
Portable Reading Assistant Headset for the Visually Impaired ...

Neurobiology of Aging xxx (2006) xxx–xxx
Impaired spatial memory in APP-overexpressing mice on ahomocysteinemia-inducing diet
Alexandra Bernardo a,b, Meghan McCord a, Aron M. Troen d,John D. Allison b,c, Michael P. McDonald a,b,c,∗
a Department of Pharmacology, Vanderbilt University, Nashville, TN 37232-0325, United Statesb Program in Neuroscience, Vanderbilt University, Nashville, TN 37232-0325, United States
c John F. Kennedy Center for Research in Human Development, Vanderbilt University, Nashville, TN 37232-0325, United Statesd USDA Human Nutrition Research Center on Aging, Tufts University, Boston, MA 02111, United States
Received 6 April 2006; received in revised form 19 May 2006; accepted 30 May 2006
Abstract
irfmntsmi©
KF
1
rodgAka
ST
0d
Consumption of a diet that significantly elevates homocysteine (homocysteinemia) induces cell death in the CA3 hippocampal subfieldn amyloid precursor protein (APP) over-expressing transgenic mice but not in wild-type controls. We assessed behavioral and other neu-opathological effects of a homocysteinemia-inducing diet in aged APP-overexpressing mice. Starting at 16–18 months of age, mice wereed either a treatment diet lacking folate, choline, and methionine, and supplemented with homocysteine, or a control diet containing nor-al amounts of folate, choline and methionine but no homocysteine. After 5 months on the experimental diets, performance on a delayed
on-matching-to-position working-memory task was unimpaired. In contrast, spatial reference memory in the water maze was impaired inransgenic mice on the treatment diet. Transgenic mice had higher homocysteine levels than wild-type mice even when fed the control diet,uggesting an effect of genotype on homocysteine metabolism. Methyl-donor deficiency did not alter amyloid deposition in the transgenicice. These results suggest that disrupted homocysteine metabolism may induce A�-associated memory impairments and neurodegeneration
n APP overexpressing mice.2006 Elsevier Inc. All rights reserved.
eywords: Alzheimer; Amyloid; Behavior; Learning; Water maze; Memory; Delayed non-matching to position; Delayed conditional discrimination; Transgenic;olate; Folic acid; Homocysteine
. Introduction
One of the primary features of Alzheimer’s disease is neu-onal cell loss in the hippocampus and cerebral cortex, areasf the brain involved in memory and cognition. As neuronsie, short-term memory fails, followed by a decline in lan-uage, reasoning, and the ability to perform everyday tasks.lthough the cause of cell death in Alzheimer’s disease is notnown, considerable evidence implicates �-amyloid (A�),39–43 amino-acid peptide cleaved from the amyloid pre-
∗ Corresponding author at: Vanderbilt University, 851 Light Hall, Mailtop #0325, Nashville, TN 37232-0325, United States.el.: +1 615 936 1082; fax: +1 615 936 1137.
E-mail address: [email protected] (M.P. McDonald).
cursor protein (APP). A� has been shown to induce DNAdamage and neuronal death in vitro, and memory deficits invivo [22,24,38,47]. When APP is overexpressed or abnor-mally cleaved, A� forms toxic oligomers that aggregate intoamyloid plaques and are associated with age-related mem-ory impairment [7,47]. This process has been modeled inseveral APP-overexpressing transgenic mouse lines bearingmutations linked to familial Alzheimer’s disease in humans[9,14,30].
Although such genetic factors are sufficient to causeAlzheimer’s disease, most cases are sporadic and otherpotentially-modifiable risk factors may be required for patho-genesis. One such factor is homocysteinemia—elevatedplasma levels of the non-protein forming, sulfur aminoacid homocysteine. Homocysteinemia is associated with
197-4580/$ – see front matter © 2006 Elsevier Inc. All rights reserved.oi:10.1016/j.neurobiolaging.2006.05.035
NBA-6564; No. of Pages 11

2 A. Bernardo et al. / Neurobiology of Aging xxx (2006) xxx–xxx
increased risk of Alzheimer’s disease and stroke [39], andmoderate elevations of plasma homocysteine in cognitivelyintact individuals are associated with increased risk of sub-sequent incident dementia [34].
The relationship of homocysteinemia to Alzheimer’s dis-ease is uncertain. At high concentrations, homocysteine isneuro- and vasotoxic. In addition, conditions that lead tohomocysteinemia have been shown to increase vulnerabilityto primary brain insults in animal models [8,40] and to inhibitthe proliferation of hippocampal neuroprogenitor cells [19].The effect of homocysteinemia on cell death has been stud-ied in 10-month-old APP-overexpressing mice bearing theSwedish double mutation (K595N, M596L). Three monthsof a methyl-donor-free diet supplemented with homocys-teine induced a reduction in the number of CA3 pyramidalcells in the APP transgenic mice but not in wild-type con-trols [18]. The CA1 subfield and soluble amyloid levels wereunaffected by the homocysteinemia-inducing diet. It is notknown whether the reduction in CA3 cell numbers in thehomocysteinemic transgenic mice was due to increased neu-ronal death or to an inhibitory effect on neural progenitors, orboth. Alzheimer’s patients have a similar loss of cells in CA3,and lesions of this hippocampal subfield induce memorydeficits in experimental animals [3,11,32,35,37]. The effectof the homocysteinemia-inducing diet on memory and neu-ropathology in APP-overexpressing mice is not known. Thepsm
2
2
bgtgtacgwrtiak
2
#d
control or treatment diets. The control diet contained 2 mgfolic acid, 14.48 g choline, and 1.70 g l-methionine per kgdiet, and no homocysteine (Dyets Inc., diet #518754). Thetreatment diet completely lacked the methyl donors folic acid,choline, and l-methionine (Dyets Inc., diet #518806) butwas supplemented with 4.5 g/kg d,l-homocysteine (Sigma#H4628). The diets did not contain sulfa drugs, thus providingconditions that permit limited folate intake via coprophagyand allowing the mice to tolerate a long-term dietary defi-ciency. Mice were maintained on their assigned experimentaldiets for 6 months. The working memory task was conductedduring the first 5 months of the diet, followed by 2 weeks ofwater–maze testing. The mice were then left undisturbed onthe experimental diets for 2 weeks before being sacrificed forhistology.
