
In situ Characterization of Catalysts Active in Partial Oxidations: TS-1 and Fe-MFI Case Studies
12
In situ characterization of catalysts active in partial oxidations: TS-1 and Fe-MFI case studies A. Zecchina a,b, *, S. Bordiga a,b , G. Spoto a,b , A. Damin a , G. Berlier a , F. Bonino a , C. Prestipino a and C. Lamberti a,b,c a Department of Inorganic, Physical and Material Chemistry, University of Turin, Via P. Giuria 7, 10125 Torino, Italy E-mail: [email protected] b INSTM Research unit of Turin University, Torino, Italy c INFM UdR di Torino Universita `, Torino, Italy A concise review of the firmly established knowledge and of the unresolved problems concerning the structure and the reactivity of Ti and Fe sites in TS-1 and Fe-MFI partial oxidation catalysts is given. Some new experimental and theoretical results are also described. KEY WORDS: TS-1; Fe-MFI; partial oxidation reactions; in situ spectroscopies 1. Introduction In recent years, two interesting classes of partial oxidation catalyst have attracted particular interest in the scientific community: (i) the Ti-based heterogeneous [1–17] and homogeneous catalysts [18–23]; and (ii) the Fe-based heterogeneous [24–31] and homogeneous cat- alysts [32–37]. In particular TS-1 [1–12], and Fe-silicalite and Fe-ZSM-5 [30,31,38] have proved their utility and selectivity in several partial oxidation reactions using hydrogen peroxide and N 2 O as oxidants. As far as TS-1 is concerned, the most relevant oxi- dation reactions [1–12] are summarized in scheme 1. Note that some of these reactions form the basis of industrial processes. As far as Fe-silicalite and Fe-ZSM- 5 are concerned, beside the oxidation of methane, they are active catalysts in the oxidation of benzene to phe- nol. Scheme 2 shows the possible future role of Fe-MFI catalysts in the adipic acid process [38]. The outstanding properties of these heterogeneous catalysts in terms of yield and selectivity, the presence of homogeneous counterparts [18–23,32–37] and the similarity of some of them (the Fe-based ones) to enzymatic catalysts [39,40] has stimulated a great deal of investigations concerning the structure, valence, coordination state, nuclearity of the catalytic sites and reaction mechanisms. In this work we give a brief overview on the most important achievements and the unresolved problems concerning these two catalysts. Due to space limitations and the broadness of the subject, this review has no presumption to be exhaustive. 2. Experimental TS-1 ðSi=Ti ¼ 50Þ has been synthesized in EniChem laboratories following a procedure described in the original patent [1]. Fe-silicalite ðSi=Fe ¼ 90Þ has been synthesized at the University of Milano following the hydrothermal method described by Ratnasamy and Kumar [41]. The structural and catalytic peculiarities of TS-1 synthesized by EniChem are well known, while those of Fe-silicalite have been thoroughly investigated in two previous papers [42,43] by Forni’s group. Ti k-edge XANES and EXAFS experiments have been performed using the EXAFS3 and EXAFS 13 beamlines of the LURE DCI storage ring using a Si(311) and Si(111) monochromator, respectively. Fe k-edge XANES and EXAFS experiments have been performed using the GILDA BM8 beamline of the ESRF storage ring using for both parts of the X-ray absorption spec- trum a Si(311) monochromator. All X-ray absorption measurements have been performed at room tempera- ture, with the only exception of the TS-1/H 2 O/H 2 O 2 system, which has been measured at liquid nitrogen temperature to stabilize the active complex. Diffuse reflectance UV–vis spectra were obtained on a Varian Cary 5 spectrometer. To limit the computational demand, in our calcu- lations, the cluster/embedded cluster ONIOM [44] scheme has been adopted on a cluster obtained by cutting a fraction of the MFI framework around the T1 site [45]. The final ONIOM energy (as such as gradients, and so on) is defined as: EðONIOMÞ¼ EðHMÞþ½EðLRÞ EðLMÞ, where E(LR) is the energy of the whole cluster calculated at (RHF/3- 21G(H,N,Si,O), 3-21G*(Ti)) low (L) level, E(LM) is the M energy at (RHF/3-21G(H,N,Si,O), 3-21G*(Ti)) and E(HM) is the M energy obtained at (B3-LYP/6- 311+G(d,p)(H,N,Si,O), ppp(Ti)) high (H) level. Here, ppp represents the basis-set employed for Ti atoms: it is composed of effective small core pseudo-potentials [46]+LANL2DZ [46]+Ahlrichs’ ‘‘p’’ polarization function [47]. *To whom correspondence should be addressed. Topics in Catalysis Vol. 21, Nos. 1–3, October 2002 (# 2002) 67 1022-5528/02/1000-0067/0 # 2002 Plenum Publishing Corporation
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of In situ Characterization of Catalysts Active in Partial Oxidations: TS-1 and Fe-MFI Case Studies

In situ characterization of catalysts active in partial oxidations: TS-1 and
Fe-MFI case studies
A. Zecchinaa,b,*, S. Bordigaa,b, G. Spotoa,b, A. Damina, G. Berliera, F. Boninoa, C. Prestipinoa and C. Lambertia,b,c
aDepartment of Inorganic, Physical and Material Chemistry, University of Turin, Via P. Giuria 7, 10125 Torino, Italy
E-mail: [email protected] Research unit of Turin University, Torino, Italy
cINFM UdR di Torino Universita, Torino, Italy
A concise review of the firmly established knowledge and of the unresolved problems concerning the structure and the reactivity
of Ti and Fe sites in TS-1 and Fe-MFI partial oxidation catalysts is given. Some new experimental and theoretical results are also
described.
KEY WORDS: TS-1; Fe-MFI; partial oxidation reactions; in situ spectroscopies
1. Introduction
In recent years, two interesting classes of partialoxidation catalyst have attracted particular interest inthe scientific community: (i) the Ti-based heterogeneous[1–17] and homogeneous catalysts [18–23]; and (ii) theFe-based heterogeneous [24–31] and homogeneous cat-alysts [32–37]. In particular TS-1 [1–12], and Fe-silicaliteand Fe-ZSM-5 [30,31,38] have proved their utility andselectivity in several partial oxidation reactions usinghydrogen peroxide and N2O as oxidants.
As far as TS-1 is concerned, the most relevant oxi-dation reactions [1–12] are summarized in scheme 1.Note that some of these reactions form the basis ofindustrial processes. As far as Fe-silicalite and Fe-ZSM-5 are concerned, beside the oxidation of methane, theyare active catalysts in the oxidation of benzene to phe-nol. Scheme 2 shows the possible future role of Fe-MFIcatalysts in the adipic acid process [38]. The outstandingproperties of these heterogeneous catalysts in terms ofyield and selectivity, the presence of homogeneouscounterparts [18–23,32–37] and the similarity of some ofthem (the Fe-based ones) to enzymatic catalysts [39,40]has stimulated a great deal of investigations concerningthe structure, valence, coordination state, nuclearity ofthe catalytic sites and reaction mechanisms.
In this work we give a brief overview on the mostimportant achievements and the unresolved problemsconcerning these two catalysts. Due to space limitationsand the broadness of the subject, this review has nopresumption to be exhaustive.
2. Experimental
TS-1 ðSi=Ti ¼ 50Þ has been synthesized in EniChemlaboratories following a procedure described in the
original patent [1]. Fe-silicalite ðSi=Fe ¼ 90Þ has beensynthesized at the University of Milano following thehydrothermal method described by Ratnasamy andKumar [41]. The structural and catalytic peculiaritiesof TS-1 synthesized by EniChem are well known,while those of Fe-silicalite have been thoroughlyinvestigated in two previous papers [42,43] by Forni’sgroup.
Ti k-edge XANES and EXAFS experiments havebeen performed using the EXAFS3 and EXAFS 13beamlines of the LURE DCI storage ring using a Si(311)and Si(111) monochromator, respectively. Fe k-edgeXANES and EXAFS experiments have been performedusing the GILDA BM8 beamline of the ESRF storagering using for both parts of the X-ray absorption spec-trum a Si(311) monochromator. All X-ray absorptionmeasurements have been performed at room tempera-ture, with the only exception of the TS-1/H2O/H2O2
system, which has been measured at liquid nitrogentemperature to stabilize the active complex. Diffusereflectance UV–vis spectra were obtained on a VarianCary 5 spectrometer.
