
Enhanced apoptotic action of trichosanthin in HIV1 infected cells
Upload
independentCategory
view
1download
0
doi: 10.1111/j.1349-7006.2009.01242.x Cancer Sci | September 2009 | vol. 100 | no. 9 | 1757–1766© 2009 Japanese Cancer Association
Blackwell Publishing Asia
Improved therapeutic efficacy against murine carcinoma by combining honokiol with gene therapy of PNAS-4, a novel pro-apoptotic geneZhu Yuan,1,5 Huanyi Liu,2,5 Fei Yan,1,5 Yongsheng Wang,1 Lantu Gou,1 Chunlai Nie,1,4 Zhenyu Ding,1 Songtao Lai,1 Yuwei Zhao,1 Xinyu Zhao,1 Jiong Li,1 Hongxin Deng,1 Yongqiu Mao,1 Lijuan Chen,1 Yuquan Wei1 and Xia Zhao3,4
1State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, West China Medical School, Sichuan University; 2Cancer Center, Chengdu Military General Hospital; 3Department of Gynecology and Obstetrics, West China Second Hospital, West China Medical School, Sichuan University, Chengdu, China
(Received December 21, 2008/Revised May 27, 2009/Accepted May 28, 2009/Online publication July 3, 2009)
PNAS-4, a novel pro-apoptotic gene activated during the earlyresponse to DNA damage, can inhibit proliferation via apoptosiswhen overexpressed in some tumor cells. Recent studies haveindicated that honokiol can induce apoptosis, inhibit angiogenesis,and suppress tumor growth. In the present study, we investigatedwhether mouse PNAS-4 (mPNAS-4) could augment the apoptosis oftumor cells induced by honokiol in vitro, and whether theantiangiogenic activity of honokiol and induction of apoptosis bymPNAS-4 could work cooperatively to improve the antitumor efficacyin vivo. In vitro, mPNAS-4 inhibited proliferation of murine colorectalcarcinoma CT26 and Lewis lung carcinoma LL2 cells through inductionof apoptosis, and significantly augmented the apoptosis of CT26and LL2 cells induced by honokiol. Compared with treatment withmPNAS-4 or honokiol alone, in vivo systemic administration of anexpression plasmid encoding mPNAS-4 and low-dose honokiolsignificantly suppressed tumor growth through the enhancedinduction of apoptosis and the augmented inhibition of angiogenesis.Our data suggest that the combined treatment with mPNAS-4 plushonokiol augments antitumor effects in vitro and in vivo, and thatthe improved antitumor activity in vivo may be associated withenhanced induction of apoptosis and augmented inhibition ofangiogenesis. The present study may provide a novel and effectivemethod for the treatment of cancer. (Cancer Sci 2009; 100: 1757–1766)
The traditional trinity of cancer therapies is comprised ofchemotherapy as the first-line treatment for a wide variety
of tumors, surgical intervention, and radiation. Althoughchemotherapy is effective in combating the growth and spreadof some tumors, dose-dependent toxicities such as bone marrowsuppression, gastrointestinal reactions, liver dysfunction, andrenal toxicity, and development of drug resistance greatly limit itstherapeutic efficacy.(1) To date, none of the treatment regimens usingconventional chemotherapy drugs such as cisplatin, adriamycin,and 5-fluorouracil is curative. Thus, continued preclinicalstudies on innovative therapeutic strategies are warranted.
Natural products have been the source of many medicallybeneficial drugs, and their importance in the prevention andtreatment of cancer is becoming increasingly apparent.(2) Honokiol,a naturally occurring compound isolated from Magnolia offici-nalis, is of medicinal use. Honokiol shows some biologicalactivities, including antioxidative activity,(3) anxiolytic potency,(4)
antiplatelet aggregation,(5) hepatoprotective effect,(6) neuroprotectiveeffect,(7) and anti-inflammatory and antimicrobial effects.(8–10)
Previous reports have shown the anticancer activities of honokiol.Honokiol induces apoptosis in various cancer cell lines(11–14) andrepresses tumor growth in nude mice.(15,16) Moreover, honokiolinhibits angiogenesis by interfering with the phosphorylationof VEGFR-2.(15) Recently, we found that liposomal honokiolsignificantly inhibits tumor-associated lymphangiogenesis and
metastasis.(17) More importantly, honokiol can overcome drugresistance in various cancer cell lines.(18–21) These findings indicatethat honokiol may be a novel, promising anticancer agent.
PNAS-4 was identified as a novel apoptosis-related protein inthe human acute promyelocytic leukemia cell line NB4. ThePNAS-4 protein contains a conserved N-terminal portionbelonging to the DUF862 family with unknown function andtwo or three hydrophobic motifs. The amino acid sequence iden-tities of PNAS-4 among various organisms are very high. Recentstudies showed that PNAS-4 is upregulated in peripheral bloodmononuclear cells that are exposed to carcinogenic agents suchas benzene,(22) in human papillomavirus 16 E6-expressing U2OScells (U2OSE64b) following mitomycin C treatment,(23) in humanpapillomavirus-infected invasive cervical cancer,(24) and inandrogen-independent prostate cancer,(25) indicating that it mightbe related to the genesis of some cancers. hPNAS-4 was identi-fied as a novel pro-apoptotic gene activated during the earlyresponse to DNA damage, and when overexpressed in osteo-sarcoma U2OS cells, it could induce significant apoptosis.(26) Inaddition, a recent report showed that the expression of porcinepPNAS-4 tended to decrease gradually during the period fordevelopment of muscle fibers, that is, the muscle fiber numberappeared to increase as a result of the decrease in pPNAS-4mRNA expression level during prenatal muscle development,implying that PNAS-4 may be involved in apoptosis.(27)
Some original investigations on the functions of PNAS-4 havebeen carried out in our laboratory. By using zebrafish as theanimal model, we observed that overexpression of PNAS-4leads to elongation of the antero-posterior body axis, and knockingdown PNAS-4 causes gastrulation defects, indicating that PNAS-4may regulate convergence and extension during vertebrate gas-trulation.(28) By searching mouse Reference mRNA sequences(refseq-rna) database from NCBI with the full-length amino acidsequence of hPNAS-4, we found a mouse PNAS-4 homolog andnamed it mPNAS-4 (GenBank accession no. NM_024282). ThemPNAS-4 cDNA encodes a protein possessing a putative DUF862domain and three hydrophobic motifs, with a calculated molecularweight of 21 434 Da. In addition, we found that overexpressionof PNAS-4 efficiently inhibited growth of LL2 cells, humanovarian cancer SKOV3 cells, and lung adenocarcinoma A549cells through induction of apoptosis.(29–31)
More recently, we have confirmed that PNAS-4 enhances thesensitivity of lung cancer to gemcitabine.(32) Similarly, wedecided to investigate whether PNAS-4 could also potentiate theantineoplastic effects of honokiol. This decision was based onthe following considerations:
4To whom correspondence should be addressed. E-mail: [email protected];[email protected] Yuan, Huan-yi Liu, and Fei Yan contributed equally to this work.

1758 doi: 10.1111/j.1349-7006.2009.01242.x© 2009 Japanese Cancer Association
(1) Honokiol has strong anticancer activities including pro-apoptotic activity, antiangiogenic activity, anti-invasive activity,antimetastatic activity, and antiproliferative activity.(11–17)
(2) More importantly, honokiol shows superiority over conven-tional chemotherapy drugs, that is, it can overcome drugresistance in some cancer cell lines.(18–21)
(3) We have carried out systematic studies on honokiol, andapplied for a Chinese patent (patent no. 200610021277.8,The preparation of liposomal honokiol and its application inthe treatment of cancer).
On the basis of these considerations and the observations thatPNAS-4 induces apoptosis when overexpressed in some tumorcells, we subsequently investigated the antitumor efficacy ofmPNAS-4 plus low-dose honokiol in vitro and in vivo, usingCT26 and LL2 murine models. The present study demonstratedthat in vitro mPNAS-4 induces apoptosis and also enhances theapoptosis of cancer cells induced by honokiol, and that theantiangiogenic activity of honokiol and the induction of apoptosisby mPNAS-4 could work cooperatively to enhance efficacyin vivo. Taken together, our results may provide a novel andeffective approach to treat cancer.
Materials and Methods
Preparation of liposome. The liposome for honokiol was pre-pared in our laboratory as described previously,(21) and the liposomefor plasmid treatment in animal experiments was a cationicliposome (DOTAP/cholesterol). The cationic liposome was preparedusing the procedure described previously.(33) Briefly, the cationiclipid DOTAP (Avanti Polar Lipids, Alabaster, AL, USA) and theneutral lipid cholesterol (Sigma, St Louis, MO, USA) weremixed at equimolar concentrations and dissolved in chloroformin a round-bottomed flask. The solution was rotated on a rotaryevaporator at 30°C for 30 min to obtain a thin lipid film. Thedried lipid film was then hydrated in 5% glucose solution to givea final concentration of 7 mM DOTAP and 7 mM cholesterol,referred to as 7 mM DOTAP:Chol. The hydrated DOTAP:Cholfilm was rotated in a water bath at 50°C for 45 min and then 35°Cfor 10 min. Then, the mixture was left overnight and sonicated atlow frequency for 5 min at 50°C. After sonication, it was transferredto a tube and heated at 50°C for 10 min. The mixture was sequentiallyextruded through polycarbonate membrane (Millipore, Billerica,MA, USA) to decrease size (five times at 0.2 μm and three timesat 0.1 μm) using syringes. The final cationic liposome (DOTAP/cholesterol) was a small multilamellar liposome in a sizerange of 100 ± 20 nm. It was stored under argon gas at 4°C.