Mice had free access to food and water throughout thestudy, except when performing the food-motivated short-termworking-memory task. During the working-memory task,mice were put on a food-restriction regimen of 8 h free accessper day, decreasing over 2 weeks to 4 h per day. Under thisregimen mice typically fall to ∼90% of their free feedingweights within the first week. Following the initial weight lossthey gain weight slowly, reaching their free-feeding weights∼1 month after initiation of the restriction regimen. Behav-ioral testing was conducted in two cohorts of mice. The firstcohort was trained on the working-memory task followedbwow
2
dmpamtDt1pttom
aTacfioa
resent experiments examined short-term working memory,patial memory, and neuropathology in Tg2576 transgenicice exposed to chronic homocysteinemia.
. Methods
.1. Study population
Female Tg2576 APP-overexpressing transgenic miceearing the Swedish double mutation (K670N, M671L) wereenerated on a B6SJLF1 hybrid background [4,14]. Wild-ype X APP transgenic matings were used to produce hemizy-ous transgenic mice (n = 14) and wild-type littermate con-rols (n = 19) for all experiments. One transgenic mouse diedfter hidden-platform training in the water maze, but beforeued-platform training. Because the retinal degeneration (rd)ene is known to segregate with the B6SJLF1 strain, all miceere screened for the presence of rd using polymerase chain
eaction (PCR), and rd-positive mice were eliminated fromhe study. Mice were housed in an AALAC-approved vivar-um, and temperature, humidity, and lighting levels were inccordance with AALAC guidelines. The colony room wasept on a 14:10 light/dark cycle, with lights on at 6 am.
.2. Experimental diets
Mice were maintained on standard lab chow (Purina5001) until 16–18 months of age, after which the mice wereivided into two groups that were fed amino-acid defined,
y water–maze testing. The second cohort was naı̈ve whenater–maze testing commenced. There were no genotype-r diet-dependent differences in body weight during the lasteek of food restriction (F’s < 0.64; p’s > .435).
.3. Delayed non-matching to position (DNMTP)
Mice were trained on a discrete-trial delayed conditionaliscrimination, a short-term working memory task com-only used with rodents and primates, including Alzheimer’s
atients [2,23,33]. We used a version of the task knowns delayed non-matching to position (DNMTP), in whichice were required to choose a spatial location different than
he one presented at the beginning of the trial [21,25,26].NMTP testing began at the age of 12–14 months, and con-
inued until water maze training began at 21–23 months. At6–18 months of age, all mice had reached criterion-levelerformance and were started on either the treatment or con-rol diet, matched for performance on the DNMTP task. Thushe mice were fed standard lab chow for the first 4 monthsf DNMTP testing, and the experimental diets for the last 5onths.The DNMTP task was conducted in commercially-
vailable operant chambers (MED Associates, Georgia VT).he operant chambers were housed within light-and sound-ttenuating cubicles, each containing a fan that providedonstant background noise (∼65 dB). Each chamber was out-tted with three nose-poke response holes, one on each sidef the front wall equidistant from the food-delivery well, andthird in the center of the rear wall. A houselight located

A. Bernardo et al. / Neurobiology of Aging xxx (2006) xxx–xxx 3
∼12 cm above the response hole on the rear wall provideda source of low-level illumination. A cue lamp inside eachresponse hole was illuminable to signal the phase of the trial.Each reinforcer consisted of 4-s access to 0.1 ml 10% sucrosesolution in distilled water, delivered to the inside of the cham-ber by an automated dipper. Mice were trained initially to emita nose-poke response in the right or left hole, and graduallytrained to perform the DNMTP contingencies. Each DNMTPtrial started with the “sample” phase, which was initiated withillumination of the sample cue stimulus inside either the leftor right nose-poke hole. A response in the illuminated holeextinguished the cue in the left or right response hole andilluminated the cue in the rear response hole, to begin the“delay” phase. A response in the unlit hole had no scheduledconsequences. The first rear response after termination of theexperimenter-imposed delay extinguished the rear responsecue and illuminated both the left and right cue lamps to signalthe “choice” phase. In order to earn a reinforcer, the mousewas required to respond in the nose-poke hole that was notthe sample stimulus, i.e., a left choice was correct with aright sample and a right choice was correct with a left sam-ple. A correct choice resulted in immediate delivery of thereinforcer; an incorrect choice resulted in immediate dark-ness for 4 s, followed by the inter-trial interval (ITI). Afterlearning this with a nominal 1-s delay to a criterion of 70%correct in three out of four consecutive sessions, longer delays(lewa6dfcm
ssmvptwtlolhaotoprF
the proportion of total errors that occurred after errors (asopposed to those that occurred after correct responses). Per-severative behavior may be indicative of a non-mnemonicimpairment, and with the correction procedure in oper-ation can result in choice accuracies significantly belowchance.
2.4. Morris water maze
At the end of DNMTP testing, mice were tested in awater maze procedure as previously described [20]. Micewere 21–23 months of age and had been fed the experimen-tal diets for 5 months when water maze testing was initiated.The experimental diets were continued for the duration ofwater–maze testing. Mice were given a block of six trials perday in a 92-cm diam. pool with an acrylic platform (10 cm2)submerged 1 cm below the surface of the water. Six trials perday were used rather than the usual four, to ensure that theAPP transgenics on the control diet were able to learn the task.Under the spaced-trial procedure, all the mice received theirfirst trial before conducting the second and subsequent trials.Sessions were captured by an overhead camera and analyzedin real time using an NIH Image macro on a Macintosh com-puter [29]. Latency to reach the platform was the variable ofinterest during acquisition (learning). To assess memory forthe platform location, a probe trial was conducted immedi-awprtbpirttaw
wwcttiodo
wflfpfw
5 and 10 s) were imposed. During all delays the rear cueamp remained illuminated to signal that the delay had not yetlapsed. Each discrete trial was separated by a 20-s ITI, duringhich the entire chamber was dark (i.e., the house lights and
ll three cue lights were off). Each daily session consisted of0 trials or 60 min, whichever came first. A correction proce-ure was implemented in which each incorrect response wasollowed by an identical trial type (i.e., either left or rightorrect), in order to prevent perseverative responding on oneanipulandum.In addition to the primary measure of choice accuracy,
everal secondary measures were assessed in the DNMTPessions in order to control for differential changes in non-nemonic sensorimotor processes [21,25,26]. A measure of
isual discrimination was derived from the mouse’s sam-le response. As described above, at the beginning of eachrial one of two nose-poke holes was randomly illuminatedhile the other remained unlit. The first response in either
he lit or unlit hole was tracked, and discrimination calcu-ated as the ratio of lit to total choices. Several measuresf slow behavior were derived from the session data. Theatency to initiate the trial by responding in the illuminatedole (the sample latency) and the latency to make a choicefter the delay (choice latency) were tracked as measuresf slow responding, which may indicate decreased motiva-ion or a sensorimotor impairment. An additional measuref slowness was the rate of responding in the rear nose-oke hole during the delay. A single rear-hole responseate across all retention intervals (delays) was calculated.inally, perseverative behavior was assessed, calculated as
tely following the final acquisition session. The probe trialas a single 60-s swim with the platform removed from theool. Platform crossings and the time spent in the target quad-ant were the two primary dependent measures derived fromhe probe trial. Platform crossings were defined as the num-er of times the mouse crossed the exact position where thelatform had been located, compared to equivalent positionsn the other three quadrants. The time in the target quad-ant is compared to the amount of time spent in the otherhree quadrants. A mouse that remembers the platform loca-ion will spend a greater amount of time in the target quadrantnd will make more platform crossings, compared to a mouseith bad memory [46].Non-mnemonic control measures were also assessed in the
ater maze, to determine whether differences in performanceere attributable to cognitive or non-cognitive factors. The
ontrol measures included speed of swimming (cm/s) andhigmotaxis. Thigmotaxis is the tendency to swim close tohe wall of the pool. This is a particular problem for micen the water maze, and can account for much of the variancebserved in water–maze acquisition [46]. Thigmotaxis wasefined as the percentage of time the mouse spent within 8 cmf the wall of the pool.
