
IL-2 immunotherapy in chronically SIV-infected Rhesus Macaques

In vivo targeting of antibody fragments to the nervous system forAlzheimer’s disease immunotherapy and molecular imaging ofamyloid plaques
Joseph F. Poduslo,*,� Muthu Ramakrishnan,* Silvina S. Holasek,* Marina Ramirez-Alvarado,�Karunya K. Kandimalla,* Emily J. Gilles,* Geoffry L. Curran* and Thomas M. Wengenack*
*Molecular Neurobiology Laboratory, Departments of Neurology, Neuroscience, and �Biochemistry/Molecular Biology, Mayo Clinic
College of Medicine, Rochester, Minnesota, USA
Abstract
Targeting therapeutic or diagnostic proteins to the nervous
system is limited by the presence of the blood–brain barrier.
We report that a F(ab¢)2 fragment of a monoclonal antibody
against fibrillar human Ab42 that is polyamine (p)-modified
has increased permeability at the blood–brain barrier, com-
parable binding to the antigen, and comparable in vitro binding
to amyloid plaques in Alzheimer’s disease (AD) transgenic
mouse brain sections. Intravenous injection of the pF(ab¢)24.1
in the AD transgenic mouse demonstrated efficient targeting
to amyloid plaques throughout the brain, whereas the
unmodified fragment did not. Removal of the Fc portion of this
antibody derivative will minimize the inflammatory response
and cerebral hemorrhaging associated with passive immun-
ization and provide increased therapeutic potential for treating
AD. Coupling contrast agents/radioisotopes might facilitate
the molecular imaging of amyloid plaques with magnetic res-
onance imaging/positron emission tomography. The efficient
delivery of immunoglobulin G fragments may also have
important applications to other neurodegenerative disorders
or for the generalized targeting of nervous system antigens.
Keywords: Alzheimer’s disease, amyloid plaques, antibody
fragments, contrast agent, molecular imaging, passive
immunization.
J. Neurochem. (2007) 102, 420–433.
A major limitation in targeting protein- or antibody-basedtherapies to the nervous system is the presence of the blood–brain barrier (BBB) or the blood–nerve barrier (BNB).Unless a protein has a specific receptor (e.g. insulin) at theluminal surface of the brain capillary endothelial cell, itstranscytosis through and exit from the endothelial cell andentry into the brain parenchyma is limited by passivediffusion (Poduslo et al. 1994). Immunoglobulin G (IgG) isone class of proteins whose permeability at the BBB is verylow [permeability coefficient · surface area product (PS) isca. 0.1 · 10)6 mL/g/s – about 240-fold less than the PS forinsulin which has receptor-mediated transport at the BBB]and hence are restricted to slow passive diffusion or fluidphase endocytosis (Poduslo et al. 1994, 2001).
The success of active or passive immunization in Alzhei-mer’s disease (AD) transgenic mice or patients is surprisingbecause of such low immunoglobulin permeability at theBBB. Yet, a number of different immunization strategies forclearing amyloid-b peptide (Ab) and amyloid plaques in thebrains of AD transgenic mice or patients have been proposed
(Schenk 2002). These include active immunization by Abpeptide injections (Schenk et al. 1999; Janus et al. 2000;Morgan et al. 2000; Lambert et al. 2001; Spooner et al.
Received September 12, 2006; revised manuscript received January 24,2007; accepted January 24, 2007.Address correspondence and reprint requests to Joseph F. Poduslo,
Molecular Neurobiology Laboratory, Departments of Neurology, Neu-roscience, and Biochemistry/Molecular Biology, Mayo Clinic College ofMedicine, 200 First Street SW, Rochester, MN 55905, USA.E-mail: [email protected] used: Ab, amyloid-b peptide; AD, Alzheimer’s disease;
APP, amyloid precursor protein; BBB, blood–brain barrier; BNB, blood–nerve barrier; BSA, bovine serum albumin; CAA, cerebral amyloid an-giopathy; CSF, cerebrospinal fluid; CVA, cerebrovascular amyloid; Gd,gadolinium; IEF, isoelectric focusing; IgG, immunoglobulin G; IH, im-munohistochemistry; MCI, mild cognitive imparement; p, polyamine-modified; PBS, phosphate-buffered saline; PBST, 0.3% Triton X-100 inPBS; PS, permeability coefficient · surface area product; PS1, humanpresenilin 1; scFv, single-chain variable fragments; SDS-PAGE, sodiumdodesyl sulfate-polyacrylamide gel electrophoresis; Vp, residual plasmavolume; WT, wild type.
Journal of Neurochemistry, 2007, 102, 420–433 doi:10.1111/j.1471-4159.2007.04591.x
420 Journal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 102, 420–433� 2007 The Authors

2002) or nasal administration with Ab peptide or phagepeptide (Frenkel et al. 2000; Weiner et al. 2000; Lemereet al. 2003), and passive immunization with monoclonal orpolyclonal antibodies by intravenous, peritoneal, intracere-bral, or ventricular injections (Bard et al. 2000; DeMattoset al. 2001; Dodart et al. 2002; Mohajeri et al. 2002;Wilcock et al. 2003). Such active or passive immunizationstrategies have generally decreased cerebral amyloid burdenand Ab levels and also attenuated neuronal dystrophy,astrogliosis, behavioral deficits, and memory loss. Usually,the success of antibody entry into the brain was attributed tolow levels of IgG that could be detected in cerebrospinalfluid, and that this level was sufficient to account for theobserved changes. Further understanding of the mechanismassociated with antibody delivery into the nervous systemhas not been rigorously pursued.
We have addressed this issue in part by evaluating thepermeability of IgG and the immune complex of IgG-Ab atthe BBB utilizing a monoclonal antibody against fibrillarhuman Ab42. We determined that the immune complexformed has a permeability that is significantly greater thanthe monoclonal antibody alone (Poduslo and Curran 2001).This suggests that Ab itself, which has a high BBBpermeability indicative of receptor-mediated transport (Podu-slo et al. 1997, 1999; Poduslo and Curran 2001) may beresponsible for transporting the antibody across the BBB.The permeability of an anti-Ab antibody at the BBB,therefore, will be critically dependent on circulating Ablevels, which are lower in AD patients compared with ADtransgenic mice and highly variable from patient to patient.Therefore, it becomes important to further understand themechanism of IgG transport at the BBB during both activeand passive immunization in AD transgenic mice and in theAD patient.
While anti-Ab active immunization has been largelysuccessful in AD transgenic mouse studies, the first clinicaltrial conducted in AD patients was problematic. An activeimmunization clinical trial in AD patients was abruptlyhalted because of meningoencphalitis in 6% of the patients(Imbimbo 2002; Schenk 2002; Nicoll et al. 2003). After thiscancellation, systemic immunization in AD transgenic mousemodels showed perivascular hemorrhages and inflammation(Pfeifer et al. 2002; Furlan et al. 2003). Active immunizationmay trigger autoinflammatory reactions and cell lysis due toT-cell activation (Grubeck-Loebenstein et al. 2000; Monso-nego et al. 2003) or induce autoimmune reactions due toantibody binding to neuronal epitopes (Rohn et al. 2000).Inflammatory responses may also be triggered by microglialactivation and phagocytosis of insoluble immune complexes(Rogers and Shen 2000). Controlling undesirable inflamma-tory reactions and minimizing cerebral hemorrhaging asso-ciated with AD immunotherapy might be realized withdomain specific antibody fragments [e.g. F(ab¢)2] which donot contain the Fc region (Tamura et al. 2005). Furthermore,
the delivery of F(ab¢)2 across the BBB might be facilitatedbecause of its reduced molecular weight.
Our ability to quantify the permeability of proteins at theBBB has allowed the evaluation of different protein modifi-cations that might be used to enhance this permeability(Poduslo et al. 1994; Poduslo and Curran 1996). In particular,dramatic increases in the BBB permeability of a number ofproteins have been observed after covalent modification withnaturally occurring polyamines, such as putrescine, spermi-dine, or spermine (Poduslo and Curran 1996; Wengenacket al. 1997; Poduslo et al. 1998, 1999). Polyamine modifica-tion of human Ab40 not only resulted in significant increasesin its BBB permeability, but also resulted in enhanced bindingto amyloid plaques in AD brain sections (Wengenack et al.2000; Poduslo et al. 2002), which in turn has led to the use ofdifferent derivatives of Ab as a contrast agent for themolecularimaging of amyloid plaques usingmagnetic resonance at high-field strength (Poduslo et al. 2004). In the present study, wecompare a monoclonal antibody against human Ab42 with itsF(ab¢)2 fragment, with and without polyamine modification,measuring their BBB permeability, in vitro plaque binding,and in vivo targeting to amyloid plaques in a transgenic mousemodel of AD after intravenous injection.
Materials and methods
Animals
The in vivo labeling experiments were performed using transgenic
mice that express two mutant human proteins associated with
familial AD and have been described in detail elsewhere (Holcomb
et al. 1998). Hemizygous transgenic mice (Tg2576) expressing
mutant human amyloid precursor protein (APP695) (Hsiao et al.1996) were mated with a second strain of hemizygous transgenic
mice (M146 L6.2) expressing mutant human presenilin 1 (PS1)
(Holcomb et al. 1998). In one experiment, an APP (Tg2576) mouse
was used. The animals were genotyped for the expression of both
transgenes by a PCR method using a sample of mouse-tail DNA. The
mice were housed in a virus-free barrier facility under a 12-hr light/
dark cycle, with ad libitum access to food and water. All procedures
performed were in accordance with NIH Guidelines for the Care andUse of Laboratory Animals using protocols approved by the Mayo
Institutional Animal Care and Use Committee. All efforts were made
to minimize both the suffering and the number of animals used.
Monoclonal antibody production
B-cell hybridomas were generated following the procedure of de
StGroth and Scheidegger (1980) in the Mayo Monoclonal Core
Facility utilizing 1 mg human Ab 1-42 that was aggregated and
fibrilized by incubating at 37�C for 24 h. Because of the large
amounts of antibody required for these studies, it would be cost-
prohibitive to use commercial antibodies. As a result, we generated
our own monoclonal antibodies. Mouse IgG was purified from
ascites using a protein G column. Positive subclones were isotyped
and cryoperserved, and later characterized by enzyme-linked
immunosorbent assay (ELISA) and immunohistochemistry (IH)
Targeting of antibody fragments to AD amyloid plaques 421
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 102, 420–433

labeling of amyloid deposits in AD transgenic mouse brain sections.