To limit the computational demand, in our calcu-lations, the cluster/embedded cluster ONIOM [44]scheme has been adopted on a cluster obtained bycutting a fraction of the MFI framework around theT1 site [45]. The final ONIOM energy (as such asgradients, and so on) is defined as: EðONIOMÞ ¼
EðHMÞ þ ½EðLRÞ � EðLMÞ�, where E(LR) is the energyof the whole cluster calculated at (RHF/3-21G(H,N,Si,O), 3-21G*(Ti)) low (L) level, E(LM) isthe M energy at (RHF/3-21G(H,N,Si,O), 3-21G*(Ti))and E(HM) is the M energy obtained at (B3-LYP/6-311+G(d,p)(H,N,Si,O), ppp(Ti)) high (H) level. Here,ppp represents the basis-set employed for Ti atoms: itis composed of effective small core pseudo-potentials[46]+LANL2DZ [46]+Ahlrichs’ ‘‘p’’ polarizationfunction [47].*To whom correspondence should be addressed.
Topics in Catalysis Vol. 21, Nos. 1–3, October 2002 (# 2002) 67
1022-5528/02/1000-0067/0 # 2002 Plenum Publishing Corporation
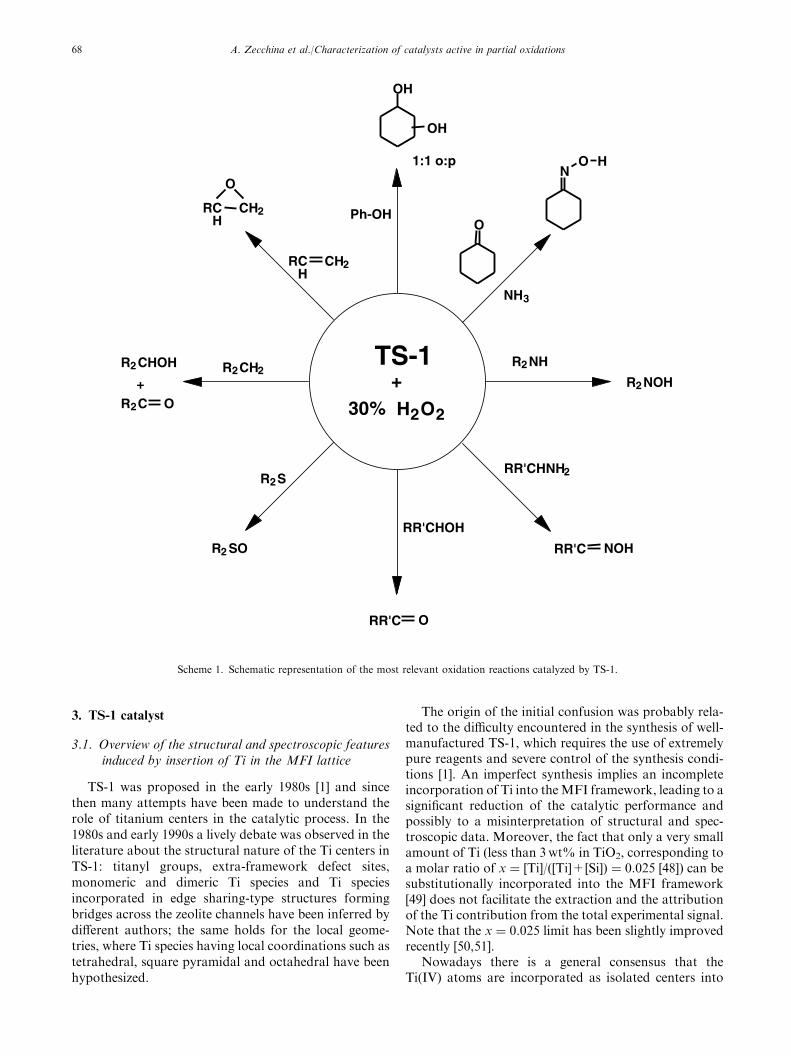
3. TS-1 catalyst
3.1. Overview of the structural and spectroscopic featuresinduced by insertion of Ti in the MFI lattice
TS-1 was proposed in the early 1980s [1] and sincethen many attempts have been made to understand therole of titanium centers in the catalytic process. In the1980s and early 1990s a lively debate was observed in theliterature about the structural nature of the Ti centers inTS-1: titanyl groups, extra-framework defect sites,monomeric and dimeric Ti species and Ti speciesincorporated in edge sharing-type structures formingbridges across the zeolite channels have been inferred bydifferent authors; the same holds for the local geome-tries, where Ti species having local coordinations such astetrahedral, square pyramidal and octahedral have beenhypothesized.
The origin of the initial confusion was probably rela-ted to the difficulty encountered in the synthesis of well-manufactured TS-1, which requires the use of extremelypure reagents and severe control of the synthesis condi-tions [1]. An imperfect synthesis implies an incompleteincorporation of Ti into theMFI framework, leading to asignificant reduction of the catalytic performance andpossibly to a misinterpretation of structural and spec-troscopic data. Moreover, the fact that only a very smallamount of Ti (less than 3wt% in TiO2, corresponding toa molar ratio of x ¼ ½Ti�/([Ti]+½Si�Þ ¼ 0:025 [48]) can besubstitutionally incorporated into the MFI framework[49] does not facilitate the extraction and the attributionof the Ti contribution from the total experimental signal.Note that the x ¼ 0:025 limit has been slightly improvedrecently [50,51].
Nowadays there is a general consensus that theTi(IV) atoms are incorporated as isolated centers into
NO H
O
OH
OH
Ph-OH
1:1 o:p
NH3
RR'CHNH2
R NOH2
NOHRR'C
RR'CHOH
R NH2
R S2
R SO2
R CH2 2
ORR'C
R CHOH2
+OR C2
+
H O2 230%
RC 2H
CH
RC 2H
CH
O
TS-1
Scheme 1. Schematic representation of the most relevant oxidation reactions catalyzed by TS-1.
68 A. Zecchina et al./Characterization of catalysts active in partial oxidations

the framework and are substituting Si atoms in the tet-rahedral positions. The model of isomorphous sub-stitution has been put forward on the basis of severalindependent characterization techniques, namely X-raydiffraction [48,52,53] or neutron diffraction [50,54,55]studies, and IR (Raman) [11,12,51,56–61], UV–vis[12,59,62,63], EXAFS and XANES [12,50,63–69] spec-troscopies. The details are given below.
(i) Diffraction data show that the unit cell volume ofsilicalite increases linearly with Ti content whenTi(IV) atoms are inserted in the framework[48,50,52–55].
(ii) The IR spectra of TS-1 in the framework stretchingregion shows an absorption band located at960 cm�1, virtually absent in perfect silicalite andimmediately identified as a fingerprint of TS-1[1,11,12,51,56–63]. A qualitative correlationbetween the intensity of the infrared band at960 cm�1 and titanium content has been observedsince the first synthesis of TS-1. Indeed, the occur-rence of that band is one of the distinctive featuresof the material cited in the original patent [1].However, the quantitative correlation has beenreported only very recently [51]. In the samework the nature of the 960 cm�1 band has beendiscussed in terms of both cluster and periodicalapproaches.
(iii) The UV–vis spectra of TS-1 in vacuo gives a simpleand clear proof of the presence of tetrahedral Ti(IV)in the zeolite framework [4,12,51,59,61–63]: in fact aTi4þO2� ! Ti3þO� ligand to metal charge transfer
(LMCT) located at 48 000cm�1 can be unam-biguously assigned to the charge transfer transitionfrom the oxygen ligand to an unoccupied orbital ofa Ti(IV) ion tetrahedrally coordinated in isolated[TiO4].
(iv) XANES spectroscopy shows that a narrow andintense pre-edge peak at 4967 eV due to the 1s!3delectronic transition involving Ti atoms in tetra-hedral coordination is present in well-manufacturedTS-1. This peak is virtually absent in the case ofoctahedrally coordinated Ti(IV). Indeed the transi-tions A1g!T2g are symmetrically forbidden in thecase of octahedral coordination of Ti(IV), but thetransition A1!T2 is allowed in the case of tetra-hedral coordination of Ti(IV), as in the case of[TiO4] units [12,51,63–65,67,69].