Following the hidden platform version of the task, miceere trained on a visible-platform version. In this task, aag was attached to the platform so that it was clearly visiblerom any point in the pool. The platform location and startingosition were changed on every trial, and mice were givenour trials per day for 5 days. All other aspects of the taskere identical to the hidden platform version.

4 A. Bernardo et al. / Neurobiology of Aging xxx (2006) xxx–xxx
2.5. Thioflavin S staining
After a brief isoflurane anesthesia, mice of the secondcohort were quickly decapitated, and brains and serum takenfor postmortem analyses. The brain was cut along the midline,and a section of the cortex and hippocampus removed. Afterparaffin embedding, 12-�m sections were cut through the tis-sue. For Thioflavin S staining the procedures of Wengenacket al. [45] were followed. Sections were first stained withhematoxylin for 5 min to block nuclear auto-fluorescence,followed by Thioflavin S for 5 min. Sections were then rinsedonce in distilled water and once in 70% ethanol before coverslips were applied. Digital images of the dorsal hippocampusand overlying cortex were taken using a fluorescent imagingmicroscope with an AxioCam high-resolution camera (CarlZeiss Microlmaging Inc., Thornwood, NY, USA) at a mag-nification of 40×. The amount of hippocampal and corticalareas occupied by plaque were determined from the imagesusing ImageJ public-domain software (National Institutes ofHealth, Bethesda, MD), by an experimenter blind to geno-type and diet condition. The areas of the dorsal hippocampusand overlying cortex were first calculated separately for threesections from each brain, corresponding to −1.60, −1.90,and −2.10 mm posterior to bregma [12]. The images wereinverted and the threshold adjusted such that only aggregatedamyloid was visible in the image. The same threshold valuewldc
2
acma−ptS
2
amasDstgwr
centage data (water–maze time-in-quadrant and search error)analyses were performed on log transformations. Follow-up analyses for all ANOVAs were conducted using factorialANOVAs or the Bonferroni–Dunn statistic.
3. Results
3.1. Short-term working memory is unaffected bygenotype or diet
All mice learned to perform the DNMTP task to cri-terion, and there was no effect of genotype on the rateof learning (data not shown). After 4 months of DNMTPtraining, mice were matched for performance and placedon the experimental diets. After 5 months on the diets,there was no effect of genotype [F(1,15) = 0.21, p = .651]or diet [F(1,15) = 1.40, p = .255] on the number of trialscompleted per session, and no Genotype X Diet interac-tion [F(1,15) = 1.02, p = .329]. This suggests that the dietsdid not differentially affect the ability or motivation to workfor the sucrose reinforcer. There was no significant effectof genotype [F(1,15) = 1.43, p = .250] or diet [F(1,15) = 0.44,p = .517] on choice accuracy in the DNMTP task (Fig. 1). Inaddition, all interactions among genotype, diet, and retentioninterval were non-significant (F’s < 0.83; p’s > .44). With theenaltihtBttstrc
3t
ce[gdtcpaw
as used for each image. The percent of plaque was calcu-ated as the number of pixels visible as aggregated amyloidivided by the total number of pixels in the hippocampal orortical area examined.
.6. Blood biochemistry
After water–maze testing, unfasted mice were sacrificeds described above, and blood was collected for biochemi-al analysis. Samples were placed on wet ice for approxi-ately 45 min before being spun in a refrigerated centrifuge
t 2500 rpm for 15 min. Plasma was extracted and frozen at80 ◦C until use. Plasma folate was measured by a 96-well
late microbial (Lactobacillus casei) assay [13], and plasmaotal homocysteine (tHcy) by HPLC according to Araki andako [1].
.7. Statistical analysis
Delayed conditional discrimination and water–mazecquisition data were analyzed using three-way repeated-easures analysis of variance (RMANOVA), with genotype
nd diet as between-subject factors and retention interval oression as repeated measures, respectively. Data used for theNMTP analyses were the averages of the last five daily ses-
ions for each mouse. Secondary measures in the DNMTPask were analyzed using two-way factorial ANOVAs, withenotype and diet as factors. Water–maze probe-trial dataere analyzed using RMANOVA within each group sepa-
ately, with quadrant as the repeated measure. Probe-trial per-
xception of sample latency, all secondary measures wereormal for all groups. Genotype [F(1,15) = 5.52, p = .0330]nd diet [F(1,15) = 8.52, p = .0106] differences in sampleatency were attributable to a single wild-type mouse onhe control diet that waited ∼4 min between trials, result-ng in group values (144.8 ± 61.1) that were significantlyigher than those of wild-type (35.6 ± 12.3; p = .0048) andransgenic (19.1 ± 5.9; p = .0042) mice on the treatment diet.ecause the Bonferroni–Dunn statistic is corrected for mul-
iple comparisons, the difference between wild type andransgenic mice on the control diet (49.2 ± 19.1) was notignificant (p = .0215; corrected α = .0083). Additional con-rol measures, such as visual discrimination, choice latency,esponse rate, and preservative responding, were not signifi-antly affected by genotype or diet (F’s < 1.96; p’s > .182).