Preparation of liposomal honokiol. Separation and purificationof honokiol were carried out according to methods describedpreviously.(34) In order to make it soluble, the purified honokiolwas encapsulated with liposome according to the methodreported previously.(21)
Plasmid construction and purification. The pcDNA3.1-mPS plasmidwas constructed as previously described.(31) pcDNA3.1 vectorwas used as a control. Both pcDNA3.1-mPS and pcDNA3.1 werepurified by two rounds of passage over EndoFree columns (Qiagen,Chatsworth, CA, USA), as reported previously.(35,36) The purifiedplasmids were mixed with liposome to form a DNA–liposomecomplex, and then used for subsequent animal experiments.
Cell culture and transfection. LL2 and CT26 cells were obtainedfrom American Type Culture Collection (Manassas, VA, USA) andcultured in DMEM (Gibco, Carlsbad, CA, USA) and RPMI-1640(Gibco), respectively, containing 10% FBS in a 37°C incubatorwith a humidified 5% CO2 atmosphere. For transfection, eachwell of six-well or 96-well plates was seeded with 2 × 105 or2 × 103 cells, respectively. Transfection was carried out usingLipofectamine 2000 (Life Technologies, Carlsbad, CA, USA)according to the manufacturer’s instructions.
Treatments of cells in the in vitro experiments. LL2 and CT26cells were classified into the following five groups, and treatedas follows:
• Control: the cells were left untreated, and when cultured for72 h, cells were harvested.
• pcDNA3.1 (empty vector) alone: the cells were first incubatedfor 24 h, then transfected with pcDNA3.1 plasmid. Forty-eighthours after transfection, cells were harvested.
• Honokiol alone: when the cells were cultured for 48 h,honokiol was added at a concentration of 10 μg/mL. Twenty-fourhours later, cells were harvested.
• mPNAS-4 alone: the cells were first incubated for 24 h,then transfected with pcDNA3.1-mPS plasmid. Forty-eighthours after transfection, cells were harvested.
• mPNAS-4 plus honokiol (combination): the cells were firstincubated for 24 h, then transfected with pcDNA3.1-mPSplasmid. Twenty-four hours after transfection, honokiolwas added at a concentration of 10 μg/mL. Forty-eight hoursafter transfection, cells were harvested.
The harvested cells above were used for MTT assay, flowcytometric analysis, agarose gel DNA electrophoresis, andmorphological analysis. In the morphological analysis, differentcell groups were treated as described above. However, in orderto identify transfected cells from non-transfected cells upongreen fluorescence, the cells were cotransfected with pcDNA3.1 orpcDNA3.1-mPS plus pEGFP-N1 plasmids (Clontech Laboratories,Mountain View, CA, USA). The concentration of pEGFP-N1 vectorwas one-fifth that of the pcDNA3.1 or pcDNA3.1-mPS vector.
Detection of mPNAS-4 expression in vitro and in vivo. For detectionof mPNAS-4 expression in vitro, LL2 cells were treated accordingto the schedules described above. One part of the harvested cellswas used to isolate RNA and then subjected to RT-PCR foramplification of the coding region of mPNAS-4, and the restwas used for the detection of protein expression by western blotanalysis according to the method described previously,(37) usingthe anti-mPNAS-4 polyclonal antibodies produced by us.(29) Fordetection of mPNAS-4 expression in vivo, when mice werekilled at the end of the experiment, the tumor tissues werecollected for detecting mPNAS-4 expression by RT-PCR. Theprimers used for amplification of mPNAS-4 (608 bp) were asfollows: 5′-GCGGATCCGCCACC ATGGCCAACCAGCCCATCATC-3′ and 5′-CCGCTCGAGCTATAGTTTTGTGTGGCGCCCAGG-3′, whereas the primers used for amplification ofGAPDH (187 bp) were designed as reported previously.(38)
MTT assay. Survival of cells after treatments was quantifiedusing the MTT assay.(39) Cells were seeded in triplicate for eachgroup and the processes were repeated three times. Media-onlytreated cells served as the indicator of 100% cell viability.
Flow cytometric analysis. Flow cytometric analysis was carriedout to identify sub-G1 cells and apoptotic cells, and to measurethe percentage of sub-G1 cells after propidium iodide staining inhypotonic buffer as previously described.(40)
Agarose gel DNA electrophoresis. The pattern of DNA cleavagewas analyzed by agarose gel electrophoresis as describedpreviously.(40)
Morphological analysis. LL2 cells were treated according to theschedules above. Apoptotic morphology was analyzed accordingto previously reported methods with a slight modification.(41,42)
Briefly, the cells were cotransfected as described above andcultured for the indicated period of time, then used for apoptoticmorphology analysis. The apoptotic morphology of tumor cellswas determined at 48 h after transfection and the percentage ofapoptotic cells was determined by the number of green cellswith apoptotic morphology divided by the total number of greencells. The images of enhanced green fluorescent protein (EGFP)expression were captured by a fluorescence microscope.
Yuan et al. Cancer Sci | September 2009 | vol. 100 | no. 9 | 1759© 2009 Japanese Cancer Association
Animal tumor models and treatment. Female mice at 6–8 weeksof age were transplanted with 5 × 105 tumor cells. The LL2 andCT26 models were established in C57BL/6 and BALB/c mice,respectively. Ten days after tumor cell inoculation, when tumorswere 5–8 mm in diameter, the mice were assigned randomly toone of the following five groups (10 mice/group), and treatedwith: (i) 100 μL PBS; (ii) 100 μg pcDNA3.1 plasmid and 300 μgliposome complexes in 100 μL PBS; (iii) 100 μL of 0.5 mgliposomal honokiol (25 mg/kg bodyweight); (iv) 100 μg pcDNA3.1-mPS plasmid and 300 μg liposome complexes in 100 μL PBS;and (v) 100 μg pcDNA3.1-mPS plasmid and 300 μg liposomecomplexes in 100 μL PBS and 100 μL of 0.3 mg liposomalhonokiol (15 mg/kg bodyweight). The mice were treated withDNA–liposome complex by intravenous administration via thetail vein twice a week, and received honokiol by intraperitonealroute every day for 4 weeks. Tumor size was measured with adial caliper at 3-day intervals, and tumor volume was calculatedby the following formula: tumor volume (mm3) = 0.52 × length(mm) × width (mm) × width (mm).(43) All studies involving micewere approved by the Institutional Animal Care and TreatmentCommittee of Sichuan University.
Alginate-encapsulated tumor cell assay. Alginate-encapsulatedtumor cell assays were carried out as described previously witha slight modification.(44) Briefly, CT26 cells were resuspended insodium alginate solution (w/v, 1.8%) and added dropwise to250 mM calcium chloride solution. One of the formed alginatebeads contained approximately 1 × 105 tumor cells. BALB/c micewere then anesthetized, and four beads were implanted subcutane-ously into an incision made on the dorsal side. Mice weresubdivided into five groups (five mice per group) and treated asdescribed above. The treatments were started 24 h after thebeads were implanted. Two weeks later, the mice were injectedi.v. with 100 μL of 100 mg/kg FITC–dextran (Sigma) solutionthrough the tail vein. Beads were removed and photographed20 min after FITC–dextran injection. The FITC–dextranuptake was measured against a standard curve of FITC–dextran.
Histological analysis. Dissected tumors were divided in half,one-half for paraffin sections fixed in 10% NBF and embedded inparaffin, and the other half frozen at –80°C. For MVD analysis,frozen sections (7 μm) were fixed in acetone, incubated, and stainedwith an antibody reactive to CD31 as reported previously.(45) MVDwas determined by counting the number of microvessels perhigh-power field as described previously.(46,47) For quantitativeassessment of apoptosis, TUNEL was carried out with an In situCell Death Detection Kit (Hoffmann-La Roche, Basel, Switzerland)following the manufacturer’s protocol.(48) For observations ofpotential side effects, the tissues (including heart, liver, spleen,lung, kidney, and brain) were fixed in 10% NBF and embeddedin paraffin. Sections (3–5 μm) were stained with H&E.(49) Sectionsin H&E staining and immunohistochemical staining were observedby two pathologists in a blinded manner.
Statistical analysis. The statistical analysis was carried outusing SPSS software (version 12.0 for Windows, SPSS UK Ltd.,Woking, Surrey, UK). All of the values were expressed as
means ± SD. Data were analyzed by one-way ANOVA, and thendifferences among the means were analyzed using Tukey–Kramermultiple comparison test. Survival curves were constructedaccording to the Kaplan–Meier method,(50) and statisticalsignificance was determined by the log-rank test.(51) Differenceswere considered significant at P < 0.05.