Subclone IgG4.1 (PC4.1) was used in the present studies.
Preparation of F(ab¢)2 fragments
The Pierce Immunopure IgG1 Fab and F(ab¢)2 preparation kit (PierceBiotechnology, Rockford, IL, USA) was used to make F(ab¢)2fragments from IgG4.1 antibody according to the instructions provided
by the manufacturer. Briefly, 1 mL of 3.5 mg/mL IgG4.1 was buffer
exchanged with mild elution buffer from the kit using PD10 desalting
column (Amersham Pharmacia, Piscataway, NJ, USA). The immobi-
lizedficin columnwas equilibratedwith thedigestion buffer containing
4 mg of cysteine HCl (or 40 mg cysteine HCl for Fab fragments). The
IgG4.1 (1 mL) was passed onto the column, and the gel bed was
incubated at 37�C for 28 h. The digested samples were eluted using
Immunopure binding buffer supplied with the kit. Undigested IgG4.1
and Fc fragmentswere removedusing aUnoQanion exchange column
(Bio-Rad, Hercules, CA, USA) leaving the F(ab¢)24.1 in the flow
through. F(ab¢)24.1 fragments were further purified from trace Fab4.1
fragments using a Hiload Superdex gel filtration column (Amersham
Pharmacia). In other experiments, Fab4.1 was prepared by papain
digestion and purified in a similar manner. Sample purity was checked
by 7.5% sodium dodesyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE), and protein concentrations were determined using the
BCA protein assay (Pierce Biotechnology).
Polyamine modification of IgG4.1 and F(ab¢)24.1
Modification of the monoclonal antibody and fragments were
performed as described in detail previously (Poduslo and Curran
1996). The polyamine putrescine was covalently attached to
carboxylic acid groups of the proteins using carbodiimide. The
polyamine modified proteins were designated pIgG4.1 and
pF(ab¢)24.1. Ionization of the carboxylic acid groups was controlled
by adjusting the pH, which in turn controlled the extent of
modification with the polyamine.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and
isoelectric focusing
Aliquots of IgG4.1, Fab4.1, and F(ab¢)24.1 were run on a SDS-
PAGE gel (15% T and 1% C) in the absence of a reducing agent and
stained with Coomassie blue using standard methods. Isoelectric
focusing (IEF) of IgG4.1 and pIgG4.1 as well as the F(ab¢)24.1 and
pF(ab¢)24.1 samples was performed using the Bio-Rad Model 111
Mini IEF Cell according to the manufacturer’s instructions. Briefly,
40% Bio-Lyte 3/10 ampholyte solution (Bio-Rad) was used with a
polyacrylamide gel (24.25% T, 0.75% C). The gel was cast between
the acrylic casting tray and gel support film to give gel dimensions
of 125 mm · 65 mm · 0.4 mm. Monomer-ampholyte polymeriza-
tion time under a fluorescent desk lamp was 45 min, and 6 min was
allowed for samples to diffuse into the gel. IEF was carried out
under constant voltage in a stepped fashion: 45 min at 100 V and
15 min at 450 V. Bio-Rad IEF standards containing nine native
proteins with isoelectric points ranging from 4.45 to 9.6, as well as
albumin (4.7) and cytochrome C (9.6), were run for comparison. All
bands were visualized with 0.04% Coomassie Brilliant Blue stain.
Enzyme-linked immunosorbent assay
Ab peptide (Ab40, Ab42, and fibrillar Ab40 and Ab42 synthesized
as described by Poduslo et al. 2004) was coated in the wells of 96-
well microtiter plates (Immulon 1B, Thermo Lab Systems, Franklin,
MA, USA) at a concentration of 1 lg/well and incubated overnight
at 23�C. Fibrils were formed by incubating Ab40 or Ab42 in pH 7.4
Tris buffer at 37�C for ca. 48 h. Wells were washed with phosphate-
buffered saline (PBS), pH 7.4 and blocked with 100 lL of PBS
containing 1% of bovine serum albumin (BSA, Sigma, St. Louis,
MO, USA) at 23�C for 60 min. The wells were rinsed with PBS,
and serially diluted samples were then added in triplicate and
included IgG4G8, IgG4.1, F(ab¢)24.1, and pF(ab¢)24.1 modified at
pH values of 6.0, 6.8, 7.4 and 7.8. After 3 h incubation at 37�C, thewells were washed with PBS and incubated with 100 ng of Fab
specific goat anti-mouse IgG secondary antibody conjugated to
alkaline phosphatase (Sigma A-1682) for 1 h at 23�C. The wells
were washed and equilibrated with diethanolamine buffer, pH 9.8,
for 5 min. 4-Nitrophenyl phosphate disodium (Sigma) was used as a
substrate to visualize the bound antibodies and fragments. After
15 min, the reaction was stopped by adding 50 lL of 1 mol/L
sodium hydroxide. Optical densities were recorded at 405 nm using
an automated plate reader (Spectramax Plus, Molecular Devices,
Sunnydale, CA, USA).
PS/Vp measurements of radioiodinated native and polyamine
modified monoclonal IgG4.1 and F(ab¢)24.1 at the BBB
Aliquots of the proteins were labeled with 125I or 131I using the
chloramine-T procedure and PS/Vp measurements were performed
as described in detail previously (Poduslo et al. 1994, 2001).
Briefly, an i.v. bolus injection of PBS containing either 125I-IgG4.1,125I-pIgG4.1, 125I-F(ab¢)24.1, or 125I-pF(ab¢)24.1 (100 lCi) was
rapidly injected into the femoral vein of an anesthetized (isoflurane,
1.5%) wild-type (WT) mouse. Serial blood samples (20 lL) werecollected from the femoral artery over the next 30–120 min, diluted
to a volume of 100 lL with saline, and centrifuged. The supernatant
was then TCA precipitated, and the precipitate was assayed for 125I
and 131I radioactivity. At 30–60 s before the end of the experiment, a
second aliquot of the same protein radiolabeled with 131I (100 lCi)was administered i.v. to serve as a Vp indicator of vascular space.
After the final blood sample, the animals were sacrificed, the brain
and meninges were removed, and the brain was dissected into
cortex, caudate-putamen (neostriatum), hippocampus, thalamus,
brain stem, and cerebellum. Tissue was lyophilized, and dry weights
were determined with a microbalance and converted to respective
wet weights with wet weight/dry weight ratios previously deter-
mined. Tissue and plasma samples were assayed for 125I and 131I
radioactivity in a two channel gamma counter (Packard COBRA II)
with radioactivity corrected for background and crossover of 131I
activity into the 125I channel. Data are presented as �x� SEM values
with statistical evaluation using ANOVA with a significance accepted
at the P < 0.05 level. The PS and Vp measurements were calculated
as described previously (Poduslo et al. 2001).
Labeling of amyloid plaques in vitro
Thirty-micron-thick, fixed, free-floating sections from a 1-year old
APP (Tg2576) transgenic and a WT, non-transgenic mouse brain
were reacted with either IgG4.1, Fab4.1, pFab4.1, F(ab¢)24.1 or
pF(ab¢)24.1. The presence of the immunoglobulin or fragments was
detected by standard immunoperoxidase methods. Briefly, sections
were first rinsed with 0.3% Triton X-100 in PBS (PBST) for 5 min.
The endogenous peroxidase activity in the tissue was blocked by
422 J. F. Poduslo et al.
Journal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 102, 420–433� 2007 The Authors

reacting with 0.5% H2O2 in PBST for 30 min. The sections were
rinsed twice in PBST for 5 min each and then blocked for 60 min
with 1.5% normal horse serum in PBST. Sections were rinsed twice
in PBST for 5 min each and then incubated with the specified
dilution of the immunoglobulin or fragment in 0.3% Triton X-100,
0.1% BSA in PBS overnight at 4�C (IgG, 2 lg/mL; Fab4.1, 2 lg/mL; pFab4.1, 2 lg/mL; F(ab¢)24.1, 2 lg/mL; pF(ab¢)24.1, 2 lg/mL). A control section was incubated in 0.3% Triton X-100, 0.1%
BSA in PBS alone without the addition of the antibody. As a
positive control, other sections were incubated with a standard anti-
Ab antibody (IgG4G8, 1 : 1000, Signet Laboratories, Dedham, MA,
USA). The second day, sections were rinsed twice with PBST for
5 min each and then incubated with biotinylated horse anti-mouse
secondary antibody (Vector Laboratories, Burlingame, CA, USA) at
a dilution of 1 : 250 for 30 min. After rinsing twice in PBST for
5 min each, sections were incubated with ABC reagent (Vectastain
Elite ABC; Vector Laboratories) for 30 min and then rinsed twice in
PBS (minus 0.3% Triton X-100) for 5 min each. Sections were
reacted with 3,3¢-diaminobenzidine peroxidase substrate (SK-4100;
Vector Laboratories) until a reaction product was visible (approxi-
mately 2-4 min) and were then rinsed three times in tap water for
5 min each. Sections were mounted on gelatin-subbed slides, dried
overnight at 37�C, and then dehydrated with successive changes of
ethanol and xylene and coverslipped. In another experiment, in vitrolabeling was repeated using a goat anti-mouse IgG Fab-specific
biotinylated secondary antibody (no. B7151, 1 : 200; Sigma
Chemical Co.). In this instance, 10% normal goat serum was used
as the blocking agent, but all other conditions were the same. As a
control for the effects of polyamine modification on the binding of
the antibodies and fragments to amyloid plaques and brain tissue, a
non-specific antibody (an anti-human Ia antigen IgG1kappa anti-
body produced by mouse B lymphocyte hybridoma L227, Catalog
no. HB-96, ATCC, Manassas, VA, USA), both native and
polyamine-modified, was reacted with APP and WT brain sections
at 2 lg/mL using the same immunohistochemical protocol as above
with the anti-Ab antibodies and fragments.