.2. Water–maze performance is impaired in APPransgenics on the treatment diet
Following the DNMTP task, water–maze testing wasonducted in two cohorts of mice. There was no differ-nce in water–maze acquisition between the two cohortsF(1,26) = 1.60, p = .217], and no interaction of cohort withenotype, diet, or training session (F’s < 2.06; p’s > .16). Thusata were pooled for subsequent analyses. During acquisi-ion, Tg2576 mice had significantly longer escape latenciesompared to wild-type littermate controls [F(1,30) = 8.40,= .0069]. The main effect of diet [F(1,30) = 4.12, p = .051]nd Genotype X Diet interaction [F(1,30) = 1.60, p = .216]ere not significant. There were no interactions between

A. Bernardo et al. / Neurobiology of Aging xxx (2006) xxx–xxx 5
Fig. 1. The treatment diet did not affect performance of a delayed conditional discrimination task. Mice were trained daily to perform a delayed non-matching-toposition (DNMTP) task, a measure of short-term working memory. After reaching a predetermined criterion mice were assigned to treatment or control diet,stratified across DNMTP performance. Five months on the experimental diet did not significantly affect working memory in mice of either genotype. Data areexpressed as mean ± S.E.M. (n = 4–9 per group).
session and either of the independent measures (F’s < 1.08,p’s > .377). Follow-up comparisons showed that the Tg2576mice on the treatment diet had significantly impaired escapelearning, relative to the wild-type mice on both the control(p = .0028) and treatment (p = .0055) diets (Fig. 2A). The dif-ference between transgenic mice on the treatment and controldiets was not statistically significant (p = .063), and trans-genic mice on the control diet were not significantly differentfrom either of the wild-type groups (p’s > .30).
Control measures were assessed in the water maze toensure that differences observed were attributable to differ-ences in cognitive abilities and not to non-mnemonic perfor-mance variables. There was no effect of genotype or diet, andno Genotype X Diet interaction, on swim speed during acqui-sition (F’s < 0.96, p’s > .336). There were also no significanteffects of genotype or interactions of genotype with diet ortraining session, on thigmotaxis during water–maze acquisi-tion (F’s < 1.54, p’s > .116). However, there was a significantmain effect of diet on thigmotaxis [Fig. 2B; F(1,30) = 5.54,p = .0253]. Specifically, mice on the treatment diet weremore thigmotactic than mice on the control diet. However,on post-hoc analysis none of the four groups was signifi-cantly different than any other group (p’s > .024; correctedα = .0083). The increased thigmotaxis in mice on the treat-ment diet was restricted to the beginning of training. Using a“hockey-stick” approach to assess initial versus asymptoticcot[ogs
One hour after the last training session mice were given a60-s probe trial with the platform removed from the pool, toassess memory for the platform location. Memory is inferredfrom the amount of time the mouse spends in each quadrantof the pool, as well as the more precise measurement of howoften the mouse swims over the former platform location.There were no effects of genotype or diet on the distribu-tion of the amount of time spent in each quadrant of the poolduring the probe trial (Fig. 3A; F’s < 0.66, p’s > .583). Micespent more time in the former target quadrant than in theother three quadrants [F(3,90) = 21.23, p < .0001]. However,there was a significant genotype effect in the distribution ofplatform crossings across quadrants [Fig. 3B; F(3,90) = 3.25,p = .0254]. When examined separately, wild-type mice onboth diets demonstrated good selective search for the for-mer platform location (F’s > 8.1, p’s < .0004). Although theTg2576 mice on the control diet did not perform as well aswild-type mice, they were also significantly more likely toswim over the target location than equivalent positions in theother three quadrants [F(3,15) = 3.81, p = .0328]. In contrast,Tg2576 mice on the treatment diet had poor memory for theplatform location, as indicated by indiscriminate swimmingduring the probe trial [Fig. 3B; F(3,21) = .79, p = .511]. Takentogether, the time-in-quadrant and platform-crossings datasuggest that Tg2576 mice on the treatment diet had a generalidea about the location of the target, but their memories wereneawtac
omponents of the acquisition curve demonstrates that micen the treatment diet were significantly more thigmotactichan mice on the control diet during the first six sessions onlyF(1,30) = 6.30, p = .0174]. There was no effect of the dietn thigmotaxis during the last six sessions, when mice of allroups had reached an asymptote of ∼10% thigmotaxis peression [F(1,30) = 2.04, p = .163].
ot as precise as those of mice in the other three groups. Toxamine this, the percentage of time spent within 10, 10–20,nd 20–30 cm (annulus size) of the former platform locationas calculated as a measure of precision during the probe
rial. There was a significant interaction between genotypend annulus size [Fig. 3C; F(2,60) = 4.94, p = .0103], indi-ating that Tg2576 mice had less accurate memory for the
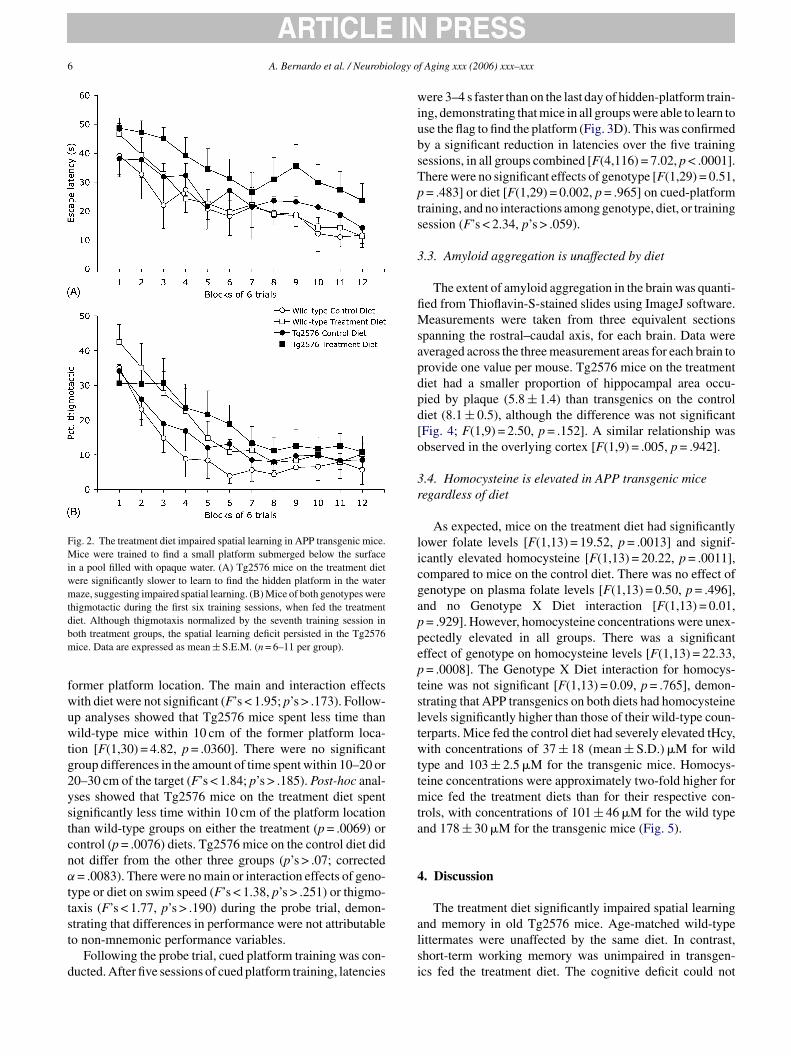
6 A. Bernardo et al. / Neurobiology of Aging xxx (2006) xxx–xxx
Fig. 2. The treatment diet impaired spatial learning in APP transgenic mice.Mice were trained to find a small platform submerged below the surfacein a pool filled with opaque water. (A) Tg2576 mice on the treatment dietwere significantly slower to learn to find the hidden platform in the watermaze, suggesting impaired spatial learning. (B) Mice of both genotypes werethigmotactic during the first six training sessions, when fed the treatmentdiet. Although thigmotaxis normalized by the seventh training session inboth treatment groups, the spatial learning deficit persisted in the Tg2576mice. Data are expressed as mean ± S.E.M. (n = 6–11 per group).
