theKis for CYP1B1were significantly lower than those for CYP1A1. Of theflavonoids that inhibited both CYP1B1 and CYP1A1to some extent, myricetin had the highest calculatedselectivity (6.2-fold) for CYP1B1 compared to CYP1A1inhibition. Isoquercetin was slightly more selective forCYP1A1 inhibition. TheKi of CYP1B1 inhibition byrutin was significantly higher than theKis of inhibitionfor all the other flavonoids with either enzyme (ANOVA,p < 0.05).
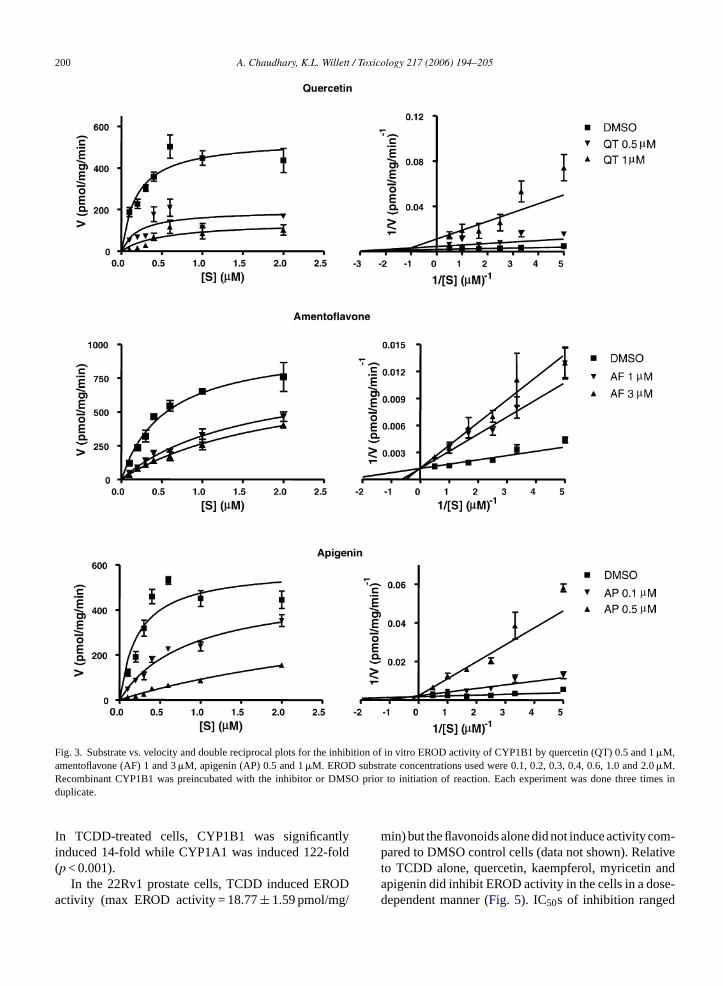
In order to investigate the mechanism of CYP1B1inhibition kinetics, quercetin, amentoflavone and api-genin were selected for further study because quercetinis the flavonoid typically found in the highest concentra-tions in both St. John’s wort and certain foods (especiallyonions and apples), amentoflavone inhibited CYP1B1but did not cause inhibition of CYP1A1 at the concentra-tions tested, and apigenin was the most potent CYP1B1inhibitor. EROD activities were determined with sub-strate concentrations ranging from 0.1 to 2.0�M.Kinetic parameters,Vmax andKm, were calculated using
Michaelis–Menten equation ([S] versusV curve) andwere used to plot double reciprocal plots (Fig. 3). Thehighest concentration of amentoflavone (3�M) signifi-cantly increasedKm from 0.56�M (DMSO) to 2.1�Mwithout significantly changingVmax (p < 0.05,Table 2).Similarly, apigenin also significantly increasedKm from0.26 to 4.1�M without significantly alteringVmax. Thissuggests that apigenin and amentoflavone were com-petitive inhibitors of CYP1B1 with calculatedKis of0.08± 0.02 and 0.42± 0.05�M, respectively. TheseKis were not statistically different than those calcu-lated by the Cheng–Prusoff equation shown inTable 1.In contrast, for quercetin bothKm and Vmax changedstatistically significantly with increasing inhibitor con-centration suggesting a mixed type of inhibition(Table 2).