In vivo targeting
Amyloid precursor protein/PS1 transgenic mice (9 months of age)
were catheterized in the femoral vein under general anesthesia
(isoflurane, 1.5%) and injected with 200 lg (2 mCi) of 125I-IgG4.1,125I-pIgG4.1, 125I-F(ab¢)24.1, or 125I-pF(ab¢)24.1. After 4 h, each
animal was given an overdose of sodium pentobarbital (200 mg/kg,
IP) and perfused with PBS, followed by neutral-buffered, 10%
formalin, and then 10% sucrose in 0.1 mol/L sodium phosphate, pH
7.2. Brains were dissected out and snap frozen in 2-methylbutane
chilled in dry ice. Frozen sections (15 lm) of each brain were cut with
a cryostat throughout the whole extent of the cerebral cortex and then
processed with anti-Ab IH and emulsion autoradiography for the
presence of radiolabeled immunoglobulins, as described previously
(Wengenack et al. 2000; Poduslo et al. 2004). The sections first
underwent IH to reveal the locations of b-amyloid deposits using a
standard anti-Ab monoclonal mouse antibody (IgG4G8, 1 : 1000,
Signet Laboratories) and standard immunoperoxidase methods
(Vectastain Elite ABC Kit; Vector Laboratories) with 3,3¢-diam-
inobenzidine as a substrate. Next, the sections were dipped in an
autoradiographic emulsion (Type NTB-3; Kodak, Rochester, NY,
USA) for direct comparison of the location of the 125I-labeled
immunoglobulins in relation to the amyloid deposits. The slides were
dipped in emulsion, exposed for various durations, and developed
according to the instructions. The sections were dehydrated with
successive changes of ethanol and xylene and then coverslipped.
Silver grains from sections exposed for 4 weeks were then
quantitated using unbiased, stereological techniques. Silver grains
were counted over plaques and adjacent parenchyma in three
sections from each animal in the retrosplenial cortex and CA1 region
of the hippocampus using a 10-lm · 10-lm dissector at 400· by an
observer blind to the antibody injected. The mean background level
of exposed silver grains was also determined for each section. The
background was sampled over the emulsion-coated, blank slide
adjacent to the tissue section in the vicinity of the retrosplenial
cortex and hippocampus. The results were expressed as the mean
number of silver grains/100 lm2 minus the background. Statistical
analysis was then performed using two-way ANOVA followed by
Student–Newman–Keuls multiple comparisons.
Results
Sodium dodecyl sulfate-polyacrylamide gel
electrophoresis analysis of IgG4.1, papain-digested
Fab4.1, and ficin-digested F(ab¢)24.1 derivatives
Aliquots of purified IgG4.1, Fab4.1, and F(ab¢)24.1 were runon SDS-PAGE gels (15% T and 1% C) in the absence of areducing agent and stained with Coomassie blue. As shownin Fig. 1, single bands were observed for IgG4.1 at amolecular weight of approximately 150 000 Da, for Fab4.1at a molecular weight of approximately 50 000 Da, andfor F(ab¢)24.1 at an approximate molecular weight of102 000 Da, indicating complete digestion of the immuno-globulin. Furthermore, single bands indicated that no con-taminants, such as residual Fc fragments were present.
Isoelectric focusing
As determined by IEF, native IgG4.1 had a pI of approximately6.8. After polyamine modification, the pI shifted to the morebasic isoelectric point of approximately 7.3 and higher,indicating complete modification of the immunoglobulin(Fig. 2a). The pI of F(ab¢)2 4.1 was 7.7–7.9 (Fig. 2b). Modi-fication of F(ab¢)24.1 with putrescine at different pH values(7.8, 7.4, 7.0, 6.8, and 6.0) resulted in shifts in the isoelectricpoints that were increasingly more basic, respectively, dem-onstrating that increasing numbers of the carboxylic acidgroups undergo different degrees of polyamine modification.The progressive shift to more basic pH levels confirms thatincreasingly more polyamine is linked to F(ab¢)24.1 as the pHof the carbodiimide reaction is reduced and more carboxylicacid groups are ionized. Complete modification with thepolyamine was demonstrated at all five pH levels, includingpH 7.8. No residual unmodified F(ab¢)2 was observed at anypH, indicating that no further chromatography separation wasnecessary to separate native F(ab¢)2 from the pF(ab¢)2.
Targeting of antibody fragments to AD amyloid plaques 423
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 102, 420–433
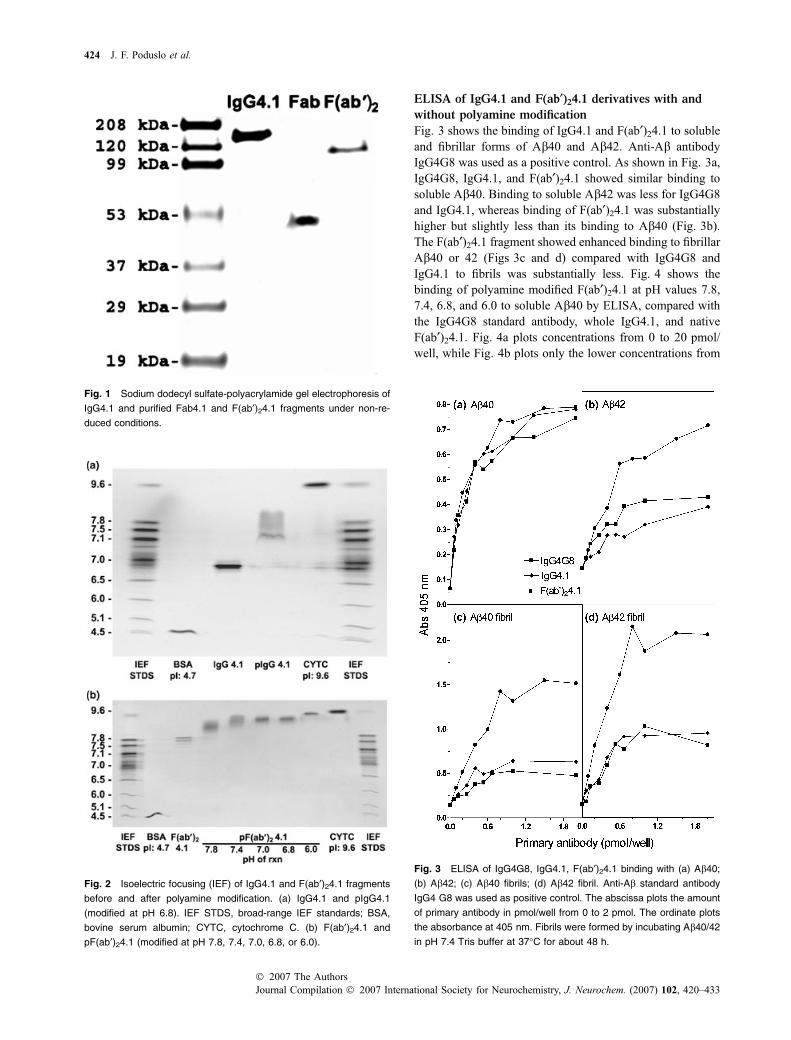
ELISA of IgG4.1 and F(ab¢)24.1 derivatives with and
without polyamine modification
Fig. 3 shows the binding of IgG4.1 and F(ab¢)24.1 to solubleand fibrillar forms of Ab40 and Ab42. Anti-Ab antibodyIgG4G8 was used as a positive control. As shown in Fig. 3a,IgG4G8, IgG4.1, and F(ab¢)24.1 showed similar binding tosoluble Ab40. Binding to soluble Ab42 was less for IgG4G8and IgG4.1, whereas binding of F(ab¢)24.1 was substantiallyhigher but slightly less than its binding to Ab40 (Fig. 3b).The F(ab¢)24.1 fragment showed enhanced binding to fibrillarAb40 or 42 (Figs 3c and d) compared with IgG4G8 andIgG4.1 to fibrils was substantially less. Fig. 4 shows thebinding of polyamine modified F(ab¢)24.1 at pH values 7.8,7.4, 6.8, and 6.0 to soluble Ab40 by ELISA, compared withthe IgG4G8 standard antibody, whole IgG4.1, and nativeF(ab¢)24.1. Fig. 4a plots concentrations from 0 to 20 pmol/well, while Fig. 4b plots only the lower concentrations from
Fig. 1 Sodium dodecyl sulfate-polyacrylamide gel electrophoresis of
IgG4.1 and purified Fab4.1 and F(ab¢)24.1 fragments under non-re-
duced conditions.
Fig. 2 Isoelectric focusing (IEF) of IgG4.1 and F(ab¢)24.1 fragments
before and after polyamine modification. (a) IgG4.1 and pIgG4.1
(modified at pH 6.8). IEF STDS, broad-range IEF standards; BSA,
bovine serum albumin; CYTC, cytochrome C. (b) F(ab¢)24.1 and
pF(ab¢)24.1 (modified at pH 7.8, 7.4, 7.0, 6.8, or 6.0).
Fig. 3 ELISA of IgG4G8, IgG4.1, F(ab¢)24.1 binding with (a) Ab40;
(b) Ab42; (c) Ab40 fibrils; (d) Ab42 fibril. Anti-Ab standard antibody
IgG4 G8 was used as positive control. The abscissa plots the amount
of primary antibody in pmol/well from 0 to 2 pmol. The ordinate plots
the absorbance at 405 nm. Fibrils were formed by incubating Ab40/42
in pH 7.4 Tris buffer at 37�C for about 48 h.