Results
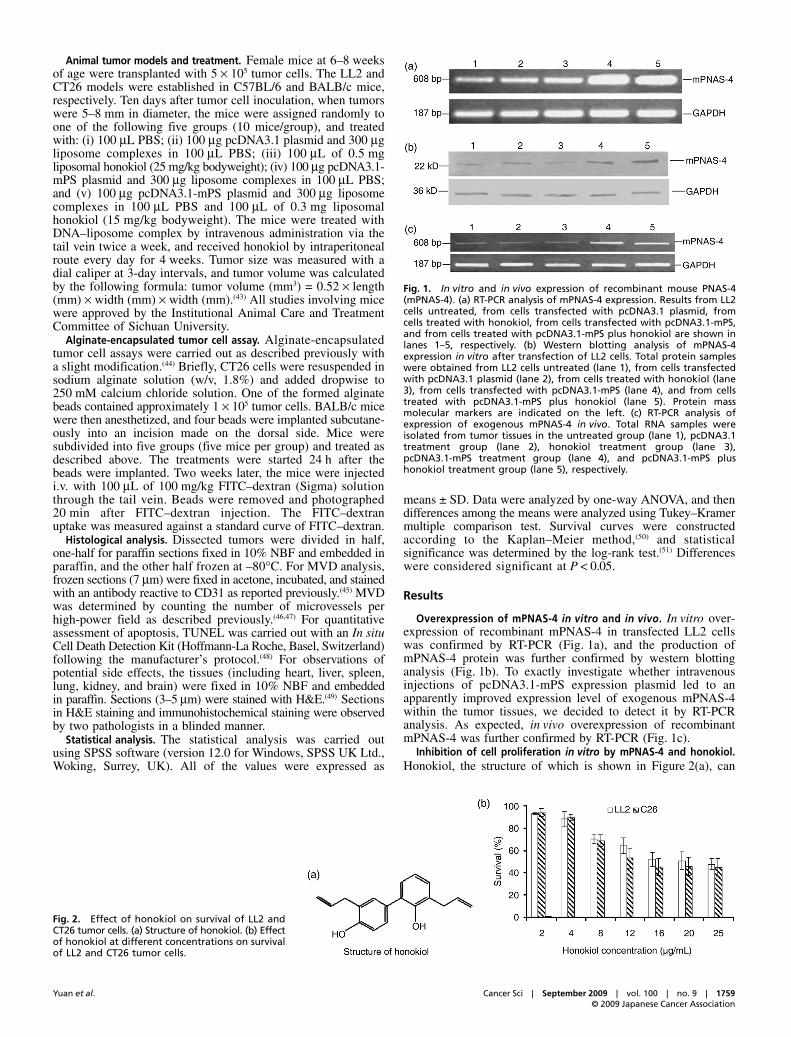
Overexpression of mPNAS-4 in vitro and in vivo. In vitro over-expression of recombinant mPNAS-4 in transfected LL2 cellswas confirmed by RT-PCR (Fig. 1a), and the production ofmPNAS-4 protein was further confirmed by western blottinganalysis (Fig. 1b). To exactly investigate whether intravenousinjections of pcDNA3.1-mPS expression plasmid led to anapparently improved expression level of exogenous mPNAS-4within the tumor tissues, we decided to detect it by RT-PCRanalysis. As expected, in vivo overexpression of recombinantmPNAS-4 was further confirmed by RT-PCR (Fig. 1c).
Inhibition of cell proliferation in vitro by mPNAS-4 and honokiol.Honokiol, the structure of which is shown in Figure 2(a), can
Fig. 1. In vitro and in vivo expression of recombinant mouse PNAS-4(mPNAS-4). (a) RT-PCR analysis of mPNAS-4 expression. Results from LL2cells untreated, from cells transfected with pcDNA3.1 plasmid, fromcells treated with honokiol, from cells transfected with pcDNA3.1-mPS,and from cells treated with pcDNA3.1-mPS plus honokiol are shown inlanes 1–5, respectively. (b) Western blotting analysis of mPNAS-4expression in vitro after transfection of LL2 cells. Total protein sampleswere obtained from LL2 cells untreated (lane 1), from cells transfectedwith pcDNA3.1 plasmid (lane 2), from cells treated with honokiol (lane3), from cells transfected with pcDNA3.1-mPS (lane 4), and from cellstreated with pcDNA3.1-mPS plus honokiol (lane 5). Protein massmolecular markers are indicated on the left. (c) RT-PCR analysis ofexpression of exogenous mPNAS-4 in vivo. Total RNA samples wereisolated from tumor tissues in the untreated group (lane 1), pcDNA3.1treatment group (lane 2), honokiol treatment group (lane 3),pcDNA3.1-mPS treatment group (lane 4), and pcDNA3.1-mPS plushonokiol treatment group (lane 5), respectively.
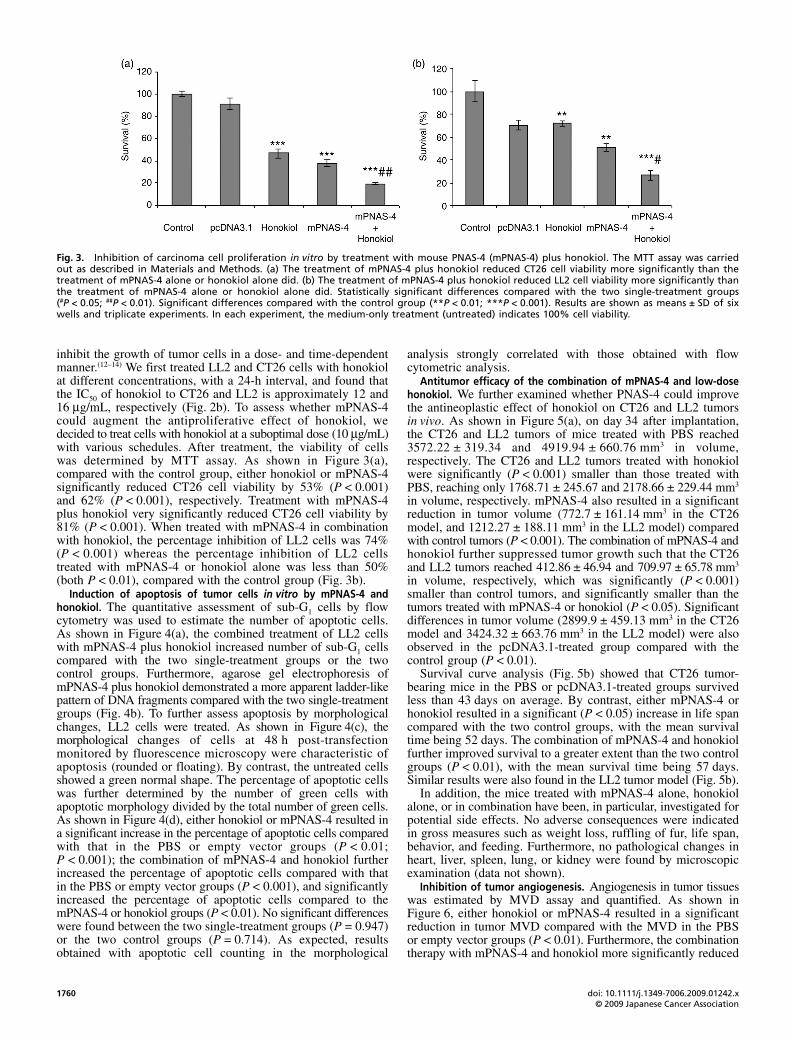
Fig. 2. Effect of honokiol on survival of LL2 andCT26 tumor cells. (a) Structure of honokiol. (b) Effectof honokiol at different concentrations on survivalof LL2 and CT26 tumor cells.
1760 doi: 10.1111/j.1349-7006.2009.01242.x© 2009 Japanese Cancer Association
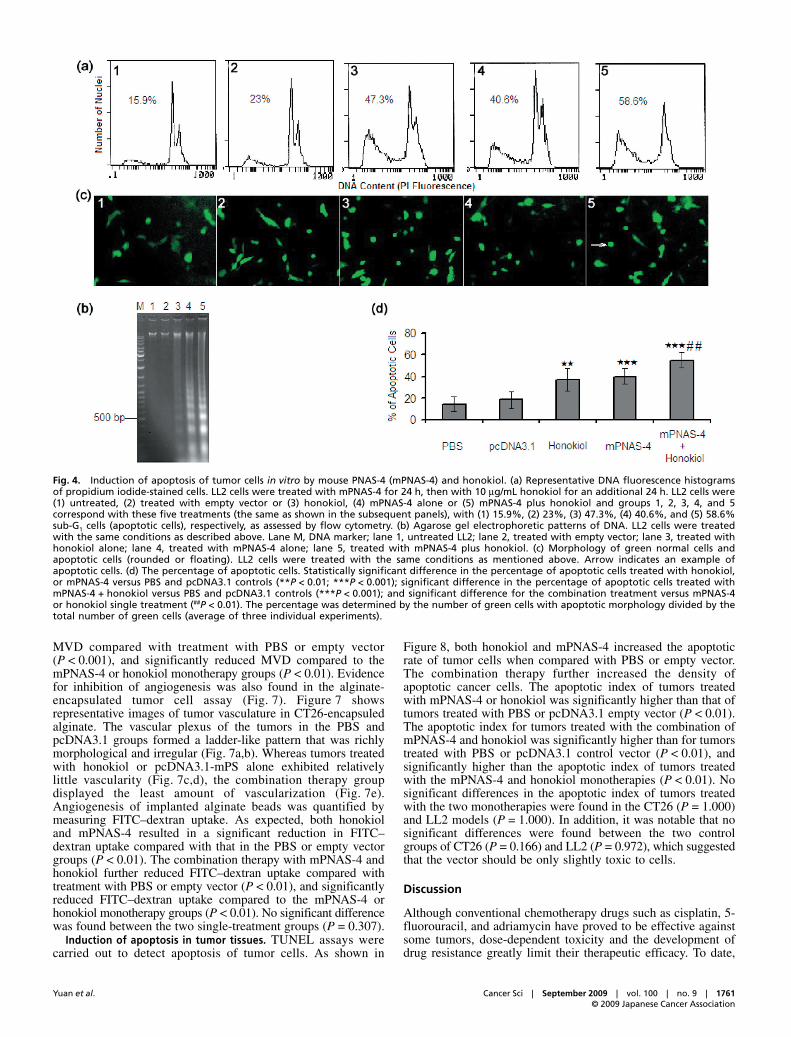
inhibit the growth of tumor cells in a dose- and time-dependentmanner.(12–14) We first treated LL2 and CT26 cells with honokiolat different concentrations, with a 24-h interval, and found thatthe IC50 of honokiol to CT26 and LL2 is approximately 12 and16 μg/mL, respectively (Fig. 2b). To assess whether mPNAS-4could augment the antiproliferative effect of honokiol, wedecided to treat cells with honokiol at a suboptimal dose (10 μg/mL)with various schedules. After treatment, the viability of cellswas determined by MTT assay. As shown in Figure 3(a),compared with the control group, either honokiol or mPNAS-4significantly reduced CT26 cell viability by 53% (P < 0.001)and 62% (P < 0.001), respectively. Treatment with mPNAS-4plus honokiol very significantly reduced CT26 cell viability by81% (P < 0.001). When treated with mPNAS-4 in combinationwith honokiol, the percentage inhibition of LL2 cells was 74%(P < 0.001) whereas the percentage inhibition of LL2 cellstreated with mPNAS-4 or honokiol alone was less than 50%(both P < 0.01), compared with the control group (Fig. 3b).