In order to further investigate the potential biologi-cal significance of flavonoid-mediated CYP1 inhibition,a prostate cancer cell line was used. To ensure that22Rv1 cells did express both CYP1A1 and CYP1B1mRNA, quantitative RT/RT-PCR was performed onDMSO and TCDD treated cells (Fig. 4). Relative con-stitutive expression of CYP1A1 and CYP1B1 mRNAwas not statistically different in the 22Rv1 cells.
Table 2Kinetic parameters,Vmax (pmol/mg/min) andKm (�M) ± standard errors (n = 3), for the inhibition of recombinant human CYP1B1 by quercetin(0.5 and 1.0�M), amentoflavone (1.0 and 3.0�M) and apigenin (0.1 and 0.5�M), determined by nonlinear regression curve fit using theMichaelis–Menten equation ([S] vs. V) plot using GraphPad Prism 4
ne
1�M
V 921±K 1.78±
Quercetin Amentoflavo
DMSO 0.5�M 1 �M DMSO
max 572 ± 77 202± 26 142± 50 1030± 220
m 0.24± 0.06 0.28± 0.06 0.71± 0.21 0.560± 0.13
Apigenin
3 �M DMSO 0.1�M 0.5�M
210 816± 76 599± 39 521± 66 493± 300.67 2.10± 0.37 0.260± 0.06 0.873± 0.17 4.14± 1.5
200 A. Chaudhary, K.L. Willett / Toxicology 217 (2006) 194–205
Fig. 3. Substrate vs. velocity and double reciprocal plots for the inhibition of in vitro EROD activity of CYP1B1 by quercetin (QT) 0.5 and 1�M,amentoflavone (AF) 1 and 3�M, apigenin (AP) 0.5 and 1�M. EROD substrate concentrations used were 0.1, 0.2, 0.3, 0.4, 0.6, 1.0 and 2.0�M.Recombinant CYP1B1 was preincubated with the inhibitor or DMSO prior to initiation of reaction. Each experiment was done three times induplicate.
In TCDD-treated cells, CYP1B1 was significantlyinduced 14-fold while CYP1A1 was induced 122-fold(p < 0.001).
In the 22Rv1 prostate cells, TCDD induced ERODactivity (max EROD activity = 18.77± 1.59 pmol/mg/
min) but the flavonoids alone did not induce activity com-pared to DMSO control cells (data not shown). Relativeto TCDD alone, quercetin, kaempferol, myricetin andapigenin did inhibit EROD activity in the cells in a dose-dependent manner (Fig. 5). IC50s of inhibition ranged

A. Chaudhary, K.L. Willett / Toxicology 217 (2006) 194–205 201
Fig. 4. TCDD induced induction of CYP1A1 and CYP1B1 mRNAexpression in 22Rv1 cells measured by quantitative RT/RT-PCR.Responses were normalized to�-actin and expressed relative to DMSOlevels. Exposures done in triplicate for 6 h and each sample analyzed intriplicate by PCR. Bars with different letters are statistically different(p < 0.05 by ANOVA, Neumann–Keuls multiple comparison tests).
Table 3IC50s (�M) for flavonoid-mediated inhibition of EROD activity inTCDD-induced 22Rv1 prostate cancer cells
Flavonoid IC50 ± S.E.M. (�M)
Myricetin 3.04± 0.57Apigenin 3.16± 0.48Kaempferol 3.76± 0.44Quercetin 4.14± 0.58Amentoflavone No inhibition
Data was probit transformed prior to IC50 calculation.
from 3.04± 0.57�M for myricetin to 4.14± 0.58�Mfor quercetin (Table 3). In contrast, amentoflavone didnot cause inhibition of EROD activity in the 22Rv1prostate cells.