424 J. F. Poduslo et al.
Journal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 102, 420–433� 2007 The Authors
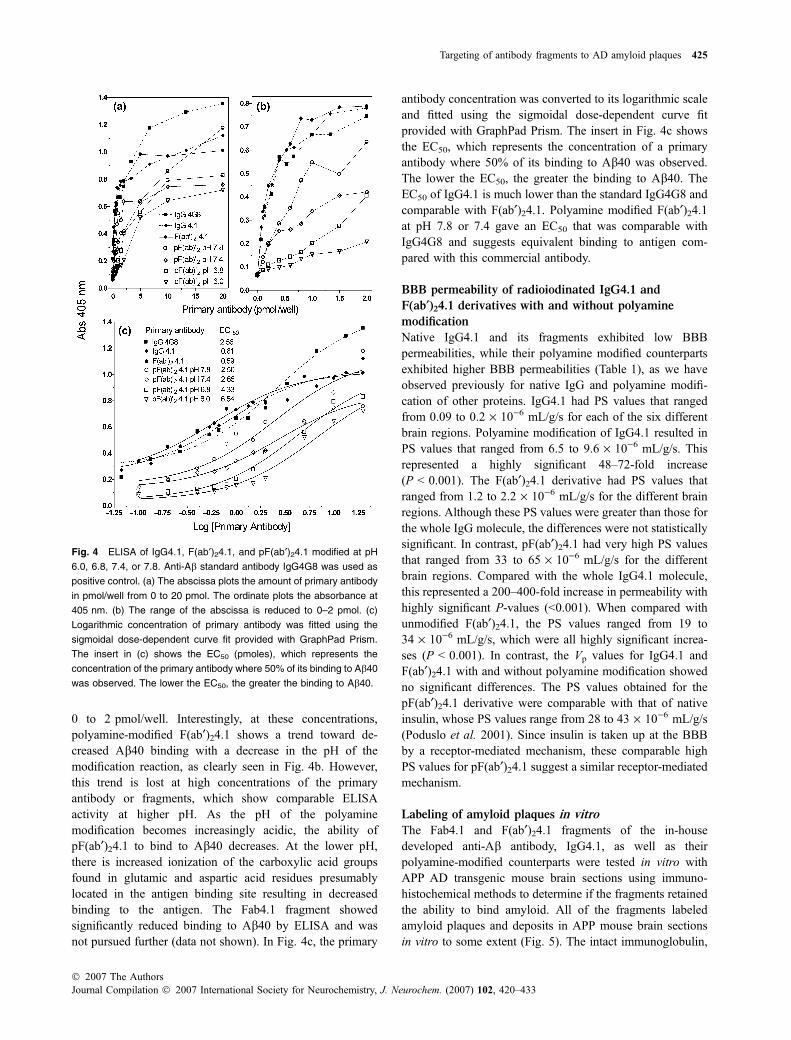
0 to 2 pmol/well. Interestingly, at these concentrations,polyamine-modified F(ab¢)24.1 shows a trend toward de-creased Ab40 binding with a decrease in the pH of themodification reaction, as clearly seen in Fig. 4b. However,this trend is lost at high concentrations of the primaryantibody or fragments, which show comparable ELISAactivity at higher pH. As the pH of the polyaminemodification becomes increasingly acidic, the ability ofpF(ab¢)24.1 to bind to Ab40 decreases. At the lower pH,there is increased ionization of the carboxylic acid groupsfound in glutamic and aspartic acid residues presumablylocated in the antigen binding site resulting in decreasedbinding to the antigen. The Fab4.1 fragment showedsignificantly reduced binding to Ab40 by ELISA and wasnot pursued further (data not shown). In Fig. 4c, the primary
antibody concentration was converted to its logarithmic scaleand fitted using the sigmoidal dose-dependent curve fitprovided with GraphPad Prism. The insert in Fig. 4c showsthe EC50, which represents the concentration of a primaryantibody where 50% of its binding to Ab40 was observed.The lower the EC50, the greater the binding to Ab40. TheEC50 of IgG4.1 is much lower than the standard IgG4G8 andcomparable with F(ab¢)24.1. Polyamine modified F(ab¢)24.1at pH 7.8 or 7.4 gave an EC50 that was comparable withIgG4G8 and suggests equivalent binding to antigen com-pared with this commercial antibody.
BBB permeability of radioiodinated IgG4.1 and
F(ab¢)24.1 derivatives with and without polyamine
modification
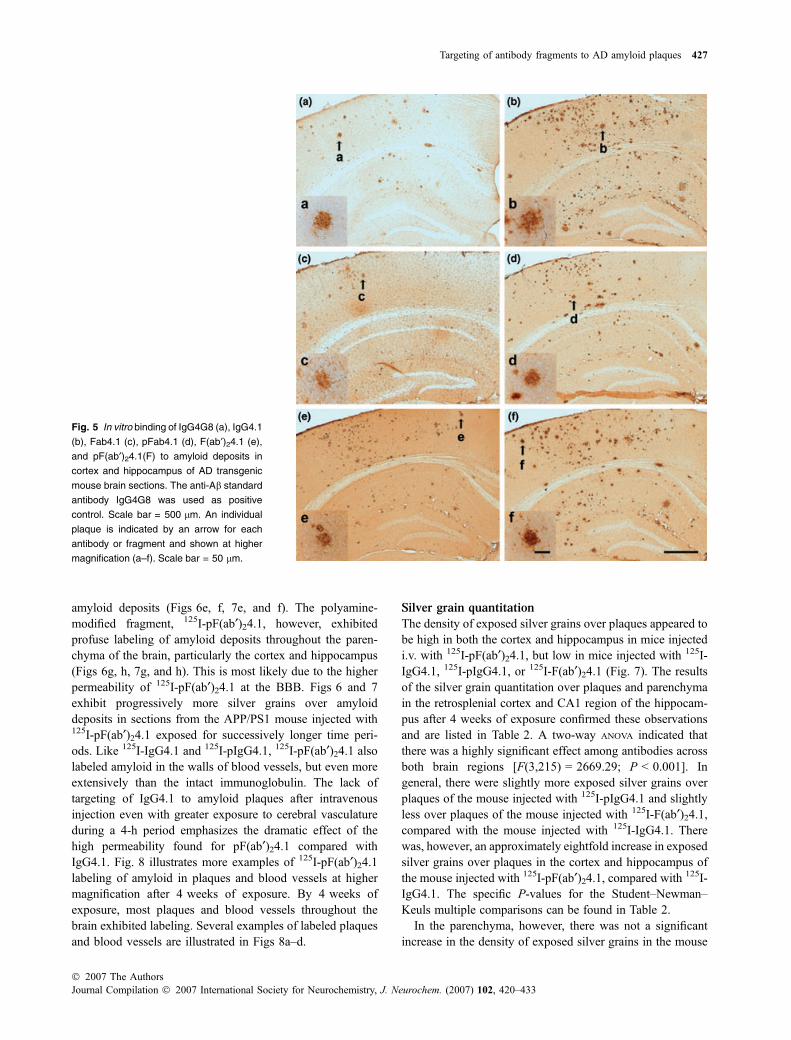
Native IgG4.1 and its fragments exhibited low BBBpermeabilities, while their polyamine modified counterpartsexhibited higher BBB permeabilities (Table 1), as we haveobserved previously for native IgG and polyamine modifi-cation of other proteins. IgG4.1 had PS values that rangedfrom 0.09 to 0.2 · 10)6 mL/g/s for each of the six differentbrain regions. Polyamine modification of IgG4.1 resulted inPS values that ranged from 6.5 to 9.6 · 10)6 mL/g/s. Thisrepresented a highly significant 48–72-fold increase(P < 0.001). The F(ab¢)24.1 derivative had PS values thatranged from 1.2 to 2.2 · 10)6 mL/g/s for the different brainregions. Although these PS values were greater than those forthe whole IgG molecule, the differences were not statisticallysignificant. In contrast, pF(ab¢)24.1 had very high PS valuesthat ranged from 33 to 65 · 10)6 mL/g/s for the differentbrain regions. Compared with the whole IgG4.1 molecule,this represented a 200–400-fold increase in permeability withhighly significant P-values (<0.001). When compared withunmodified F(ab¢)24.1, the PS values ranged from 19 to34 · 10)6 mL/g/s, which were all highly significant increa-ses (P < 0.001). In contrast, the Vp values for IgG4.1 andF(ab¢)24.1 with and without polyamine modification showedno significant differences. The PS values obtained for thepF(ab¢)24.1 derivative were comparable with that of nativeinsulin, whose PS values range from 28 to 43 · 10)6 mL/g/s(Poduslo et al. 2001). Since insulin is taken up at the BBBby a receptor-mediated mechanism, these comparable highPS values for pF(ab¢)24.1 suggest a similar receptor-mediatedmechanism.
Labeling of amyloid plaques in vitro
The Fab4.1 and F(ab¢)24.1 fragments of the in-housedeveloped anti-Ab antibody, IgG4.1, as well as theirpolyamine-modified counterparts were tested in vitro withAPP AD transgenic mouse brain sections using immuno-histochemical methods to determine if the fragments retainedthe ability to bind amyloid. All of the fragments labeledamyloid plaques and deposits in APP mouse brain sectionsin vitro to some extent (Fig. 5). The intact immunoglobulin,
Fig. 4 ELISA of IgG4.1, F(ab¢)24.1, and pF(ab¢)24.1 modified at pH
6.0, 6.8, 7.4, or 7.8. Anti-Ab standard antibody IgG4G8 was used as
positive control. (a) The abscissa plots the amount of primary antibody
in pmol/well from 0 to 20 pmol. The ordinate plots the absorbance at
405 nm. (b) The range of the abscissa is reduced to 0–2 pmol. (c)
Logarithmic concentration of primary antibody was fitted using the
sigmoidal dose-dependent curve fit provided with GraphPad Prism.
The insert in (c) shows the EC50 (pmoles), which represents the
concentration of the primary antibody where 50% of its binding to Ab40
was observed. The lower the EC50, the greater the binding to Ab40.