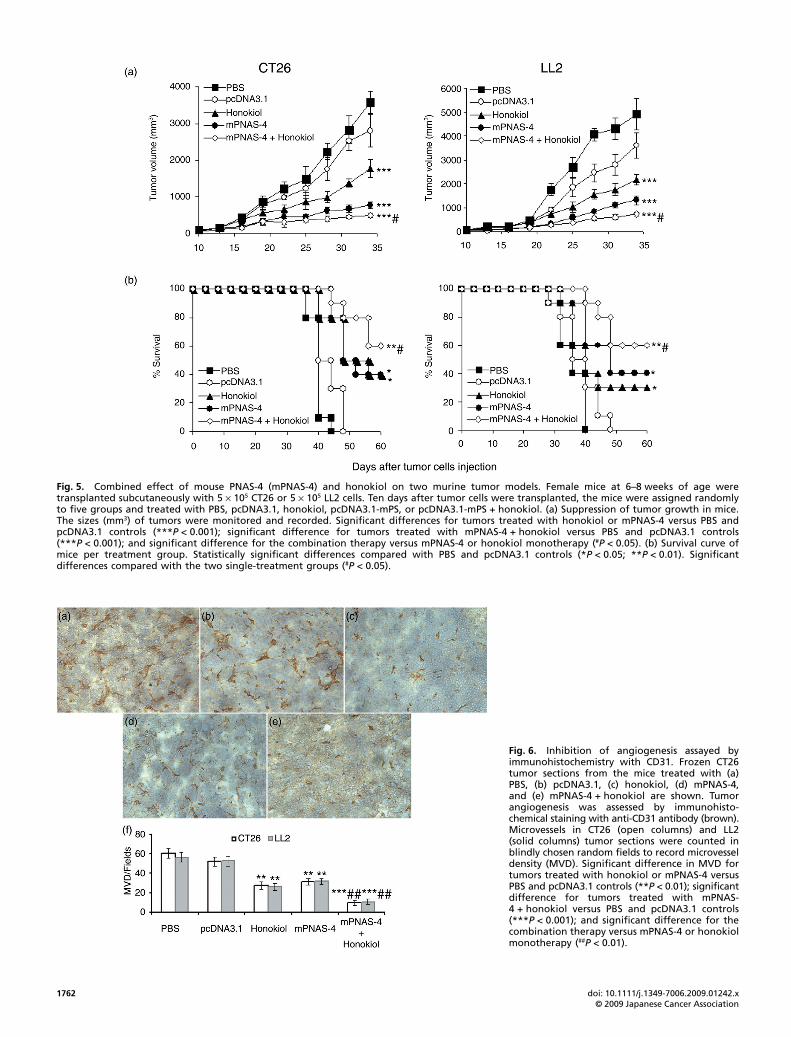
Induction of apoptosis of tumor cells in vitro by mPNAS-4 andhonokiol. The quantitative assessment of sub-G1 cells by flowcytometry was used to estimate the number of apoptotic cells.As shown in Figure 4(a), the combined treatment of LL2 cellswith mPNAS-4 plus honokiol increased number of sub-G1 cellscompared with the two single-treatment groups or the twocontrol groups. Furthermore, agarose gel electrophoresis ofmPNAS-4 plus honokiol demonstrated a more apparent ladder-likepattern of DNA fragments compared with the two single-treatmentgroups (Fig. 4b). To further assess apoptosis by morphologicalchanges, LL2 cells were treated. As shown in Figure 4(c), themorphological changes of cells at 48 h post-transfectionmonitored by fluorescence microscopy were characteristic ofapoptosis (rounded or floating). By contrast, the untreated cellsshowed a green normal shape. The percentage of apoptotic cellswas further determined by the number of green cells withapoptotic morphology divided by the total number of green cells.As shown in Figure 4(d), either honokiol or mPNAS-4 resulted ina significant increase in the percentage of apoptotic cells comparedwith that in the PBS or empty vector groups (P < 0.01;P < 0.001); the combination of mPNAS-4 and honokiol furtherincreased the percentage of apoptotic cells compared with thatin the PBS or empty vector groups (P < 0.001), and significantlyincreased the percentage of apoptotic cells compared to themPNAS-4 or honokiol groups (P < 0.01). No significant differenceswere found between the two single-treatment groups (P = 0.947)or the two control groups (P = 0.714). As expected, resultsobtained with apoptotic cell counting in the morphological
analysis strongly correlated with those obtained with flowcytometric analysis.
Antitumor efficacy of the combination of mPNAS-4 and low-dosehonokiol. We further examined whether PNAS-4 could improvethe antineoplastic effect of honokiol on CT26 and LL2 tumorsin vivo. As shown in Figure 5(a), on day 34 after implantation,the CT26 and LL2 tumors of mice treated with PBS reached3572.22 ± 319.34 and 4919.94 ± 660.76 mm3 in volume,respectively. The CT26 and LL2 tumors treated with honokiolwere significantly (P < 0.001) smaller than those treated withPBS, reaching only 1768.71 ± 245.67 and 2178.66 ± 229.44 mm3
in volume, respectively. mPNAS-4 also resulted in a significantreduction in tumor volume (772.7 ± 161.14 mm3 in the CT26model, and 1212.27 ± 188.11 mm3 in the LL2 model) comparedwith control tumors (P < 0.001). The combination of mPNAS-4 andhonokiol further suppressed tumor growth such that the CT26and LL2 tumors reached 412.86 ± 46.94 and 709.97 ± 65.78 mm3
in volume, respectively, which was significantly (P < 0.001)smaller than control tumors, and significantly smaller than thetumors treated with mPNAS-4 or honokiol (P < 0.05). Significantdifferences in tumor volume (2899.9 ± 459.13 mm3 in the CT26model and 3424.32 ± 663.76 mm3 in the LL2 model) were alsoobserved in the pcDNA3.1-treated group compared with thecontrol group (P < 0.01).
Survival curve analysis (Fig. 5b) showed that CT26 tumor-bearing mice in the PBS or pcDNA3.1-treated groups survivedless than 43 days on average. By contrast, either mPNAS-4 orhonokiol resulted in a significant (P < 0.05) increase in life spancompared with the two control groups, with the mean survivaltime being 52 days. The combination of mPNAS-4 and honokiolfurther improved survival to a greater extent than the two controlgroups (P < 0.01), with the mean survival time being 57 days.Similar results were also found in the LL2 tumor model (Fig. 5b).
In addition, the mice treated with mPNAS-4 alone, honokiolalone, or in combination have been, in particular, investigated forpotential side effects. No adverse consequences were indicatedin gross measures such as weight loss, ruffling of fur, life span,behavior, and feeding. Furthermore, no pathological changes inheart, liver, spleen, lung, or kidney were found by microscopicexamination (data not shown).
Inhibition of tumor angiogenesis. Angiogenesis in tumor tissueswas estimated by MVD assay and quantified. As shown inFigure 6, either honokiol or mPNAS-4 resulted in a significantreduction in tumor MVD compared with the MVD in the PBSor empty vector groups (P < 0.01). Furthermore, the combinationtherapy with mPNAS-4 and honokiol more significantly reduced
Fig. 3. Inhibition of carcinoma cell proliferation in vitro by treatment with mouse PNAS-4 (mPNAS-4) plus honokiol. The MTT assay was carriedout as described in Materials and Methods. (a) The treatment of mPNAS-4 plus honokiol reduced CT26 cell viability more significantly than thetreatment of mPNAS-4 alone or honokiol alone did. (b) The treatment of mPNAS-4 plus honokiol reduced LL2 cell viability more significantly thanthe treatment of mPNAS-4 alone or honokiol alone did. Statistically significant differences compared with the two single-treatment groups(#P < 0.05; ##P < 0.01). Significant differences compared with the control group (**P < 0.01; ***P < 0.001). Results are shown as means ± SD of sixwells and triplicate experiments. In each experiment, the medium-only treatment (untreated) indicates 100% cell viability.