4. Discussion
As described in recent reviews (McFadyen et al.,2004; Chun and Kim, 2003), CYP1B1 has become anincreasingly popular potential target for cancer therapyand prevention. Our goal was to characterize for thefirst time the CYP1B1 inhibition potential of a series offlavonoids found in herbal supplements and food. Theaim to inhibit CYP1B1 stems from the fact that it isup-regulated in cancer cells, it bioactivates PAHs andestrogen to DNA reactive metabolites, and it can inacti-vate chemotherapeutic drugs.Doostdar et al. (2000)pre-viously reported that flavones (acacetin and diosmetin)were better inhibitors of human CYP1 EROD activitythan the flavanones (eriodictyol, hesperetin, homoeriod-ictyol and naringenin). Additionally, they reported thathomoeriodictyol selectively inhibited human CYP1B1with an IC50 of 0.24�M which was very similar to theIC50s that we found for apigenin, kaempferol, quercetinand myricetin. Other compounds with reported selec-tivity for CYP1B1 inhibition include 2-ethynylpyrene,�-naphthoflavone and 2,4,3′,5′-tetramethoxystilbene(Chun and Kim, 2003). With the exception of rutin,none of the flavonoids tested in this study significantlyinhibited CYP1B1 relative to CYP1A1. However, certainflavonoids were still relatively potent CYP1 inhibitorsand may be effective in cancer chemoprevention.
The most potent CYP1B1 inhibitor was apigenin witha Ki of 60 nM. In our system, apigenin also inhibitedCYP1A1 withKi of 0.2�M which closely matched the
l.Des,
andB1
ppear
Fig. 5. Effects of increasing concentration of myricetin, apigenin, kaem activity inintact 22Rv1 human prostate cancer cells. Following 24 h TCDD exposu dfor 30 min prior to the EROD measurement. Each experiment was done fo an–Keulspost hoc test was used to determine treatment effects. Within treatment ifferent fromone another (p < 0.05).
Ki (0.3�M) previously reported byPastrakuljic et a(1997). While apigenin was potent in inhibiting EROactivity in both the cells and recombinant microsomamentoflavone was not inhibitory in the cell systemwas∼17-fold less potent than apigenin in the CYP1microsomes. Therefore, amentoflavone does not a
pferol, quercetin, and amentoflavone on TCDD-induced ERODre, media was removed from the cells and flavonoids (0.5–5�M) were addeur times in triplicate. Global one-way ANOVA with Student–Newms, bars not sharing a common letter (a, b, c, d) are significantly d

202 A. Chaudhary, K.L. Willett / Toxicology 217 (2006) 194–205
to break down into apigenin monomers in cell media,because its activity was not similar to apigenin.
Consistent with our work,Schwarz and Roots (2003)compared the abilities of apigenin, myricetin, quercetinand kaempferol to inhibit both CYP1A1-mediatedEROD activity and epoxidation of 7,8-dihydrodiolbenzo(a)pyrene. For most of the polyphenols that theytested, excepting resveratrol and (−)-epigallo catechingallate, the EROD assay gave a qualitatively similarresult compared to the measure of procarcinogen acti-vation. Their IC50s of CYP1A1 inhibition (0.2–0.5�M)were lower than those reported in this study (5–38�M),possibly due to different expression systems. Morerecently, Schwarz et al. (2005)found that quercetininhibited epoxidation of 7,8-dihydrodiol benzo(a)pyreneby CYP1A1 allelic variants with IC50s between 1.6and 7�M and Kis between 2 and 9.3�M with mixed-type inhibition. Our study suggests that quercetin isalso a mixed-type inhibitor of CYP1B1. With respect toP450 interactions, quercetin inhibits CYP2C9, CYP1A2,CYP3A (Von Moltke et al., 2004) and CYP1A1 as previ-ously discussed (Schwarz et al., 2003, 2005). Tsyrlov etal. (1994)found using mouse cDNA-expressed CYP1A1that quercetin also inhibited EROD (IC50: 7.45�M) andMROD (IC50: 3.7�M) activities. In our study, quercetininhibited recombinant CYP1B1 and CYP1A1 with IC50sof 0.55 and 6�M, respectively.
Compared to the other flavonoids tested, rutin was theleast effective inhibitor of CYP1B1 or CYP1A1 activity.Similarly, it did not inhibit CYP1-mediated 7,8-diol-BaP
e
sub-bittedandhirdxylfor
sti-by
aredion.si-
onl,
andtoesetion.
Previous work bySchwarz and Roots (2003)reportedthat apigenin, myricetin, kaempferol, and rutin didnot significantly change the cytochromec reductionactivity of P450 reductase, however at 20�M quercetincaused a 50% inhibition in reductase. Their datasuggests that at the concentrations used in this study(e.g.≤10�M), P450 reductase inhibition is likely nota major contributor.