Targeting of antibody fragments to AD amyloid plaques 425
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 102, 420–433
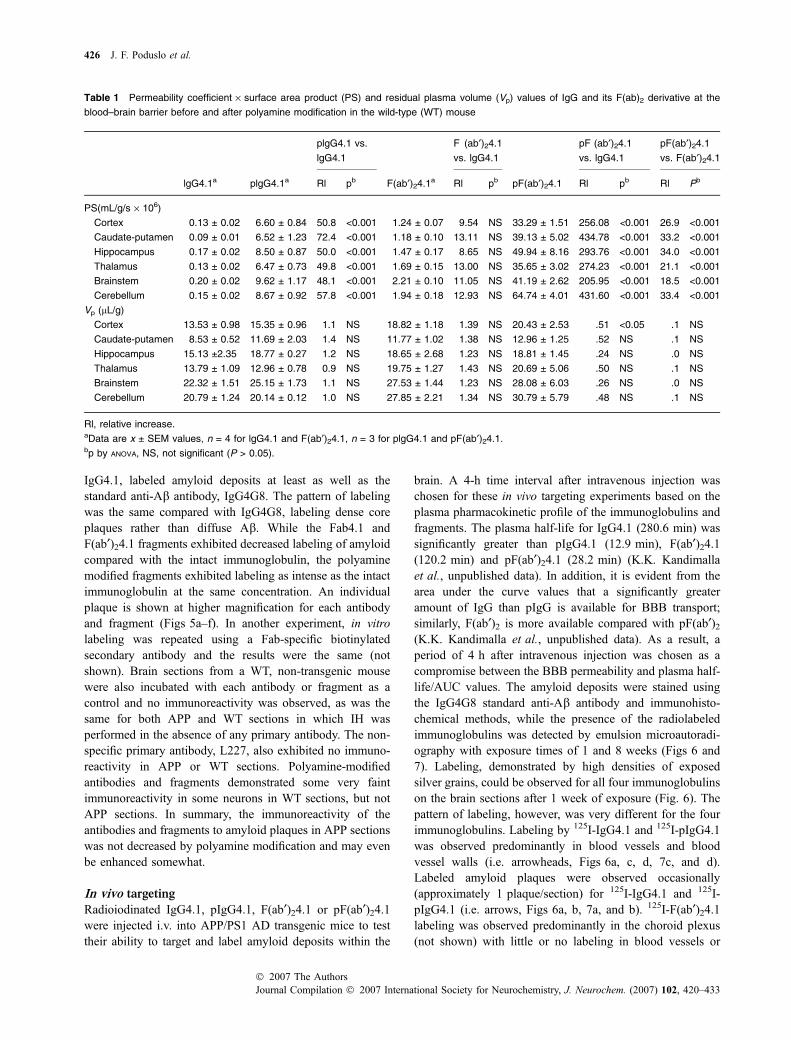
IgG4.1, labeled amyloid deposits at least as well as thestandard anti-Ab antibody, IgG4G8. The pattern of labelingwas the same compared with IgG4G8, labeling dense coreplaques rather than diffuse Ab. While the Fab4.1 andF(ab¢)24.1 fragments exhibited decreased labeling of amyloidcompared with the intact immunoglobulin, the polyaminemodified fragments exhibited labeling as intense as the intactimmunoglobulin at the same concentration. An individualplaque is shown at higher magnification for each antibodyand fragment (Figs 5a–f). In another experiment, in vitrolabeling was repeated using a Fab-specific biotinylatedsecondary antibody and the results were the same (notshown). Brain sections from a WT, non-transgenic mousewere also incubated with each antibody or fragment as acontrol and no immunoreactivity was observed, as was thesame for both APP and WT sections in which IH wasperformed in the absence of any primary antibody. The non-specific primary antibody, L227, also exhibited no immuno-reactivity in APP or WT sections. Polyamine-modifiedantibodies and fragments demonstrated some very faintimmunoreactivity in some neurons in WT sections, but notAPP sections. In summary, the immunoreactivity of theantibodies and fragments to amyloid plaques in APP sectionswas not decreased by polyamine modification and may evenbe enhanced somewhat.
In vivo targeting
Radioiodinated IgG4.1, pIgG4.1, F(ab¢)24.1 or pF(ab¢)24.1were injected i.v. into APP/PS1 AD transgenic mice to testtheir ability to target and label amyloid deposits within the
brain. A 4-h time interval after intravenous injection waschosen for these in vivo targeting experiments based on theplasma pharmacokinetic profile of the immunoglobulins andfragments. The plasma half-life for IgG4.1 (280.6 min) wassignificantly greater than pIgG4.1 (12.9 min), F(ab¢)24.1(120.2 min) and pF(ab¢)24.1 (28.2 min) (K.K. Kandimallaet al., unpublished data). In addition, it is evident from thearea under the curve values that a significantly greateramount of IgG than pIgG is available for BBB transport;similarly, F(ab¢)2 is more available compared with pF(ab¢)2(K.K. Kandimalla et al., unpublished data). As a result, aperiod of 4 h after intravenous injection was chosen as acompromise between the BBB permeability and plasma half-life/AUC values. The amyloid deposits were stained usingthe IgG4G8 standard anti-Ab antibody and immunohisto-chemical methods, while the presence of the radiolabeledimmunoglobulins was detected by emulsion microautoradi-ography with exposure times of 1 and 8 weeks (Figs 6 and7). Labeling, demonstrated by high densities of exposedsilver grains, could be observed for all four immunoglobulinson the brain sections after 1 week of exposure (Fig. 6). Thepattern of labeling, however, was very different for the fourimmunoglobulins. Labeling by 125I-IgG4.1 and 125I-pIgG4.1was observed predominantly in blood vessels and bloodvessel walls (i.e. arrowheads, Figs 6a, c, d, 7c, and d).Labeled amyloid plaques were observed occasionally(approximately 1 plaque/section) for 125I-IgG4.1 and 125I-pIgG4.1 (i.e. arrows, Figs 6a, b, 7a, and b). 125I-F(ab¢)24.1labeling was observed predominantly in the choroid plexus(not shown) with little or no labeling in blood vessels or
Table 1 Permeability coefficient · surface area product (PS) and residual plasma volume (Vp) values of IgG and its F(ab)2 derivative at the
blood–brain barrier before and after polyamine modification in the wild-type (WT) mouse
plgG4.1 vs.
lgG4.1
F (ab¢)24.1
vs. lgG4.1
pF (ab¢)24.1
vs. lgG4.1
pF(ab¢)24.1
vs. F(ab¢)24.1
lgG4.1a plgG4.1a Rl pb F(ab¢)24.1a Rl pb pF(ab¢)24.1 Rl pb Rl Pb
PS(mL/g/s · 106)
Cortex 0.13 ± 0.02 6.60 ± 0.84 50.8 <0.001 1.24 ± 0.07 9.54 NS 33.29 ± 1.51 256.08 <0.001 26.9 <0.001
Caudate-putamen 0.09 ± 0.01 6.52 ± 1.23 72.4 <0.001 1.18 ± 0.10 13.11 NS 39.13 ± 5.02 434.78 <0.001 33.2 <0.001
Hippocampus 0.17 ± 0.02 8.50 ± 0.87 50.0 <0.001 1.47 ± 0.17 8.65 NS 49.94 ± 8.16 293.76 <0.001 34.0 <0.001
Thalamus 0.13 ± 0.02 6.47 ± 0.73 49.8 <0.001 1.69 ± 0.15 13.00 NS 35.65 ± 3.02 274.23 <0.001 21.1 <0.001
Brainstem 0.20 ± 0.02 9.62 ± 1.17 48.1 <0.001 2.21 ± 0.10 11.05 NS 41.19 ± 2.62 205.95 <0.001 18.5 <0.001
Cerebellum 0.15 ± 0.02 8.67 ± 0.92 57.8 <0.001 1.94 ± 0.18 12.93 NS 64.74 ± 4.01 431.60 <0.001 33.4 <0.001
Vp (lL/g)
Cortex 13.53 ± 0.98 15.35 ± 0.96 1.1 NS 18.82 ± 1.18 1.39 NS 20.43 ± 2.53 .51 <0.05 .1 NS
Caudate-putamen 8.53 ± 0.52 11.69 ± 2.03 1.4 NS 11.77 ± 1.02 1.38 NS 12.96 ± 1.25 .52 NS .1 NS
Hippocampus 15.13 ±2.35 18.77 ± 0.27 1.2 NS 18.65 ± 2.68 1.23 NS 18.81 ± 1.45 .24 NS .0 NS
Thalamus 13.79 ± 1.09 12.96 ± 0.78 0.9 NS 19.75 ± 1.27 1.43 NS 20.69 ± 5.06 .50 NS .1 NS
Brainstem 22.32 ± 1.51 25.15 ± 1.73 1.1 NS 27.53 ± 1.44 1.23 NS 28.08 ± 6.03 .26 NS .0 NS
Cerebellum 20.79 ± 1.24 20.14 ± 0.12 1.0 NS 27.85 ± 2.21 1.34 NS 30.79 ± 5.79 .48 NS .1 NS
Rl, relative increase.aData are x ± SEM values, n = 4 for lgG4.1 and F(ab¢)24.1, n = 3 for plgG4.1 and pF(ab¢)24.1.bp by ANOVA, NS, not significant (P > 0.05).
426 J. F. Poduslo et al.
Journal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 102, 420–433� 2007 The Authors
amyloid deposits (Figs 6e, f, 7e, and f). The polyamine-modified fragment, 125I-pF(ab¢)24.1, however, exhibitedprofuse labeling of amyloid deposits throughout the paren-chyma of the brain, particularly the cortex and hippocampus(Figs 6g, h, 7g, and h). This is most likely due to the higherpermeability of 125I-pF(ab¢)24.1 at the BBB. Figs 6 and 7exhibit progressively more silver grains over amyloiddeposits in sections from the APP/PS1 mouse injected with125I-pF(ab¢)24.1 exposed for successively longer time peri-ods. Like 125I-IgG4.1 and 125I-pIgG4.1, 125I-pF(ab¢)24.1 alsolabeled amyloid in the walls of blood vessels, but even moreextensively than the intact immunoglobulin. The lack oftargeting of IgG4.1 to amyloid plaques after intravenousinjection even with greater exposure to cerebral vasculatureduring a 4-h period emphasizes the dramatic effect of thehigh permeability found for pF(ab¢)24.1 compared withIgG4.1. Fig. 8 illustrates more examples of 125I-pF(ab¢)24.1labeling of amyloid in plaques and blood vessels at highermagnification after 4 weeks of exposure. By 4 weeks ofexposure, most plaques and blood vessels throughout thebrain exhibited labeling. Several examples of labeled plaquesand blood vessels are illustrated in Figs 8a–d.