Yuan et al. Cancer Sci | September 2009 | vol. 100 | no. 9 | 1761© 2009 Japanese Cancer Association
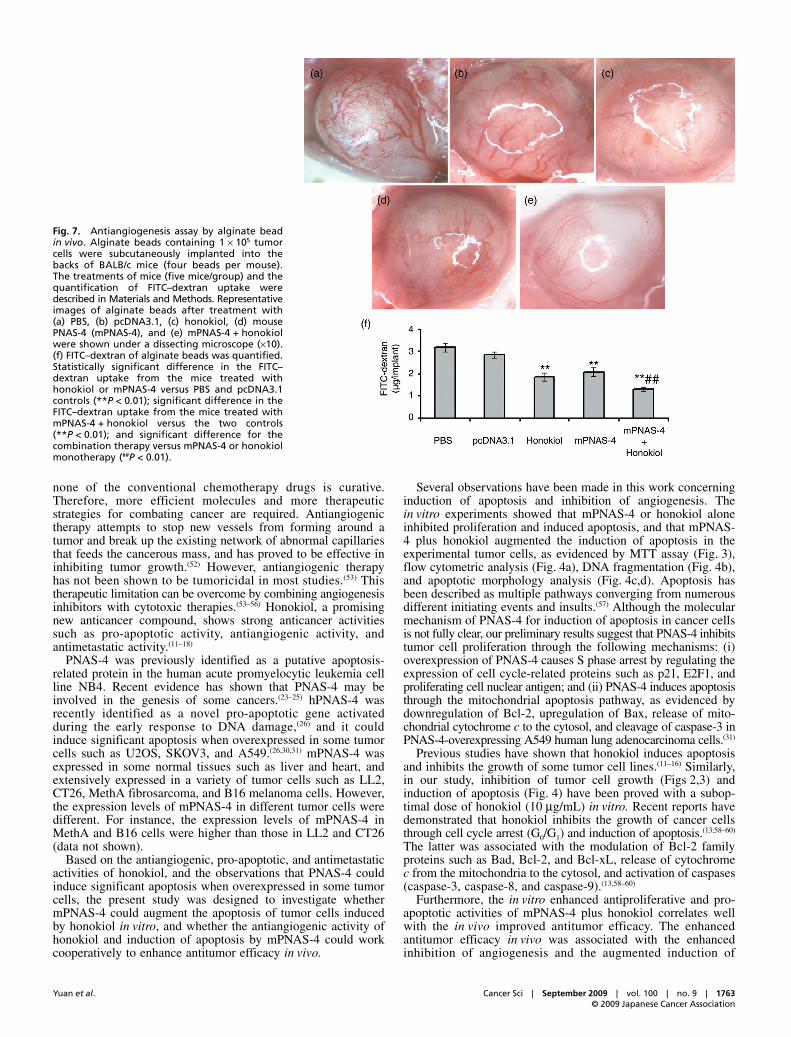
MVD compared with treatment with PBS or empty vector(P < 0.001), and significantly reduced MVD compared to themPNAS-4 or honokiol monotherapy groups (P < 0.01). Evidencefor inhibition of angiogenesis was also found in the alginate-encapsulated tumor cell assay (Fig. 7). Figure 7 showsrepresentative images of tumor vasculature in CT26-encapsuledalginate. The vascular plexus of the tumors in the PBS andpcDNA3.1 groups formed a ladder-like pattern that was richlymorphological and irregular (Fig. 7a,b). Whereas tumors treatedwith honokiol or pcDNA3.1-mPS alone exhibited relativelylittle vascularity (Fig. 7c,d), the combination therapy groupdisplayed the least amount of vascularization (Fig. 7e).Angiogenesis of implanted alginate beads was quantified bymeasuring FITC–dextran uptake. As expected, both honokioland mPNAS-4 resulted in a significant reduction in FITC–dextran uptake compared with that in the PBS or empty vectorgroups (P < 0.01). The combination therapy with mPNAS-4 andhonokiol further reduced FITC–dextran uptake compared withtreatment with PBS or empty vector (P < 0.01), and significantlyreduced FITC–dextran uptake compared to the mPNAS-4 orhonokiol monotherapy groups (P < 0.01). No significant differencewas found between the two single-treatment groups (P = 0.307).
Induction of apoptosis in tumor tissues. TUNEL assays werecarried out to detect apoptosis of tumor cells. As shown in
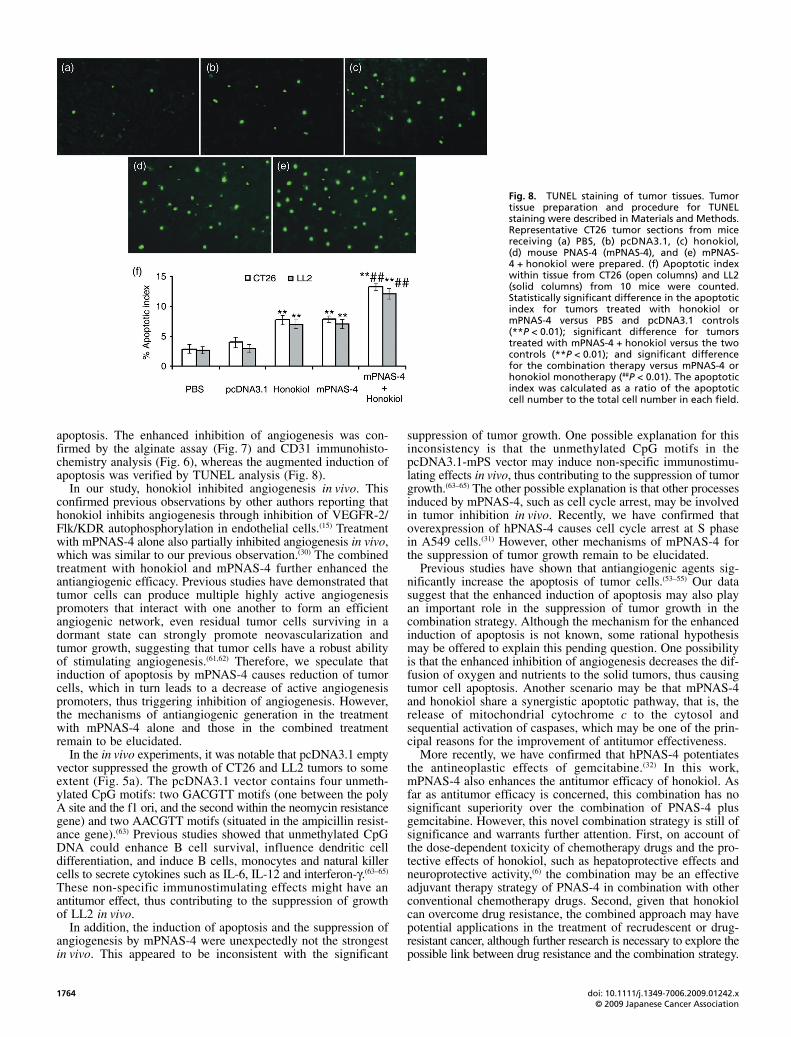
Figure 8, both honokiol and mPNAS-4 increased the apoptoticrate of tumor cells when compared with PBS or empty vector.The combination therapy further increased the density ofapoptotic cancer cells. The apoptotic index of tumors treatedwith mPNAS-4 or honokiol was significantly higher than that oftumors treated with PBS or pcDNA3.1 empty vector (P < 0.01).The apoptotic index for tumors treated with the combination ofmPNAS-4 and honokiol was significantly higher than for tumorstreated with PBS or pcDNA3.1 control vector (P < 0.01), andsignificantly higher than the apoptotic index of tumors treatedwith the mPNAS-4 and honokiol monotherapies (P < 0.01). Nosignificant differences in the apoptotic index of tumors treatedwith the two monotherapies were found in the CT26 (P = 1.000)and LL2 models (P = 1.000). In addition, it was notable that nosignificant differences were found between the two controlgroups of CT26 (P = 0.166) and LL2 (P = 0.972), which suggestedthat the vector should be only slightly toxic to cells.
Discussion
Although conventional chemotherapy drugs such as cisplatin, 5-fluorouracil, and adriamycin have proved to be effective againstsome tumors, dose-dependent toxicity and the development ofdrug resistance greatly limit their therapeutic efficacy. To date,
Fig. 4. Induction of apoptosis of tumor cells in vitro by mouse PNAS-4 (mPNAS-4) and honokiol. (a) Representative DNA fluorescence histogramsof propidium iodide-stained cells. LL2 cells were treated with mPNAS-4 for 24 h, then with 10 μg/mL honokiol for an additional 24 h. LL2 cells were(1) untreated, (2) treated with empty vector or (3) honokiol, (4) mPNAS-4 alone or (5) mPNAS-4 plus honokiol and groups 1, 2, 3, 4, and 5correspond with these five treatments (the same as shown in the subsequent panels), with (1) 15.9%, (2) 23%, (3) 47.3%, (4) 40.6%, and (5) 58.6%sub-G1 cells (apoptotic cells), respectively, as assessed by flow cytometry. (b) Agarose gel electrophoretic patterns of DNA. LL2 cells were treatedwith the same conditions as described above. Lane M, DNA marker; lane 1, untreated LL2; lane 2, treated with empty vector; lane 3, treated withhonokiol alone; lane 4, treated with mPNAS-4 alone; lane 5, treated with mPNAS-4 plus honokiol. (c) Morphology of green normal cells andapoptotic cells (rounded or floating). LL2 cells were treated with the same conditions as mentioned above. Arrow indicates an example ofapoptotic cells. (d) The percentage of apoptotic cells. Statistically significant difference in the percentage of apoptotic cells treated with honokiol,or mPNAS-4 versus PBS and pcDNA3.1 controls (**P < 0.01; ***P < 0.001); significant difference in the percentage of apoptotic cells treated withmPNAS-4 + honokiol versus PBS and pcDNA3.1 controls (***P < 0.001); and significant difference for the combination treatment versus mPNAS-4or honokiol single treatment (##P < 0.01). The percentage was determined by the number of green cells with apoptotic morphology divided by thetotal number of green cells (average of three individual experiments).