Our particular research interest is in prostate can-cer chemoprevention, and several studies have impli-cated CYP1B1 in prostate cancer progression (Carnellet al., 2004; Chang et al., 2003; Tanaka et al., 2002).Accordingly, we selected a prostate cancer cell-lineto further investigate the inhibitory potential of fiveselected flavonoids. The advantage of using the 22Rv1prostate cell-line was that unlike the LNCaP, PC-3, andDU145 prostate cells, 22Rv1 cells are derived froma primary prostate carcinoma instead of a metastasis,and the only steroid receptor that they express is theandrogen receptor (Hartel et al., 2003). They respondto dihydroxytestosterone, epidermal growth factor, andexpress prostate specific antigen and, hence, are a goodin vitro system to study prostate cancer (Sramkoski etal., 1999). We, for the first time, found that CYP1A1and CYP1B1 mRNAs are constitutively expressed andTCDD-inducible in 22Rv1 cells. Previous studies haveshown TCDD inducibility of CYP1 in LNCaP (Jana etal., 2002) and high passage PC3 and DU145 prostatecancer cells (Schaufler et al., 2002). In the 22Rv1 cells,EROD activity was inhibited by quercetin, kaempferol,
entnentaliortiongu-y,B1DDcur-theA1
iso-otever,ort
by
soy-
rels
epoxidation (Schwarz et al., 2003) or protect from thmutagenicity of aromatic amines (Lee et al., 1994). Rutinwas the only flavonoid tested with the disaccharidestituent; the bulk or polarity of this moiety may prohiinteraction with P450s. All eight of the flavonoids teswere flavones which had hydroxyl groups at fifthseventh position. Substitution differed only at the tposition and in the number and position of hydrogroups on the phenyl ring. The potency of apigeninCYP1B1 inhibition may be related to the lack of subtution at the third position. Substitution at position 3a sugar group (isoquercetin, rutin, quercitrin) compto a hydroxy group generally reduced CYP1 inhibitPhenyl ring substitution with differing number and potion of hydroxyl groups had relatively little effectCYP1B1 inhibition considering theKis for kaempferoquercetin, and myricetin ranged from only 0.10± 0.03to 0.23± 0.06�M.
P450 reductase is critical for electron transferCYP1 catalytic activity. Therefore, it is importantconsider whether the inhibitory effects seen in thexperiments are due to P450 reductase inhibi
myricetin and apigenin. Myricetin was the most potEROD inhibitor in the cell system while amentoflavowas ineffective. It should be noted that the experimedesign (i.e. incubating cells with inhibitor 30 min prto the EROD assay) predominantly reflects inhibiat the enzyme level and not, for example, down relation of CYP1 mRNA or protein levels. Additionallwe did not test the relative extent to which CYP1and CYP1A1 proteins were induced by the 24 h TCexposure. These molecular aspects of inhibition arerently under investigation in our laboratory. BecauseEROD assay is not CYP1 isoform specific and CYP1has higher substrate affinity, the extent of individualform inhibition of CYP1A1 versus CYP1B1 could nbe determined in the prostate cell experiments. HowCYP1 inhibition in the cell system does further suppthe potential for in vivo relevance of CYP1 inhibitionflavonoids.
Previous studies with flavonoids have found thatisoflavones (Hussain et al., 2003) green tea polyphenols (Saleem et al., 2003), PC-SPES/biacalin mixtu(Ikezoe et al., 2001) and wine antioxidant polypheno

A. Chaudhary, K.L. Willett / Toxicology 217 (2006) 194–205 203
(Kampa et al., 2000) have activities consistent with pos-sible prostate cancer chemoprotection (Le Marchand,2002). The mechanisms by which flavonoids act havebeen reviewed (Brownson et al., 2002; Galati et al.,2000) and include, for example, disassembly of micro-tubules (Takagi et al., 1998) inhibition of proliferation(Manthey and Guthrie, 2002; Kampa et al., 2000), cellcycle arrest (Kobayashi et al., 2002), and inhibition ofandrogen receptor expression (Xing et al., 2001). Sili-binin has shown such promising efficacy it has enteredphase I clinical trials in prostate cancer patients (Singhand Agarwal, 2004).