Silver grain quantitation
The density of exposed silver grains over plaques appeared tobe high in both the cortex and hippocampus in mice injectedi.v. with 125I-pF(ab¢)24.1, but low in mice injected with 125I-IgG4.1, 125I-pIgG4.1, or 125I-F(ab¢)24.1 (Fig. 7). The resultsof the silver grain quantitation over plaques and parenchymain the retrosplenial cortex and CA1 region of the hippocam-pus after 4 weeks of exposure confirmed these observationsand are listed in Table 2. A two-way ANOVA indicated thatthere was a highly significant effect among antibodies acrossboth brain regions [F(3,215) = 2669.29; P < 0.001]. Ingeneral, there were slightly more exposed silver grains overplaques of the mouse injected with 125I-pIgG4.1 and slightlyless over plaques of the mouse injected with 125I-F(ab¢)24.1,compared with the mouse injected with 125I-IgG4.1. Therewas, however, an approximately eightfold increase in exposedsilver grains over plaques in the cortex and hippocampus ofthe mouse injected with 125I-pF(ab¢)24.1, compared with 125I-IgG4.1. The specific P-values for the Student–Newman–Keuls multiple comparisons can be found in Table 2.
In the parenchyma, however, there was not a significantincrease in the density of exposed silver grains in the mouse
Fig. 5 In vitro binding of IgG4G8 (a), IgG4.1
(b), Fab4.1 (c), pFab4.1 (d), F(ab¢)24.1 (e),
and pF(ab¢)24.1(F) to amyloid deposits in
cortex and hippocampus of AD transgenic
mouse brain sections. The anti-Ab standard
antibody IgG4G8 was used as positive
control. Scale bar = 500 lm. An individual
plaque is indicated by an arrow for each
antibody or fragment and shown at higher
magnification (a–f). Scale bar = 50 lm.
Targeting of antibody fragments to AD amyloid plaques 427
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 102, 420–433

injected with 125I-pF(ab¢)24.1 compared with the mouseinjected with 125I-IgG4.1. While a two-way ANOVA indicatedthat there was a statistically significant overall effect acrossantibodies [F(3,23) = 19.64; P < 0.001], Student–Newman–Keuls multiple comparisons revealed that this was due to aslight, but statistically significant decrease in the density ofexposed silver grains in the parenchyma of the mouseinjected with 125I-F(ab¢)24.1. There was not a statisticallysignificant difference between the densities of exposed silvergrains in the parenchyma of the mouse injected with 125I-IgG4.1 and the mouse injected with 125I-pF(ab¢)24.1. Thespecific P-values for the Student–Newman–Keuls multiple
comparisons can be found in Table 2. This indicates that the125I-pF(ab¢)24.1 specifically targeted plaques without in-creased background or non-specific labeling of the paren-chyma.
Discussion
The results of this study provide support for the developmentof pF(ab¢)24.1 as a potential molecular imaging agent forAD, as well as an attractive alternative to conventionalantibody-based therapies for AD. Polyamine modificationincreased the BBB permeability of pF(ab¢)24.1 compared
Fig. 6 In vivo labeling of amyloid deposits
in Alzheimer’s disease transgenic mouse
brain following i.v. injection of 125I-IgG4.1,125I-pIgG4.1, 125I-F(ab¢)24.1, or 125I-pF(ab¢)24.1.
Photomicrographs of histological sections
processed for IgG4G8 anti-Ab immuno-
histochemistry and emulsion autoradiogra-
phy with 1 week of exposure. 125I-IgG4.1
labeling in cortex (a). Arrows indicate
labeled plaques. Arrowheads indicate
labeled blood vessels. Scale bar = 50 lm.125I-IgG4.1 labeling in hippocampus (b).125I-pIgG4.1 labeling in cortex (c) and hip-
pocampus (d). 125I-F(ab¢)24.1 labeling in cor-
tex (e) and hippocampus (f). 125I-pF(ab¢)24.1
labeling in cortex (g) and hippocampus (h).
428 J. F. Poduslo et al.
Journal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 102, 420–433� 2007 The Authors

with IgG4.1, pIgG4.1, and F(ab¢)24.1 without reducing itsantigen binding in vitro as demonstrated by either ELISA orIH on AD transgenic mouse brain sections. Furthermore,following i.v. injection, 125I-pF(ab¢)24.1 extensively labeledamyloid deposits in AD transgenic mouse brain parenchyma,as detected by IgG4G8 IH and emulsion autoradiography. Incontrast, 125I-IgG4.1, 125I-pIgG4.1, and 125I-F(ab¢)24.1 labe-led predominantly blood vessels and cerebrovascular amy-loid (CVA) and very few amyloid deposits. 125I-pF(ab¢)24.1demonstrated even more extensive labeling of CVA than 125I-IgG4.1, 125I-pIgG4.1, or 125I-F(ab¢)24.1. These results sup-port the concept that IgG permeability at the BBB is very lowwith only limited delivery to the brain parenchyma. Clearly,
polyamine modification significantly enhances that permeab-ility.
These results are not the first report of the use of labeledantibodies or antibody fragments as potential diagnosticimaging agents. Numerous articles exist in the cancerliterature that report the imaging of tumors in the brain andperiphery of animals and humans using radiolabeled anti-bodies, as well as F(ab¢)2 and Fab fragments (e.g. Lamonicaet al. 2002; Vicente et al. 2004). The imaging of CVA in ADpatients has also been attempted unsuccessfully with aradiolabeled anti-Ab Fab fragment (Friedland et al. 1997).The successful imaging of brain tumors, but not CVA withantibodies and antibody fragments is most likely due to the
Fig. 7 In vivo labeling of amyloid deposits
in Alzheimer’s disease transgenic mouse
brain following i.v. injection of 125I-IgG4.1,125I-pIgG4.1, 125I-F(ab¢)24.1, or 125I-pF(ab¢)24.1.
Photomicrographs of histological sections
processed for IgG4G8 anti-Ab immunohist-
ochemistry and emulsion autoradiography
with 8 weeks of exposure. 125I-IgG4.1
labeling in cortex (a). Arrows indicate labe-
led plaques. Scale bar = 50 lm. 125I-IgG4.1
labeling in hippocampus (b). 125I-pIgG4.1
labeling in cortex (c) and hippocampus (d).125I-F(ab¢)24.1 labeling in cortex (e) and
hippocampus (f). 125I-pF(ab¢)24.1 labeling in
cortex (g) and hippocampus (h).
Targeting of antibody fragments to AD amyloid plaques 429
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 102, 420–433

BBB. Brain tumors have little or no BBB and can be imagedby magnetic resonance imaging (MRI) using commerciallyavailable gadolinium-labelled contrast agent, which has verylow BBB permeability. Although historically a topic of muchdebate, it has become generally thought that the BBBremains largely intact in early AD and we have reported hereand elsewhere that antibodies and their fragments have lowBBB permeability (Poduslo et al. 1994; Poduslo and Curran1996, 2001).
Another possible explanation for the failure of earlierantibody probes to image AD amyloid is low spatialresolution. Many of the previous studies were performedusing SPECT and radionuclides such as Tc-99 m withrelatively short half-lives. MicroSPECT, and microPET as
well, have a maximum spatial resolution of approximately2-5 mm. The use of an antibody fragment-based contrastagent with MRI has a higher probability of success inimaging AD amyloid, particularly individual plaques, due tothe higher resolution of MRI (30–50 lm). Our laboratory hasimaged individual amyloid deposits in AD transgenic mousebrain ex vivo with MRI following i.v. injection of apolyamine modified Ab peptide-based contrast agent (Podu-slo et al. 2002). While we have successfully MRI individualamyloid plaques with a diameter of 35 lm in vivo in ADtransgenic mice using a T2-weighted spin echo sequencewithout the aid of a contrast agent (Jack et al. 2004, 2005),thus far, specific labeling and enhancement of plaques usinga polyamine modified Ab peptide-based contrast agent
Fig. 8 In vivo labeling of amyloid deposits
in Alzheimer’s disease transgenic mouse
brain cortex following i.v. injection of125I-pF(ab¢)24.1 at higher magnification.
Photomicrographs of histological sections
processed for IgG4G8 anti-Ab immuno-
histochemistry and emulsion autoradio-
graphy with 4 weeks of exposure. P
indicates labeled amyloid plaques. BV
indicates labeled blood vessels. (a) Scale
bar = 200 lm. (b) Scale bar = 100 lm. (c)
Scale bar = 50 lm. (d) Scale bar = 50 lm.
Table 2 Quantitation of exposed silver grains in autoradiographic emulsion on brain sections of amyloid precursor protein (APP)/human pres-
enilin 1 (PS1) mice injected i.v. with 125I-IgG4.1, 125I-pIgG4.1, 125I-F(ab¢)24.1, or 125I-pF(ab¢)24.1
Plaquesa Parenchymab
Cortex Hippocampus Cortex Hippocampus
Antibody �x ± SEM p vs. IgG �x ± SEM p vs. IgG �x ± SEM p vs. IgG �x ± SEM p vs. IgG
lgG4.1 17.26 ± 1.27 – 21.79 ± 2.42 – 1.57 ± 0.07 – 1.66 ± 0.11 –
plgG4.1 24.46 ± 1.37 ** 24.76 ± 1.63 ns 2.16 ± 0.17 ns 2.14 ± 0.35 ns
F(ab¢)24.1 9.35 ± 0.62 ** 12.05 ± 0.78 *** 0.74 ± 0.19 * 0.64 ± 0.21 **
pF(ab¢)24.1 152.89 ± 1.90 *** 143.91 ± 2.99 *** 2.09 + 0.12 ns 1.70 ± 0.26 ns
aMean (±SEM) exposed silver grains per 100 lm2 over amyloid plaques in cortex or hippocampus. ANOVA (two-way: antibody vs. brain region):
antibody – [F(3,215) = 2669.29; P < 0.001]; brain region – [F(1,215) = 0.08; ns]; antibody · brain region – [F (3,215) = 5.63; P < 0.001] followed
by Student–Newman–Keuls multiple comparisons (p vs. lgG4.1: ns, not significant; **P < 0.01; ***P < 0.001).bMean (±SEM) exposed silver grains per 100 lm2 over parenchyma in cortex or hippocampus. ANOVA (two-way: antibody vs. brain region):
antibody – [F(3,23) = 19.64; P < 0.001]; brain region – [F(1,23) = 0.54; ns]; antibody · brain region – [F(3,23) = 0.53; ns] followed by Student–
Newman–Keuls multiple comparisons (p vs. lgG4.1: ns, not significant; *P < 0.05; **P < 0.01).