(v) EXAFS spectroscopy [12,51,63–65,66,69] reportsthat, within experimental error, Ti(IV) species arecoordinated with four oxygen atoms at a Ti–O dis-tance (1.79–1.81 A) which is much greater than thetypical Si–O distance of [SiO4] units in zeolites(1.59–1.60 A). EXAFS spectroscopy hasexplained, on a local basis, the origin of the volumeunit increase measured with diffraction methods, seepoint (i).
It has also been shown that the catalytic activity of aTi site, in several partial oxidation reactions, is related tothe fraction of tetrahedral Ti atoms incorporated in TS-1 [10,70]. Moreover, theoretical studies have shown thatthe isomorphous insertion of Ti atoms in the MFI latticeis energetically favored [71–77].
H2 air+
OHO
HNO3
N2O + OHOH
OO
2 H 2
N2O
OH
Adipic Acid Process (current)
New step-change proposal
Scheme 2. Schematic representation of the possible future role of Fe-MFI catalysts in the adipic acid process, as suggested by Panov et al. [38].
A. Zecchina et al./Characterization of catalysts active in partial oxidations 69
The picture reported in points (i)–(v) holds fordehydrated TS-1 only. Such studies have reportedimportant proofs witnessing the nature of the Ti activecenter. It is, however, evident that the next generation ofexperiments must be designed in order to investigate thecatalyst under reactive conditions. In this brief overviewwe move along this direction by focusing attention oncomputational and experimental studies on the interac-tion of TS-1 with H2O, NH3 and H2O2. This choice isnot accidental. In fact, interaction with water is impor-tant since the catalyst works in aqueous solution. Theinterest in the study of NH3 is related to the fact thatammonia is a reactant in the ammoximation of cyclo-hexanone to give cyclohexanone oxime (see scheme 1),while the role of H2O2 is fundamental, being the oxi-dizing agent.
It is evident that the tetrahedral coordination assumedby Ti(IV) is somehow unusual and such four-coordinatedframework Ti species have a pronounced tendency toexpand their coordination upon interacting with mole-cules. It is a matter of fact that a pure tetrahedral coor-dination is present only in outgassed TS-1, completelydeprived of the adsorbed water acting as additionalligand, while on highly hydrated samples an increase ofthe coordination from four (tetrahedral) up to six(octahedral) passing through a coordination of five (tri-gonal bipyramidal), according to the extent of hydration,is observed. The progressive increase of the coordinationof Ti heteroatoms with an increasing amount of adsorbedwater is witnessed by a gradual transition of the spec-troscopic properties toward those of octahedral Ti(IV) asreported either by experiment [4,11,12,56–59,63,65,67,78–80] or theory [11,12,74,76, 81,82].
3.2. In situ UV–vis, XANES and EXAFS spectroscopy ofTS-1 interacting with H2O, NH3 and H2O2
On a historical basis UV–vis spectroscopy has playeda key role in the elucidation of the structure of Ti(IV)species in TS-1, both in vacuo and in the presence ofadsorbates or in the presence of water or in dilutedwater solutions of different molecules [4,12,51,59,61–63,78].
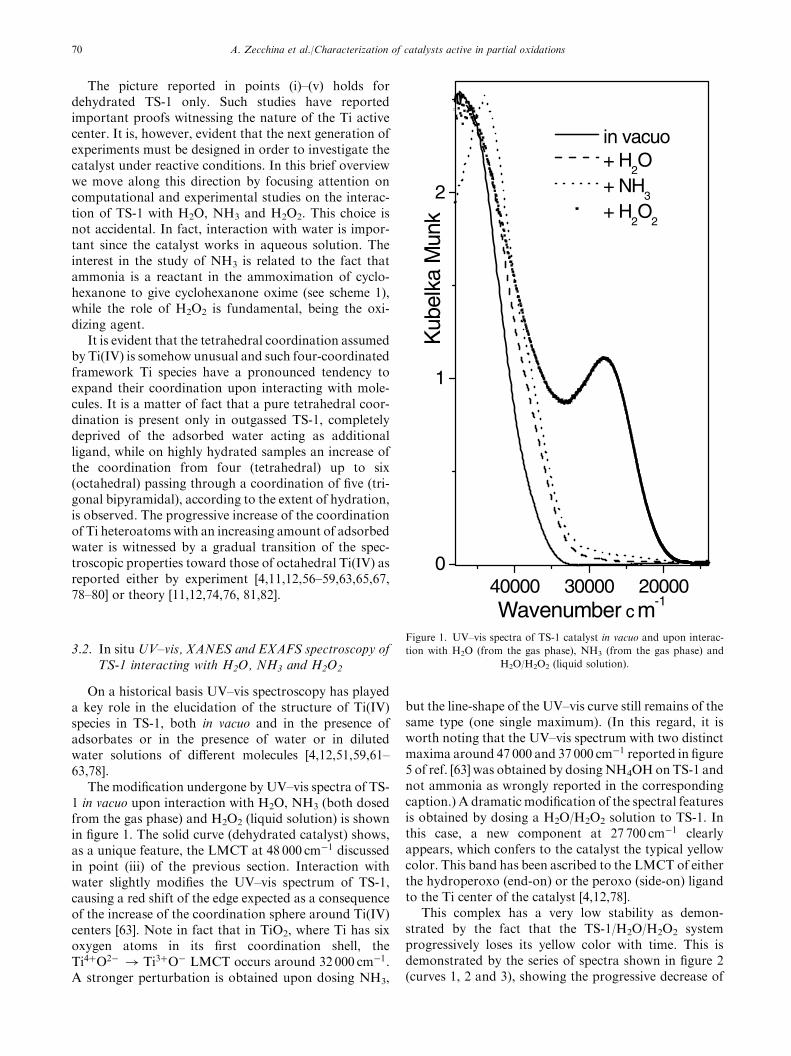
The modification undergone by UV–vis spectra of TS-1 in vacuo upon interaction with H2O, NH3 (both dosedfrom the gas phase) and H2O2 (liquid solution) is shownin figure 1. The solid curve (dehydrated catalyst) shows,as a unique feature, the LMCT at 48 000 cm�1 discussedin point (iii) of the previous section. Interaction withwater slightly modifies the UV–vis spectrum of TS-1,causing a red shift of the edge expected as a consequenceof the increase of the coordination sphere around Ti(IV)centers [63]. Note in fact that in TiO2, where Ti has sixoxygen atoms in its first coordination shell, theTi4þO2� ! Ti3þO� LMCT occurs around 32 000 cm�1.A stronger perturbation is obtained upon dosing NH3,
but the line-shape of the UV–vis curve still remains of thesame type (one single maximum). (In this regard, it isworth noting that the UV–vis spectrum with two distinctmaxima around 47 000 and 37 000 cm�1 reported in figure5 of ref. [63] was obtained by dosingNH4OHonTS-1 andnot ammonia as wrongly reported in the correspondingcaption.) A dramatic modification of the spectral featuresis obtained by dosing a H2O/H2O2 solution to TS-1. Inthis case, a new component at 27 700 cm�1 clearlyappears, which confers to the catalyst the typical yellowcolor. This band has been ascribed to the LMCT of eitherthe hydroperoxo (end-on) or the peroxo (side-on) ligandto the Ti center of the catalyst [4,12,78].
This complex has a very low stability as demon-strated by the fact that the TS-1/H2O/H2O2 systemprogressively loses its yellow color with time. This isdemonstrated by the series of spectra shown in figure 2(curves 1, 2 and 3), showing the progressive decrease of
40000 30000 200000
1
2
in vacuo + H
2O
+ NH3
+ H2O
2
Kub
elka
Mun
k
Wavenumber c m-1
Figure 1. UV–vis spectra of TS-1 catalyst in vacuo and upon interac-
tion with H2O (from the gas phase), NH3 (from the gas phase) and
H2O/H2O2 (liquid solution).
70 A. Zecchina et al./Characterization of catalysts active in partial oxidations

the 27 700 cm�1 component. The important new findingobtained in the experiment shown in figure 2 is that theintensity of the 27 700 cm�1 component is nearlyrestored upon adding pure H2O to the system. Thishighlights the fundamental role of water in the stabili-zation of the hydroperoxo or peroxo complex in TS-1.