St. John’s wort has become popular among some can-cer patients because of its antidepressant activity. Fur-thermore, many cancer patients surveyed are currently“self-medicating” with vitamins and herbal supplements(Eng et al., 2003; Richardson et al., 2000). A com-mon dose of St. John’s wort is 900 mg/day (Barnes etal., 2001), and flavonoids represent from 2 to 12% ofthe fresh plant (Greeson et al., 2001). The estimatedsteady state plasma concentration of hyperforin was100 ng/ml (∼0.18�M) when 3× 300 mg/day extract ofHypericum perforatum was administered to human vol-unteers (Biber et al., 1998). Studies are not availablethat report plasma concentrations of flavonoids follow-ing intake of St. John’s wort, however the flavonoidsreported in this study are also present in a number offoods. Normal doses of St. John’s wort may not inhibitCYP1s based on the IC50s reported here, however whenconsidering the dietary intake of flavonoids from foods( par-t ec-t calr ther-l ffl 2I etine )p(t er ingi ent)p( rrm rptiond aryfl sig-n
rol,m in
prostate cancer cells. These flavonoids, based on theirpotency, are particularly attractive for further study.Clearly, potent EROD inhibition is not enough evidenceto definitively conclude that a substance is chemoprotec-tive. Therefore, further studies are required to addresseffects on CYP1 enzymes at the message and proteinlevels and determine the in vivo bioavailability of thesecompounds.
Acknowledgements
The authors would like to thank Drs. Robert Speth,John Matthews and Larry Walker (University of Mis-sissippi) for helpful suggestions in the preparation ofthis manuscript and the inhibition kinetics analyses. Thisresearch was supported by funding from American Asso-ciation of Colleges of Pharmacy and by NIH Grant Num-ber RR016476 from the MFGN INBRE Program of theNational Center for Research Resources.
References
Barnes, J., Anderson, L.A., Phillipson, J.D., 2001. St. John’s wort(Hypericum perforatum L.): a review of its chemistry, pharma-cology and clinical properties. J. Pharm. Pharmacol. 53, 583–600.
Biber, A., Fischer, H., Romer, A., Chatterjee, S.S., 1998. Oral bioavail-ability of hyperforin from hypericum extracts in rats and humanvolunteers. Pharmacopsychiatry 31, 36–43.
Bournique, B., Lemarie, A., 2002. Docetaxel (Taxotere) is not metab-olized by recombinant human CYP1B1 in vitro, but acts as aneffector of this isozyme. Drug Metab. Dispos. 30, 1149–1152.
S.F.,. 132,
P.J.,tionteine. Int.
gan,es inh 4-
of
ro-and
lysis.
.A.,acs,asso-r 89,
itors–668.lec-and
onions, soy, apples, citrus, berries), tea and wine,icularly if a person was trying to intake chemoprotive quantities, CYP1 inhibition could be physiologielevant. For example, studies in the US and The Neands estimated average daily intakes of∼20 mg/day oavonoids (Hertog et al., 1993; Sampson et al., 200).n human subjects given fried onions (68 mg quercquivalents, 225�mol) or apples (325�mol quercetineak plasma concentrations were 0.74 and 0.30�MHollman et al., 1997). The IC50 for CYP1B1 inhibi-ion by quercetin (0.55�M) is, therefore, within thange reported in human plasma. Similarly, followngestion of endive soup (9 mg kaempferol equivaleak plasma concentrations ranged from 0.05 to 0.1�MDuPont et al., 2004), only ∼5–10-fold lower than oueported CYP1B1 IC50 (0.48�M). Importantly, muchore research is necessary to understand the absoistribution, accumulation, and elimination of dietavonoids to gauge their true potential physiologicalificance.
We found that flavonoids (quercetin, kaempfeyricetin, and apigenin) inhibited EROD activity
,
Brownson, D.M., Azios, N.G., Fuqua, B.K., Dharmawardhane,Mabry, T.J., 2002. Flavonoid effects relevant to cancer. J. Nutr3482S–3489S.
Carnell, D.M., Smith, R.E., Daley, F.M., Barber, P.R., Hoskin,Wilson, G.D., Murray, G.I., Everett, S.A., 2004. Target validaof cytochrome P450 CYP1b1 in prostate carcinoma with proexpression in associated hyperplastic and premalignant tissuH. Tadiat. Oncol. Biol. Phys. 58, 500–509.
Cavalieri, E.L., Devanesan, P., Bosland, M.C., Badawi, A.F., RoE.G., 2002. Catechol estrogen metabolites and conjugatdifferent regions of the prostate of Noble rats treated withydroxyestradiol: implications for estrogen-induced initiationprostate cancer. Carcinogenesis 23, 329–333.