430 J. F. Poduslo et al.
Journal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 102, 420–433� 2007 The Authors

in vivo has had limited success. T2-weighted images,however, are susceptible to many artifacts such as bloodvessels and white matter tracts due to their high iron content,which has high T2 relaxivity. The use of a Gd-labeledcontrast agent and a T1-weighted spin echo sequence wouldminimize artifacts due to endogenous iron, since Gd has highT1 relaxivity while iron has low T1 relaxivity. The results ofthe present study are promising for the development ofpF(ab¢)24.1 as an in vivo MRI contrast agent becausepF(ab¢)24.1 exhibits even more extensive labeling of amyloiddeposits in AD transgenic mouse brain following i.v.injection as demonstrated in the present study comparedwith the polyamine modified Ab peptide-based contrastagent (Poduslo et al. 2004).
The ability of pF(ab¢)24.1 to bind to CVA as well as amyloidplaques may make this a useful MRI contrast agent fordistinguishing patients with mild cognitive imparement /ADwhere binding to vessels as well as plaques might be observedfrom those with cerebral amyloid angiopathy (CAA) whereonly binding to vessels might be seen. There also may beimplications for other types of amyloid angiopathy outside thecentral nervous system. A recent report (Pichelmayer et al.2005) described the use of a monoclonal antibody againstvascular primary cardiac amyloidosis that had severe dyspneaand underwent repeated thoracenteses for pleural effusions.VEGF inhibitors are typically considered antiangiogenesisagents and their use in constricting blood supply to coloncancer has been documented. The damage caused by theamyloid in blood vessels in primary amyloidosis is not wellunderstood, but based on this report by Pichelmayer et al.(2005), it is suggested that anti-VEGF may help constrict theamyloid-infested blood-vessels, ameliorating the ‘leakiness’effect that the amyloidmay cause on the vessels and improvingthe condition of the patients. By having a method to detectamyloid in blood vessels, the opportunities for promptintervention will increase. The development of antibodyfragments against other forms of amyloid may ultimately beuseful as contrast agents for the MRI of amyloid in bloodvessels to facilitate such intervention.
The attractiveness of using pF(ab¢)24.1 for passiveimmunization therapy in AD is attributed to its high BBBpermeability and the extensive targeting to amyloid plaquesthroughout the brain parenchyma following i.v. injection.This was not observed for either F(ab¢)24.1 or the wholeimmunoglobulin, IgG4.1 or pIgG4.1. Because of this lowimmunoglobulin permeability at the BBB, the success ofactive or passive immunization in AD transgenic mice stillremains surprising. The reduction of amyloid burden as aresult of passive immunization is clearly due in part to theuniqueness of the antibody used since some monoclonalantibodies effectively reduce amyloid plaque and clearsoluble species of Ab, whereas others do not. The notionthat N-terminal, mid-domain, or C-terminal antibodies willhave unique group specific effects that are distinct from one
another is likely wrong (Morgan and Gitter 2004). Sucheffects are likely antibody idiosyncratic. Alternatively, thesuccess or failure of different antibodies for clearing plaquesand/or reducing soluble Ab oligomers may operate at amechanistic level. Perhaps the successful antibodies haveuniquely high permeabilities at the BBB which allowefficient entry into the brain parenchyma. It would be ofinterest, therefore, to compare the BBB permeabilities ofthese antibodies compared with the unsuccessful antibodiesor even to antibody m266, which appears to work only in theperipheral circulation by acting as a sink to remove Ab fromthe brain parenchyma (DeMattos et al. 2001). Such anantibody might have an even lower BBB permeability.Unfortunately, many of the successful antibodies in use todayare not commercially available and are not being madeavailable by the pharmaceutical industry, presumablybecause they are in clinical trials, even though their usehas been documented in the scientific literature. Of course,the ability of the different antibodies to form immunecomplexes with circulating Ab may directly affect its BBBpermeability (Poduslo and Curran 2001; Kandimalla et al.2005). We have previously reported that IgG–Ab immunecomplexes have higher BBB permeability than IgG alone,suggesting that the success of passive immunization therapymay be facilitated by receptor-mediated transport into thebrain by an Ab transporter (Poduslo and Curran 2001).
The polyamine modified F(ab¢)24.1 should be furtherinvestigated for passive immunization therapy in AD. Theuse of the immunoglobulin fragment, F(ab¢)24.1 eliminatespotential problems related to the interaction of the Fc regionof the whole antibody with effector molecules such ascomplement in serum or Fc receptors expressed on a varietyof immune cells in the periphery, as well as inflammatorycells in the central nervous system, such as microglia. This isparticularly important in AD immunotherapy as the Fc regionhas been implicated in undesirable inflammatory reactionsand cerebral hemorrhaging. The Fc fragment reacts withdistinct Fcc receptors to mediate pro- and anti-inflammatoryactivities of whole IgG. It has recently been shown thatdifferent Fc sialylation provides a switch from innate anti-inflammatory activity to generate pro-inflammatory effectsafter antigenic challenge (Kaneko et al. 2006). F(ab¢)2fragments have been demonstrated to clear amyloid depositsas well as the full length antibody (Bacskai et al. 2002).Moreover, Wilcock et al. (2004) demonstrated that anti-AbF(ab¢)2 fragments failed to activate microglia, were efficientat clearing diffuse Ab deposits, but were less efficient inremoving fibrillar amyloid.
Single-chain variable fragments (scFv) against Ab havebeen shown to react with Ab peptide (Tammer et al. 2002)and inhibit Ab aggregation and prevent Ab-induced neuro-toxicity in vitro (Liu et al. 2004). Such scFv’s were isolatedfrom phage display libraries, and while two scFv’s to non-amino terminal regions of Ab inhibited both AD aggregation
Targeting of antibody fragments to AD amyloid plaques 431
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 102, 420–433

and cytotoxicity, they were not tested further for their abilityto reduce amyloid plaques in AD transgenic mice (Liu et al.2004). Clearly, the development of humanized scFv’s is animportant direction for immunotherapy in AD patients.Furthermore, the production of high avidity scFv multimers,diabodies, and triabodies (Hudson and Kortt 1999) mightallow such multifunction fragments to first bind to differentAb species and plaques and then secondly affect theirefficient clearance from the brain. Of course, polyaminemodification of these multifunction fragments would facili-tate their delivery into the brain, as well as enhance theirbinding to different Ab species and amyloid plaques.
In summary, pF(ab¢)24.1 exhibits increased permeability atthe BBB and preserved antigen binding in vitro. Moreover,125I-pF(ab¢)24.1 extensively labeled amyloid deposits in ADtransgenic mouse brain parenchyma following i.v. injection.Coupling contrast agents or appropriate radioisotopes topolyamine-modified F(ab¢)24.1 might facilitate the molecularimaging of amyloid deposits using MRI or micro-PET.Polyamine-modified F(ab¢)24.1 might also increase theeffectiveness of AD immunotherapy. This technique notonly has potential applications for diagnostic imaging andimmunotherapy in AD, but possibly many other neurologicaldisorders or for the generalized targeting of nervous systemantigens to which an antibody can be raised.
Acknowlegements
The authors would like to thank Dr Karen Duff for the PS1
transgenic mouse line and Robert Sikkink, Dawn Gregor, and
Jennifer Scott for their expert technical assistance. Support was
provided by NIH AG22034 and the Mayo Foundation.
References
Bacskai B. J., Kajdasz S. T., McLellan M. E., Games D., Seubert P.,Schenk D. and Hyman B. T. (2002) Non-Fc-mediated mechanismsare involved in clearance of amyloid-beta in vivo by immuno-therapy. J. Neurosci. 22, 7873–7878.
Bard F., Cannon C., Barbour R. et al. (2000) Peripherally administeredantibodies against amyloid beta-peptide enter the central nervoussystem and reduce pathology in a mouse model of Alzheimerdisease. Nat. Med. 6, 916–919.
DeMattos R. B., Bales K. R., Cummins D. J., Dodart J. C., Paul S. M.and Holtzman D. M. (2001) Peripheral anti-A beta antibody altersCNS and plasma A beta clearance and decreases brain A betaburden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad.Sci. U. S. A. 98, 8850–8855.
Dodart J. C., Bales K. R., Gannon K. S. et al. (2002) Immunizationreverses memory deficits without reducing brain Abeta burden inAlzheimer’s disease model. Nat. Neurosci. 5, 452–457.
Frenkel D., Katz O. and Solomon B. (2000) Immunization againstAlzheimer’s beta -amyloid plaques via EFRH phage administra-tion. Proc. Natl. Acad. Sci. U. S. A. 97, 11455–11459.
Friedland R. P., Kalaria R., Berridge M., Miraldi F., Hedera P., Reno J.,Lyle L. and Marotta C. A. (1997) Neuroimaging of vessel amyloidin Alzheimer’s disease. Ann. N. Y. Acad. Sci. 826, 242–247.
Furlan R., Brambilla E., Sanvito F., Roccatagliata L., Olivieri S., Ber-gami A., Pluchino S., Uccelli A., Comi G. and Martino G. (2003)Vaccination with amyloid-beta peptide induces autoimmuneencephalomyelitis in C57/BL6 mice. Brain 126, 285–291.
Grubeck-Loebenstein B., Blasko I., Marx F. K. and Trieb I. (2000)Immunization with beta-amyloid: could T-cell activation have aharmful effect? Trends Neurosci. 23, 114.
Holcomb L., Gordon M. N., McGowan E. et al. (1998) AcceleratedAlzheimer-type phenotype in transgenic mice carrying both mutantamyloid precursor protein and presenilin 1 transgenes. Nat. Med. 4,97–100.
Hsiao K., Chapman P., Nilsen S., Eckman C., Harigaya Y., Younkin S.,Yang F. and Cole G. (1996) Correlative memory deficits, Abetaelevation, and amyloid plaques in transgenic mice. Science 274,99–102.
Hudson P. J. and Kortt A. A. (1999) High avidity scFv multimers;diabodies and triabodies. J. Immunol. Methods 231, 177–189.
Imbimbo B. P. (2002) Toxicity of beta-amyloid vaccination in patientswith Alzheimer’s disease. Ann. Neurol. 51, 794.
Jack C. R., Jr, Garwood M., Wengenack T. M., Borowski B., Curran G.L., Lin J., Adriany G., Grohn O. H., Grimm R. and Poduslo J. F.(2004) In vivo visualization of Alzheimer’s amyloid plaques bymagnetic resonance imaging in transgenic mice without a contrastagent. Magn. Reson. Med. 52, 1263–1271.