former platform location. The main and interaction effectswith diet were not significant (F’s < 1.95; p’s > .173). Follow-up analyses showed that Tg2576 mice spent less time thanwild-type mice within 10 cm of the former platform loca-tion [F(1,30) = 4.82, p = .0360]. There were no significantgroup differences in the amount of time spent within 10–20 or20–30 cm of the target (F’s < 1.84; p’s > .185). Post-hoc anal-yses showed that Tg2576 mice on the treatment diet spentsignificantly less time within 10 cm of the platform locationthan wild-type groups on either the treatment (p = .0069) orcontrol (p = .0076) diets. Tg2576 mice on the control diet didnot differ from the other three groups (p’s > .07; correctedα = .0083). There were no main or interaction effects of geno-type or diet on swim speed (F’s < 1.38, p’s > .251) or thigmo-taxis (F’s < 1.77, p’s > .190) during the probe trial, demon-strating that differences in performance were not attributableto non-mnemonic performance variables.
Following the probe trial, cued platform training was con-ducted. After five sessions of cued platform training, latencies
were 3–4 s faster than on the last day of hidden-platform train-ing, demonstrating that mice in all groups were able to learn touse the flag to find the platform (Fig. 3D). This was confirmedby a significant reduction in latencies over the five trainingsessions, in all groups combined [F(4,116) = 7.02, p < .0001].There were no significant effects of genotype [F(1,29) = 0.51,p = .483] or diet [F(1,29) = 0.002, p = .965] on cued-platformtraining, and no interactions among genotype, diet, or trainingsession (F’s < 2.34, p’s > .059).
3.3. Amyloid aggregation is unaffected by diet
The extent of amyloid aggregation in the brain was quanti-fied from Thioflavin-S-stained slides using ImageJ software.Measurements were taken from three equivalent sectionsspanning the rostral–caudal axis, for each brain. Data wereaveraged across the three measurement areas for each brain toprovide one value per mouse. Tg2576 mice on the treatmentdiet had a smaller proportion of hippocampal area occu-pied by plaque (5.8 ± 1.4) than transgenics on the controldiet (8.1 ± 0.5), although the difference was not significant[Fig. 4; F(1,9) = 2.50, p = .152]. A similar relationship wasobserved in the overlying cortex [F(1,9) = .005, p = .942].
3.4. Homocysteine is elevated in APP transgenic miceregardless of diet
licgappeptsltwttmta
4
alsi
As expected, mice on the treatment diet had significantlyower folate levels [F(1,13) = 19.52, p = .0013] and signif-cantly elevated homocysteine [F(1,13) = 20.22, p = .0011],ompared to mice on the control diet. There was no effect ofenotype on plasma folate levels [F(1,13) = 0.50, p = .496],nd no Genotype X Diet interaction [F(1,13) = 0.01,= .929]. However, homocysteine concentrations were unex-ectedly elevated in all groups. There was a significantffect of genotype on homocysteine levels [F(1,13) = 22.33,= .0008]. The Genotype X Diet interaction for homocys-
eine was not significant [F(1,13) = 0.09, p = .765], demon-trating that APP transgenics on both diets had homocysteineevels significantly higher than those of their wild-type coun-erparts. Mice fed the control diet had severely elevated tHcy,ith concentrations of 37 ± 18 (mean ± S.D.) �M for wild
ype and 103 ± 2.5 �M for the transgenic mice. Homocys-eine concentrations were approximately two-fold higher for
ice fed the treatment diets than for their respective con-rols, with concentrations of 101 ± 46 �M for the wild typend 178 ± 30 �M for the transgenic mice (Fig. 5).
. Discussion
The treatment diet significantly impaired spatial learningnd memory in old Tg2576 mice. Age-matched wild-typeittermates were unaffected by the same diet. In contrast,hort-term working memory was unimpaired in transgen-cs fed the treatment diet. The cognitive deficit could not

A. Bernardo et al. / Neurobiology of Aging xxx (2006) xxx–xxx 7
Fig. 3. The treatment diet impaired spatial memory in APP transgenic mice. Following hidden-platform training on the water maze, a 60-s probe trial wasconducted, followed by visible-platform training. (A) Using the broad measure of quadrant time, mice in all groups showed good selective search for the formerplatform location. (B) The more precise measure of platform crossings revealed a significant spatial memory impairment in the Tg2576 mice on the treatmentdiet. (C) A specific measure of precision shows that Tg2576 mice on the treatment diet had significantly impaired accuracy when attempting to find the hiddenplatform. (D) Mice in all four groups quickly learned to find the visible platform, demonstrating that they had the motivation and sensorimotor capabilities toescape from the pool proficiently. Data are expressed as mean ± S.E.M. (n = 6–11 per group).
be attributed to greater amyloid aggregation, as the treat-ment diet did not significantly affect plaque load in Tg2576mice. Thus the combination of A� overexpression with long-term dietary metabolic stress that results in homocysteinemiaappears sufficient to impair spatial memory.
Although only transgenic mice fed the treatment diet hadimpaired memory, both diet and genotype had robust, inde-pendent effects on homocysteine levels. As expected, thetreatment diet increased plasma tHcy concentrations abovethose observed in the respective control-fed groups. Both
F n in Tgv n stainih picted.
ig. 4. The treatment diet did not significantly affect amyloid aggregatioisualization of aggregated amyloid. There was no significant difference iippocampus (A, B) or the neocortex (C, D). Representative sections are de
2576 mice. Brains were sectioned and stained with Thioflavin S for theng between Tg2576 mice on the treatment and control diets, in either theScale bar = 500 �m.