Chaib, H., Cockrell, E.K., Rubin, M.A., Macoska, J.A., 2001. Pfiling and verification of gene expression patterns in normalmalignant human prostate tissues by cDNA microarray anaNeoplasia 3, 43–52.
Chang, B.L., Zheng, S.L., Isaacs, S.D., Turner, A., Hawkins, GWiley, K.E., Bleecker, E.R., Walsh, P.C., Meyers, D.A., IsaW.B., Xu, J., 2003. Polymorphisms in the CYP1B1 gene areciated with increased risk of prostate cancer. Br. J. Cance1524–1529.
Chun, Y., Kim, S., 2003. Discovery of cytochrome P450 1B1 inhibas new promising anti-cancer agents. Med. Res. Rev. 23, 657
Doostdar, H., Burke, M.D., Mayer, R.T., 2000. Bioflavonoids: setive substrates and inhibitors for cytochrome P450 CYP1ACYP1B1. Toxicology 144, 31–38.

204 A. Chaudhary, K.L. Willett / Toxicology 217 (2006) 194–205
DuPont, M.S., Day, A.J., Bennett, R.N., Mellon, F.A., Kroon,P.A., 2004. Absorption of kaempferol from endive, a sourceof kaempferol-3-glucuronids, in humans. Eur. J. Clin. Nutr. 58,947–954.
Eng, J., Ramsum, D., Verhoef, M., Guns, E., Davison, J., Gallagher, R.,2003. A population-based survey of complementary and alterna-tive medicine use in men recently diagnosed with prostate cancer.Integr. Cancer Ther. 2, 212–216.
Finnstrom, N., Bjelfman, C., Soderstrom, T.G., Smith, G., Egevad, L.,Norlen, B.J., Wolf, C.R., Rane, A., 2001. Detection of cytochromeP450 mRNA transcripts in prostate samples by RT-PCR. Eur. J.Clin. Invest. 31, 880–886.
Galati, G., Teng, S., Moridani, M.Y., Chan, T.S., O’Brien, P.J., 2000.Cancer chemoprevention and apoptosis mechanisms induced bydietary polyphenolics. Drug Metabol. Drug Interact. 17, 311–349.
Greeson, J.M., Sanford, B., Monti, D.A., 2001. St. John’s wort (Hyper-icum perforatum): a review of the current pharmacological, toxico-logical, and clinical literature. Psychopharmacology 153, 402–414.
Hanna, I.H., Dawling, S., Roodi, N., Guengerich, F.P., Parl, F.F., 2000.Cytochrome P450 1B1 (CYP1B1) pharmacogenetics: associationof polymorphisms with functional differences in estrogen hydrox-ylation activity. Cancer Res. 60, 3440–3444.
Hartel, A., Didier, A., Pfaffl, M.W., Meyer, H.H.D., 2003. Charac-terization of gene expression patterns in 22Rv1 cells for determi-nation of environmental androgenic/antiandrogenic compounds. J.Steroid Biochem. Mol. Biol. 84, 231–238.
Hayes, C.L., Spink, D.C., Spink, B.C., Cao, J.Q., Walker, N.J., Sut-ter, T.R., 1996. 17�-Estradiol hydroxylation catalyzed by humancytochrome P450 1B1. Proc. Natl. Acad. Sci. 93, 9776–9781.
Hertog, M.G., Hollman, P.C., Katan, M.B., Kromhout, D., 1993. Intakeof potentially anticarcinogenic flavonoids and their determinantsin adults in The Netherlands. Nutr. Cancer 20, 21–29.
Hollman, P.C., van Trijp, J.M.P., Buysman, M.N., Van der Gaag, M.S.,Mengelers, M.J.B., de Bries, J.H.M., Katan, M.B., 1997. Relativebioavailability of the antioxidant flavonoid quercetin from various
.N.,ood,state
001.theycle
Sone,o-
roidNatl.
gou,000.an
s on176,
tivity-450
ino-
s—a
Lin, P., Hu, S., Chang, T., 2003. Correlation between gene expres-sion of aryl hydrocarbon receptor (AhR), hydrocarbon receptornuclear translocator (Arnt), cytochromes P4501A (CYP1A1) and1B1 (CYP1B1), and inducibility of CYP1A1 and CYP1B1 inhuman lymphocytes. Toxicol. Sci. 71, 20–26.