Jack C. R. Jr, Wengenack T. M., Reyes D. A. et al. (2005) In vivomagnetic resonance microimaging of individual amyloid plaques inAlzheimer’s transgenic mice. J. Neurosci. 25, 10041–10048.
Janus C., Pearson J., McLaurin J. et al. (2000) A beta peptide immun-ization reduces behavioural impairment and plaques in a model ofAlzheimer’s disease. Nature 408, 979–982.
Kandimalla K. K., Curran G. L., Holasek S. S., Gilles E. J., WengenackT. M. and Poduslo J. F. (2005) Pharmacokinetic analysis of theblood–brain barrier transport of 125I-amyloid beta protein 40 inwild-type and Alzheimer’s disease transgenic mice (APP,PS1) andits implications for amyloid plaque formation. J. Pharmacol. Exp.Ther. 313, 1370–1378.
Kaneko Y., Nimmerjahn F. and Ravetch J. V. (2006) Anti-inflammatoryactivity of immunoglobulin G resulting from Fc sialylation. Sci-ence 313, 670–673.
Lambert M. P., Viola K. L., Chromy B. A., Chang L., Morgan T. E., YuJ., Venton D. L., Krafft G. A., Finch C. E. and Klein W. L. (2001)Vaccination with soluble Abeta oligomers generates toxicity-neutralizing antibodies. J. Neurochem. 79, 595–605.
Lamonica D., Czuczman M., Nabi H., Klippenstein D. and Grossman Z.(2002) Radioimmunoscintigraphy (RIS) with bectumomab(Tc99 m labeled IMMU-LL2, Lymphoscan) in the assessment ofrecurrent non-Hodgkin’s lymphoma (NHL). Cancer Biother.Radiopharm. 17, 689–697.
Lemere C. A., Spooner E. T., LaFrancois J. et al. (2003) Evidence forperipheral clearance of cerebral Abeta protein following chronic,active Abeta immunization in PSAPP mice. Neurobiol. Dis. 14,10–18.
Liu R., Yuan B., Emadi S., Zameer A., Schulz P., McAllister C.,Lyubchenko Y., Goud G. and Sierks M. R. (2004) Single chainvariable fragments against beta-amyloid (Abeta) can inhibit Abetaaggregation and prevent abeta-induced neurotoxicity. Biochemistry43, 6959–6967.
Mohajeri M. H., Saini K., Schultz J. G., Wollmer M. A., Hock C. andNitsch R. M. (2002) Passive immunization against beta-amyloidpeptide protects central nervous system (CNS) neurons from in-creased vulnerability associated with an Alzheimer’s disease-causing mutation. J. Biol. Chem. 277, 33012–33017.
Monsonego A., Imitola J., Zota V., Oida T. and Weiner H. L. (2003)Microglia-mediated nitric oxide cytotoxicity of T cells following
432 J. F. Poduslo et al.
Journal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 102, 420–433� 2007 The Authors

amyloid beta-peptide presentation to Th1 cells. J. Immunol. 171,2216–2224.
Morgan D. and Gitter B. D. (2004) Evidence supporting a role for anti-Abeta antibodies in the treatment of Alzheimer’s disease. Neuro-biol. Aging 25, 605–608.
Morgan D., Diamond D. M., Gottschall P. E. et al. (2000) A beta peptidevaccination prevents memory loss in an animal model of Alzhei-mer’s disease. Nature 408, 982–985.
Nicoll J. A., Wilkinson D., Holmes C., Steart P., Markham H. and WellerR. O. (2003) Neuropathology of human Alzheimer disease afterimmunization with amyloid-beta peptide: a case report. Nat. Med.9, 448–452.
Pfeifer M., Boncristiano S., Bondolfi L., Stalder A., Deller T., StaufenbielM., Mathews P. M. and Jucker M. (2002) Cerebral hemorrhage afterpassive anti-Abeta immunotherapy. Science 298, 1379.
Pichelmayer O., Zielinski C. and Raderer M. (2005) Response of anonmalignant pleural effusion to bevacizumab. N. Engl. J. Med.353, 740–741.
Poduslo J. F. and Curran G. L. (1996) Polyamine modification increasesthe permeability of proteins at the blood–nerve and blood–brainbarriers. J. Neurochem. 66, 1599–1609.
Poduslo J. F. and Curran G. L. (2001) Amyloid beta peptide as a vaccinefor Alzheimer’s disease involves receptor-mediated transport at theblood–brain barrier. Neuroreport 12, 3197–3200.
Poduslo J. F., Curran G. L. and Berg C. T. (1994) Macromolecularpermeability across the blood–nerve and blood–brain barriers.Proc. Natl. Acad. Sci. U. S. A. 91, 5705–5709.
Poduslo J. F., Curran G. L., Haggard J. J., Biere A. L. and Selkoe D. J.(1997) Permeability and residual plasma volume of human, Dutchvariant, and rat amyloid beta-protein 1-40 at the blood–brain bar-rier. Neurobiol. Dis. 4, 27–34.
Poduslo J. F., Curran G. L. and Gill J. S. (1998) Putrescine-modifiednerve growth factor: bioactivity, plasma pharmacokinetics, blood–brain/nerve barrier permeability, and nervous system biodistribu-tion. J. Neurochem. 71, 1651–1660.
Poduslo J. F., Curran G. L., Sanyal B. and Selkoe D. J. (1999) Receptor-mediated transport of human amyloid beta-protein 1-40 and 1-42 atthe blood–brain barrier. Neurobiol. Dis. 6, 190–199.
Poduslo J. F., Curran G. L., Wengenack T. M., Malester B. and Duff K.(2001) Permeability of proteins at the blood–brain barrier in thenormal adult mouse and double transgenic mouse model of Alz-heimer’s disease. Neurobiol. Dis. 8, 555–567.
Poduslo J. F., Wengenack T. M., Curran G. L., Wisniewski T., SigurdssonE. M., Macura S. I., Borowski B. J., Jack C. R., Jr. (2002) Moleculartargeting of Alzheimer’s amyloid plaques for contrast-enhancedmagnetic resonance imaging. Neurobiol. Dis. 11, 315–329.
Poduslo J. F., Curran G. L., Peterson J. A., McCormick D. J., Fauq A.H., Khan M. A. and Wengenack T. M. (2004) Design and chemicalsynthesis of a magnetic resonance contrast agent with enhanced invitro binding, high blood–brain barrier permeability, and in vivotargeting to Alzheimer’s disease amyloid plaques. Biochemistry 43,6064–6075.
Rogers J. and Shen Y. (2000) A perspective on inflammation in Alz-heimer’s disease. Ann. N. Y. Acad. Sci. 924, 132–135.
Rohn T. T., Ivins K. J., Bahr B. A., Cotman C. W. and Cribbs D. H.(2000) A monoclonal antibody to amyloid precursor proteininduces neuronal apoptosis. J. Neurochem. 74, 2331–2342.
Schenk D. (2002) Amyloid-beta immunotherapy for Alzheimer’sdisease: the end of the beginning. Nat. Rev. Neurosci. 3,824–828.
Schenk D., Barbour R., Dunn W. et al. (1999) Immunization withamyloid-beta attenuates Alzheimer-disease-like pathology in thePDAPP mouse. Nature 400, 173–177.
Spooner E. T., Desai R. V., Mori C., Leverone J. F. and Lemere C. A.(2002) The generation and characterization of potentially thera-peutic Abeta antibodies in mice: differences according to strain andimmunization protocol. Vaccine 21, 290–297.
de StGroth S. F. and Scheidegger D. (1980) Production of mono-clonal antibodies: strategy and tactics. J. Immunol. Methods 35,1–21.
Tammer A. H., Coia G., Cappai R., Fuller S., Masters C. L., Hudson P.and Underwood J. R. (2002) Generation of a recombinant Fabantibody reactive with the Alzheimer’s disease-related Abetapeptide. Clin. Exp. Immunol. 129, 453–463.
Tamura Y., Hamajima K., Matsui K., Yanoma S., Narita M., Tajima N.,Xin K. Q., Klinman D. and Okuda K. (2005) The F(ab’)2 fragmentof an Abeta-specific monoclonal antibody reduces Abeta depositsin the brain. Neurobiol. Dis. 20, 541–549.
Vicente A. G., Almoguera M., Alonso J. C., Heffernan A. J., Gomez A.,Contreras P. I. and Martin-Comin J. (2004) Diagnosis of orthopedicinfection in clinical practice using Tc-99 m sulesomab (antigra-nulocyte monoclonal antibody fragment Fab’2). Clin. Nucl. Med.29, 781–785.
Weiner H. L., Lemere C. A., Maron R., Spooner E. T., Grenfell T. J.,Mori C., Issazadeh S., Hancock W. W. and Selkoe D. J. (2000)Nasal administration of amyloid-beta peptide decreases cerebralamyloid burden in a mouse model of Alzheimer’s disease. Ann.Neurol. 48, 567–579.
Wengenack T. M., Curran G. L., Olson E. E. and Poduslo J. F. (1997)Putrescine-modified catalase with preserved enzymatic activityexhibits increased permeability at the blood–nerve and blood–brainbarriers. Brain Res. 767, 128–135.
Wengenack T. M., Curran G. L. and Poduslo J. F. (2000) Targetingalzheimer amyloid plaques in vivo. Nat. Biotechnol. 18, 868–872.
Wilcock D. M., DiCarlo G., Henderson D., Jackson J., Clarke K., UgenK. E., Gordon M. N. and Morgan D. (2003) Intracraniallyadministered anti-Abeta antibodies reduce beta-amyloid depositionby mechanisms both independent of and associated with microglialactivation. J. Neurosci. 23, 3745–3751.
Wilcock D. M., Munireddy S. K., Rosenthal A., Ugen K. E., Gordon M.N. and Morgan D. (2004) Microglial activation facilitates Abetaplaque removal following intracranial anti-Abeta antibody admin-istration. Neurobiol. Dis. 15, 11–20.
Targeting of antibody fragments to AD amyloid plaques 433
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 102, 420–433
Copyright © 2022 FDOKUMEN




