8 A. Bernardo et al. / Neurobiology of Aging xxx (2006) xxx–xxx
Fig. 5. APP transgenic mice on both diets have elevated homocysteine lev-els. Serum was collected postmortem, and folate and homocysteine weremeasured. (A) There was a significant reduction in serum folate in mice ofboth genotypes on the treatment diet. (B) Mice of both genotypes also hadsignificantly elevated homocysteine levels on the treatment diet. However,there was also a significant effect of genotype that was independent of diet.Tg2576 mice had significantly higher homocysteine levels than wild-typemice, regardless of the diet condition. Data are expressed as mean ± S.E.M.(n = 3–4 per group).
wild-type and transgenic mice responded to treatment withan approximately two-fold increase above control levels.Plasma tHcy was significantly higher in Tg2576 mice inde-pendent of diet. However, we found unexpected severe homo-cysteinemia in all groups, including in wild type mice fedthe control diets, indicating that some aspect of homocys-teine metabolism was disrupted in these mice. Althoughthe mice were not fasted before blood collection, plasmatHcy in folate-deficient rodents tends to remain stable inthe fed state (Troen, unpublished observations), so it isunlikely that the observed severe homocysteinemia in allgroups was a post-prandial effect. In humans, comparablefasting tHcy concentrations are found in individuals withcongenital defects of homocysteine metabolism or renaldisease. A comparably severe homocysteinemia can beeninduced in mice through dietary or genetic means; how-
ever, in these cases the severity of homocysteinemia reflectsthe severity of the primary metabolic disruption (Fig. 5)[5,41,43,44].
Spontaneous homocysteinemia in older mice of thisgenetic background has not been previously reported. In10-month-old APP transgenic and wild-type C57BL/6J con-trol mice, Kruman et al. [18] found tHcy concentrations of<5 �M for mice fed control diets and moderately-elevatedtHcy (∼27 �M) for mice fed treatment diets, with no diet-dependent differences in homocysteine levels between geno-types [18]. In humans, plasma homocysteine levels increasewith age, and are higher in males than in females [16]. Itis possible that mice of the advanced age and/or geneticbackground used in the present study can develop homo-cysteinemia spontaneously. Alternatively, it is possible thatintake of an artificial, amino-acid defined diet for 6 monthsproduced an unanticipated metabolic stress that was exacer-bated by A� overexpression.
Spatial memory impairments were only observed inTg2576 mice fed the treatment diet. These same mice also hadthe highest tHcy levels. It is possible, however unlikely, thatthat the severity of metabolic disruption and homocysteine-mia in this group exceeded a threshold required to producesuch behavioral deficits. If homocysteine were inherentlytoxic and homocysteinemia is independently damaging to theCNS, then the prolonged exposure experienced by the miceilptrartls
icWfdttgttmb[tTtDdi
n this study would have been expected to induce deficits in ateast the treatment-fed wild-type mice as well. Alternatively,erformance of all groups of mice may have been poorerhan if their homocysteine levels had been within the normalange. In either case, the behavioral impairments cannot bettributed to homocysteinemia per se, and this may equallyelate to the primary metabolic disruption. Nevertheless,he pathological and behavioral benefits of homocysteine-owering strategies in these mice may be of interest in futuretudies.
Transgenics on the treatment diet were significantlympaired in learning the escape location in the hippo-ampally-mediated hidden-target version of the water maze.ith the exception of thigmotaxis, there were no group dif-
erences on any of the non-mnemonic control measures. Thisemonstrates that the Tg2576 mice understood the rules ofhe task and had the motivation and sensorimotor abilitieso escape quickly from the pool. Interestingly, mice of bothenotypes fed the treatment diet were thigmotactic duringhe first half of training, compared to their counterparts onhe control diet (Fig. 2B). It is not known whether thig-
otaxis is a process independent of spatial learning, or aehavior suggesting an inability to learn spatial associations42]. Under most situations it cannot be discerned becausehe two are confounded. However, in our data wild-type andg2576 mice on the treatment diet were equally thigmotac-
ic but differed in their ability to find the hidden platform.espite being thigmotactic, wild-type mice on the treatmentiet performed as well as wild-types on the control diet dur-ng the first six acquisition sessions (Fig. 2A). In contrast,

A. Bernardo et al. / Neurobiology of Aging xxx (2006) xxx–xxx 9
learning in the Tg2576 mice on the treatment diet was sig-nificantly impaired over the same period. By the seventhsession and throughout the rest of training, the treatment-diet groups were no more thigmotactic than mice on thecontrol diet. Nevertheless, the learning deficit persisted inthe Tg2576 mice on the treatment diet. Thus, although it isnot clear why the treatment diet may have induced thigmo-taxis, we can be confident that it did not interfere with ourability to detect a spatial learning deficit in the transgenicmice.
In contrast to performance on the water maze, performanceon the DNMTP task was unimpaired by administration ofthe treatment diet. Although the DNMTP task can be con-sidered a spatial task, it involves discrimination of discretevisual stimuli at two stable locations within the operant cham-ber. In this sense the DNMTP task is more similar to thevisible-platform version of the water maze, i.e., it requiresdiscrimination of discrete, proximal stimuli as opposed to adistal configuration of spatial cues [31]. Consistent with this,Tg2576 mice on the treatment diet were unimpaired on thevisible-platform version of the water–maze task. The visible-platform task is considered a control for the sensorimotorand motivational aspects of water–maze learning. Althoughthere is a spatial component to solving the visible-platformtask, it does not involve learning about extra-maze room cuesand thus is not thought to require an intact hippocampus[ist
TttsiasgwhomtnAafetomrtia
homocysteine metabolism and/or subsequent homocysteine-mia interact with the Tg2576 genotype or with A� to pro-duce such regionally specific neuronal loss remain to beelucidated.
We have shown that Tg2576 mice fed a homocysteinemia-inducing diet exhibit specific deficits in spatial learning andmemory, but not in short term working memory. This isconsistent with reports of cell loss in the CA3 hippocam-pal subfield, which in turn may be due to cell death ordecreased neurogenesis associated with homocysteinemia[18,19]. The observation of elevated homocysteine in agedTg2576 mice independent of diet condition is consistentwith reports of low folate and increased homocysteine lev-els in Alzheimer’s patients [6,28], and positive correlationsof plasma A� and homocysteine in aged humans [15]. Theobserved deterioration of memory function by disruptedhomocysteine metabolism in an in vivo model of Alzheimer-type neuropathology is consistent with the association ofhomocysteinemia with dementia and Alzheimer’s disease asobserved in human populations [6,28,34]. Further researchis needed to determine how A�, homocysteine, and otherpathological processes interact to produce such cognitivedeficits.