1762 doi: 10.1111/j.1349-7006.2009.01242.x© 2009 Japanese Cancer Association
Fig. 5. Combined effect of mouse PNAS-4 (mPNAS-4) and honokiol on two murine tumor models. Female mice at 6–8 weeks of age weretransplanted subcutaneously with 5 × 105 CT26 or 5 × 105 LL2 cells. Ten days after tumor cells were transplanted, the mice were assigned randomlyto five groups and treated with PBS, pcDNA3.1, honokiol, pcDNA3.1-mPS, or pcDNA3.1-mPS + honokiol. (a) Suppression of tumor growth in mice.The sizes (mm3) of tumors were monitored and recorded. Significant differences for tumors treated with honokiol or mPNAS-4 versus PBS andpcDNA3.1 controls (***P < 0.001); significant difference for tumors treated with mPNAS-4 + honokiol versus PBS and pcDNA3.1 controls(***P < 0.001); and significant difference for the combination therapy versus mPNAS-4 or honokiol monotherapy (#P < 0.05). (b) Survival curve ofmice per treatment group. Statistically significant differences compared with PBS and pcDNA3.1 controls (*P < 0.05; **P < 0.01). Significantdifferences compared with the two single-treatment groups (#P < 0.05).
Fig. 6. Inhibition of angiogenesis assayed byimmunohistochemistry with CD31. Frozen CT26tumor sections from the mice treated with (a)PBS, (b) pcDNA3.1, (c) honokiol, (d) mPNAS-4,and (e) mPNAS-4 + honokiol are shown. Tumorangiogenesis was assessed by immunohisto-chemical staining with anti-CD31 antibody (brown).Microvessels in CT26 (open columns) and LL2(solid columns) tumor sections were counted inblindly chosen random fields to record microvesseldensity (MVD). Significant difference in MVD fortumors treated with honokiol or mPNAS-4 versusPBS and pcDNA3.1 controls (**P < 0.01); significantdifference for tumors treated with mPNAS-4 + honokiol versus PBS and pcDNA3.1 controls(***P < 0.001); and significant difference for thecombination therapy versus mPNAS-4 or honokiolmonotherapy (##P < 0.01).
Yuan et al. Cancer Sci | September 2009 | vol. 100 | no. 9 | 1763© 2009 Japanese Cancer Association
none of the conventional chemotherapy drugs is curative.Therefore, more efficient molecules and more therapeuticstrategies for combating cancer are required. Antiangiogenictherapy attempts to stop new vessels from forming around atumor and break up the existing network of abnormal capillariesthat feeds the cancerous mass, and has proved to be effective ininhibiting tumor growth.(52) However, antiangiogenic therapyhas not been shown to be tumoricidal in most studies.(53) Thistherapeutic limitation can be overcome by combining angiogenesisinhibitors with cytotoxic therapies.(53–56) Honokiol, a promisingnew anticancer compound, shows strong anticancer activitiessuch as pro-apoptotic activity, antiangiogenic activity, andantimetastatic activity.(11–18)
PNAS-4 was previously identified as a putative apoptosis-related protein in the human acute promyelocytic leukemia cellline NB4. Recent evidence has shown that PNAS-4 may beinvolved in the genesis of some cancers.(23–25) hPNAS-4 wasrecently identified as a novel pro-apoptotic gene activatedduring the early response to DNA damage,(26) and it couldinduce significant apoptosis when overexpressed in some tumorcells such as U2OS, SKOV3, and A549.(26,30,31) mPNAS-4 wasexpressed in some normal tissues such as liver and heart, andextensively expressed in a variety of tumor cells such as LL2,CT26, MethA fibrosarcoma, and B16 melanoma cells. However,the expression levels of mPNAS-4 in different tumor cells weredifferent. For instance, the expression levels of mPNAS-4 inMethA and B16 cells were higher than those in LL2 and CT26(data not shown).
Based on the antiangiogenic, pro-apoptotic, and antimetastaticactivities of honokiol, and the observations that PNAS-4 couldinduce significant apoptosis when overexpressed in some tumorcells, the present study was designed to investigate whethermPNAS-4 could augment the apoptosis of tumor cells inducedby honokiol in vitro, and whether the antiangiogenic activity ofhonokiol and induction of apoptosis by mPNAS-4 could workcooperatively to enhance antitumor efficacy in vivo.
Several observations have been made in this work concerninginduction of apoptosis and inhibition of angiogenesis. Thein vitro experiments showed that mPNAS-4 or honokiol aloneinhibited proliferation and induced apoptosis, and that mPNAS-4 plus honokiol augmented the induction of apoptosis in theexperimental tumor cells, as evidenced by MTT assay (Fig. 3),flow cytometric analysis (Fig. 4a), DNA fragmentation (Fig. 4b),and apoptotic morphology analysis (Fig. 4c,d). Apoptosis hasbeen described as multiple pathways converging from numerousdifferent initiating events and insults.(57) Although the molecularmechanism of PNAS-4 for induction of apoptosis in cancer cellsis not fully clear, our preliminary results suggest that PNAS-4 inhibitstumor cell proliferation through the following mechanisms: (i)overexpression of PNAS-4 causes S phase arrest by regulating theexpression of cell cycle-related proteins such as p21, E2F1, andproliferating cell nuclear antigen; and (ii) PNAS-4 induces apoptosisthrough the mitochondrial apoptosis pathway, as evidenced bydownregulation of Bcl-2, upregulation of Bax, release of mito-chondrial cytochrome c to the cytosol, and cleavage of caspase-3 inPNAS-4-overexpressing A549 human lung adenocarcinoma cells.(31)
Previous studies have shown that honokiol induces apoptosisand inhibits the growth of some tumor cell lines.(11–16) Similarly,in our study, inhibition of tumor cell growth (Figs 2,3) andinduction of apoptosis (Fig. 4) have been proved with a subop-timal dose of honokiol (10 μg/mL) in vitro. Recent reports havedemonstrated that honokiol inhibits the growth of cancer cellsthrough cell cycle arrest (G0/G1) and induction of apoptosis.(13,58–60)
The latter was associated with the modulation of Bcl-2 familyproteins such as Bad, Bcl-2, and Bcl-xL, release of cytochromec from the mitochondria to the cytosol, and activation of caspases(caspase-3, caspase-8, and caspase-9).(13,58–60)
Furthermore, the in vitro enhanced antiproliferative and pro-apoptotic activities of mPNAS-4 plus honokiol correlates wellwith the in vivo improved antitumor efficacy. The enhancedantitumor efficacy in vivo was associated with the enhancedinhibition of angiogenesis and the augmented induction of
Fig. 7. Antiangiogenesis assay by alginate beadin vivo. Alginate beads containing 1 × 105 tumorcells were subcutaneously implanted into thebacks of BALB/c mice (four beads per mouse).The treatments of mice (five mice/group) and thequantification of FITC–dextran uptake weredescribed in Materials and Methods. Representativeimages of alginate beads after treatment with(a) PBS, (b) pcDNA3.1, (c) honokiol, (d) mousePNAS-4 (mPNAS-4), and (e) mPNAS-4 + honokiolwere shown under a dissecting microscope (×10).(f) FITC–dextran of alginate beads was quantified.Statistically significant difference in the FITC–dextran uptake from the mice treated withhonokiol or mPNAS-4 versus PBS and pcDNA3.1controls (**P < 0.01); significant difference in theFITC–dextran uptake from the mice treated withmPNAS-4 + honokiol versus the two controls(**P < 0.01); and significant difference for thecombination therapy versus mPNAS-4 or honokiolmonotherapy (##P < 0.01).
1764 doi: 10.1111/j.1349-7006.2009.01242.x© 2009 Japanese Cancer Association
apoptosis. The enhanced inhibition of angiogenesis was con-firmed by the alginate assay (Fig. 7) and CD31 immunohisto-chemistry analysis (Fig. 6), whereas the augmented induction ofapoptosis was verified by TUNEL analysis (Fig. 8).
In our study, honokiol inhibited angiogenesis in vivo. Thisconfirmed previous observations by other authors reporting thathonokiol inhibits angiogenesis through inhibition of VEGFR-2/Flk/KDR autophosphorylation in endothelial cells.(15) Treatmentwith mPNAS-4 alone also partially inhibited angiogenesis in vivo,which was similar to our previous observation.(30) The combinedtreatment with honokiol and mPNAS-4 further enhanced theantiangiogenic efficacy. Previous studies have demonstrated thattumor cells can produce multiple highly active angiogenesispromoters that interact with one another to form an efficientangiogenic network, even residual tumor cells surviving in adormant state can strongly promote neovascularization andtumor growth, suggesting that tumor cells have a robust abilityof stimulating angiogenesis.(61,62) Therefore, we speculate thatinduction of apoptosis by mPNAS-4 causes reduction of tumorcells, which in turn leads to a decrease of active angiogenesispromoters, thus triggering inhibition of angiogenesis. However,the mechanisms of antiangiogenic generation in the treatmentwith mPNAS-4 alone and those in the combined treatmentremain to be elucidated.