Luo, J., Dunn, T., Ewing, C., Sauvageot, J., Chen, Y., Trent, J., Isaacs,W., 2002. Gene expression signature of benign prostatic hyperpla-sia revealed by cDNA microarray analysis. Prostate 51, 189–200.
Manthey, J.A., Guthrie, N., 2002. Antiproliferative activities of cit-rus flavonoids against six human cancer cell lines. J. Agric. FoodChem. 50, 5837–5843.
McFadyen, M.C.E., McLeod, H.L., Jackson, F.C., Melvin, W.T.,Doehmer, J., Murray, G.I., 2001. Cytochrome P450 CYP1B1 pro-tein expression: a novel mechanism of anticancer drug resistance.Biochem. Pharmacol. 62, 207–212.
McFadyen, M.C.E., Melvin, W.T., Murray, G.I., 2004. CytochromeP450 enzymes: novel options for cancer therapeutics. Mol. CancerTher. 3, 363–371.
Murray, G.I., Melvin, W.T., Greenlee, W.F., Burke, M.D., 2001. Regu-lation, function and tissue-specific expression of cytochrome P450CYP1B1. Annu. Rev. Pharmacol. Toxicol. 41, 297–316.
Obach, R.S., 2000. Inhibition of human cytochrome P450 enzymes byconstituents of St. John’s Wort, an herbal preparation used in thetreatment of depression. J. Pharmacol. Exp. Ther. 294, 88–95.
Pastrakuljic, A., Tang, B.K., Roberts, E.A., Kalow, W., 1997. Distinc-tion of CYP1A1 and CYP1A2 activity by selective inhibition usingfluvoxamine and isosafrole. Biochem. Pharmacol. 53, 531–538.
Richardson, M.A., Sanders, T., Palmer, J.L., Greisinger, A., Singletary,S.E., 2000. Complementary/alternative medicine use in a compre-hensive cancer center and the implications for the oncology. J. Clin.Oncol. 18, 2505–2514.
Rochat, B., Morsman, J.M., Murray, G.I., Figg, W.D., McLeod, H.L.,2001. Human CYP1B1 and anticancer agent metabolism: mecha-nism for tumor-specific drug inactivation? J. Pharmacol. Exp. Ther.296, 537–541.
eaview.
.B.,ls. J.
mer,o-C 3
d. 9,
s andfor-1.
ctivecer
byens
303,
uen-
ntdro-
foods in man. FEBS Lett. 418, 152–156.Hussain, M., Banerjee, M., Sarkar, F.H., Djuric, Z., Pollak, M
Doerge, D., Fontana, J., Chinni, S., Davis, J., Forman, J., WD.P., Kucuk, O., 2003. Soy isoflavones in the treatment of procancer. Nutr. Cancer 47, 111–117.
Ikezoe, T., Chen, S.S., Heber, D., Taguchi, H., Koeffler, H.P., 2Baicalin is a major component of PC-SPES which inhibitsproliferation of human cancer cells via apoptosis and cell carrest. Prostate 49, 285–292.
Jana, N.R., Sarkar, S., Ishizuka, M., Yonemoto, J., Tohyama, C.,H., 2002. Comparative effects of 2,3,7,8-tetrachlorodibenzp-dioxin on MCF-7, RL95-2, and LNCaP cells: role of target stehormones in cellular responsiveness to CYP1A1 induction. J.Cancer Inst. 94, 1247–1249.
Kampa, M., Hatzoglou, A., Notas, G., Damianaki, A., BakogeorE., Gemetzi, C., Kouroumalis, E., Martin, P.M., Castanas, E., 2Wine antioxidant polyphenols inhibit the proliferation of humprostate cancer cell lines. Nutr. Cancer 37, 223–233.
Kobayashi, T., Nakata, T., Kuzumaki, T., 2002. Effect of flavonoidcell cycle progression in prostate cancer cells. Cancer Lett.17–23.
Lee, H., Wang, H.W., Su, H.Y., Hao, N.J., 1994. The structure–acrelationships of flavonoids as inhibitors of cytochrome Penzymes in rat liver microsomes and the mutagenicity of 2-am3-methyl-imidazo[4,5-f]quinoline. Mutagenesis 9, 101–106.
Le Marchand, L., 2002. Cancer preventive effects of flavonoidreview. Biomed. Pharmacother. 56, 296–301.