In order to obtain more information about thereactivity of the Ti(IV) center towards H2O2, ab initiocluster calculations have been made. Employing a sui-table model of the Ti(IV) center in TS-1, we haveinvestigated the interaction of one H2O2 molecule withsuch a center. Bare H2O2 interacts with the Ti center togive a weakly bonded complex at 2.34 A, which cannotexplain the experimentally observed adsorption band at
27 700 cm�1. On a chemical basis, the formation ofeither the hydroperoxo or the peroxo complex impliesthe partial or total deprotonation of H2O2. To investi-gate this we have evaluated the energetics of the partialdeprotonation via the hydrolysis of one framework Ti–O–Si linkage. This process, at least for a bare H2O2
molecule, is energetically unfavored by 65 kJ/mol. Thisseems to be in line with the UV–vis spectra shown infigure 2 which underlines the key role played by thepresence of water. New computations are being carriedout in order to verify the cooperative effect of watermolecules to the stability of hydroperoxo or peroxospecies.
As was the case for UV–vis spectroscopy (figure 1),XANES spectroscopy is also a powerful technique indetermining the modification undergone by Ti(IV) cen-ters of TS-1 upon contact with reagents. Figure 3 showsthe XANES spectra of TS-1 in vacuo, upon interaction
40000 30000 200000.0
1.5
3.0
4
3
2
1
Kub
elka
Mun
k
Wavenumber cm-1
Figure 2. Evolution of the UV–vis spectra of TS-1 catalyst contacted
with an aqueous solution of H2O2 as a function of time: 1min, 4 h and
8 h (curves 1, 2 and 3, respectively). Curve 4 shows the effect of H2O
dosage on catalyst sample after acquisition of spectrum 3. The Kubelka
Munk intensities reported in figures 1 and 2 cannot be quantitatively
compared since they refer to different experimental conditions.
4975 5000 50250
1
in vacuo + H
2O
+ NH3
+ H2O
2
no
rmal
ieze
d x
Photon Energy (eV)Figure 3. XANES spectra of TS-1 catalyst in vacuo and upon inter-
action with H2O (from the liquid phase), NH3 (from the gas phase) and
H2O/H2O2 (liquid solution).
A. Zecchina et al./Characterization of catalysts active in partial oxidations 71
with H2O (from the liquid phase), with NH3 (from thegas phase) and with H2O/H2O2 (liquid solution). Theincrease of the Ti(IV) coordination sphere together withthe progressive evolution from a tetrahedral- to anoctahedral-like geometry is evidenced by: (i) the reduc-tion of the pre-edge peak (from 0.79 to 0.09, see table 1);(ii) its broadening (from 1.4 to 3.4 eV); and (iii) theincrease of the white line intensity (from 1.14 to 1.46). Itis worth noting that the increase of the white lineintensity is accompanied by a parallel red shift of thewhite line maximum (from 5004 to 4984 eV). Note thatin the case of the TS-1/H2O/H2O2 system we observe adouble peak at 4984 and 4995 eV with nearly the sameintensity. Shifts of the pre-edge peak (up to 1 eV) havealso been observed; however, being the instrumentalresolution of the spectra collected at LURE of the samemagnitude, such shifts have not been reported. High-resolution XANES spectra can be obtained at theESRF, where we have shown that the pre-edge peak ofTS-1 in vacuo is as high as 1.05 and as narrow as 0.8 eV[51]. The quality of the XANES spectra for Fe-silicaliteacquired under such conditions is discussed in section4.2.
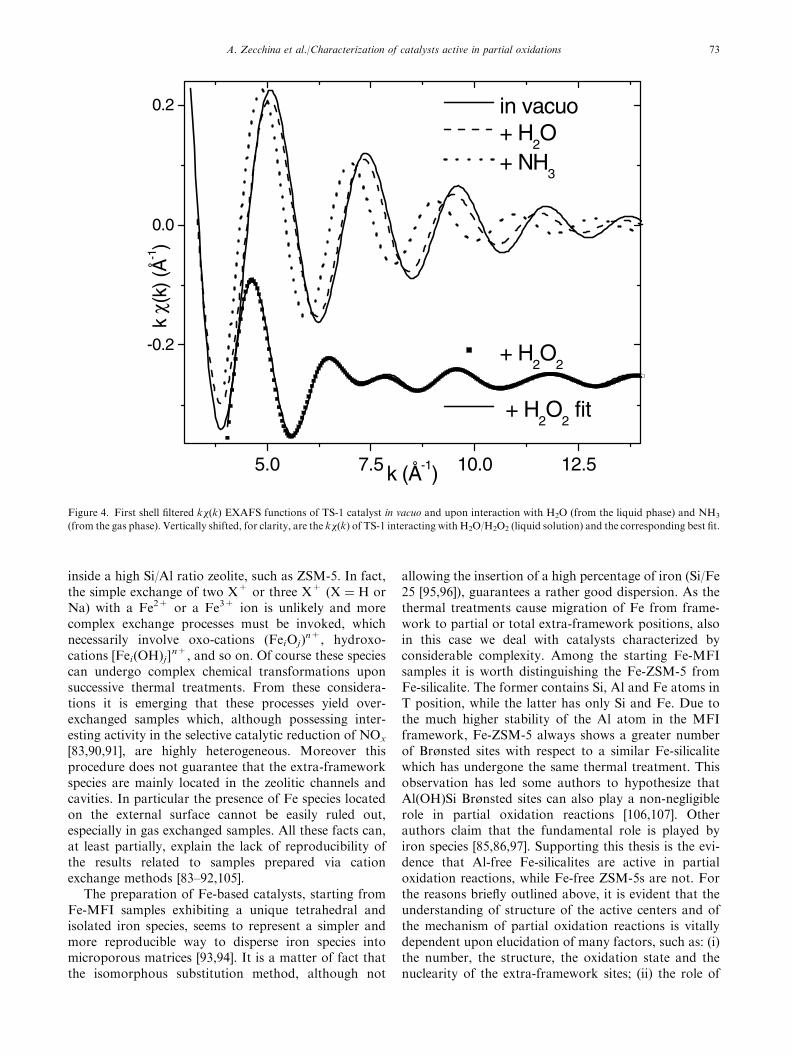
Considering EXAFS spectroscopy, the first shell fil-tered k�(k) functions of TS-1 in vacuo and after inter-actions with the three reagents are shown in figure 4,while the structural outputs obtained in the corre-sponding first shell fits are summarized in table 1. Weobserve that the interaction with water and ammoniahas the effect of increasing the framework Ti�O bondlength of 0.03 and 0.05 A, respectively. The small per-turbation caused by water adsorption is evidenced bythe small increase of the period of the EXAFS oscilla-tions (compare solid and dashed curves in figure 4). Asthoroughly debated elsewhere [79,80], a higher pertur-bation is caused by ammonia adsorption, but we are stilldealing with a sinusoidal-like signal (dotted line curve infigure 4). This does not hold any more in the case of theTS-1/H2O/H2O2 system where the first shell filteredk�(k) function is dominated by strong beats caused by
the interference between two distinct EXAFS signals.The former is that of the ‘‘residual’’ oxygen frameworkatoms now located at 1.83 A. We speak about ‘‘resi-dual’’ because there was no way to fit this contributionwith four oxygen atoms, even by increasing the corre-sponding Debye–Waller factor to unreasonable values.The O�O moiety is strongly adsorbed (low Debye–Waller factor) in a side-on geometry where both Oatoms are located at a Ti–O distance of 2.01 A (see table1). This implies the formation of a side-on peroxocomplex, accompanied by the hydrolysis of one, or eventwo, Ti�O�Si bond, suggesting a total deprotonationof H2O2. This preliminary result will be improved with ahigher shell study, performed in the frame of the mul-tiple scattering approach, which is in progress.
4. Fe-silicalite and Fe-ZSM-5 catalysts
4.1. Overview of the different methodologies adopted toinsert Fe species inside the MFI framework and ontheir heterogeneity
Due to the high industrial interest of this process, aremarkable number of scientific works have appearedconcerning iron-containing MFI systems. Two mainpreparation procedures have been employed to disperseiron species inside the zeolite channels: (i) post-synthesisinsertion via ion exchange in solution and in the gasphase [83–92]; and (ii) controlled migration of iron intoextra-framework positions starting from MFI contain-ing isomorphous Fe3+ [67,93–104]. Until now only theFe-based systems prepared following the second methodhave been shown to possess sufficient stability andselectivity to be proposed as catalysts in partial oxida-tion reactions of industrial interest (scheme 2).