In the in vivo experiments, it was notable that pcDNA3.1 emptyvector suppressed the growth of CT26 and LL2 tumors to someextent (Fig. 5a). The pcDNA3.1 vector contains four unmeth-ylated CpG motifs: two GACGTT motifs (one between the polyA site and the f1 ori, and the second within the neomycin resistancegene) and two AACGTT motifs (situated in the ampicillin resist-ance gene).(63) Previous studies showed that unmethylated CpGDNA could enhance B cell survival, influence dendritic celldifferentiation, and induce B cells, monocytes and natural killercells to secrete cytokines such as IL-6, IL-12 and interferon-γ.(63–65)
These non-specific immunostimulating effects might have anantitumor effect, thus contributing to the suppression of growthof LL2 in vivo.
In addition, the induction of apoptosis and the suppression ofangiogenesis by mPNAS-4 were unexpectedly not the strongestin vivo. This appeared to be inconsistent with the significant
suppression of tumor growth. One possible explanation for thisinconsistency is that the unmethylated CpG motifs in thepcDNA3.1-mPS vector may induce non-specific immunostimu-lating effects in vivo, thus contributing to the suppression of tumorgrowth.(63–65) The other possible explanation is that other processesinduced by mPNAS-4, such as cell cycle arrest, may be involvedin tumor inhibition in vivo. Recently, we have confirmed thatoverexpression of hPNAS-4 causes cell cycle arrest at S phasein A549 cells.(31) However, other mechanisms of mPNAS-4 forthe suppression of tumor growth remain to be elucidated.
Previous studies have shown that antiangiogenic agents sig-nificantly increase the apoptosis of tumor cells.(53–55) Our datasuggest that the enhanced induction of apoptosis may also playan important role in the suppression of tumor growth in thecombination strategy. Although the mechanism for the enhancedinduction of apoptosis is not known, some rational hypothesismay be offered to explain this pending question. One possibilityis that the enhanced inhibition of angiogenesis decreases the dif-fusion of oxygen and nutrients to the solid tumors, thus causingtumor cell apoptosis. Another scenario may be that mPNAS-4and honokiol share a synergistic apoptotic pathway, that is, therelease of mitochondrial cytochrome c to the cytosol andsequential activation of caspases, which may be one of the prin-cipal reasons for the improvement of antitumor effectiveness.
More recently, we have confirmed that hPNAS-4 potentiatesthe antineoplastic effects of gemcitabine.(32) In this work,mPNAS-4 also enhances the antitumor efficacy of honokiol. Asfar as antitumor efficacy is concerned, this combination has nosignificant superiority over the combination of PNAS-4 plusgemcitabine. However, this novel combination strategy is still ofsignificance and warrants further attention. First, on account ofthe dose-dependent toxicity of chemotherapy drugs and the pro-tective effects of honokiol, such as hepatoprotective effects andneuroprotective activity,(6) the combination may be an effectiveadjuvant therapy strategy of PNAS-4 in combination with otherconventional chemotherapy drugs. Second, given that honokiolcan overcome drug resistance, the combined approach may havepotential applications in the treatment of recrudescent or drug-resistant cancer, although further research is necessary to explore thepossible link between drug resistance and the combination strategy.
Fig. 8. TUNEL staining of tumor tissues. Tumortissue preparation and procedure for TUNELstaining were described in Materials and Methods.Representative CT26 tumor sections from micereceiving (a) PBS, (b) pcDNA3.1, (c) honokiol,(d) mouse PNAS-4 (mPNAS-4), and (e) mPNAS-4 + honokiol were prepared. (f) Apoptotic indexwithin tissue from CT26 (open columns) and LL2(solid columns) from 10 mice were counted.Statistically significant difference in the apoptoticindex for tumors treated with honokiol ormPNAS-4 versus PBS and pcDNA3.1 controls(**P < 0.01); significant difference for tumorstreated with mPNAS-4 + honokiol versus the twocontrols (**P < 0.01); and significant differencefor the combination therapy versus mPNAS-4 orhonokiol monotherapy (##P < 0.01). The apoptoticindex was calculated as a ratio of the apoptoticcell number to the total cell number in each field.

Yuan et al. Cancer Sci | September 2009 | vol. 100 | no. 9 | 1765© 2009 Japanese Cancer Association
In conclusion, our data suggest that PNAS-4, a novel pro-apoptotic gene, can inhibit proliferation of tumor cells viaapoptosis and augment the apoptosis of tumor cells induced byhonokiol in vitro. Our data also demonstrate that the combina-tion of mPNAS-4 and honokiol enhances the antitumor efficacyin vivo, and that the improved therapeutic efficacy may beassociated with the enhanced induction of apoptosis and theaugmented inhibition of angiogenesis. Our results may providea novel and effective approach to treat cancer.
Acknowledgments
The authors thank Dr Tong Aiping and Dr Wei Haiyan for their advice,discussion, and critical review of the manuscript, and Dr Wan Yang forhis technical support. This work was supported by the National KeyBasic Research Program of China (2004CB518800, 2005CB522506).
Abbreviations
FBS fetal bovine serumhPNAS-4 human PNAS-4IL interleukinmPNAS-4 mouse PNAS-4MVD microvessel densityNBF neutral buffered formalinpcDNA3.1-mPS pcDNA3.1 plasmid encoding mouse PNAS-4 genepPNAS-4 porcine PNAS-4TUNEL terminal deoxynucleotidyltransferase-mediated dUTP
nick end labelingVEGFR vascular endothelial growth factor receptor
Disclosure Statement
No potential conflicts of interest were disclosed.
References
1 Kuo MT. Redox regulation of multidrug resistance in cancer chemotherapy:molecular mechanisms and therapeutic opportunities. Antioxid Redox Signal2009; 11: 99–133.
2 Pezzuto JM. Plant-derived anticancer agents. Biochem Pharmacol 1997; 53:121–33.
3 Park EJ, Zhao YZ, Na M et al. Protective effects of honokiol and magnololon tertiary butyl hydroperoxide- or d-galactosamine-induced toxicity in ratprimary hepatocytes. Planta Med 2003; 69: 33–7.
4 Kuribara H, Stavinoha WB, Maruyama Y. Honokiol, a putative anxiolyticagent extracted from magnolia bark, has no diazepam-like side-effects inmice. J Pharm Pharmacol 1999; 51: 97–103.
5 Pyo MK, Lee Y, Yun-choi HS. Anti-platelet effect of the constituentsisolated from the barks and fruits of magnolia obovata. Arch Pharm Res2002; 25: 325–8.
6 Cao AH, Vo LT, King RG. Honokiol protects against carbon tetrachlorideinduced liver damage in the rat. Phytother Res 2005; 19: 932–7.
7 Lin YR, Chen HH, Ko CH, Chan MH. Neuroprotective activity of honokioland magnolol in cerebellar granule cell damage. Eur J Pharmaco 2006; 537:64–9.
8 Shin TY, Kim DK, Chae BS, Lee EJ. Antiallergic action of magnoliaofficinalis on immediate hypersensitivity reaction. Arch Pharm Res 2001; 24:249–55.
9 Chang B, Lee Y, Ku Y, Bae K, Chung C. Antimicrobial activity of magnololand honokiol against periodontopathic microorganisms. Planta Med 1998;64: 367–9.
10 Park J, Lee J, Jung E et al. In vitro antibacterial and anti-inflammatoryeffects of honokiol and magnolol against propionibacterium sp. Eur JPharmacol 2004; 496: 189–95.
11 Hirano T, Gotoh M, Oka K. Natural flavonoids and lignans are potentcytostatic agents against human leukemic HL-60 cells. Life Sci 1994; 55:1061–9.
12 Hibasami H, Achiwa Y, Katsuzaki H et al. Honokiol induces apoptosis inhuman lymphoid leukemia Molt 4B cells. Int J Mol Med 1998; 2: 671–3.
13 Yang SE, Hsieh MT, Tsai TH, Hsu SL. Downmodulation of Bcl-XL, releaseof cytochrome c and sequential activation of caspases during honokiol-induced apoptosis in human squamous lung cancer CH27 cells. BiochemPharmacol 2002; 63: 1641–51.
14 Wang T, Chen F, Chen Z et al. Honokiol induces apoptosis throughp53-independent pathway in human colorectal cell line RKO. World JGastroenterol 2004; 10: 2205–8.
15 Bai X, Cerimele F, Ushio-Fukai M et al. Honokiol, a small molecular weightnatural product, inhibits angiogenesis in vitro and tumor growth in vivo. JBiol Chem 2003; 278: 35 501–7.
16 Chen F, Wang T, Wu YF et al. Honokiol: a potent chemotherapy candidatefor human colorectal carcinoma. World J Gastroenterol 2004; 10: 3459–63.
17 Wen J, Fu AF, Chen LJ et al. Liposomal honokiol inhibits VEGF-D-inducedlymphangiogenesis and metastasis in xenograft tumor model. Int J Cancer2009; 124: 2709–18.
18 Ishitsuka K, Hideshima T, Hamasaki M et al. Honokiol overcomesconventional drug resistance in human multiple myeloma by induction ofcaspase-dependent and -independent apoptosis. Blood 2005; 106: 1794–800.
19 Liu H, Zang C, Emde A et al. Anti-tumor effect of honokiol alone and incombination with other anti-cancer agents in breast cancer. Eur J Pharmacol2008; 591: 43–51.
20 Xu D, Lu Q, Hu X. Down-regulation of P-glycoprotein expression in MDRbreast cancer cell MCF-7/ADR by honokiol. Cancer Lett 2006; 243: 274–80.
21 Luo H, Zhong Q, Chen LJ et al. Liposomal honokiol, a promising agent fortreatment of cisplatin-resistant human ovarian cancer. J Cancer Res ClinOncol 2008; 134: 937–45.