Saleem, M., Adhami, V.M., Siddiqui, I.A., Mukhtar, H., 2003. Tbeverage in chemoprevention of prostate cancer: a mini-reNutr. Cancer 47, 13–23.
Sampson, L., Rimm, E., Hollman, P.C., de Vries, J.H., Katan, M2002. Flavonol and flavone intakes in US health professionaAm. Diet Assoc. 102, 1414–1420.
Schaufler, K., Haslmayer, P., Jager, W., Pec, M., ThalhamT., 2002. The environmental toxin 2,3,7,8-tetrachlorodibenzp-dioxin induces cytochrome P450 activity in high passage Pand DU 145 human prostate cancer cell lines. Int. J. Mol. Me411–416.
Schwarz, D., Kisselev, P., Roots, I., 2003. St. John’s wort extractsome of their constituents potently inhibit ultimate carcinogenmation from benzo[a]pyrene-7,8-dihydrodiol by human CYP1ACancer Res 63, 8062–8068.
Schwarz, D., Kisselev, P., Roots, I., 2005. CYP1A1 genotype-seleinhibition of benzo[a]pyrene activation by quercetin. Eur. J. Can41, 151–158.
Schwarz, D., Roots, I., 2003. In vitro assessment of inhibitionnatural polyphenols of metabolic activation of procarcinogby human CYP1A1. Biochem. Biophys. Res. Commun.902–907.
Shimada, T., Gillam, E.M., Oda, Y., Tsumura, F., Sutter, T.R., Ggerich, F.P., Inoue, K., 1999. Metabolism of benzo(a)pyrene totrans-7,8-dihydroxy-7,8-dihydrobenzo(a)pyrene by recombinahuman cytochrome P450 1B1 and purified liver epoxide hylase. Chem. Res. Toxicol. 12, 623–629.

A. Chaudhary, K.L. Willett / Toxicology 217 (2006) 194–205 205
Singh, R.P., Agarwal, R., 2004. Prostate cancer prevention by silibinin.Curr. Cancer Drug Targets 4, 1–11.
Sramkoski, R.M., Pretlow Jr., T.G., Giaconia, J.M., Pretlow, T.P.,Schwartz, S., Sy, M.S., Marengo, S.R., Rhim, J.S., Zhang, D.,Jacobberger, J.W., 1999. A new human prostate carcinoma cellline, 22Rv1. In Vitro Cell Dev. Biol. 35, 403–409.
Takagi, T., Takekoshi, S., Okabe, T., Nagata, H., Honma, T., Watan-abe, K., 1998. Quercetin, a flavonol, promotes disassembly ofmicrotubules in prostate cancer cells: possible mechanism ofits antitumor activity. Acta Histochem. Cytochem. 31, 435–445.
Tanaka, Y., Sasaki, M., Kaneuchi, M., Shiina, H., Igawa, M., Dahiya,R., 2002. Polymorphisms of the CYP1B1 gene have higher riskfor prostate cancer. Biochem. Biophys. Res. Commun. 296, 820–826.
Tsyrlov, I.B., Mikhailenko, V.M., Gelboin, H.V., 1994. Isozyme- andspecies-specific susceptibility of cDNA-expressed CYP1A P-450sto different flavonoids. Biochim. Biophys. Acta 1205, 325–335.
Vogel, G., 2001. How the body’s ‘garbage disposal’ may inactivatedrugs. Science 291, 36–37.
Von Moltke, L.L., Weemhoff, J.L., Bedir, E., Khan, I.A., Harmatz,J.S., Goldman, P., Greenblatt, D.J., 2004. Inhibition of humancytochromes P450 by components ofGinkgo biloba. J. Pharm.Pharmacol. 56, 1039–1044.
Willett, K.L., Randerath, K., Zhou, G.D., Safe, S.H., 1998. Inhibitionof CYP1A1-dependent activity by the polynuclear aromatic hydro-carbon (PAH) fluoranthene. Biochem. Pharmacol. 55, 831–839.
Xing, N., Chen, Y., Mitchel, S.H., Young, C.Y., 2001. Quercetininhibits the expression and function of the androgen receptor inLNCaP prostate cancer cells. Carcinogenesis 22, 409–414.
Copyright © 2022 FDOKUMEN




