As far as the former method is concerned, which givesonly Fe species in extra-framework positions, we shallrecall the intrinsic difficulty of the achievement of a‘‘proper’’ cation exchange of di- or even trivalent ions
Table 1
Summary of the pre-edge and white line data (XANES) and of the first shell EXAFS results of TS-1 catalyst in vacuo and in the
presence of the different reagents. Shifts of the pre-edge peak (up to 1 eV) have also been observed; however, being the instrumental
resolution of the same magnitude, such shifts have not been reported.
TS-1 Pre-edge Pre-edge White line White line Shell R (A) N
conditions intensity FWHM intensity position (eV) ð0:01Þ ð20%Þ
In vacuo 0.79 1.4 1.14 5004 Framework 1.79 4
oxygen
+H2O 0.40 2.7 1.24 4997 Framework 1.82 4
oxygen
+NH3 0.22 2.9 1.32 4998 Framework 1.84 4
oxygen
+H2O/H2O2 0.09 3.4 1.46 (1.45) 4984 (4995) Framework 1.83 2.5
oxygen
Peroxo 2.01 2
oxygen
72 A. Zecchina et al./Characterization of catalysts active in partial oxidations
inside a high Si/Al ratio zeolite, such as ZSM-5. In fact,the simple exchange of two X+ or three X+
ðX ¼ H orNa) with a Fe2+ or a Fe3+ ion is unlikely and morecomplex exchange processes must be invoked, whichnecessarily involve oxo-cations (FeiOj)
n+, hydroxo-cations [Fei(OH)j ]
n+, and so on. Of course these speciescan undergo complex chemical transformations uponsuccessive thermal treatments. From these considera-tions it is emerging that these processes yield over-exchanged samples which, although possessing inter-esting activity in the selective catalytic reduction of NOx
[83,90,91], are highly heterogeneous. Moreover thisprocedure does not guarantee that the extra-frameworkspecies are mainly located in the zeolitic channels andcavities. In particular the presence of Fe species locatedon the external surface cannot be easily ruled out,especially in gas exchanged samples. All these facts can,at least partially, explain the lack of reproducibility ofthe results related to samples prepared via cationexchange methods [83–92,105].
The preparation of Fe-based catalysts, starting fromFe-MFI samples exhibiting a unique tetrahedral andisolated iron species, seems to represent a simpler andmore reproducible way to disperse iron species intomicroporous matrices [93,94]. It is a matter of fact thatthe isomorphous substitution method, although not
allowing the insertion of a high percentage of iron (Si/Fe25 [95,96]), guarantees a rather good dispersion. As thethermal treatments cause migration of Fe from frame-work to partial or total extra-framework positions, alsoin this case we deal with catalysts characterized byconsiderable complexity. Among the starting Fe-MFIsamples it is worth distinguishing the Fe-ZSM-5 fromFe-silicalite. The former contains Si, Al and Fe atoms inT position, while the latter has only Si and Fe. Due tothe much higher stability of the Al atom in the MFIframework, Fe-ZSM-5 always shows a greater numberof Brønsted sites with respect to a similar Fe-silicalitewhich has undergone the same thermal treatment. Thisobservation has led some authors to hypothesize thatAl(OH)Si Brønsted sites can also play a non-negligiblerole in partial oxidation reactions [106,107]. Otherauthors claim that the fundamental role is played byiron species [85,86,97]. Supporting this thesis is the evi-dence that Al-free Fe-silicalites are active in partialoxidation reactions, while Fe-free ZSM-5s are not. Forthe reasons briefly outlined above, it is evident that theunderstanding of structure of the active centers and ofthe mechanism of partial oxidation reactions is vitallydependent upon elucidation of many factors, such as: (i)the number, the structure, the oxidation state and thenuclearity of the extra-framework sites; (ii) the role of
5.0 7.5 10.0 12.5
-0.2
0.0
0.2
k (k
) (Å
-1)
in vacuo + H
2O
+ NH3
+ H2O
2
+ H2O
2 fit
k (Å-1)
χ
Figure 4. First shell filtered k�(k) EXAFS functions of TS-1 catalyst in vacuo and upon interaction with H2O (from the liquid phase) and NH3
(from the gas phase). Vertically shifted, for clarity, are the k�(k) of TS-1 interacting with H2O/H2O2 (liquid solution) and the corresponding best fit.
A. Zecchina et al./Characterization of catalysts active in partial oxidations 73

Fe and Al concentration on the cluster nuclearity; (iii)the role of thermal treatments in determining the sitestructure and location; and (iv) the propensity of the Fesites to undergo redox processes.
4.2. In situ XANES and EXAFS spectroscopy oftemplate burning and interaction of Fe-silicalite withN2O
The remarkable success of EXAFS in the character-ization of TS-1 [12,51,63–65,66,69] (see section 3.2) ismainly related to the fact that Ti active sites in TS-1 areframework species characterized by a homogeneouslocal environment, which behaves in the same way forall sites upon interactions with reactants. This impliesthat all Ti sites give rise to the same contribution whichthus adds constructively to the overall EXAFS signal.We will see that Fe-silicalite behaves in a different way.
Figure 5(a) shows the k3-weighted, phase uncorrectedFourier transform (FT) of the EXAFS functions of Fe-silicalite in the presence of a template and after activa-tion at 773 and 973K. Even a superficial view of theexperimental data allows one to note a dramaticreduction of the EXAFS signal upon activation atincreasing temperatures. This impressive phenomenon isrelated to the high coordinative unsaturation of extra-
framework species, but has also a second origin. When afraction of iron atoms migrates into extra-frameworkpositions they form a complex variety of isolated andclustered species. Such heterogeneity implies that thelocal environment of Fe atoms has a consistent spread inboth Fe–O bond distances, dynamic Debye–Wallerfactors and coordination numbers (note that with O wemean oxygen atoms of both oxidic nanocluster andzeolitic framework at the anchoring site). As a con-sequence, the EXAFS signal coming from extra-frame-work iron species is affected by a large Debye–Wallerfactor (of static origin) so as to become strongly dumped[101,108–110].
Figure 5(b) shows the k3-weighted, phase uncorrectedFT of the EXAFS spectra collected on Fe-silicaliteactivated at 773K before and after oxidation with N2O.EXAFS spectroscopy indicates that a considerablefraction of iron ions has undergone an importantmodification of its coordination sphere, with the inser-tion of an oxygen ligand at a rather short distance [110].This ligand should be the adsorbed oxygen atom, pro-duced upon N2O decomposition, indicated by Panov etal. to be the active site in partial oxidation reactions[100].
Fe-MFI being a redox catalyst, the in situ monitoringof the Fe3+ÐFe2+ interplay can provide informationof fundamental importance. High-resolution XANES
1.0 1.5 2.0 2.51.0 1.5 2.0 2.5
5 Å-4
R (Å)
(a)
R (Å)
(b)
2 Å-4
Figure 5. (a) k3-weighted, phase uncorrected Fourier transform of the EXAFS functions of Fe-silicalite in the presence of template (full line), and
after activation at 773 and 973K (dotted and dashed lines, respectively). (b) As (a) for Fe-silicalite activated at 773K (shown again for sake of
comparison, dotted line) and for the same catalyst after subsequent oxidation in N2O at 523K. Note that the two parts have a different ordinate
scale.
74 A. Zecchina et al./Characterization of catalysts active in partial oxidations

spectroscopy is one of the most promising techniques toachieve this goal. Figure 6(a) shows the effect of tem-plate burning at increasing temperature on an Fe-sili-calite sample. The shift of the edge position is evident,being at 7123.6 eV for the sample measured with atemplate and at 7122.4 and 7120.6 eV for the samplesactivated at 773 and 973K, respectively. These dataconfirm that iron in the as-prepared sample is present inform of ferric species. After thermal activation ferricspecies undergo a progressive reduction to ferrous spe-cies. The intensity of the 1s!3d pre-edge peak (figure6(c)) is even greater than that of FePO4 model com-pound (not reported for brevity) indicating that the localsymmetry of Fe3+ is closer to the ideal tetrahedral thanthat exhibited in FePO4. In fact EXAFS indicates that inthis case we are dealing with four equivalent Fe–Obonds at 1.86 A. The thermal treatment results in aconsistent reduction of the 1s!3d peak intensity,demonstrating the evident migration of iron speciesfrom the framework tetrahedral positions. In thisregard, figure 6(c) shows the appearance of a low-energyshoulder in the 1s!3d feature, the intensity of whichprogressively increases with increasing activation tem-perature. The energy position of this new component(around 7111.8 eV) is close to that observed for Fe2+
compounds [110]. However, the very large full-widthindicates the presence of more than one Fe2+ species, inagreement with the high heterogeneity of extra-frame-work Fe species probed by EXAFS. Note that thesample with template shows a white line intensity similarto that of FePO4 (1.31 vs. 1.35 [110]), much lower than
that observed for six-fold coordinated model com-pounds (which is in the range 1.52–1.60 [110]), reflectingthe four-fold coordination of iron in the as-preparedsample. Migration to extra-framework positions causesa progressive reduction of the white line intensity from1.23 to 1.20 for samples activated at 773 and 973K,respectively (table 2). This further reduction of the whiteline intensity discounts the presence of a considerablefraction of iron species in aggregated clusters suggestingthe presence of isolated Fe species exhibiting a highcoordinative unsaturation. This evidence is confirmedby IR spectroscopy of adsorbed NO, showing that mostof the Fe2+ extra-framework species are able to coor-dinate up to three NO molecules [104,110].