Acknowledgements
Aao9w(
R
10,23,27]. Unimpaired DNMTP and visible-platform learn-ng, together with impaired hidden-platform learning, is con-istent with a homocysteine-induced lesion that is restrictedo the hippocampus.
Probe-trial performance was significantly impaired in theg2576 mice on the treatment diet, confirming the spa-
ial memory deficit observed during acquisition. Althoughhe transgenics on the treatment diet exhibited proficientearch strategies as measured by time in quadrant, they werempaired on the more precise measures of platform crossingsnd annulus time. This lack of precision is consistent with thepecific CA3 hippocampal damage exhibited by APP trans-enic mice after 3 months on the treatment diet [18]. Ratsith specific kainate-induced lesions of the CA3 subfieldad impaired water–maze acquisition compared to sham-perated controls [36]. Like our Tg2576 mice on the treat-ent diet, CA3-lesioned rats were unimpaired on the probe
rial with respect to time in quadrant; however, they were sig-ificantly impaired when platform crossings was examined.similar deficit in spatial learning and memory was reported
fter transection of recurrent CA3–CA3 fibers and Schaf-er collaterals projecting from CA3 to CA1 [35]. In a studyxamining working-and reference-memory performance onhe radial maze, CA3-lesioned rats had intact working mem-ry but were significantly impaired learning the reference-emory component of the task [17]. Taken together, these
esults suggest that the impaired spatial learning and probe-rial performance of the Tg2576 mice on the treatment diets consistent with the CA3 cell loss reported by Kruman etl. [18]. The in vivo mechanisms through which disrupted
This work was supported by the National Institute ofging (AG022439), the National Institute of Child Health
nd Development (HD015052), and the U.S. Departmentf Agriculture under cooperative agreement No. 58-1950--001. Some of the behavioral experiments were conductedithin the Murine Neurobehavioral Laboratory core facility
www.medschool.mc.vanderbilt.edu/mnl).
eferences
[1] Araki A, Sako Y. Determination of free and total homocysteine inhuman plasma by high-performance liquid chromatography with flu-orescence detection. J Chromatogr 1987;422:43–52.
[2] Blaizot X, Meguro K, Millien I, Baron JC, Chavoix C. Correlationsbetween visual recognition memory and neocortical and hippocampalglucose metabolism after bilateral rhinal cortex lesions in the baboon:implications for Alzheimer’s disease. J Neurosci 2002;22:9166–70.
[3] Bobinski M, Wegiel J, Tarnawski M, Bobinski M, Reisberg B,de Leon MJ, et al. Relationships between regional neuronal lossand neurofibrillary changes in the hippocampal formation and dura-tion and severity of Alzheimer disease. J Neuropathol Exp Neurol1997;56:414–20.
[4] Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Mar-shall VJ, Irizarry M, et al. Impaired synaptic plasticity and learningin aged amyloid precursor protein transgenic mice. Nat Neurosci1999;2:271–6.
[5] Chen Z, Karaplis AC, Ackerman SL, Pogribny IP, Melnyk S, Lussier-Cacan S, et al. Mice deficient in methylenetetrahydrofolate reductaseexhibit hyperhomocysteinemia and decreased methylation capacity,with neuropathology and aortic lipid deposition. Hum Mol Genet2001;10:433–43.

10 A. Bernardo et al. / Neurobiology of Aging xxx (2006) xxx–xxx
[6] Clarke R, Smith AD, Jobst KA, Refsum H, Sutton L, Ueland PM.Folate, vitamin B12, and serum total homocysteine levels in con-firmed Alzheimer disease. Arch Neurol 1998;55:1449–55.
[7] Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, KuskowskiMA, Selkoe DJ, et al. Natural oligomers of the amyloid-beta pro-tein specifically disrupt cognitive function. Nat Neurosci 2005;8:79–84.
[8] Duan W, Ladenheim B, Cutler RG, Kruman II, Cadet JL, Matt-son MP. Dietary folate deficiency and elevated homocysteine levelsendanger dopaminergic neurons in models of Parkinson’s disease. JNeurochem 2002;80:101–10.
[9] Games D, Adams D, Alessandrini R, Barbour R, Berthelette P,Blackwell C, et al. Alzheimer-type neuropathology in transgenicmice overexpressing V717F beta-amyloid precursor protein. Nature1995;373:523–7.
[10] Gerlai R. Behavioral tests of hippocampal function: simpleparadigms complex problems. Behav Brain Res 2001;125:269–77.
[11] Ho L, Purohit D, Haroutunian V, Luterman JD, Willis F, NaslundJ, et al. Neuronal cyclooxygenase 2 expression in the hippocampalformation as a function of the clinical progression of Alzheimerdisease. Arch Neurol 2001;58:487–92.
[12] Hof PR, Young WG, Bloom FE, Belichenko PV, Celio MR. Com-parative cytoarchitectonic atlas of the C57BL6 and 129Sv mousebrains. New York: Elsevier; 2000.
[13] Home DW, Patterson D. Lactobacillus casei microbiological assayof folic acid derivatives in 96-well microtiter plates. Clin Chem1988;34:2357–9.
[14] Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, YounkinS, et al. Correlative memory deficits, Abeta elevation, and amyloidplaques in transgenic mice. Science 1996;274:99–102.
[15] Irizarry MC, Gurol ME, Raju S, Diaz-Arrastia R, Locascio JJ,
[
[
[
[
[
[
[
[
[
[
[26] McDonald MP, Willard LB, Wenk GL, Crawley JN. Coadminis-tration of galanin antagonist M40 with a muscarinic M1 agonistimproves delayed nonmatching to position choice accuracy in ratswith cholinergic lesions. J Neurosci 1998;18:5078–85.
[27] McDonald RJ, White NM. Parallel information processing in thewater maze: evidence for independent memory systems involv-ing dorsal striatum and hippocampus. Behav Neural Biol 1994;61:260–70.
[28] Miller JW. Homocysteine and Alzheimer’s disease. Nutr Rev 1999;57:126–9.
[29] Miyakawa T, Yared E, Pak JH, Huang FL, Huang KP, CrawleyJN. Neurogranin null mutant mice display performance deficits onspatial learning tasks with anxiety related components. Hippocampus2001;11:763–75.
[30] Moechars D, Dewachter I, Lorent K, Reverse D, Baekelandt V, NaiduA, et al. Early phenotypic changes in transgenic mice that overex-press different mutants of amyloid precursor protein in brain. J BiolChem 1999;274:6483–92.