22 Forrest MS, Lan Q, Hubbard AE et al. Discovery of novel biomarkers bymicroarray analysis of peripheral blood mononuclear cell gene expression inbenzene-exposed workers. Environ Health Perspect 2005; 113: 801–7.
23 Filippov V, Filippova M, Duerksen-Hughes PJ. The early response to DNAdamage can lead to activation of alternative splicing activity resulting inCD44 splice pattern changes. Cancer Res 2007; 67: 7621–30.
24 Santin AD, Zhan F, Bignotti E et al. Gene expression profiles of primaryHPV16- and HPV18-infected early stage cervical cancers and normalcervical epithelium: identification of novel candidate molecular markers forcervical cancer diagnosis and therapy. Virology 2005; 331: 269–91.
25 Best CJ, Gillespie JW, Yi YJ et al. Molecular alterations in primary prostatecancer after androgen ablation therapy. Clin Cancer Res 2005; 11: 6823–34.
26 Filippov V, Filippova M, Sinha D, Duerksen-Hughes PJ. PNAS-4: a novelpro-apoptotic gene activated during the early response to DNA damage. ProcAm Assoc Cancer Res 2005; 46: 717.
27 Mo DL, Zhu ZM, te Pas MFW et al. Characterization, expression profiles,intracellular distribution and association analysis of porcine PNAS-4 genewith production traits. BMC Genetics 2008; 9: 40.
28 Yao SH, Xie LF, Qian ML et al. Pnas4 is a novel regulator for convergenceand extension during vertebrate gastrulation. FEBS Lett 2008; 582: 2325–32.
29 Zhang P, Wang CT, Yan F et al. Prokaryotic expression of a novel mousepro-apoptosis protein PNAS-4 and application of its polyclonal antibodies.Braz J Med Biol Res 2008; 41: 504–11.
30 Yang F, Li ZY, Deng HX et al. Efficient inhibition of ovarian cancer growthand prolonged survival by transfection with a novel pro-apoptotic gene,hPNAS-4, in a mouse model. Oncology 2008; 75: 137–44.
31 Yan F, Gou LT, Yang JL et al. A novel pro-apoptosis gene PNAS4 thatinduces apoptosis in A549 human lung adenocarcinoma cells and inhibitstumor growth in mice. Biochimie 2009; 91: 502–7.
32 Hou S, Zhao Z, Yan F et al. Genetic transfer of PNAS-4 induces apoptosis andenhances sensitivity to gemcitabine in lung cancer. Cell Biol Int 2009; 33: 276–82.
33 Templeton NS, Lasic DD, Frederik PM, Strey HH, Roberts DD, PavlakisGN. Improved DNA: liposome complexes for increased systemic deliveryand gene expression. Nature Biotech 1997; 15: 647–52.
34 Chen LJ, Zhang Q, Yang GL et al. Sutherland: rapid purification and scale-up of honokiol and magnolol using high performance countercurrentchromatography (HPCCC). J Chromatograph A 2007; 1142: 115–22.
35 Krieg AM, Wu T, Weeratna R et al. Sequence motifs in adenoviral DNAblock immune activation by stimulatory CpG motifs. Proc Natl Acad SciUSA 1998; 95: 12 631–6.
36 Su JM, Wei YQ, Tian L et al. Active immunogene therapy of cancer withvaccine on the basis of chicken homologous matrix metalloproteinase-2.Cancer Res 2003; 63: 600–7.
37 Zhang R, Tian L, Chen LJ et al. Combination of MIG (CXCL9) chemokinegene therapy with low-dose cisplatin improves therapeutic efficacy againstmurine carcinoma. Gene Ther 2006; 13: 1263–71.
38 Furihata T, Hosokawa M, Satoh T, Chiba K. Synergistic role of specificityproteins and upstream stimulatory factor 1 in transactivation of the mousecarboxylesterase 2/microsomal acylcarnitine hydrolase gene promoter.Biochem J 2004; 384: 101–10.
39 Blezinger P, Wang J, Gondo M et al. Systemic inhibition of tumor growthand tumor metastases by intramuscular administration of the endostatin gene.Nat Biotechnol 1999; 17: 343–8.
40 Wei YQ, Zhao X, Kariya Y et al. Induction of apoptosis by quercetin:involvement of heat shock protein. Cancer Res 1994; 54: 4952–7.

1766 doi: 10.1111/j.1349-7006.2009.01242.x© 2009 Japanese Cancer Association
41 Wu SY, Loke HN, Rehemtulla A. Ultraviolet radiation-induced apoptosis ismediated by daxx. Neoplasia 2002; 4: 486–92.
42 Sanna MG, Correia J, Ducrey O et al. IAP suppression of apoptosis involvesdistinct mechanisms: the TAK1/JNK1 signaling cascade and caspaseinhibition. Mol Cell Biol 2002; 22: 1754–66.
43 Sauter BV, Martinet O, Zhang WJ, Mandeli J, Woo SLC. Adenovirus-mediated gene transfer of endostatin in vivo results in high level of transgeneexpression and inhibition of tumor growth and metastases. Proc Natl AcadSci USA 2000; 97: 4802–7.
44 Hoffmann J, Schirner M, Menrad A, Schneider MR. A highly sensitivemodel for quantification of in vivo tumor angiogenesis induced by alginate-encapsulated tumor cells. Cancer Res 1997; 57: 3847–51.
45 Wei YQ, Huang MJ, Yang L et al. Immunogene therapy of tumors withvaccine based on Xenopus homologous vascular endothelial growth factor asa model antigen. Proc Natl Acad Sci USA 2001; 98: 11 545–50.
46 O’reilly MS, Boehm T, Shing Y et al. Endostatin: an endogenous inhibitorof angiogenesis and tumor growth. Cell 1997; 88: 277–85.
47 Wild R, Ramakrishnan S, Sedgewick J, Griffioen AW. Quantitativeassessment of angiogenesis and tumor vessel architecture by computer-assisted digital image analysis: effects of VEGF-toxin conjugate on tumormicrovessel density. Microvasc Res 2000; 59: 368–76.
48 Mukherjee P, El-abbadi MM, Kasperzyk JL, Ranes MK, Seyfried TN.Dietary restriction reduces angiogenesis and growth in an orthotopic mousebrain tumor model. Br J Cancer 2002; 86: 1615–21.
49 Wei YQ, Wang QR, Zhao X et al. Immunotherapy of tumors withxenogeneic endothelial cells as a vaccine. Nat Med 2000; 6: 1160–6.
50 Kaplan EL, Meier P. Nonparametric estimation from incompleteobservations. J Am Stat Assoc 1958; 53: 457–81.
51 Peto R, Pelo J. Asymptotically efficient rank invariant test procedures. J RoyStat Soc Series a Gener 1972; 135: 185–206.
52 Marx J. Angiogenesis: a boost for tumor starvation. Science (Wash DC)2003; 301: 452–4.
53 Reimer CL, Agata N, Tammam JG et al. Antineoplastic effects ofchemotherapeutic agents are potentiated by NM-3, an inhibitor ofangiogenesis. Cancer Res 2002; 62: 789–95.
54 Schiller JH, Bittner G. Potentiation of platinum antitumor effects in humanlung tumor xenografts by the angiogenesis inhibitor squalamine: effects ontumor neovascularization. Clin Cancer Res 1999; 5: 4287–94.
55 Browder T, Butterfield CE, Kraling BM et al. Antiangiogenic scheduling ofchemotherapy improves efficacy against experimental drug-resistant cancer.Cancer Res 2000; 60: 1878–86.
56 Morioka H, Weissbach L, Vogel T et al. Antiangiogenesis treatmentcombined with chemotherapy produces chondrosarcoma necrosis. ClinCancer Res 2003; 9: 1211–17.
57 Eastman A, Rigas JR. Modulation of apoptosis signaling pathways and cellcycle regulation. Semin Oncol 1999; 26 (Suppl 16): 7–16.
58 Wolf I, O’Kelly J, Wakimoto N et al. Honokiol, a natural biphenyl, inhibitsin vitro and in vivo growth of breast cancer through induction of apoptosisand cell cycle arrest. Int J Oncol 2007; 30: 1529–37.
59 Park EJ, Min HY, Chung HJ et al. Down-regulation of c-Src/EGFR-mediated signaling activation is involved in the honokiol-induced cell cyclearrest and apoptosis in MDA-MB-231 human breast cancer cells. CancerLett 2009; 277: 133–40.
60 Battle TE, Arbiser J, Frank DA. The natural product honokiol inducescaspase-dependent apoptosis in B-cell chronic lymphocytic leukemia (B-CLL) cells. Blood 2005; 106: 690–7.
61 Yancopoulos GD, Davis S, Gale NW et al. Vascular-specific growth factorsand blood vessel formation. Nature (Lond.) 2000; 407: 242–8.
62 Boehm T, Folkman J, Browder T, O’Reilly MS. Antiangiogenic therapy ofexperimental cancer does not induce acquired drug resistance. Nature(Lond.) 1997; 390: 404–7.
63 Verfailliea T, Coxa E, Goddeeris BM. Immunostimulatory capacity of DNAvaccine vectors in porcine PBMC: a specific role for CpG-motifs? VetImmunol Immunopathol 2005; 103: 141–51.
64 Krieg AM. CpG motifs in bacterial DNA and their immune effects. AnnuRev Immunol 2002; 20: 709–60.
65 Ballas ZK, Krieg AM, Warren T et al. Divergent therapeutic andimmunologic effects of oligodeoxynucleotides with distinct CpG motifs. JImmunol 2001; 167: 4878–86.
Copyright © 2022 FDOKUMEN




