The effect of a successive oxidation treatment withN2O at 523K for 1 h has been investigated by XANESspectroscopy (figure 6(b) and 6(d)) for the sample pre-viously activated at 973K. The edge position shifts from7120.6 to 7123.1 eV indicating that an effective oxidationprocess has been achieved resulting in a nearly completerestoration of ferric species. The same conclusion can bereached by observing the total erosion of the 7111.8 eVcomponent of the 1s!3d pre-edge peak ascribed toFe2+ species (figure 6(d)). A similar trend has beenobserved for the sample activated at 773K and succes-sively oxidized, in this case the phenomenon is lesspronounced because of the low fraction of the Fe2+
species that migrated to extra-framework positions. Wefinally observe that the oxidation process causes anincrease of the white line intensity, passing from 1.20 to1.24 (table 2). This increase indicates the addition of a
7120 7130 7140Energy (eV)
(a)
0.25
Energy (eV)
Nor
mal
ised
x
Energy (eV)
Fe2+
0.05
7120 7130 7140
(d)
(c)(b)
0.25
7112 7116
Fe3+
0.05
Figure 6. (a, b) As corresponding parts of figure 5 for XANES spectroscopy. (c, d) Magnification of the pre-edge peak of (a) and (b), respectively.
A. Zecchina et al./Characterization of catalysts active in partial oxidations 75

new ligand to the coordination sphere of the extra-framework iron species. Also the ability of iron speciesto coordinate NO molecules is strongly reduced uponoxidation treatment with N2O, as monitored by IRspectroscopy [110].
5. Summary, and future developments of TS-1 and
Fe-MFI systems
We have observed that TS-1 is a rather hydrophobicmaterial since UV–vis, XANES and EXAFS featuresare weakly affected by interaction with water (dosed byboth gas and liquid phases). This picture is confirmed byparallel IR, microcalorimetric experimental evidenceand by ab initio simulations (results not reported herefor sake of brevity). Interaction with ammonia has astronger effect; however, it does not cause the breakingof framework Ti�O�Si bonds: the adsorption of twoNH3 molecules per Ti site causes the elongation of theTi�O bonds and the red shift of the UV–vis edge.Conversely, interaction with the H2O/H2O2 solutiondramatically modifies all spectroscopic features (UV–vis, XANES and EXAFS). UV–vis experiments showclearly the formation of a new optically active species(peroxo or hydroperoxo), and highlight the importanceof the presence of water in the formation of such species.Preliminary ab initio calculations, showing the forma-tion of a complex, confirm this picture, since the partialdeprotonation of a bare H2O2 molecule via the hydro-lysis of one framework Ti–O–Si linkage is energeticallyunfavored by 65 kJ/mol. However, it is not yet clearwhich mechanism is at the base of such process. Themodel emerging from EXAFS experiments suggests theformation of a strongly bound peroxo species, accom-panied by the hydrolysis of up to two of the four Ti–O–Si linkages. Finally, all these results underline thenecessity to include water molecules in ab initio calcu-lations in order to take into account their cooperativerole demonstrated by experiments.
Considering Fe-silicalite, it has been shown that aftersynthesis Fe3+ species occupies framework sites being
tetrahedrally coordinated to four oxygen atoms at1.86 A, as is the case for Ti(IV) in TS-1. However, suchferric species are not as stable as Ti and undergo areductive dislodgment from framework positions upontemplate burning at increasing temperatures. The Fe2+
extra-framework species so formed exhibits a high het-erogeneity and a remarkably high coordinative unsa-turation, which is probably the origin of the highreactivity of such species. N2O is able to oxidize suchextra-framework species, resulting in the insertion of anoxygen ligand at rather short distance. This ligandshould be the adsorbed oxygen atom, produced uponN2O decomposition, indicated by Panov to be the activesite in partial oxidation reactions.
In the future it should be of great interest to report aparallel study on both Fe-silicalite and Fe-ZSM-5 sys-tems, in order to understand why Fe-ZSM-5 catalystsare always more active than Fe-silicalite with the sameiron content. Such a study should be aimed at under-standing the interplay between framework Al(III) spe-cies and extra-framework iron species. Finally, due tothe heterogeneity of extra-framework Fe species, aconvincing model of the active site has yet to be pro-duced. This represents a handicap because it preventsthe systematic use of ab initio methods, which haveshown their usefulness in the case of TS-1.
Acknowledgements
This study has been supported by MURSTCOFIN2000 ‘‘Structure and reactivity of catalytic cen-ters in zeolitic materials’’. The authors are indebted toR. Buzzoni (EniChem Novara) and L. Forni (Universityof Milano) for the synthesis of TS-1 and Fe-silicalites,respectively. Acknowledgments are also due to the staffof the EXAFS 13 beamline of the LURE DCI storagering (particularly to F. Villain and F. Bouamrane) andthe staff of the GILDA BM8 beamline of the ESRFstorage ring (particularly to F. D’Acapito) where the Tiand the Fe k-edge X-ray absorption measurements wereperformed.
Table 2
Position and normalized intensity of the 1s 3d pre-edge peak and of the white line of the
XANES spectra shown in figure 2 for Fe-silicalite after different treatments ðs ¼ shoulder).
Fe-silicalite Pre-edge peak White line
conditions ————————————— —————————————
Position (eV) Intensity Position (eV) Intensity
With template 7114.2 0.205 7138.1 1.31
Activated at 773K 7114.5 0.142 7136.7 1.23
Activated at 973K 7111.8 (s) 0.037 7136.7 1.20
7114.5 0.128
Oxidized with 7114.3 0.137 7137.6 1.24
N2O at 523K
76 A. Zecchina et al./Characterization of catalysts active in partial oxidations

References
[1] M. Taramasso, G. Perego and B. Notari, US Patent. 4410501
(1983).
[2] G.M. Clerici, Appl. Catal. 68 (1991) 249.
[3] G.M. Clerici, G. Bellussi, and U. Romano, J. Catal. 129 (1991)
159.
[4] A. Zecchina, G. Spoto, S. Bordiga, F. Geobaldo, G. Petrini, G.
Leofanti, M. Padovan, M. Mantegazza and P. Roffia, in: New
Frontiers in Catalysis, eds. L. Guczi, F. Solymosi and P. Tetenyi
(Elsevier, Amsterdam, 1993) p. 719.
[5] G. Bellussi, A. Carati, G.M. Clerici, G. Maddinelli and R.
Millini, J. Catal. 133 (1992) 220.
[6] W. Holderich, M. Messe and F. Naumann, Angew. Chem. Int.
Ed. Engl. 27 (1988) 26.
[7] B. Notari, Adv. Catal. 41 (1996) 253, and references therein.
[8] P. Roffia, G. Leofanti, A. Cesana, M.A. Mantegazza, M.
Padovan, G. Petrini, S. Tonti and P. Gervasutti, Stud. Surf. Sci.
Catal. 55 (1990) 543.
[9] M.A. Mantegazza, G. Leofanti, G. Petrini, M. Padovan, A.
Zecchina and S. Bordiga, Stud. Surf. Sci. Catal. 82 (1994) 541.
[10] M.A. Mantegazza, G. Petrini, G. Spano, R. Bagatin and F.
Rivetti, J. Mol. Catal. A 146 (1999) 223.
[11] G. Tozzola, M.A. Mantegazza, G. Ranghino, G. Petrini, S.