[31] Reiserer RS, Harrison FE, Syverud DC, McDonald MP. Impairedspatial learning in the APPSwe + PSEN1�E9 bigenic mouse modelof Alzheimer’s disease. Genes, Brain Behav 2006. doi:10.1111/J.1601-183X.2006.00221.x.
[32] Roozendaal B, Phillips RG, Power AE, Brooke SM, Sapolsky RM,McGaugh JL. Memory retrieval impairment induced by hippocampalCA3 lesions is blocked by adrenocortical suppression. Nat Neurosci2001;4:1169–71.
[33] Sahgal A, Galloway PH, McKeith IG, Lloyd S, Cook JH, Ferrier IN,et al. Matching-to-sample deficits in patients with senile dementias ofthe Alzheimer and Lewy body types. Arch Neurol 1992;49:1043–6.
[34] Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH,D’Agostino RB, et al. Plasma homocysteine as a risk factor
[
[
[
[
[
[
[
[
[
[
Tennis M, et al. Association of homocysteine with plasma amy-loid beta protein in aging and neurodegenerative disease. Neurology2005;65:1402–8.
16] Jacques PF, Bostom AG, Wilson PW, Rich S, Rosenberg IH, SelhubJ. Determinants of plasma total homocysteine concentration in theFramingham offspring cohort. Am J Clin Nutr 2001;73:613–21.
17] Jarrard LE. Selective hippocampal lesions and behavior: effects ofkainic acid lesions on performance of place and cue tasks. BehavNeurosci 1983;97:873–89.
18] Kruman II, Kumaravel TS, Lohani A, Pedersen WA, Cutler RG,Kruman Y, et al. Folic acid deficiency and homocysteine impairDNA repair in hippocampal neurons and sensitize them to amyloidtoxicity in experimental models of Alzheimer’s disease. J Neurosci2002;22:1752–62.
19] Kruman II, Mouton PR, Emokpae Jr R, Cutler RG, Mattson MP.Folate deficiency inhibits proliferation of adult hippocampal progen-itors. Neuroreport 2005;16:1055–9.
20] Lijam N, Paylor R, McDonald MP, Crawley JN, Deng CX, HerrupK, et al. Social interaction and sensorimotor gating abnormalities inmice lacking Dvl1. Cell 1997;90:895–905.
21] McDonald MP, Crawley JN. Galanin receptor antagonist M40 blocksgalanin-induced choice accuracy deficits on a delayed-nonmatching-to-position task. Behav Neurosci 1996;110:1025–32.
22] McDonald MP, Dahl EE, Overmier JB, Mantyh P, Cleary J. Effects ofan exogenous beta-amyloid peptide on retention for spatial learning.Behav Neural Biol 1994;62:60–7.
23] McDonald MP, Overmier JB. Present imperfect: a critical review ofanimal models of the mnemonic impairments in Alzheimer’s disease.Neurosci Biobehav Rev 1998;22:99–120.
24] McDonald MP, Overmier JB, Bandyopadhyay S, Babcock D, ClearyJ. Reversal of beta-amyloid-induced retention deficit after exposureto training and state cues. Neurobiol Learn Mem 1996;65:35–47.
25] McDonald MP, Wenk GL, Crawley JN. Analysis of galanin and thegalanin antagonist M40 on delayed non-matching-to-position per-formance in rats lesioned with the cholinergic immunotoxin 192IgG-saporin. Behav Neurosci 1997;111:552–63.
for dementia and Alzheimer’s disease. N Engl J Med 2002;346:476–83.
35] Steffenach HA, Sloviter RS, Moser El, Moser MB. Impaired reten-tion of spatial memory after transection of longitudinally orientedaxons of hippocampal CA3 pyramidal cells. Proc Natl Acad SciUSA 2002;99:3194–8.
36] Stubley-Weatherly L, Harding JW, Wright JW. Effects of discretekainic acid-induced hippocampal lesions on spatial and contextuallearning and memory in rats. Brain Res 1996;716:29–38.
37] Su JH, Deng G, Cotman CW. Transneuronal degeneration in thespread of Alzheimer’s disease pathology: immunohistochemical evi-dence for the transmission of tau hyperphosphorylation. NeurobiolDis 1997;4:365–75.
38] Sweeney WA, Luedtke J, McDonald MP, Overmier JB. Intrahip-pocampal injections of exogenous beta-amyloid induce postdelayerrors in an eight-arm radial maze. Neurobiol Learn Mem 1997;68:97–101.
39] Troen A, Rosenberg I. Homocysteine and cognitive function. SeminVase Med 2005;5:209–14.
40] Troen AM. The central nervous system in animal models ofhyperhomocysteinemia. Prog Neuropsychopharmacol Biol Psychia-try 2005;29:1140–51.
41] Troen AM, Lutgens E, Smith DE, Rosenberg IH, Selhub J. Theatherogenic effect of excess methionine intake. Proc Natl Acad SciUSA 2003;100:15089–94.
42] Vyssotski AL, DeN’Omo G, Poletaeva II, Vyssotsk DL, MinichielloL, Klein R, et al. Long-term monitoring of hippocampus-dependentbehavior in naturalistic settings: mutant mice lacking neurotrophinreceptor TrkB in the forebrain show spatial learning but impairedbehavioral flexibility. Hippocampus 2002;12:27–38.
43] Wang L, Chen X, Tang B, Hua X, Klein-Szanto A, Kruger WD.Expression of mutant human cystathionine beta-synthase rescuesneonatal lethality but not homocystinuria in a mouse model. HumMol Genet 2005;14:2201–8.
44] Watanabe M, Osada J, Aratani Y, Kluckman K, Reddick R, Mali-now MR, et al. Mice deficient in cystathionine beta-synthase: animal

A. Bernardo et al. / Neurobiology of Aging xxx (2006) xxx–xxx 11
models for mild and severe homocyst(e)inemia. Proc Natl Acad SciUSA 1995;92:1585–9.
[45] Wengenack TM, Whelan S, Curran GL, Duff KE, PodusloJF. Quantitative histological analysis of amyloid deposition inAlzheimer’s double transgenic mouse brain. Neuroscience 2000;101:939–44.
[46] Wolfer DP, Stagljar-Bozicevic M, Errington ML, Lipp HP. Spatialmemory and learning in transgenic mice: fact or artifact? News Phys-iol Sci 1998;13:118–23.
[47] Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxiceffects of amyloid beta protein: reversal by tachykinin neuropeptides.Science 1990;250:279–82.
Copyright © 2022 FDOKUMEN




