Bordiga, G. Ricchiardi, C. Lamberti, R. Zulian and A.
Zecchina, J. Catal. 179 (1998) 64.
[12] C. Zecchina, S. Bordiga, C. Lamberti, G. Ricchiardi, D.
Scarano, G. Petrini, G. Leofanti and M. Mantegazza, Catal.
Today 32 (1996) 97.
[13] M.P. Coles, C.G. Lugmair, K.W. Terry and T.D. Tilly, Chem.
Mater. 12 (2000) 122.
[14] W. Adam, A. Corma, T. I. Reddy and M. Renz, J. Org. Chem.
62 (1997) 3631.
[15] T. Blasco, M.A. Camblor, A. Corma, P. Esteve, J.M. Guil, A.
Martınez, J.A. Perdigon-Melon and S. Valencia, J. Phys. Chem.
B 102 (1998) 75.
[16] R. Raja, G. Sankar and J. M. Thomas, J. Am. Chem. Soc. 123
(2001) 8153.
[17] G. Sankar and J.M. Thomas, Acc. Chem. Res. 34 (2001) 571.
[18] T. Katsuki and K.B. Sharpless, J. Am. Chem. Soc. 102 (1980)
5976.
[19] P. Pitchen, E. Dunach, M.N. Deshmukh and H.B. Kagan, J.
Am. Chem. Soc. 106 (1984) 8188.
[20] Y. Gao, R.M. Hanson, J.M. Klunder, S.Y. Ko, H. Masamune
and K.B. Sharpless, J. Am. Chem. Soc. 109 (1987) 5765.
[21] S. Superchi and C. Rosini, Tetrahedron Asymm. 8 (1997) 349.
[22] M. Bonchio, S. Calloni, F. Di Furia, G. Licini, G. Modena, S.
Moro and W.A. Nugent, J. Am. Chem. Soc. 119 (1997) 6935.
[23] M. Bonchio, G. Licini, G. Modena, O. Bortolini, S. Moro and
W. A. Nugent, J. Am. Chem. Soc. 121 (1999) 6258.
[24] R.L. Garten, W.N. Delgass and M. Boudart, J. Catal. 18 (1970)
90.
[25] K.-I. Segawa, Y. Chen, J.E. Kubsh, W.N. Delgass, J.A.
Dumesic and W.K. Hall, J. Catal. 76 (1982) 112.
[26] J.W. Jermyn, T.J. Johnson, E.F. Vansant and J.H. Lunsford, J.
Phys. Chem. 77 (1973) 2964.
[27] D. Ballivet-Tkatchenko and G. Coudurier, Inorg. Chem. 18
(1979) 558.
[28] T. Bein and P.A. Jacobs, J. Chem. Soc. Faraday Trans. 80
(1984) 1391.
[29] J.B. Nagy, M. Van Eeno and E.G. Derouane, J. Catal. 58 (1979)
230.
[30] G.I. Panov, V.I. Sobolev, K.A. Dubkov, V.N. Parmon, N.S.
Ovanesyan, A.E. Shilov and A.A. Shteinman, React. Kin.
Catal. Lett. 61 (1997) 251.
[31] K.A. Dubkov, V.I. Sobolev, E.P. Talsi, M.A. Rodkin, N.H.
Watkins, A.A. Shteinman and G.I. Panov, J. Mol. Catal. A 123
(1997) 155.
[32] A. Stassinopoulos, G. Schulte, G.C. Papaefthymiou and J.P.
Caradonna, J. Am. Chem. Soc. 113 (1991) 8686.
[33] H.C. Tung, C. Kang and D.T. Sawyer, J. Am. Chem. Soc. 114
(1992) 3445.
[34] S. Menage, E.C. Wilkinson, L. Que, Jr., and M. Fontecave,
Angew. Chem. Int. Ed. Engl. 34 (1995) 203.
[35] J. Kim, E. Larka, E.C. Wilkinson and L. Que, Jr., Angew.
Chem. Int. Ed. Engl. 34 (1995) 2048.
[36] S. Menage, J.B. Galey, J. Dumats, G. Hussler, M. Seite, I.
Gautier Luneau, G. Chottard and M. Fontecave, J. Am. Chem.
Soc. 120 (1998) 13370.
[37] H. Zheng, S.J. Yoo, E. Munck and L. Que, Jr., J. Am. Chem.
Soc. 122 (2000) 3789.
[38] A.K. Uriarte, M.A. Rodkin, M.J. Gross, A.S. Kharitonov and
G.I. Panov, Stud. Surf. Sci. Catal. 110 (1997) 857.
[39] J. Green and H. Dalton, Biochem. J. 259 (1989) 167.
[40] L. Shu, J.C. Neisheim, K. Kauffmann, E. Munck, J.D.
Lipscomb and L. Que, Jr., Science 275 (1997) 515.
[41] P. Ratnasamy and R. Kumar, Catal. Today 9 (1991) 329.
[42] E. Selli, A. Isernia and L. Forni, Phys. Chem. Chem. Phys. 2
(2000) 3301.
[43] R. Leanza, I. Rossetti, I. Mazzola and L. Forni, Appl. Catal. A
205 (2001) 9.
[44] S. Humbel, S. Sieber and K. Morokuma, J. Chem. Phys. 105
(1996) 1959.
[45] S. Bordiga, P. Ugliengo, A. Damin, C. Lamberti, G. Spoto, A.
Zecchina, G. Spano, R. Buzzoni, L. Dalloro and F. Rivetti,
Top. Catal. 15 (2001) 43.
[46] P.J. Hay and W.R. Wadt, J. Chem. Phys. 82 (1985) 299.
[47] A. Schaefer, C. Huber and R. Ahlrichs, J. Chem. Phys. 100
(1994) 5829.
[48] R. Millini, E. Previdi Massara, G. Perego and G. Bellussi, J.
Catal. 137 (1992) 497.
[49] G. Perego, G. Bellussi, C. Corno, M. Taramasso, F. Buonuomo
and A. Esposito, Stud. Surf. Sci. Catal. 28 (1987) 129.
[50] C. Lamberti, S. Bordiga, A. Zecchina, G. Artioli, G. L. Marra
and G. Spano, J. Am. Chem. Soc. 123 (2001) 2204.
[51] G. Ricchiardi, A. Damin, S. Bordiga, C. Lamberti, G. Spano,
F. Rivetti and A. Zecchina, J. Am. Chem. Soc. 123 (2001)
11409.
[52] C. Lamberti, S. Bordiga, A. Zecchina, A. Carati, A.N. Fitch, G.
Artioli, G. Petrini, M. Salvalaggio and G.L. Marra, J. Catal.
183 (1999) 222.
[53] G. L. Marra, G. Artioli, A.N. Fitch, M. Milanesio and C.
Lamberti, Microporous Mesoporous Mater. 40 (2000) 85.
[54] C.A. Hijar, R.M. Jacubinas, J. Eckert, N.J. Henson, PJ. Hay
and K.C. Ott, J. Phys. Chem. B 104 (2000) 12157.
[55] P.F. Henry, M.T. Weller and C.C. Wilson, J. Phys. Chem. B
105 (2001) 7452.
[56] M.R. Boccuti, K.M. Rao, A. Zecchina, G. Leofanti and G.
Petrini, Stud. Surf. Sci. Catal. 48 (1989) 133.
[57] A. Zecchina, G. Spoto, S. Bordiga, M. Padovan and G.
Leofanti, Stud. Surf. Sci. Catal. 65 (1991) 671.
[58] D. Scarano, A. Zecchina, S. Bordiga, F. Geobaldo, G. Spoto,
G. Petrini, G. Leofanti, M. Padovan and G. Tozzola, J. Chem.
Soc. Faraday Trans. 89 (1993) 4123.
[59] A. Zecchina, G. Spoto, S. Bordiga, A. Ferrero, G. Petrini, M.
Padovan and G. Leofanti, Stud. Surf. Sci. Catal. 69 (1991) 251.
[60] G. Deo, A.M. Turek, I.E. Wachs, D.R.C. Huybrechts and P.A.
Jacobs, Zeolites 13 (1993) 365.
[61] C. Li, G. Xiong, Q. Xin, J. Liu, P. Ying, Z. Feng, J. Li, W.
Yang, Y. Wang, G. Wang, X. Liu, M. Lin, X. Wang and E.
Min, Angew. Chem. Int. Ed. 38 (1999) 2220.
[62] T. Blasco, M. Camblor, A. Corma and J. Perez-Parriente, J.
Am. Chem. Soc. 115 (1993) 11806.
[63] S. Bordiga, S. Coluccia, C. Lamberti, L. Marchese, A. Zecchina,
F. Boscherini, F. Buffa, F. Genoni, G. Leofanti, G. Petrini and
G. Vlaic, J. Phys. Chem. 98 (1994) 4125.
A. Zecchina et al./Characterization of catalysts active in partial oxidations 77

[64] S. Pei, G.W. Zajac, J.A. Kaduk, J. Faber, B.I. Boyanov, D.
Duck, D. Fazzini, T.I. Morrison and D.S. Yang, Catal. Lett. 21
(1993) 333.
[65] S. Bordiga, F. Boscherini, S. Coluccia, F. Genoni, C. Lamberti,
G. Leofanti, L. Marchese, G. Petrini, G. Vlaic and A. Zecchina,
Catal. Lett. 26 (1994) 195.
[66] L. Le Noc, D. Trong On, S. Solomykina, B. Echchahed, F.
Beland, C. Cartier dit Moulin and L. Bonneviot, Stud. Surf. Sci.
Catal. 101 (1996) 611.
[67] S. Bordiga, F. Geobaldo, C. Lamberti, A. Zecchina, F.
Boscherini, F. Genoni, G. Leofanti, G. Petrini, M. Padovan,
S. Geremia and G. Vlaic, Nucl. Instr. Meth. B 97 (1995) 23.
[68] C. Lamberti, S. Bordiga, D. Arduino, A. Zecchina, F.
Geobaldo, G. Spano, F. Genoni, G. Petrini, A. Carati, F.
Villain and G. Vlaic, J. Phys. Chem. B 102 (1998) 6382.
[69] D. Gleeson, G. Sankar, C.R.A. Catlow, J.M. Thomas, G.
Spano, S. Bordiga, A. Zecchina, and C. Lamberti, Phys. Chem.
Chem. Phys. 2 (2000) 4812.
[70] A. Thangaraj, R. Kumar, S.P. Mirajkar and B. Ratnasami, J.
Catal. 130 (1991) 1.
[71] A. Jentys and C.R.A. Catlow, Catal. Lett. 22 (1993) 251.
[72] R. Millini, G. Perego and K. Seiti, Stud. Surf. Sci. Catal. 84
(1994) 2123.
[73] A.J.M. de Mann and J.Sauer, J. Phys. Chem. 100 (1996) 5025.
[74] P.E. Sinclair, G. Sankar, C.R.A. Catlow, J.M. Thomas and T.
Maschmeyer, J. Phys. Chem. B 101 (1997) 4237.
[75] P.E. Sinclair and C.R.A. Catlow, J. Phys. Chem. B 103 (1999)
1084.
[76] C.M. Zicovich-Wilson, R. Dovesi and A. Corma, J. Phys.
Chem. B 103 (1999) 988.
[77] G. Ricchiardi, A. de Man and J. Sauer, Phys. Chem. Chem.
Phys. 2 (2000) 2195.
[78] F. Geobaldo, S. Bordiga, A. Zecchina, E. Giamello, G. Leofanti
and G. Petrini, Catal. Lett. 16 (1992) 109.
[79] V. Bolis, S. Bordiga, C. Lamberti, A. Zecchina, A. Carati, G.
Petrini, F. Rivetti and G. Spano, Microporous Mesoporous
Mater. 30 (1999) 67.
[80] V. Bolis, S. Bordiga, C. Lamberti, A. Zecchina, A. Carati, F.
Rivetti, G. Spano and G. Petrini, Langmuir 15 (1999) 5753.
[81] H. Munakata, Y. Oumi and A. Miyamoto, J. Phys. Chem. B
105 (2001) 3493.
[82] G.N. Vayssilov and R.A. van Santenn, J. Catal. 175 (1998) 170.
[83] X. Feng and W. K. Hall, J. Catal. 166 (1997) 368.
[84] W.K. Hall, X. Feng, J. Dumesic and R. Watwe, Catal. Lett. 52
(1998) 173.
[85] A.M. Vodolin, V.I. Sobolev, and G.M. Zhidomirov, Kin. Catal.
39 (1998) 775.
[86] A. Dubkov, N.S. Ovanesyan, A.A. Shteinman, K.A. Dubkov,
V.I. Sobolev and G.I. Panov, Kin. Catal. 39 (1998) 792.
[87] H.-Y. Chen and W.M.H. Sachtler, Catal. Today 42 (1998) 73.
[88] R. Joyner and M. Stockenhuber, J. Phys. Chem. B 103 (1999)
5963.
[89] M. Kogel, R. Monning, W. Schwieger, A. Tissler and T.Turek,
J. Catal. 182 (1999) 470.
[90] P. Marturano, A. Kogelbauer and P. Prins, J. Catal. 190 (2000)
460.
[91] P. Marturano, L. Drozdova, A. Kogelbauer and R. Prins, J.
Catal. 192 (2000) 236.
[92] H.-Y. Chen, El-M. El-Malki, X. Wang, R.A. van Santen and
W.M.H. Sachtler, J. Mol. Catal. A 162 (2000) 159.
[93] S. Bordiga, R. Buzzoni, F. Geobaldo, C. Lamberti, E.
Giamello, A. Zecchina, G. Leofanti, G. Petrini, G. Tozzola
and G. Vlaic, J. Catal. 158 (1996) 486.
[94] A. Zecchina, F. Geobaldo, C. Lamberti, S. Bordiga, G. Turnes
Palomino and C. Otero Arean, Catal. Lett. 42 (1996) 25.
[95] A. Meagher, V. Nair and R. Szostak, Zeolites 8 (1988) 3.
[96] K. Yogo, S. Tanaka, T. Ono, T. Mikami and E. Kikuchi,
Micro. Mater. 3 (1994) 39.
[97] P. Feies, J.B. Nagy, J. Halasz and A. Oszko, Appl. Catal. 61
(1990) 85.
[98] A. Ribera, I.W.C.E. Arends, S. de Vries, J. Perez-Ramırez and
R.A. Sheldon, J. Catal. 195 (2000) 287.
[99] V.L. Zholobenko, I.N. Senchenya, L.M. Kustov and V.B.
Kazanskii, Kinet. Katal. 32 (1991) 132.
[100] V.I. Sobolev, G.I. Panov, A.S. Kharitonov, V.N. Romannikov,
A.M. Volodin and K.G. Ione, J. Catal. 139 (1993) 435.
[101] C. Lamberti, G. Turnes Palomino, S. Bordiga, D. Arduino, A.
Zecchina and G. Vlaic, Jpn. J. Appl. Phys. 38 (1999) 55.
[102] S.A. Axon, K.K. Fox, S.W. Carr and J. Klinowski, Chem. Phys.
Lett. 189 (1992) 1.
[103] D.W. Lewis, R.A. Catlow, J. Sankar and S.W. Carr, J. Phys.
Chem. 99 (1995) 2377.
[104] G. Spoto, A. Zecchina, G. Berlier, S. Bordiga, M.G. Clerici and
L. Basini, J. Mol. Catal. A 158 (2000) 107.
[105] L. Brabec, M. Jeschke, R. Klik, J. Novakova, L. Kubelkova
and J. Meusinger, Appl. Catal. A 170 (1998) 105.
[106] E. Suzuki, K. Nakashiro and Y. Ono, Chem. Lett. (1998) 953.
[107] P.P. Notte, Topics in Catal. 13 (2000) 387.
[108] C. Lamberti, S. Bordiga, A. Zecchina, M. Salvalaggio, F.
Geobaldo and C. Otero Arean, J. Chem. Soc. Faraday Trans.
94 (1998) 1519.
[109] C. Lamberti, G. Turnes Palomino, S. Bordiga, A. Zecchina, G.
Spano and C. Otero Arean, Catal. Lett. 63 (1999) 213.
[110] G. Berlier, G. Spoto, S. Bordiga, G. Ricchiardi, P. Fisicaro, A.
Zecchina, I. Rossetti, E. Selli, L. Forni, E. Giamello and C.
Lamberti, J. Catal. (submitted).
78 A. Zecchina et al./Characterization of catalysts active in partial oxidations




















